
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 19 November 2020
Sec. Psychopharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.548505
This article is part of the Research TopicPharmacotherapy of Anxiety Disorders: Promises and PitfallsView all 5 articles
 Madeeha Nasir1*
Madeeha Nasir1* Daniel Trujillo1
Daniel Trujillo1 Jessica Levine1
Jessica Levine1 Jennifer B. Dwyer1,2
Jennifer B. Dwyer1,2 Zachary W. Rupp1,3Michael H. Bloch1,4
Zachary W. Rupp1,3Michael H. Bloch1,4Serotonin reuptake inhibitors and benzodiazepines are evidence-based pharmacological treatments for Anxiety Disorders targeting serotonin and GABAergic systems, respectively. Although clearly effective, these medications fail to improve anxiety symptoms in a significant proportion of patients. New insights into the glutamate system have directed attention toward drugs that modulate glutamate as potential alternative treatments for anxiety disorders. Here we summarize the current understanding of the potential role of glutamate neurotransmission in anxiety disorders and highlight specific glutamate receptors that are potential targets for novel anxiety disorder treatments. We also review clinical trials of medications targeting the glutamate system in DSM-5 anxiety disorders. Understanding the role of the glutamate system in the pathophysiology of anxiety disorder may aid in developing novel pharmacological agents that are effective in treating anxiety disorders.
- Serotonin Reuptake Inhibitors and benzodiazepines are evidence-based pharmacological treatments for Anxiety Disorders. These primarily target serotonin and GABA systems, respectively.
- Although clearly effective, these medications do not help improve anxiety symptoms in many patients and have significant adverse effects.
- Preclinical studies and H-MRS studies show anxiolytic potential in targeting glutamate systems.
- While some clinical studies have shown some promise, most have been largely underpowered or not used placebo groups. Larger placebo-controlled trials need to be conducted in order to verify their utility in the management of anxiety disorders.
Anxiety disorders affect ~18% of adults making them one of the most common psychiatric disorders (1). The Diagnostic Statistical Manual 5 (DSM-5) lists separation anxiety, generalized anxiety disorder (GAD), social anxiety disorder (SAD), specific phobia, panic disorder, and agoraphobia under the anxiety disorders (2). While the DSM-5 recently removed obsessive-compulsive disorder (OCD) and post-traumatic disorder (PTSD) from the category of anxiety disorders, anxiety is still considered a prominent and distressing symptom in these and other psychiatric disorders such as autism spectrum disorders (ASD) and depression. Co-morbidity of anxiety disorders with other psychiatric disorders is associated with a more chronic, treatment resistant course and with elevated suicide risk (3, 4). Additionally, patients with anxiety disorders have increased risk of substance abuse and certain medical disorders such as hypertension and irritable bowel syndrome (5). Thus, anxiety's link to impairment is well established across a broad constellation of domains and disorders. The high prevalence of anxiety as a primary or comorbid disorder and its significant functional disability result in a high personal, social and economic cost (6).
Given the high impairment and cost of anxiety disorders, an array of psychotherapeutic approaches and medications have been assessed and validated for the treatment of anxiety disorders. Both pharmacological and psychotherapeutic treatments are currently the recommended first-line treatments for DSM-5 anxiety disorders (7–9). The combination of pharmacological and behavioral treatments for anxiety disorders is likely more effective than either intervention alone, especially in patients with moderate-to-severe anxiety symptoms (10, 11). Cognitive behavioral therapy is currently the recommended first-line psychotherapeutic treatment for DSM-5 anxiety disorders (7–9), although several other psychotherapeutic approaches have some demonstrated efficacy in the treatment of anxiety disorders (10). However, access and successful dissemination of CBT and other psychotherapies remains a major treatment challenge. Furthermore, 46.4% of the patients who are lucky enough to have access to gold-standard psychotherapeutic treatment for anxiety disorders, do not respond to therapy (12). Others still have substantial residual anxiety despite having a good response to therapy, or are resistant to engaging in therapy due to concerns about cost, time, stigma or fear of engaging in exposures (13). Due to these reasons, pharmacological treatments remains integral to the care of patients with anxiety disorders, despite the fact that psychotherapeutic treatments are as or more effective than any pharmacological treatment for anxiety currently available (10).
First-line pharmacological treatment for DSM-5 anxiety disorders primarily consists of antidepressants that modulate serotonin neurotransmission such as selective serotonin reuptake inhibitors (SSRIs) and serotonin norepinephrine reuptake inhibitors (SNRIs) (10). Both SSRI and SNRI medications have demonstrated significant benefits compared to placebo in treating anxiety disorders in meta-analysis of randomized controlled trials (RCTs) (10). Although current treatment guidelines recommend treating patients for at least 8 weeks at the maximally tolerated dose of SRI medication, longitudinal meta-analysis found that it takes only four weeks for SSRIs (SMD = 0.60) and two weeks for SNRIs (SMD = 0.51) to attain a statistical significant benefit compared with placebo (14). However, the maximal treatment benefit of SSRI and SNRI medications for anxiety is fully observed until 12 weeks after the initiation of treatment (14). This delay in therapeutic benefit of serotonergic antidepressants can be exacerbated in clinical practice where dose titration is typically more conservative than that adhered to in clinical trials (14, 15). Serotonergic antidepressants are also associated with significant sexual side-effects that can limit tolerability and impact adherence (16–19). Although typically quite well-tolerated, serotonergic antidepressants are associated with other common adverse effects include nausea, diarrhea, diaphoresis, headaches, tremor, asthenia, insomnia, and somnolence (20–25). Finally, sudden discontinuation of these serotonergic antidepressants can result in a withdrawal syndrome which can include worsening of anxiety symptoms, panic attacks, dizziness, nausea that can be quite substantial and may take several few weeks to resolve without treatment (26). In summation, serotonergic antidepressants are only partially effective (leaving many patients who do not respond or only partially respond to treatment) and when beneficial take a long time to become fully effective and have a slew of adverse effects.
Benzodiazepines are another evidence-based pharmacological treatment that are commonly utilized and have strong evidence of efficacy for the treatment of DSM-5 anxiety disorders. Benzodiazepines act as positive allosteric modulators at y-aminobutyric acid (GABA)-A receptors, enhancing overall GABAergic activity. GABA functions as the primary inhibitory neurotransmitter in the central nervous system. Benzodiazepines can rapidly improve anxiety symptoms, unlike the serotonergic antidepressants, which take more time for benefits to accrue. However, benzodiazepines have the potential for abuse and dependence and are associated with greater adverse effects than serotonergic antidepressants. Common side-effects associated with acute benzodiazepine treatment include ataxia or incoordination, fatigue, slurred speech, drowsiness, sexual dysfunction (decreased libido, or anorgasmia), xerostomia, constipation, and light-headedness (27–29). Long-term use of benzodiazepines can be associated with dependency (and thus reduced efficacy at a given dose), rebound anxiety, a discontinuation syndrome and memory impairment (15). Therefore, although quite effective in the acute treatment of anxiety disorders, benzodiazepine use is often reserved for as needed short-term relief on anxiety symptoms, in patients refractory to serotonergic antidepressants and psychotherapy, and as an adjunct when starting serotonergic antidepressants to bridge the gap of their “therapeutic delay” and prevent worsening of anxiety at the beginning of treatment (15).
Although there exist several effective psychotherapeutic and pharmacological treatments currently in our armamentarium for the treatment of DSM-5 anxiety disorders, there still exists a substantial need for novel treatments, as currently available treatments either do not help a substantial minority of patients or often leave substantial residual symptoms. Additionally, they have bothersome side-effects and typically take several months to reach full efficacy.
Here we will review the current evidence-base of pharmacotherapies that target glutamate and GABA neurotransmission for the treatment of DSM-5 anxiety disorders. Glutamate systems in corticolimbic circuits interact with GABAergic, serotonin, dopamine, and other systems involved in the stress response, which implies a potential role in the circuitry underlying anxiety disorders (30–32). Consistent with this hypothesized role, investigational drugs that target glutamate receptors have shown promise as treatment strategies for anxiety disorders (33–35). This review aims to summarize the characteristics and potential pharmacological targets of the glutamate system. In addition, clinical trials of the main glutamatergic drugs for anxiety disorders as classified under the DSM-5 will be reviewed.
Glutamate is the chief excitatory neurotransmitter in the mature central nervous system (CNS). Its projections take part in effectively all neural circuits, interconnecting cortical, and subcortical systems (36). It is prevalent throughout the CNS and has been found to play important physiological roles in neurodevelopment, cognition, and learning and memory formation (37, 38). It plays a part in a variety of cellular activities in both neurons and glia beyond glutaminergic neurotransmission. It has a role in protein synthesis, energy metabolism, as a part of the tricarboxylic acid cycle, is a precursor for both glutathione, which mitigates oxidative stress, and GABA, an inhibitory neurotransmitter as depicted by Figure 1 (39).
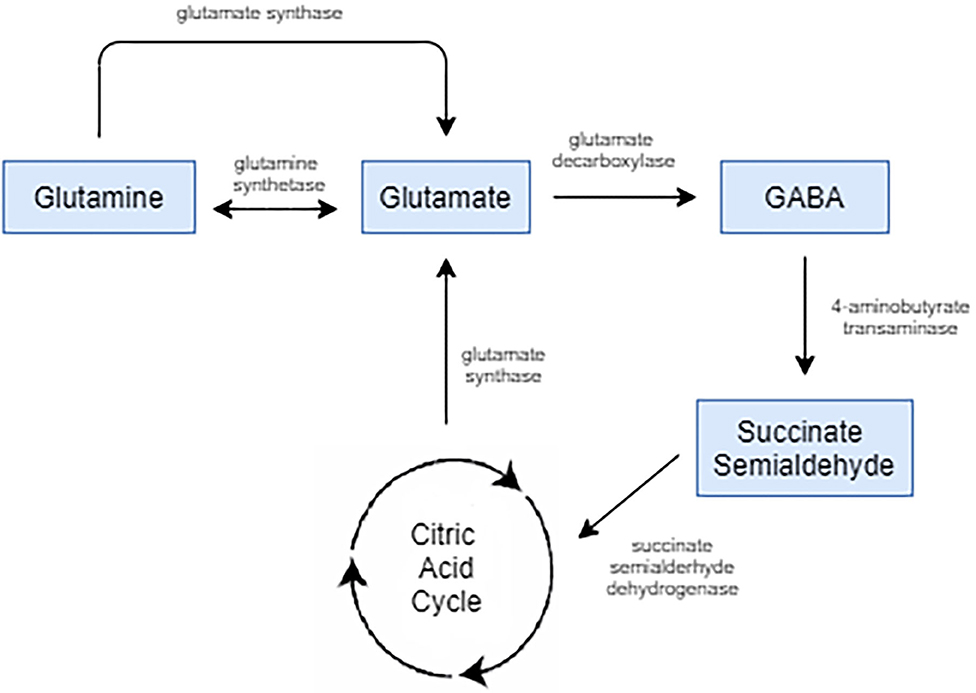
Figure 1. Glutamate is the biological precursor for GABA. Glutamate is synthesized from the nonessential amino acid glutamine, and glutamate is converted into GABA by the enzyme glutamate decarboxylase.
Glutamate has the characteristics of a typical neurotransmitter (36). It is synthesized and packed into vesicles at pre-synaptic terminals of glutamatergic neurons, by vesicular glutamate transporters (vGluT) (40). On arrival of an action potential, the vesicles fuse with the pre-synaptic membrane and release glutamate into the synaptic cleft. Once released, glutamate binds with both the postsynaptic receptors, which transmit the signal onto the next neuron, and presynaptic receptors, which prevent further release of glutamate resulting in negative feedback.
Glutamate receptors can either be ionotropic receptors, which are ligand gated cation channels, or metabotropic receptors, which are G-protein coupled transmembrane proteins. Ionotropic receptors are of three types: N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and kainite receptors. Metabotropic receptors have eight types termed mGluR1−8 (41).
Heteromeric NMDA receptors are present in high densities in the hippocampal region, frontal cortex and other limbic areas of the brain. They are comprised of two conserved NR1 subunits and two or three regionally specific NR2 subunits. Activation requires three conditions: occupation of glutamate binding site on the NR2 subunit, occupation of the co-agonist glycineB (GLYB) site on the NR1 subunit, and membrane depolarization (30). Thus, both pre- and post-synaptic neurons need to be depolarized in order to activate NMDA receptors, adding an additional level of regulation to glutamate transmission (42). Once activated, a Mg2+ ion that blocks the central pore is released, allowing influx of Na+, and importantly, Ca2+ into the cell, which plays a crucial part in synaptic plasticity and memory formation (43).
In contrast, the channels on the AMPA receptors open when glutamate molecules bind onto a minimum of two out of four available binding sites, allowing influx of Na+, K+, and Ca2+ ions (44). While similar in structure, kainate receptors open for a shorter time than AMPA receptors. Unlike the NMDA and AMPA receptors which transmit the action potential, kainate receptors are believed to act as pre- and post-synaptic modulators (45).
mGLuR are transmembrane receptors, coupled to G-proteins, that when activated by an extracellular ligand trigger intracellular cascades that modulate neuronal activity. Different subtypes are expressed differentially in specific brain areas. For example, mGlu1 and mGlu5 are predominantly in the amygdala, hippocampus and thalamus, whereas mGlu3 is located primarily on glia (38).
It is not only the receptor subtype that determines function within a circuit, but also the location of those receptors. Extra-synaptic glutamate receptors have all together distinct effects compared to synaptic receptors. For example, while synaptic NMDA receptor activation transmits action potentials across neurons and promotes neuronal growth and plasticity, extra-synaptic NMDA receptors inhibit these processes and can result in neuronal damage (46). Significant physiologic energy is devoted to containing glutamate within the synapse with tight regulation of spill over via high affinity glutamate transporters, primarily on glial cells (mainly astrocytes). Activation of extra-synaptic receptors occurs when these reuptake mechanisms are overwhelmed and contributes to excitotoxic cascades and cell death (47). Table 1 gives a list of drugs acting on the glutamate system based on their mechanisms of action and Figure 2 depicts a glutamatergic synapse and displays the receptor sites at which the agents discussed in this review act.

Table 1. Describes the mechanisms with which drugs affect glutamate neurotransmission.
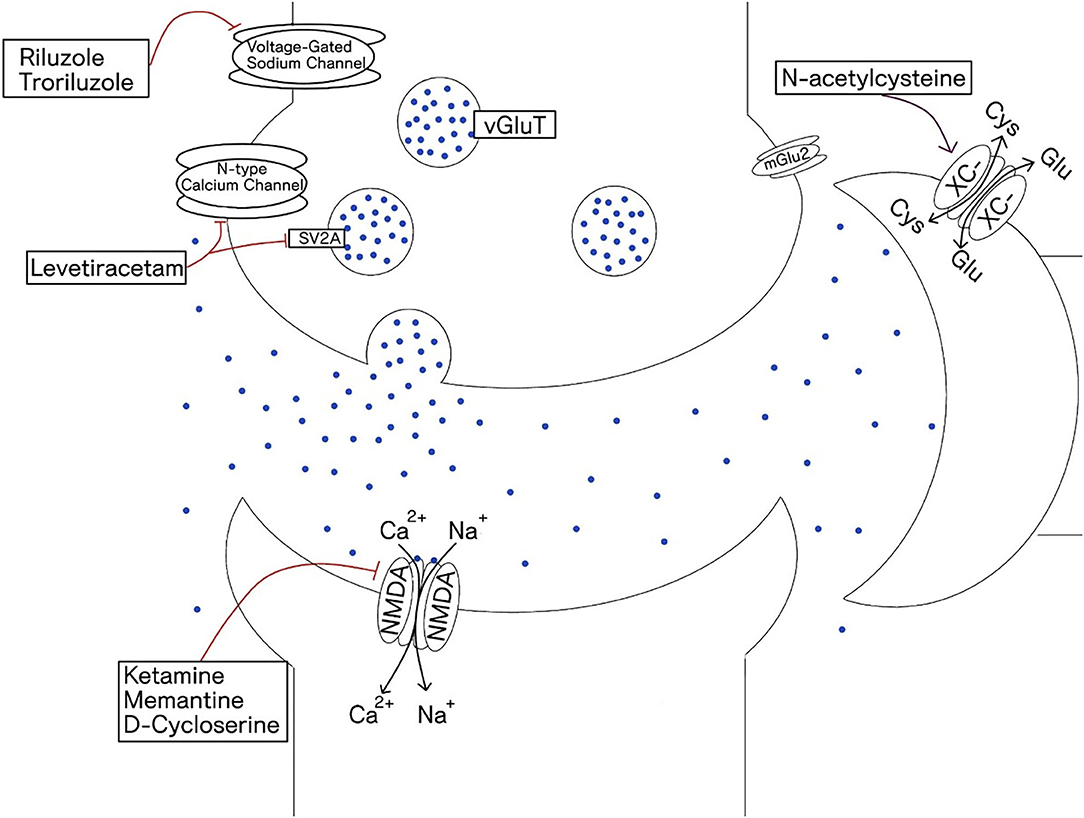
Figure 2. Depicts glutaminergic synapse and the receptors that are the site of action of glutaminergic drugs. Glutamate is packed into vesicles by vesicular glutamate transporter (vGluT). It binds to both ionotropic receptors and metabotropic receptors. Glial cells play a primary role in glutamate reuptake thus terminating the glutamate synaptic signal. Steady-state extra synaptic glutamate levels are also regulated by the glial cystine-glutamate antiporter (XC-).
H-MRS is an imaging technique that quantifies endogenous brain metabolites in vivo. Most previous H-MRS studies in anxiety disorders have focused on the CNS specific metabolite, N-acetylaspartate (NAA), since it has a prominent signal in MRS with levels correlating to neuronal integrity (48–52). Some studies, however, have investigated region specific changes in glutamate levels in patients with anxiety disorders.
Glutamate (Glu) and glutamine (Gln) have similar molecular structures which give rise to similar magnetic spectra and thus have traditionally been considered to constitute one pool denoted Glx. Glx levels have been found to be higher (53) or unchanged in OCD (54) and lower in social anxiety disorder (55). These inconsistent results could be due to a variety of different reasons. For example, measuring Glx levels does not aid in studying them separately or in conditions where Glu and Gln concentrations are altered in opposing directions. Another complication is that since glutamate has multiple roles outside of neurotransmission, it is difficult to distinguish between an increase in extracellular glutamate seen due to microdialysis vs. an increase in synaptic glutamate release. Additionally, imaging studies often suffer from low power and familywise error due multiple inferential statistical tests done on the same dataset, resulting in a high rate of type 1 errors, variation within studies, and uncertainty. The studies reviewed here are pilot studies and are to the same limitations and thus require larger multisite replications.
Newer MRS techniques can now distinguish between Glu and Gln peaks, which has allowed rapid increase in our understanding of the dynamics between Glu and Gln (56). Higher frontal cortex Glu levels have been detected in healthy subjects with high trait state anxiety compared to low trait state anxiety (57). Patients with social anxiety were found to have a 13.2% higher Glu levels in the anterior cingulate cortex (ACC) compared to controls. This increase was correlated with the severity of social anxiety symptoms (58).
Glutamate has been implicated in depression, OCD and PTSD (33, 59, 60), which have overlapping symptomatology or are often comorbid with anxiety disorders. Post-mortem studies on patients with depression have found altered glutamate levels in plasma, cerebrospinal fluid and brain tissue as well as differences in NMDA receptor expression and affinities (61). Blood glutamate scavengers, such as oxaloacetate and pyruvate inactivate glutamate by converting it into 2-ketoglutarate leading to a decrease in glutamate blood levels. This decrease in blood levels causes glutamate to shift down its concentration gradient from the brain to the blood resulting in lower brain glutamate levels. Animal studies have shown potential benefits in the treatment of post-stroke depression (62). Genetics studies also suggest links between genes associated with glutamatergic transmission and depression (63, 64). Likewise, OCD patients have shown aberrant levels of glutamate in cerebrospinal fluid (60, 65). Like in depression, there have been some findings of elevated Glx levels in OCD patients, however, they have also been inconsistent (42, 66, 67). While there have been associations between several genes related to the glutamatergic system and OCD, they too have not been consistent. The best replicated finding is the correlation of OCD risk with variants of the glutamate transporter EAAT3, coded by SLC1A1 gene (68–75). The causative mutations and their effects have not been elucidated (42). Variants of this gene were also associated with greater risk of PTSD after trauma and greater symptom severity (64). Another study with PTSD patients also found increased Glx levels in the rostral ACC when compared to healthy controls and those in remission (76). Finally, medications that target the glutamate system have been utilized to treat these disorders with some success, thus, reinforcing the argument for investigating glutamatergic agents for treatment of anxiety disorders (42, 59, 61).
Preclinical studies have provided a significant scientific rationale for the potential of glutamate modulators in the management of anxiety disorders (30, 33, 77). Stress is a key factor in the development of anxiety disorders and this is simulated using a variety of animal stress models. Stressing a rat has been shown to stimulate glutamate release in the prefrontal cortex of the its brain (78, 79). In contrast to acute stress which has shown to increase glutaminergic transmission in the prefrontal cortex and other limbic regions, chronic stress has been associated with a decrease in glutamate receptors resulting in lower glutamate transmission (80). The glutamate system also plays a major role in the extinction process in fear learning and extinction paradigms (81). As discussed below, the results of stress and fear studies on animal models are in line with this theory (82).
Animal models do not reflect all the complexities of specific anxiety disorders instead, they aim to create a state of anxiety-like behavior that can be generalized to these disorders (83). Unconditioned anxiety models rely on creating situations where the rats face opposite motivational forces to explore or to hide in novel situations (e.g., the elevated plus maze and the social interaction test) or can be predator based (e.g., cat and rat exposure test). Animal anxiety models can also involve classical conditioning (e.g., the fear-potentiated startle response and place aversion test) or operant conditioning (e.g., Geller-Seifter test and Vogel conflict test) (84). Finally, there are pathophysiological models which utilize chronic immobility or stress and trauma paradigms (84, 85).
These models have been used to assess the anxiolytic activity of drugs acting on NMDA, AMPA, kainite, and mGLuR receptors. Injecting the NMDA receptor blocker, DL-2 amino-5-phosphonopentanoic acid (AP5), into the pontine reticular nucleus of rates attenuated the fear potentiated startle response in a dose dependent manner (86). When injected into the amygdala, it inhibited the acquisition of the fear potentiated startle response, but not the expression of previously acquired fear responses (87).
The effects of ketamine (an NMDA antagonist) on anxiety have also been studied in rat models, with one finding no significant differences in anxiety levels as measured by the elevated plus maze test between rats exposed to subanesthetic ketamine doses (30 mg/kg) and saline-injected controls (88); another study found that a single anesthetic ketamine dose (100 mg/kg) caused rats to exhibit higher anxiety as measured by performance in the open field test (89). In another study, the systemic administration of intra-amygdala infusions of D-Cycloserine (DCS), a partial NMDA receptor agonist that can antagonize the NMDA receptor at high doses (90) resulted in the dose dependent facilitation of fear extinction (91–94). Thus, NMDA blockade in rats via administration of different pharmacological agents has demonstrated mixed effects on anxiety levels, with more studies required to elucidate the cause of these differences.
Both kainic acid and topiramate are AMPA/kainite receptor agonists and have shown to decrease the fear potentiated startle response and stress induced startles responses in rats (95, 96). Administrating methyl-6-(phenlythynyl)-pyridine (MPEP), a mGluR5 antagonist, resulted in decreased in fear potentiated startle (97) and increased punished responding (decreased avoidance of painful shock in order to obtain reward) (98). Unlike mGluR5 which are postsynaptic at the glutamatergic synapse and coupled with Gq-proteins, mGluR2/3 exist at the presynaptic end and are coupled with inhibitory Gi/Go proteins (41). The anxiolytic effect of activating these receptors is seen across several rat anxiety models, after systemic and oral administration of LY354740, a mGluR2/3 agonist, resulting in decreased fear potentiated startle responses (99, 100), decreased lactate-induced panic (101) and an increase exploration time in the exposed arms of an elevated plus maze (102). Finally, lamotrigine which inhibits glutamate response by blocking sodium channels and has shown to decrease postsynaptic NMDA receptor mediated excitatory postsynaptic potential in rat amygdala neurons (103). Similarly, rilozule also blocks voltage gated sodium channels, in addition to blocking excitatory amino acid receptors and various calcium channels (103). When injected in rates, both these drugs showed anxiolysis and increased conditioned emotional response rates during the presentation of food with a light which was previously paired with shock (103). Taken together these data indicate a general trend toward activation of the glutamate system and fear startle response acquisition and its blockade with facilitating fear response extinction and anxiolytic-like effect. However, this may not be a universal rule as it is contradicted by the fact that AMPA/kainite receptor agonists reduced potentiated startle response. The mixed pharmacology of most drugs makes it challenging to solely attribute the anxiolysis to one target. Additionally, alternative justifications for this anxiolytic-like effect cannot be ruled out, for example, that it instead is the result of drug impaired memory and attention.
This decade has seen increasing support for the utilization of glutamate modulating drugs in psychiatric conditions. Medications already in clinical use for conditions like epilepsy, neurodegenerative diseases, and alcohol abuse were found to have glutamatergic mechanisms of actions (39). The FDA approved indications of the drugs are listed in Table 2. There have been several trials to test their use as treatments for anxiety disorders (see Table 3).
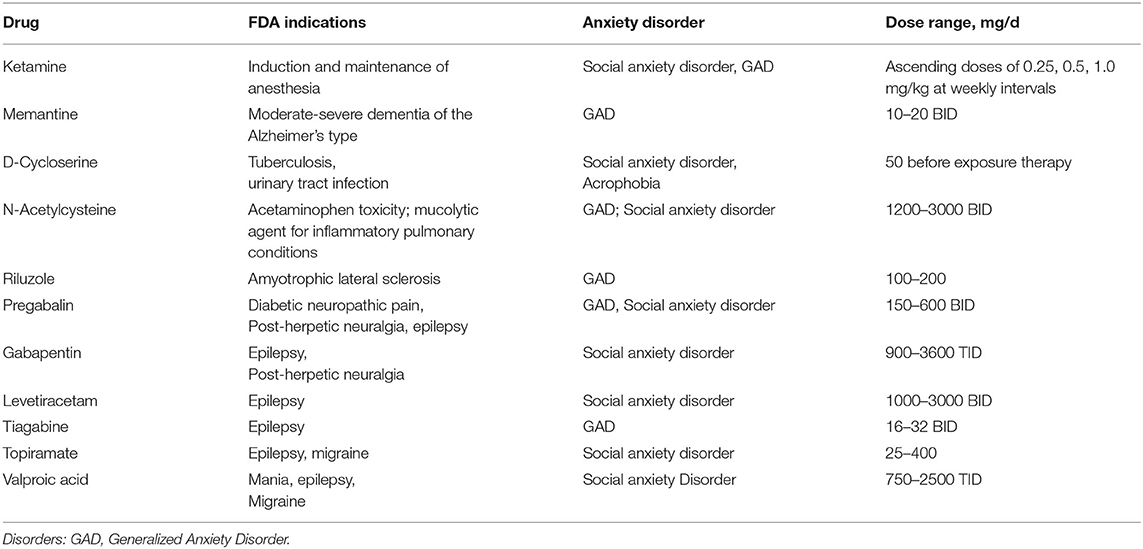
Table 2. Lists the FDA approved indications of the agents and the anxiety disorders they have been tested for.
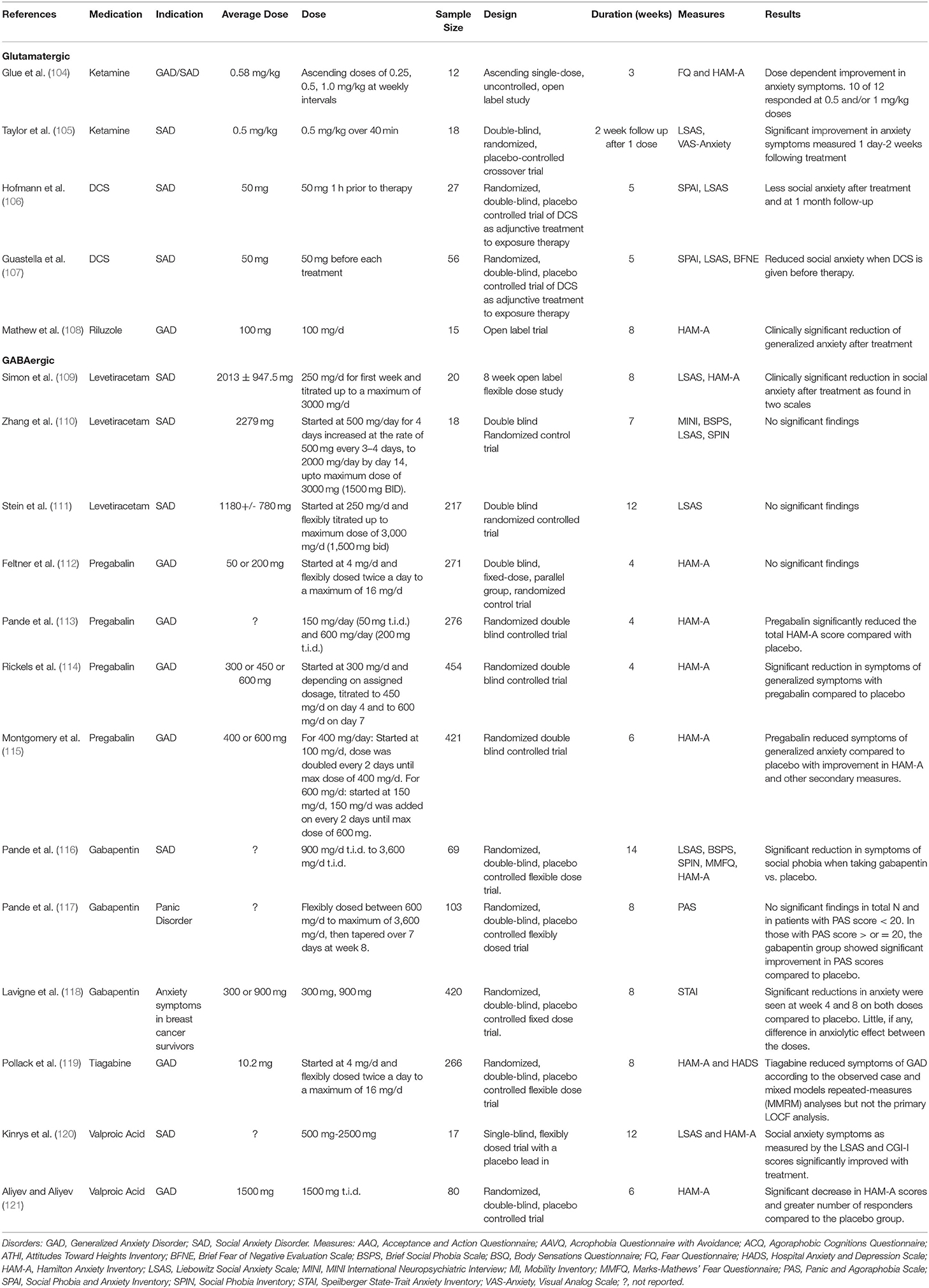
Table 3. Summary of glutamate psychopharmacology trials for anxiety disorders.
Originally used as a rapid-acting intravenous anesthetic, in the last 20 years ketamine has been the subject of much research for its potential use in a variety of psychiatric disorders. In contrast to the delayed therapeutic effects of antidepressants acting on serotonin receptors ketamine has a fast acting mechanism of action the specifics of which remain largely debated. Ketamine acts as a noncompetitive antagonist of NMDA receptors, thus inhibiting downstream neuronal activation pathways (122), although more complex glutamatergic actions have been recently identified (123). Subsequent AMPA receptor activation is suggested to play a role since after 24 h ketamine has been metabolized to a point where direct NMDA antagonism could no longer be an ongoing mechanism of action and non-ketamine NMDA receptor antagonists do not exhibit robust ketamine like antidepressant effects (124).
Ketamine's efficacy in depression and mood disorders was first demonstrated in 2000 (125), and has subsequently been demonstrated in multiple clinical trials. A recent meta-analysis that included 9 RCTs comparing one-time infusion of ketamine to placebo showed a rapid and significant reduction in depressive symptoms (126). Ketamine has also shown efficacy in PTSD, with a midazolam-controlled trial showing that a single dose of 0.5 mg/kg of ketamine improved symptoms as measured by Impact of Event Scale-Revised scores at 24 h post-infusion; (127, 128), and in OCD, with an RCT showing improvement in symptoms (as measured by Y-BOCS scores) after a single dose of 0.5 mg/g of ketamine compared to placebo (129).
More recently, ketamine has also begun to be studied in anxiety disorders (130). One small open-label trial looking at the efficacy of ketamine in treatment-refractory generalized anxiety and/or social anxiety disorders (104) found that ten out of twelve patients responded (i.e., achieved ≥50% reduction in HAM-A scores and/or Fear Questionnaire scores) after three ascending doses of once weekly subcutaneous ketamine (0.25, 0.5, and 1 mg/kg); a subsequent trial from the same group further supported the safety and efficacy of ketamine (1 mg/kg) for this same population with once or twice weekly subcutaneous administrations over a period of three months (131). Furthermore, a double-blind randomized controlled crossover study enrolled 18 patients with SAD and found intravenous ketamine (0.5 mg/kg) resulted in significantly greater response rate (as measured by a reduction in Liebowitz Social Anxiety Scale scores of at least 35%) in the first two weeks post-infusion compared to placebo (105). Midazolam-controlled, parallel-group trials, examining the efficacy of repeated dosing of ketamine (e.g., twice-weekly for 2–4 weeks) are needed to examine the sustained effects of ketamine on both anxiety disorders and OCD.
Memantine is commonly used in Alzheimer's disease to improve cognitive symptoms (132), and similar to ketamine, it acts as an antagonist of ionotropic NMDA receptors. The differences in therapeutic profile between ketamine and memantine may be partially explained by their differing affinity for NMDA receptor subunits, as well as differences in subunit expression among synaptic and extra-synaptic receptors, and throughout the brain (133).
Given the hypothesized role of glutamatergic hyperactivity in the expression of obsessive and anxiety symptoms, one study compared the efficacy of memantine for OCD vs. GAD via a 12 week uncontrolled trial of 10 mg BID; the OCD group experienced a mean 40.6% reduction in YBOCS scores, while the GAD group only saw a reduction of 22.4% in HARS scores, suggesting potentially that memantine may have larger treatment benefits in OCD vs. GAD (134). A recent meta-analysis including 8 trials involving 125 participants with OCD patients found that 20 mg daily memantine augmentation to SRI pharmacotherapy for OCD suggested that after at least 8 weeks memantine augmentation was associated with a 3.6-fold greater likelihood of treatment response than placebo and an average 11-point improvement in Y-BOCS score (135). However, much of the promising data regarding memantine in OCD involves uncontrolled trials or small, single-site trials that are highly prone to bias. Larger, adequately powered, multi-site trials are needed to truly measure the treatment effect of memantine in anxiety and OCD.
DCS is an analog of D-alanine that inhibits enzymes involved in bacterial peptidoglycan formation, and it is approved for use in resistant tuberculosis and select urinary tract infections (136). However, unlike alanine –which demonstrates full agonistic effects on the glycine-binding site of NMDA receptors–, DCS acts as a partial agonist, demonstrating in vivo agonistic effects in low concentrations and antagonistic effects in higher concentrations; this may be partly due to its high affinity for NMDA receptor subtype NR1/NR2C (where it exerts agonistic action), and low affinity for subtypes NR1/NR2A and NR1/NR2B (where it is antagonistic) (137).
Studies looking into the use of DCS in anxiety disorders have mostly been focused on its use as an augmentative agent to behavioral therapy, to enhance fear extinction. One meta-analysis from 2017 which included 21 studies found a statistically significant advantage of DCS over placebo in the augmentation of exposure-based CBT for anxiety-related disorders (including anxiety, obsessive-compulsive, and posttraumatic stress disorders), but the overall effect size was small (138). Another meta-analysis from the same year included 23 studies and found the overall effect of DCS to be almost non-existent among patients with anxiety and obsessive-compulsive disorder; slightly larger effect sizes were found among social anxious patients (139). A more recent study from 2019 recruited 81 patients with social anxiety disorder, who underwent a fear conditioning and extinction paradigm measuring skin conductance response to conditioned stimuli and shock expectancy ratings; DCS was not found to have a moderating influence on study outcome measures and there was no effect on the retention of extinction learning under the study paradigm (140). In totality, these data suggest that DCS at best has minimal benefit as an augmentation agent to behavioral therapy in anxiety disorders.
NAC has classically been used in the context of acetaminophen toxicity to reduce hepatic injury (141), as well as a mucolytic agent for inflammatory pulmonary conditions (142). More recently, evidence has emerged supporting the potential use of NAC in a variety of neuropsychiatric disorders, including addiction and substance use disorders (143), neurodegenerative diseases such as spinocerebellar degeneration, Parkinson's disease, and others (144), schizophrenia (145), stroke (146), traumatic brain injury (147), obsessive-compulsive and related disorders (148), and depressive disorders (149), among other pathologies (150). NAC is a precursor to glutathione, a major antioxidant in the body, and its therapeutic properties are thought to be due its role in restoring cellular and mitochondrial redox imbalance, and reducing subsequent inflammation often seen with in neuropsychiatric conditions (150, 151). Additionally, NAC causes dose-dependent increases in extracellular glutamate via its action on cystine-glutamate antiporters found on glial cells; free glutamate then activates mGluR2/3 on the presynaptic nerve terminals and ultimately reduces synaptic glutamate release via negative feedback (150, 151).
Despite some promising initial data suggesting possible benefits of NAC in anxiety-related disorders such as OCD and depressive disorders, research examining its efficacy in anxiety disorders is quite sparse. Clinical trials specifically looking at the effects of NAC on anxiety are lacking. One case report showed improvement on generalized anxiety disorder and social phobia symptoms after adjunctive NAC (600 mg BID) was added to sertraline monotherapy, with a further improvement seen after increasing to 1200 mg (152). Another 16-week trial evaluating the efficacy of 3,000 mg NAC augmentation in forty OCD patients found a significant (p = 0.02) decrease in the Beck Anxiety Inventory (BAI) compared to placebo (153). However, when anxiety symptoms were analyzed in placebo-controlled trials looking at major depressive disorder (154) and trichotillomania (155), no significant differences were found in anxiety scores as measured by Hamilton Anxiety Rating Scale (HAM-A). Evidence examining the effects of NAC is both sparse and mixed at best. Larger, multi-site trials with anxiety as the primary measure are needed to estimate the effects of NAC for the treatment of anxiety disorders.
Riluzole was originally developed as an anticonvulsant, though it only has FDA approval for amyotrophic lateral sclerosis. It likely acts upon the glutamatergic system via multiple mechanisms including blockade of presynaptic voltage-gated sodium and calcium channels to block glutamate release, potentiating glial reuptake of glutamate, and modulating trophic and toxic effects of glutamate (156).
The potential use of riluzole has been studied for a variety of psychiatric disorders with mixed success (157). In the setting of treatment-refractory OCD, for example, one RCT found modest improvement in Y-BOCS scores, though results were not statistically significant (158). For generalized anxiety disorder, an eight-week open trial was conducted on the effects of 100 mg/d of riluzole on HAM-A scores, with 80% of patients responding to the treatment (reduction in scores by 50% or more); further, 53% of these patients met remission criteria, with HAM-A scores becoming equal to or <7 (108). There are currently ongoing, mutli-site trials examining the effects of troriluzole, a riluzole prodrug, in the treatment of generalized anxiety disorder (NCT03829241) and OCD (NCT03299166).
In contrast to glutamate, GABA is the primary inhibitory neurotransmitter of the nervous system. Anxiety is thought to stem from an imbalance between the excitatory and inhibitory systems resulting in dysregulation (159). Thus, medications which increase GABA activity will counterbalance the excitatory action of glutamate resulting in anxiolysis (160). Several medications that modulate ionotropic (GABAA) and metabotropic (GABAB) receptors are widely used in the treatment of disorders such as anxiety, epilepsy, insomnia, spasticity (161). Benzodiazepines act on GABAA receptors which are highly complex due to their structural heterogeneity, their many connected binding sites and the numerous chemically distinct ligands that can bind to them (161). Thus, there is a growing interest in the development and application of subtype-selective drugs that will achieve specific therapeutic benefits without undesirable side effects.
Levetiracetam is an antiepileptic that acts by binding to the synaptic vesicle protein SVA2, interfering with neurotransmitter exocytosis, and via inhibition of N-type calcium channels; it also increases tissue concentrations of GABA (162). Studies on the efficacy of levetiracetam in psychiatric disorders are relatively limited. There is some evidence that suggests potential benefit in the setting of treatment-refractory PTSD (120), and a small case series involving patients with alcohol dependence found improvement of anxiety symptoms with levetiracetam use (163). An open-label, flexible-dose study of levetiracetam (starting at 250 mg per day, up to 1500 mg twice per day) included 20 patients with SAD, and found a clinically significant 20.5-point decrease in Liebowitz Social Anxiety Scale (LSAS) scores in the intent-to-treat, last-observation-carried forward analysis; only 13 patients remained at the 8-week endpoint, with attrition attributed to side effects (n = 3), lack of efficacy (n = 1), and loss of follow-up (n = 3) (109). In a 7-week double-blind placebo controlled study involving 18 patients, 44% of the levetiracetam group showed response based on BSPS scores compared to 14% improvement in the placebo group levetiracetam, though results were not statistically significant using last-observation-carried-forward analysis; higher powered RCTs need to be conducted to gain a more decisive understanding of the utility of levetiracetam as a potential treatment for SAD (110). For generalized anxiety disorder, one 12-week double-blind randomized controlled trial of levetiracetam (starting dose 250 mg per day, flexibly titrated up to 1500 mg twice per day) involving 217 adult outpatients failed to demonstrate differences compared to placebo, with mean reductions in LSAS scores and response rates using levetiracetam equaling 24.4 and 41.3% respectively, and placebo demonstrating 28.7 point reductions and 46.6% response rate (111).
Although gabapentin and pregabalin are chemically similar to GABA, they actually bind and inhibit central nervous system voltage-gated α2-δ calcium channels and do not act via GABA receptors; this mechanism is thought to contribute to antiepileptic, antinociceptive, and anxiolytic effects (164). Notably, pregabalin exhibits greater potency as an α2-δ ligand compared to gabapentin, and it is also absorbed more rapidly and has greater bioavailability across dosing ranges.
Pregabalin has consistently been seen to improve anxiety symptoms among patients with GAD and has been effective both as monotherapy and as adjunct therapy (164, 165), and a meta-analysis from 2016 including 8 studies found pregabalin to be significantly superior to placebo in reducing HAM-A scores among patients with GAD (166). One early double blinded trial evaluated the effect of pregabalin at doses of 150 and 600 mg, lorazepam at 6 mg, and placebo on patients with GAD over a ten-week period; the drop in HAM-A scores seen in both the pregabalin low dose (-9.2) and high dose (-10.3) groups were comparable to the lorazepam group (-12.0) and significantly higher than the placebo (113). Follow up randomized control trials have shown that pregabalin has higher compliance rates than benzodiazepines therefore implying better tolerance of the drug. In addition, its efficacy was similar to the two to three times daily doses of benzodiazepines (112, 114).
Another double-blind trial compared the effects of pregabalin at 400 mg/d and 600 mg/d, the SNRI venlafaxine at 75 mg/d and a placebo on GAD. Both pregabalin groups showed an equivalent reduction in HAM-A scores with respect to the venlafaxine group. Pregabalin however had a faster onset of action of within 1 week (compared to the 2 weeks of the venlafaxine) and a much higher compliance rate (94–87% vs. 80% for venlafaxine). This implies that pregabalin may be preferable to SNRIs in patients with GAD (115). As an adjunctive therapy in patients with partial response to SSRIs or SNRIs, one randomized, double-blind, placebo-controlled trial showed superior efficacy of pregabalin (150–600 mg/d) compared to placebo after 8 weeks of combination treatment as demonstrated by statistically greater reduction in HAM-A scores (-7.6 vs. 6.4) and response rates (47.5 vs. 35.2%) (167).
The evidence for the efficacy of gabapentin is less robust and clinical trials are limited; one study found improvement in anxiety among breast cancer survivors (118), another in social phobia symptoms (116), while a third trial found gabapentin to more beneficial than placebo only among more severely affected patients and not in milder anxiety (117).
Tiagabine is an anti-epileptic drug, that selectively inhibits GABA reuptake, thereby decreasing glutamatergic activity. Common adverse effects seen in trials include dizziness, asthenia, somnolence, accidental injury, infection, headache, nausea, and nervousness. These were usually mild to moderate in severity and did not need medical intervention (168).
The largest tiagabine anxiety trial was an eight-week randomized placebo-controlled trial, on 266 subjects with GAD. The tiagabine treatment of upto 16 mg/d was associated with significantly lower HAM- A scores compared to placebo (119). An open-label trial with 54 patients demonstrated improvement in the Lieboweitz social anxiety scale, social phobia inventory and Sheehan Disability Scale after being administered 4-16 mg/d for 12 weeks (169).
Other trials on the use of tiagabine in anxiety disorders have been largely underpowered due to small sample sizes or lack of placebo groups or blinding. One open label trial on 28 subjects with panic disorder found statistically but not clinically significant reductions in panic disorder scales and HAM-A when they were administered 2-20 mg/day for 10 weeks (170). A later four-week RCT was conducted on 19 panic disorder patients in which the tiagabine group was started at 5 mg/d which was increased to a maximum of 30 mg/d depending on adverse effects. They found no difference between the tiagabine group and the placebo group in the clinical ratings. However, they also conducted panic challenges with CCK-4 in which the tiagabine treated subjects showed decreased sensitivity to experimentally induced panic (171). These mixed results may be disentangled by running larger studies that are better powered to pick up any possible effect. Another eight week, open-label study investigated the utility of tiagabine as an augmentation therapy for patients with anxiety that remained symptomatic despite appropriate anxiety drug trials. Tiagabine was started at 4 mg/d and flexibly increased to a maximum of 20 mg/d to optimize efficacy and tolerability, with a resulting mean dose of 13 mg/d. A response was reflected in the HAM-A scores were seen in thirteen (76%) of the subjects. This implies that patients who do not respond to the traditional first and second line anxiety may respond to tiagabine augmentation. However, this study only had eighteen participants and more adequate RCTs need to be conducted to confirm the results (172).
Topiramate is an anti-epileptic that is also FDA approved as a migraine prophylactic. It has several mechanisms through which it alters neuronal function. It regulates Na+ and Ca2+ channel opening, potentiates GABA, and acts as an antagonist on AMPA and kainate receptors (173).
The results of a sixteen-week, open label trial with 23 subjects suggests its efficacy in the treatment of SAD. A flexible dose ranging from 25 to 75 mg/d was given to the active group. Seventy five percent of patients in the active group showed reduction in Leibowitz Social Anxiety Scale scores (174). Further, double-blind placebo-controlled trials are needed to determine the true efficacy of this agent.
Valproic acid is believed to exert its antiepileptic effects by reducing neuronal activity via inhibition of voltage-gated ion channels, as well as by increasing concentrations of GABA levels by reducing its degradation (175).
Its effectiveness within anxiety disorders has not been thoroughly explored, and clinical trials are lacking. A six week RCT with eighty GAD patients found significantly more treatment responders (i.e., at least 50% improvement in Hamilton Anxiety Scale scores) and significantly greater symptom improvement (p < 0.001) among the group receiving 1500 mg/day of valproic acid compared to placebo (121). Benefits have also been reported in social anxiety disorder according to a 12 week open label flexible dose (500–2500 mg/day) study which showed significantly greater improvement in the Liebowitz Social Anxiety Scale (LSAS) in the treatment group (120). Another similar flexible dose open label study was conducted on ten participants with panic disorder. After a week-long placebo wash out period patients were given an ascending dose of valproic acid starting at 500 mg to a maximum of 2250 mg resulting in a significant improvement (p < 0.05) in the panic factor of the Symptom Checklist (SCL-90) (176). However, both these studies had a small sample size and a placebo lead in instead of a placebo group, thus, higher powered placebo-controlled trials would be needed to confirm these findings.
Despite the presence of effective pharmacological and behavioral treatments, anxiety disorders remain a significant source of morbidity for many patients across the lifespan. Currently evidence-based pharmacological agents available to treat anxiety disorders include serotinergic antidepressants and benzodiazepines. Serotonergic antidepressants have a delayed onset of action, are only partially effective, leaving a substantial portion of patients unimproved or with significant residual symptoms after treatment, and have side-effects that somewhat limit adherence and tolerability. Benzodiazepines, although fast-acting and effective in treating anxiety symptoms, are not ideal because of the potential for patients to build tolerance or develop dependence with regular long-term use and also possible side-effects. Psychotherapy for anxiety disorder, although also quite effective, presents significant obstacles in terms of dissemination, access and acceptability to many patients. Thus, novel treatments for anxiety disorders are needed.
Drugs altering the glutamate and GABA systems have been increasingly studied as potential novel treatments for anxiety disorders. Evidence that abnormalities in the glutamate system are inherent to the pathophysiology of anxiety and other related-disorders strengths the rationale for studying these agents in clinical trials. Although breakthroughs have emerged regarding the benefits of glutamatergic agents such as ketamine and esketamine in the treatment of comorbid disorders such as Major Depression and possibly PTSD, strong evidence regarding the efficacy of glutamate modulating agents for anxiety disorders is currently lacking. Evidence regarding the efficacy of glutamatergic agents for anxiety disorder have largely been confined to uncontrolled and single-site studies. Large, adequately-powered, multi-site studies examining the efficacy of agents affecting glutamatergic neurotransmission in anxiety disorders are lacking and are needed.
The order of the author list reflects the amount of effort per person with MB and JD as senior authors.
MB receives research support from Therapix Biosciences, Neurocrine Biosciences, Janssen Pharmacueticals, Emalex Pharmaceuticals and Biohaven Pharmaceuticals. MB gratefully acknowledges additional research support from NIH, NARSAD, and the Patterson Foundation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month dSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry. (2005) 62:617–27. doi: 10.1001/archpsyc.62.6.617
3. Nepon J, Belik SL, Bolton J, Sareen J. The relationship between anxiety disorders and suicide attempts: findings from the national epidemiologic survey on alcohol and related conditions. Depress Anxiety. (2010) 27:791–8. doi: 10.1002/da.20674
4. Schaffer A, McIntosh D, Goldstein BI, Rector NA, McIntyre RS, Beaulieu S, et al. The CANMAT task force recommendations for the management of patients with mood disorders and comorbid anxiety disorders. Ann Clin Psychiatry. (2012) 24:6–22.
5. Marciniak M, Lage MJ, Landbloom RP, Dunayevich E, Bowman L. Medical and productivity costs of anxiety disorders: case control study. Depress Anxiety. (2004) 19:112–20. doi: 10.1002/da.10131
6. Erickson SR, Guthrie S, VanEtten-Lee M, Himle J, Hoffman J, Santos SF, et al. Severity of anxiety and work-related outcomes of patients with anxiety disorders. Depress Anxiety. (2009) 26:1165–71. doi: 10.1002/da.20624
7. Bernstein GA, Shaw K. Practice parameters for the assessment and treatment of anxiety disorders. J Am Acad Child Adolesc Psychiatry. (1993) 32:1089–98. doi: 10.1097/00004583-199309000-00044
8. National Collaborating Centre for Chronic Conditions. Tuberculosis: Clinical Diagnosis and Management of Tuberculosis, and Measures for its Prevention and Control. London: Royal College of Physicians (2006).
9. Stein MB, Marcia Goin CK, Pollack MH, Roy-Byrne P, Sareen J, Simon NM, et al. Practice Guideline For The Treatment Of Patients With Panic Disorder Second Edition Work Group On Panic Disorder Steering Committee On Practice Guidelines. (2010). Retrieved from: http://www.psychiatryonline.com/pracGuide/pracGuideTopic_9.aspx.
10. Bandelow B, Reitt M, Röver C, Michaelis S, Görlich Y, Wedekind D. Efficacy of treatments for anxiety disorders: a meta-analysis. Int Clin Psychopharmacol. (2015) 30:183–92. doi: 10.1097/YIC.0000000000000078
11. Taylor JH, Lebowitz ER, Jakubovski E, Coughlin CG, Silverman WK, Bloch MH. Monotherapy insufficient in severe anxiety? Predictors and moderators in the child/adolescent anxiety multimodal study. J Clin Child Adolesc Psychol. (2018) 47:266–81. doi: 10.1080/15374416.2017.1371028
12. Loerinc AG, Meuret AE, Twohig MP, Rosenfield D, Bluett EJ, Craske MG. (2015) Response rates for cBT for anxiety disorders: need for standardized criteria. Clinical Psychology Review. Elsevier Inc. doi: 10.1016/j.cpr.2015.08.004
13. Ringle VA, Read KL, Edmunds JM, Brodman DM, Kendall PC, Barg F, et al. Barriers to and facilitators in the implementation of cognitive-behavioral therapy for youth anxiety in the community. Psychiatric Services. (2015) 66:938–45. doi: 10.1176/appi.ps.201400134
14. Jakubovski E, Johnson JA, Nasir M, Müller-Vahl K, Bloch MH. Systematic review and meta-analysis: dose-response curve of sSRIs and sNRIs in anxiety disorders. Depress Anxiety. (2019) 36:198–212. doi: 10.1002/da.22854
15. Quagliato LA, Freire RC, Nardi AE. Risks and benefits of medications for panic disorder: a comparison of sSRIs and benzodiazepines. Expert Opinion on Drug Safety. (2018) 17:315–24. doi: 10.1080/14740338.2018.1429403
16. Segraves RT, Balon R. Antidepressant-induced sexual dysfunction in men. Pharm Biochem Behav. (2014) 121:132–7. doi: 10.1016/j.pbb.2013.11.003
17. Lorenz T, Rullo J, Faubion S. Antidepressant-Induced female sexual dysfunction. Mayo Clinic Proc. (2016) 91:1280–6. doi: 10.1016/j.mayocp.2016.04.033
18. Clayton AH, El Haddad S, Iluonakhamhe J.-P., Ponce Martinez C, Schuck AE. Sexual dysfunction associated with major depressive disorder and antidepressant treatment. Expert Opin Drug Safety. (2014) 13:1361–74. doi: 10.1517/14740338.2014.951324
19. Kronstein PD, Ishida E, Khin NA, Chang E, Hung HMJ, Temple RJ, et al. Summary of findings from the fDA regulatory science forum on measuring sexual dysfunction in depression trials. J Clin Psychiatry. (2015) 76:1050–9. doi: 10.4088/JCP.14r09699
20. Asnis GM, Hameedi FA, Goddard AW, Potkin SG, Black D, Jameel M, et al. Fluvoxamine in the treatment of panic disorder: a multi-center, double-blind, placebo-controlled study in outpatients. Psychiatry Res. (2001) 103:1–4. doi: 10.1016/S0165-1781(01)00265-7
21. Ballenger JC, Wheadon DE, Steiner M, Bushnell W, Gergel IP. Double-blind, fixed-dose, placebo-controlled study of paroxetine in the treatment of panic disorder. Am J Psychiatry. (1998) 155:36–42. doi: 10.1176/ajp.155.1.36
22. Londborg PD, Wolkow R, Smith WT, DuBoff E, England D, Ferguson J, et al. Sertraline in the treatment of panic disorder. A multi-site, double- blind, placebo-controlled, fixed-dose investigation. Br J Psychiatry. (1998) 173:54–60. doi: 10.1192/bjp.173.1.54
23. Michelson D, Allgulander C, Dantendorfer K, Knezevic A, Maierhofer D, Micev V, et al. Efficacy of usual antidepressant dosing regimens of fluoxetine in panic disorder. Randomised, placebo-controlled trial. Br J Psychiatry. (2001) 179:514–8. doi: 10.1192/bjp.179.6.514
24. Murrough JW, Yaqubi S, Sayed S, Charney DS. Emerging drugs for the treatment of anxiety. Exp Opin Emerg Drugs. (2015) 20:393–406. doi: 10.1517/14728214.2015.1049996
25. Pohl RB, Wolkow RM, Clary CM. Sertraline in the treatment of panic disorder: a double-blind multicenter trial. Am J Psychiatry. (1998) 155:1189–95. doi: 10.1176/ajp.155.9.1189
26. Black K, Shea C, Dursun S, Kutcher S. Selective serotonin reuptake inhibitor discontinuation1 syndrome: proposed diagnostic criteria. J Psychiatry Neurosci. (2000) 25:255.
27. Nardi E, Machado A, Ferreira Almada S, Paes L, Cardoso Silva F, Jose Marques A, et al. Clonazepam for the treatment of panic disorder. Current Drug Targets. (2013) 14:353–64. doi: 10.2174/1389450111314030007
28. Nardi AE, Valença AM, Nascimento I, Lopes FL, Mezzasalma MA, Freire RC, et al. A three-year follow-up study of patients with the respiratory subtype of panic disorder after treatment with clonazepam. Psychiatry Res. (2005) 137:61–70. doi: 10.1016/j.psychres.2005.05.011
29. Rosenbaum JF. Attitudes toward benzodiazepines over the years. J Clin Psychiatry. (2005) 66(Suppl 2):4–8.
30. Millan MJ. N-methyl-D-aspartate receptor-coupled glycineB receptors in the pathogenesis and treatment of schizophrenia: a critical review. Curr Drug Targets. CNS Neurol Disord. (2002) 1:191–213. doi: 10.2174/1568007024606258
31. Moghaddam B. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem. (1993) 60:1650–7. doi: 10.1111/j.1471-4159.1993.tb13387.x
32. Schwendt M, Jezová D. Gene expression of two glutamate receptor subunits in response to repeated stress exposure in rat hippocampus. Cell Mol Neurobiol. (2000) 20:319–29. doi: 10.1023/a:1007062109386
33. Krystal JH, Mathew SJ, Dsouza DC, Garakani A, Gunduz-Bruce H, Charney DS. Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists. CNS Drugs. (2010) 24:669–93. doi: 10.2165/11533230-000000000-00000
34. Palucha A, Pilc A. Metabotropic glutamate receptor ligands as possible anxiolytic and antidepressant drugs. Pharmacol Ther. (2007) 115:116–47. doi: 10.1016/j.pharmthera.2007.04.007
35. Wierońska JM, Pilc A. Metabotropic glutamate receptors in the tripartite synapse as a target for new psychotropic drugs. Neurochem Int. (2009) 55:85–97. doi: 10.1016/j.neuint.2009.02.019
36. Shepherd GM. The Synaptic Organization of the Brain. New Haven, CT: Oxford University Press. (2004). doi: 10.1093/acprof:oso/9780195159561.001.1
37. Barkus C, McHugh SB, Sprengel R, Seeburg PH, Rawlins JNP, Bannerman DM. Hippocampal NMDA receptors and anxiety: At the interface between cognition and emotion. Eur J Pharm. (2010) 626:49–56. doi: 10.1016/j.ejphar.2009.10.014
38. Kew JNC, Kemp JA. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology. (2005) 179:4–29. doi: 10.1007/s00213-005-2200-z
39. Amiel JM, Mathew SJ. Glutamate and anxiety disorders. Curr Psychiatry Rep. (2007) 9:278–83. doi: 10.1007/s11920-007-0033-7
40. Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, et al. The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci. (2001) 21:22. doi: 10.1523/jneurosci.21-22-j0001.2001
41. Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Dis. (2005) 4:131–44. doi: 10.1038/nrd1630
42. Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. (1999) 51:7–62.
43. Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal cA1 nMDA receptor-dependent synaptic plasticity in spatial memory. Cell. (1996) 87:1327–38. doi: 10.1016/S0092-8674(00)81827-9
44. Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharm Rev. (2010) 62:405–96. doi: 10.1124/pr.109.002451
45. Mayer ML. Crystal structures of the gluR5 and gluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron. (2005) 45:539–52. doi: 10.1016/j.neuron.2005.01.031
46. Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. (2010) 11:682–96. doi: 10.1038/nrn2911
47. Haroon E, Miller AH, Sanacora G. Inflammation, glutamate, and glia: a Trio of trouble in mood disorders. Neuropsychopharmacology. (2017) 42:193–215. doi: 10.1038/npp.2016.199
48. Davidson JR, Krishnan KR, Charles HC, Boyko O, Potts NL, Ford SM, et al. Magnetic resonance spectroscopy in social phobia: preliminary findings. J Clin Psychiatry. (1993). 54:19–25.
49. De Bellis MD, Matcheri S, Keshavan S, Spencer BSJ, Hall BA. N-Acetylaspartate concentration in the anterior cingulate of maltreated children and adolescents with pTSD. Am J Psychiatry. (2000) 157:1175–7.
50. Freeman TW, Cardwell D, Karson CN, Komoroski RA. In vivo proton magnetic resonance spectroscopy of the medial temporal lobes of subjects with combat-related posttraumatic stress disorder. Magn Reson Med. (1998) 40:66–71. doi: 10.1002/mrm.1910400110
51. Mathew SJ, Mao X, Coplan JD, Smith ELP, Sackeim HA, Gorman JM, et al. Dorsolateral prefrontal cortical pathology in generalized anxiety disorder: a proton magnetic resonance spectroscopic imaging study. Am J Psychiatry. (2004) 161:1119–21. doi: 10.1176/appi.ajp.161.6.1119
52. Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AMA. N-Acetylaspartate in the cNS: From Neurodiagnostics to Neurobiology. Progress in Neurobiology. Bethesda, MD: NIH Public Access. (2007). doi: 10.1016/j.pneurobio.2006.12.003
53. Whiteside SP, Port JD, Deacon BJ, Abramowitz JS. A magnetic resonance spectroscopy investigation of obsessive-compulsive disorder and anxiety. Psychiatry Res. (2006) 146:137–47. doi: 10.1016/j.pscychresns.2005.12.006
54. Bédard MJ, Chantal S. Brain magnetic resonance spectroscopy in obsessive-compulsive disorder: the importance of considering subclinical symptoms of anxiety and depression. Psychiatry Res. (2011) 192:45–54. doi: 10.1016/j.pscychresns.2010.10.008
55. Howells FM, Hattingh CJ, Syal S, Breet E, Stein DJ, Lochner C. 1H-magnetic resonance spectroscopy in social anxiety disorder. Prog Neuro-Psychopharmacol Biol Psychiatry. (2015) 58:97–104. doi: 10.1016/j.pnpbp.2014.12.008
56. Ramadan S, Lin A, Stanwell P. Glutamate and glutamine: a review of in vivo mRS in the human brain. NMR Biomed. (2013). doi: 10.1002/nbm.3045
57. Grachev ID, Vania Apkarian. A. Chemical mapping of anxiety in the brain of healthy humans: an in vivo 1H-MRS study on the effects of sex, age, brain region. Hum Brain Mapp. (2000) 11:261–72. doi: 10.1002/1097-0193(200012)11:4<261::AID-HBM30>3.0.CO;2-6
58. Phan K, Luan CA, Fitzgerald DA, Cortese BM, Seraji-Bozorgzad N, Tancer ME, Moore GJ. Anterior cingulate neurochemistry in social anxiety disorder: 1H-MRS at 4 tesla | ovid. Neur Rep. (2005) 16:183–6. doi: 10.1097/00001756-200502080-00024
59. Averill LA, Purohit P, Averill CL, Boesl MA, Krystal JH, Abdallah CG. Glutamate dysregulation and glutamatergic therapeutics for PTSD: Evidence from human studies. Neurosci Lett. (2017) 649:147–55. doi: 10.1016/j.neulet.2016.11.064
60. Chakrabarty K, Bhattacharyya S, Christopher R, Khanna S. Glutamatergic dysfunction in oCD. Neuropsychopharmacology. (2005) 30:1735–40. doi: 10.1038/sj.npp.1300733
61. Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel,improved therapeutics for mood disorders. Nat Rev Drug Dis. (2008) 7:426–37. doi: 10.1038/nrd2462
62. Frank D, Kuts R, Tsenter P, Gruenbaum BF, Grinshpun Y, Zvenigorodsky V, et al. The effect of pyruvate on the development and progression of post-stroke depression: a new therapeutic approach. Neuropharmacology. (2019) 155:173–84. doi: 10.1016/j.neuropharm.2019.05.035
63. Lee PH, Perlis RH, Jung JY, Byrne EM, Rueckert E, Siburian R, et al. Multi-locus genome-wide association analysis supports the role of glutamatergic synaptic transmission in the etiology of major depressive disorder. Transl Psychiatry. (2012) 2:95. doi: 10.1038/tp.2012.95
64. Smoller JW. The genetics of stress-Related disorders: pTSD, depression, anxiety disorders. Neuropsychopharmacology. (2016) 41:297–319. doi: 10.1038/npp.2015.266
65. Bhattacharyya S, Khanna S, Chakrabarty K, Mahadevan A, Christopher R, Shankar SK. Anti-brain autoantibodies and altered excitatory neurotransmitters in obsessive-compulsive disorder. Neuropsychopharmacology. (2009) 34:2489–96. doi: 10.1038/npp.2009.77
66. Bartha R, Stein MB, Williamson PC, Drost DJ, Neufeld RWJ, Carr TJ, et al. A short echo 1H spectroscopy and volumetric mRI study of the corpus striatum in patients with obsessive-compulsive disorder and comparison subjects. Am J Psychiatry. (1998) 155:1584–91. doi: 10.1176/ajp.155.11.1584
67. Rosenberg DR, MacMaster FP, Keshavan MS, Fitzgerald KD, Stewart CM, Moore GJ. Decrease in caudate glutamatergic concentrations in pediatric obsessive-compulsive disorder patients taking paroxetine. J Am Acad Child Adoles Psychiatry. (2000) 39:1096–103. doi: 10.1097/00004583-200009000-00008
68. Arnold PD, Sicard T, Burroughs E, Richter MA, Kennedy JL. Glutamate transporter gene sLC1A1 associated with obsessive-compulsive disorder. Arch Gen Psychiatry. (2006) 63:769–76. doi: 10.1001/archpsyc.63.7.769
69. Dickel DE, Veenstra-VanderWeele J, Cox NJ, Wu X, Fischer DJ, Van Etten-Lee M, et al. Association testing of the positional and functional candidate gene sLC1A1/EAAC1 in early-onset obsessive-compulsive disorder. Arch Gen Psychiatry. (2006) 63:778–85. doi: 10.1001/archpsyc.63.7.778
70. Kwon JS, Joo YH, Nam HJ, Lim M, Cho EY, Jung MH, et al. Association of the glutamate transporter gene sLC1A1 with atypical antipsychotics-induced obsessive-compulsive symptoms. Arch Gen Psychiatry. (2009) 66:1233–41. doi: 10.1001/archgenpsychiatry.2009.155
71. Liang KY, Wang Y, Yin YS, Grados M, Fyer AJ, Rauch S, et al. Evidence for potential relationship between sLC1A1 and a putative genetic linkage region on chromosome 14q to obsessive-compulsive disorder with compulsive hoarding. Am J Med Gen. (2008) 147:1000–2. doi: 10.1002/ajmg.b.30713
72. Shugart YY, Wang Y, Samuels JF, Grados MA, Greenberg BD, Knowles JA, et al. A family-based association study of the glutamate transporter gene sLC1A1 in obsessive-compulsive disorder in 378 families. Am J Med Gen. (2009) 150:886–92. doi: 10.1002/ajmg.b.30914
73. Stewart SE, Fagerness JA, Platko J, Smoller JW, Scharf JM, Illmann C, et al. Association of the sLC1A1 glutamate transporter gene and obsessive-compulsive disorder. Am J Med Gen. (2007) 144:1027–33. doi: 10.1002/ajmg.b.30533
74. Wang Y, Adamczyk A, Shugart YY, Samuels JF, Grados MA, Greenberg BD, et al. A screen of sLC1A1 for oCD-related alleles. Am J Med Gen. (2010) 153:675–9. doi: 10.1002/ajmg.b.31001
75. Wendland JR, Moya PR, Timpano KR, Anavitarte AP, Kruse MR, Wheaton MG, et al. A haplotype containing quantitative trait loci for sLC1A1 gene expression and its association with obsessive-compulsive disorder. Arch Gen Psychiatry. (2009) 66:408–16. doi: 10.1001/archgenpsychiatry.2009.6
76. Yang ZY, Quan H, Peng ZL, Zhong Y, Tan ZJ, Gong QY. Proton magnetic resonance spectroscopy revealed differences in the glutamate + glutamine/creatine ratio of the anterior cingulate cortex between healthy and pediatric post-traumatic stress disorder patients diagnosed after 2008 wenchuan earthquake. Psychiatry Clin Neurosci. (2015) 69:782–90. doi: 10.1111/pcn.12332
77. Bergink V, Van Megen HJGM, Westenberg HGM. Glutamate and anxiety. Eur Neuropsychopharmacol. (2004) 14:175–83. doi: 10.1016/S0924-977X(03)00100-7
78. Harvey BH, McEwen BS, Stein DJ. Neurobiology of antidepressant withdrawal: Implications for the longitudinal outcome of depression. Biol Psychiatry. (2003) 54:1105–17. doi: 10.1016/S0006-3223(03)00528-6
79. Moghaddam B. Stress activation of glutamate neurotransmission in the prefrontal cortex: implications for dopamine-associated psychiatric disorders. Biol Psychiatry. (2002) 51:775–87. doi: 10.1016/S0006-3223(01)01362-2
80. Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. (2012). doi: 10.1038/nrn3138
81. Myers KM, Carlezon WA, Davis M. Glutamate receptors in extinction and extinction-based therapies for psychiatric illness. Neuropsychopharmacol. (2011) 36:274–93. doi: 10.1038/npp.2010.88
82. Harvey BH, Shahid M. Metabotropic and ionotropic glutamate receptors as neurobiological targets in anxiety and stress-related disorders: focus on pharmacology and preclinical translational models. Pharmacol Biochem Behav. (2012). doi: 10.1016/j.pbb.2011.06.014
83. Kharade SM, Khetmar SS, Desai PS, Lokhande RS, Patil SS. Evaluation of anxiolytic activity of Carum copticum by using elevated plus maze and open field method. Int Res J Phar. (2011) 2:165–8.
84. Campos AC, Fogaça MV, Aguiar DC, Guimarães FS. Animal models of anxiety disorders and stress. Revista Brasileira de Psiquiatria. (2013) 35:S101–S11. doi: 10.1590/1516-4446-2013-1139
85. Kvetnansky R, Mikulaj L. Adrenal and urinary catecholamines in rats during adaptation to repeated immobilization stress. Endocrinology. (1970) 87:738–43. doi: 10.1210/endo-87-4-738
86. Fendt M, Koch M, Schnitzler HU. NMDA receptors in the pontine brainstem are necessary for fear potentiation of the startle response. Eur J Pharmacol. (1996) 318:1–6. doi: 10.1016/s0014-2999(96)00749-2
87. Campeau S, Miserendino MJD, Davis M. Intra-Amygdala infusion of the n-Methyl-D-Aspartate receptor antagonist aP5 blocks acquisition but not expression of fear-Potentiated startle to an auditory conditioned stimulus. Behav Neurosci. (1992) 106:569–74. doi: 10.1037/0735-7044.106.3.569
88. Becker A, Peters B, Schroeder H, Mann T, Huether G, Grecksch G. Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry. (2003) 27:687–700. doi: 10.1016/S0278-5846(03)00080-0
89. Pitsikas N, Georgiadou G, Delis F, Antoniou K. Effects of anesthetic ketamine on anxiety-Like behaviour in rats. Neurochem Res. (2019) 44:829–38. doi: 10.1007/s11064-018-02715-y
90. Viu E, Zapata A, Capdevila JL, Fossom LH, Skolnick P, Trullas R. Glycine site antagonists and partial agonists inhibit n-methyl-D-aspartate receptor-mediated [3H]arachidonic acid release in cerebellar granule cells. J Pharmacol Exp Ther. (1998) 285:527–32.
91. Ledgerwood L, Richardson R, Cranney J. Effects of d-cycloserine on extinction of conditioned freezing. Behav Neurosci. (2003) 117:341–9. doi: 10.1037/0735-7044.117.2.341
92. Ledgerwood L, Richardson R, Cranney J. D-cycloserine and the facilitation of extinction of conditioned fear: consequences for reinstatement. Behav Neurosci. (2004) 118:505–13. doi: 10.1037/0735-7044.118.3.505
93. Parnas AS, Weber M, Richardson R. Effects of multiple exposures to d-cycloserine on extinction of conditioned fear in rats. Neurobiol Learn Mem. (2005) 83:224–31. doi: 10.1016/j.nlm.2005.01.001
94. Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of d-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. (2002) 22:2343–51. doi: 10.1523/jneurosci.22-06-02343.2002
95. Fendt M. Expression and conditioned inhibition of fear-potentiated startle after stimulation and blockade of aMPA/Kainate and gABA(A) receptors in the dorsal periaqueductal gray. Brain Res. (2000) 880:1–10. doi: 10.1016/S0006-8993(00)02665-2
96. Khan S, Liberzon I. Topiramate attenuates exaggerated acoustic startle in an animal model of pTSD. Psychopharmacology. (2004) 172:225–9. doi: 10.1007/s00213-003-1634-4
97. Schulz B, Fendt M, Gasparini F, Lingenhöhl K, Kühn R, Koch M. The metabotropic glutamate receptor antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) blocks fear conditioning in rats. Neuropharmacology. (2001) 41:1–7. doi: 10.1016/S0028-3908(01)00036-3
98. Ballard TM, Woolley ML, Prinssen E, Huwyler J, Porter R, Spooren W. The effect of the mGlu5 receptor antagonist mPEP in rodent tests of anxiety and cognition: a comparison. Psychopharmacology. (2005) 179:218–29. doi: 10.1007/s00213-005-2211-9
99. Helton DR, Tizzano JP, Monn JA, Schoepp DD, Kallman MJ. Anxiolytic and side-effect profile of lY354740: a potent, highly selective, orally active agonist for group iI metabotropic glutamate receptors. J Pharmacol Exp Ther. (1998) 284:651–60.
100. Walker DL, Rattiner LM, Davis M. Group iI metabotropic glutamate receptors within the amygdala regulate fear as assessed with potentiated startle in rats. Behav Neurosci. (2002) 116:1075–83. doi: 10.1037//0735-7044.116.6.1075
101. Shekhar A, Keim SR. LY354740, a potent group iI metabotropic glutamate receptor agonist prevents lactate-induced panic-like response in panic-prone rats. Neuropharmacology. (2000) 39:1139–46. doi: 10.1016/S0028-3908(99)00215-4
102. Linden AM, Greene SJ, Bergeron M, Schoepp DD. Anxiolytic activity of the mGLU2/3 receptor agonist lY354740 on the elevated plus maze is associated with the suppression of stress-induced c-Fos in the hippocampus and increases in c-Fos induction in several other stress-sensitive brain regions. Neuropsychopharmacology. (2004) 29:502–13. doi: 10.1038/sj.npp.1300321
103. Mirza NR, Bright JL, Stanhope KJ, Wyatt A, Harrington NR. Lamotrigine has an anxiolytic-like profile in the rat conditioned emotional response test of anxiety: a potential role for sodium channels? Psychopharmacology. (2005) 180:159–68. doi: 10.1007/s00213-005-2146-1
104. Glue P, Medlicott NJ, Harland S, Neehoff S, Anderson-Fahey B, Le Nedelec M, et al. Ketamine's dose-related effects on anxiety symptoms in patients with treatment refractory anxiety disorders. J Psychopharmacol. (2017) 31:1302–5. doi: 10.1177/0269881117705089
105. Taylor JH, Landeros-Weisenberger A, Coughlin C, Mulqueen J, Johnson JA, Gabriel D, et al. Ketamine for social anxiety disorder: a randomized, placebo-controlled crossover trial. Neuropsychopharmacology. (2018) 43:325–33. doi: 10.1038/npp.2017.194
106. Hofmann SG, Meuret AE, Smits JAJ, Simon NM, Pollack MH, Eisenmenger K, et al. Augmentation of exposure therapy with d-cycloserine for social anxiety disorder. Arch Gen Psychiatry. (2006) 63:298–304. doi: 10.1001/archpsyc.63.3.298
107. Guastella AJ, Richardson R, Lovibond PF, Rapee RM, Gaston JE, Mitchell P, et al. A randomized controlled trial of d-Cycloserine enhancement of exposure therapy for social anxiety disorder. Biol Psychiatry. (2008) 63:544–9. doi: 10.1016/j.biopsych.2007.11.011
108. Mathew SJ, Amiel JM, Coplan JD, Fitterling HA, Sackeim HA, Gorman JM. Open-label trial of riluzole in generalized anxiety disorder. Am J Psychiatry. (2005) 162:2379–81. doi: 10.1176/appi.ajp.162.12.2379
109. Simon NM, Worthington JJ, Doyle AC, Hoge EA, Kinrys G, Fischmann D, et al. An open-label study of levetiracetam for the treatment of social anxiety disorder. J Clin Psychiatry. (2004) 65:1219–22. doi: 10.4088/jcp.v65n0909
110. Zhang W, Connor KM, Davidson JRT. Levetiracetam in social phobia: a placebo controlled pilot study. J Psychopharmacol. (2005) 19:551–3. doi: 10.1177/0269881105056526
111. Stein MB, Ravindran LN, Simon NM, Liebowitz MR, Khan A, Brawman-Mintzer O, et al. Levetiracetam in generalized social anxiety disorder: a double-blind, randomized controlled trial. J Clin Psychiatry. (2010) 71:627–31. doi: 10.4088/JCP.08m04949gre
112. Feltner DE, Crockatt JG, Dubovsky SJ, Cohn CK, Shrivastava RK, Targum SD, et al. A randomized, double-blind, placebo-controlled, fixed-dose, multicenter study of pregabalin in patients with generalized anxiety disorder. J Clin Psychopharmacol. (2003) 23:240–9. doi: 10.1097/01.jcp.0000084032.22282.ff
113. Pande AC, Crockatt JG, Feltner DE, Janney CA, Smith WT, Weisler R, et al. Pregabalin in generalized anxiety disorder: a placebo-controlled trial. Am J Psychiatry. (2003) 160:533–40. doi: 10.1176/appi.ajp.160.3.533
114. Rickels K, Pollack MH, Feltner DE, Lydiard RB, Zimbroff DL, Bielski RJ, et al. Pregabalin for treatment of generalized anxiety disorder: a 4-week, multicenter, double-blind, placebo-controlled trial of pregabalin and alprazolam. Arch Gen Psychiatry. (2005) 62:1022–30. doi: 10.1001/archpsyc.62.9.1022
115. Montgomery SA, Tobias K, Zornberg GL, Kasper S, Pande AC. Efficacy and safety of pregabalin in the treatment of generalized anxiety disorder: a 6-week, multicenter, randomized, double-blind, placebo-controlled comparison of pregabalin and venlafaxine. J Clin Psychiatry. (2006) 67:771–82. doi: 10.4088/JCP.v67n0511
116. Pande AC, Davidson JRT, Jefferson JW, Janney CA, Katzelnick DJ, Weisler RH, et al. Treatment of social phobia with gabapentin: a placebo-controlled study. J Clin Psychopharmacol. (1999) 19:341–8. doi: 10.1097/00004714-199908000-00010
117. Pande AC, Pollack MH, Crockatt J, Greiner M, Chouinard G, Lydiard RB, et al. Placebo-controlled study of gabapentin treatment of panic disorder. J Clin Psychopharmacol. (2000) 20:467–71. doi: 10.1097/00004714-200008000-00011
118. Lavigne JE, Heckler C, Mathews JL, Palesh O, Kirshner JJ, Lord R, et al. A randomized, controlled, double-blinded clinical trial of gabapentin 300 versus 900 mg versus placebo for anxiety symptoms in breast cancer survivors. Breast Cancer Res Treat. (2012) 136:479–86. doi: 10.1007/s10549-012-2251-x
119. Pollack MH, Roy-Byrne PP, Van Ameringen M, Snyder H, Brown C, Ondrasik J, et al. The selective gABA reuptake inhibitor tiagabine for the treatment of generalized anxiety disorder: results of a placebo-controlled study. J Clin Psychiatry. (2005) 66:1401–8. doi: 10.4088/JCP.v66n1109
120. Kinrys G, Pollack MH, Simon NM, Worthington JJ, Nardi AE, Versiani M. Valproic acid for the treatment of social anxiety disorder. Int Clin Psychopharmacol. (2003) 18:169–72. doi: 10.1097/01.yic.0000064261.66765.9f
121. Aliyev NA, Aliyev ZN. Valproate (depakine-chrono) in the acute treatment of outpatients with generalized anxiety disorder without psychiatric comorbidity: randomized, double-blind placebo-controlled study. Eur Psychiatry. (2008) 23:109–14. doi: 10.1016/j.eurpsy.2007.08.001
122. Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, et al. Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev. (2018) 70:621–60. doi: 10.1124/pr.117.015198
123. Lumsden EW, Troppoli TA, Myers SJ, Zanos P, Aracava Y, Kehr J, et al. Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block nMDA receptor function. Proc Natl Acad Sci USA. (2019) 116:5160–9. doi: 10.1073/pnas.1816071116
124. Yang C, Yang J, Luo A, Hashimoto K. Molecular and cellular mechanisms underlying the antidepressant effects of ketamine enantiomers and its metabolites. Transl Psychiatry. (2019) 9:1. doi: 10.1038/s41398-019-0624-1
125. Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. (2000) 47:351–4. doi: 10.1016/S0006-3223(99)00230-9
126. Kishimoto T, Chawla JM, Hagi K, Zarate CA, Kane JM, Bauer M, et al. Single-dose infusion ketamine and non-ketamine n-methyl-d-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychol Med. (2016) 46:1459–72. doi: 10.1017/S0033291716000064
127. Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry. (2014) 71:681–8. doi: 10.1001/jamapsychiatry.2014.62
128. Liriano F, Hatten C, Schwartz TL. Ketamine as Treatment for Post-Traumatic Stress Disorder: A Review. Drugs in Context. Syracuse, NY: Bioexcel Publishing LTD. doi: 10.7573/dic.212305
129. Rodriguez CI, Kegeles LS, Levinson A, Feng T, Marcus SM, Vermes D, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. (2013) 38:2475–83. doi: 10.1038/npp.2013.150
130. Banov MD, Young JR, Dunn T, Szabo ST. Efficacy and safety of ketamine in the management of anxiety and anxiety spectrum disorders: a review of the literature. CNS Spectrums. (2019) 25:331–42. doi: 10.1017/S1092852919001238
131. Glue P, Neehoff SM, Medlicott NJ, Gray A, Kibby G, McNaughton N. Safety and efficacy of maintenance ketamine treatment in patients with treatment-refractory generalised anxiety and social anxiety disorders. J Psychopharmacol. (2018) 32:663–7. doi: 10.1177/0269881118762073
132. Van Marum RJ. Update on the use of memantine in Alzheimer's disease. Neuropsych Dis Treat. (2009) 5:237. doi: 10.2147/ndt.s4048
133. Glasgow NG, Povysheva NV, Azofeifa AM, Johnson JW. Memantine and ketamine differentially alter nMDA receptor desensitization. J Neurosci. (2017) 37:9686–704. doi: 10.1523/JNEUROSCI.1173-17.2017
134. Feusner JD, Kerwin L, Saxena S, Bystritsky A. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: An open-label trial. Psychopharmacol Bull. (2009) 42:81–93.
135. Modarresi A, Chaibakhsh S, Koulaeinejad N, Koupaei SR. A systematic review and meta-analysis: memantine augmentation in moderate to severe obsessive-compulsive disorder. Psychiatry Res. (2019) 282:112602. doi: 10.1016/j.psychres.2019.112602
136. Sondack DL, Gainer FE, Wesselman HJ. GLC of cycloserine. J Pharm Sci. (1973) 62:1344–6. doi: 10.1002/jps.2600620826
137. Danysz W, Parsons CG. Glycine and n-methyl-D-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol Rev. (1998) 50:597–664.
138. Mataix-Cols D, De La Cruz LF, Monzani B, Rosenfield D, Andersson E, Pérez-Vigil A, et al. D-cycloserine augmentation of exposure-based cognitive behavior therapy for anxiety, obsessive-compulsive, and posttraumatic stress disorders a systematic review and meta-analysis of individual participant data. JAMA Psychiatry. (2017). doi: 10.1001/jamapsychiatry.2016.3955
139. Bürkner PC, Bittner N, Holling H, Buhlmann U. D-cycloserine augmentation of behavior therapy for anxiety and obsessive-compulsive disorders: a meta-analysis. PLoS ONE. (2017) 12:e0173660. doi: 10.1371/journal.pone.0173660
140. Hofmann SG, Papini S, Carpenter JK, Otto MW, Rosenfield D, Dutcher CD, et al. Effect of d-cycloserine on fear extinction training in adults with social anxiety disorder. PLoS ONE. (2019) 14:e0223729. doi: 10.1371/journal.pone.0223729
141. Flanagan RJ, Meredith TJ. Use of n-acetylcysteine in clinical toxicology. Am J Med. (1991) 91:131S−9S. doi: 10.1016/0002-9343(91)90296-a
142. Dekhuijzen PN, van Beurden WJ. The role for n-acetylcysteine in the management of cOPD. Int J Chron Obstruct Pulm Dis. (2006) 1:99. doi: 10.2147/copd.2006.1.2.99
143. Duailibi MS, Cordeiro Q, Brietzke E, Ribeiro M, LaRowe S, Berk M, et al. N-acetylcysteine in the treatment of craving in substance use disorders: Systematic review and meta-analysis. Am J Addict. (2017) 26:660–6. doi: 10.1111/ajad.12620
144. Arakawa M, Ito Y. N-acetylcysteine and neurodegenerative diseases: basic and clinical pharmacology. Cerebellum. (2007) 6:308–314. doi: 10.1080/14734220601142878
145. Chen AT, Chibnall JT, Nasrallah HA. Placebo-controlled augmentation trials of the antioxidant NAC in schizophrenia: a review. Ann Clin Psychiatry. (2016) 28:190–6.
146. Uemura T, Watanabe K, Ko K, Higashi K, Kogure N, Kitajima M, et al. Protective effects of brain infarction by n-Acetylcysteine derivatives. Stroke. (2018) 49:1727–33. doi: 10.1161/STROKEAHA.118.021755
147. Chen G, Shi J, Hu Z, Hang C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of n-Acetylcysteine. Med Inflamm. (2008) 2008:716458. doi: 10.1155/2008/716458
148. Oliver G, Dean O, Camfield D, Blair-West S, Ng C, Berk M, et al. N-acetyl cysteine in the treatment of obsessive compulsive and related disorders: a systematic review. Clin Psychopharmacol Neurosci. (2015) 13:12. doi: 10.9758/cpn.2015.13.1.12
149. Fernandes BS, Dean OM, Dodd S, Malhi GS, Berk M. N-acetylcysteine in depressive symptoms and functionality: A systematic review and meta-analysis. J Clin Psychiatry. (2016) 77:457–66. doi: 10.4088/JCP.15r09984
150. Deepmala Slattery J, Kumar N, Delhey L, Berk M, Dean O, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev. (2015) 55:294–321. doi: 10.1016/j.neubiorev.2015.04.015
151. Vargus-Adams JN, McMahon MA, Michaud LJ, Bean J, Vinks AA. Pharmacokinetics of amantadine in children with impaired consciousness due to acquired brain injury: preliminary findings using a sparse-sampling technique. PM&R. (2010) 2:37–42.
152. Strawn JR, Saldaña SN. Treatment with adjunctive n-acetylcysteine in an adolescent with selective serotonin reuptake inhibitor-resistant anxiety. J Child Adolesc Psychopharmacol. (2012) 22:472–3. doi: 10.1089/cap.2012.0066
153. Costa DLC, Diniz JB, Requena G, Joaquim MA, Pittenger C, Bloch MH, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. (2017) 78:e766–e73. doi: 10.4088/JCP.16m11101
154. Bush AI, Cotton SM, Berk M, Malhi GS, Schapkaitz I, Moss K, et al. The efficacy of adjunctive n -Acetylcysteine in major depressive disorder. J Clin Psychiatry. (2014) 75:628–36. doi: 10.4088/jcp.13m08454
155. Grant JE, Odlaug BL, Suck WK. N-acetylcysteine, a glutamate modulator, in the treatment of trichotillomania: a double-blind, placebo-controlled study. Arch Gen Psychiatry. (2009) 66:756–63. doi: 10.1001/archgenpsychiatry.2009.60
156. Pittenger C, Vladimir C, Banasr M, Bloch M, Krystal JH, Sanacora G. Riluzole in the treatment of mood and anxiety disorders. CNS Drugs. (2008) 22:761–86. doi: 10.2165/00023210-200822090-00004
157. De Boer JN, Vingerhoets C, Hirdes M, McAlonan GM, Amelsvoort TV, Zinkstok JR. Efficacy and tolerability of riluzole in psychiatric disorders: a systematic review and preliminary meta-analysis. Psychiatry Res. (2019) 278:294–302. doi: 10.1016/j.psychres.2019.06.020
158. Pittenger C, Bloch MH, Wasylink S, Billingslea E, Simpson R, Jakubovski E, et al. Riluzole augmentation in treatment-refractory obsessive-compulsive disorder: a pilot randomized placebo-controlled trial. J Clin Psychiatry. (2015) 76:1075–84. doi: 10.4088/JCP.14m09123
159. Möhler H. The gABA system in anxiety and depression and its therapeutic potential. Neuropharmacol. (2012) 62:42–53. doi: 10.1016/j.neuropharm.2011.08.040
161. Jembrek M, Vlainic J. GABA receptors: pharmacological potential and pitfalls. Curr Pharm Design. (2015) 21:4943–59. doi: 10.2174/1381612821666150914121624
162. Hansen CC, Ljung H, Brodtkorb E, Reimers A. Mechanisms underlying aggressive behavior induced by antiepileptic drugs: focus on topiramate, levetiracetam, and perampanel. Behav Neurol. (2018) 2018:2064027. doi: 10.1155/2018/2064027
163. Mariani JJ, Levin FR. Levetiracetam for the treatment of co-occurring alcohol dependence and anxiety: case series and review. Am J Drug Alcohol Abuse. (2008) 34:683–91. doi: 10.1080/00952990802308213
164. Greenblatt HK, Greenblatt DJ. Gabapentin and pregabalin for the treatment of anxiety disorders. Clin Pharm Drug Dev. (2018). doi: 10.1002/cpdd.446
165. Baldwin DS, den Boer JA, Lyndon G, Emir B, Schweizer E, Haswell H. Efficacy and safety of pregabalin in generalised anxiety disorder: a critical review of the literature. J Psychopharmacol. (2015) 29:1047–60. doi: 10.1177/0269881115598411
166. Generoso MB, Trevizol AP, Kasper S, Cho HJ, Cordeiro Q, Shiozawa P. Pregabalin for generalized anxiety disorder: an updated systematic review and meta-analysis. Int Clin Psychopharmacol. (2017) 32:49–55. doi: 10.1097/YIC.0000000000000147
167. Rickels K, Shiovitz TM, Ramey TS, Weaver JJ, Knapp LE, Miceli JJ. Adjunctive therapy with pregabalin in generalized anxiety disorder patients with partial response to sSRI or sNRI treatment. Int Clin Psychopharmacol. (2012) 27:142–50. doi: 10.1097/YIC.0b013e328350b133
168. Schachter SC. Pharmacology and clinical experience with tiagabine. Exp Opin Pharmacother. (2001) 2:179–87. doi: 10.1517/14656566.2.1.179
169. Dunlop BW, Papp L, Garlow SJ, Weiss PS, Knight BT, Ninan PT. Tiagabine for social anxiety disorder. Hum Psychopharmacol. (2007) 22:241–4. doi: 10.1002/hup.846
170. Sheehan DV, Sheehan KH, Raj BA, Janavs J. An open-label study of tiagabine in panic disorder. Psychopharmacol Bull. (2007) 40:32–40.
171. Zwanzger P, Eser D, Nothdurfter C, Baghai TC, Möller H.-J., et al. Effects of the gABA-reuptake inhibitor tiagabine on panic and anxiety in patients with panic disorder. Pharmacopsychiatry. (2009) 42:266–9. doi: 10.1055/s-0029-1241798
172. Schwartz TL, Azhar N, Husain J, Nihalani N, Simionescu M, Coovert D, et al. An open-label study of tiagabine as augmentation therapy for anxiety. Ann Clin Psychiatry. (2005) 17:167–72. doi: 10.1080/10401230591002138
173. Langtry HD, Gillis JC, Davis R. Topiramate. A review of its pharmacodynamic and pharmacokinetic properties and clinical efficacy in the management of epilepsy. Drugs. (1997) 54:752–73. doi: 10.2165/00003495-199754050-00009
174. Van Ameringen M, Mancini C, Pipe B, Oakman J, Bennett M. An open trial of topiramate in the treatment of generalized social phobia. J Clin Psychiatry. (2004) 65:1674–8. doi: 10.4088/JCP.v65n1213
175. Ghodke-Puranik Y, Thorn CF, Lamba JK, Leeder JS, Song W, Birnbaum AK, et al. Valproic acid pathway: pharmacokinetics and pharmacodynamics. Pharm Genom. (2013) 23:236–41. doi: 10.1097/FPC.0b013e32835ea0b2
Keywords: glutamate, anxiety, pharmacology, psychiatry, clinical trials, preclinical trials
Citation: Nasir M, Trujillo D, Levine J, Dwyer JB, Rupp ZW and Bloch MH (2020) Glutamate Systems in DSM-5 Anxiety Disorders: Their Role and a Review of Glutamate and GABA Psychopharmacology. Front. Psychiatry 11:548505. doi: 10.3389/fpsyt.2020.548505
Received: 02 April 2020; Accepted: 12 October 2020;
Published: 19 November 2020.
Edited by:
Rafael Christophe Freire, Queen's University, CanadaReviewed by:
Alan Lars Pehrson, Montclair State University, United StatesCopyright © 2020 Nasir, Trujillo, Levine, Dwyer, Rupp and Bloch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Madeeha Nasir, bWFkZWVoYS5uYXNpckB5YWxlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.