
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Psychiatry, 17 September 2020
Sec. Mood Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.569248
This article is part of the Research TopicStress Induced Neural Changes in Emotional DisordersView all 19 articles
 Fiona Limanaqi1†
Fiona Limanaqi1† Carla Letizia Busceti2†
Carla Letizia Busceti2† Francesca Biagioni2
Francesca Biagioni2 Francesco Fornai1,2*
Francesco Fornai1,2* Stefano Puglisi-Allegra2*
Stefano Puglisi-Allegra2*Stressful events, similar to abused drugs, significantly affect the homeostatic balance of the catecholamine brain systems while activating compensation mechanisms to restore balance. In detail, norepinephrine (NE)- and dopamine (DA)-containing neurons within the locus coeruleus (LC) and ventral tegmental area (VTA), are readily and similarly activated by psychostimulants and stressful events involving neural processes related to perception, reward, cognitive evaluation, appraisal, and stress-dependent hormonal factors. Brain catecholamine response to stress results in time-dependent regulatory processes involving mesocorticolimbic circuits and networks, where LC-NE neurons respond more readily than VTA-DA neurons. LC-NE projections are dominant in controlling the forebrain DA-targeted areas, such as the nucleus accumbens (NAc) and medial pre-frontal cortex (mPFC). Heavy and persistent coping demand could lead to sustained LC-NE and VTA-DA neuronal activity, that, when persisting chronically, is supposed to alter LC-VTA synaptic connections. Increasing evidence has been provided indicating a role of autophagy in modulating DA neurotransmission and synaptic plasticity. This alters behavior, and emotional/cognitive experience in response to drug abuse and occasionally, to psychological stress. Thus, relevant information to address the role of stress and autophagy can be drawn from psychostimulants research. In the present mini-review we discuss the role of autophagy in brain catecholamine response to stress and its dysregulation. The findings here discussed suggest a crucial role of regulated autophagy in the response and adaptation of LC-NE and VTA-DA systems to stress.
Stress is one consequence of challenges to the organism produced by events known as stressors that are usually identified with stimuli (or conditions) that, by definition, need to be unpredictable, uncontrollable and of forecasting uncertainty. These external or internal stimuli promote classic stress responses aimed at adaptation according to physiological and/or psychological compensation (1). Stress-associated adaptive changes may increase the resistance to pathological outcomes, thus favoring resilience, at best, or, at worst, causing dysfunctional coping that increases “allostatic load” (1, 2), leading to a disease state instead. In mammals, including humans, the brain norepinephrine (NE) and dopamine (DA) systems, originating from the locus coeruleus (LC) and the ventral tegmental area (VTA) respectively, produce spread brain networks with cortical and subcortical projections (3). Both NE and DA brainstem neurons are targeted by stress hormones of the hypothalamus-pituitary axis (HPA) (4, 5). NE-LC and DA-VTA neurons are readily activated by stressful events involving neural processes related to perception, cognitive evaluation, appraisal, and stress-dependent hormonal factors. These systems operate in parallel and in synergism, allowing to implement neural adaptations and behavioral strategies aimed at supporting resilience and overcoming stressful events (6–9). In fact, both systems are crucially involved in reward and in efforts to support motivation and coping (3, 10–12).
Brain catecholamine response to stress results in time-dependent regulatory processes involving mesocorticolimbic circuits and networks (13–16). In this context, LC-NE neurons and their projections to the cortical, thalamic, basal forebrain, and brainstem regions, including the VTA, as well as forebrain DA-targeted areas, such as the medial prefrontal cortex (mpFC) and the nucleus accumbens (NAc), appear dominant in controlling DA-dependent responses to stress (2, 17–23). In fact, stressful stimuli activate LC-NE more readily than VTA-DA neurons, which is evident by the powerful release of NE within the mPFC surpassing at large that of DA, and by the increase in tyrosine hydroxylase (TH) and Fos expression occurring within LC but not VTA neurons (13, 14, 18, 24–28).
In detail, NE in the medial pre-frontal cortex (mPFC) may produce opposite effects on DA responses, inhibiting cortical DA transmission while increasing the accumbal DA outflow being induced by first exposure to motivationally salient stimuli (17, 18). Fluctuations of accumbal DA during novel uncontrollable/unavoidable stressful experiences are tightly controlled by the opposing influences of mPFC DA and NE. Enhanced DA release in the NAc is determined by NE release on pre-frontal cortical alpha 1-adrenoceptors (α1-ARs) in a condition of low mesocortical DA activation. Instead, inhibition of NAc DA release is promoted by the return of NE to basal levels and by a sustained increase of mesocortical DA release (18, 19), as occurs in long-lasting acute or in repeated/chronic stress (2, 29, 30). mPFC NE and DA might activate two different pathways to regulate mesoaccumbens DA release in opposite ways; an ‘‘activating pathway’’ provided by indirect glutamatergic (GLUT) projections onto VTA-DA cells (31) and an ‘‘inhibitory pathway’’ provided by prefrontal GLUT efferents to VTA-GABAergic interneurons or striato-mesencephalic neurons (32–34).
Again, stress-induced LC-NE over-activation, by potentiating DA outflow in the midline thalamus, leads to rapid and persistent decrease of GABAA-mediated inhibitory transmission within the NAc-projecting neurons of the posterior paraventricular nucleus of the thalamus (pPVT) (23). This, in turn, promotes disinhibition of NAc-projecting neurons of the pPVT, which increases sensitivity to stress while potentiating DA efflux in the NAc shell (23, 35).
Sustained activity of LC neurons may directly potentiate the firing rate of VTA-DA neurons, mostly through α1-ARs (36–40). In addition to this direct excitation of VTA-DA neurons by LC-NE transmission acting on post-synaptic α1-ARs (36–40), an indirect stimulation is provided by inhibiting or stimulating VTA-GABA and VTA- GLUT terminals, respectively (41, 42). In fact, LC-NE may indirectly activate VTA-DA neurons by acting on pre-synaptic α1-ARs within VTA-GABA and VTA-GLUT terminals, and also on DA and GLUT terminals within the NAc (41–43). The regulation of VTA-DA neuronal activity by LC-NE inputs is quite complex. In fact, NE-induced excitation of VTA-DA neurons is followed by a long-lasting inhibition (36). This is in line with studies showing that selective lesion of LC may paradoxically increase the firing of VTA-DA neurons, though the underpinning mechanisms remain to be clarified (44). This may explain the apparent discrepancy which is present in experimental studies suggesting that stress-induced over-activity of LC-NE may exert either inhibitory or excitatory control of VTA-DA neurons (6, 45). In fact, LC-NE activity may reduce the vulnerability to emotional stress through opposite effects on VTA-DA neurons (6, 45), which may depend on the brain region and the time window of α1-AR stimulation. Such an issue remains under debate and needs experimental clarifications.
Noteworthy, stress is likely to affect catecholamine metabolism and neuroplasticity in a way which is reminiscent of the effects produced by abused substances (7, 46–48). In fact, stressful events are often reported to cause neuropsychiatric disorders going from depression to substance abuse, up to neurodegenerative insults where brain catecholamine-containing neurons and/or their projections are involved (4, 5). Chronic or heavy stress, similar to substance abuse, produces catecholamine-driven behavioral effects ranging from depression to addiction, and schizophrenia-like phenotypes. Such behavioral outcomes involving catecholamine systems are due to plastic phenomena underlying “neuronal sensitization” which in turn, is bound to alterations in the responsivity of type 1- or 2-like DA receptors (D1/D2-like DRs), and alpha/beta adrenergic receptors (α/β-ARs) (6, 16, 19, 49, 50). The occurrence of neural adaptation/maladaptation leads to specific stress-induced alterations of emotion, motivation, cognitive ability and coping.
In the latter decades, substantial attention has been paid to the role of the autophagy machinery in the physiology of catecholamine brain systems when insulted by pharmacological and neurotoxic agents (51–54). Autophagy has been recently connected with stress- and substance abuse-related disorders such as depression and addiction (51, 53, 55, 56). In keeping with this, the beneficial effects of several antidepressants and mood stabilizers are bound to autophagy activation (57–60) and autophagy inducers counteract behavioral sensitization induced by abused drugs (51, 56, 61). This wide and prolific research produced results that strongly suggest a crucial role of autophagy in response and adaptation of LC-NE and VTA-DA systems to stress. This enlightens the mechanisms by which the functional balance of the nervous system and the related behavioral and cognitive capacities are guaranteed. A few studies directly explored the connection between stress and autophagy (62–64); most information can be drawn indirectly from psychostimulants research. In the present review we discuss the role of autophagy in brain catecholamine response to stress and those factors which may lead to dysregulation.
Autophagy is a phylogenetically conserved eukaryotic cell-clearing system that plays a primordial role in cell homeostasis (65). It is generally distinguished into macroautophagy (hereafter referred to as “autophagy”), microautophagy, and chaperone-mediated autophagy, which all promote lysosome-dependent substrate degradation (66). Beyond removing altered protein substrates, autophagy targets mitochondria, pathogens, ribosomes, portions of endoplasmic reticulum or synaptic vesicles, which are conventionally designated as “mitophagy”, “xenophagy”, “ribophagy”, “reticulophagy”, and “vesiculophagy”, respectively (66, 67). Moreover, autophagy modulates key cell functions ranging from synapse development to neurotransmitter release, and synaptic plasticity, as well as neuro-inflammation and –immunity (68, 69). A complex machinery including more than 30 autophagy-related-gene (Atg) products governs autophagy progression, starting from the biogenesis and maturation of autophagosomes up to their fusion with lysosomes. In particular, conversion of Atg8 (LC3 in mammals) into LC3-I, its ubiquitination-like enzymatic lipidation into LC3-II isoform, and eventually the incorporation of LC3-II into the phagophore membrane are critical for autophagosome assembly (59). In line with this, LC3-II is widely employed as a marker for monitoring autophagy at the morphological, ultrastructural, and biochemical level (52). However, since increased LC3-II levels may witness for either an increase or a decrease of the autophagy flux due to accumulation of stagnant vacuoles, assessment of LC3-II levels through semi-quantitative techniques can lead to results misinterpretation unless it is coupled with other autophagy markers or ultrastructural immune-labeling (52). Various additional Atg proteins ranging from Atg3 to Atg16 participate in autophagy progression via the processing and conjugation of Atg8/LC3 to the growing autophagosome membrane lipids (65). For instance, during Atg8 lipidation, Atg7 directly binds to and activates Atg8 fostering its transfer to the E2 enzyme Atg3. At the same time, Atg7 binds to Atg12 fostering its binding to Atg5. This leads to the formation of the Atg12-Atg5 conjugate complex, which then recruits Atg16 (65). The Atg12-Atg5/Atg16 complex localizes to the expanding phagopore where its acts as an E3 ligase mediating the final transfer of Atg8 to its lipid target phosphotidylethanolamine (PE).
The best-known autophagy-modulating pathway consists of the mTOR complex1 (mTORC1), a downstream substrate of the phosphatidylinositol-3-kinase (PI3K)/phosphatase and tensin homolog (PTEN)/AKT axis, which conveys extracellular and environmental stimuli to control cell growth, proliferation, protein synthesis and metabolism in response to bioenergetics and nutritional requests (70). Other well-known pathways that foster autophagy initiation consist of the activation of 5′ AMP-activated Protein Kinase (AMPK), and transcription factor EB (TFEB) or inhibition of glycogen synthase kinase 3 beta (GSK3-β) (58, 71).
A number of CNS disorders are characterized by dysregulated autophagy and related synaptic alterations, and/or oxidative and inflammatory processes connected with neuronal loss (51, 69). In line with this, autophagy provides neuroprotection in general, and for catecholamine neurons, which are mostly susceptible to oxidative-related alterations, in particular (49, 52, 53, 68). In fact, autophagy grants the survival of both DA- and NE-containing neurons during a variety of stressful conditions (49, 51, 52, 68, 70). Autophagy alterations are associated with the effects of abused substances (amphetamine, methamphetamine, cocaine, ethanol) on brain catecholamine neurons concerning their morphology, neuroplasticity, as well as neurotoxic and behavioral effects (51, 52, 56, 61). This is evident by a variety of behavioral effects produced by psychostimulants based on autophagy-dependent alterations (56, 61). Psychostimulants alter neuroplasticity of DA and NE neurons through receptor sensitization/desensitization while affecting their activity and metabolism (7, 49). In keeping with this, autophagy orchestrates the turnover and responsivity of various neurotransmitter receptors by intermingling with the proteasome system and intracellular trafficking and secretory pathways (66, 67). This is key to modulate neurotransmitter release while promoting either desensitization or recycling of neurotransmitter receptors to the plasma membrane. In this context it is worth noting that alterations in autophagy-dependent modulation of vesicular DA trafficking and amount of DA release contribute to maladaptive plastic changes underlying various behavioral disorders (51, 68). Conversely, autophagy induction via mTOR or GSK3β inhibition improves early psychomotor and cognitive alterations by rescuing neurotransmission defects in various DA-related disorders (50, 51, 59, 61, 68, 72–74).
This is not surprising since behavioral alterations are related to intracellular pathways being placed downstream of neurotransmitter receptors, which are bound to the autophagy machinery. For instance, D1/D2-like DRs, including D1DR and D5DR act as negative regulators of autophagy via mTOR activation (75). In detail, D1/5-DR silencing in cells lines increases LC3-II levels while attenuating mTOR activity as evident by the decrease in the levels of its downstream substrate phospho-p70-S6K, indicating activation of autophagy (75). Opposite results are obtained following D1/5-DR overexpression (75). Remarkably, when D1/5-DR silencing is combined with administration of the autophagy flux blockers bafilomycin/chloroquine, the latter are less effective in inhibiting autophagy compared with negative controls, as shown by higher LC3-II and lower phospho-p70-S6K levels (75). This suggests that D1/D5-DRs may exert a powerful negative control on the autophagy machinery. Opposite results were obtained for D2-likeDRs, indicating that they act as autophagy stimulators through AMPK activation and mTOR inhibition (75). This was confirmed in several cells lines, including TH-positive primary midbrain neurons, where DRD2 and DRD3 activation by pramipexole and quinpirole promotes beclin 1-depedent autophagy activation (76). This is associated with neuroprotection and inhibition of alpha-synuclein/SNCA accumulation both in rotenone-treated catecholamine-containing cells that overexpress wild-type or mutant alpha-synuclein and in SNCA transgenic mice (76). More recently, D3DRs were shown to be specifically responsible for autophagy activation via AMPK stimulation and mTOR inhibition (77).
Despite such an evidence suggesting that D1-likeDRs may block while D2-likeDRs may promote the autophagy flux, further in vivo confirmatory studies are needed.
Autophagy is also variously altered by the signaling pathways cyclic AMP (cAMP)/protein kinase A (PKA)/protein kinase C (PKC) and TFEB/peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PCG-α) which are triggered downstream of ARs (78–83). In particular, β2-ARs may induce autophagy, which is associated with NE-related protection (79, 82, 83). For instance, agonist-induced β2‐ARs activation prevents disruption of autophagy flux in skeletal muscle of mice with neurogenic myopathy, which is associated with improved skeletal muscle proteostasis and contractility properties (83). Autophagy blockade through either chloroquine or skeletal muscle‐specific deletion of Atg7 abolishes the beneficial effects of β2‐ARs activation. Similarly, administration of the β2-agonist clenbuterol stimulates the autophagy flux in hepatic cells, while the β2-antagonist propranolol produces opposite effects (79). These effects were confirmed by co-administering chloroquine and through both biochemical (LC3II and p62 quantification) and ultrastructural analyses (79). Nonetheless, the potentially beneficial effects of NE-induced autophagy remain to be confirmed.
Abused psychoactive substances and stress engage shared DA and NE neural mechanisms, as shown for instance by numerous studies pointing to a reciprocal cross-sensitization (84, 85). Stress, similar to abused substances, strongly stimulates LC-NE and VTA-DA transmission, producing an activity overload that brings into play adaptation mechanisms based on feedback circuits between connected neural systems and molecular adjustments in the cells. When stress persists, these compensatory mechanisms fail to restore an ante-stress balance and foster neurodegeneration (6, 82, 86). In fact, during chronic/prolonged stress or drug abuse, a dysregulation of LC-VTA connectivity may occur, decreasing catecholamine release due to diminished LC and VTA activities (9, 86–92). While early being associated with apathy and depression, stress- or drug-induced alterations within LC and VTA may predispose to cognitive decline and neurodegeneration (86, 93). Here we consider available evidence to cast the hypotheses that (i) stress or drug-induced LC-NE overload may alter VTA-DA neurons activity, plasticity and metabolism; (ii) DA overload may in turn lead to a progressive reduction of LC activity that occurs during chronic stress; (iii) a reduction of LC activity may occlude the neurotrophic and neuroprotective effects of NE in LC projecting areas, including the VTA. These functional links between DA and NE brain systems suggest a crucial role for the catecholamine network in adaptive behavior and in stress demands to which the organism has to cope with. At the same time, some contradictory findings may be explained by the double-faceted effects of NE in the brain. While at physiological levels NE exerts neuro-protection, abnormally increased NE levels may induce apoptosis and neurodegeneration (94). In this frame, the autophagy machinery, as a major pathway that regulates both neuronal proteostasis and synaptic plasticity, may be involved in various steps of catecholamine systems’ response to stress and drug intake/administration.
Brain DA and NE systems are connected through cortical-subcortical circuitry as well as through direct bidirectional LC–VTA pathways. The two-way communication between LC and VTA is key for drug-induced reward and reinforcement underlying maladaptive synaptic plasticity in striatal, limbic and cortical brain areas (7, 95). In detail, NE from the LC potently regulates drug-induced reward and reinforcement by stimulating DA release mostly within the ventral striatum (96). This may occur either directly or indirectly since NE-containing axons of the LC project to both VTA, and likely, the NAc shell (97). While DRD1-expressing medium spiny neurons of the NAc medial shell directly inhibit mesolimbic VTA-DA neurons, NAc lateral shell neurons mainly project to the VTA-GABA neurons to disinhibit VTA-DA neurons (98–100). These in turn, fire back to both the NAc lateral shell (98) and the LC (101). Selective stimulation of NAc lateral terminals in the VTA induces a potent reward phenotype, which is likely caused by a disinhibition of VTA-DA neurons (98). Acute exposure to stress, similar to drugs of abuse, alters inhibitory plasticity which may increase VTA excitability (42, 84, 100, 102). This occurs by blocking the induction of long-term potentiation at GABAA synapses while increasing GLUT release on VTA neurons (42, 43, 84, 100, 102).
In this scenario, autophagy is a key by acting at the level of both GABA and DA systems (103–106). Conditional deletion of Atg7 in GABA inhibitory or excitatory neurons, similar to what observed in Unc-51 Like Autophagy Activating Kinase 2 heterozygous (Ulk2+/-) mice, leads to autistic-like behavioral abnormalities including social deficits, increased distress and anxiety along with cognitive alterations (103, 104). In detail, autophagy deficiency within GABA neurons brings to hyper-excitability due to reduced membrane expression of GABAA receptors. In fact, these receptors are entrapped within SQSTM1/p62-positive aggregates (103, 104). Autophagy activation replaces GABAA receptors on the plasma membrane thus reducing abnormal hyperexcitability (104). At behavioral level this is evident by rescuing behavioral deficits in Ulk2+/- mice (104). Again, mice lacking Atg7 specifically within DA neurons display increased evoked striatal DA secretion along with decreased DA re-uptake (105, 106). In line with this, activation of mTOR-dependent autophagy decreases evoked DA release in wild-type but not in transgenic mice (105). Thus, an impairment of mTOR-dependent autophagy at the synapse fosters unrestrained DA release (105). Consistently with this, abused substances activate mTOR signaling in the mesolimbic reward circuit while administration of the mTORC1 inhibitor and autophagy activator rapamycin reverses drug-induced relapse and reinforcement (61, 107–110). For instance, systemic treatment with rapamycin, similar to the infusion of lentivirus-expressing mTOR-shRNA into the NAc shell, suppresses the induction of methamphetamine-induced sensitization while rescuing morphological alterations in the NAc’s dendritic spines (61). Again, selective deletion of mTOR within mouse VTA counteracts drug addiction by decreasing DA release in the NAc through potentiation of VTA-GABAergic neuron firing (110). Thus, mTOR-dependent autophagy regulates drug action by modulating both DA and GABA signaling within the VTA and subsequent DA release within target brain areas. This indicates that an autophagy impairment within the VTA may strengthen the feedback loop in which the VTA fires to the NAc, and back to the LC. In this way, the LC would then feedback into the VTA via α1-ARs to further evoke DA release in the NAc, potentially sustaining behavioral sensitization (7, 39).
When dealing with the multiple effects of α1-ARs, we may summarize that LC-NE transmission increases VTA-DA neurons activity both directly and indirectly by acting on α1-ARs within i) VTA-DA neurons, ii) GABA and GLUT terminals within the VTA, iii) DA and GLUT terminals in the NAc and mPFC (36–63). A crucial role of α1-ARs stimulation in the VTA by LC-NE is documented for the neurochemical and reward processes of abused substances, which leads to an increase in DA release through LC projections to VTA and NAc shell (7, 39, 47, 97). Overlapping with the effects of acute stress and drug exposure, activation of presynaptic α1-AR within the VTA depresses GABA while enhancing GLUT release and increasing AMPAR/NMDAR ratios within VTA-DA neurons (41, 42, 84, 111). Although different brain nuclei being targeted by LC-NE are known to serve as a source of GLUT to the VTA (including the prefrontal cortex and the bed nucleus of stria terminalis), α1-AR -induced GLUT inputs into the VTA seems to derive mostly from local GLUT neurons (41, 112, 113). While contributing to stress- and/or drug-induced behavioral alterations (41, 84, 111–113), an excess of α1-ARs-induced GLUT release onto VTA may increase its vulnerability to Ca2+-related excitotoxicity. In fact, stress- and drug-induced GLUT release onto VTA neurons is coupled to the calcium (Ca2+)-related PKC signaling pathway (41), which produces amphetamine-related oxidative damage going along with autophagy impairment (114, 115). In this context, autophagy is implicated in both GLUT-dependent synaptic plasticity and excitotoxicity. Transient exposure to low doses of NMDA induces autophagy through PI3K/AKT/mTOR pathway inhibition, which is key to promote AMPAR degradation in cultured rat hippocampal neurons and in rodent models of auditory fear reconsolidation (116, 117). On the other hand, neuroprotection against GLUT excitotoxicity is achieved by administering either mTOR-dependent or -independent autophagy inducers rapamycin or trehalose (118). This is in line with evidence showing that NMDAR antagonists may rescue autophagy flux and mitophagy to confer neuroprotection (119). These findings suggest that an autophagy failure being bound to either impaired degradation of plasma membrane AMPA receptors or NMDAR-mediated Ca2+ signaling, may be implicated in the responsivity of VTA neurons to α1-AR-induced GLUT release.
Again, the coupling of α1-AR signaling and the stress hormone corticotrophin-releasing factor (CRF) produces social stress enhancement of drug conditioning via NMDAR-mediated GLUT transmission within the VTA (8). Intriguingly, this is coupled to an amplification of IP3-Ca2+signaling, which is known to impinge on the autophagy pathway (57, 59). Within LC neurons, constitutive overexpression of CRF increases NE activity and redistributes beta-amyloid (Aβ) peptides from synapses to somato-dendritic processes, which occurs along with altered distribution and morphology of autophagy-related vacuoles (120). Again, CRF was recently shown to inhibit the autophagy pathway in vitro (121), suggesting that α1-AR and CRF stimulation following abnormal NE release may synergize to alter autophagy within VTA.
These findings are also in line with evidence on a deleterious role of high NE levels, which similar to stress/drug exposure, do occur in REM sleep deprivation (94, 122–124). In ex vivo and in vivo models of REM sleep deprivation, high NE levels lead to iron and calcium-related oxidative damage within neurons and glia via α1-ARs, which is accompanied by mitochondrial failure and altered levels of AKT (94, 122–124). This occurs in various brain regions including the LC itself, though the VTA remains to be examined. In this frame, it is likely that stress- and drug-induced catecholamine alterations may increase the susceptibility of LC and VTA to neuronal damage by increasing the formation of highly oxidative DA- and NE-derived metabolites, which are known to impair neuronal proteostasis (125, 126). This would explain why catecholamine-containing neurons are particularly susceptible to degeneration associated with an autophagy failure (127–129).
These findings support a correlation between early potentiation of NE-DA activity and autophagy-related alterations within the LC-VTA network following stressful stimuli or exposure to addictive substances (Figure 1).
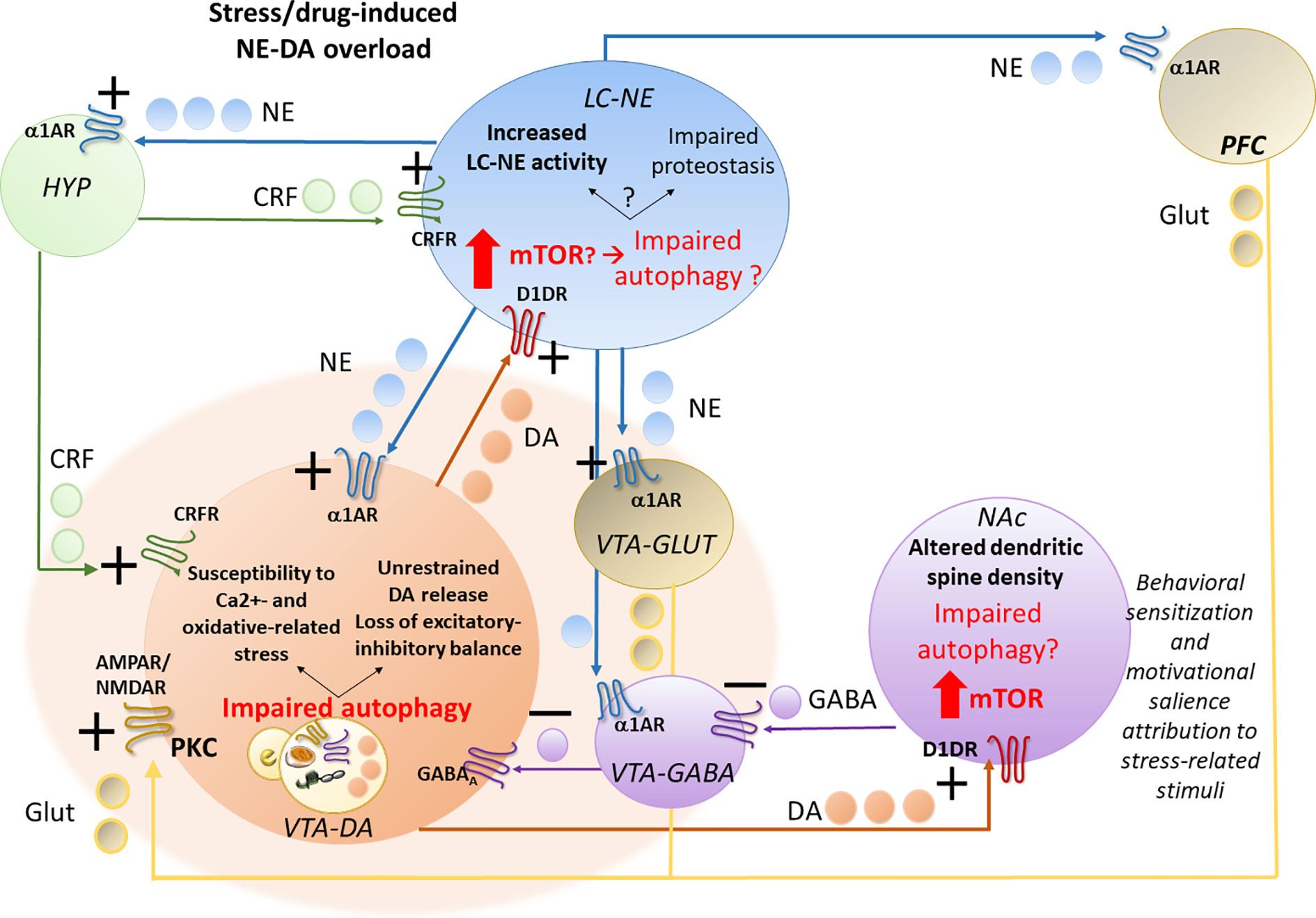
Figure 1 Acute stress- or drug-induced potentiation of locus coeruleus–norepinephrine (LC-NE) and ventral tegmental area–dopamine (VTA-DA) activity and potential role of autophagy. Stress, similar to abused drugs, readily activates LC-NE and VTA-DA neurons, leading to a sustained NE transmission and increased NE release in the hypothalamic CRF-producing neurons, in the forebrain DA-targeted areas pre-frontal cortex (PFC) and nucleus accumbens (NAc) and also directly on VTA neurons. In fact, LC-NE excites VTA-DA neurons either directly through post-synaptic α1-ARs, or indirectly, through inhibition of VTA-GABA neurons and activation of VTA-GLUT neurons via stimulation of pre-synaptic α1-ARs. Again, NE in the PFC, through α1-ARs stimulation, might activate DA release on the NAc through GLUT cortical projections to VTA-DA cells. While altering the excitatory-inhibitory balance and increasing the metabolic rates within VTA-DA neurons, these NE-mediated events may predispose to oxidative stress, and glutamate-related alterations, which may in turn contribute to overwhelming autophagy. While increasing VTA neurons susceptibility to oxidative and Ca2+-related alterations, autophagy impairment within VTA neurons promotes an empowering of DA release to the NAc and likely, also back to the LC. In this way, through stimulation of post-synaptic D1DRs, which is known to promote mTOR activation, DA overload may in turn impair autophagy within the NAc, where it alters dendritic spine density while contributing to drug-induced behavioral sensitization and likely, altered motivational salience attribution to stress-related stimuli. VTA-DA overload, through stimulation of post-synaptic D1DRs, may also impair autophagy within the LC. This may lead to either potentiation of NE release or a progressive impairment of proteostasis impinging on LC neuronal integrity, which remains to be investigated.
Dysregulations of NE activity during exposure to prolonged/chronic stress or abused drugs may include either an increase or a decrease of LC activity. In fact, cumulative cocaine self-administration in rats leads to functional reductions in the LC (92), and a decrease in baseline LC neuron activity and NE release occurs in rodent models of post-traumatic stress disorder - single prolonged stress (SPS), chronic unpredictable mild stress (CUMS), as well as chronic social defeat stress (CSDS) (9, 86, 92). The activation state of VTA-projecting LC-NE neurons, and the amount of NE released into the VTA are critical for determining the vulnerability to emotional stress (45). The loss of NE neurons projecting to the VTA leads to a potentiation of VTA-DA firing conferring an increased susceptibility to stress induced by social defeat compared with resilient mice (45). This is in line with evidence showing that selective lesion of LC increases the firing activity VTA-DA neurons (44). Since VTA-DA excites LC-NE neurons (101), it is expected that exaggerated firing of VTA neurons occurring during acute/repeated stress or abused substance intake/administration, may progressively impinge on LC thus altering this nucleus. In this context it would be worth investigating whether LC alterations are bound to VTA-DA-induced stimulation of D1DRs within the LC, which is essential for ethanol-triggered reinforcement behavior (48). This would be key in the light of evidence indicating that D1DRs that occur within LC neurons (48), may impair autophagy (75). Thus, similar to NE-dependent alterations affecting the VTA discussed in 3.2, autophagy may be implicated in DA-dependent alterations within the LC. In this scenario, a protective role for NE, which may act as an autophagy inducer (79, 82, 83), is substantiated by evidence showing that LC-NE dysfunction may predispose to degenerative phenomena involving various LC-NE-targeted brain areas (82, 86). Here we hypothesize that these may include DA-containing neurons, where reduction of LC-NE release could contribute to undermining intracellular protection mechanisms.
Neuroprotective effects of the LC-NE on DA neurons in vivo and in vitro have been documented emphasizing the effects of NE as a neurotrophic factor and its ability to stimulate the expression of other neurotrophic factors (28, 130–134). This is the case of brain-derived neurotrophic factor (BDNF), which is synthesized within glial cells or neurons mainly through β1/β2-ARs (133, 134). In fact, β2AR agonists reverse DA neurotoxicity in vitro and in vivo (130, 133). As shown in mice models, this occurs through the inhibition of microglial activation rather than exerting a direct effect on VTA-DA neurons, which in fact lack β2-ARs (133). NE-induced neuroprotection of DA neurons depends on the presence of β2AR complexed to β-arrestin (133), which intriguingly, mediates neuroprotection in experimental cerebral ischemia through coordination of BECN1-dependent autophagy (135).
In line with this, there is evidence indicating that NE induces protective autophagy mostly through β2-AR stimulation (79, 82, 83), and again, the protective effects of BDNF are bound to autophagy activation. In detail, BDNF works through inhibition of either mTOR (136) or GSK3β pathway (137), which are main upstream regulators of autophagy. In vivo, BDNF enhances autophagy flux and promotes mitophagy through the HIF-1α/BNIP3 pathway (138). BDNF and related autophagy ameliorate stress-induced behavioral and emotional alterations, suggesting that a direct association exists between autophagy impairment and BDNF deficiency (136, 138). This is not surprising since autophagy has been implicated in the regulation of neurogenesis, which is altered by psychological stress and represents a risk factor for the development of mood/neuropsychiatric disorders (63). In line with this, the administration of either antidepressant drugs or the naturally occurring autophagy inducer resveratrol alleviates depressive-like behavior in mice models of CUMS or post-partum depression by increasing BDNF and autophagy-associated proteins (136, 138). This goes along with reduced HPA axis hyperactivity, CRF and pro-inflammatory cytokines levels (136, 138). In line with this, blockade of autophagy by chloroquine abrogates whereas the autophagy inducer rapamycin protects against the pro-inflammatory effects of CRH in other mice tissues besides the brain (139). These findings suggest that VTA-DA neurons may benefit from glial β2-AR stimulation and BDNF/autophagy induction through reduction of CRF and microglial-mediated inflammation. Autophagy failure is expected to occlude the neurotrophic and anti-inflammatory effects of NE. In fact, microglia-specific Atg5-deficient mice show higher inflammation levels, reduced BDNF expression, and exacerbated depressive-like behavior compared with wild-type mice (138). However, the link between autophagy, NE levels and stress/drug-related behavior remains to be investigated.
As discussed above, an impairment of autophagy within VTA-DA neurons is expected to occlude neurotrophic and neuroprotective mechanisms depending in part on LC-NE activity. Remarkably, an early loss of VTA-DA neurons, which precedes LC neuronal degeneration, has been documented in models of Alzheimer’s disease (93). The potential mechanisms underlying such a temporal dissociation between LC and VTA degeneration will not be dealt herewith. Here, we wish to point out that different patterns of stress exposure might induce hypo-DAergic states (88–90). In detail, acute stress readily activates whereas chronic stress exposure may lead to a compensatory downregulation of the DA system (88–90). In this context, also the timing of stress is critical since contrarily to adolescent stress, adult stress induces a depression-like hypo-DAergic state (140). Similarly, chronic drug intake/administration is associated with a decrease in DA release mostly within the striatum, which may explain the decreased sensitivity to natural rewards and the compulsive drug use as a means to temporarily compensate for this deficit (87). Such an effect is associated with reduced striatal levels of D2-/D3-DRs (87, 141, 142). In the light of an interdependency between autophagy, substance abuse, and D2-/D3-Rs activity discussed in section 3, a potential link deserves to be investigated in stress-related disorders. A reduction of DA activity may be the outcome of either abnormally increased LC-NE release predisposing VTA-DA cells to excitotoxicity, or LC neuronal loss occluding the protective effects of NE upon VTA-DA neurons, as discussed in sections 3.2 and 4.2, respectively. This may be explained by the double-faceted effects of NE in the brain, exerting neuro-protection at physiological levels, while inducing apoptosis at high concentrations (94, 122–124).
Chronic exposure to psychosocial stressors in adults seems to be associated with reduced striatal DA synthesis, mostly within the ventral striatum (90). Autophagy and stress-related VTA-DA alterations may be reminiscent of the molecular effects produced by psychostimulants, which alter autophagy vacuoles (50, 143). This hypothesis remains to be confirmed and may represent the starting point to explore the effect of stress at subcellular level within VTA-DA cells. Despite appearing in contrast with evidence indicating that autophagy blunts DA transmission, these findings suggest that autophagy regulation may be a finely-tuned and context-dependent process, depending on specific patterns of neuronal activity and metabolic demands. In fact, an autophagy impairment within different cell compartments may have different effects, for instance, driving neuropathological changes at the soma while producing striatal-driven behavioral changes by increasing the extracellular availability of DA at the synapse (106).
The evidence here discussed points to the remarkable action of autophagy in NE-LC and VTA-DA connections and its role in NE-dependent neuroprotection, which is crucial for the organism adaptive response to stress and allostatic load. In fact, heavy and/or chronic stress is known to induce or foster neurodegeneration in brain catecholamine neurons involved in a number of psychiatric and neurodegenerative diseases. We proposed here the autophagy machinery as a relevant mechanism of regulation and dysregulation of catecholamine neurons. As for NE-containing neurons, autophagy is seminal for the survival of DA neurons and remarkably, it plays a central role in DA release. The role of autophagy in brainstem NE and DA neurons and their projections in response to psychostimulants indicates adaptive mechanisms that can be activated by stress following its impact on neurotransmission. LC and VTA are both crucial in the response of the organism to stressors and both orchestrate brain systems involved in the appraisal and in the management of psychological stress. Lessons from the effects of abused substances suggest that autophagy, with its role in catecholamine synapse and in neuronal protection, is crucial in modulating the effects of psychological stress on emotional and cognitive driven behavior to foster adaptive emotional outcomes aimed to wellbeing.
At cellular level, psychological stress may translate into intracellular stress-responsive events, such as ER- and oxidative- stress and inflammation (144), which are known to promptly recruit autophagy in the attempt to restore homeostasis. However, under conditions of chronic/persistent stress, when alterations in neurotransmitter activity translate into maladaptive neuronal changes, autophagy may be consistently affected, fostering progressive synaptic deterioration up to neurodegeneration. In the light of an interdependency between autophagy and the mechanisms governing neurotransmission and neuronal homeostasis, we strongly believe that the role of autophagy deserves to be further investigated in the context of catecholamine response to psychological stress specifically.
The possibility of relating stress-related emotional and cognitive experiences to functional aspects through molecular pathways, may facilitate the discovery of potential biomarkers identifying an early risk. Individual differences in the response to stress and autophagy activity, which can make some individuals more or less susceptible than others, should also be addressed by future studies.
SP-A, FL, and FF drafted and wrote the manuscript. FL, CB, and FB contributed to literature review, manuscript editing, and art-work. SP-A and FF are coordinators of the paper; they critically revised the article for important intellectual content.
The present research was funded by Ministero della Salute (Ricerca Corrente 2020).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. McEwen BS, Akil H. Revisiting the Stress Concept: Implications for Affective Disorders. J Neurosci (2020) 40:12–21. doi: 10.1523/JNEUROSCI.0733-19.2019
2. Cabib S, Puglisi-Allegra S. The mesoaccumbens dopamine in coping with stress. Neurosci Biobehav Rev (2012) 36:79–89. doi: 10.1016/j.neubiorev.2011.04.012
3. Sara SJ. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci (2009) 10:211–23. doi: 10.1038/nrn2573
4. Douma EH, de Kloet ER. Stress-induced plasticity and functioning of ventral tegmental dopamine neurons. Neurosci Biobehav Rev (2019) 108:48–77. doi: 10.1016/j.neubiorev.2019.10.015
5. Hupalo S, Bryce CA, Bangasser DA, Berridge CW, Valentino RJ, Floresco SB. Corticotropin-Releasing Factor (CRF) circuit modulation of cognition and motivation. Neurosci Biobehav Rev (2019) 103:50–9. doi: 10.1016/j.neubiorev.2019.06.010
6. Zhang H, Chaudhury D, Nectow AR, Friedman AK, Zhang S, Juarez B, et al. α(1)- and β(3)-Adrenergic Receptor-Mediated Mesolimbic Homeostatic Plasticity Confers Resilience to Social Stress in Susceptible Mice. Biol Psychiatry (2019) 85:226–36. doi: 10.1016/j.biopsych.2018.08.020
7. Ferrucci M, Limanaqi F, Ryskalin L, Biagioni F, Busceti CL, Fornai F. The Effects of Amphetamine and Methamphetamine on the Release of Norepinephrine, Dopamine and Acetylcholine From the Brainstem Reticular Formation. Front Neuroanat (2019) 13:48. doi: 10.3389/fnana.2019.00048
8. Tovar-Díaz J, Pomrenze MB, Kan R, Pahlavan B, Morikawa H. Cooperative CRF and α1 Adrenergic Signaling in the VTA Promotes NMDA Plasticity and Drives Social Stress Enhancement of Cocaine Conditioning. Cell Rep (2018) 22:2756–66. doi: 10.1016/j.celrep.2018.02.039
9. George SA, Knox D, Curtis AL, Aldridge JW, Valentino RJ, Liberzon I. Altered locus coeruleus-norepinephrine function following single prolonged stress. Eur J Neurosci (2013) 37:901–9. doi: 10.1111/ejn.12095
10. Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev (1998) 28:309–69. doi: 10.1016/S0165-0173(98)00019-8
11. Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron (2010) 68:815–34. doi: 10.1016/j.neuron.2010.11.022
12. Lammel S, Lim BK, Malenka RC. Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology (2014) Pt B:351–9. doi: 10.1016/j.neuropharm.2013.03.019
13. Feenstra MG, Botterblom MH, Mastenbroek S. Dopamine and noradrenaline efflux in the prefrontal cortex in the light and dark period: effects of novelty and handling and comparison to the nucleus accumbens. Neuroscience (2000) 100:741–8. doi: 10.1016/S0306-4522(00)00319-5
14. Latagliata EC, Valzania A, Pascucci T, Campus P, Cabib S, Puglisi-Allegra S. Stress-induced activation of ventral tegmental mu-opioid receptors reduces accumbens dopamine tone by enhancing dopamine transmission in the medial pre- frontal cortex. Psychopharmacol (Berl) (2014) 231:4099–108. doi: 10.1007/s00213-014-3549-7
15. Cabib S, Puglisi-Allegra S. Stress, depression and the mesolimbic dopamine system. Psychopharmacology (1996) 128:331–42. doi: 10.1007/s002130050142
16. Laruelle M. The role of endogenous sensitization in the pathophysiology of schizophrenia: implications from recent brain imaging studies. Brain Res Brain Res Rev (2000) 31:371–84. doi: 10.1016/S0165-0173(99)00054-5
17. Tassin J-P. Norepinephrine-dopamine interactions in the prefrontal cortex and the ventral tegmental area: relevance to mental diseases. Adv Pharmacol (1998) 42:712–6. doi: 10.1016/S1054-3589(08)60847-9
18. Pascucci T, Ventura R, Latagliata EC, Cabib S, Puglisi-Allegra S. The medial prefrontal cortex determines the accumbens dopamine response to stress through the opposing influences of norepinephrine and dopamine. Cereb Cortex (2007) 17:2796–804. doi: 10.1093/cercor/bhm008
19. Nicniocaill B, Gratton A. Medial prefrontal cortical alpha1 adrenoreceptor modulation of the nucleus accumbens dopamine response to stress in Long–Evans rats. Psychopharmacol (Berl) (2007) 191:835–42. doi: 10.1007/s00213-007-0723-1
20. Zhu MY. Noradrenergic Modulation on Dopaminergic Neurons. Neurotox Res (2018) 34:848–59. doi: 10.1007/s12640-018-9889-z
21. Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev (2003) 42:33–84. doi: 10.1016/S0165-0173(03)00143-7
22. McCormick DA, Pape HC, Williamson A. Actions of norepinephrine in the cerebral cortex and thalamus: implications for function of the central noradrenergic system. Prog Brain Res (1991) 88:293–305. doi: 10.1016/s0079-6123(08)63817-0
23. Beas BS, Wright BJ, Skirzewski M, Leng Y, Hyun JH, Koita O, et al. The locus coeruleus drives disinhibition in the midline thalamus via a dopaminergic mechanism. Nat Neurosci (2018) 21:963–73. doi: 10.1038/s41593-018-0167-4
24. Smith MA, Brady LS, Glowa J, Gold PW, Herkenham M. Effects of stress and adrenalectomy on tyrosine hydroxylase mRNA levels in the locus ceruleus by in situ hybridization. Brain Res (1991) 544:26–32. doi: 10.1016/0006-8993(91)90881-U
25. Ishida Y, Hashiguchi H, Takeda R, Ishizuka Y, Mitsuyama Y, Kannan H, et al. Conditioned-fear stress increases Fos expression in monoaminergic and GABAergic neurons of the locus coeruleus and dorsal raphe nuclei. Synapse (2002) 45:46–51. doi: 10.1002/syn.10086
26. McDevitt RA, Szot P, Baratta MV, Bland ST, White SS, Maier SF, et al. Stress-induced activity in the locus coeruleus is not sensitive to stressor controllability. Brain Res (2009) 1285:109–18. doi: 10.1016/j.brainres.2009.06.017
27. Finlay JM, Zigmond MJ, Abercrombie ED. Increased dopamine and norepinephrine release in medial prefrontal cortex induced by acute and chronic stress: effects of diazepam. Neuroscience (1995) 64:619–28. doi: 10.1016/0306-4522(94)00331-X
28. Fiore VG, Mannella F, Mirolli M, Latagliata EC, Valzania A, Cabib S, et al. Corticolimbic catecholamines in stress: a computational model of the appraisal of controllability. Brain Struct. Funct (2015) 220:1339–53. doi: 10.1007/s00429-014-0727-7
29. Cabib S, Puglisi-Allegra S. Different effects of repeated stressful experiences on mesocortical and mesolimbic dopamine metabolism. Neuroscience (1996) 73:375–80. doi: 10.1016/0306-4522(96)00750-6
30. Jedema HP, Sved AF, Zigmond MJ, Finlay JM. Sensitization of norepinephrine release in medial prefrontal cortex: effect of different chronic stress protocols. Brain Res (1999) 830:211–7. doi: 10.1016/s0006-8993(99)01369-4
31. Omelchenko N, Sesack SR. Laterodorsal tegmental projections to identified cell populations in the rat ventral tegmental area. J Comp Neurol (2005) 483:217–35. doi: 10.1002/cne.20417
32. Carr DB, O’Donnell P, Card JP, Sesack SR. Dopamine terminals in the rat prefrontal cortex synapse on pyramidal cells that project to the nucleus accumbens. J Neurosci (1999) 1999) 19:11049–60. doi: 10.1523/JNEUROSCI.19-24-11049.1999
33. Carr DB, Sesack SR. Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci (2000) 20:3864–73. doi: 10.1523/JNEUROSCI.20-10-03864.2000
34. Sesack SR, Carr DB. Selective prefrontal cortex inputs to dopamine cells: implications for schizophrenia. Physiol Behav (2002) 77:513–7. doi: 10.1016/S0031-9384(02)00931-9
35. Parsons MP, Li S, Kirouac GJ. Functional and anatomical connection between the paraventricular nucleus of the thalamus and dopamine fibers of the nucleus accumbens. J Comp Neurol (2007) 500:1050–63. doi: 10.1002/cne.21224
36. Grenhoff J, North RA, Johnson SW. Alpha 1-adrenergic effects on dopamine neurons recorded intracellularly in the rat midbrain slice. Eur J Neurosci (1995) 7:1707–13. doi: 10.1111/j.1460-9568.1995.tb00692.x
37. Grenhoff J, Svensson TH. Prazosin modulates the firing pattern of dopamine neurons in rat ventral tegmental area. Eur J Pharmacol (1993) 233:79–84. doi: 10.1016/0014-2999(93)90351-H
38. Linnér L, Endersz H, Ohman D, Bengtsson F, Schalling M, Svensson TH. Reboxetine modulates the firing pattern of dopamine cells in the ventral tegmental area and selectively increases dopamine availability in the prefrontal cortex. J Pharmacol Exp Ther (2001) 297:540–6.
39. Solecki WB, Kielbinski M, Karwowska K, Zajda K, Wilczkowski M, Rajfur Z, et al. Alpha(1)-adrenergic receptor blockade in the ventral tegmental area modulates conditional stimulus-induced cocaine seeking. Neuropharmacology (2019) 158:107680. doi: 10.1016/j.neuropharm.2019.107680
40. Solecki WB, Szklarczyk K, Klasa A, Pradel K, Dobrzański G, Przewłocki R. Alpha(1)-adrenergic receptor blockade in the VTA modulates fear memories and stress responses. Eur Neuropsychopharmacol (2017) 27:782–94. doi: 10.1016/j.euroneuro.2017.05.008
41. Velásquez-Martinez MC, Vázquez-Torres R, Jiménez-Rivera CA. Activation of alpha1-adrenoceptors enhances glutamate release onto ventral tegmental area dopamine cells. Neuroscience (2012) 216:18–30. doi: 10.1016/j.neuroscience.2012.03.056
42. Velásquez-Martínez MC, Vázquez-Torres R, Rojas LV, Sanabria P, Jiménez-Rivera CA. Alpha-1 adrenoreceptors modulate GABA release onto ventral tegmental area dopamine neurons. Neuropharmacology (2015) 88:110–21. doi: 10.1016/j.neuropharm.2014.09.002
43. Mitrano DA, Schroeder JP, Smith Y, Cortright JJ, Bubula N, Vezina P, et al. α-1 Adrenergic receptors are localized on presynaptic elements in the nucleus accumbens and regulate mesolimbic dopamine transmission. Neuropsychopharmacology (2012) 37:2161–72. doi: 10.1038/npp.2012.68
44. Guiard BP, El Mansari M, Merali Z, Blier P. Functional interactions between dopamine, serotonin and norepinephrine neurons: an in-vivo electrophysiological study in rats with monoaminergic lesions. Int J Neuropsychopharmacol (2008) 11:625–39. doi: 10.1017/S1461145707008383
45. Isingrini E, Perret L, Rainer Q, Amilhon B, Guma E, Tanti A, et al. Resilience to chronic stress is mediated by noradrenergic regulation of dopamine neurons. Nat Neurosci (2016) 19:560–3. doi: 10.1038/nn.4245
46. Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron (2011) 69:628–49. doi: 10.1016/j.neuron.2011.02.010
47. Drouin C, Darracq L, Trovero F, Blanc G, Glowinski J, Cotecchia S, et al. Alpha1b-adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J Neurosci (2002) 22:2873–84. doi: 10.1523/JNEUROSCI.22-07-02873.2002
48. Shelkar GP, Kumar S, Singru PS, Subhedar NK, Kokare DM. Noradrenergic inputs from locus coeruleus to posterior ventral tegmental area are essential to support ethanol reinforcement. Addict Biol (2017) 22:291–302. doi: 10.1111/adb.12321
49. Limanaqi F, Gambardella S, Biagioni F, Busceti C, Fornai F. Epigenetic effects induced by methamphetamine and methamphetamine-dependent oxidative stress. Oxid Med Cell Longev (2018) 2018:28. doi: 10.1155/2018/4982453
50. Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol (2009) 49:327–47. doi: 10.1146/annurev.pharmtox.011008.145634
51. Ryskalin L, Limanaqi F, Frati A, Busceti CL, Fornai F. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. Int J Mol Sci (2018) 19:E2226. doi: 10.3390/ijms19082226
52. Lazzeri G, Biagioni F, Fulceri F, Busceti CL, Scavuzzo MC, Ippolito C, et al. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid Med Cell Longev (2018) 2018:6124745. doi: 10.1155/2018/6124745
53. Pasquali L, Lazzeri G, Isidoro C, Ruggieri S, Paparelli A, Fornai F. Role of autophagy during methamphetamine neurotoxicity. Ann N Y Acad Sci (2008) 1139:191–6. doi: 10.1196/annals.1432.016
54. Chauhan AK, Mallick BN. Association between autophagy and rapid eye movement sleep loss-associated neurodegenerative and patho-physio-behavioral changes. Sleep Med (2019) 63:29–37. doi: 10.1016/j.sleep.2019.04.019
55. Gassen NC, Hartmann J, Schmidt MV, Rein T. FKBP5/FKBP51 enhances autophagy to synergize with antidepressant action. Autophagy (2015) 11:578–80. doi: 10.1080/15548627.2015.1017224
56. Lu HF, Xiao W, Deng SL, Cheng XL, Zheng HL, Chen JG, et al. Activation of AMPK-dependent autophagy in the nucleus accumbens opposes cocaine-induced behaviors of mice. Addict Biol (2020) 25:e12736. doi: 10.1111/adb.12736
57. Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol (2005) 170:1101–11. doi: 10.1083/jcb.200504035
58. Pasquali L, Busceti CL, Fulceri F, Paparelli A, Fornai F. Intracellular pathways underlying the effects of lithium. Behav Pharmacol (2010) 21:473–92. doi: 10.1097/FBP.0b013e32833da5da
59. Limanaqi F, Biagioni F, Ryskalin L, Busceti CL, Fornai F. Molecular Mechanisms Linking ALS/FTD and Psychiatric Disorders, the Potential Effects of Lithium. Front Cell Neurosci (2019) 13:450. doi: 10.3389/fncel.2019.00450
60. Gassen NC, Rein T. Is There a Role of Autophagy in Depression and Antidepressant Action? Front Psychiatry (2019) 10:337. doi: 10.3389/fpsyt.2019.00337
61. Huang SH, Wu WR, Lee LM, Huang PR, Chen JC. mTOR signaling in the nucleus accumbens mediates behavioral sensitization to methamphetamine. Prog Neuropsychopharmacol Biol Psychiatry (2018) 86:331–9. doi: 10.1016/j.pnpbp.2018.03.017
62. Jung S, Choe S, Woo H, Jeong H, An HK, Moon H, et al. Autophagic death of neural stem cells mediates chronic stress-induced decline of adult hippocampal neurogenesis and cognitive deficits. Autophagy (2020) 16:512–30. doi: 10.1080/15548627.2019.1630222
63. Puri D, Subramanyam D, Stress -. (self) eating: Epigenetic regulation of autophagy in response to psychological stress. FEBS J (2019) 286:2447–60. doi: 10.1111/febs.14826
64. Xiao X, Shang X, Zhai B, Zhang H, Zhang T. Nicotine alleviates chronic stress-induced anxiety and depressive-like behavior and hippocampal neuropathology via regulating autophagy signaling. Neurochem Int (2018) 114:58–70. doi: 10.1016/j.neuint.2018.01.004
65. Tooze SA, Schiavo G. Liaisons dangereuses: Autophagy, neuronal survival and neurodegeneration. Curr Opin Neurobiol (2008) 18:504–15. doi: 10.1016/j.conb.2008.09.015
66. Okamoto K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J Cell Biol (2014) 205:435–45. doi: 10.1083/jcb.201402054
67. Binotti B, Pavlos NJ, Riedel D, Wenzel D, Vorbrüggen G, Schalk AM, et al. The GTPase Rab26 links synaptic vesicles to the autophagy pathway. Elife (2015) 4:e05597. doi: 10.7554/eLife.05597
68. Limanaqi F, Biagioni F, Gambardella S, Ryskalin L, Fornai F. Interdependency Between Autophagy and Synaptic Vesicle Trafficking: Implications for Dopamine Release. Front Mol Neurosci (2018b) 11:299. doi: 10.3389/fnmol.2018.00299
69. Limanaqi F, Biagioni F, Busceti CL, Ryskalin L, Soldani P, Frati A, et al. Cell Clearing Systems Bridging Neuro-Immunity and Synaptic Plasticity. Int J Mol Sci (2019a) 20:2197. doi: 10.3390/ijms20092197
70. Zoncu R, Efeyan A, Sabatini DM. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol (2011) 12:21–35. doi: 10.1038/nrm3025
71. Weikel KA, Cacicedo JM, Ruderman NB, Ido Y. Knockdown of GSK3β increases basal autophagy and AMPK signalling in nutrient-laden human aortic endothelial cells. Biosci Rep (2016) 36:e00382. doi: 10.1042/BSR20160174
72. Kara NZ, Flaisher-Grinberg S, Anderson GW, Agam G, Einat H. Mood-stabilizing effects of rapamycin and its analog temsirolimus: relevance to autophagy. Behav Pharmacol (2018) 29:379–84. doi: 10.1097/FBP.0000000000000334
73. Masini D, Bonito-Oliva A, Bertho M, Fisone G. Inhibition of mTORC1 signaling reverts cognitive and affective deficits in a mouse model of Parkinson’s disease. Front Neurol (2018) 9:208. doi: 10.3389/fneur.2018.00208
74. Tramutola A, Lanzillotta C, Barone E, Arena A, Zuliani I, Mosca L, et al. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl Neurodegener (2018) 7:28. doi: 10.1186/s40035-018-0133-9
75. Wang D, Ji X, Liu J, Li Z, Zhang X. Dopamine Receptor Subtypes Differentially Regulate Autophagy. Int J Mol Sci (2018) 19:E1540. doi: 10.3390/ijms19051540
76. Wang JD, Cao YL, Li Q, Yang YP, Jin M, Chen D, et al. A pivotal role of FOS-mediated BECN1/Beclin 1 upregulation in dopamine D2 and D3 receptor agonist-induced autophagy activation. Autophagy (2015) 11:2057–73. doi: 10.1080/15548627.2015.1100930
77. Barroso-Chinea P, Luis-Ravelo D, Fumagallo-Reading F, Castro-Hernandez J, Salas-Hernandez J, Rodriguez-Nuñez J, et al. DRD3 (dopamine receptor D3) but not DRD2 activates autophagy through MTORC1 inhibition preserving protein synthesis. Autophagy (2020) 16:1279–95. doi: 10.1080/15548627.2019.1668606
78. Aránguiz-Urroz P, Canales J, Copaja M, Troncoso R, Vicencio JM, Carrillo C, et al. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim Biophys Acta (2011) 1812:23–31. doi: 10.1016/j.bbadis.2010.07.003
79. Farah BL, Sinha RA, Wu Y, Singh BK, Zhou J, Bay BH, et al. β-Adrenergic agonist and antagonist regulation of autophagy in HepG2 cells, primary mouse hepatocytes, and mouse liver. PloS One (2014) 9:e98155. doi: 10.1371/journal.pone.0098155
80. Zhi X, Li B, Li Z, Zhang J, Yu J, Zhang L, et al. Adrenergic modulation of AMPK‑dependent autophagy by chronic stress enhances cell proliferation and survival in gastric cancer. Int J Oncol (2019) 54:1625–38. doi: 10.3892/ijo.2019.4753
81. Khan MM, Lustrino D, Silveira WA, Wild F, Straka T, Issop Y, et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc Natl Acad Sci U.S.A. (2016) 113:746–50. doi: 10.1073/pnas.1524272113
82. Giorgi FS, Ryskalin L, Ruffoli R, Biagioni F, Limanaqi F, Ferrucci M, et al. The Neuroanatomy of the Reticular Nucleus Locus Coeruleus in Alzheimer’s Disease. Front Neuroanat (2017) 11:80. doi: 10.3389/fnana.2017.00080
83. Campos JC, Baehr LM, Ferreira ND, Bozi LHM, Andres AM, Ribeiro MAC, et al. β2 -adrenoceptor activation improves skeletal muscle autophagy in neurogenic myopathy. FASEB J (2020) 34:5628–41. doi: 10.1096/fj.201902305R
84. Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron (2003) 37:577–82. doi: 10.1016/s0896-6273(03)00021-7
85. Booij L, Welfeld K, Leyton M, Dagher A, Boileau I, Sibon I, et al. Dopamine cross-sensitization between psychostimulant drugs and stress in healthy male volunteers. Transl Psychiatry (2016) 6:e740. doi: 10.1038/tp.2016.6
86. Zhang Q, Hu DX, He F, Li CY, Qi GJ, Cai HW, et al. Locus coeruleus-CA1 projections are involved in chronic depressive stress-induced hippocampal vulnerability to transient global ischaemia. Nat Commun (2019) 10:2942. doi: 10.1038/s41467-019-10795-9
87. Volkow ND, Wang GJ, Fowler JS, Tomasi D, Telang F, Baler R. Addiction: decreased reward sensitivity and increased expectation sensitivity conspire to overwhelm the brain’s control circuit. Bioessays (2010) 32:748–55. doi: 10.1002/bies.201000042
88. Moreines JL, Owrutsky ZL, Grace AA. Involvement of Infralimbic Prefrontal Cortex but not Lateral Habenula in Dopamine Attenuation After Chronic Mild Stress. Neuropsychopharmacology (2017) 42:904–13. doi: 10.1038/npp.2016.249
89. Holly EN, Miczek KA. Ventral tegmental area dopamine revisited: effects of acute and repeated stress. Psychopharmacol (Berl) (2016) 233:163–86. doi: 10.1007/s00213-015-4151-3
90. Bloomfield MA, McCutcheon RA, Kempton M, Freeman TP, Howes O. The effects of psychosocial stress on dopaminergic function and the acute stress response. Elife (2019) 8:e46797. doi: 10.7554/eLife.46797
91. Wang B, Wang Y, Wu Q, Huang HP, Li S. Effects of α2A Adrenoceptors on Norepinephrine Secretion from the Locus Coeruleus during Chronic Stress-Induced Depression. Front Neurosci (2017) 11:243. doi: 10.3389/fnins.2017.00243
92. Calipari ES, Beveridge TJ, Jones SR, Porrino LJ. Withdrawal from extended-access cocaine self-administration results in dysregulated functional activity and altered locomotor activity in rats. Eur J Neurosci (2013) 38:3749–57. doi: 10.1111/ejn.12381
93. Nobili A, Latagliata EC, Viscomi MT, Cavallucci V, Cutuli D, Giacovazzo G, et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat Commun (2017) 8:14727. doi: 10.1038/ncomms14727
94. Singh A, Das G, Kaur M, Mallick BN. Noradrenaline Acting on Alpha1 Adrenoceptor as well as by Chelating Iron Reduces Oxidative Burden on the Brain: Implications With Rapid Eye Movement Sleep. Front Mol Neurosci (2019) 12:7. doi: 10.3389/fnmol.2019.00007
95. Mejías-Aponte CA, Drouin C, Aston-Jones G. Adrenergic and noradrenergic innervation of the midbrain ventral tegmental area and retrorubral field: prominent inputs from medullary homeostatic centers. J Neurosci (2009) 29:3613–26. doi: 10.1523/JNEUROSCI.4632-08.2009
96. Pan WH, Sung JC, Fuh SM. Locally application of amphetamine into the ventral tegmental area enhances dopamine release in the nucleus accumbens and the medial prefrontal cortex through noradrenergic neurotransmission. J Pharmacol Exp Ther (1996) 278:725–31.
97. Berridge CW, Stratford TL, Foote SL, Kelley AE. Distribution of dopamine beta hydroxylase-like immunoreactive fibers within the shell subregion of the nucleus accumbens. Synapse (1997) 27:230–41. doi: 10.1002/(SICI)1098-2396(199711)27:3<230::AID-SYN8>3.0.CO;2-E
98. Yang H, de Jong JW, Tak Y, Peck J, Bateup HS, Lammel S. Nucleus Accumbens Subnuclei Regulate Motivated Behavior via Direct Inhibition and Disinhibition of VTA Dopamine Subpopulations. Neuron (2018) 97:434–449.e4. doi: 10.1016/j.neuron.2017.12.022
99. Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, de Roo M, et al. Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science (2013) 341:1521–5. doi: 10.1126/science.1237059
100. Edwards NJ, Tejeda HA, Pignatelli M, Zhang S, McDevitt RA, Wu J, et al. Circuit specificity in the inhibitory architecture of the VTA regulates cocaine-induced behaviour. Nat Neurosci (2017) 20:438–48. doi: 10.1038/nn.4482
101. Deutch AY, Goldstein M, Roth RH. Activation of the locus coeruleus induced by selective stimulation of the ventral tegmental area. Brain Res (1986) 363:307–14. doi: 10.1016/0006-8993(86)91016-4
102. Niehaus JL, Murali M, Kauer JA. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur J Neurosci (2010) 32:108–17. doi: 10.1111/j.1460-9568.2010.07256.x
103. Hui KK, Tanaka M. Autophagy links MTOR and GABA signaling in the brain. Autophagy (2019) 15:1848–9. doi: 10.1080/15548627.2019.1637643
104. Sumitomo A, Yukitake H, Hirai K, Horike K, Ueta K, Chung Y, et al. Ulk2 controls cortical excitatory-inhibitory balance via autophagic regulation of p62 and GABAA receptor trafficking in pyramidal neurons. Hum Mol Genet (2018) 27:3165–76. doi: 10.1093/hmg/ddy219
105. Hernandez D, Torres CA, Setlikm W, Cebrián C, Mosharov EV, Tang G, et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron (2012) 74:277–84. doi: 10.1016/j.neuron.2012.02.020
106. Hunn BHM, Vingill S, Threlfell S, Alegre-Abarrategui J, Magdelyns M, Deltheil T, et al. Impairment of Macroautophagy in Dopamine Neurons Has Opposing Effects on Parkinsonian Pathology and Behavior. Cell Rep (2019) 29:920–31. doi: 10.1016/j.celrep.2019.09.029
107. Dayas CV, Smith DW, Dunkley PR. An emerging role for the mammalian target of rapamycin in “pathological” protein translation: relevance to cocaine addiction. Front Pharmacol (2012) 3:13. doi: 10.3389/fphar.2012.00013
108. Barak S, Liu F, Ben Hamida S, Yowell QV, Neasta J, Kharazia V, et al. Disruption of alcohol-related memories by mTORC1 inhibition prevents relapse. Nat Neurosci (2013) 16:1111–7. doi: 10.1038/nn.3439
109. Neasta J, Barak S, Hamida SB, Ron D. mTOR complex 1: a key player in neuroadaptations induced by drugs of abuse. J Neurochem (2014) 130:172–84. doi: 10.1111/jnc.12725
110. Liu X, Li Y, Yu L, Vickstrom CR, Liu QS. VTA mTOR Signaling Regulates Dopamine Dynamics, Cocaine-Induced Synaptic Alterations, and Reward. Neuropsychopharmacology (2018) 43:1066–77. doi: 10.1038/npp.2017.247
111. Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature (2001) 411:583–7. doi: 10.1038/35079077
112. Cecchi M, Khoshbouei H, Javors M, Morilak DA. Modulatory effects of norepinephrine in the lateral bed nucleus of the stria terminalis on behavioral and neuroendocrine responses to acute stress. Neuroscience (2002) 112:13–21. doi: 10.1016/s0306-4522(02)00062-3
113. Ramos BP Arnsten AF. Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol Ther (2007) 113:523–36. doi: 10.1016/j.pharmthera.2006.11.006
114. Lin M, Chandramani-Shivalingappa P, Jin H, Ghosh A, Anantharam V, Ali S, et al. Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase C delta in dopaminergic neuronal cells. Neuroscience (2012) 210:308–32. doi: 10.1016/j.neuroscience.2012.03.004
115. Huang SP, Chien JY, Tsai RK. Ethambutol induces impaired autophagic flux and apoptosis in the rat retina. Dis Model Mech (2015) 8:977–87. doi: 10.1242/dmm.019737
116. Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci (2012) 32:10413–22. doi: 10.1523/JNEUROSCI.4533-11.2012
117. Shehata M, Abdou K, Choko K, Matsuo M, Nishizono H, Inokuchi K. Autophagy Enhances Memory Erasure through Synaptic Destabilization. J Neurosci (2018) 38:3809–22. doi: 10.1523/JNEUROSCI.3505-17.2018
118. Kulbe JR, Mulcahy Levy JM, Coultrap SJ, Thorburn A, Bayer KU. Excitotoxic glutamate insults block autophagic flux in hippocampal neurons. Brain Res (2014) 1542:12–9. doi: 10.1016/j.brainres.2013.10.032
119. Hirano K, Fujimaki M, Sasazawa Y, Yamaguchi A, Ishikawa KI, Miyamoto K, et al. Neuroprotective effects of memantine via enhancement of autophagy. Biochem Biophys Res Commun (2019) 518:161–70. doi: 10.1016/j.bbrc.2019.08.025
120. Ross JA, Alexis R, Reyes BAS, Risbrough V, Van Bockstaele EJ. Localization of amyloid beta peptides to locus coeruleus and medial prefrontal cortex in corticotropin releasing factor overexpressing male and female mice. Brain Struct Funct (2019) 224:2385–405. doi: 10.1007/s00429-019-01915-8
121. Jin L, Qian Y, Zhou J, Dai L, Cao C, Zhu C, et al. Activated CRH receptors inhibit autophagy by repressing conversion of LC3BI to LC3BII. Cell Signal (2019) 58:119–30. doi: 10.1016/j.cellsig.2019.03.001
122. Mallick BN, Adya HV, Faisal M. Norepinephrine-stimulated increase in Na+, K+-ATPase activity in the rat brain is mediated through alpha1A-adrenoceptor possibly by dephosphorylation of the enzyme. J Neurochem (2000) 74:1574–8. doi: 10.1046/j.1471-4159.2000.0741574.x
123. Biswas S, Mishra P, Mallick BN. Increased apoptosis in rat brain after rapid eye movement sleep loss. Neuroscience (2006) 142:315–31. doi: 10.1016/j.neuroscience.2006.06.026
124. Somarajan BI, Khanday MA, Mallick BN. Rapid eye movement sleep deprivation induces neuronal apoptosis by noradrenaline acting on α1 adrenoceptor and by triggering mitochondrial intrinsic pathway. Front Neurol (2016) 7:25. doi: 10.3389/fneur.2016.00025
125. da Luz MH, Peres IT, Santos TG, Martins VR, Icimoto MY, Lee KS. Dopamine induces the accumulation of insoluble prion protein and affects autophagic flux. Front Cell Neurosci (2015) 9:12. doi: 10.3389/fncel.2015.00012
126. Kang SS, Liu X, Ahn EH, Xiang J, Manfredsson FP, Yang X, et al. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J Clin Invest (2020) 130:422–37. doi: 10.1172/JCI130513
127. Du D, Hu L, Wu J, Wu Q, Cheng W, Guo Y, et al. Neuroinflammation contributes to autophagy flux blockage in the neurons of rostral ventrolateral medulla in stress-induced hypertension rats. J Neuroinflammation (2017) 14:169. doi: 10.1186/s12974-017-0942-2
128. Wei ZB, Yuan YF, Jaouen F, Ma MS, Hao CJ, Zhang Z, et al. SLC35D3 increases autophagic activity in midbrain dopaminergic neurons by enhancing BECN1-ATG14-PIK3C3 complex formation. Autophagy (2016) 12:1168–79. doi: 10.1080/15548627.2016.1179402
129. Sato S, Hattori N. Dopaminergic Neuron-Specific Autophagy-Deficient Mice. Methods Mol Biol (2018) 1759:173–5. doi: 10.1007/7651_2018_156
130. Zhu MY, Raza MU, Zhan Y, Fan Y. Norepinephrine upregulates the expression of tyrosine hydroxylase and protects dopaminegic neurons against 6-hydrodopamine toxicity. Neurochem Int (2019) 131:104549. doi: 10.1016/j.neuint.2019.104549
131. Counts SE, Mufson EJ. Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J Neurochem (2010) 113:649–60. doi: 10.1111/j.1471-4159.2010.06622.x
132. Juric DM, Loncar D, Carman-Krzan M. Noradrenergic stimulation of BDNF synthesis in astrocytes: mediation via alpha1- and beta1/beta2-adrenergic receptors. Neurochem Int (2008) 52:297–306. doi: 10.1016/j.neuint.2007.06.035
133. Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L, et al. β2-adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. J Immunol (2011) 186:4443–54. doi: 10.4049/jimmunol.1002449
134. Wang P, Xu TY, Wei K, Guan YF, Wang X, Xu H, et al. ARRB1/β-arrestin-1 mediates neuroprotection through coordination of BECN1-dependent autophagy in cerebral ischemia. Autophagy (2014) 10:1535–48. doi: 10.4161/auto.29203
135. Zheng Z, Zhang L, Qu Y, Xiao G, Li S, Bao S, et al. Mesenchymal Stem Cells Protect Against Hypoxia-Ischemia Brain Damage by Enhancing Autophagy Through Brain Derived Neurotrophic Factor/Mammalin Target of Rapamycin Signaling Pathway. Stem Cells (2018) 36:1109–21. doi: 10.1002/stem.2808
136. Yang XH, Song SQ, Xu Y. Resveratrol ameliorates chronic unpredictable mild stress-induced depression-like behavior: involvement of the HPA axis, inflammatory markers, BDNF, and Wnt/β-catenin pathway in rats. Neuropsychiatr Dis Treat (2017) 13:2727–36. doi: 10.2147/NDT.S150028
137. Jin H, Zhu Y, Li Y, Ding X, Ma W, Han X, et al. BDNF-mediated mitophagy alleviates high-glucose-induced brain microvascular endothelial cell injury. Apoptosis (2019) 24:511–28. doi: 10.1007/s10495-019-01535-x
138. Tan X, Du X, Jiang Y, Botchway BOA, Hu Z, Fang M. Inhibition of Autophagy in Microglia Alters Depressive-Like Behavior via BDNF Pathway in Postpartum Depression. Front Psychiatry (2018) 9:434. doi: 10.3389/fpsyt.2018.00434
139. Wang SL, Shao BZ, Zhao SB, Chang X, Wang P, Miao CY, et al. Intestinal autophagy links psychosocial stress with gut microbiota to promote inflammatory bowel disease. Cell Death Dis (2019) 10:391. doi: 10.1038/s41419-019-1634-x
140. Gomes FV, Zhu X, Grace AA. The pathophysiological impact of stress on the dopamine system is dependent on the state of the critical period of vulnerability. Mol Psychiatry (2019) 10.1038/s41380-019-0514-1. doi: 10.1038/s41380-019-0514-1
141. Lee B, London ED, Poldrack RA, Farahi J, Nacca A, Monterosso JR, et al. Striatal dopamine d2/d3 receptor availability is reduced in methamphetamine dependence and is linked to impulsivity. Version 2. J Neurosci (2009) 29:14734–40. doi: 10.1523/JNEUROSCI.3765-09.2009
142. Thanos PK, Taintor NB, Rivera SN, Umegaki H, Ikari H, Roth G, et al. DRD2 gene transfer into the nucleus accumbens core of the alcohol preferring and nonpreferring rats attenuates alcohol drinking. Alcohol Clin Exp Res (2004) 28:720–8. doi: 10.1097/01.alc.0000125270.30501.08
143. Tallóczy Z, Martinez J, Joset D, Ray Y, Gácser A, Toussi S, et al. Methamphetamine inhibits antigen processing, presentation, and phagocytosis. Version 2. PloS Pathog (2008) 4:e28. doi: 10.1371/journal.ppat.0040028
Keywords: locus coeruleus, ventral tegmental area, dopamine, norepinephrine, drug addiction, sensitization, corticotrophin-releasing factor, brain-derived neurotrophic factor
Citation: Limanaqi F, Busceti CL, Biagioni F, Fornai F and Puglisi-Allegra S (2020) Autophagy-Based Hypothesis on the Role of Brain Catecholamine Response During Stress. Front. Psychiatry 11:569248. doi: 10.3389/fpsyt.2020.569248
Received: 04 June 2020; Accepted: 31 August 2020;
Published: 17 September 2020.
Edited by:
Jason H. Huang, Baylor Scott and White Health, United StatesReviewed by:
Yanhua Huang, University of Pittsburgh, United StatesCopyright © 2020 Limanaqi, Busceti, Biagioni, Fornai and Puglisi-Allegra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Fornai, ZnJhbmNlc2NvLmZvcm5haUBuZXVyb21lZC5pdA==; ZnJhbmNlc2NvLmZvcm5haUBtZWQudW5pcGkuaXQ=; Stefano Puglisi-Allegra, c3RlZmFuby5wdWdsaXNpYWxsZWdyYUBuZXVyb21lZC5pdA==; c3RlZmFuby5wdWdsaXNpLWFsbGVncmFAdW5pcm9tYTEuaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.