
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 14 May 2020
Sec. Psychopharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fpsyt.2020.00369
This article is part of the Research TopicDecreasing the Impact of Treatment Resistance in Schizophrenia: Identifying Novel Molecular Targets/ Pathways to Increase Treatment EfficacyView all 16 articles
 Andrea de Bartolomeis1*
Andrea de Bartolomeis1* Mirko Manchia2,3
Mirko Manchia2,3 Federica Marmo1
Federica Marmo1 Licia Vellucci1
Licia Vellucci1 Felice Iasevoli1
Felice Iasevoli1 Annarita Barone1
Annarita Barone1Treatment-resistant schizophrenia (TRS) or suboptimal response to antipsychotics affects almost 30% of schizophrenia (SCZ) patients, and it is a relevant clinical issue with significant impact on the functional outcome and on the global burden of disease. Among putative novel treatments, glycine-centered therapeutics (i.e. sarcosine, glycine itself, D-Serine, and bitopertin) have been proposed, based on a strong preclinical rationale with, however, mixed clinical results. Therefore, a better appraisal of glycine interaction with the other major players of SCZ pathophysiology and specifically in the framework of dopamine – glutamate interactions is warranted. New methodological approaches at cutting edge of technology and drug discovery have been applied to study the role of glycine in glutamate signaling, both at presynaptic and post-synaptic level and have been instrumental for unveiling the role of glycine in dopamine-glutamate interaction. Glycine is a non-essential amino acid that plays a critical role in both inhibitory and excitatory neurotransmission. In caudal areas of central nervous system (CNS), such as spinal cord and brainstem, glycine acts as a powerful inhibitory neurotransmitter through binding to its receptor, i.e. the Glycine Receptor (GlyR). However, glycine also works as a co-agonist of the N-Methyl-D-Aspartate receptor (NMDAR) in excitatory glutamatergic neurotransmission. Glycine concentration in the synaptic cleft is finely tuned by glycine transporters, i.e. GlyT1 and GlyT2, that regulate the neurotransmitter's reuptake, with the first considered a highly potential target for psychosis therapy. Reciprocal regulation of dopamine and glycine in forebrain, glycine modulation of glutamate, glycine signaling interaction with postsynaptic density proteins at glutamatergic synapse, and human genetics of glycinergic pathways in SCZ are tackled in order to highlight the exploitation of this neurotransmitters and related molecules in SCZ and TRS.
Schizophrenia (SCZ) is a chronic and debilitating severe mental disorder affecting approximately 0.3–0.7% of the population worldwide (1). It is characterized by a pleomorphic symptomatology including hallucinations, delusions (“positive symptoms”), social withdrawal, avolition and anhedonia (“negative symptoms”), and deficits in multiple executive functions (cognitive symptoms). SCZ is nowadays conceptualized at molecular level as a disorder of the synaptic plasticity (2) and of abnormal cortical-subcortical connectivity (3–5). Most of the individuals affected by SCZ develop their illness in adolescence and early adulthood with about 15% showing a chronic and unremitting clinical course (6). The long-term, if not lifelong, illness trajectory, the associated high mortality, mostly determined by the elevated rates of medical comorbidities and suicide (7), and the low levels of recovery (8), make this disease a major psychiatric disorder with a great need of significant therapeutic innovation. Furthermore, the treatment response to antipsychotics, the mainstay of SCZ treatment, remains suboptimal (9). A recent study, in which analysis of 16 randomized controlled trials (RCT) were pooled together, showed that the percentage of short-term non-response ranged from 20 to 87% depending on the threshold applied, with a non-remission rate of 67% (9). In addition, a not-negligible proportion (up to 20%) of SCZ patients who are resistant to standard antipsychotic treatment, does not respond even to clozapine (10), which is the gold standard in this scenario. In this context, the identification of clinically novel effective and safe pharmacological treatments is crucial.
The interest for the role of glycine, a co-agonist with glutamate at N-methyl-D-aspartate receptor (NMDAR) in the framework of dopamine-glutamate interactions for SCZ pathophysiology and treatment, stems from the following evidence: 1) increased dopamine release in the striatum is one of the most replicated in vivo findings in SCZ pathophysiology (11–14); 2) all antipsychotics block or occupy dopamine D2 receptor (D2R) with no exception (15–18); 3) dopamine release is controlled, among other mechanisms, by NMDARs modulation (19); 4) NMDAR hypofunction is believed to be one of the putative pathogenetic mechanism of the disease (19, 20); 5) glutamatergic dysfunction, moreover, has been implicated even in those cases of SCZ that are not characterized by dopamine excess in subcortical regions and are not responsive to conventional antipsychotics (21–25); 6) multiple lines of evidence indicate a reciprocal modulation of both dopamine and glutamate by glycine (26); 7) over time different pharmacological glycine-centered approaches for treatment-resistant schizophrenia (TRS) have been proposed with strong preclinical rationale but mixed clinical results (27, 28).
New methodological approaches at the cutting edge of the technology such as long-timescale molecular dynamics simulations (29, 30) and single molecule fluorescence resonance energy transfer (smFRET) (31) have unveiled the very specific details of the structural and functional mutual interaction between glycine and glutamate system at preclinical level. At clinical level novel human genetic findings and imaging genetics studies link glycine signaling to SCZ. Given the extensive evidence that glycine is deeply involved in regulating glutamatergic neurotransmission, glycine reveals itself as promising potential candidate for drug discovery and safe novel pharmacological treatments. Glycine binding site on GluN1 and GluN3A NMDAR subunits is a major determinant in regulating NMDAR delivery on cell surface and furthermore influencing significantly the NMDAR activity (32).
Here, we aim to selectively review the preclinical and clinical evidence demonstrating that glycine, as well as the components of its signaling pathway, might be suitable targets for the identification of novel treatment strategies in severe psychiatric disorder and particularly in SCZ. The following research questions have led our dissertation:
1. Which is the role of glycine in the general framework of dopamine–glutamate interaction and SCZ pathophysiology?
2. How does glycine affect dynamics of post-synaptic proteins?
3. How and to what extent can glycine neurotransmission and its interaction with NMDAR be exploited to unveil novel treatments for TRS?
First, therein we describe glycine transmission, focusing on the characteristics of its components, namely receptors and transporters and their relevance in the brain circuits relevant for SCZ clinics and pathophysiology. Then, we detail the role of glycine in regulating dopamine-glutamate interaction, as well as its involvement in SCZ molecular pathophysiology. Furthermore, we review the evidence on the associations of genes encoding for elements of the glycine pathway with SCZ. Finally, a critical appraisal of potential role of glycinergic agents in treatment of psychiatric diseases is addressed.
Traditional models of SCZ focused on dopaminergic dysfunction to explain key symptoms of the disorder. This hypothesis holds that hyperactivity of dopamine transmission is responsible for positive symptoms (1) and it was formulated in the 1960s, after the discovery of antipsychotic action of chlorpromazine (33), and further endorsed by the correlation between clinical response to antipsychotic drugs and their potency to block D2 receptors (34, 35). Neuroimaging studies using positron emission (PET) or single photon emission (SPECT) demonstrated that, after acute amphetamine administration, patients with SCZ showed greater levels of dopamine release in subcortical regions (particularly in the striatum) compared to healthy subjects, and displayed a transient worsening of positive symptoms (11, 36), supporting the idea that hyperfunction of dopaminergic neurons is a relevant, albeit not unique, component in SCZ pathogenesis.
Administration of phencyclidine (PCP), ketamine, and other NMDAR antagonists has been known to reproduce those thought disorders observed in SCZ, such as poverty of speech, circumstantiality, and loss of goal (37). Furthermore, NMDAR antagonists may affect widespread neuropsychological domains: working memory, response inhibition, and executive processing, resulting in cognitive symptoms that are also described in SCZ, suggesting the involvement of glutamatergic neurotransmission in the pathogenetic mechanism underlying psychotic and cognitive abnormalities (38). Therefore, glutamatergic neurotransmission has been proposed as the major initial aberration in the pathophysiology of SCZ. Subcortical dopaminergic dysregulation itself might be a result of impairment in glutamatergic neurons projecting from prefrontal cortex (PFC) to midbrain dopaminergic neurons, therefore exerting control on their firing (19, 39). Indeed, in animals and humans, it has been demonstrated that NMDAR antagonist administration results in an increase of amphetamine-induced dopamine release (19, 40). These data support the hypothesis of a deficiency of glutamatergic control on dopamine neuronal activity that might underlie the increase in amphetamine-induced dopamine release.
The activation of GABAergic interneurons by glutamatergic projections is mediated by NMDAR, and NMDAR-hypofunction may specifically affect corticolimbic GABAergic parvalbumin-positive (PV+) interneurons, reducing their excitability and expression of specific molecular markers such as somatostatin and vasoactive intestinal peptide (VIP), as well as increasing oxidative stress (41). Transgenic mice with selective NMDAR deletion in cortical and hippocampal GABAergic interneurons showed specific SCZ-like phenotypes (42), supporting the so-called “GABAergic origin hypothesis” of SCZ (41).
Since NMDAR dysfunctions account for both dopaminergic and GABAergic dysregulation, it can be assumed that NMDAR dysfunction could represent the final common pathway leading from pathogenesis to symptoms (43). Glycine is deeply involved in regulating the glutamatergic transmission, acting as a co-agonist of NMDAR, allowing for its activation and enhancing excitatory glutamatergic tone (44, 45). Glycine is also involved in the regulation of dopamine transmission, exerting a multimodal action depending on its concentration and possibly inhibiting dopamine release in the striatum when administered at high doses (presumably by modulating the dopaminergic hyperfunction associated to SCZ) (46).
Potential implication of glycine signaling in the pathophysiology of SCZ is supported by a number of recent studies exploring genetic abnormalities within glycinergic system associated with SCZ as well as by the evidence of potential pro-cognitive and antipsychotic phenotype exhibited by animal models of Glycine Transporter type 1 (GlyT1) functional inhibition both by recombinant knock out (47) and pharmacological treatments (48, 49). Finally, the recent finding of elevated brain glycine and glutamate levels in patients with first-episode psychosis, measured in vivo by means of echo time–averaged proton magnetic resonance spectroscopy (MRS) at 4 Tesla, further confirm the relevant role of glycine in the framework of multiple interacting neurotransmitters in SCZ pathophysiology (50).
Glycine is widely distributed in the mammalian central nervous system (CNS), functioning as an inhibitory or excitatory neurotransmitter, depending on its localization. Glycine is the main neurotransmitter in inhibitory interneurons of the spinal cord, brainstem, and in some other brain regions involved in the processing of sensorimotor information and locomotor behavior (51). In the CNS, glycine is synthesized through the catalysis of serine by the isoenzyme serine hydroxymethyltransferase (SHMT), and it is largely degraded by the glycine cleavage system, also known as glycine decarboxylase complex (GDC) (52). Glycine is released by Renshaw interneurons and regulates motoneurons' excitability, exerting negative feedback through recurrent inhibition (53). Glycinergic inhibitory interneurons are involved also in the spinal reflex coordination, mediating reciprocal inhibition in stretch reflex circuits and regulating the coordination of opposing muscles (54). The anatomical distribution of glycine immunoreactive (IR) cell bodies points to the cochlear nuclei, the superior olivary complex, the medial nuclei of the trapezoid body, the cerebellar cortex, the deep cerebellar nuclei, the area postrema, and the thalamus of adult rats as main localizations (55, 56). Moreover, glycine-IR fibers are localized in the hypothalamus and basal forebrain, distant from their glycine IR cell bodies (56). Glycine receptors (GlyR) have been found enriched in the spinal cord, in apical dendrites of pyramidal neurons in the cerebral cortex (57), in the limbic system, and in the hippocampus of humans and rats (58), where they are involved in synaptic plasticity (59) and in a variety of physiological processes, especially in mediating inhibitory neurotransmission.
Glycine can activate two classes of distinct ligand-gated ion channels: chloride-permeable inhibitory GlyRs, and cation selective excitatory NMDARs. GlyRs are ligand-gated anionic channels and belong to the pentameric Cys-loop receptor superfamily (60). Electrophysiological, immunocytochemical, and in situ hybridization studies have shown that GlyRs are prominent in the brainstem and spinal cord (61, 62) and detectable also in the following brain regions: prefrontal cortex, hippocampus, amygdala, hypothalamus, cerebellum, nucleus accumbens, ventral tegmental area, and substantia nigra (63–65). GlyRs exist either in homomeric or heteromeric forms and are composed by five subunits arranged symmetrically in a ring around a central Cl− permeable pore. Heteromeric GlyRs are localized at the synapses and consist of three α and two β subunits, forming a pentameric receptor complex. The homomeric forms are composed of five α subunits and are located extra-synaptically. The β subunits colocalize with receptor-associated protein gephyrin, that anchors the GlyR complex at the synaptic locus, thus providing a cluster of heteroligomeric GlyRs within synapses (66, 67). The expression of α subunits changes during neurodevelopment and it is regionally specific, whereas β subunits are transcribed in all developmental stages in several regions. Recent studies have detected functional GlyRs even in absence of the glycinergic terminals in dopaminergic neurons of the juvenile immature substantia nigra pars compacta and in developing cortical neurons, but the function of these non-synaptic GlyRs remains unclear (68, 69). Overall, a variety of functions may be performed by GlyR, depending on the major subunit of the receptor and its oligomerization. Moreover, the pattern of GlyR expression seems to be relevant during critical stages of brain development in cortical and subcortical brain regions that have attracted the attention for the animal modeling of SCZ pathophysiology.
As an excitatory neurotransmitter, glycine acts as a co-agonist of NMDAR, allowing for depolarization, removal of the magnesium blockade and Na+/Ca2+ passage through the channel, which ultimately enhances the glutamatergic excitatory tone that is critical for learning and neuronal plasticity (44, 45). While glutamate binds to a bi-lobulated cavity in NMDAR GluN2 subunit, glycine binds to a cavity located in GluN1 or GluN3, the so-called glycine-B site or strychnine-insensitive receptor (51). Glycine may be released at excitatory sites from at least two different sources: i.e., neuronal cells via alanine–serine–cysteine transporter-1 (Asc-1) (70) and astroglial cells via the functional reversal of GlyT1 (26, 71, 72). Moreover, since it colocalizes with NMDAR at post-synaptic level (73, 74), GlyT1 is believed to modulate the excitability of NMDAR by reducing glycine levels in the synaptic cleft, thus preventing saturation of the glycine-B site (75–78).
Noteworthy, the affinity of glycine for NMDARs is significantly higher than that of GlyRs (EC50 = 134 nM vs. EC50 = 270 μM) (63, 79), thus, under physiological conditions endogenous glycine may exert mainly an excitatory effect in the hippocampus, where both GlyRs and NMDARs are expressed. On the other hand, excessive glycine produced in pathological conditions, such as ischemia and epilepsy (80, 81), may spillover into extra-synaptic sites to activate inhibitory GlyRs in order to counteract the excitotoxic damage. In these conditions, GlyR-mediated inhibitory activity may be stronger than NMDAR-mediated excitatory one, resulting in a net effect of depression of excitatory post-synaptic currents (EPSCs) (82). Therefore, levels of glycine could be the major determinants in setting the polarity of glycine's role either in brain damage, either in correcting unwanted synaptic plasticity (82, 83)
Multiple lines of evidence suggest a relevant crosstalk between glycine and D-amino acids during the neurodevelopmental stages that are critical to SCZ pathophysiology. D-Serine is synthetized in the neurons starting from astrocytic L-serine by serine racemase (SR), according to the “serine shuttle” hypothesis formulated by Wolosker, (84) and its levels in the synaptic cleft are controlled by Asc-1 transporter (70, 85, 86). Among D-amino acids, D-serine seems to be a crucial player in synaptic plasticity, such as long term potentiation (LTP) (87–91), and it has been considered the putative endogenous ligand at NMDAR glycine B-site (92, 93), since it appears to be functionally up to 100-fold more effective than glycine at potentiating NMDAR activity. The role of D-serine in the activation of NMDAR is confirmed by the reduction of synaptic transmission by treatment with D-amino acid oxidase (DAAO), which depletes endogenous D-serine but not glycine (93). Noteworthy, NMDAR responses do not seem to be fully reversed by DAAO and a “DAAO-insensitive fraction” has been shown in rat hippocampus, that accounts up to 30–50% of receptor activity (93), presumably because the remainder of the sites may be already occupied by glycine, which therefore may act in some parts of the brain and at certain stages of the neurodevelopment as the major ligand. Immunohistochemical studies comparing D-serine, glycine, and NMDARs pattern of distribution in rat brain, showed that D-serine and NMDARs overlap each other and have the highest concentration in telencephalon and developing cerebellum; conversely, glycine immunoreactivity does not correspond to NMDARs localization (except in the brainstem, where it parallels the distribution of NMDARs) but seems to prevail over D-serine in the adult cerebellum, hindbrain, and olfactory bulb (94). Papouin and colleagues performed an electrophysiological study, in order to assess the specific contribution of glycine and D-serine at synaptic and extra-synaptic NMDAR sites in CA1 region of hippocampus. Using specific enzymes that degrade either D-serine or glycine, they provide supporting evidence for assuming that D-serine may be the co-agonist at synaptic receptors, whereas glycine may act at extra-synaptic NMDARs, which have little or no role in synaptic plasticity (90). Nonetheless, Yan Li and colleagues proposed that the identity of the endogenous ligand might be determined by the level of synaptic activity, thus emphasizing the contribution of glycine in LTP induction process (91), and extending previous in vitro reports supporting the involvement of glycine in LTP enhancement (95). They showed that tonic activation of NMDARs in the amygdala under resting conditions may be achieved by D-serine, whereas glycine may be released from astrocytes in response to afferent impulses. Therefore, ambient D-serine may act as major ligand in absence of evoked synaptic events, while activity-dependent release of glycine may be involved in LTP-related NMDAR activation in the context of fear conditioning pathways (91). Rosenberg et al. also proposed that D-serine would not be the sole co-agonist at synaptic NMDAR sites: glycine and D-serine may have partial overlapping roles in regulating synaptic activity at NMDARs, and specific glycine effects may be revealed by deleting serine racemase (SR), the enzyme that synthesized D-serine (70). In fact, in an electrophysiological experiment, they demonstrated that the synaptic NMDAR responses were essentially unaltered in adult SR-KO mice (70). Moreover, it has been demonstrated that even the GlyT1 inhibitor bitopertin increases the magnitude of LTP in rat hippocampal CA1 pyramidal cells, and this effect likely results from an increase in the extracellular levels of glycine (96). Direct application of glycine seems to exert the same effects on LTP induction, as well as increases the amplitude of NMDAR currents of approximately 50% (96). Nonetheless, it has been reported that application of high concentrations of glycine, exceeding the synaptic concentration of the endogenous Glycine B-site agonist, produce opposite effects on NMDAR currents amplitude and LTP, consistent with the internalization of a percentage of NMDARs primed by glycine (96). Glycine and glycine inhibitors may therefore display an inverted U-shape concentration-response profile on LTP induction, for whom higher glycine B-site occupancies may lead to a lack of efficacy (49). However, taken together, these findings suggest that two endogenous co-agonists, namely glycine and D-serine, may regulate distinct populations of NMDARs, with one or the other prevailing at a given synapse, finely tuning excitatory transmission in order to diversify a wide ranging repertoire of biological effects (97).
Interestingly, several lines of evidence disclose the role of D-serine for inflammation, excitotoxicity, and epileptogenesis. Inflammatory factors (such as amyloid β and lipopolysaccharide) stimulates the astrocytes and microglia to express SR (98, 99), thus these cells become the primary source of D-serine in inflammatory conditions (100). The amount of D-serine obtained by this way promotes excitotoxic damage and synaptic dysfunction through the activation of NR2 subunit at extra-synaptic sites (101). Transgenic mouse models for amyotrophic lateral sclerosis (ALS) exhibit several-fold higher levels of D-serine in spinal cord, and the elevation positively correlates with disease progression (102). Moreover, D-serine increase in spinal cord was observed even in sporadic postmortem human ALS cases or ALS relatives (102). Furthermore, it may be of interest that SR knockout mice were protected against cerebral ischemia and excitotoxic damage (103). These findings suggest that D-serine, rather than glycine, may be a key determinant for NMDAR-mediated neurotoxicity.
Correlation between D-serine levels and SCZ is demonstrated by multiple studies (104–107) that showed the decrease of its levels in CSF and serum of schizophrenic patients. Despite convergent lines of evidence pointing to the potential of D-serine in treating SCZ, it displays a low oral bioavailability, being largely metabolized by DAAO (104). Past clinical trials have demonstrated benefits of adding D-serine to the antipsychotic therapy in SCZ and bipolar disorder (108), but these results were not unequivocally replicated (109–111), leaving aside the fact that the high doses required may provoke peripheral neuropathies and nephrotoxic effects (112–114). Rather than therapeutic agent for SCZ symptoms, D-serine has recently been proposed as promising biomarker to antidepressant response to ketamine (112), with low plasma levels of D-serine predicting ketamine efficacy.
In summary, D-serine action at NMDAR may be more relevant than originally thought, and may have a pivotal role for synaptic plasticity and cognitive functions, as well as neurodegeneration and excitotoxicity.
Finally, even if it is not the topic of this review, the role of other D-amino acids in NMDAR modulation should be acknowledged, and among all the one of D-aspartate. This amino acid has been implicated in brain development (115, 116), a feature that is specifically appealing for SCZ, that is conceptualized as a pathology of the neurodevelopment with abnormal synaptic pathophysiology and altered brain connectivity. Moreover, multiple lines of evidence from animal modeling (117, 118) to postmortem brain abnormal gene expression and epigenetics (119) indicate a potential role of D-aspartate in SCZ pathophysiology, and lay the foundation for a potential use of D-aspartate as adjunctive therapy in those cases poorly responding to conventional antipsychotics (120).
Several recent studies have focused on changes in glycinergic signaling and expression pattern of glycinergic markers, such as glycine transporters and glycine receptors during the development (68, 121) making the neurotransmitter of interest for a disease believed to be of putative neurodevelopmental origin, such as SCZ. It can be hypothesized that the synaptic release of glycine is involved in the proper development of many motor and sensory circuits (i.e., auditory, visual, respiratory, and nociceptive) (122, 123). GlyRs expression is specifically regulated in terms of subunit composition during the development and throughout the CNS. Homomeric α2 subunits are mainly expressed during the fetal period. Thus, a developmental switch from α2 homomeric GlyRs to α1β heteromeric GlyRs takes place between the birth and the third postnatal week in rats (69). Indeed, several studies demonstrated that α1 is the most abundantly expressed subunit in adult rats and, since β subunit interaction with gephyrin is essential for the clustering of GlyRs in the synapses, it is plausible that the α1β heteromeric form of GlyRs is the most common subtype within synapses (124).
Different subtypes of GlyRs might fulfill opposite roles during the development. Since intracellular chloride concentrations are high in embryonic neurons, homomeric α2 GlyRs expressed during the fetal period might be excitatory, mediating the depolarizing chloride flux and the subsequent inward calcium flux. Therefore, GlyR activation may exert an excitatory action in immature neurons, whereas it mediates inhibitory neurotransmission in adult CNS, by increasing Cl- permeability and leading to a membranal hyperpolarization (125). Precisely this latter subtype, α2 GlyR, seems to be involved in pathophysiology of the autism spectrum disorder (ASD), condition that shares many clinical features and biomarkers with SCZ (126). In ASD is assumed to be an imbalance between glutamate and glycine in favor of an increased activity of glutamatergic neurotransmission early in neuromotor development. As highlighted by genetic and functional studies, suggesting the potential role of α2 GlyRs in synaptic plasticity, as well as learning and memory, glycinergic signaling might be linked to social and cognitive abnormalities in ASD (127). In summary, glycine neurotransmission seems to be highly modulated during brain development. This is in line with a potential crucial involvement of this neurotransmitter in pathophysiology of a disease, such as SCZ, strongly associated to a neurodevelopmental dysregulation.
NMDARs have unique functional characteristics: voltage dependence, calcium permeability, slow kinetics, and complex modulatory processes (128). NMDAR alone requires for an efficient gating the binding of both glutamate and a co-agonist, identified as glycine in the 1987 by Johnson and Asher (129). NMDARs are hetero-oligomeric proteins composed by a combination of different subunits called GluN1, GluN2, and GluN3. Notably, while GluN1 subunit is mandatory, different subunit composition leads to different receptor properties. The GluN1 subunit forms the glycine binding site, whereas the GluN2 subunit provides part of the glutamate binding site; moreover, the two sites appear to be allosterically coupled (130). Mice that express reduced levels of GluN1 subunits display a lowered glycine affinity and a variety of cognitive and learning defects, including hyperactivity, increased stereotyped behavior, disruptions of nest building activity, and poor performance in the Morris water maze, a measure of cued learning (131, 132). The behavioral phenotypes of these glycine-insensitive mutant mice may resemble in certain respects the positive and negative symptoms of SCZ, consistent with NMDAR hypofunction hypothesis. This evidence further emphasizes the role of glycinergic signaling in the pathophysiology of SCZ.
Glycine regulates glutamatergic neurotransmission at different levels; however, glutamate affects glycine concentration too. In fact, in vitro studies showed that elevated extracellular glutamate concentration reduces glycine release and high-frequency trains of stimulation decrease glycinergic inhibitory post-synaptic currents (IPSC) (133).
Glycine transporters (GlyT) are membrane-bound proteins belonging to the Na+/Cl− dependent neurotransmitter transporters family involved in the reuptake of glycine from synaptic cleft. Two glycine transporters, encoded by different genes, are known: GlyT1 and GlyT2. They share an amino acid sequence identity of approximately 50%, but differ in their expression pattern, subcellular localization, and functional properties (125).
GlyT1 works in a bidirectional fashion with a stoichiometry of 2Na+/Cl−/Gly, regulating glycine availability in the extracellular space, and terminates glycine signaling (134), significantly modulating the glutamatergic neurotransmission. GlyT1 has long been considered as exclusively expressed by glial cells, since early immunohistochemical studies did not recognize GlyT1 neuronal forms, presumably due to epitope occlusion of neuronal protein (135). However, there is an increasing evidence that GlyT1 is also expressed in neurons throughout the brain, where it is closely associated with the glutamatergic pathway (136). In glutamatergic neurons, GlyT1 is localized in both pre-synaptic membrane and postsynaptic density, where it interacts with the scaffold protein PSD-95 (136, 137).
GlyT1 plays a pivotal role in neurodevelopment as well as in cognitive processes of adult brain, as shown by the phenotype of GlyT1 mutant mice. Homozygous GlyT1−/− mutant mice appeared normal but unexpectedly died on the first day of birth, showing severe motor-sensory deficits, suggesting a vital role for GlyT1 that, even if dispensable for embryonic development, it is crucial for postnatal survival (138). Heterozygous GlyT1+/− mice, on the other hand, survive and show promnesic phenotypes, as well as a resistance to amphetamine disruptive effect on prepulse inhibition (PPI) (47, 139, 140). PPI of the acoustic startle reflex is an operational measure of the pre-attentive filtering process known as sensorimotor gating (141, 142) that is disrupted in SCZ as well as after stimulants administration even in healthy subjects, whereas antipsychotic drugs commonly are able to reverse PPI disruption (143–145). Therefore, it has been suggested that a radical reduction in expression of GlyT1 may be responsible for sensori-motor gating deficits due to the hyperactive inhibitory glycine-mediated signaling (146), whereas a mild reduction in expression of GlyT1 might enhance NMDAR function and memory retention, and might be protective against amphetamine-induced sensorimotor gating deficits, suggesting that drugs which inhibit GlyT1 might exert procognitive and antipsychotic effects (47).
Conversely to GlyT1, GlyT2 is exclusively expressed by glycinergic neurons and localized in presynaptic terminals adjacent to the active zones. Within the glycinergic bouton, GlyT2 appears associated with the plasma membrane or as discrete clumps and it seems to be excluded from the active site of the synapse and synaptic cleft (147). Unlike GlyT1, GlyT2 is coupled to electrochemical movement of 3Na+, maintaining the high concentration gradient on the presynaptic terminals and refilling presynaptic vesicles with glycine (148). Further, its expression is restricted to regions with glycinergic transmission, such as the cerebellum, brainstem, and the spinal cord (135). Homozygous GlyT2 −/− knockout mice also die in the second postnatal week, however, they show a phenotype entirely different from GlyT1 knockout mice, developing a lethal motor deficiency, reminiscent of severe forms of human hyperekplexia (hereditary startle disease), characterized by muscular spasticity, tremor, and impaired motor coordination (149).
GlyT1 function is thought to be closely regulated by several molecular mechanisms, e.g., inhibition by arachidonic acid, a second messenger released following phospholipase A2 activation (150). Moreover, intracellular pH value also modulates GlyT1 activity. Low doses of Zn++, which is released with glutamate by different types of excitatory neurons, induce GlyT1 inhibition but have no effect on GlyT2 (151). Activation of protein kinase C (PKC), induced by sustained intracellular Ca++ influx, decreases GlyT1 and GlyT2 expression on the neuron surface (152). Probably PKC does not affect directly GlyTs, but intermediate substrate proteins and additional kinases such as MEK1/2 kinases or PI3-kinase and CaMKII are involved in this mechanism (152). In addition, several proteins interacting with GlyTs regulate their trafficking and recycling at the pre-synaptic terminal. Particularly, Ca++ influx induced by depolarization promotes the GlyT2 expression on plasma membrane surrounding the active zone and this process is thought to be regulated by the interaction between GlyT2 and syntaxin-1 (153).
In summary, it has been hypothesized that GlyTs have a pivotal role in the regulation of neurotransmission both in glycinergic and in glutamatergic synapses and several lines of research suggest that changes in the activity, density, and localization of GlyTs in glial and nerve terminals are involved in synaptic efficacy and neuronal plasticity.
Glutamate effects on dopamine regulation have been extensively recognized, since the evidence that NMDAR-antagonists increase dopamine release in the striatum dates back to 1998. It is also well known that glycine acts as a co-agonist of NMDAR, but less is known regarding the effect of glycine on dopamine release both in cortex and in striatum. Consistent with its ability to reverse PCP-induced hyperactivity and psychotic-like symptoms, it is conceivable that glycine may decrease NMDAR-mediated dopamine release. Nevertheless, evidence in this respect is controversial and glycine seems to be able to increase, decrease, or have no effect on dopamine release.
Earlier studies on rat striatal slices unexpectedly showed a net effect of glycine to potentiate dopamine release in the striatum (154), and these results were replicated in other studies (155, 156); later this phenomenon was observed even in freely moving rats, to whom glycine was administered via a microdialysis probe in the anterior striatum, and who exhibited increased local release of dopamine and its metabolites (157). A recent study also shows that glycine may potentiate the excitability of dopaminergic neurons in substantia nigra pars compacta (SNc) by amplifying NMDAR-dependent signals. In fact, exogenous applications of glycine on midbrain slices of juvenile rats may regulate dopaminergic firing, leading to a switch from tonic spontaneous firing to the bursting activity, and then increase dopamine release at post-synaptic sites (158). If that were true, glycine effect on dopamine in the striatum would not be advantageous for treating psychosis; nevertheless, glycine could rather be involved in nigral information processing and locomotor behavior. As shown by a recent study, glycine binding site stimulants might be helpful in alleviating antipsychotic-induced EPS in treated patients rather than their psychotic symptomatology, potentially by mitigating the reduced dopamine function in the nigrostriatal pathway (159).
On the other hand, according to an in vitro study on mouse striatal tissue, low-dose of glycine seems to potentiate basal dopamine release from presynaptic dopamine terminals, whereas high-dose glycine showed to significantly inhibit striatal dopamine release, which would be expected to be therapeutically beneficial in SCZ (160). The same authors have later demonstrated that a GlyT1 inhibitor, ALX5311, potentiates NMDA-dependent GABA-release, and that this effect leads to significant inhibition of striatal dopamine release, supporting a model in which NMDARs have dual excitatory/inhibitory function within striatum (46). Therefore, glycine might exert an excitatory effect by acting on NMDARs located on pre-synaptic dopamine terminal or conversely, an inhibitory effect by acting on NMDARs located on GABAergic interneurons, increasing or decreasing, respectively, striatal dopamine release (46).
In summary, glycine seems to exert a multimodal effect on regulation of dopamine release, depending on the brain region in which the action is considered, glycine concentration, pre-synaptic or post-synaptic action, as well as dopaminergic functional state. This multimodal action should be taken into account to explain some inconsistent clinical effects of glycinergic agents in SCZ therapy.
Another way in which glycine modulates dopamine neurotransmission is through GlyRs. GlyRs-mediated regulation of dopaminergic firing seems to involve other neurotransmitters such as GABA (161) and acetylcholine in ventral tegmental area (162), and glutamate in nucleus accumbens (163). The role of GlyRs in the regulation of mesolimbic dopaminergic neurotransmission is confirmed by a study in which accumbal perfusion of strychnine (the GlyRs antagonist) was found to decrease dopamine levels in rats, and this effect was reverted by glycine (164). Moreover, ethanol may produce its reinforcing and dopamine-elevating effects precisely via GlyRs: Molander and Söderpalm showed that accumbal perfusion of strychnine decreased dopamine levels per se, as well as prevented further dopamine increase after ethanol administration (165). Accumbal GlyRs seem to be involved not only in dopamine elevations induced by ethanol, but also may contribute also to dopamine elevations induced by cannabinoids and nicotine (166), thus showing important implications for mechanisms related to alcoholism, other addictions and dopamine-related psychiatric disorders such as psychosis.
Glycine may affect dopaminergic output indirectly, acting on presynaptic GlyRs expressed on GABAergic terminals. Noteworthy, at birth both GABA (167) and glycine (168, 169) are excitatory neurotransmitters, but during the development, they became inhibitory ones. Ye and coauthors demonstrate that, despite these differences during the neurodevelopment, the net effect of glycine on dopamine, through GABA, in ventral tegmental area consists of a strengthening of the dopamine firing (161). Furthermore, if glycine may affect dopamine release, it is also relevant the role of dopamine in regulation of glycine release. An intriguing modality by which dopamine could regulate glycine release has been recently proposed by Shibasaki et al. (26) The authors demonstrated that dopamine may increase glycine release from cortical astrocytes by reversing the GlyT1. According to this view stimulation of dopamine receptors may change the intracellular metabolic milieu inducing glycolysis and oxidative phosphorylation (170), resulting in increased intracellular glycine levels. This increase in glycine concentration may reverse transport by GlyT1 (171).
Overall, in the framework of SCZ pathophysiology, a bidirectional regulation of glycine on dopamine should be conceived: in fact, changes in dopamine release could modulate glycine concentration, and in turn modify the response of NMDAR via Glyt1 potential reverse activity, especially at cortical level.
Therefore, an “inverse” and different mechanism from the canonical one, linking NMDAR and dopamine could be suggested in regulating dopaminergic balance, which is a crucial issue in SCZ.
Postsynaptic density (PSD) is an electron-dense structure composed of glutamate receptors (NMDARs, AMPARs, mGluRs), proteins involved in signal transduction (disrupted in schizophrenia 1, activity-regulated cytoskeleton-associated protein, calcium/calmodulin-dependent protein kinase II, Ras GTPase, and ion channels), scaffold proteins (post-synaptic density protein 95, Shank, Homers), and cytoskeletal structures (tubulin, septin, and others) localized at the distal tip of dendritic spines at excitatory synapses (172–174). The type and the number of the proteins highly influence PSD architecture, therefore significantly impacting the synaptic plasticity and dendritic shape (172–174). PSD has been implicated in pathophysiology of psychiatric disorders, including SCZ, and their treatment (175–178).
In several studies postsynaptic density protein 95 (PSD-95) has been found reduced in cortical and subcortical regions of postmortem brain samples from patients affected by SCZ (179–181). Considering that PSD-95 is physically and functionally linked to NMDARs and Homer proteins (182), all believed to be involved in SCZ, it will be of interest to understand how glycine may affect and interact with PSD-95. Immunohistochemical studies showed that in hippocampus and dentate gyrus GlyT1 is localized at the PSD of asymmetric glutamatergic synapses, belonging to a protein complex including NMDAR. Therefore, recent studies have highlighted that PSD-95 physically interacts with GlyT1 in the rat brain, stabilizing its localization at post-synaptic membrane and suppressing its internalization from cell surface, thereby increasing glycine uptake (136, 137). It has been hypothesized that PSD-95 could act as a link between GlyT1 and NMDAR, regulating glycine concentration in the micro-environment of NMDAR at glutamatergic synapses (Figure 1) (137). In heterozygote mutant GlyT1 +/− mice, who display increased concentrations of glycine, higher levels of GluN2 and increased expression of PSD-95 have been found, suggesting that PSD-95 may anchor GluN2-containing-NMDAR at synapses (that is the NMDAR subtype mostly involved in learning and synaptic plasticity) (183), preventing their internalization (184). Other studies examined how heterozygous GlyT1+/− mice display an enriched composition of PSD, showing concomitant increased levels of GluN1/2A NMDAR subunits and GluA1/2 AMPAR subunits, since an increase in NMDARs may probably cause an elevation of synaptic AMPARs (185). Nevertheless, other recent studies have found that the AMPAR/NMDAR ratio was decreased in mutants compared to wild-type mice displaying the complexity and variability of synaptic adaptation to altered GlyT1 function (140).
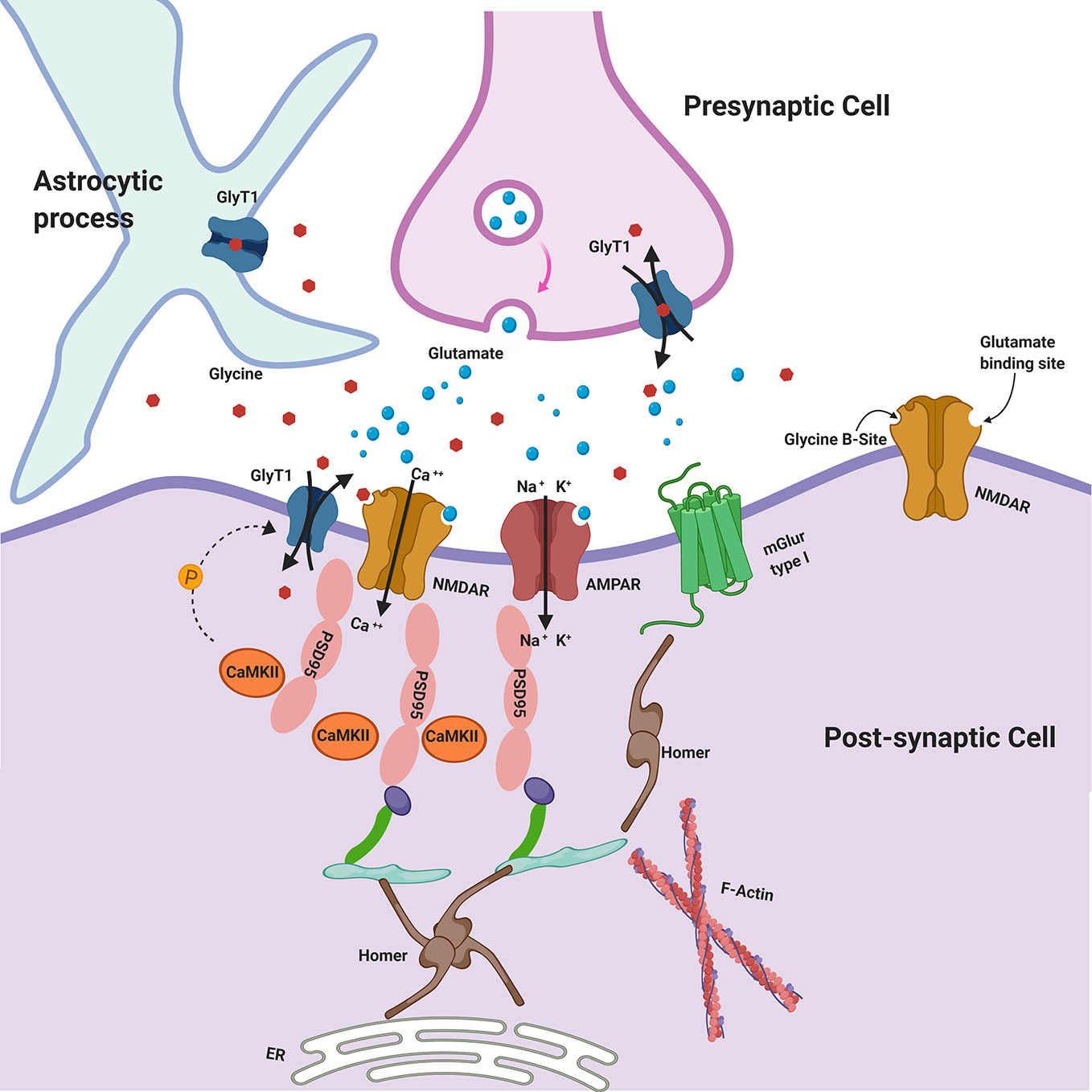
Figure 1 Glycine Transporters and the Postsynaptic Density (PSD). GlyT1 is localized at the PSD of asymmetric glutamatergic synapses, belonging to a protein complex including NMDAR. PSD-95 physically interacts with GlyT1, stabilizing its localization at postsynaptic membrane. CaMKII may regulate GlyT1 activity via indirect phosphorylation mechanisms. Glycine may be released in the synaptic cleft also by astroglial cells via functional reversal of GlyT1. GlyT1: Glycine Receptor Transporter 1; NMDAR: N-Methyl-D-aspartate receptors; PSD-95: postsynaptic density protein 95; CaMKII: Ca2+/calmodulin-dependent protein kinase; AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; mGluR type I: metabotropic glutamate receptors type I.
Calcium/calmodulin-dependent protein kinase II (CaMKII) is a core component of the PSD that may regulate GlyT1 activity including indirect phosphorylation mechanisms (e.g., activation of a signaling cascade or the phosphorylation of cytoskeletal protein involved in trafficking of GlyT1) and there is evidence that GlyT1 is inhibited by CaMKII inhibitors (Figure 1) (186). Sequence analysis of SLC6 family transporters, including GlyT1, revealed multiple consensus sites for phosphorylation by kinases. It is plausible that PKCα/β also could play a regulatory role in glycine transport by phosphorylating GlyT1 (152).
The role of glycine neurotransmission in the modulation of Homer gene expression has been explored in reference to the action of antipsychotics, alone or with the adjunction of glycine B-site agonists such as D-cycloserine (187). Multiple lines of evidence suggest an involvement of Homer (long and short forms, and their splicing variants) in antipsychotics action at level of PSD (188–191). Polese et al. have shown how add-on D-cycloserine to typical (haloperidol) and atypical (clozapine) antipsychotic treatment, modulate the expression of Homer 1a, in a negative trend in caudate-putamen (187). These data and the “dominant negative” function of Homer 1a can be predictive of a relative increase of mGluR surface clustering. It could be interpreted as a mechanism of mGluR activity enhancement, mediated by NMDAR glycine site agonists.
Among proteins located at PSD, disrupted in schizophrenia 1 (DISC-1) has attracted the attention of SCZ scholars in the last decade, since human genetic, imaging genetics, and preclinical models indicate a pivotal role of this protein in SCZ pathophysiology. Several antipsychotics, as well as compounds modulating the glutamate signaling, have been demonstrated to affect disk1 expression and function. Within the scope of this review, is noteworthy that rapastinel (formerly GLYX-13), an amidated tetrapeptide (threonine-proline-proline-threonine-amide) acting as allosteric partial agonist of glycine B-site (192) has been shown to counterbalance prepulse inhibition (PPI) disruption, hyperlocomotion, and memory deficits, induced by MK-801 administration in mice as well as to revert the associated decrease in disk-1 and GluN2B proteins. It is possible that this “antipsychotic-like” effect can be mediated by GluN2B expression, since GLYX-13 seems to be ineffective in GluN2B-knockdown mice (193).
Finally, although brain derived neurotrophic factor (BDNF) cannot be strictly considered a constitutive component of PSD, it has been isolated from this structure and directly affects its function. It has been demonstrated that in rat hippocampus BDNF reduces glycine reuptake by affecting the insertion of GlyT2 in cell membrane (194). Despite the intriguing relationship between BDNF and glycine as underlined by the previous finding, the role of BDNF in vivo in regulating glycine pathways relevant for SCZ pathophysiology is still controversial. In fact, patients treated with sarcosine, an agonist at glycine B-site of NMDAR, display no changes in plasma levels of BDNF (195).
A number of studies have explored the role of variation within genes encoding for elements of the glycinergic pathway in SCZ, with conflicting results in some cases. Deng et al. (196) found that a polymorphism within the SLC6A5 gene, encoding for GlyT2, was significantly associated with SCZ. Further, although indirect, support for a role of genetic variation within the glycinergic system in SCZ came from the study of Ohnuma et al. (197). These authors found that polymorphisms within the gene encoding for D-amino acid oxidase (DAO), which partially mediates the degradation of D-serine, a component of the glutamatergic transmission and an endogenous ligand for the glycine B-site on NMDAR (121), were statistically significantly associated with SCZ in a case-control study (197). More recently, a whole genome sequencing study in subjects with a high familial loading for psychotic disorders over three generations (198) found a frameshift mutation (rs10666583) in the GRIN3B gene, which codes for the GluN3B subunit of the NMDAR. This mutation was present in all family members with a psychotic disorder, but not in healthy relatives (198). The authors conclude that, given that this mutation induces an amino acid shift that degrades the S1/S2 glycine binding domain of the GluN3B subunit of the NMDAR, which subsequently affects the permeability of the channel pore to calcium ions, a decreased glycine affinity for the GluN3B subunit might cause impaired functional capability of the NMDAR (198). Of interest, a recent genetic association study in subjects with schizotypal traits showed statistically significant associations between the minor allele of three SNPs, rs2915885, rs11167557, and rs1428159, all positioned within the glycine receptor α1 subunit (GLRA1) gene, and dimensional schizotypy, specifically with the disorganized symptoms cluster (199). Further support for a role of the glycinergic system comes from a genetic-metabolomic study (200), which showed that 5-oxoproline, aspartate, and glutamate, known to affect NMDAR function, were significantly elevated in patients with rare variants in genes encoding for the glycine cleavage system. Finally, a groundbreaking study showed that genetically informed pharmacological treatment targeted at the glycinergic/glutamatergic signaling could improve significantly clinical response in patients with psychosis (201). These authors found several CNVs spanning 9p24.1 in a proband and his mother, who had diagnoses of schizoaffective disorder and bipolar disorder with psychotic features, respectively. Among the genes involved, the gene encoding for the glycine decarboxylase was of particular interest given its role in the catabolism of glycine (201). The authors performed two proof-of-principle clinical trials with glycine and d-cycloserine obtaining an additional 20 to 26% reduction in symptom severity with the former and 13 to 30% reduction with the latter (201)
Conversely, a series of negative studies do not appear to support a role for glycine transmission and signaling in SCZ molecular pathophysiology, at least on genetics ground. (202–206). The study of Feng et al. (202) explored the role of genetic mutations within the glycine receptor α2 subunit gene (GLRA2) in SCZ, using a sequencing approach. These authors detected three silent mutations in the coding region, C894T in exon 5, C1134T in exon 7, and C1476T in exon 9 (202), highlighting that a role of these variants in the pathogenesis of SCZ is unlikely. Similarly, subsequent case-controls studies confirmed these negative findings (203–206). Another negative finding derived from a gene expression analysis in post-mortem dorsolateral prefrontal cortex and cerebellum brain samples (207). Indeed, Burnet et al. did not find alterations of GlyT1 expression levels in these brain areas in 18 SCZ patients compared to 20 healthy controls (207).
Taken together, there is some discrepancy in the genetic findings of glycine and related signaling in SCZ. However, it should be noted that most of the early, negative, studies used case-control approaches often with inadequately powered sample sizes. The more recent translational evidence points to a role of genetic determinants in the pathophysiology of SCZ and contributes to the hypothesis that a subgroup of affected patients might take advantage of treatments targeted at the glycinergic/glutamatergic pathway.
Several studies indicate that cognitive processes may be regulated by glycine levels at glutamatergic synapses (47, 208, 209). Glycine concentration, in turn, is regulated by GlyT1, even if the glycine-B site would be tonically saturated (210). There is consensus, however, that GlyT1 prevents saturation of the glycine binding site on NMDARs and that further glycine increase can enhance NMDAR activation (155, 211), thus representing a potential target to modulate excitatory synapses.
Recent studies in GlyT1+/− mice showing NMDAR hyperfunction, highlighted the presence of an increased number of dendritic branching in the CA1 region of the hippocampus, an enhanced synaptogenesis (184), as well as higher density of excitatory glutamatergic synapses, and an increased expression of PSD-95 compared to wild type. In summary, these results suggest that glycine contributes to the regulation of synaptic plasticity, dendritic maturation, and glutamate-induced spinogenesis in the CNS (212).
Concerning behavioral phenotype, heterozygote GlyT1 +/− mice displayed improved memory retention during spatial learning task (47), and deletion of GlyT1 in the forebrain neurons resulted in a pro-cognitive profile characterized by facilitated associative learning, working memory, reference memory, and reversal learning (208, 209). Indeed, pharmacological blockade of GlyT1 exerted pro-cognitive effects in a preclinical model of SCZ, as also showed by a GlyT1 inhibitor, PF-3463275, that has been found to reverse ketamine-induced working memory deficits (213). Furthermore, SSR-504734, another GlyT1 inhibitor, facilitated cognitive flexibility, as assessed in the attentional set-shifting task in rats (214). These compounds probably may improve cognitive function and memory by increasing NMDAR signaling (214, 215); moreover, they may increase long term potentiation (LTP) that is one of the most studied manifestations of neuroplasticity. Therefore, it might be possible that GlyT1 inhibitors can reduce psychotic symptoms by improving neuroplasticity (216).
Deletion of the GlyT1 gene causes a divergent effect on PPI, depending on regional specificity. In fact, complete GlyT1 deletion in cortex confers resistance to PPI disruption induced by the NMDAR antagonist MK-801 (217), and may lead to a “psychosis-resistant” phenotype. On the contrary, deletion of GlyT1 in the striatum provokes a relevant PPI deficit, resembling a SCZ endophenotype, and the animals remain sensitive to the PPI-disruptive effect of MK-801 (217). Hence, there is no unequivocal support to the antipsychotic potential of GlyT1 inhibition and much more remains to be discovered.
Despite the complexity of the issue, in animal models of SCZ, such as neonatal lesion of the hippocampus (218) or acute NMDAR blockade (219, 220), GlyT1 inhibitors, such as ALX-5407, sarcosine, ORG 24598, SSR504734, and SSR103800 were effective in reverting PPI disruption, thus displaying an attractive antipsychotic-like activity (217).
The therapeutic strategies based on glycine neurotransmission have yielded contrasting results with significant improvement of SCZ symptoms in some clinical trials, as well as inconclusive results or no effect at all in other studies (221) (Table 1).
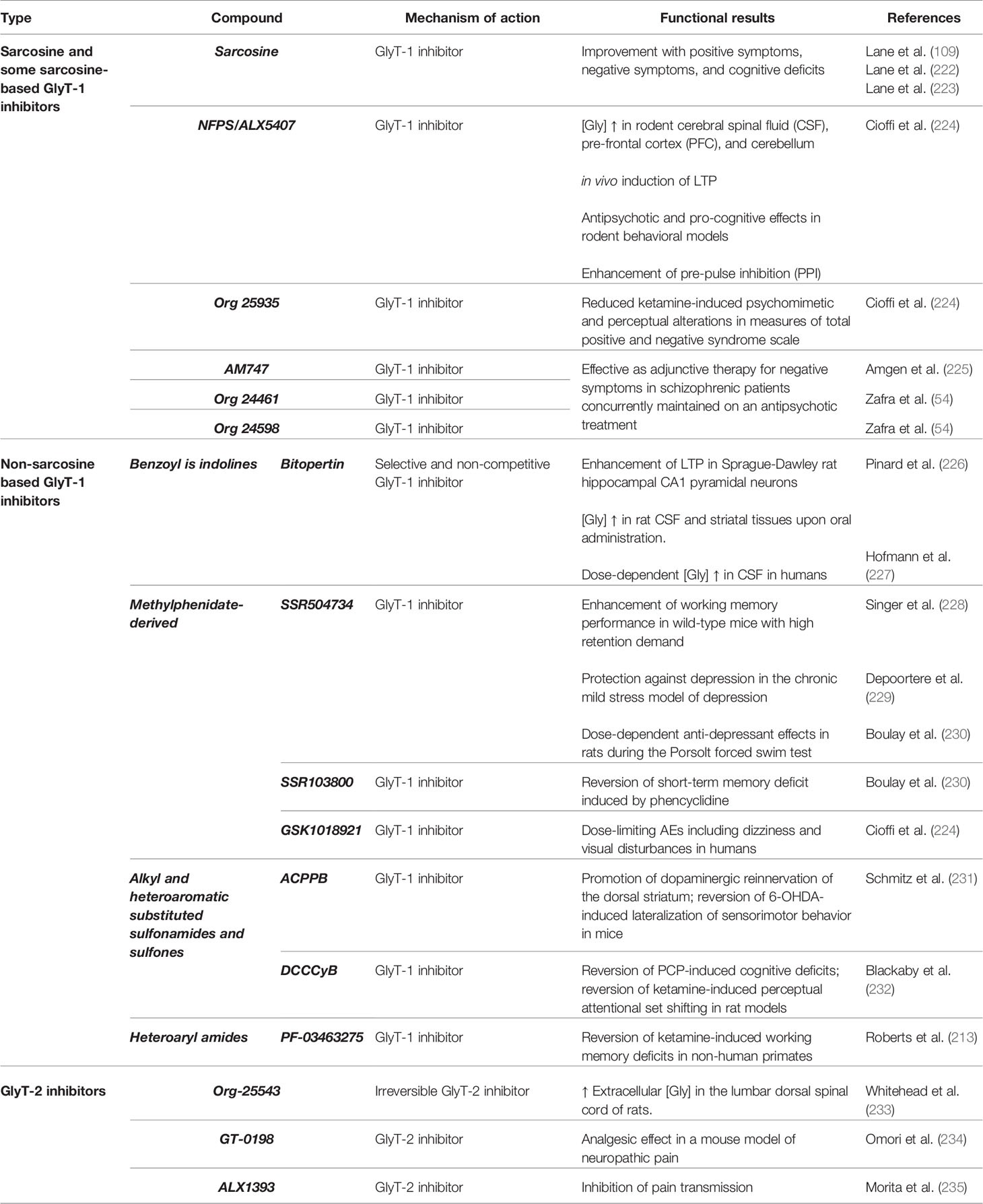
Table 1 Summary of the GlyT1 and GlyT2 inhibitors and their clinical and pre-clinical effects.
To date, more than 70 placebo-controlled clinical trials of agonists or partial agonists acting at NMDAR glycine modulatory site in SCZ (including glycine, D-serine, D-cycloserine, and D-alanine) have been reported in medical literature, with controversial results (236). An alternative approach to increase glycine availability is to block glycine reuptake via GlyT1. However, even though several GlyT1 inhibitors seemed to be efficacious in animal models, clinical studies in humans have been disappointing at least for major endpoints. The most advanced compound tested with the highest accuracy in terms of sample size and duration of the trials, is the non-competitive GlyT1 antagonist bitopertin, which also failed to reach its endpoints in Phase III trials (236).
With respect to the failure of GlyT1 inhibitors in clinical trials, potential reasons are herein explored, in order to better understand why a promising pharmacological strategy should not be abandoned.
One aspect that could be critical in evaluating the effect of GlyT1 inhibitors, included bitopertin, is the possibility that this mechanism of action may lead to an increase in glycine levels merely at extra-synaptic sites, therefore being less effective in potentiating NMDAR synaptic function. Nonetheless, the experiments of Martina and colleagues (96), confirming the ability of bitopertin to increase the activity of synaptic NMDARs, as well as to induce LTP processes, headed in a different direction. Perhaps, bitopertin may correct a certain degree of NMDAR hypofunction, being still unable to restore completely the glutamatergic transmission. Beyond the degree of activity, NMDAR dysfunction of SCZ may lay in the decreased number of receptors (237), abnormal coupling with PSD proteins (238), altered phosphorylation status (239), and other non-neurotransmitter cues that can impact synaptic efficacy. Not all these alterations potentially occurring in SCZ can be reverted or counterbalanced merely by increasing NMDAR activity.
Moreover, we should observe that GlyT1 inhibitors display an inverted U-shape concentration-response profile of action and this element, taking into account wide inter-individual differences in drug metabolism and pharmacokinetics, may be responsible for conflicting clinical results. In fact, the inverted U-shape dose-response curve displayed by bitopertin in LTP induction processes, as well as the partial receptor occupancy needed for efficacy (<50%) (49), maybe made further complicated a successful translation from animals to patients in terms of dose-finding issues.
Another point that should be raised is that, despite the extensive preclinical evidence supporting the role of bitopertin in treating SCZ, chronic administration has never been tested in animal models, in order to exclude a potential loss of effect in prolonged treatments.
In the original protocol of the clinical study, bitopertin was co-administered with routine antipsychotic treatment (240), but the class of antipsychotic was variable within the sample. Therefore, a question remains to be answered about the possibility that a certain combination would be less effective than others, resulting in an overall lack of efficacy of bitopertin intervention. Noteworthy, preclinical studies showed that glycinergic agents, when combined with antipsychotics with different receptor profile, may exert a wide ranging molecular pattern of responses (187).
Placebo effects observed in bitopertin phase III trials were larger than in phase II, and placebo response rate was assessed at 56–61% both in DayLyte and FlashLyte studies, which could explain the failure to detect any significant difference between arms. The magnitude of symptoms improvement in the placebo groups of RCTs (even those testing antipsychotics) is considerably growing over time, as described by several reviews (241, 242), making it even more difficult for the active medication groups to separate in a statistically significant way from placebo.
Finally, it is also important to remember that bitopertin has been tested for negative symptoms of SCZ, that are not always easy to be assessed reliably, as well as are difficult to be distinguished between primary and secondary ones. Despite the validity of the assessment instruments, negative symptoms are often not a focus of assessment or treatment in clinical practice, because they are rarely responsible for acute crisis or hospitalizations (243). Noteworthy, to date, antipsychotic medications remarkably effective in treating negative symptoms are few. Trying to understand the reason of this granitic-like resistance, Velligan et al. proposed a negative symptom maintenance loop theory, wherein decreased initiation and withdrawal lead to a series of self-perpetuating outcomes (i.e., reduced responsiveness to social stimuli, low interest in relationships, and decreased reinforcements from the social context) (244). In this perspective, pharmacological treatment might struggle to break the cycle, and although they may motivate patients to increase their social drive, patients may still lack the ability to interact due to previous chronic isolation. Therefore, to achieve a tangible improvement of negative symptoms, adjunctive behavioral training may be required.
TRS is a major clinical and therapeutic challenge in the management of SCZ patients, representing also a crucial mental health issue for the social implications and for care costs (245, 246). Therefore, the search for new compounds alone or in combination with the available antipsychotics is warranted, especially when the gold standard (i.e., clozapine) therapy fails. Ongoing research suggests that the multidimensional symptoms of SCZ may arise from dysregulation in multiple signaling pathways that may revolve around glutamatergic neurotransmission. NMDAR may represent a converging point of environmental hits and genetic factors, leading to downstream neurochemical dysfunctions that may account for positive, negative, and cognitive symptoms. Therefore, it can be hypothesized that pharmacological augmentation of NMDAR transmission through glycine signaling enhancement might restore the function of prefrontal cortex to control dopamine release, offering a potentially useful strategy in SCZ treatment. Glycine-based treatments for SCZ have their rationale first of all for the potential of this amino acid to regulate glutamate signaling and to modulate in a reciprocal interplay dopamine release, interacting, indeed, with two neurotransmitters shown to be among the major players in SCZ pathophysiology. Moreover, it should be remarked that TRS, at least for those cases that are not fully responsive to dopamine antagonists or partial agonists, is believed to be linked to aberrant glutamatergic signaling. Several lines of evidence suggest a glutamatergic mechanism of action even for clozapine that, coincidentally, is significantly effective in TRS. This superior efficacy is presumably due to an additional mechanism to D2 receptor occupancy and possibly to a pro-glutamatergic action. Indeed, it has been hypothesized that clozapine may have an intrinsic agonist or partial agonist activity at the glycine B-site, that may contribute to its unique clinical effects (24).
It is clear that, despite the relevance of the issue and the strong neurobiological rationale, glycine-based pharmacological interventions are still inconclusive but, at the same time, strongly suggestive of the high therapeutic potential, especially for the severe form of TRS.
How the utilization of glycine enhancers or modulators can be improved for SCZ therapy? A first level of analysis should clearly separate the strategies based on potentiation of transmission at NMDAR glycine B-site from the ones based on GlyT1 inhibition. Comparing the outcomes of the two types of strategies may be important to try to figure what kind of targets are respectively achieved in terms of clinical improvement, therefore a focused (meta?) analysis is needed.
From the perspective of clinical trial methodology, some trials with compounds active at NMDAR glycine B-site have shown positive results; however, larger sample size and more homogeneous subsets of patients, separating those with prevalent positive or negative symptoms and longer duration of treatment, should be required. A number of measures should be considered in order to minimize the placebo response, including reducing the number of collaborating study sites and recruiting patients preferably from academic ones (241). A better evaluation is needed to determine which patients should be treated only with glycine-based pharmacological intervention, and in which ones these agents should be administered in augmentation with canonical antipsychotics. Finally, clinical trials using glycinergic agents are not always designed specifically for TRS patients, therefore an effort for including this class of patients should be done.
Furthermore, for more TRS “tuned” treatment based on glycine signaling, a better knowledge of the major kinetic steps responsible for the activation of glutamate-bound NMDAR by glycine is paramount to elucidate the pharmacodynamics of glycinergic compounds (247). Therefore, how ambient glycine levels regulate NMDAR function under a pattern of multiple stimulations, how glycine transporters interact with multiple PSD proteins, and how glycine affect overall dopamine–glutamate interaction are key questions for the development of new compounds.
In conclusion, despite the mismatch between the significant advance of our knowledge of glycine signaling in the modeling of SCZ pathophysiology and the results of clinical trials, glycine–based pharmacological therapy, alone or in combination with available antipsychotics is still worth to be explored and refined.
AdB conceived the project of the manuscript. AdB, FM, AB, and LV conceived the strategy for literature retrieval and selection. AdB, AB, MM, LV, and FM participated in the writing process of the first draft of the manuscript. AdB, MM, and FI revised the final version of the manuscript. All authors have read and approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Carlsson A, Lindqvist M. Effect Of Chlorpromazine Or Haloperidol On Formation Of 3methoxytyramine And Normetanephrine In Mouse Brain. Acta Pharmacol Toxicol (Copenh) (1963) 20:140–4. doi: 10.1111/j.1600-0773.1963.tb01730.x
2. Stephan KE, Friston KJ, Frith CD. Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull (2009) 35(3):509–27. doi: 10.1093/schbul/sbn176
3. Rolls ET, Cheng W, Gilson M, Gong W, Deco G, Lo CZ, et al. Beyond the disconnectivity hypothesis of schizophrenia. Cereb Cortex (2019) 30(3):1213–33. doi: 10.1093/cercor/bhz161
4. Friston KJ. Dysfunctional connectivity in schizophrenia. World Psychiatry: Off J World Psychiatr Assoc (WPA) (2002) 1(2):66–71.
5. Horga G, Abi-Dargham A. An integrative framework for perceptual disturbances in psychosis. Nat Rev Neurosci (2019) 20(12):763–78. doi: 10.1038/s41583-019-0234-1
6. Millan MJ, Andrieux A, Bartzokis G, Cadenhead K, Dazzan P, Fusar-Poli P, et al. Altering the course of schizophrenia: progress and perspectives. Nat Rev Drug Discovery (2016) 15(7):485–515. doi: 10.1038/nrd.2016.28
7. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry (2007) 64(10):1123–31. doi: 10.1001/archpsyc.64.10.1123
8. Jaaskelainen E, Juola P, Hirvonen N, McGrath JJ, Saha S, Isohanni M, et al. A systematic review and meta-analysis of recovery in schizophrenia. Schizophr Bull (2013) 39(6):1296–306. doi: 10.1093/schbul/sbs130
9. Samara MT, Nikolakopoulou A, Salanti G, Leucht S. How Many Patients With Schizophrenia Do Not Respond to Antipsychotic Drugs in the Short Term? An Analysis Based on Individual Patient Data From Randomized Controlled Trials. Schizophr Bull (2018) 45(3):639–45. doi: 10.1093/schbul/sby095
10. Siskind D, Siskind V, Kisely S. Clozapine Response Rates among People with Treatment-Resistant Schizophrenia: Data from a Systematic Review and Meta-Analysis. Can J Psychiatry (2017) 62(11):772–7. doi: 10.1177/0706743717718167
11. Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci U S A (1997) 94(6):2569–74. doi: 10.1073/pnas.94.6.2569
12. Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry (1998) 155(6):761–7. doi: 10.1176/ajp.155.6.761
13. Mizrahi R, Addington J, Rusjan PM, Suridjan I, Ng A, Boileau I, et al. Increased stress-induced dopamine release in psychosis. Biol Psychiatry (2012) 71(6):561–7. doi: 10.1016/j.biopsych.2011.10.009
14. Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry (2010) 67(3):231–9. doi: 10.1001/archgenpsychiatry.2010.10
15. Amato D, Vernon AC, Papaleo F. Dopamine, the antipsychotic molecule: A perspective on mechanisms underlying antipsychotic response variability. Neurosci Biobehav Rev (2018) 85:146–59. doi: 10.1016/j.neubiorev.2017.09.027
16. de Bartolomeis A, Tomasetti C, Iasevoli F. Update on the Mechanism of Action of Aripiprazole: Translational Insights into Antipsychotic Strategies Beyond Dopamine Receptor Antagonism. CNS Drugs (2015) 29(9):773–99. doi: 10.1007/s40263-015-0278-3
17. Uchida H, Takeuchi H, Graff-Guerrero A, Suzuki T, Watanabe K, Mamo DC. Dopamine D2 receptor occupancy and clinical effects: a systematic review and pooled analysis. J Clin Psychopharmacol (2011) 31(4):497–502. doi: 10.1097/JCP.0b013e3182214aad
18. Nord M, Farde L. Antipsychotic occupancy of dopamine receptors in schizophrenia. CNS Neurosci Ther (2011) 17(2):97–103. doi: 10.1111/j.1755-5949.2010.00222.x
19. Kegeles LS, Abi-Dargham A, Zea-Ponce Y, Rodenhiser-Hill J, Mann JJ, Van Heertum RL, et al. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry (2000) 48(7):627–40. doi: 10.1016/S0006-3223(00)00976-8
20. Rujescu D, Bender A, Keck M, Hartmann AM, Ohl F, Raeder H, et al. A pharmacological model for psychosis based on N-methyl-D-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol Psychiatry (2006) 59(8):721–9. doi: 10.1016/j.biopsych.2005.08.029
21. Tuominen HJ, Tiihonen J, Wahlbeck K. Glutamatergic drugs for schizophrenia. Cochrane Database Syst Rev (2006) (2):Cd003730. doi: 10.1002/14651858.CD003730.pub2
22. Millan MJ. N-Methyl-D-aspartate receptors as a target for improved antipsychotic agents: novel insights and clinical perspectives. Psychopharmacol (Berl) (2005) 179(1):30–53. doi: 10.1007/s00213-005-2199-1
23. Coyle JT. Substance use disorders and Schizophrenia: a question of shared glutamatergic mechanisms. Neurotox Res (2006) 10(3-4):221–33. doi: 10.1007/BF03033359
24. Heresco-Levy U. N-Methyl-D-aspartate (NMDA) receptor-based treatment approaches in schizophrenia: the first decade. Int J Neuropsychopharmacol (2000) 3(3):243–58. doi: 10.1017/S1461145700001978
25. Heresco-Levy U. Glutamatergic neurotransmission modulation and the mechanisms of antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry (2003) 27(7):1113–23. doi: 10.1016/j.pnpbp.2003.09.007
26. Shibasaki K, Hosoi N, Kaneko R, Tominaga M, Yamada K. Glycine release from astrocytes via functional reversal of GlyT1. J Neurochem (2017) 140(3):395–403. doi: 10.1111/jnc.13741
27. Millan MJ. N-methyl-D-aspartate receptor-coupled glycineB receptors in the pathogenesis and treatment of schizophrenia: a critical review. Curr Drug Targets CNS Neurol Disord (2002) 1(2):191–213. doi: 10.2174/1568007024606258
28. Heresco-Levy U, Javitt DC. Comparative effects of glycine and D-cycloserine on persistent negative symptoms in schizophrenia: a retrospective analysis. Schizophr Res (2004) 66(2-3):89–96. doi: 10.1016/S0920-9964(03)00129-4
29. Yu A, Alberstein R, Thomas A, Zimmet A, Grey R, Mayer ML, et al. Molecular lock regulates binding of glycine to a primitive NMDA receptor. Proc Natl Acad Sci U S A (2016) 113(44):E6786–e95. doi: 10.1073/pnas.1607010113
30. Yu A, Lau AY. Glutamate and Glycine Binding to the NMDA Receptor. Structure (2018) 26(7):1035–43.e2. doi: 10.1016/j.str.2018.05.004
31. Dolino DM, Cooper D, Ramaswamy S, Jaurich H, Landes CF, Jayaraman V. Structural dynamics of the glycine-binding domain of the N-methyl-D-aspartate receptor. J Biol Chem (2015) 290(2):797–804. doi: 10.1074/jbc.M114.605436
32. Skrenkova K, Hemelikova K, Kolcheva M, Kortus S, Kaniakova M, Krausova B, et al. Structural features in the glycine-binding sites of the GluN1 and GluN3A subunits regulate the surface delivery of NMDA receptors. Sci Rep (2019) 9(1):12303. doi: 10.1038/s41598-019-48845-3
33. Delay J, Deniker P, Harl JM. [Therapeutic use in psychiatry of phenothiazine of central elective action (4560 RP)]. Annales Medico-Psychol (1952) 110(2 1):112–7.
34. Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science (1976) 192(4238):481–3. doi: 10.1126/science.3854
35. Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science (1975) 188(4194):1217–9. doi: 10.1126/science.1145194
36. Laruelle M, Abi-Dargham A. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. J Psychopharmacol (1999) 13(4):358–71. doi: 10.1177/026988119901300405
37. Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D, et al. Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry (1999) 156(10):1646–9. doi: 10.1176/ajp.156.10.1646
38. Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry (1995) 52(12):998–1007. doi: 10.1001/archpsyc.1995.03950240016004
39. Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia–implications for schizophrenia and Parkinson's disease. Trends Neurosci (1990) 13(7):272–6. doi: 10.1016/0166-2236(90)90108-M
40. Miller DW, Abercrombie ED. Effects of MK-801 on spontaneous and amphetamine-stimulated dopamine release in striatum measured with in vivo microdialysis in awake rats. Brain Res Bull (1996) 40(1):57–62. doi: 10.1016/0361-9230(95)02144-2
41. Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology (2012) 62(3):1574–83. doi: 10.1016/j.neuropharm.2011.01.022
42. Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci (2010) 13(1):76–83. doi: 10.1038/nn.2447
43. Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull (2010) 83(3-4):108–21. doi: 10.1016/j.brainresbull.2010.04.006
44. Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci (2004) 5(5):361–72. doi: 10.1038/nrn1385
45. Collingridge GL, Volianskis A, Bannister N, France G, Hanna L, Mercier M, et al. The NMDA receptor as a target for cognitive enhancement. Neuropharmacology (2013) 64:13–26. doi: 10.1016/j.neuropharm.2012.06.051
46. Javitt DC, Hashim A, Sershen H. Modulation of striatal dopamine release by glycine transport inhibitors. Neuropsychopharmacology (2005) 30(4):649–56. doi: 10.1038/sj.npp.1300589
47. Tsai G, Ralph-Williams RJ, Martina M, Bergeron R, Berger-Sweeney J, Dunham KS, et al. Gene knockout of glycine transporter 1: characterization of the behavioral phenotype. Proc Natl Acad Sci U S A (2004) 101(22):8485–90. doi: 10.1073/pnas.0402662101
48. Cioffi CL, Guzzo PR. Inhibitors of Glycine Transporter-1: Potential Therapeutics for the Treatment of CNS Disorders. Curr Top Med Chem (2016) 16(29):3404–37. doi: 10.2174/1568026616666160405113340
49. Pinard E, Borroni E, Koerner A, Umbricht D, Alberati D, Glycine Transporter Type I. (GlyT1) Inhibitor, Bitopertin: A Journey from Lab to Patient. Chimia (Aarau) (2018) 72(7):477–84. doi: 10.2533/chimia.2018.477
50. Kim SY, Kaufman MJ, Cohen BM, Jensen JE, Coyle JT, Du F, et al. In Vivo Brain Glycine and Glutamate Concentrations in Patients With First-Episode Psychosis Measured by Echo Time-Averaged Proton Magnetic Resonance Spectroscopy at 4T. Biol Psychiatry (2018) 83(6):484–91. doi: 10.1016/j.biopsych.2017.08.022
51. Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature (2002) 415(6873):793–8. doi: 10.1038/nature715
52. Sato K, Yoshida S, Fujiwara K, Tada K, Tohyama M. Glycine cleavage system in astrocytes. Brain Res (1991) 567(1):64–70. doi: 10.1016/0006-8993(91)91436-5
53. Curtis DR, Game CJ, Lodge D, McCulloch RM. A pharmacological study of Renshaw cell inhibition. J Physiol (1976) 258(1):227–42. doi: 10.1113/jphysiol.1976.sp011416
54. Zafra F, Ibanez I, Bartolome-Martin D, Piniella D, Arribas-Blazquez M, Gimenez C. Glycine Transporters and Its Coupling with NMDA Receptors. Adv Neurobiol (2017) 16:55–83. doi: 10.1007/978-3-319-55769-4_4
55. Aoki E, Semba R, Keino H, Kato K, Kashiwamata S. Glycine-like immunoreactivity in the rat auditory pathway. Brain Res (1988) 442(1):63–71. doi: 10.1016/0006-8993(88)91432-1
56. Rampon C, Luppi PH, Fort P, Peyron C, Jouvet M. Distribution of glycine-immunoreactive cell bodies and fibers in the rat brain. Neuroscience (1996) 75(3):737–55. doi: 10.1016/0306-4522(96)00278-3
57. Naas E, Zilles K, Gnahn H, Betz H, Becker CM, Schroder H. Glycine receptor immunoreactivity in rat and human cerebral cortex. Brain Res (1991) 561(1):139–46. doi: 10.1016/0006-8993(91)90758-N
58. Keck T, White JA. Glycinergic inhibition in the hippocampus. Rev Neurosci (2009) 20(1):13–22. doi: 10.1515/REVNEURO.2009.20.1.13
59. Dudeck O, Lubben S, Eipper S, Knorle R, Kirsch M, Honegger J, et al. Evidence for strychnine-sensitive glycine receptors in human amygdala. Naunyn Schmiedebergs Arch Pharmacol (2003) 368(3):181–7. doi: 10.1007/s00210-003-0786-4
60. Gielen M, Thomas P, Smart TG. The desensitization gate of inhibitory Cys-loop receptors. Nat Commun (2015) 6:6829. doi: 10.1038/ncomms7829
61. Altschuler RA, Betz H, Parakkal MH, Reeks KA, Wenthold RJ. Identification of glycinergic synapses in the cochlear nucleus through immunocytochemical localization of the postsynaptic receptor. Brain Res (1986) 369(1-2):316–20. doi: 10.1016/0006-8993(86)90542-1
62. Alvarez FJ, Dewey DE, Harrington DA, Fyffe RE. Cell-type specific organization of glycine receptor clusters in the mammalian spinal cord. J Comp Neurol (1997) 379(1):150–70. doi: 10.1002/(SICI)1096-9861(19970303)379:1<150::AID-CNE10>3.0.CO;2-T
63. Chattipakorn SC, McMahon LL. Pharmacological characterization of glycine-gated chloride currents recorded in rat hippocampal slices. J Neurophysiol (2002) 87(3):1515–25. doi: 10.1152/jn.00365.2001
64. McCool BA, Botting SK. Characterization of strychnine-sensitive glycine receptors in acutely isolated adult rat basolateral amygdala neurons. Brain Res (2000) 859(2):341–51. doi: 10.1016/S0006-8993(00)02026-6
65. Ye JH, Ren J, Liu PL, McArdle JJ. Glycine-activated chloride currents of neurons freshly isolated from the ventral tegmental area of rats. Brain Res (1998) 796(1-2):53–62. doi: 10.1016/S0006-8993(98)00317-5
66. Legendre P. The glycinergic inhibitory synapse. Cell Mol Life Sci (2001) 58(5-6):760–93. doi: 10.1007/PL00000899
67. Meyer G, Kirsch J, Betz H, Langosch D. Identification of a gephyrin binding motif on the glycine receptor beta subunit. Neuron (1995) 15(3):563–72. doi: 10.1016/0896-6273(95)90145-0
68. Flint AC, Liu X, Kriegstein AR. Nonsynaptic glycine receptor activation during early neocortical development. Neuron (1998) 20(1):43–53. doi: 10.1016/S0896-6273(00)80433-X
69. Mangin JM, Guyon A, Eugene D, Paupardin-Tritsch D, Legendre P. Functional glycine receptor maturation in the absence of glycinergic input in dopaminergic neurones of the rat substantia nigra. J Physiol (2002) 542(Pt 3):685–97. doi: 10.1113/jphysiol.2002.018978
70. Rosenberg D, Artoul S, Segal AC, Kolodney G, Radzishevsky I, Dikopoltsev E, et al. Neuronal D-serine and glycine release via the Asc-1 transporter regulates NMDA receptor-dependent synaptic activity. J Neurosci Off J Soc Neurosci (2013) 33(8):3533–44. doi: 10.1523/JNEUROSCI.3836-12.2013
71. Snyder SH, Kim PM. D-Amino Acids as Putative Neurotransmitters: Focus on D-Serine. Neurochem Res (2000) 25(5):553–60. doi: 10.1023/A:1007586314648
72. Harsing LG Jr., Matyus P. Mechanisms of glycine release, which build up synaptic and extrasynaptic glycine levels: the role of synaptic and non-synaptic glycine transporters. Brain Res Bull (2013) 93:110–9. doi: 10.1016/j.brainresbull.2012.12.002
73. Cubelos B, Leite C, Gimenez C, Zafra F. Localization of the glycine transporter GLYT1 in glutamatergic synaptic vesicles. Neurochem Int (2014) 73:204–10. doi: 10.1016/j.neuint.2013.09.002
74. Smith KE, Borden LA, Hartig PR, Branchek T, Weinshank RL. Cloning and expression of a glycine transporter reveal colocalization with NMDA receptors. Neuron (1992) 8(5):927–35. doi: 10.1016/0896-6273(92)90207-T
75. Wilcox KS, Fitzsimonds RM, Johnson B, Dichter MA. Glycine regulation of synaptic NMDA receptors in hippocampal neurons. J Neurophysiol (1996) 76(5):3415–24. doi: 10.1152/jn.1996.76.5.3415
76. Bergeron R, Meyer TM, Coyle JT, Greene RW. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc Natl Acad Sci U S A (1998) 95(26):15730–4. doi: 10.1073/pnas.95.26.15730
77. Chen L, Muhlhauser M, Yang CR. Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J Neurophysiol (2003) 89(2):691–703. doi: 10.1152/jn.00680.2002
78. Martina M, Krasteniakov NV, Bergeron R. D-Serine differently modulates NMDA receptor function in rat CA1 hippocampal pyramidal cells and interneurons. J Physiol (2003) 548(Pt 2):411–23. doi: 10.1113/jphysiol.2002.037127
79. Vyklický L Jr., Benveniste M, Mayer ML. Modulation of N-methyl-D-aspartic acid receptor desensitization by glycine in mouse cultured hippocampal neurones. J Physiol (1990) 428:313–31. doi: 10.1113/jphysiol.1990.sp018214
80. Globus MY, Busto R, Martinez E, Valdés I, Dietrich WD, Ginsberg MD. Comparative effect of transient global ischemia on extracellular levels of glutamate, glycine, and gamma-aminobutyric acid in vulnerable and nonvulnerable brain regions in the rat. J Neurochem (1991) 57(2):470–8. doi: 10.1111/j.1471-4159.1991.tb03775.x
81. Lasley SM. Roles of neurotransmitter amino acids in seizure severity and experience in the genetically epilepsy-prone rat. Brain Res (1991) 560(1-2):63–70. doi: 10.1016/0006-8993(91)91215-M
82. Chen R-Q, Wang S-H, Yao W, Wang J-J, Ji F, Yan J-Z, et al. Role of glycine receptors in glycine-induced LTD in hippocampal CA1 pyramidal neurons. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol (2011) 36(9):1948–58. doi: 10.1038/npp.2011.86
83. Roux MJ, Supplisson S. Neuronal and glial glycine transporters have different stoichiometries. Neuron (2000) 25(2):373–83. doi: 10.1016/S0896-6273(00)80901-0
84. Yoshikawa M, Nakajima K, Takayasu N, Noda S, Sato Y, Kawaguchi M, et al. Expression of the mRNA and protein of serine racemase in primary cultures of rat neurons. Eur J Pharmacol (2006) 548(1–3):74–6. doi: 10.1016/j.ejphar.2006.07.029
85. Sason H, Billard JM, Smith GP, Safory H, Neame S, Kaplan E, et al. Asc-1 Transporter Regulation of Synaptic Activity via the Tonic Release of d-Serine in the Forebrain. Cereb Cortex (New York NY: 1991) (2017) 27(2):1573–87. doi: 10.1093/cercor/bhv350
86. Martineau M, Parpura V, Mothet JP. Cell-type specific mechanisms of D-serine uptake and release in the brain. Front Synaptic Neurosci (2014) 6:12.
87. Le Bail M, Martineau M, Sacchi S, Yatsenko N, Radzishevsky I, Conrod S, et al. Identity of the NMDA receptor coagonist is synapse specific and developmentally regulated in the hippocampus. Proc Natl Acad Sci United States America (2015) 112(2):E204–E13. doi: 10.1073/pnas.1416668112
88. Panatier A, Theodosis DT, Mothet J-P, Touquet B, Pollegioni L, Poulain DA, et al. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell (2006) 125(4):775–84. doi: 10.1016/j.cell.2006.02.051
89. Fossat P, Turpin FR, Sacchi S, Dulong J, Shi T, Rivet J-M, et al. Glial D-serine gates NMDA receptors at excitatory synapses in prefrontal cortex. Cereb Cortex (New York NY: 1991) (2012) 22(3):595–606. doi: 10.1093/cercor/bhr130
90. Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, et al. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell (2012) 150(3):633–46. doi: 10.1016/j.cell.2012.06.029
91. Li Y, Sacchi S, Pollegioni L, Basu AC, Coyle JT, Bolshakov VY. Identity of endogenous NMDAR glycine site agonist in amygdala is determined by synaptic activity level. Nat Commun (2013) 4:1760. doi: 10.1038/ncomms2779
92. Tsujimura K, Abematsu M, Kohyama J, Namihira M, Nakashima K. Neuronal differentiation of neural precursor cells is promoted by the methyl-CpG-binding protein MeCP2. Exp Neurol (2009) 219(1):104–11. doi: 10.1016/j.expneurol.2009.05.001
93. Mothet JP, Parent AT, Wolosker H, Brady RO Jr., Linden DJ, Ferris CD, et al. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci United States America (2000) 97(9):4926–31. doi: 10.1073/pnas.97.9.4926
94. Schell MJ, Brady RO Jr., Molliver ME, Snyder SH. D-serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci Off J Soc Neurosci (1997) 17(5):1604–15. doi: 10.1523/JNEUROSCI.17-05-01604.1997
95. Tauck DL, Ashbeck GA. Glycine synergistically potentiates the enhancement of LTP induced by a sulfhydryl reducing agent. Brain Res (1990) 519(1-2):129–32. doi: 10.1016/0006-8993(90)90070-R
96. Martina M, Gorfinkel Y, Halman S, Lowe JA, Periyalwar P, Schmidt CJ, et al. Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J Physiol (2004) 557(Pt 2):489–500. doi: 10.1113/jphysiol.2004.063321
97. Henneberger C, Bard L, King C, Jennings A, Rusakov DA. NMDA receptor activation: two targets for two co-agonists. Neurochem Res (2013) 38(6):1156–62. doi: 10.1007/s11064-013-0987-2
98. Martineau M, Baux G, Mothet J-P. D-serine signalling in the brain: friend and foe. Trends Neurosci (2006) 29(8):481–91. doi: 10.1016/j.tins.2006.06.008
99. Wu S, Barger SW. Induction of serine racemase by inflammatory stimuli is dependent on AP-1. Ann New York Acad Sci (2004) 1035:133–46. doi: 10.1196/annals.1332.009
100. Ivanov AD, Mothet J-P. The plastic d-serine signaling pathway: Sliding from neurons to glia and vice-versa. Neurosci Lett (2019) 689:21–5. doi: 10.1016/j.neulet.2018.05.039
101. Perez EJ, Tapanes SA, Loris ZB, Balu DT, Sick TJ, Coyle JT, et al. Enhanced astrocytic d-serine underlies synaptic damage after traumatic brain injury. J Clin Invest (2017) 127(8):3114–25. doi: 10.1172/JCI92300
102. Sasabe J, Chiba T, Yamada M, Okamoto K, Nishimoto I, Matsuoka M, et al. D-serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J (2007) 26(18):4149–59. doi: 10.1038/sj.emboj.7601840
103. Mustafa AK, Ahmad AS, Zeynalov E, Gazi SK, Sikka G, Ehmsen JT, et al. Serine racemase deletion protects against cerebral ischemia and excitotoxicity. J Neurosci Off J Soc Neurosci (2010) 30(4):1413–6. doi: 10.1523/JNEUROSCI.4297-09.2010
104. Hashimoto A, Nishikawa T, Konno R, Niwa A, Yasumura Y, Oka T, et al. Free d-serine, d-aspartate and d-alanine in central nervous system and serum in mutant mice lacking d-amino acid oxidase. Neurosci Lett (1993) 152(1):33–6. doi: 10.1016/0304-3940(93)90476-2
105. Ohnuma T, Sakai Y, Maeshima H, Hatano T, Hanzawa R, Abe S, et al. Changes in plasma glycine, L-serine, and D-serine levels in patients with schizophrenia as their clinical symptoms improve: results from the Juntendo University Schizophrenia Projects (JUSP). Prog Neuropsychopharmacol Biol Psychiatry (2008) 32(8):1905–12. doi: 10.1016/j.pnpbp.2008.07.022
106. El-Tallawy HN, Saleem TH, El-Ebidi AM, Hassan MH, Gabra RH, Farghaly WM, et al. Clinical and biochemical study of d-serine metabolism among schizophrenia patients. Neuropsychiatr Dis Treat (2017) 13:1057–63. doi: 10.2147/NDT.S126979
107. Liu R, Dang W, Du Y, Zhou Q, Liu Z, Jiao K. Correlation of functional GRIN2A gene promoter polymorphisms with schizophrenia and serum D-serine levels. Gene (2015) 568(1):25–30. doi: 10.1016/j.gene.2015.05.011
108. Tsai G, Yang P, Chung LC, Lange N, Coyle JT. D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry (1998) 44(11):1081–9. doi: 10.1016/S0006-3223(98)00279-0
109. Lane HY, Chang YC, Liu YC, Chiu CC, Tsai GE. Sarcosine or D-serine add-on treatment for acute exacerbation of schizophrenia: a randomized, double-blind, placebo-controlled study. Arch Gen Psychiatry (2005) 62(11):1196–204. doi: 10.1001/archpsyc.62.11.1196
110. Weiser M, Heresco-Levy U, Davidson M, Javitt DC, Werbeloff N, Gershon AA, et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J Clin Psychiatry (2012) 73(6):e728–e34. doi: 10.4088/JCP.11m07031
111. Iwata Y, Nakajima S, Suzuki T, Keefe RSE, Plitman E, Chung JK, et al. Effects of glutamate positive modulators on cognitive deficits in schizophrenia: a systematic review and meta-analysis of double-blind randomized controlled trials. Mol Psychiatry (2015) 20(10):1151–60. doi: 10.1038/mp.2015.68
112. Moaddel R, Luckenbaugh DA, Xie Y, Villaseñor A, Brutsche NE, Machado-Vieira R, et al. D-serine plasma concentration is a potential biomarker of (R,S)-ketamine antidepressant response in subjects with treatment-resistant depression. Psychopharmacology (2015) 232(2):399–409. doi: 10.1007/s00213-014-3669-0
113. Choi S-R, Roh D-H, Yoon S-Y, Choi H-S, Kang S-Y, Han H-J, et al. Astrocyte D-serine modulates the activation of neuronal NOS leading to the development of mechanical allodynia in peripheral neuropathy. Mol Pain (2019) 15:1744806919843046–. doi: 10.1177/1744806919843046
114. Kantrowitz JT, Malhotra AK, Cornblatt B, Silipo G, Balla A, Suckow RF, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res (2010) 121(1-3):125–30. doi: 10.1016/j.schres.2010.05.012
115. Errico F, Mothet JP, Usiello A. D-Aspartate: An endogenous NMDA receptor agonist enriched in the developing brain with potential involvement in schizophrenia. J Pharm Biomed Anal (2015) 116:7–17. doi: 10.1016/j.jpba.2015.03.024
116. Errico F, Nuzzo T, Carella M, Bertolino A, Usiello A. The Emerging Role of Altered d-Aspartate Metabolism in Schizophrenia: New Insights From Preclinical Models and Human Studies. Front Psychiatry (2018) 9:559. doi: 10.3389/fpsyt.2018.00559
117. Krashia P, Ledonne A, Nobili A, Cordella A, Errico F, Usiello A, et al. Persistent elevation of D-Aspartate enhances NMDA receptor-mediated responses in mouse substantia nigra pars compacta dopamine neurons. Neuropharmacology (2016) 103:69–78. doi: 10.1016/j.neuropharm.2015.12.013
118. Errico F, D'Argenio V, Sforazzini F, Iasevoli F, Squillace M, Guerri G, et al. A role for D-aspartate oxidase in schizophrenia and in schizophrenia-related symptoms induced by phencyclidine in mice. Trans Psychiatry (2015) 5(2):e512. doi: 10.1038/tp.2015.2
119. Keller S, Punzo D, Cuomo M, Affinito O, Coretti L, Sacchi S, et al. DNA methylation landscape of the genes regulating D-serine and D-aspartate metabolism in post-mortem brain from controls and subjects with schizophrenia. Sci Rep (2018) 8(1):10163. doi: 10.1038/s41598-018-28332-x
120. Errico F, Rossi S, Napolitano F, Catuogno V, Topo E, Fisone G, et al. D-aspartate prevents corticostriatal long-term depression and attenuates schizophrenia-like symptoms induced by amphetamine and MK-801. J Neurosci Off J Soc Neurosci (2008) 28(41):10404–14. doi: 10.1523/JNEUROSCI.1618-08.2008
121. Becker CM, Hoch W, Betz H. Glycine receptor heterogeneity in rat spinal cord during postnatal development. EMBO J (1988) 7(12):3717–26. doi: 10.1002/j.1460-2075.1988.tb03255.x
122. López-Corcuera B, Geerlings A, Aragón C. Glycine neurotransmitter transporters: an update. Mol Membr Biol (2001) 18(1):13–20. doi: 10.1080/09687680010028762
123. Harvey RJ, Depner UB, Wässle H, Ahmadi S, Heindl C, Reinold H, et al. GlyR alpha3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science (2004) 304(5672):884–7. doi: 10.1126/science.1094925
124. Dutertre S, Becker C-M, Betz H. Inhibitory glycine receptors: an update. J Biol Chem (2012) 287(48):40216–23. doi: 10.1074/jbc.R112.408229
125. Betz H, Gomeza J, Armsen W, Scholze P, Eulenburg V. Glycine transporters: essential regulators of synaptic transmission. Biochem Soc Trans (2006) 34(Pt 1):55–8. doi: 10.1042/BST0340055
126. Prata J, Santos SG, Almeida MI, Coelho R, Barbosa MA. Bridging Autism Spectrum Disorders and Schizophrenia through inflammation and biomarkers - pre-clinical and clinical investigations. J Neuroinflammation (2017) 14(1):179. doi: 10.1186/s12974-017-0938-y
127. Pilorge M, Fassier C, Le Corronc H, Potey A, Bai J, De Gois S, et al. Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol Psychiatry (2016) 21(7):936–45. doi: 10.1038/mp.2015.139
128. Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol (2001) 11(3):327–35. doi: 10.1016/S0959-4388(00)00215-4
129. Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature (1987) 325(6104):529–31. doi: 10.1038/325529a0
131. Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell (1999) 98(4):427–36. doi: 10.1016/S0092-8674(00)81972-8
132. Ballard TM, Pauly-Evers M, Higgins GA, Ouagazzal AM, Mutel V, Borroni E, et al. Severe impairment of NMDA receptor function in mice carrying targeted point mutations in the glycine binding site results in drug-resistant nonhabituating hyperactivity. J Neurosci (2002) 22(15):6713–23. doi: 10.1523/JNEUROSCI.22-15-06713.2002
133. Ishibashi H, Yamaguchi J, Nakahata Y, Nabekura J. Dynamic regulation of glycine-GABA co-transmission at spinal inhibitory synapses by neuronal glutamate transporter. J Physiol (2013) 591(16):3821–32. doi: 10.1113/jphysiol.2012.250647
134. Supplisson S, Roux MJ. Why glycine transporters have different stoichiometries. FEBS Lett (2002) 529(1):93–101. doi: 10.1016/S0014-5793(02)03251-9
135. Zafra F, Aragon C, Olivares L, Danbolt NC, Gimenez C, Storm-Mathisen J. Glycine transporters are differentially expressed among CNS cells. J Neurosci (1995a) 15(5 Pt 2):3952–69. doi: 10.1523/JNEUROSCI.15-05-03952.1995
136. Cubelos B, Gimenez C, Zafra F. Localization of the GLYT1 glycine transporter at glutamatergic synapses in the rat brain. Cereb Cortex (2005) 15(4):448–59. doi: 10.1093/cercor/bhh147
137. Cubelos B, Gonzalez-Gonzalez IM, Gimenez C, Zafra F. The scaffolding protein PSD-95 interacts with the glycine transporter GLYT1 and impairs its internalization. J Neurochem (2005) 95(4):1047–58. doi: 10.1111/j.1471-4159.2005.03438.x
138. Gomeza J, Hulsmann S, Ohno K, Eulenburg V, Szoke K, Richter D, et al. Inactivation of the glycine transporter 1 gene discloses vital role of glial glycine uptake in glycinergic inhibition. Neuron (2003a) 40(4):785–96. doi: 10.1016/S0896-6273(03)00672-X
139. Gabernet L, Pauly-Evers M, Schwerdel C, Lentz M, Bluethmann H, Vogt K, et al. Enhancement of the NMDA receptor function by reduction of glycine transporter-1 expression. Neurosci Lett (2005) 373(1):79–84. doi: 10.1016/j.neulet.2004.09.064
140. Martina M, BT ME, Halman S, Tsai G, Tiberi M, Coyle JT, et al. Reduced glycine transporter type 1 expression leads to major changes in glutamatergic neurotransmission of CA1 hippocampal neurones in mice. J Physiol (2005) 563(Pt 3):777–93. doi: 10.1113/jphysiol.2004.080655
141. Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry (1990) 47(2):181–8. doi: 10.1001/archpsyc.1990.01810140081011
142. Geyer MA, Braff DL. Startle habituation and sensorimotor gating in schizophrenia and related animal models. Schizophr Bull (1987) 13(4):643–68. doi: 10.1093/schbul/13.4.643
143. Geyer MA, McIlwain KL, Paylor R. Mouse genetic models for prepulse inhibition: an early review. Mol Psychiatry (2002) 7(10):1039–53. doi: 10.1038/sj.mp.4001159
144. Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacol (Berl) (2008) 199(3):331–88. doi: 10.1007/s00213-008-1072-4
145. Mansbach RS, Geyer MA, Braff DL. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacol (Berl) (1988) 94(4):507–14. doi: 10.1007/BF00212846
146. Glycine transporters. Br J Pharmacol (2009) 158(Suppl. 1):S195–6. doi: 10.1111/j.1476-5381.2009.00505_7.x
147. Spike RC, Watt C, Zafra F, Todd AJ. An ultrastructural study of the glycine transporter GLYT2 and its association with glycine in the superficial laminae of the rat spinal dorsal horn. Neuroscience (1997) 77(2):543–51. doi: 10.1016/S0306-4522(96)00501-5
148. Rousseau F, Aubrey KR, Supplisson S. The glycine transporter GlyT2 controls the dynamics of synaptic vesicle refilling in inhibitory spinal cord neurons. J Neurosci (2008) 28(39):9755–68. doi: 10.1523/JNEUROSCI.0509-08.2008
149. Gomeza J, Ohno K, Hulsmann S, Armsen W, Eulenburg V, Richter DW, et al. Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron (2003b) 40(4):797–806. doi: 10.1016/S0896-6273(03)00673-1
150. Pearlman RJ, Aubrey KR, Vandenberg RJ. Arachidonic acid and anandamide have opposite modulatory actions at the glycine transporter, GLYT1a. J Neurochem (2003) 84(3):592–601. doi: 10.1046/j.1471-4159.2003.01549.x
151. Ju P, Aubrey KR, Vandenberg RJ. Zn2+ inhibits glycine transport by glycine transporter subtype 1b. J Biol Chem (2004) 279(22):22983–91. doi: 10.1074/jbc.M312484200
152. Vargas-Medrano J, Castrejon-Tellez V, Plenge F, Ramirez I, Miranda M. PKCβ-dependent phosphorylation of the glycine transporter 1. Neurochem Int (2011) 59(8):1123–32. doi: 10.1016/j.neuint.2011.08.006
153. Geerlings A, Lopez-Corcuera B, Aragon C. Characterization of the interactions between the glycine transporters GLYT1 and GLYT2 and the SNARE protein syntaxin 1A. FEBS Lett (2000) 470(1):51–4. doi: 10.1016/S0014-5793(00)01297-7
154. Ransom RW, Deschenes NL. Glycine modulation of NMDA-evoked release of [3H]acetylcholine and [3H]dopamine from rat striatal slices. Neurosci Lett (1989) 96(3):323–8. doi: 10.1016/0304-3940(89)90399-6
155. Krebs MO, Kemel ML, Gauchy C, Desban M, Glowinski J. Glycine potentiates the NMDA-induced release of dopamine through a strychnine-insensitive site in the rat striatum. Eur J Pharmacol (1989) 166(3):567–70. doi: 10.1016/0014-2999(89)90378-6
156. Giorguieff-Chesselet MF, Kemel ML, Wandscheer D, Glowinski J. Glycine stimulates the spontaneous release of newly synthesized 3H-dopamine in rat striatal slices. Eur J Pharmacol (1979) 60(1):10u–4. doi: 10.1016/0014-2999(79)90057-8
157. Yadid G, Pacak K, Golomb E, Harvey-White JD, Lieberman DM, Kopin IJ, et al. Glycine stimulates striatal dopamine release in conscious rats. Br J Pharmacol (1993) 110(1):50–3. doi: 10.1111/j.1476-5381.1993.tb13770.x
158. Destreel G, Seutin V, Engel D. Subsaturation of the N-methyl-D-aspartate receptor glycine site allows the regulation of bursting activity in juvenile rat nigral dopamine neurons. Eur J Neurosci (2019) 50(9):3454–71. doi: 10.1111/ejn.14491
159. Shimizu S, Sogabe S, Yanagisako R, Inada A, Yamanaka M, Iha HA, et al. Glycine-Binding Site Stimulants of NMDA Receptors Alleviate Extrapyramidal Motor Disorders by Activating the Nigrostriatal Dopaminergic Pathway. Int J Mol Sci (2017) 18(7):1416. doi: 10.3390/ijms18071416
160. Javitt DC, Sershen H, Hashim A, Lajtha A. Inhibition of striatal dopamine release by glycine and glycyldodecylamide. Brain Res Bull (2000) 52(3):213–6. doi: 10.1016/S0361-9230(00)00258-6
161. Ye J-H, Wang F, Krnjevic K, Wang W, Xiong Z-G, Zhang J. Presynaptic glycine receptors on GABAergic terminals facilitate discharge of dopaminergic neurons in ventral tegmental area. J Neurosci Off J Soc Neurosci (2004) 24(41):8961–74. doi: 10.1523/JNEUROSCI.2016-04.2004
162. Söderpalm B, Löf E, Ericson M. Mechanistic studies of ethanol's interaction with the mesolimbic dopamine reward system. Pharmacopsychiatry (2009) 42 Suppl 1:S87–94. doi: 10.1055/s-0029-1220690
163. Chau P, Söderpalm B, Ericson M. The mGluR5 antagonist MPEP elevates accumbal dopamine and glycine levels; interaction with strychnine-sensitive glycine receptors. Addict Biol (2011) 16(4):591–9. doi: 10.1111/j.1369-1600.2011.00343.x
164. Molander A, Söderpalm B. Glycine receptors regulate dopamine release in the rat nucleus accumbens. Alcohol Clin Exp Res (2005) 29(1):17–26. doi: 10.1097/01.ALC.0000150006.17168.F7
165. Molander A, Söderpalm B. Accumbal strychnine-sensitive glycine receptors: an access point for ethanol to the brain reward system. Alcohol Clin Exp Res (2005) 29(1):27–37. doi: 10.1097/01.ALC.0000150012.09608.81
166. Jonsson S, Adermark L, Ericson M, Söderpalm B. The involvement of accumbal glycine receptors in the dopamine-elevating effects of addictive drugs. Neuropharmacology (2014) 82:69–75. doi: 10.1016/j.neuropharm.2014.03.010
167. Cherubini E, Gaiarsa JL, Ben-Ari Y. GABA: an excitatory transmitter in early postnatal life. Trends Neurosci (1991) 14(12):515–9. doi: 10.1016/0166-2236(91)90003-D
168. Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa JL. GABAA, NMDA and AMPA receptors: a developmentally regulated ‘ménage à trois'. Trends Neurosci (1997) 20(11):523–9. doi: 10.1016/S0166-2236(97)01147-8
169. Laube B, Maksay G, Schemm R, Betz H. Modulation of glycine receptor function: a novel approach for therapeutic intervention at inhibitory synapses? Trends Pharmacol Sci (2002) 23(11):519–27. doi: 10.1016/S0165-6147(02)02138-7
170. Requardt RP, Wilhelm F, Rillich J, Winkler U, Hirrlinger J. The biphasic NAD(P)H fluorescence response of astrocytes to dopamine reflects the metabolic actions of oxidative phosphorylation and glycolysis. J Neurochem (2010) 115(2):483–92. doi: 10.1111/j.1471-4159.2010.06940.x
171. Huang H, Barakat L, Wang D, Bordey A. Bergmann glial GlyT1 mediates glycine uptake and release in mouse cerebellar slices. J Physiol (2004) 560(Pt 3):721–36. doi: 10.1113/jphysiol.2004.067801
172. Yamauchi T. Molecular constituents and phosphorylation-dependent regulation of the post-synaptic density. Mass Spectrom Rev (2002) 21(4):266–86. doi: 10.1002/mas.10033
173. Boeckers TM. The postsynaptic density. Cell Tissue Res (2006) 326(2):409–22. doi: 10.1007/s00441-006-0274-5
174. Gold MG. A frontier in the understanding of synaptic plasticity: solving the structure of the postsynaptic density. Bioessays (2012) 34(7):599–608. doi: 10.1002/bies.201200009
175. de Bartolomeis A, Buonaguro EF, Iasevoli F, Tomasetti C. The emerging role of dopamine-glutamate interaction and of the postsynaptic density in bipolar disorder pathophysiology: Implications for treatment. J Psychopharmacol (2014) 28(6):505–26. doi: 10.1177/0269881114523864
176. Grabrucker AM. A role for synaptic zinc in ProSAP/Shank PSD scaffold malformation in autism spectrum disorders. Dev Neurobiol (2014) 74(2):136–46. doi: 10.1002/dneu.22089
177. Grabrucker S, Proepper C, Mangus K, Eckert M, Chhabra R, Schmeisser MJ, et al. The PSD protein ProSAP2/Shank3 displays synapto-nuclear shuttling which is deregulated in a schizophrenia-associated mutation. Exp Neurol (2014) 253:126–37. doi: 10.1016/j.expneurol.2013.12.015
178. Grabrucker S, Jannetti L, Eckert M, Gaub S, Chhabra R, Pfaender S, et al. Zinc deficiency dysregulates the synaptic ProSAP/Shank scaffold and might contribute to autism spectrum disorders. Brain (2014) 137(Pt 1):137–52. doi: 10.1093/brain/awt303
179. Coley AA, Gao WJ. PSD95: A synaptic protein implicated in schizophrenia or autism? Prog Neuropsychopharmacol Biol Psychiatry (2018) 82:187–94. doi: 10.1016/j.pnpbp.2017.11.016
180. de Bartolomeis A, Latte G, Tomasetti C, Iasevoli F. Glutamatergic postsynaptic density protein dysfunctions in synaptic plasticity and dendritic spines morphology: relevance to schizophrenia and other behavioral disorders pathophysiology, and implications for novel therapeutic approaches. Mol Neurobiol (2014) 49(1):484–511. doi: 10.1007/s12035-013-8534-3
181. Funk AJ, Mielnik CA, Koene R, Newburn E, Ramsey AJ, Lipska BK, et al. Postsynaptic Density-95 Isoform Abnormalities in Schizophrenia. Schizophr Bull (2017) 43(4):891–9. doi: 10.1093/schbul/sbw173
182. Iasevoli F, Tomasetti C, Buonaguro EF, de Bartolomeis A. The glutamatergic aspects of schizophrenia molecular pathophysiology: role of the postsynaptic density, and implications for treatment. Curr Neuropharmacol (2014) 12(3):219–38. doi: 10.2174/1570159X12666140324183406
183. Foster KA, McLaughlin N, Edbauer D, Phillips M, Bolton A, Constantine-Paton M, et al. Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J Neurosci Off J Soc Neurosci (2010) 30(7):2676–85. doi: 10.1523/JNEUROSCI.4022-09.2010
184. Bakkar W, Ma CL, Pabba M, Khacho P, Zhang YL, Muller E, et al. Chronically saturating levels of endogenous glycine disrupt glutamatergic neurotransmission and enhance synaptogenesis in the CA1 region of mouse hippocampus. Synapse (2011) 65(11):1181–95. doi: 10.1002/syn.20956
185. Balu DT, Coyle JT. Glutamate receptor composition of the post-synaptic density is altered in genetic mouse models of NMDA receptor hypo- and hyperfunction. Brain Res (2011) 1392:1–7. doi: 10.1016/j.brainres.2011.03.051
186. Gadea A, Lopez E, Hernandez-Cruz A, Lopez-Colome AM. Role of Ca2+ and calmodulin-dependent enzymes in the regulation of glycine transport in Muller glia. J Neurochem (2002) 80(4):634–45. doi: 10.1046/j.0022-3042.2001.00735.x
187. Polese D, de Serpis AA, Ambesi-Impiombato A, Muscettola G, de Bartolomeis A. Homer 1a gene expression modulation by antipsychotic drugs: involvement of the glutamate metabotropic system and effects of D-cycloserine. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol (2002) 27(6):906–13. doi: 10.1016/S0893-133X(02)00371-8
188. Iasevoli F, Ambesi-Impiombato A, Fiore G, Panariello F, Muscettola G, de Bartolomeis A. Pattern of acute induction of Homer1a gene is preserved after chronic treatment with first- and second-generation antipsychotics: effect of short-term drug discontinuation and comparison with Homer1a-interacting genes. J Psychopharmacol (2011) 25(7):875–87. doi: 10.1177/0269881109358199
189. de Bartolomeis A, Buonaguro EF, Latte G, Rossi R, Marmo F, Iasevoli F, et al. Immediate-Early Genes Modulation by Antipsychotics: Translational Implications for a Putative Gateway to Drug-Induced Long-Term Brain Changes. Front Behav Neurosci (2017) 11:240. doi: 10.3389/fnbeh.2017.00240
190. de Bartolomeis A, Iasevoli F, Marmo F, Buonaguro EF, Avvisati L, Latte G, et al. Nicotine and caffeine modulate haloperidol-induced changes in postsynaptic density transcripts expression: Translational insights in psychosis therapy and treatment resistance. Eur Neuropsychopharmacol (2018) 28(4):538–59. doi: 10.1016/j.euroneuro.2018.01.006
191. Buonaguro EF, Iasevoli F, Marmo F, Eramo A, Latte G, Avagliano C, et al. Re-arrangements of gene transcripts at glutamatergic synapses after prolonged treatments with antipsychotics: A putative link with synaptic remodeling. Prog Neuropsychopharmacol Biol Psychiatry (2017) 76:29–41. doi: 10.1016/j.pnpbp.2017.02.012
192. Moskal JR, Burgdorf JS, Stanton PK, Kroes RA, Disterhoft JF, Burch RM, et al. The Development of Rapastinel (Formerly GLYX-13); A Rapid Acting and Long Lasting Antidepressant. Curr Neuropharmacol (2017) 15(1):47–56. doi: 10.2174/1570159X14666160321122703
193. Zhou D, Lv D, Wang Z, Zhang Y, Chen Z, Wang C. GLYX-13 Ameliorates Schizophrenia-Like Phenotype Induced by MK-801 in Mice: Role of Hippocampal NR2B and DISC1. Front Mol Neurosci (2018) 11:121. doi: 10.3389/fnmol.2018.00121
194. Aroeira RI, Vaz SH, Sebastiao AM, Valente CA. BDNF modulates glycine uptake in hippocampal synaptosomes by decreasing membrane insertion of glycine transporter 2. Neurochem Int (2016) 99:94–102. doi: 10.1016/j.neuint.2016.06.007
195. ClinicalTrials.gov. (2012). Identifier: NCT01503359 - Effect of Sarcosine on Symptomatology, Quality of Life, Oxidative Stress and Glutamatergic Parameters in Schizophrenia (PULSAR). Available from: https://clinicaltrialsgov/ct2/show/NCT01503359.
196. Deng X, Sagata N, Takeuchi N, Tanaka M, Ninomiya H, Iwata N, et al. Association study of polymorphisms in the neutral amino acid transporter genes SLC1A4, SLC1A5 and the glycine transporter genes SLC6A5, SLC6A9 with schizophrenia. BMC Psychiatry (2008) 8:58. doi: 10.1186/1471-244X-8-58
197. Ohnuma T, Shibata N, Maeshima H, Baba H, Hatano T, Hanzawa R, et al. Association analysis of glycine- and serine-related genes in a Japanese population of patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2009) 33(3):511–8. doi: 10.1016/j.pnpbp.2009.02.004
198. Hornig T, Gruning B, Kundu K, Houwaart T, Backofen R, Biber K, et al. GRIN3B missense mutation as an inherited risk factor for schizophrenia: whole-exome sequencing in a family with a familiar history of psychotic disorders. Genet Res (Camb) (2017) 99:e1. doi: 10.1017/S0016672316000148
199. Vora AK, Fisher AM, New AS, Hazlett EA, McNamara M, Yuan Q, et al. Dimensional Traits of Schizotypy Associated With Glycine Receptor GLRA1 Polymorphism: An Exploratory Candidate-Gene Association Study. J Pers Disord (2018) 32(3):421–32. doi: 10.1521/pedi_2017_31_303
200. Yoshikawa A, Nishimura F, Inai A, Eriguchi Y, Nishioka M, Takaya A, et al. Mutations of the glycine cleavage system genes possibly affect the negative symptoms of schizophrenia through metabolomic profile changes. Psychiatry Clin Neurosci (2018) 72(3):168–79. doi: 10.1111/pcn.12628
201. Bodkin JA, Coleman MJ, Godfrey LJ, Carvalho CMB, Morgan CJ, Suckow RF, et al. Targeted Treatment of Individuals With Psychosis Carrying a Copy Number Variant Containing a Genomic Triplication of the Glycine Decarboxylase Gene. Biol Psychiatry (2019) 86(7):523–35. doi: 10.1016/j.biopsych.2019.04.031
202. Feng J, Craddock N, Jones IR, Cook EH Jr., Goldman D, Heston LL, et al. Systematic screening for mutations in the glycine receptor alpha2 subunit gene (GLRA2) in patients with schizophrenia and other psychiatric diseases. Psychiatr Genet (2001) 11(1):45–8. doi: 10.1097/00041444-200103000-00009
203. Tsai SJ, Cheng CY, Hong CJ, Liao DL, Hou SJ, Yen FC, et al. Association study of polymorphisms in glycine transporter with schizophrenia. J Neural Transm (Vienna) (2006) 113(10):1545–9. doi: 10.1007/s00702-006-0438-1
204. Jamra RA, Villela AW, Klein K, Becker T, Schulze TG, Schmael C, et al. No association between genetic variants at the GLYT2 gene and bipolar affective disorder and schizophrenia. Psychiatr Genet (2006) 16(3):91. doi: 10.1097/01.ypg.0000199450.07786.ab
205. Tsai SJ, Cheng CY, Hong CJ, Liao DL, Liou YJ. Polymorphisms in glycine transporter with schizophrenia. Neuropsychopharmacol Hung (2006) 8(1):17–21.
206. Merk W, Kucia K, Medrala T, Kowalczyk M, Owczarek A, Kowalski J. Association study of the excitatory amino acid transporter 2 (EAAT2) and glycine transporter 1 (GlyT1) gene polymorphism with schizophrenia in a Polish population. Neuropsychiatr Dis Treat (2019) 15:989–1000. doi: 10.2147/NDT.S194924
207. Burnet PW, Hutchinson L, von Hesling M, Gilbert EJ, Brandon NJ, Rutter AR, et al. Expression of D-serine and glycine transporters in the prefrontal cortex and cerebellum in schizophrenia. Schizophr Res (2008) 102(1-3):283–94. doi: 10.1016/j.schres.2008.02.009
208. Yee BK, Balic E, Singer P, Schwerdel C, Grampp T, Gabernet L, et al. Disruption of glycine transporter 1 restricted to forebrain neurons is associated with a procognitive and antipsychotic phenotypic profile. J Neurosci (2006) 26(12):3169–81. doi: 10.1523/JNEUROSCI.5120-05.2006
209. Singer P, Boison D, Mohler H, Feldon J, Yee BK. Deletion of glycine transporter 1 (GlyT1) in forebrain neurons facilitates reversal learning: enhanced cognitive adaptability? Behav Neurosci (2009) 123(5):1012–27. doi: 10.1037/a0016676
210. Kemp JA, Foster AC, Leeson PD, Priestley T, Tridgett R, Iversen LL, et al. 7-Chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyl-D-aspartate receptor complex. Proc Natl Acad Sci United States America (1988) 85(17):6547–50. doi: 10.1073/pnas.85.17.6547
211. Martínez-Fong D, Rosales MG, Góngora-Alfaro JL, Hernández S, Aceves J. NMDA receptor mediates dopamine release in the striatum of unanesthetized rats as measured by brain microdialysis. Brain Res (1992) 595(2):309–15. doi: 10.1016/0006-8993(92)91065-M
212. Kwon HB, Sabatini BL. Glutamate induces de novo growth of functional spines in developing cortex. Nature (2011) 474(7349):100–4. doi: 10.1038/nature09986
213. Roberts BM, Shaffer CL, Seymour PA, Schmidt CJ, Williams GV, Castner SA. Glycine transporter inhibition reverses ketamine-induced working memory deficits. Neuroreport (2010) 21(5):390–4. doi: 10.1097/WNR.0b013e3283381a4e
214. Nikiforuk A, Kos T, Rafa D, Behl B, Bespalov A, Popik P. Blockade of glycine transporter 1 by SSR-504734 promotes cognitive flexibility in glycine/NMDA receptor-dependent manner. Neuropharmacology (2011) 61(1-2):262–7. doi: 10.1016/j.neuropharm.2011.04.010
215. D'Souza DC, Carson RE, Driesen N, Johannesen J, Ranganathan M, Krystal JH. Dose-Related Target Occupancy and Effects on Circuitry, Behavior, and Neuroplasticity of the Glycine Transporter-1 Inhibitor PF-03463275 in Healthy and Schizophrenia Subjects. Biol Psychiatry (2018) 84(6):413–21. doi: 10.1016/j.biopsych.2017.12.019
216. Krystal JH, Tolin DF, Sanacora G, Castner SA, Williams GV, Aikins DE, et al. Neuroplasticity as a target for the pharmacotherapy of anxiety disorders, mood disorders, and schizophrenia. Drug Discovery Today (2009) 14(13-14):690–7. doi: 10.1016/j.drudis.2009.05.002
217. Singer P, Boison D, Möhler H, Feldon J, Yee BK. Modulation of sensorimotor gating in prepulse inhibition by conditional brain glycine transporter 1 deletion in mice. Eur Neuropsychopharmacol J Eur Coll Neuropsychopharmacol (2011) 21(5):401–13. doi: 10.1016/j.euroneuro.2010.06.014
218. Le Pen G, Kew J, Alberati D, Borroni E, Heitz MP, Moreau JL. Prepulse inhibition deficits of the startle reflex in neonatal ventral hippocampal-lesioned rats: reversal by glycine and a glycine transporter inhibitor. Biol Psychiatry (2003) 54(11):1162–70. doi: 10.1016/S0006-3223(03)00374-3
219. Lipina T, Labrie V, Weiner I, Roder J. Modulators of the glycine site on NMDA receptors, D-serine and ALX 5407, display similar beneficial effects to clozapine in mouse models of schizophrenia. Psychopharmacol (Berl) (2005) 179(1):54–67. doi: 10.1007/s00213-005-2210-x
220. Yang SY, Hong CJ, Huang YH, Tsai SJ. The effects of glycine transporter I inhibitor, N-methylglycine (sarcosine), on ketamine-induced alterations in sensorimotor gating and regional brain c-Fos expression in rats. Neurosci Lett (2010) 469(1):127–30. doi: 10.1016/j.neulet.2009.11.058
221. Palmer C, Ellis KA, O'Neill BV, Croft RJ, Leung S, Oliver C, et al. The cognitive effects of modulating the glycine site of the NMDA receptor with high-dose glycine in healthy controls. Hum Psychopharmacol (2008) 23(2):151–9. doi: 10.1002/hup.904
222. Lane HY, Lin CH, Huang YJ, Liao CH, Chang YC, Tsai GE. A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and D-serine add-on treatment for schizophrenia. Int J Neuropsychopharmacol (2010) 13(4):451–60. doi: 10.1017/S1461145709990939
223. Lane HY, Liu YC, Huang CL, Chang YC, Liau CH, Perng CH, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry (2008) 63(1):9–12. doi: 10.1016/j.biopsych.2007.04.038
224. Cioffi CL. Glycine transporter-1 inhibitors: a patent review (2011-2016). Expert Opin Ther Pat (2018) 28(3):197–210. doi: 10.1080/13543776.2018.1429408
226. Pinard E, Alanine A, Alberati D, Bender M, Borroni E, Bourdeaux P, et al. Selective GlyT1 inhibitors: discovery of [4-(3-fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-methanesulfonyl-2-((S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone (RG1678), a promising novel medicine to treat schizophrenia. J Med Chem (2010) 53(12):4603–14. doi: 10.1021/jm100210p
227. Hofmann C, Pizzagalli F, Boetsch C, Alberati D, Ereshefsky L, Jhee S, et al. Effects of the glycine reuptake inhibitors bitopertin and RG7118 on glycine in cerebrospinal fluid: results of two proofs of mechanism studies in healthy volunteers. Psychopharmacol (Berl) (2016) 233(13):2429–39. doi: 10.1007/s00213-016-4317-7
228. Singer P, Feldon J, Yee BK. The glycine transporter 1 inhibitor SSR504734 enhances working memory performance in a continuous delayed alternation task in C57BL/6 mice. Psychopharmacol (Berl) (2009) 202(1-3):371–84. doi: 10.1007/s00213-008-1286-5
229. Depoortere R, Dargazanli G, Estenne-Bouhtou G, Coste A, Lanneau C, Desvignes C, et al. Neurochemical, electrophysiological and pharmacological profiles of the selective inhibitor of the glycine transporter-1 SSR504734, a potential new type of antipsychotic. Neuropsychopharmacology (2005) 30(11):1963–85. doi: 10.1038/sj.npp.1300772
230. Boulay D, Pichat P, Dargazanli G, Estenne-Bouhtou G, Terranova JP, Rogacki N, et al. Characterization of SSR103800, a selective inhibitor of the glycine transporter-1 in models predictive of therapeutic activity in schizophrenia. Pharmacol Biochem Behav (2008) 91(1):47–58. doi: 10.1016/j.pbb.2008.06.009
231. Schmitz Y, Castagna C, Mrejeru A, Lizardi-Ortiz JE, Klein Z, Lindsley CW, et al. Glycine transporter-1 inhibition promotes striatal axon sprouting via NMDA receptors in dopamine neurons. J Neurosci Off J Soc Neurosci (2013) 33(42):16778–89. doi: 10.1523/JNEUROSCI.3041-12.2013
232. Blackaby WP, Lewis RT, Thomson JL, Jennings AS, Goodacre SC, Street LJ, et al. Identification of an Orally Bioavailable, Potent, and Selective Inhibitor of GlyT1. ACS Med Chem Lett (2010) 1(7):350–4. doi: 10.1021/ml1001085
233. Whitehead KJ, Pearce SM, Walker G, Sundaram H, Hill D, Bowery NG. Positive N-methyl-D-aspartate receptor modulation by selective glycine transporter-1 inhibition in the rat dorsal spinal cord in vivo. Neuroscience (2004) 126(2):381–90. doi: 10.1016/j.neuroscience.2004.04.006
234. Omori Y, Nakajima M, Nishimura K, Takahashi E, Arai T, Akahira M, et al. Analgesic effect of GT-0198, a structurally novel glycine transporter 2 inhibitor, in a mouse model of neuropathic pain. J Pharmacol Sci (2015) 127(3):377–81. doi: 10.1016/j.jphs.2015.02.010
235. Morita K, Motoyama N, Kitayama T, Morioka N, Kifune K, Dohi T. Spinal antiallodynia action of glycine transporter inhibitors in neuropathic pain models in mice. J Pharmacol Exp Ther (2008) 326(2):633–45. doi: 10.1124/jpet.108.136267
236. Balu DT. The NMDA Receptor and Schizophrenia: From Pathophysiology to Treatment. Adv Pharmacol (San Diego Calif) (2016) 76:351–82. doi: 10.1016/bs.apha.2016.01.006
237. Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biol Psychol (2016) 116:57–67. doi: 10.1016/j.biopsycho.2015.10.013
238. Buonaguro EF, Tomasetti C, Chiodini P, Marmo F, Latte G, Rossi R, et al. Postsynaptic density protein transcripts are differentially modulated by minocycline alone or in add-on to haloperidol: Implications for treatment resistant schizophrenia. J Psychopharmacol (2017) 31(4):406–17. doi: 10.1177/0269881116658987
239. Ogundele OM, Lee CC. CaMKIIα expression in a mouse model of NMDAR hypofunction schizophrenia: Putative roles for IGF-1R and TLR4. Brain Res Bull (2018) 137:53–70. doi: 10.1016/j.brainresbull.2017.11.007
240. Bugarski-Kirola D, Wang A, Abi-Saab D, Blättler T. A phase II/III trial of bitopertin monotherapy compared with placebo in patients with an acute exacerbation of schizophrenia - results from the CandleLyte study. Eur Neuropsychopharmacol (2014) 24(7):1024–36. doi: 10.1016/j.euroneuro.2014.03.007
241. Dold M, Kasper S. Increasing placebo response in antipsychotic trials: a clinical perspective. Evid Based Ment Health (2015) 18(3):77–9. doi: 10.1136/eb-2015-102098
242. Alphs L, Benedetti F, Fleischhacker WW, Kane JM. Placebo-related effects in clinical trials in schizophrenia: what is driving this phenomenon and what can be done to minimize it? Int J Neuropsychopharmacol (2012) 15(7):1003–14. doi: 10.1017/S1461145711001738
243. Sarkar S, Hillner K, Velligan DI. Conceptualization and treatment of negative symptoms in schizophrenia. World J Psychiatry (2015) 5(4):352–61. doi: 10.5498/wjp.v5.i4.352
244. Velligan D, Maples N, Roberts DL, Medellin EM. Integrated Psychosocial Treatment for Negative Symptoms. Am J Psychiatr Rehabil (2014) 17(1):1–19. doi: 10.1080/15487768.2013.873370
245. Iasevoli F, Avagliano C, Altavilla B, Barone A, D'Ambrosio L, Matrone M, et al. Disease Severity in Treatment Resistant Schizophrenia Patients Is Mainly Affected by Negative Symptoms, Which Mediate the Effects of Cognitive Dysfunctions and Neurological Soft Signs. Front Psychiatry (2018) 9:553. doi: 10.3389/fpsyt.2018.00553
246. Kennedy JL, Altar CA, Taylor DL, Degtiar I, Hornberger JC. The social and economic burden of treatment-resistant schizophrenia: a systematic literature review. Int Clin Psychopharmacol (2014) 29(2):63–76. doi: 10.1097/YIC.0b013e32836508e6
Keywords: N-methyl-d-aspartate, glutamate, dopamine, glycine transporter 1, PSD-95, Homer, disk-1, antipsychotics
Citation: de Bartolomeis A, Manchia M, Marmo F, Vellucci L, Iasevoli F and Barone A (2020) Glycine Signaling in the Framework of Dopamine-Glutamate Interaction and Postsynaptic Density. Implications for Treatment-Resistant Schizophrenia. Front. Psychiatry 11:369. doi: 10.3389/fpsyt.2020.00369
Received: 22 January 2020; Accepted: 14 April 2020;
Published: 14 May 2020.
Edited by:
Maria Rosaria Anna Muscatello, University of Messina, ItalyReviewed by:
Francisco Zafra, Autonomous University of Madrid, SpainCopyright © 2020 de Bartolomeis, Manchia, Marmo, Vellucci, Iasevoli and Barone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea de Bartolomeis, YWRlYmFydG9AdW5pbmEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.