
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 11 August 2020
Sec. Molecular Psychiatry
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.00781
This article is part of the Research TopicCognitive Dysfunctions in Psychiatric Disorders: Brain-Immune Interaction Mechanisms and Integrative Therapeutic ApproachesView all 14 articles
 Paria Baharikhoob1,2,3
Paria Baharikhoob1,2,3 Nathan J. Kolla1,2,3,4,5*
Nathan J. Kolla1,2,3,4,5*According to the stress-diathesis model of suicidal behavior, completed suicide depends on the interaction between psychosocial stressors and a trait-like susceptibility. While there are likely multiple biological processes at play in suicidal behavior, recent findings point to over-activation of microglia, the resident macrophages of the central nervous system, as implicated in stress-induced suicidal behavior. However, it remains unclear how microglial dysregulation can be integrated into a clinical model of suicidal behavior. Therefore, this narrative review aims to (1) examine the findings from human post-mortem and neuroimaging studies that report a relationship between microglial activation and suicidal behavior, and (2) update the clinical model of suicidal behavior to integrate the role of microglia. A systematic search of SCOPUS, PubMed, PsycINFO, and Embase databases revealed evidence of morphological alterations in microglia and increased translocator protein density in the brains of individuals with suicidality, pointing to a positive relationship between microglial dysregulation and suicidal behavior. The studies also suggested several pathological mechanisms leading to suicidal behavior that may involve microglial dysregulation, namely (1) enhanced metabolism of tryptophan to quinolinic acid through the kynurenine pathway and associated serotonin depletion; (2) increased quinolinic acid leading to excessive N-methyl-D-aspartate-signaling, resulting in potential disruption of the blood brain barrier; (3) increased quinolinic acid resulting in higher neurotoxicity, and; (4) elevated interleukin 6 contributing to loss of inhibition of glutamatergic neurons, causing heightened glutamate release and excitotoxicity. Based on these pathways, we reconceptualized the stress-diathesis theory of suicidal behavior to incorporate the role of microglial activity.
Understanding the complex, multifactorial phenomena of suicidal behavior (SB) has long been a research priority. In 1999, Mann and colleagues were the first to introduce a stress-diathesis approach to suicide risk, which they denoted as the clinical model of SB. This model postulates that state-dependent “stress” in the form of psychosocial stressors and psychiatric illness(es) in combination with a trait-like “diathesis” to suicidal tendencies could result in SB (1). The stress component refers to the proximal risk factors that affect the timing and probability of suicidal acts, while the diathesis component describes distal risk factors influencing SB and may be considered independent of a psychiatric illness (2). This model is highly influential and has formed the framework for much of the contemporary research in suicidology; however, the model is not without limitations. Most importantly, it is difficult to discern the neurobiological or molecular underpinnings that influence the stress component and its interactions with the diathesis component.
The clinical model of SB has since been modified by the fields of neurobiology and genetics in order to incorporate a neurobiological basis. For example, studies have demonstrated that early life stressors or trauma, such as physical or sexual abuse, decrease serotonergic neurotransmission (3–6), which is associated with increased impulsivity and aggressive behavior in adults (7). Epigenetic regulation of genes involved with hypothalamic pituitary adrenal (HPA) axis responsiveness have also been implicated in the model, which results in a blunted cortisol response and reduced resilience in managing stress (8–15). These changes likely contribute to emotion dysregulation, high anxiety, impulsive/aggressive behavior, as well as impaired problem-solving and decision-making. Furthermore, early life stressors are also found to be associated with reduced cerebrospinal fluid (CSF) oxytocin levels (16). Neuropeptides, such as opioids, oxytocin, and vasopressin, play an important role in emotion regulation and are disrupted in individuals with borderline personality disorder, who are highly prone to engaging in SB (17).
Though the aforementioned findings regarding the neurobiological and genetic dimensions of suicide risk have undoubtedly been useful in improving the clinical model of SB, the most updated stress-diathesis orientated model that integrates and illustrates these findings (18) is by no means exhaustive. This is because there have been other molecular abnormalities described in relation to the risk of suicide that have not yet been incorporated in the model. In particular, several studies have recently suggested a relationship between microglial activation and SB. Specifically, the over-activation of microglia, which are the resident immune cells of the central nervous system (CNS), is posited to contribute to SB. In addition to traumatic and immune stimuli, microglia can be activated as a result of psychosocial stress (19–21). Chronic microglial activation may, therefore, also influence the interaction between the stress and diathesis components of the stress-diathesis model of SB, including contributing to an increased vulnerability to suicidal tendencies. The role of microglial dysregulation has not been discussed within the context of a stress-diathesis model. In this paper, we review the results from the original immunohistochemistry and neuroimaging studies that discuss microglial activity in relation to SB. We then incorporate the role of microglial activity in an updated stress-diathesis theory of SB.
Microglia are the resident tissue macrophages of the CNS, representing 5 to 20% of the total glial cell population (22). In a healthy brain, “resting” or “quiescent” microglia have a small cell body with fine, ramified, and highly dynamic processes. This increased surface area allows microglia to continually scan their local environment for signs of potential threats. Upon recognition of harmful stimuli or infectious agents within the micro-environment, microglia rapidly become activated. This leads to a retraction of microglia protrusions and hypertrophy of their soma, giving the microglia an amoeboid-like shape (20). When activated, microglia release different inflammatory mediators depending on the state of activation.
Activated microglia can be divided into two states, the pro-inflammatory (M1) and anti-inflammatory (M2) pathways. The M1 pathway is triggered after neuronal injury has occurred and results in the release of a number of pro-inflammatory cytokines by microglia, including interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and nitric oxide (NO) (23). This is followed by the secondary response of the M2 pathway, which leads to the release of anti-inflammatory signals by microglia, namely IL-10, insulin-like growth factor 1, transforming growth factor beta, as well as various neurotrophic factors. The anti-inflammatory response of the M2 pathway is meant to restore homeostasis through the clearing of debris, reconstruction of the extracellular matrix, tissue repair, and angiogenesis. In a healthy immune system, both the M1 and M2 pathways are necessary, and the balance between the two states is part of a functional immune response (19, 23).
The nomenclature of the M1 and M2 pathways has, nevertheless, been challenged. This is because the states were defined based on the exposure of macrophages to stimuli in vitro and not in vivo. Thus, several scholars argue that the differentiation of the two states for activated microglia may be too reductionistic and instead hypothesize that microglial activation exists along a continuum (20, 24). With this point in mind, we will refer to microglial activation in general terms rather than specifying the M1 or M2 pathways for the purposes of this review.
According to several studies involving animal models, prolonged psychosocial stress can activate microglia (19). In fact, as evident in Table 1, at least 16 animal studies have shown that in the prefrontal cortex, exposure to different psychosocial stressors gives rise to increases in ionized calcium-binding adaptor molecule 1 (Iba-1) immunoreactivity, a specific marker for the morphological changes and expansion of microglia. These psychosocial stressors involve repeated social defeat (SD), varying unpredictable stress, prenatal stress, social isolation, and chronic restraint (25–37). Similar effects from psychosocial stressors were also evident in other areas of the brain, including the amygdala (25, 26), hippocampus (25–28, 30, 36, 38, 39, 41–47), nucleus accumbens (36, 37) and paraventricular nucleus (25, 26). The SD paradigm, which is described below, appears to have the most marked effect on Iba-1 immunoreactivity.

Table 1 Effect of psychosocial stress on microglial activity in prefrontal cortex.
SD is considered an animal model of depression as it induces depressive-like behavior in animals following prolonged psychosocial stress. The paradigm is typically initiated when a male rodent is presented to the domicile of an older, more dominant and aggressive male. The less dominant intruder rodent is rapidly attacked and forced into submission, consistently exhibiting signs of distress. These signs involve elevated glucocorticoid activity, hyperthermia, and tachycardia (48). Chronic psychosocial stress induced by repeated exposure to this situation has also been shown to impose long-term, depressive-like behavioral changes in the intruder, including sensitivity to other stressors and signs of social avoidance, anhedonia, locomotor hypomotility, and metabolic changes (49–51).
Unlike in other animal models of depression, chronic, but not acute, administration of anti-inflammatory medications to rodents can be effective in reducing depressive-like manifestations. Four-week treatment with minocycline, a tetracycline antibiotic which inhibits the increase and activation of microglia, successfully reduces the depressive-like behavior of rats exposed to chronic stress. Minocycline normalizes Iba-1 immunoreactivity in the hippocampus and reduces the increase of mRNA levels of pro-inflammatory cytokines, such as IL-1β, IL-18, and IL-6 (34, 52). These results suggest that microglial activation may play a crucial role in the regulation of stress-induced depressive-like behavior.
These findings are supported by a number of other animal studies, which have shown that stress-induced depressive-like outcomes by SD may, in part, be mediated by microglial activation (40, 47, 53–56). Stein and colleagues, who reviewed these studies in depth, posit that chronic stress contributes to the over-activated states of microglia. The over-activation of microglia, as a result, leads to highly increased levels of pro-inflammatory mediators, cytotoxins, and reactive oxygen species (ROS), which may produce excessively pruned synaptic connections, causing cellular dystrophy and loss or reduced functioning of neuronal activity. The cellular damage as well as impaired neuronal activity may subsequently induce the development of depressive-like outcomes (57).
Depressive-like behavior, which is part of the stress component of the clinical model of SB, can be emulated by animal models, such as the aforementioned SD paradigm; however, the outcome, SB, cannot be reproduced by animals as there are no convincing animal models of SB. This is partially due to the notion that animals cannot arguably execute SB due to their inability to possess a will to die, to perceive the ultimate consequences of their suicidal acts, or to consider the emotional reactions of those who would survive their conceivable death (58, 59). Therefore, for this review, we turn our attention to microglial dysregulation in relation to suicidality in human studies.
Interestingly, in human post-mortem studies, microglial over-activation also appears to be implicated in the pathophysiology of SB. According to Steiner and colleagues, suicide victims with a prior history of either major depressive disorder (MDD) or paranoid schizophrenia (SCZ) have elevated levels of microglial activity in the dorsolateral prefrontal cortex (DLPFC), anterior cingulate cortex (ACC) and mediodorsal thalamus (MDT) (60). Moreover, in a study that measured Iba-1 immunoreactivity similar to the animal models above, the results demonstrated a significantly higher degree of microglial priming in the dorsal ACC white matter of depressed suicide victims (61). Other studies have theorized, likewise to Stein and investigators (57), that microglial dysregulation results in elevated levels of pro-inflammatory cytokines, such as IL-1β, IL-6, TNF-α, cytotoxins, and NOX2-derived ROS. These studies also propose that such mechanisms contribute to neurotoxicity, excitotoxicity, and heightened glutamate release, alterations of which may then lead to depressive-like behavior apposite to suicidality (62–64). The aforementioned literature, alongside other human studies evaluating microglial dysregulation in SB, will be described in detail later in this review.
Potentially relevant studies were identified using the four electronic databases, SCOPUS, PubMed, PsycINFO, and Embase. Studies eligible for review included those concerning microglial activation and suicidal and self-harming acts in humans. They included human post-mortem and neuroimaging studies. Searches were performed between January 2006 and December 2019. We chose 2006 as the lower limit because it marked the year that the first paper addressing a possible relationship between microglial dysregulation and suicidality in humans was published (65). The following keywords were used for searches for each database: “microglia”, “microglial activation”, “microglial dysregulation”, “suicidality”, “suicidal behavior”, “suicide”, “suicide attempt”, “suicidal ideation”, “parasuicidal behavior”, “self-harming behavior”, and “self-harm”.
Considering the different databases used for identifying studies, the abstracts and titles identified from the database searches were initially screened for eligibility using the following inclusion criteria: (1) original articles published between January 1, 2006, and December 31, 2019. Following this step, 44 were excluded from an initial 277 studies. This led to a remaining 233 studies, which were then screened using the following inclusion criteria: (1) studies that involved human subjects, (2) studies that were in English, and (3) studies that were not review articles. 121 studies were eliminated after this stage of screening, resulting in 112 articles.
The remaining 112 articles that met the aforementioned eligibility criteria were extracted to the reference management software, Zotero (Version 5.0.79). The full text of these articles was reviewed and included if they reported microglial dysregulation in the context of SB. All duplicate articles were subsequently deleted. This step in the screening process eliminated 103 articles, constituting a total of 9 studies that were included in this review.
The lead author was responsible for reviewing titles and abstracts following the database searches. The full text of articles that passed the first set of eligibility criteria were also reviewed by the lead author. If the relevance of an article was deemed questionable by the lead author, then the second author was responsible for reviewing the full text of the article and concluding its appropriateness for inclusion. The study selection process is summarized in Figure 1.
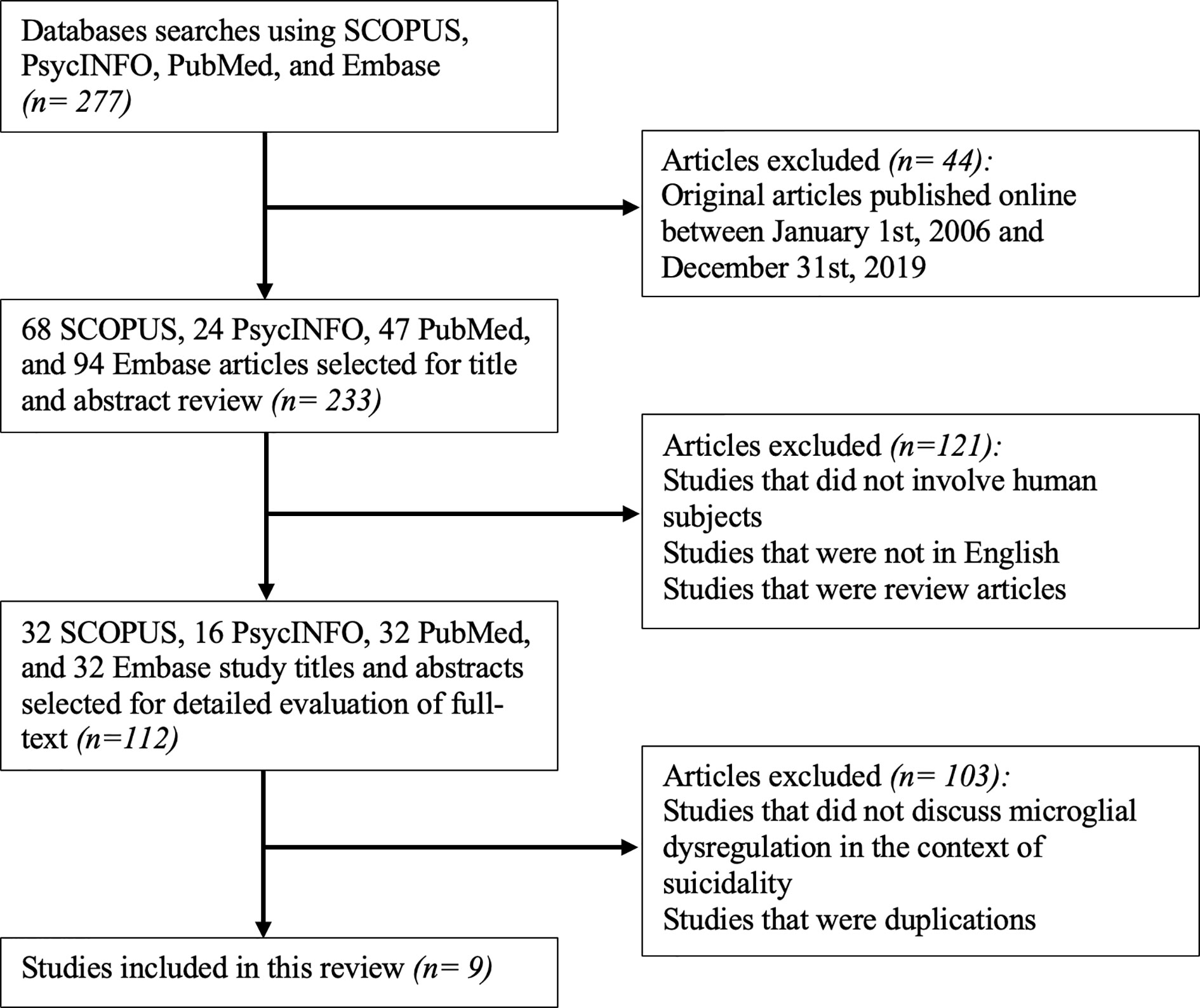
Figure 1 Flowchart for the inclusion of appropriate original articles. This flowchart showcases the selection process for the inclusion of articles for review according to the PRISMA statement (66).
Performing immunohistochemical analyses from post-mortem brain samples is an important technique for furthering our understanding of the neurochemical dimensions of complex psychiatric phenomena, including SB (67). Eight separate post-mortem studies utilized this technique to investigate potentially dysregulated microglial activity in SB, which can be observed in Table 2. Findings from four studies pointed to significant microglial activity in the brains of suicide victims compared with healthy control (HC) subjects (60–62, 65); two studies observed no significant differences in activated microglia between suicide victims and HC subjects or found a minor percentage of microglia changes (63, 64); one study observed decreased microglial activity in their non-suicide group (69); and, a final study found decreased microglial activity in suicide victims compared to HC subjects (68).
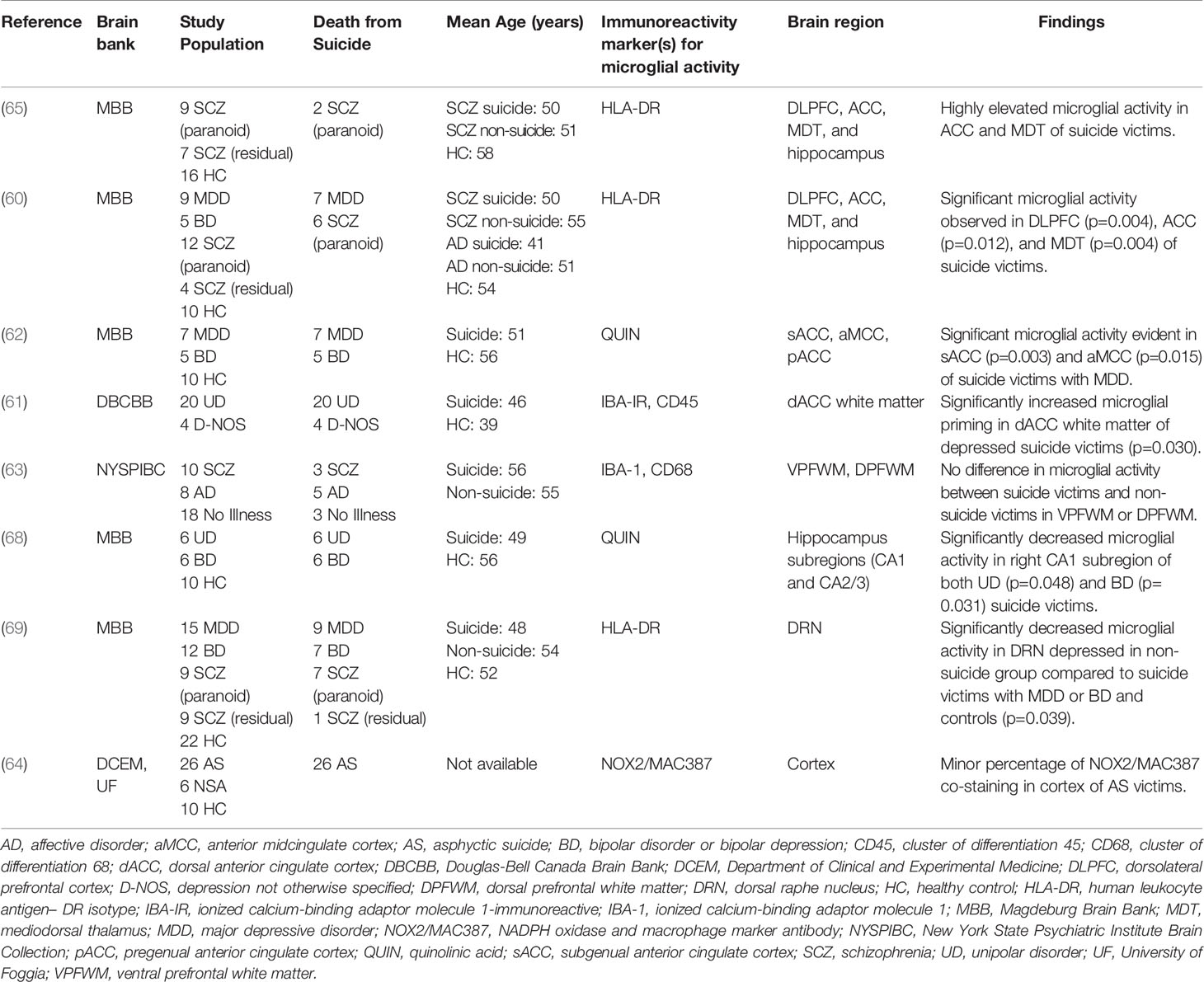
Table 2 Post-mortem studies of microglial activity in SB.
The results from four studies point to a positive association between increased microglial activity and SB. To begin, in 2006, Steiner et al. published the first study with an incidental observation of increased microglial densities in the ACC and MDT of 2 paranoid SCZ suicide victims (65). A similar group followed up on this finding and went on to provide the first significant evidence, using HLA-DR staining, of higher microglial density in the DLPFC, ACC, and MDT of suicide victims who had MDD or paranoid SCZ (60). This positive relationship was similarly replicated in a later study using the endogenous modulator, quinolinic acid (QUIN), as an immunoreactivity signature. The authors from this study observed significantly increased density of QUIN-positive cells in the subgenual ACC and anterior midcingulate cortex of suicide victims with MDD (62). Furthermore, another paper observed increased microglial priming in the dorsal ACC white matter of depressed suicide victims through the use of Iba-1–immunoreactive and Cluster of Differentiation (CD45) staining (61). Altogether, these results suggest that increased microglial activity may be correlated with death by suicide and that mechanisms relating to increased QUIN production by activated microglia may underlie this association.
Two studies presented unclear conclusions that neither support nor refute the a priori hypothesis of a positive relationship between the degree of microglial over-activation and SB. A team examining Iba-1 with CD68 discovered no significant differences in the ventral prefrontal white matter or dorsal prefrontal white matter between suicide victims and non-suicidal victims after adjusting for multiple statistical tests. There was an effect of suicide on the density of activated phagocytes in the ventral prefrontal white matter compared with the dorsal prefrontal white matter. Nevertheless, considering that activated phagocytes in their study comprised both activated microglia and macrophages, making any conclusions about the exclusivity of microglial activity is difficult (63). Comparatively, a study utilizing NADPH oxidase and macrophage marker antibody (NOX2/MAC387) co-staining found a small percentage of this staining in the cortex of victims of asphyxia suicide. It remains unclear, however, whether this percentage was significantly different from the percentage of those who died by non-suicidal asphyxia or in HC subjects (64). These results preclude firm conclusions; however, the authors speculated that disruption of the blood brain barrier (BBB) due to higher QUIN levels as well as increased IL-6 production could be mechanisms associated with SB.
Finally, two studies found evidence of unchanged or reduced microglial activity in suicide cases compared to HC subjects. A paper from 2017 identified significantly decreased microglial activity, using HLA-DR staining, in the dorsal raphe nucleus of the non-suicidal depressed group in comparison to suicide victims with either MDD or bipolar disorder (BD), as well as HC subjects (69). Similarly, a study by Busse and colleagues revealed decreased microglial QUIN-immunoreactivity after post hoc tests in the right hippocampal CA1 field of suicidal patients with MDD or BD. These subjects, nevertheless, had an unchanged hippocampal volume size (68). The findings of these studies suggest that local anti-inflammatory and/or neuroprotective responses in these subjects may have influenced the extent of the microglial activation.
In summary, four of the eight post-mortem studies described above provided evidence of morphological alterations, predominantly increased cell density, in the microglia of the brains of suicide victims. These results support the theory of a positive relationship between microglial dysregulation and SB regardless of psychiatric diagnosis, implying that microglial-induced SB may be independent of psychiatric illness(es). However, the remaining four post-mortem studies had inconclusive findings, for example, unchanged or reduced microglial activity in subjects with SB. With this information in mind, more extensive post-mortem or immunohistochemistry investigations are required to make any definitive statements regarding neuroinflammation, specifically activated microglia, in suicide victims.
There are some limitations to the post-mortem studies reviewed that must be mentioned. First, the microglial markers HLA-DR, Iba-1, and NOX2/MAC387 were limiting in that there was no suggestion of a molecular mechanism for which increased microglial activity may be related to SB. Moreover, with regards to the microglial marker, QUIN, the presence of released or secreted QUIN in the extracellular space and/or of protein expression or activity of the kynurenine (KYN) pathway enzyme QUIN phosphoribosyltransferease may have also influenced glutamatergic neurotransmission. These other sources of QUIN thereby eliminate our ability to conclude that strictly microglial QUIN influences neuromodulation and ultimately behavioral manifestations. Second, all of the studies had small sample sizes, especially with consideration to the number of suicide victims. Third, for many of the studies, brain samples were obtained from different brain banks and comprised patients who had differing lengths of psychiatric illnesses, causes of death, exposure to psychopharmacological interventions, and potentially other age-related brain pathologies.
The imaging modality positron emission tomography (PET) with the radioligand, [11C]-(R)-PK11195, is another useful tool for visualizing microglial activity in the CNS in various psychiatric disorders. This is because [11C]-(R)-PK11195 binds selectively to the translocator protein (TSPO) 18 kDa, a protein that is upregulated in the mitochondria of activated microglia. Binding of [11C]-(R)-PK11195 or similar tracers may, therefore, be a biomarker of microglial activation as it is expected to be higher in areas of the brain with significant inflammation or neuronal damage (70).
Using [11C]-(R)-PK11195 PET imaging, Holmes and colleagues (71) published the first in vivo study examining microglial activation and suicidal thinking. The authors observed increased TSPO density in the individuals with MDD compared to HC subjects, with the most pronounced finding in the ACC. They also found that TSPO expression was significantly elevated in the ACC and insula in patients with suicidal thoughts compared to patients without suicidal thoughts. The authors concluded that the greater TSPO binding in people with MDD and suicidal thoughts supported what four of the eight post-mortem studies had observed regarding a positive relationship between microglial activation and SB. It was also postulated that there was a higher specificity of microglial activation for suicide over psychiatric diagnoses.
The authors, however, were unable to distinguish between the resting and activated phenotypes of microglia and, therefore, could not infer any underlying mechanisms. Currently, TSPO PET imaging is considered the only valid modality for examining microglial activity in vivo; however, there are some additional drawbacks to this imaging technique that must be discussed. Though TSPO binding is thought to exclusively reflect microglial activation (72), some argue that under certain circumstances, it can be expressed when astrocytes are activated as well (73). Moreover, the radioligand, [11C]-(R)-PK11195, used in the above PET study is limited due to the short half-life of carbon-11, high nonspecific binding, high plasma protein binding, and low brain penetration (74). [11C]-(R)-PK11195 PET imaging, in addition, requires a brain region with no specific binding of TSPO as a reference region in order to quantify the in vivo binding. Nevertheless, no region is completely devoid of TSPO expression, as it is thought to be present throughout the entire brain (70, 75).
Due to these problems, second generation TSPO radiotracers have been synthesized to offer a superior quality for quantifying in vivo TSPO expression. These second-generation radioligands include [18F]-FEPPA (76), [18F]-PBR06 (77), [18F]-FEDAA1106 (78), [11C]-DAA1106 (79), [11C]-DPA-713 (80), [18F]-PBR111 (81), [18F]-DPA-714 (82), and [11C]-PBR28 (83). [18F]-FEPPA is promising, for example, given that it displays an appropriate metabolic profile, a high affinity for TSPO, good pharmacokinetics, and high brain penetration (74). However, given the sensitivity of second-generation tracers to a single nucleotide polymorphism in the TSPO gene, third-generation TSPO radiotracers are also being developed that would be insensitive to this polymorphism. Examples of third-generation radioligands are [18F]-GE180 and [11C]-ER176; nonetheless, the safety and efficacy of these compounds in humans have not yet been established (84, 85).
In sum, TSPO PET imaging, specifically [11C]-(R)-PK11195 PET, has several limitations that may affect the findings of studies using this modality, including the aforementioned study. More recent TSPO second-generation radiotracers are recommended for future studies investigating microglial activity in vivo.
The most recent neurobiologically inclined stress-diathesis model proposed that life stressors or trauma lead to neurobiological alternations, such as deceased serotonergic neurotransmission, dysregulated HPA axis responsiveness, and reduced CSF oxytocin levels that may then result in behavioral alterations (18). This reconceptualized model is undoubtedly helpful in shedding additional light on the mechanisms by which early stressors contribute to the vulnerability to SB in later adolescence and adulthood. The model does not, however, include the growing evidence regarding the association between microglial dysregulation and SB. To address this, we evaluated eight immunohistochemistry studies and one TSPO PET imaging study that directly discuss microglial over-activation in relation to SB. Now, for this section, we further explore the neurobiological mechanisms that were proposed by some of these studies and also provide an updated stress-diathesis model of SB.
In 1969, a study introduced the “serotonin hypothesis”, a theory which proposes that dysregulation in the metabolic pathway of the essential amino acid, tryptophan (TRY), is a significant risk factor for depression. Specifically, this theory suggests that there is a dysregulated shift, using TRY as a precursor, from serotonin synthesis to KYN synthesis (86). The dysregulated shift is theorized to occur due to the up-regulation of indoleamine 2,3-dioxygenase (IDO1). This enzyme becomes activated due to increased hormones and/or proinflammatory cytokines, including IL-1β, IL-6, TNF-α, and interferon gamma (IFN-γ) (87–89). By consistently stimulating the shift from serotonin synthesis to increased KYN synthesis via the KYN pathway, systematic IDO1 activation ultimately leads to depressive-like behavior (87). Nevertheless, the exact mechanisms that underlie how this dysregulation leads to depressive-like outcomes and/or SB remain unclear.
Steiner and collaborators (62) primarily tackle this conundrum by proposing that increased KYN contributes to serotonin depletion and a higher production of the excitotoxic metabolite QUIN, both of which result in behavioral changes. Both this team and others (63) suggest that depleted serotonin levels may be caused by activated microglia that have enhanced metabolism of TRY to QUIN through the KYN pathway (62, 63). Given that previous literature has found low serotonergic functioning to be associated with increased impulsivity and aggressive behaviors in adults (6), the finding that activated microglia decrease serotonin levels provides some understanding as to how microglial activation influences depressive-like behavior pertinent to SB. Moreover, a recent study has shown that reduced serotonin level is associated with an increased risk for SB despite adjustments for depression severity, further suggesting that serotonin depletion via KYN dysregulation may impact suicidality directly, regardless of a depression diagnosis (90).
Beyond serotonin reduction, the shift from TRY to QUIN is also posited to detrimentally affect the metabolism of kynurenic acid (KYNA), an N-methyl-D-aspartic acid (NMDA) receptor antagonist that is considered neuroprotective due to its inhibitory effect on glutamate neurotransmission (91, 92). In fact, reduced concentrations of KYNA have been correlated with excitotoxicity by QUIN (93). QUIN is considered excitotoxic because it is an agonist of the NMDA receptor; therefore, an increase in QUIN can lead to excessive NMDA signaling or glutamatergic dysregulation (94, 95). These mechanisms, as postulated by Schnieder and colleagues (63), can potentially disrupt the BBB. As a result, QUIN can heighten the stimulation of glutamatergic neurons, particularly in the ventral prefrontal cortex, causing a decrease in normal neuron connectivity within the limbic system and, subsequently, reduced impulse control. In addition to being a direct NMDA receptor agonist, QUIN has pro-oxidant properties that worsen the neurotoxic effects by corticosterone and pro-inflammatory cytokines, as well as pro-inflammatory capabilities due to enhancement of the IFN-γ/IL-10 ratio (68). The adverse consequences of elevated QUIN are theorized to result in depressive features and/or SB. While astrocytes appear to predominantly catabolize KYNA from KYN, microglia instead transform KYN into 3-hydroxy kynurenine and QUIN (96, 97). This suggests that the above-mentioned mechanisms by which QUIN negatively affect the CNS may primarily be attributed to activated microglia.
Furthermore, an increase in IL-6, which is produced by microglia, is predicted by Schiavone and investigators (64) to enhance expression of NOX2 and NOX2-derived ROS production in GABAergic neurons. This subsequently causes altered GABAergic neurotransmission and thereby a loss of inhibition of glutamatergic neurons. The loss of inhibitory tone on the glutamatergic neurons contributes to excitotoxicity from increased glutamate release. This mechanism is hypothesized to also result in behavioral manifestations corresponding with SB; however, the authors do not specify which behavioral alterations may be affected.
Based on the aforementioned mechanisms, we propose that psychosocial stress, one of the stress components of the stress-diathesis theory, induces microglial activation, which increases the release of pro-inflammatory cytokines, shifting from serotonin to KYN synthesis through the induction of IDO1 (62). Subsequently, the catalyzation of TRY by IDO1 produces QUIN, which disrupts glutamatergic neurotransmission, disrupting the BBB and ultimately leading to reduced impulse control through reduced neuronal connectivity within the limbic system (63). Production of QUIN by activated microglia has pro-inflammatory and pro-oxidant properties that additionally contribute to increased neurotoxicity (68). Microglial activation also, in part, leads to enhanced IL-6 release, which causes elevated NOX2 expression and NOX2-derived ROS production, resulting in a loss of inhibition in glutamatergic neurons. These series of reactions affect glutamatergic neurotransmission by increasing glutamate release. The increased excitotoxicity due to the higher glutamate release then contribute to the altered behavior that is associated with SB (64). Thus, the changes in serotonergic and glutamatergic neurotransmission mentioned above lead to behavioral changes. These altered behavioral traits in suicide victims do not appear to be diagnosis specific (64, 68), thereby constituting a diagnosis-independent diathesis for SB. Figure 2 illuminates these mechanisms and the interactions between the stress and diathesis components of the clinical model of SB.
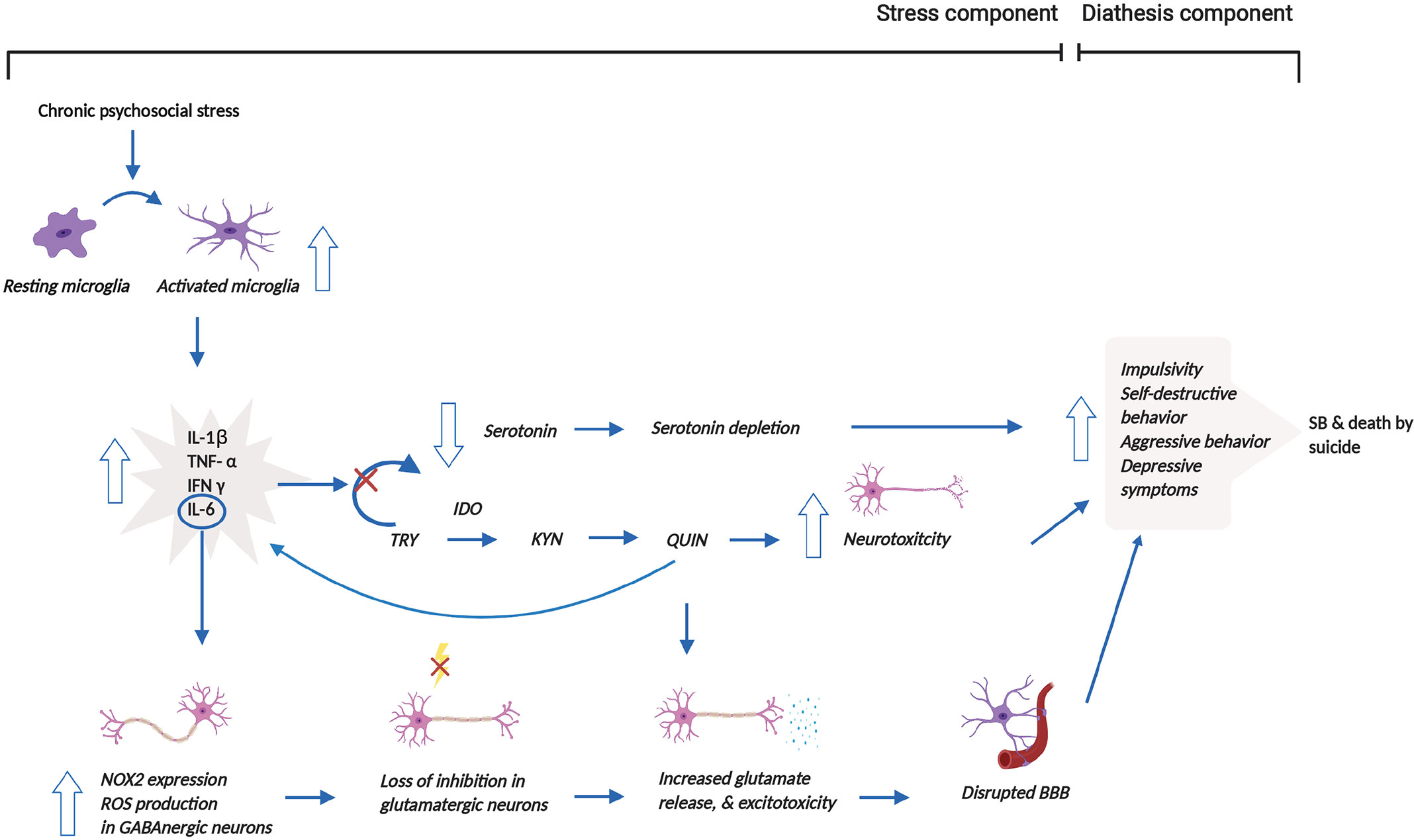
Figure 2 Reconceptualized clinical model of SB incorporating microglial dysregulation. This model proposes that prolonged psychosocial stress leads to the over-activation of microglia and associated release of pro-inflammatory cytokines. The release of pro-inflammatory cytokines stimulates the shift from serotonin to KYN synthesis through the induction of IDO1. This shift results in the depletion of serotonin. The production of QUIN from KYN synthesis, in addition, increases neurotoxicity. Moreover, increased QUIN production directly leads to heightened glutamate release and associated excitotoxicity but also indirectly causes this elevation by increasing IL-6 levels. Increased glutamate release and excitotoxicity contribute to the disruption of the BBB. Altogether, all these mechanisms contribute to behavioral manifestations, which may then result in SB and/or death by suicide. This figure was created with BioRender.com. BBB, blood brain barrier; IDO1, indoleamine 2,3-dioxygenase; IFN-γ, interferon gamma; IL-1β, interleukin-1β; IL-6, interleukin-6; NOX2, NADPH oxidase; KYN, kynurenine; QUIN, quinolinic acid; ROS, reactive oxygen species; SB, suicidal behavior; TNF-α, tumor necrosis factor-α; TRY, tryptophan.
A number of human studies have also investigated the relationship between metabolites of the KYN pathway and depressive features and/or SB by measuring these metabolites in the blood or CSF. Eleven studies of the KYN pathway metabolite, KYN, provided no convincing evidence of differences in blood KYN levels between depressed patients without SB relative to healthy controls (91, 98–107). On the other hand, seven studies reported lower blood KYN concentrations in the depressed group (108–114). A meta-analysis, which evaluated these studies, similarly concluded that blood KYN levels were lower in depressed patients compared to HC (115). Moreover, the study by the Setoyama group found that blood KYN levels were negatively correlated with suicidal ideation in depressed patients (114). By contrast, Sublette and colleagues demonstrated that blood KYN concentrations were elevated in suicide attempters compared to non-suicidal patients (107). Another study reported a higher blood KYN/TRP ratio in MDD subjects with SB versus MDD subjects without SB or HC. Results indicated that the KYN/TRP ratio was positively correlated with the severity of suicidality in patients with histories of SB (116). Brundin et al. (117) additionally identified a higher ratio of CSF KYN/TRP in patients with prior suicide attempts relative to HC. Altogether, these results suggest that higher concentrations of KYN have a greater association with SB compared to the relationship with depression.
Twelve studies examining KYNA, another metabolite of the KYN pathway, found no significant differences in blood KYNA concentrations between patients with depression without SB and HC (91, 99, 100–102, 105, 106, 108–110, 118, 119). One study, in addition, reported no differences in levels of CSF KYNA among depressed patients and HC (120). Four studies, however, provided evidence of reduced levels of KYNA concentrations in depressed patients relative to HC, with three studies reporting the result based on KYNA levels in blood (98, 111, 121) and one from KYNA levels in the CSF (120). Also, a meta-analysis that reviewed the above-mentioned studies measuring blood and CSF KYNA likewise concluded that concentrations of this metabolite were reduced in patients with depression in comparison to HC (115). On the other hand, one study reported that psychiatric patients who made previous suicide attempts rather than simply endorsing an MDD diagnosis had reduced levels of KYNA in the CSF when compared to HC (122). These studies evaluating KYNA showed largely no differences between study and control groups for either depressive-like behaviors or SB.
Several studies have also measured levels of the KYN pathway metabolite, QUIN, in the blood or CSF in patients with depression. Among these investigations, six studies identified no group differences in blood QUIN concentrations between depressed patients and HC (99, 100, 102, 105, 106, 115), while three found elevated levels of QUIN in patients with depression, with two reporting the finding after measuring QUIN levels in blood (91, 98) and another from measuring QUIN levels in CSF (120). A meta-analysis, which also reviewed the above studies, additionally revealed that blood QUIN levels were higher in the depressed group, although the effect size was small (115). Moreover, Bay-Richter and colleagues reported greater concentrations of CSF QUIN in patients with prior histories of suicide attempts compared to HC (122). A more recent study found a decreased ratio of CSF picolinic acid (PIC), an additional neuroprotective metabolite of the KYN pathway, to QUIN in subjects who attempted suicide relative to HC (117). These results indicate that a lower PIC/QUIN ratio could potentially represent a marker of suicidality; however, these results remain preliminary and await further research. Although the majority of investigations examining QUIN relevant to depression found no significant results, the studies that analyzed QUIN in relation to SB conversely reported findings that were significant when compared with HC.
To summarize, there have been many studies of blood or CSF markers of the KYN pathway that may relate to depression and/or SB. Of these studies, a number of them investigated metabolites of the KYN pathway (e.g., KYN, KYNA, QUIN) in relation to depression; however, most studies did not find significant results. Among studies that sampled patients with SB, results predominantly showed significant results, such as elevated levels of blood or CSF KYN/TRP ratio and CSF QUIN, indicating perhaps that the upregulation of the KYN pathway may be associated with suicidality. However, the number of studies evaluating this relationship was limited and more research is needed.
In the past decades, several theories have been proposed to conceptualize the etiology of SB; particularly, in recent years, many studies have focused on investigating the neurobiological aspects of this complex, multifactorial phenomenon. Through this review, we examined the original immunohistochemistry and neuroimaging studies that analyzed the relationship between microglial dysregulation and SB. Based on their results and proposed mechanisms, we were then able to construct a reconceptualized stress-diathesis theory of SB that incorporates the role of microglial activity. In our updated model, we suggest that chronic microglial activation may play a mediating role in the etiology of SB. That is, microglial dysregulation may be triggered by environmental challenges, namely psychosocial stressors, and then mediate several pathological neurobiological pathways that lead to behavioral changes and ultimately the outcome of SB.
The neurobiological pathways illustrated in this model predominantly involve dysfunction in the KYN pathway. As previously discussed, the results from studies, which evaluated the concentrations and/or ratios of various metabolites of the KYN pathway, also showed that SB may be associated with KYN dysregulation. Such findings are consistent with the results and hypothesized mechanisms from studies of microglial over-activation in suicidality that were examined in this review; nevertheless, it should be noted that the number of studies measuring peripheral blood and/or CSF metabolite levels of the KYN pathway in patients with SB are highly limited at this time. Research that further investigates these metabolites in suicidal patients is, therefore, necessary. It would also behoove investigators to test the efficacy of currently available pharmacotherapeutics in normalizing KYN dysregulation in suicidal individuals.
We synthesized our updated clinical model of SB by evaluating the specific hypothesized neurobiological pathways discussed in the studies that were included in this review. However, advances in the fields of neurobiology and epigenetics have revealed that other mediating factors, namely HPA axis responsiveness, blunted cortisol, and oxytocin are also related to the outcome of SB (9, 11–14, 16, 17). These factors were not incorporated in our reconceptualized model but may be related to microglial dysregulation as well, thereby representing another area for which to conduct original research.
Also, it remains unclear whether microglial cells become primed due to early adverse experiences, thus making them more susceptible to prolonged activation in response to subsequent stressors later in life. In this case, we may speculate that chronic microglial activation would not only assume a role in instigating the diathesis for SB, which we outlined in Figure 1, but might provoke, later in time, the outcome of SB. In this case, future research, ideally from prospective or longitudinal study designs, is needed to definitively establish whether microglial dysregulation exclusively impacts the risk for developing the diathesis traits leading to SB or whether it also perpetuates SB at a later time in life.
All studies that were included in this review consisted of subjects who were either victims of suicide or participants who had suicidal thinking. None of the papers discussed the over-activation of microglia in relation to self-harming behavior, despite our inclusion of the keywords, “parasuicidal behavior”, “self-harming behavior”, and “self-harm”. Considering this paucity of information, it would be interesting to determine, likely through neuroimaging research, whether there is also a positive relationship between microglial activity and self-harm, as well as to inform through an updated stress-diathesis model how self-harm subsequently relates to the risk for death by suicide.
In this review, we observed an overarching number of immunohistochemistry studies in comparison to the singular neuroimaging study. It appears that PET studies that can detect microglial activity, such as the aforementioned TSPO PET imaging, are in their infancy and require further research and refinements. The field of suicidology would benefit greatly by enhanced radioligands that have improved signal-to-noise ratio and lower nonspecific binding. Second- and third-generation radiotracers show promise for future research investigations as they do display these favorable properties. Accordingly, using such radiotracers can allow for more definitive conclusions to be made from studies that evaluate microglial cells through TSPO PET imaging. It would also enable scientists to perform clinical trials that test anti-inflammatory psychopharmacological medications, such as minocycline, on humans with SB in vivo. This work would be valuable for elucidating the role of neuroinflammation in SB as well as prospectively providing an accessible treatment option to individuals who are at high risk of death by suicide. To reiterate, however, as SB is a highly complex phenomenon, extensive future research is warranted to further refine our understanding of the neurobiology of these behaviors before a targeted psychopharmacological intervention may be established.
NJK conceived and designed the study, reviewed the full-text of three articles that were deemed questionable for eligibility by PB, and made revisions to the manuscript drafts. PB reviewed the titles and abstracts of studies following database searches as well as the full-text of select articles, drafted the manuscript, and made edits to the manuscript drafts. All authors contributed to the article and approved the submitted version.
This review article was funded by a Canadian Institutes of Health Research Clinician Scientist Salary Award and the Centre for Addiction and Mental Health Alternative Funding Plan, both awarded to NJK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACC, Anterior Cingulate Cortex; AD, affective disorder; AS, Asphyctic Suicide; BD, Bipolar Disorder or Bipolar Depression; BBB, Blood Brain Barrier; CSF, Cerebrospinal Fluid; CD45, Cluster of Differentiation 45; CD68, Cluster of Differentiation 68; CNS, Central Nervous System; dACC, Dorsal Anterior Cingulate Cortex; DBCBB, Douglas-Bell Canada Brain Bank; DCEM, Department of Clinical and Experimental Medicine; DLPFC, Dorsolateral Prefrontal Cortex; D-NOS, Depression Not Otherwise Specified; DPFWM, Dorsal Prefrontal White Matter; DRN, Dorsal Raphe Nucleus; HC, Healthy Control; HLA-DR, Human Leukocyte Antigen–DR isotype; HPA, Hypothalamic Pituitary Adrenal; Iba-IR, Ionized Calcium-binding Adaptor Molecule 1-immunoreactive; Iba-1, Ionized Calcium-binding Adaptor Molecule 1; IDO1, Indoleamine 2,3-dioxygenase; IFN-γ, Interferon Gamma; IL-1β, Interleukin-1β; IL-6, Interleukin-6; IL-10, Interleukin-10; IL-18, Interleukin-18; KYN, Kynurenine; KYNA, Kynurenic Acid; M1, Pro-inflammatory Pathway; M2, Anti-inflammatory Pathway; MBB, Magdeburg Brain Bank; MDT, Mediodorsal Thalamus; MDD, Major Depressive Disorder; NMDA, N-methyl-D-aspartic Acid; NO, Nitric Oxide; NOX2/MAC387, NADPH Oxidase and Macrophage Marker Antibody; NYSPIBC, New York State Psychiatric Institute Brain Collection; pACC, Pregenual Anterior Cingulate Cortex; PET, Positron Emission Tomography; PIC, Picolinic Acid; QUIN, Quinolinic acid; ROS, Reactive Oxygen Species; SB, Suicidal Behavior; SCZ, Schizophrenia; TNF-α, Tumor Necrosis Factor-α; TRY, Tryptophan; TSPO, Translocator Protein; UD, Unipolar Disorder; UF, University of Foggia; VPFWM, Ventral Prefrontal White Matter.
1. Mann JJ, Waternaux C, Haas GL, Malone KM. Toward a clinical model of suicidal behavior in psychiatric patients. Am J Psychiatry (1999) 156(2):181–9. doi: 10.1176/ajp.156.2.181
2. Mann JJ. Neurobiology of suicidal behaviour. Nat Rev Neurosci (2003) 4(10):819–28. doi: 10.1038/nrn1220
3. Benedetti F, Riccaboni R, Poletti S, Radaelli D, Locatelli C, Lorenzi C, et al. The serotonin transporter genotype modulates the relationship between early stress and adult suicidality in bipolar disorder. Bipolar Disord (2014) 16(8):857–66. doi: 10.1111/bdi.12250
4. Gibb BE, McGeary JE, Beevers CG, Miller IW. Serotonin transporter (5-HTTLPR) genotype, childhood abuse, and suicide attempts in adult psychiatric inpatients. Suicide Life Threat Behav (2006) 36(6):687–93. doi: 10.1521/suli.2006.36.6.687
5. Roy A, Hu X-Z, Janal MN, Goldman D. Interaction between childhood trauma and serotonin transporter gene variation in suicide. Neuropsychopharmacology (2007) 32(9):2046–52. doi: 10.1038/sj.npp.1301331
6. Stanley M, Mann JJ. Increased serotonin-2 binding sites in frontal cortex of suicide victims. Lancet (1983) 321(8318):214–6. doi: 10.1016/S0140-6736(83)92590-4
7. Brodsky BS, Oquendo M, Ellis SP, Haas GL, Malone KM, Mann JJ. The relationship of childhood abuse to impulsivity and suicidal behavior in adults with major depression. Am J Psychiatry (2001) 158(11):1871–7. doi: 10.1176/appi.ajp.158.11.1871
8. Steinberg LJ, Mann JJ. Abnormal stress responsiveness and suicidal behavior: A risk phenotype. Biomark Neuropsychiat (2020) 10:100011. doi: 10.1016/j.bionps.2020.100011
9. O’Connor DB, Green JA, Ferguson E, O’Carroll RE, O’Connor RC. Effects of childhood trauma on cortisol levels in suicide attempters and ideators. Psychoneuroendocrinology (2018) 88:9–16. doi: 10.1016/j.psyneuen.2017.11.004
10. O’Connor DB, Green JA, Ferguson E, O’Carroll RE, O’Connor RC. Cortisol reactivity and suicidal behavior: Investigating the role of hypothalamic-pituitary-adrenal axis responses to stress in suicide attempters and ideators. Psychoneuroendocrinology (2017) 75:183–91. doi: 10.1016/j.psyneuen.2016.10.019
11. Melhem NM, Keilp JG, Porta G, Oquendo MA, Burke A, Stanley B, et al. Blunted HPA axis activity in suicide attempters compared to those at high risk for suicidal behavior. Neuropsychopharmacology (2016) 41(6):1447–56. doi: 10.1038/npp.2015.309
12. Roy A, Gorodetsky E, Yuan Q, Goldman D, Enoch M-A. Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology (2010) 35(8):1674–83. doi: 10.1038/npp.2009.236
13. McGirr A, Diaconu G, Berlim MT, Pruessner JC, Sablé R, Cabot S, et al. Dysregulation of the sympathetic nervous system, hypothalamic–pituitary–adrenal axis and executive function in individuals at risk for suicide. J Psychiatry Neurosci Jpn (2010) 35(6):399–408. doi: 10.1503/jpn.090121
14. Pfennig A, Kunzel HE, Kern N, Ising M, Majer M, Fuchs B, et al. Hypothalamus-pituitary-adrenal system regulation and suicidal behavior in depression. Biol Psychiatry (2005) 57(4):336–42. doi: 10.1016/j.biopsych.2004.11.017
15. Turecki G, Ota VK, Belangero SI, Jackowski A, Kaufman J. Early life adversity, genomic plasticity, and psychopathology. Lancet Psychiatry (2014) 1(6):461–6. doi: 10.1016/S2215-0366(14)00022-4
16. Chatzittofis A, Nordström P, Uvnäs-Moberg K, Asberg M, Jokinen J. CSF and plasma oxytocin levels in suicide attempters, the role of childhood trauma and revictimization. Neuro Endocrinol Lett (2014) 35(3):213–7.
17. Stanley B, Siever LJ. The interpersonal dimension of borderline personality disorder: Toward a neuropeptide model. Am J Psychiatry (2010) 167(1):24–39. doi: 10.1176/appi.ajp.2009.09050744
18. Brodsky BS. Early childhood environment and genetic interactions: the diathesis for suicidal behavior. Curr Psychiatry Rep (2016) 18(9):86. doi: 10.1007/s11920-016-0716-z
19. Calcia MA, Bonsall DR, Bloomfield PS, Selvaraj S, Barichello T, Howes OD. Stress and neuroinflammation: a systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacol (Berl) (2016) 233(9):1637–50. doi: 10.1007/s00213-016-4218-9
20. Mondelli V, Vernon AC, Turkheimer F, Dazzan P, Pariante CM. Brain microglia in psychiatric disorders. Lancet Psychiatry (2017) 4(7):563–72. doi: 10.1016/S2215-0366(17)30101-3
21. Tay TL, Béchade C, D’Andrea I, St-Pierre M-K, Henry MS, Roumier A, et al. Microglia gone rogue: Impacts on psychiatric disorders across the lifespan. Front Mol Neurosci (2017) 10:421. doi: 10.3389/fnmol.2017.00421
22. Yang I, Han SJ, Kaur G, Crane C, Parsa AT. The role of microglia in central nervous system immunity and glioma immunology. J Clin Neurosci Off J Neurosurg Soc Australas (2010) 17(1):6–10. doi: 10.1016/j.jocn.2009.05.006
23. Howes OD, McCutcheon R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: a reconceptualization. Transl Psychiatry (2017) 7(2):e1024–4. doi: 10.1038/tp.2016.278
24. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci (2016) 19(8):987–91. doi: 10.1038/nn.4338
25. Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci (2011) 31(17):6277–88. doi: 10.1523/JNEUROSCI.0450-11.2011
26. Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology (2012) 37(9):1491–505. doi: 10.1016/j.psyneuen.2012.02.003
27. Ferle V, Repouskou A, Aspiotis G, Raftogianni A, Chrousos G, Stylianopoulou F, et al. Synergistic effects of early life mild adversity and chronic social defeat on rat brain microglia and cytokines. Physiol Behav (2020) 215:112791. doi: 10.1016/j.physbeh.2019.112791
28. Bian Y, Pan Z, Hou Z, Huang C, Li W, Zhao B. Learning, memory, and glial cell changes following recovery from chronic unpredictable stress. Brain Res Bull (2012) 88(5):471–6. doi: 10.1016/j.brainresbull.2012.04.008
29. Horchar MJ, Wohleb ES. Glucocorticoid receptor antagonism prevents microglia-mediated neuronal remodeling and behavioral despair following chronic unpredictable stress. Brain Behav Immun (2019) 81:329–40. doi: 10.1016/j.bbi.2019.06.030
30. Ślusarczyk J, Trojan E, Głombik K, Budziszewska B, Kubera M, Lasoń W, et al. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front Cell Neurosci (2015) 9:82. doi: 10.3389/fncel.2015.00082
31. Couch Y, Anthony DC, Dolgov O, Revischin A, Festoff B, Santos AI, et al. Microglial activation, increased TNF and SERT expression in the prefrontal cortex define stress-altered behaviour in mice susceptible to anhedonia. Brain Behav Immun (2013) 29:136–46. doi: 10.1016/j.bbi.2012.12.017
32. Kopp BL, Wick D, Herman JP. Differential effects of homotypic vs. heterotypic chronic stress regimens on microglial activation in the prefrontal cortex. Physiol Behav (2013) 122:246–52. doi: 10.1016/j.physbeh.2013.05.030
33. Wohleb ES, Terwilliger R, Duman CH, Duman RS. Stress-induced neuronal colony stimulating factor 1 provokes microglia-mediated neuronal remodeling and depressive-like behavior. Biol Psychiatry (2018) 83(1):38–49. doi: 10.1016/j.biopsych.2017.05.026
34. Hinwood M, Morandini J, Day TA, Walker FR. Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex (2012) 22(6):1442–54. doi: 10.1093/cercor/bhr229
35. Hinwood M, Tynan RJ, Charnley JL, Beynon SB, Day TA, Walker FR. Chronic stress induced remodeling of the prefrontal cortex: Structural re-organization of microglia and the inhibitory effect of minocycline. Cereb Cortex (2013) 23(8):1784–97. doi: 10.1093/cercor/bhs151
36. Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, et al. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun (2010) 24(7):1058–68. doi: 10.1016/j.bbi.2010.02.001
37. Schiavone S, Sorce S, Dubois-Dauphin M, Jaquet V, Colaianna M, Zotti M, et al. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry (2009) 66(4):384–92. doi: 10.1016/j.biopsych.2009.04.033
38. Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science (2013) 339(6123):1095–9. doi: 10.1126/science.1228261
39. Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV, et al. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry (2014) 19(6):699–709. doi: 10.1038/mp.2013.155
40. Tong L, Gong Y, Wang P, Hu W, Wang J, Chen Z, et al. Microglia loss contributes to the development of major depression induced by different types of chronic stresses. Neurochem Res (2017) 42(10):2698–711. doi: 10.1007/s11064-017-2270-4
41. Brevet M, Kojima H, Asakawa A, Atsuchi K, Ushikai M, Ataka K, et al. Chronic foot-shock stress potentiates the influx of bone marrow-derived microglia into hippocampus. J Neurosci Res (2010) 88(9):1890–7. doi: 10.1002/jnr.22362
42. Diz-Chaves Y, Astiz M, Bellini MJ, Garcia-Segura LM. Prenatal stress increases the expression of proinflammatory cytokines and exacerbates the inflammatory response to LPS in the hippocampal formation of adult male mice. Brain Behav Immun (2013) 28:196–206. doi: 10.1016/j.bbi.2012.11.013
43. Diz-Chaves Y, Pernía O, Carrero P, Garcia-Segura LM. Prenatal stress causes alterations in the morphology of microglia and the inflammatory response of the hippocampus of adult female mice. J Neuroinflamm (2012) 9(1):71. doi: 10.1186/1742-2094-9-71
44. Kojo A, Yamada K, Kubo K-Y, Yamashita A, Yamamoto T. Occlusal disharmony in mice transiently activates microglia in hippocampal CA1 region but not in dentate gyrus. Tohoku J Exp Med (2010) 221(3):237–43. doi: 10.1620/tjem.221.237
45. Park JH, Yoo K-Y, Lee CH, Kim IH, Shin BN, Choi JH, et al. Comparison of glucocorticoid receptor and ionized calcium-binding adapter molecule 1 immunoreactivity in the adult and aged gerbil hippocampus following repeated restraint stress. Neurochem Res (2011) 36(6):1037–45. doi: 10.1007/s11064-011-0444-z
46. Yoo K-Y, Lee CH, Park JH, Hwang IK, Park OK, Kwon S-H, et al. Antioxidant enzymes are differently changed in experimental ischemic hippocampal CA1 region following repeated restraint stress. J Neurol Sci (2011) 302(1):33–42. doi: 10.1016/j.jns.2010.12.006
47. McKim DB, Niraula A, Tarr AJ, Wohleb ES, Sheridan JF, Godbout JP. Neuroinflammatory dynamics underlie memory impairments after repeated social defeat. J Neurosci (2016) 36(9):2590–604. doi: 10.1523/JNEUROSCI.2394-15.2016
48. Hollis F, Kabbaj M. Social defeat as an animal model for depression. ILAR J (2014) 55(2):221–32. doi: 10.1093/ilar/ilu002
49. Hollis F, Wang H, Dietz D, Gunjan A, Kabbaj M. The effects of repeated social defeat on long-term depressive-like behavior and short-term histone modifications in the hippocampus in male Sprague–Dawley rats. Psychopharmacol (Berl) (2010) 211(1):69–77. doi: 10.1007/s00213-010-1869-9
50. Rygula R, Abumaria N, Flügge G, Fuchs E, Rüther E, Havemann-Reinecke U. Anhedonia and motivational deficits in rats: Impact of chronic social stress. Behav Brain Res (2005) 162(1):127–34. doi: 10.1016/j.bbr.2005.03.009
51. Toyoda A. Social defeat models in animal science: What we have learned from rodent models. Anim Sci J (2017) 88(7):944–52. doi: 10.1111/asj.12809
52. Wang Y-L, Han Q-Q, Gong W-Q, Pan D-H, Wang L-Z, Hu W, et al. Microglial activation mediates chronic mild stress-induced depressive- and anxiety-like behavior in adult rats. J Neuroinflamm (2018) 15(1):21. doi: 10.1186/s12974-018-1054-3
53. Lehmann ML, Cooper HA, Maric D, Herkenham M. Social defeat induces depressive-like states and microglial activation without involvement of peripheral macrophages. J Neuroinflamm (2016) 13(1):224. doi: 10.1186/s12974-016-0672-x
54. Ramirez K, Niraula A, Sheridan JF. GABAergic modulation with classical benzodiazepines prevent stress-induced neuro-immune dysregulation and behavioral alterations. Brain Behav Immun (2016) 51:154–68. doi: 10.1016/j.bbi.2015.08.011
55. Ramirez K, Sheridan JF. Antidepressant imipramine diminishes stress-induced inflammation in the periphery and central nervous system and related anxiety- and depressive- like behaviors. Brain Behav Immun (2016) 57:293–303. doi: 10.1016/j.bbi.2016.05.008
56. Brachman RA, Lehmann ML, Maric D, Herkenham M. Lymphocytes from chronically stressed mice confer antidepressant-like effects to naive mice. J Neurosci (2015) 35(4):1530–8. doi: 10.1523/JNEUROSCI.2278-14.2015
57. Stein DJ, Vasconcelos MF, Albrechet-Souza L, Ceresér KMM, de Almeida RMM. Microglial over-activation by social defeat stress contributes to anxiety- and depressive-like behaviors. Front Behav Neurosci (2017) 11:207. doi: 10.3389/fnbeh.2017.00207
58. Preti A. Suicide among animals: A review of evidence. Psychol Rep (2007) 101(3):831–48. doi: 10.2466/pr0.101.3.831-848
59. Preti A. Animal model and neurobiology of suicide. Prog Neuropsychopharmacol Biol Psychiatry (2011) 35(4):818–30. doi: 10.1016/j.pnpbp.2010.10.027
60. Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, et al. Immunological aspects in the neurobiology of suicide: Elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res (2008) 42(2):151–7. doi: 10.1016/j.jpsychires.2006.10.013
61. Torres-Platas SG, Cruceanu C, Chen GG, Turecki G, Mechawar N. Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun (2014) 42:50–9. doi: 10.1016/j.bbi.2014.05.007
62. Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein H-G, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflamm (2011) 8(1):1–9. doi: 10.1016/j.npbr.2012.02.043
63. Schnieder TP, Trencevska I, Rosoklija G, Stankov A, Mann JJ, Smiley J, et al. Microglia of prefrontal white matter in suicide. J Neuropathol Exp Neurol (2014) 73(9):880–90. doi: 10.1097/NEN.0000000000000107
64. Schiavone S, Neri M, Mhillaj E, Morgese MG, Cantatore S, Bove M, et al. The NADPH oxidase NOX2 as a novel biomarker for suicidality: Evidence from human post mortem brain samples. Transl Psychiatry (2016) 6(5):e813. doi: 10.1038/tp.2016.76
65. Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein H-G, et al. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol (Berl) (2006) 112(3):305–16. doi: 10.1007/s00401-006-0090-8
66. Moher D. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Ann Intern Med (2009) 151(4):264. doi: 10.7326/0003-4819-151-4-200908180-00135
67. Ferrer I, Martinez A, Boluda S, Parchi P, Barrachina M. Brain banks: benefits, limitations and cautions concerning the use of post-mortem brain tissue for molecular studies. Cell Tissue Bank (2008) 9(3):181. doi: 10.1007/s10561-008-9077-0
68. Busse M, Busse S, Myint AM, Gos T, Dobrowolny H, Müller UJ, et al. Decreased quinolinic acid in the hippocampus of depressive patients: evidence for local anti-inflammatory and neuroprotective responses? Eur Arch Psychiatry Clin Neurosci (2015) 265(4):321–9. doi: 10.1007/s00406-014-0562-0
69. Brisch R, Steiner J, Mawrin C, Krzyżanowska M, Jankowski Z, Gos T. Microglia in the dorsal raphe nucleus plays a potential role in both suicide facilitation and prevention in affective disorders. Eur Arch Psychiatry Clin Neurosci (2017) 267(5):403–15. doi: 10.1007/s00406-017-0774-1
70. Turkheimer FE, Rizzo G, Bloomfield PS, Howes O, Zanotti-Fregonara P, Bertoldo A, et al. The methodology of TSPO imaging with positron emission tomography. Biochem Soc Trans (2015) 43(4):586–92. doi: 10.1042/BST20150058
71. Holmes SE, Hinz R, Conen S, Gregory CJ, Matthews JC, Anton-Rodriguez JM, et al. Elevated translocator protein in anterior cingulate in major depression and a role for inflammation in suicidal thinking: A positron emission tomography study. Biol Psychiatry (2018) 83(1):61–9. doi: 10.1016/j.biopsych.2017.08.005
72. Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discovery (2010) 9(12):971–88. doi: 10.1038/nrd3295
73. Lavisse S, Guillermier M, Herard A-S, Petit F, Delahaye M, Van Camp N, et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci (2012) 32(32):10809–18. doi: 10.1523/JNEUROSCI.1487-12.2012
74. Mizrahi R, Rusjan PM, Kennedy J, Pollock B, Mulsant B, Suridjan I, et al. Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [18F]-FEPPA. J Cereb Blood Flow Metab (2012) 32(6):968–72. doi: 10.1038/jcbfm.2012.46
75. Chauveau F, Boutin H, Van Camp N, Dollé F, Tavitian B. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imag (2008) 35(12):2304–19. doi: 10.1007/s00259-008-0908-9
76. Wilson AA, Garcia A, Parkes J, McCormick P, Stephenson KA, Houle S, et al. Radiosynthesis and initial evaluation of [18F]-FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl Med Biol (2008) 35(3):305–14. doi: 10.1016/j.nucmedbio.2007.12.009
77. Fujimura Y, Zoghbi SS, Simèon FG, Taku A, Pike VW, Innis RB, et al. Quantification of translocator protein (18 kDa) in the human brain with PET and a novel radioligand, 18F-PBR06. J Nucl Med (2009) 50(7):1047–53. doi: 10.2967/jnumed.108.060186
78. Fujimura Y, Ikoma Y, Yasuno F, Suhara T, Ota M, Matsumoto R, et al. Quantitative analyses of 18F-FEDAA1106 binding to peripheral benzodiazepine receptors in living human brain. J Nucl Med (2006) 47(1):43–50.
79. Ikoma Y, Yasuno F, Ito H, Suhara T, Ota M, Toyama H, et al. Quantitative analysis for estimating binding potential of the peripheral benzodiazepine receptor with [11C]DAA1106. J Cereb Blood Flow Metab (2007) 27(1):173–84. doi: 10.1038/sj.jcbfm.9600325
80. Boutin H, Chauveau F, Thominiaux C, Grégoire M-C, James ML, Trebossen R, et al. 11C-DPA-713: A novel peripheral benzodiazepine receptor PET ligand for in vivo imaging of neuroinflammation. J Nucl Med (2007) 48(4):573–81. doi: 10.2967/jnumed.106.036764
81. Fookes CJR, Pham TQ, Mattner F, Greguric I, Loc’h C, Liu X, et al. Synthesis and biological evaluation of substituted [18F]Imidazo[1,2-a]pyridines and [18F]Pyrazolo[1,5-a]pyrimidines for the study of the peripheral benzodiazepine receptor using positron emission tomography. J Med Chem (2008) 51(13):3700–12. doi: 10.1021/jm7014556
82. Arlicot N, Vercouillie J, Ribeiro M-J, Tauber C, Venel Y, Baulieu J-L, et al. Initial evaluation in healthy humans of [18F]DPA-714, a potential PET biomarker for neuroinflammation. Nucl Med Biol (2012) 39(4):570–8. doi: 10.1016/j.nucmedbio.2011.10.012
83. Fujita M, Imaizumi M, Zoghbi SS, Fujimura Y, Farris AG, Suhara T, et al. Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential biomarker for inflammation. NeuroImage (2008) 40(1):43–52. doi: 10.1016/j.neuroimage.2007.11.011
84. Best L, Ghadery C, Pavese N, Tai YF, Strafella AP. New and old TSPO PET radioligands for imaging brain microglial activation in neurodegenerative Disease. Curr Neurol Neurosci Rep (2019) 19(5):24. doi: 10.1007/s11910-019-0934-y
85. Dupont A-C, Largeau B, Santiago Ribeiro MJ, Guilloteau D, Tronel C, Arlicot N. Translocator protein-18 kDa (TSPO) positron emission tomography (PET) imaging and its clinical impact in neurodegenerative diseases. Int J Mol Sci (2017) 18(4):785. doi: 10.3390/ijms18040785
86. Lapin IP, Oxenkrug GF. Intensification of the central serotonergic processes as a possible determinant of the thymoleptic effect. Lancet (1969) 18293(7586):132–6. doi: 10.1016/S0140-6736(69)91140-4
87. Oxenkrug GF. Tryptophan–kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: the serotonin hypothesis revisited 40 years later. Isr J Psychiatry Relat Sci (2010) 47(1):56–63 doi: 10.3410/f.13356987.14726252
88. Bryleva EY, Brundin L. Kynurenine pathway metabolites and suicidality. Neuropharmacology (2017) 112:324–30. doi: 10.1016/j.neuropharm.2016.01.034
89. Dantzer R, Walker AK. Is there a role for glutamate-mediated excitotoxicity in inflammation-induced depression? J Neural Transm (2014) 121(8):925–32. doi: 10.1007/s00702-014-1187-1
90. Achtyes E, Keaton SA, Smart L, Burmeister AR, Heilman PL, Krzyzanowski S, et al. Inflammation and kynurenine pathway dysregulation in post-partum women with severe and suicidal depression. Brain Behav Immun (2020) 83:239–47. doi: 10.1016/j.bbi.2019.10.017
91. Savitz J, Drevets WC, Wurfel BE, Ford BN, Bellgowan PSF, Victor TA, et al. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav Immun (2015) 46:55–9. doi: 10.1016/j.bbi.2015.02.007
92. Brundin L, Bryleva EY, Thirtamara Rajamani K. Role of inflammation in suicide: From mechanisms to treatment. Neuropsychopharmacology (2017) 42(1):271–83. doi: 10.1038/npp.2016.116
93. Schwarcz R, Guidetti P, Sathyasaikumar KV, Muchowski PJ. Of mice, rats and men: Revisiting the quinolinic acid hypothesis of Huntington’s disease. Prog Neurobiol (2010) 90(2):230–45. doi: 10.1016/j.pneurobio.2009.04.005
94. Guillemin GJ. Quinolinic acid, the inescapable neurotoxin. FEBS J (2012), 279(8):1356–65. doi: 10.1111/j.1742-4658.2012.08485.x
95. Brundin L, Erhardt S, Bryleva EY, Achtyes ED, Postolache TT. The role of inflammation in suicidal behaviour. Acta Psychiatr Scand (2015) 132(3):192–203. doi: 10.1111/acps.12458
96. Dantzer R, O’Connor JC, Lawson MA, Kelley KW. Inflammation-associated depression: From serotonin to kynurenine. Psychoneuroendocrinology (2011) 36(3):426–36. doi: 10.1016/j.psyneuen.2010.09.012
97. Stone TW, Forrest CM, Darlington LG. Kynurenine pathway inhibition as a therapeutic strategy for neuroprotection. FEBS J (2012) 279(8):1386–97. doi: 10.1111/j.1742-4658.2012.08487.x
98. Young KD, Drevets WC, Dantzer R, Teague TK, Bodurka J, Savitz J. Kynurenine pathway metabolites are associated with hippocampal activity during autobiographical memory recall in patients with depression. Brain Behav Immun (2016) 56:335–42. doi: 10.1016/j.bbi.2016.04.007
99. Cho HJ, Savitz J, Dantzer R, Teague TK, Drevets WC, Irwin MR. Sleep disturbance and kynurenine metabolism in depression. J Psychosom Res (2017) 99:1–7. doi: 10.1016/j.jpsychores.2017.05.016
100. Dahl J, Andreassen OA, Verkerk R, Malt UF, Sandvik L, Brundin L, et al. Ongoing episode of major depressive disorder is not associated with elevated plasma levels of kynurenine pathway markers. Psychoneuroendocrinology (2015) 56:12–22. doi: 10.1016/j.psyneuen.2015.02.011
101. Maes M, Galecki P, Verkerk R, Rief W. Somatization, but not depression, is characterized by disorders in the tryptophan catabolite (TRYCAT) pathway, indicating increased indoleamine 2,3-dioxygenase and lowered kynurenine aminotransferase activity. Neuroendocrinol Lett (2011) 32(3):264–73.
102. Meier TB, Drevets WC, Wurfel BE, Ford BN, Morris HM, Victor TA, et al. Relationship between neurotoxic kynurenine metabolites and reductions in right medial prefrontal cortical thickness in major depressive disorder. Brain Behav Immun (2016) 53:39–48. doi: 10.1016/j.bbi.2015.11.003
103. Møller SE, Kirk L, Honoré P. Tryptophan tolerance and metabolism in endogenous depression. Psychopharmacol (Berl) (1982) 76(1):79–83. doi: 10.1007/BF00430761
104. Myint A-M, Kim YK, Verkerk R, Scharpé S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: Evidence of impaired neuroprotection. J Affect Disord (2007) 98(1):143–51. doi: 10.1016/j.jad.2006.07.013
105. Savitz J, Dantzer R, Meier TB, Wurfel BE, Victor TA, McIntosh SA, et al. Activation of the kynurenine pathway is associated with striatal volume in major depressive disorder. Psychoneuroendocrinology (2015) 62:54–8. doi: 10.1016/j.psyneuen.2015.07.609
106. Savitz J, Drevets WC, Smith CM, Victor TA, Wurfel BE, Bellgowan PS, et al. Putative neuroprotective and neurotoxic kynurenine pathway metabolites are associated with hippocampal and amygdalar volumes in subjects with major depressive disorderr. Neuropsychopharmacology (2015) 40(2):463–71. doi: 10.1038/npp.2014.194
107. Sublette ME, Galfalvy HC, Fuchs D, Lapidus M, Grunebaum MF, Oquendo MA, et al. Plasma kynurenine levels are elevated in suicide attempters with major depressive disorder. Brain Behav Immun (2011) 25(6):1272–8. doi: 10.1016/j.bbi.2011.05.002
108. Hu L-J, Li X-F, Hu J-Q, Ni X-J, Lu H-Y, Wang J-J, et al. A simple HPLC–MS/MS method for determination of tryptophan, kynurenine and kynurenic acid in human serum and its potential for monitoring antidepressant therapy. J Anal Toxicol (2017) 41(1):37–44. doi: 10.1093/jat/bkw071
109. Krause D, Myint A-M, Schuett C, Musil R, Dehning S, Cerovecki A, et al. High kynurenine (a tryptophan metabolite) predicts remission in patients with major depression to add-on treatment with celecoxib. Front Psychiatry (2017) 8:16. doi: 10.3389/fpsyt.2017.00016
110. Veen C, Myint AM, Burgerhout KM, Schwarz MJ, Schütze G, Kushner SA, et al. Tryptophan pathway alterations in the postpartum period and in acute postpartum psychosis and depression. J Affect Disord (2016) 189:298–305. doi: 10.1016/j.jad.2015.09.064
111. Nikkheslat N, Zunszain PA, Horowitz MA, Barbosa IG, Parker JA, Myint A-M, et al. Insufficient glucocorticoid signaling and elevated inflammation in coronary heart disease patients with comorbid depression. Brain Behav Immun (2015) 48:8–18. doi: 10.1016/j.bbi.2015.02.002
112. Hennings A, Schwarz MJ, Riemer S, Stapf TM, Selberdinger VB, Rief W. Exercise affects symptom severity but not biological measures in depression and somatization – Results on IL-6, neopterin, tryptophan, kynurenine and 5-HIAA. Psychiatry Res (2013) 210(3):925–33. doi: 10.1016/j.psychres.2013.09.018
113. Quak J, Doornbos B, Roest AM, Duivis HE, Vogelzangs N, Nolen WA, et al. Does tryptophan degradation along the kynurenine pathway mediate the association between pro-inflammatory immune activity and depressive symptoms? Psychoneuroendocrinology (2014) 45:202–10. doi: 10.1016/j.psyneuen.2014.03.013
114. Setoyama D, Kato TA, Hashimoto R, Kunugi H, Hattori K, Hayakawa K, et al. Plasma metabolites predict severity of depression and suicidal ideation in psychiatric patients-a multicenter pilot analysis. PloS One (2016) 11(12):e0165267. doi: 10.1371/journal.pone.0165267
115. Ogyu K, Kubo K, Noda Y, Iwata Y, Tsugawa S, Omura Y, et al. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci Biobehav Rev (2018) 90:16–25. doi: 10.1016/j.neubiorev.2018.03.023
116. Bradley KAL, Case JAC, Khan O, Ricart T, Hanna A, Alonso CM, et al. The role of the kynurenine pathway in suicidality in adolescent major depressive disorder. Psychiatry Res (2015) 227(2):206–12. doi: 10.1016/j.psychres.2015.03.031
117. Brundin L, Sellgren CM, Lim CK, Grit J, Pålsson E, Landén M, et al. An enzyme in the kynurenine pathway that governs vulnerability to suicidal behavior by regulating excitotoxicity and neuroinflammation. Transl Psychiatry (2016) 6(8):e865–5. doi: 10.1038/tp.2016.133
118. Clark SM, Pocivavsek A, Nicholson JD, Notarangelo FM, Langenberg P, McMahon RP, et al. Reduced kynurenine pathway metabolism and cytokine expression in the prefrontal cortex of depressed individuals. J Psychiatry Neurosci (2016) 41(6):386–94. doi: 10.1503/jpn.150226
119. Krause DL, Riedel M, Müller N, Weidinger E, Schwarz MJ, Myint A-M. Effects of antidepressants and cyclooxygenase-2 inhibitor on cytokines and kynurenines in stimulated in vitro blood culture from depressed patients. Inflammopharmacology (2012) 20(3):169–76. doi: 10.1007/s10787-011-0112-6
120. Erhardt S, Lim CK, Linderholm KR, Janelidze S, Lindqvist D, Samuelsson M, et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology (2013) 38(5):743–52. doi: 10.1038/npp.2012.248
121. Schwieler L, Samuelsson M, Frye MA, Bhat M, Schuppe-Koistinen I, Jungholm O, et al. Electroconvulsive therapy suppresses the neurotoxic branch of the kynurenine pathway in treatment-resistant depressed patients. J Neuroinflamm (2016) 13(1):51. doi: 10.1186/s12974-016-0517-7
Keywords: suicide, stress-diathesis model, microglia, neuroinflammation, post-mortem, positron emission tomography
Citation: Baharikhoob P and Kolla NJ (2020) Microglial Dysregulation and Suicidality: A Stress-Diathesis Perspective. Front. Psychiatry 11:781. doi: 10.3389/fpsyt.2020.00781
Received: 04 May 2020; Accepted: 22 July 2020;
Published: 11 August 2020.
Edited by:
Bart Ellenbroek, Victoria University of Wellington, New ZealandReviewed by:
Gilles J. Guillemin, Macquarie University, AustraliaCopyright © 2020 Baharikhoob and Kolla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathan J. Kolla, bmF0aGFuLmtvbGxhQGNhbWguY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.