 Zahra Choudhury
Zahra Choudhury Belinda Lennox
Belinda Lennox
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Psychiatry, 17 February 2021
Sec. Molecular Psychiatry
Volume 12 - 2021 | https://doi.org/10.3389/fpsyt.2021.585742
This article is part of the Research TopicThe Crossroads between Immunological Disorders and Neuropsychiatric Diseases. A Case for SchizophreniaView all 12 articles
Schizophrenia is a complex neurodevelopmental disorder affecting around 19. 8 million people worldwide. The etiology of the disorder is due to many interacting genetic and environmental factors, with no one element causing the full spectrum of disease symptoms. Amongst these factors, maternal immune activation (MIA) acting during specific gestational timings has been implicated in increasing schizophrenia risk in offspring. Epidemiological studies have provided the rationale for this link with prevalence of maternal infection correlating to increased risk, but these studies have been unable to prove causality due to lack of control of confounding factors like genetic susceptibility and inability to identify specific cellular and molecular mechanisms. Animal models have proved significantly more useful in establishing the extent to which MIA can predispose an individual to schizophrenia, displaying how maternal infection alone can directly result in behavioral abnormalities in rodent offspring. Alongside information from genome wide association studies (GWAS), animal models have been able to identify the role of complement proteins, particularly C4, and display how alterations in this system can cause development of schizophrenia-associated neuropathology and behavior. This article will review the current literature in order to assess whether schizophrenia can, therefore, be viewed as an immune priming disorder.
Schizophrenia is a long-term mental health disorder with an etiology characterized by a range of positive symptoms (e.g., delusions and hallucinations), negative symptoms (e.g., loss of motivation) and cognitive deficits (e.g., working memory deficits) during early adulthood (1). Heritability is estimated to be 80% (2, 3), but susceptibility of developing schizophrenia is determined by complex interactions between genes and the environment (4, 5). Risk of developing the clinical phenotype is generally modeled on the “two-hit hypothesis” where the “first hit” is an early priming event disrupting neurodevelopment and establishing increased vulnerability to a “second hit” which tends to be environmental insults occurring later in life (6, 7). Genetic abnormalities are widely accepted as the “first hit,” but MIA may also serve this function (6).
Ecological studies were first to show the possible link between MIA via infection during pregnancy and increased schizophrenia risk in offspring. Following the 1957 influenza type A2 epidemic in Finland, maternal viral infection resulted in increased schizophrenia rates per 1,000 live births recorded in Helsinki (8). Other similar studies have attempted to replicate these findings, with some showing positive associations whilst others finding no association (9). Due to widely contradictory results, MIA animal models have been necessary to establish a causal relationship between MIA and schizophrenia-related abnormalities (10–13). These models have been pivotal in identifying molecular mechanisms that affect normal neurodevelopment in early life (10), most significantly changes to the complement system (4, 14). These changes cause neuronal connectivity and synaptic pruning dysfunction which may contribute to pathological characteristics seen in post-mortem schizophrenic brains (e.g., decreased cortical gray matter thickness), leading to social deficits observed in schizophrenic patients (14–16).
MIA falls under the category of prenatal maternal stress which broadly encompasses maternal infection/inflammation, obstetric complications, nutritional deficiencies and psychosocial stress. In particular, maternal psychological stress is documented to be a well-characterized early priming event resulting in both physiological and behavioral deficits in offspring, such as cardiovascular abnormalities, enhanced anxiety-like behavior and increased risk of schizophrenia and other psychotic disorders (17–19). Increased glucocorticoid secretion due to stimulation of the hypothalamic-pituitary-adrenal (HPA) axis may play a part, resulting in schizophrenia-like phenotypes, as seen with pregnant rats exposed to variable stress paradigms (20). Although maternal psychological stressors can have a large impact on schizophrenia risk in offspring, this essay will be focused on reviewing the evidence linking MIA in particular to schizophrenia in regard to epidemiological studies, MIA animal models and the complement system in order to evaluate whether schizophrenia can be classed as an immune priming disorder.
Epidemiological studies have linked MIA to increased schizophrenia risk via infectious agents like influenza, herpes simplex virus 1 (HSV-1) and Toxoplasma gondii (T. gondii) (21–23). Ecological studies, primarily based on maternal influenza infection, identified this link, but there were many limitations associated with these. A notable limitation was diagnostic misclassification of influenza, with exposure in early studies being based upon maternal recall or whether the mother was in gestation during the epidemic, meaning around 70% of those in gestation during the 1957 influenza epidemic would have been misclassified as being infected whilst, in reality, being unexposed (8, 22, 24, 25).
A new approach was required to overcome this, leading to an increase in birth cohort studies using serological evidence, as summarized in Table 1. These studies used well-characterized birth cohorts and measured biomarkers in individual pregnancies using archived biological specimens obtained during pregnancy or early life of offspring (22). Results showed increased schizophrenia risk associated with maternal influenza infection in the first trimester (9), rather than the second trimester as found in ecological studies (8), and elevated levels of maternal antibodies against T. gondii (26, 27, 31, 32), as well as impaired cognitive functions displayed by HSV-1 seropositive offspring (23, 28, 29).
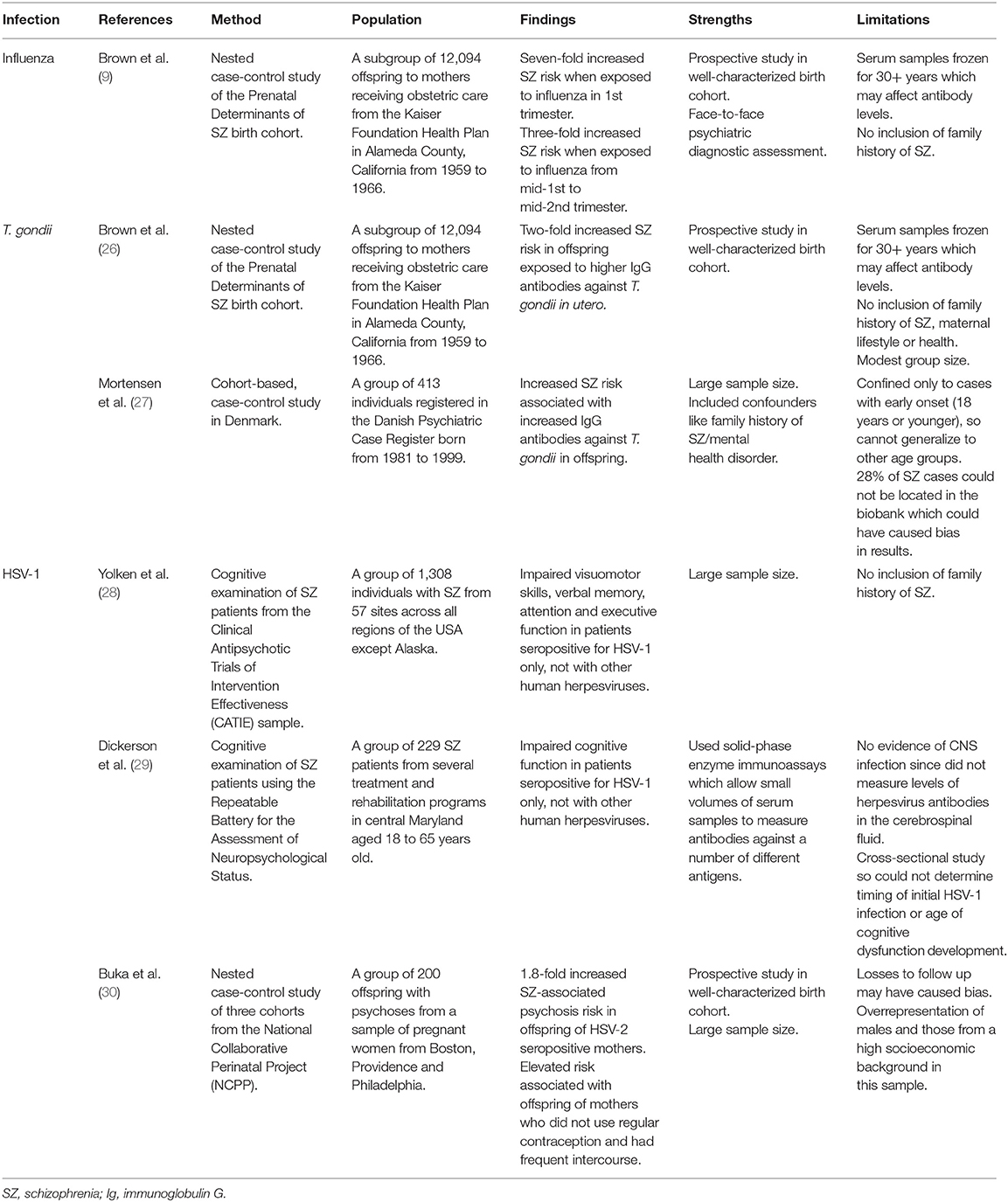
Table 1. Table reviewing serological evidence linking various infections to increased schizophrenia risk.
The mechanisms by which infection increases schizophrenia risk are only speculative and may either be infection-specific or work by a common causal mechanism (22). For example, the genome of T. gondii contains two genes encoding tyrosine hydroxylase which catalyzes L-DOPA production. L-DOPA is the precursor to dopamine, the dysregulation of which is well-established in schizophrenia pathology, thereby providing a potential infection-specific mechanism for increasing schizophrenia risk (33). However, there is currently limited evidence identifying T. gondii infection as a risk factor for schizophrenia, so larger birth cohort studies would be required to establish a more definitive association. Using human neurons derived from human induced pluripotent stem cells may also be useful in studying molecular mechanisms involved in T. gondii infection that lead to schizophrenia-associated pathogenesis. On the other hand, infections may act through common mechanisms by causing a pro-inflammatory state, increasing cytokine levels like IL-6 which has been shown to play a key role in behavioral abnormalities (22). A single injection of IL-6 during mid-pregnancy in rodents caused pre-pulse inhibition (PPI) and latent inhibition deficits in adult offspring, translating to sensorimotor gating deficits seen in schizophrenic patients, which can be prevented through administration of anti-IL-6 antibodies (34).
The prospective nature of serological studies has been powerful in establishing a link between MIA and increased schizophrenia risk through the use of qualitative measures of pathogenic markers in prenatal life. However, these studies are limited since they do not have the capacity to identify cellular and molecular mechanisms which affect neurodevelopment. Research in animal models provide a platform to overcome these limitations and potentially establish causality for these associations.
Initially, MIA was modeled using experimental mouse models of prenatal exposure to human influenza, where pregnant mice were infused with a mouse-adapted human influenza strain (35). Neuropathological signs were observed in the offspring, such as corticogenesis deficits, decreased hippocampal volume, decreased expression of γ-aminobutyric acid (GABA) markers, e.g., reelin, and behavioral abnormalities linked to schizophrenia (35). These results were extended to rhesus monkeys who showed decreased gray and white matter in cortical and parietal-cortical brain regions of neonates, verifying the relevance of rodent findings since corticogenesis in primates is more advanced and similar to humans (36). Viral MIA immune models were useful since they produced the full spectrum of immune responses caused by infections, but due to limitations associated with viral models such as the need for strict biosafety provisions and decreased control of immune response intensity and duration, animal MIA models moved onto using other immunogens such as polyinosinic:polycytidylic acid (polyI:C) to mimic viral infection, lipopolysaccharides to mimic bacterial infections and turpentine to mimic local inflammation (10). This article will particularly focus on polyI:C animal models due to the strength of their results, with Table 2 summarizing the main contributions these models have made to the study of schizophrenia pathophysiology.

Table 2. Main contributions made by polyI:C rodent models to the study of schizophrenia pathophysiology.
In viral infection, double stranded RNA (dsRNA) is an intermediate in single stranded RNA replication or during symmetrical transcription of DNA viruses (10). Recognition of dsRNA by toll-like receptor 3 (TLR3) causes increased pro-inflammatory cytokine expression (10, 13). PolyI:C acts as a synthetic analog of viral dsRNA and mimics the acute phase response to viral infection (10, 13). The most significant finding from polyI:C rodent models would be how gestational timing of MIA exposure can influence the phenotype displayed by offspring (9, 11, 12). Pregnant mice were injected with polyI:C at either gestational day (GD) 9 or GD17 (39, 40, 47), translating to mid-first trimester and early second trimester, respectively, in humans (48). GD9 offspring were found to have phenotypes consistent with positive symptoms such as PPI deficits, latent inhibition deficits, decreased dopamine 1 (D1) and D2 receptor density in the prefrontal cortex (PFC) and increased tyrosine hydroxylase levels. Conversely, GD17 offspring showed phenotypes consistent with negative and cognitive symptoms such as N-methyl-D-aspartic acid (NMDA) receptor alterations, impaired reversal learning, working memory deficits, increased hyperlocomotion to MK-801 (an NMDA receptor antagonist) and decreased hippocampal expression of the N1 subunit of NMDA receptors (12). A limitation of this study is that these results were observed only in the C57 mouse strain. Therefore, further studies were undertaken in rats to provide evidence that these results were generalizable across species (12).
Pregnant Wistar rats were injected with polyI:C at GD10 or GD19 and their offspring undertook behavioral tests such as the acoustic startle response, delayed non-match to position testing and drug-induced locomotor activity assessments (12). Results from the rat study partially replicated findings from mouse studies; offspring exposed to late MIA showed working memory deficits and hyperlocomotion in response to MK-801, and offspring from both groups exhibited hyperlocomotion in response to amphetamine (12). Unlike the mouse study, the rat study showed decreased PPI in male offspring exposed to MIA at any gestational timing (12). This may be because the estrous cycle was not controlled for in female rats, meaning estrogen may have prevented the PPI disruption (12). On the contrary, a later study found female rats exposed to MIA in late gestation also exhibited sensorimotor gating deficits (11). These contradictory results suggest GD10 and GD19 may not be key sensitive periods of gestation for rats. Studies based on different gestational days with larger sample sizes should be undertaken to compare results.
Gestational timing of MIA is of great importance due to neuronal migration in neurodevelopment. A recent study reported decreased cortical and striatal, but not hippocampal, somatostatin (SST) mRNA in polyI:C adult rat offspring (11). This may be related to the distinct origins of these inhibitory neurons and the different timings involved in their migration (11). Cortical SST-containing inhibitory neurons derive from the medial ganglionic eminence (MGE), whilst 40% of hippocampal SST-containing inhibitory neurons derive from the caudal ganglionic eminence (CGE) (11). The MGE has an early dominant period of neurogenesis between GD9.5-GD13.5, whilst the CGE has a later period between GD12.5-16.5 (11). This explains why the hippocampus, and not the cortex, was spared when MIA was inflicted during GD10 and GD19, highlighting the great impact the specific timing of MIA can have on neuronal distribution (11). Further experiments should be conducted with MIA being inflicted during the key neurogenesis period of the hippocampus. If polyI:C offspring expressed characteristics associated with hippocampal dysfunction, such as memory and attention deficits, it would confirm whether MIA affects neurodevelopment through alterations in neuronal migration.
MIA animal model studies have also played a prominent role in identifying a link between complement system dysregulation and increased risk of schizophrenia-like phenotypes. The complement system is a key mediator of immunity, meaning that dysregulation of this system directly implicates the immune system in the etiology of schizophrenia. Complement proteins are involved in neurogenesis, neuronal migration and synaptic formation (4, 14), with C3 knockout mice showing synaptic elimination deficits (49) and impaired migration of neuroblasts (50). Early studies implicating complement system dysfunction in schizophrenia used hemolytic activity assays to measure overall complement component function in the blood (51–53). Results were varied, with some studies finding decreased total hemolytic activity in patients (52), whilst others found no differences between patients and controls (53). Complement system abnormalities are more reliably observed during infection; mothers of schizophrenic patients with increased fetal antibodies against adenovirus and HSV-2 also had higher C1q levels, indicating an early neurodevelopmental role for the complement cascade that may be due to MIA (54). Western blot analysis of C1q in different brain regions of polyI:C adult rodent offspring showed increased C1q expression in the PFC compared to controls (55). Although increased C1q likely contributes to behavioral abnormalities, this hypothesis has not specifically been assessed, meaning further behavioral tests are required to confirm this.
A recent GWAS of schizophrenia confirmed a robust genetic association with the major histocompatibility (MHC) locus, strongly implicating immune dysfunction as a potential pathological mechanism in schizophrenia (56). More specifically, through the use of digital droplet polymerase chain reactions and analysis of single nucleotide polymorphism (SNP) data, overexpression of C4 (originating from the MHC locus), particularly the C4A gene, has been linked with increased schizophrenia risk, with C4A mRNA being elevated in post-mortem brain tissue from schizophrenic patients (15). Immunohistochemistry on brain sections showed majority of C4-positive cells to be in the hippocampus, and co-immunostaining with pre-synaptic (vesicular glutamate transporters 1 and 2) and post-synaptic (post-synaptic density protein 95) markers showed much of the C4 was localized at synaptic puncta (15). Additionally, C4 knockout mice showed reduced synaptic pruning, suggesting C4 overexpression may contribute to decreased synapse numbers observed in schizophrenia (15). A limitation of this investigation is that C4 is encoded for by the C4A and C4B genes in humans, whilst encoded by a single C4b gene in mice, meaning these results may not directly translate to humans (15).
The involvement of C4 in synaptic pruning was rigorously studied by Comer et al. through C4 overexpression in mouse models (16). Results show dendritic spine abnormalities in C4-overexpressing mice which is consistent with post-mortem studies of schizophrenic patients (57). Changes in dendritic spines were most prominent in L2/3 of the PFC, but abnormalities have also been observed in other regions like the frontal lobe, temporal lobe and hippocampus (57). Therefore, targeting different areas with in utero electroporation (IUE) would be required to see if C4 overexpression decreased dendritic spine density in other areas also (16). Additionally, filopodia play a large role in synaptogenesis (58); decreases in the number of filopodia seen in these mice mean C4 may cause dysregulation of filopodia-dependent synapse formation in early development, resulting in abnormal synaptic elimination and circuitry that is seen in schizophrenia (16). Findings of decreased excitatory synaptic drive show how these structural abnormalities in dendritic spine density result in functional changes in connectivity (16). They also found excessive microglial engulfment of synaptic material was driven by C4 overexpression (16). Previous studies have showed that phagocytosis of synaptic material by microglia to be a necessary aspect of normal brain wiring (59), meaning the complement system may mediate brain circuitry, resulting in pathology when dysregulated. As well as genetic and molecular associations between C4 overexpression and schizophrenia, Comer et al. have found C4 overexpression in the frontal cortex to be sufficient in changing social interactions of both juvenile and adult mice, with results suggesting that overexpression of C4 can lead to long-term changes in PFC circuitry (16).
These results are the first to have shown a direct causal link between increased C4 levels and PFC dysfunction, and how this can lead to hypoconnectivity and abnormal synaptic functioning that drives expression of behavioral phenotypes seen in schizophrenia. Although it is recognized that schizophrenia is caused by a wide range of etiological factors, strong genetic, molecular and behavioral evidence has been provided by Comer et al. that altering the expression of only the C4 gene is sufficient in mice to cause schizophrenia-like pathology. Future studies should focus on identifying whether changes in C4 expression in other brain areas can cause other schizophrenia-related phenotypes such as memory deficits.
Additionally, Comer et al. identified a critical developmental time period, around E16 in mice, during which PFC circuits are more vulnerable to changes in C4, highlighting the importance in the timing of gestational insults. This is further strengthened by a study using polyI:C MIA rodent models which, through qPCR, found a significant increase in C4 gene expression in the cingulate cortex of offspring infected at GD19 only. This provides direct evidence, paired with results from Comer et al., that late MIA can cause schizophrenia-like deficits (46). Future studies could utilize transcriptomic and proteomic technology to delineate the molecular pathways underlying the relationship between late gestational MIA and C4 overexpression.
After review of epidemiological studies and animal models of MIA, it can be argued that schizophrenia can be regarded as an immune priming disorder, with MIA increasing the vulnerability of an individual to neuropathological effects of other environmental insults. Animal models have provided a wealth of information highlighting interactions between early neurodevelopmental disturbances and psychosis, showing strong face, construct and predictive validity (60). In particular, polyI:C models have been able to model behavioral, neuroanatomical and neurochemical changes seen in schizophrenic patients, including increased mesencephalic dopamine neuronal density and decreased NMDA receptor function in the PFC, consistent with the dopaminergic and glutamatergic hypotheses of schizophrenia (13). The obvious limitation of animal models is that many clinical manifestations of schizophrenia, e.g., hallucinations and delusions, are impossible to detect in animals (60). Therefore, behavioral, anatomical and physiological characteristics must be examined through translational models rather than modeling the whole syndrome (60). Although polyI:C models cannot mimic the full spectrum of immune responses, the relevance of this model in preclinical studies is not undermined since it does not influence its capacity to mimic schizophrenia-like behaviors and neuropathology (13, 60).
Animal models have provided strong evidence for the role of MIA in schizophrenia pathology, emphasizing the importance of gestational timing of infection in neurodevelopment and behavior. More specifically, MIA in late gestation causes increased C4 expression seen in schizophrenic patients. Subsequently, C4 overexpression has been proven to cause hypoconnectivity in the PFC and social deficits that are characteristic of schizophrenia, displaying how MIA is sufficient in causing schizophrenia-like phenotypes. Future studies utilizing modern functional imaging techniques are required to elucidate this relationship in humans and further confirm the contributing factor of C4 overexpression in schizophrenia development.
Although MIA may predispose individuals to schizophrenia, it is neither sufficient, nor necessary by itself for the development of the disease. Instead MIA can be viewed as a “priming” event, increasing vulnerability to the disorder, acting as a “first hit” toward the development of the disorder. The role of complement proteins in MIA is simply one aspect, among many, of immune dysfunction schizophrenia. Furthermore, the effect may also likely to be non-specific. Maternal stress is also a significant factor resulting in increased risk of different psychiatric disorders amongst offspring, potentially due to its effect on immune molecules like cytokines, with elevated maternal pro-inflammatory cytokines being associated with abnormal neurodevelopment (61–63). Taken together, a “multi-hit threshold model” encompassing different aspects of neurodevelopment would more accurately represent the dynamic interaction between genes and the environment in schizophrenia development (64).
Understanding the neuropathological origins of schizophrenia is important for detecting increased disease risk at an early stage and the development of preventative therapies. Detection of significant immune molecules, such as C4, may allow development of medication that could target these molecules or their receptors. Additionally, the identification of a key window of vulnerability provides a critical time period where such preventative therapies would be most efficacious. However, preventative therapies for neurodevelopmental disorders are controversial and would require extensive evaluation on both a scientific and ethical basis before being implemented in a clinical setting. Nevertheless, further investigation into the immune basis of schizophrenia could provide many potential therapeutic opportunities to improve treatment of this disorder.
ZC prepared the manuscript and constructed the accompanying tables. BL provided guidance with the structure of the manuscript and contributed to revision and proofreading. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Owen MJ, Sawa A, Mortensen PB. Schizophrenia. Lancet. (2016) 388:86–97. doi: 10.1016/S0140-6736(15)01121-6
2. van de Leemput J, Hess JL, Glatt SJ, Tsuang MT. Advances in Genetics Vol. 96 eds Theodore Friedmann, Jay C. Dunlap, & Stephen F. Goodwin. San Diego, CA: Academic Press (2016). p.99–141. doi: 10.1016/bs.adgen.2016.08.001
3. Janoutová J, Janácková P, Serý O, Zeman T, Ambroz P, Kovalová M, et al. Epidemiology and risk factors of schizophrenia. Neuro Endocrinol Lett. (2016) 37:1−8.
4. Nimgaonkar VL, Prasad KM, Chowdari KV, Severance EG, Yolken RH. The complement system: a gateway to gene–environment interactions in schizophrenia pathogenesis. Mol Psychiatry. (2017) 22:1554–61. doi: 10.1038/mp.2017.151
5. Assary E, Vincent JP, Keers R, Pluess M. Gene-environment interaction and psychiatric disorders: review and future directions. Semin Cell Dev Biol. (2018) 77:133–43. doi: 10.1016/j.semcdb.2017.10.016
6. Feigenson KA, Kusnecov AW, Silverstein SM. Inflammation and the two-hit hypothesis of schizophrenia. Neurosci Biobehav Rev. (2014) 38:72–93. doi: 10.1016/j.neubiorev.2013.11.006
7. Maynard TM, Sikich L, Lieberman JA, LaMantia AS. Neural development, cell-cell signaling, and the “two-hit” hypothesis of schizophrenia. Schizophr Bull. (2001) 27:457–76. doi: 10.1093/oxfordjournals.schbul.a006887
8. Mednick SA, Machon RA, Huttunen MO, Bonett D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry. (1988) 45:189–92. doi: 10.1001/archpsyc.1988.01800260109013
9. Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, et al. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. (2004) 61:774–80. doi: 10.1001/archpsyc.61.8.774
10. Meyer U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry. (2014) 75:307–15. doi: 10.1016/j.biopsych.2013.07.011
11. Rahman T, Weickert CS, Harms L, Meehan C, Schall U, Todd J, et al. Effect of immune activation during early gestation or late gestation on inhibitory markers in adult male rats. Sci Reports. (2020) 10:1982. doi: 10.1038/s41598-020-58449-x
12. Meehan C, Harms L, Frost JD, Barreto R, Todd J, Schall U, et al. Effects of immune activation during early or late gestation on schizophrenia-related behaviour in adult rat offspring. Brain Behav Immunity. (2017) 63:8–20. doi: 10.1016/j.bbi.2016.07.144
13. Macêdo DS, Araújo DP, Sampaio LR, Vasconcelos SM, Sales PM, Sousa FC, et al. Animal models of prenatal immune challenge and their contribution to the study of schizophrenia: a systematic review. Braz J Med Biol Res. (2012) 45:179–86. doi: 10.1590/S0100-879X2012007500031
14. Magdalon J, Mansur F, Teles e Silva AL, de Goes VA, Reiner O, Sertié AL. Complement system in brain architecture and neurodevelopmental disorders. Front Neurosci. (2020) 14:4–8. doi: 10.3389/fnins.2020.00023
15. Sekar A, Bialas AR, De Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. (2016) 530:177–83. doi: 10.1038/nature16549
16. Comer AL, Jinadasa T, Sriram B, Phadke RA, Kretsge LN, Nguyen TPH, et al. Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PLoS Biol. (2020) 18:e3000604. doi: 10.1371/journal.pbio.3000604
17. Beydoun H, Saftlas AF. Physical and mental health outcomes of prenatal maternal stress in human and animal studies: a review of recent evidence. Paediatric Perinatal Epidemiol. (2008) 22:438–66. doi: 10.1111/j.1365-3016.2008.00951.x
18. de Kloet ER, Sibug RM, Helmerhorst FM, Schmidt M. Stress, genes and the mechanism of programming the brain for later life. Neurosci Biobehav Rev. (2005) 29:271–81. doi: 10.1016/j.neubiorev.2004.10.008
19. Weinstock M. The long-term behavioural consequences of prenatal stress. Neurosci Biobehav Rev. (2008) 32:1073–86. doi: 10.1016/j.neubiorev.2008.03.002
20. Koenig JI, Elmer GI, Shepard PD, Lee PR, Mayo C, Joy B, et al. Prenatal exposure to a repeated variable stress paradigm elicits behavioral and neuroendocrinological changes in the adult offspring: potential relevance to schizophrenia. Behav Brain Res. (2005) 156:251–61. doi: 10.1016/j.bbr.2004.05.030
21. Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science. (2016) 353:772–7. doi: 10.1126/science.aag3194
22. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. (2010) 167:261–80. doi: 10.1176/appi.ajp.2009.09030361
23. Prasad KM, Watson AM, Dickerson FB, Yolken RH, Nimgaonkar VL. Exposure to herpes simplex virus type 1 and cognitive impairments in individuals with schizophrenia. Schizophr Bull. (2012) 38:1137–48. doi: 10.1093/schbul/sbs046
24. Adams J, Faux SF, Nestor PG, Shenton M, Marcy B, Smith S, et al. ERP abnormalities during semantic processing in schizophrenia. Schizophr Res. (1993) 10:247–57. doi: 10.1016/0920-9964(93)90059-R
25. Limosin F, Rouillon F, Payan C, Cohen JM, Strub N. Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta Psychiatr Scand. (2003) 107:331–5. doi: 10.1034/j.1600-0447.2003.00052.x
26. Brown AS, Schaefer CA, Quesenberry CPJr, Liu L, Babulas VP, Susser ES. Maternal exposure to toxoplasmosis and risk of schizophrenia in adult offspring. Am J Psychiatry. (2005) 162:767–73. doi: 10.1176/appi.ajp.162.4.767
27. Mortensen PB, Nørgaard-Pedersen B, Waltoft BL, Sørensen TL, Hougaard D, Torrey EF, et al. Toxoplasma gondii as a risk factor for early-onset schizophrenia: analysis of filter paper blood samples obtained at birth. Biol Psychiatry. (2007) 61:688–93. doi: 10.1016/j.biopsych.2006.05.024
28. Yolken RH, Torrey EF, Lieberman JA, Yang S, Dickerson FB. Serological evidence of exposure to Herpes Simplex Virus type 1 is associated with cognitive deficits in the CATIE schizophrenia sample. Schizophr Res. (2011) 128:61–5. doi: 10.1016/j.schres.2011.01.020
29. Dickerson FB, Boronow JJ, Stallings C, Origoni AE, Ruslanova I, Yolken RH. Association of serum antibodies to herpes simplex virus 1 with cognitive deficits in individuals with schizophrenia. Arch Gen Psychiatry. (2003) 60:466–72. doi: 10.1001/archpsyc.60.5.466
30. Buka SL, Cannon TD, Torrey EF, Yolken RH. Maternal exposure to herpes simplex virus and risk of psychosis among adult offspring. Biol Psychiatry. (2008) 63:809–15. doi: 10.1016/j.biopsych.2007.09.022
31. Torrey EF, Bartko JJ, Yolken RH. Toxoplasma gondii and other risk factors for schizophrenia: an update. Schizophr Bull. (2012) 38:642–7. doi: 10.1093/schbul/sbs043
32. Mortensen PB, Nørgaard-Pedersen B, Waltoft BL, Sørensen TL, Hougaard D, Yolken RH. Early infections of Toxoplasma gondii and the later development of schizophrenia. Schizophr Bull. (2007) 33:741–4. doi: 10.1093/schbul/sbm009
33. Gaskell EA, Smith JE, Pinney JW, Westhead DR, McConkey GA. A unique dual activity amino acid hydroxylase in Toxoplasma gondii. PLoS ONE. (2009) 4:e4801. doi: 10.1371/journal.pone.0004801
34. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. (2007) 27:10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
35. Kneeland RE, Fatemi SH. Viral infection, inflammation and schizophrenia. Progress Neuro Psychopharmacol Biol Psychiatry. (2013) 42:35–48. doi: 10.1016/j.pnpbp.2012.02.001
36. Short SJ, Lubach GR, Karasin AI, Olsen CW, Styner M, Knickmeyer RC, et al. Maternal influenza infection during pregnancy impacts postnatal brain development in the rhesus monkey. Biol Psychiatry. (2010) 67:965–73. doi: 10.1016/j.biopsych.2009.11.026
37. Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. (2005) 29:913–47. doi: 10.1016/j.neubiorev.2004.10.012
38. Meyer U, Feldon J, Schedlowski M, Yee BK. Immunological stress at the maternal-foetal interface: a link between neurodevelopment and adult psychopathology. Brain Behav Immun. (2006) 20:378–88. doi: 10.1016/j.bbi.2005.11.003
39. Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. (2008) 22:469–86. doi: 10.1016/j.bbi.2007.09.012
40. Li Q, Cheung C, Wei R, Hui ES, Feldon J, Meyer U, et al. Prenatal immune challenge is an environmental risk factor for brain and behavior change relevant to schizophrenia: evidence from MRI in a mouse model. PLoS ONE. (2009) 4:e6354. doi: 10.1371/journal.pone.0006354
41. Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology. (2010) 35:2462–78. doi: 10.1038/npp.2010.129
42. De Miranda J, Yaddanapudi K, Hornig M, Villar G, Serge R, Lipkin WI. Induction of Toll-like receptor 3-mediated immunity during gestation inhibits cortical neurogenesis and causes behavioral disturbances. mBio. (2010). doi: 10.1128/mBio.00176-10
43. Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, et al. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry. (2010) 68:1172–81. doi: 10.1016/j.biopsych.2010.09.022
44. Coiro P, Padmashri R, Suresh A, Spartz E, Pendyala G, Chou S, et al. Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain Behav Immunity. (2015) 50:249–58. doi: 10.1016/j.bbi.2015.07.022
45. Duchatel RJ, Jobling P, Graham BA, Harms LR, Michie PT, Hodgson DM, et al. Increased white matter neuron density in a rat model of maternal immune activation - implications for schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. (2016) 65:118–26. doi: 10.1016/j.pnpbp.2015.09.006
46. Duchatel RJ, Meehan CL, Harms LR, Michie PT, Bigland MJ, Smith DW, et al. Increased complement component 4. (C4) gene expression in the cingulate cortex of rats exposed to late gestation immune activation. Schizophrenia Res. (2018) 199:442–4. doi: 10.1016/j.schres.2018.03.035
47. Meyer U, Nyffeler M, Schwendener S, Knuesel I, Yee BK, Feldon J. Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology. (2008) 33:441–56. doi: 10.1038/sj.npp.1301413
48. Workman AD, Charvet CJ, Clancy B, Darlington RB, Finlay BL. Modeling transformations of neurodevelopmental sequences across mammalian species. J Neurosci. (2013) 33:7368. doi: 10.1523/JNEUROSCI.5746-12.2013
49. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
50. Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson A, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J. (2006) 25:1364–74. doi: 10.1038/sj.emboj.7601004
51. Spivak B, Radwan M, Brandon J, Baruch Y, Stawski M, Tyano S, et al. Reduced total complement haemolytic activity in schizophrenic patients. Psychol Med. (1993) 23:315–8. doi: 10.1017/S0033291700028397
52. Spivak B, Radwan M, Elimelech D, Baruch Y, Avidan G, Tyano S. A study of the complement system in psychiatric patients. Biol Psychiatry. (1989) 26:640–2. doi: 10.1016/0006-3223(89)90091-7
53. Sasaki T, Nanko S, Fukuda R, Kawate T, Kunugi H, Kazamatsuri H. Changes of immunological functions after acute exacerbation in schizophrenia. Biol Psychiatry. (1994) 35:173–8. doi: 10.1016/0006-3223(94)91149-5
54. Severance EG, Gressitt KL, Buka SL, Cannon TD, Yolken RH. Maternal complement C1q and increased odds for psychosis in adult offspring. Schizophrenia Re. (2014) 159:14–9. doi: 10.1016/j.schres.2014.07.053
55. Han M, Zhang JC, Hashimoto K. Increased levels of C1q in the prefrontal cortex of adult offspring after maternal immune activation: prevention by 7,8-Dihydroxyflavone. Clin Psychopharmacol Neurosci. (2017) 15:64–7. doi: 10.9758/cpn.2017.15.1.64
56. Schizophrenia Psychiatric Genome-Wide Association Study C. Genome-wide association study identifies five new schizophrenia loci. Nat Genetics. (2011) 43:969–76. doi: 10.1038/ng.940
57. DeLisi LE. The concept of progressive brain change in schizophrenia: implications for understanding schizophrenia. Schizophr Bull. (2008) 34:312–21. doi: 10.1093/schbul/sbm164
58. Ozcan AS. Filopodia: a rapid structural plasticity substrate for fast learning. Front Synaptic Neurosci. (2017) 9:12. doi: 10.3389/fnsyn.2017.00012
59. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
60. Meyer U, Feldon J. To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology. (2012) 62:1308–21. doi: 10.1016/j.neuropharm.2011.01.009
61. Khashan AS, Abel KM, McNamee R, Pedersen MG, Webb RT, Baker PN, et al. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch Gen Psychiatry. (2008) 65:146–52. doi: 10.1001/archgenpsychiatry.2007.20
62. Andersson NW, Li Q, Mills CW, Ly J, Nomura Y, Chen J. Influence of prenatal maternal stress on umbilical cord blood cytokine levels. Arch Womens Ment Health. (2016) 19:761–7. doi: 10.1007/s00737-016-0607-7
63. Gayle DA, Beloosesky R, Desai M, Amidi F, Nuñez SE, Ross MG. Maternal LPS induces cytokines in the amniotic fluid and corticotropin releasing hormone in the fetal rat brain. Am J Physiol Regul Integr Comp Physiol. (2004) 286:R1024–1029. doi: 10.1152/ajpregu.00664.2003
Keywords: schizophrenia, maternal, immune, complement, neurodevelopment
Citation: Choudhury Z and Lennox B (2021) Maternal Immune Activation and Schizophrenia–Evidence for an Immune Priming Disorder. Front. Psychiatry 12:585742. doi: 10.3389/fpsyt.2021.585742
Received: 22 July 2020; Accepted: 25 January 2021;
Published: 17 February 2021.
Edited by:
Marion Leboyer, Université Paris-Est Créteil Val de Marne, FranceReviewed by:
Mira Jakovcevski, Max Planck Institute of Psychiatry (MPI), GermanyCopyright © 2021 Choudhury and Lennox. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zahra Choudhury, emFocmEuY2hvdWRodXJ5MTk5OEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.