 L. Sanjay Nandam1*
L. Sanjay Nandam1* Matthew Brazel
Matthew Brazel Mei Zhou
Mei Zhou Dhanisha J. Jhaveri
Dhanisha J. Jhaveri- 1Mental Health Unit, Prince Charles Hospital, Brisbane, QLD, Australia
- 2Department of Psychiatry, Royal Hobart Hospital, Hobart, TAS, Australia
- 3Mater Research Institute, The University of Queensland, Brisbane, QLD, Australia
- 4Queensland Brain Institute, The University of Queensland, Brisbane, QLD, Australia
Major depressive disorder (MDD) is a global problem for which current pharmacotherapies are not completely effective. Hypothalamic–pituitary–adrenal (HPA) axis dysfunction has long been associated with MDD; however, the value of assessing cortisol as a biological benchmark of the pathophysiology or treatment of MDD is still debated. In this review, we critically evaluate the relationship between HPA axis dysfunction and cortisol level in relation to MDD subtype, stress, gender and treatment regime, as well as in rodent models. We find that an elevated cortisol response to stress is associated with acute and severe, but not mild or atypical, forms of MDD. Furthermore, the increased incidence of MDD in females is associated with greater cortisol response variability rather than higher baseline levels of cortisol. Despite almost all current MDD treatments influencing cortisol levels, we could find no convincing relationship between cortisol level and therapeutic response in either a clinical or preclinical setting. Thus, we argue that the absolute level of cortisol is unreliable for predicting the efficacy of antidepressant treatment. We propose that future preclinical models should reliably produce exaggerated HPA axis responses to acute or chronic stress a priori, which may, or may not, alter baseline cortisol levels, while also modelling the core symptoms of MDD that can be targeted for reversal. Combining genetic and environmental risk factors in such a model, together with the interrogation of the resultant molecular, cellular, and behavioral changes, promises a new mechanistic understanding of MDD and focused therapeutic strategies.
Introduction
Major depressive disorder (MDD) is a complex, multifactorial, and heterogenous clinical syndrome that currently affects at least 120 million people worldwide and by 2030 will be the single highest contributor to the global burden of disease (1). Existing therapies are not efficacious for all patients and over the past five decades few, if any, truly novel treatments for MDD have emerged that go beyond the monoamine theory of depression first presented in the 1960s (2). Although there is growing evidence that multiple other neurotransmitter systems (3), inflammatory processes (4), and dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis are involved in MDD, these insights have not yet led to new treatments due to our limited understanding of their molecular mechanisms (5).
Progress in therapeutics has been hindered by the inadequacy of preclinical animal models to fully recapitulate the heterogeneous nature of the human condition (5). To improve this, researchers have sought to develop models that maintain adequate face, predictive, etiological, and construct validity with MDD (3). Specifically, candidate models need to demonstrate similarity between the behavioral phenotype and clinical symptoms (face validity), reversal of behavior with known or proposed antidepressants (predictive validity), and behavior inducible by factors associated with human disease (etiological validity), as well as displaying putative MDD biomarkers (construct validity) (3). In meeting these criteria, a major goal of preclinical animal models has been to emulate MDD to provide a framework for investigating neurobiological cause and effect mechanisms beyond those available from clinical studies.
The seemingly causal relationship between stress and cortisol in MDD, first described in the 1950s (6), promised just such a mechanism that was both independent of the monoamine theory and potentially able to deliver new therapeutic approaches. Accordingly, rodent models using environmental and hormonal stressors, addressing etiological and construct validity, respectively, have been developed. Quantitative changes in behavioral phenotypes, particularly those recapitulating core symptoms of major depression such as increased anxiety, anhedonia, and cognitive deficits, have been used for face validity. Multiple drug studies have subsequently explored the predictive validity of these models for new and established antidepressants. Although this approach has provided some new biological insights and increased the knowledge base, it has not yet produced the needed therapeutic breakthroughs.
In this review, we critically evaluate the validity of modelling HPA dysregulation in preclinical studies as a key driver of MDD pathophysiology. We do this by contrasting animal data with acute and chronic clinical studies that have examined stress, MDD treatment, and cortisol levels. We conclude by discussing the advantages and disadvantages of the current rodent models of MDD and HPA dysregulation, and how these might be used in combination with genetic risk factors and modern neuroscience tools to probe the neural circuitry of MDD and, ultimately, improve its treatment.
Cortisol and MDD
In MDD, glucocorticoid receptor (GR) signaling is abnormal (7) and associated with chronic hypersecretion from the corticotrophin releasing hormone (CRH) neurons of the HPA axis (Figure 1) (8). This hypersecretion is thought to shift HPA activity toward ever higher set points, resulting in the persistently elevated HPA activity seen in some MDD patients (9, 10). Many studies of MDD patients have reported abnormalities of cortisol suppression in response to pharmacological and psychological challenge (11–13), and probing the mechanisms underlying this effect has remained a popular line of inquiry for both clinical and translational research. However, the association between cortisol and MDD in humans is complex, and appears dependent on stage of illness, severity, and type of challenge employed. For example, with respect to stage of illness, HPA responsiveness does not appear to be affected in chronic MDD (symptoms of more than 2 years duration), with patients and controls not showing any difference in salivary and serum cortisol levels following dexamethasone suppression testing (DST), even after controlling for variability in dexamethasone metabolism (14). In contrast, remitted MDD patients generate higher cortisol levels than controls following exposure to a visual stress cue (15). In those with remitted MDD, persisting hyperresponsiveness to dexamethasone can predict relapse at 6 months (16), whereas cortisol levels do not provide prognostic information in chronic MDD (14).
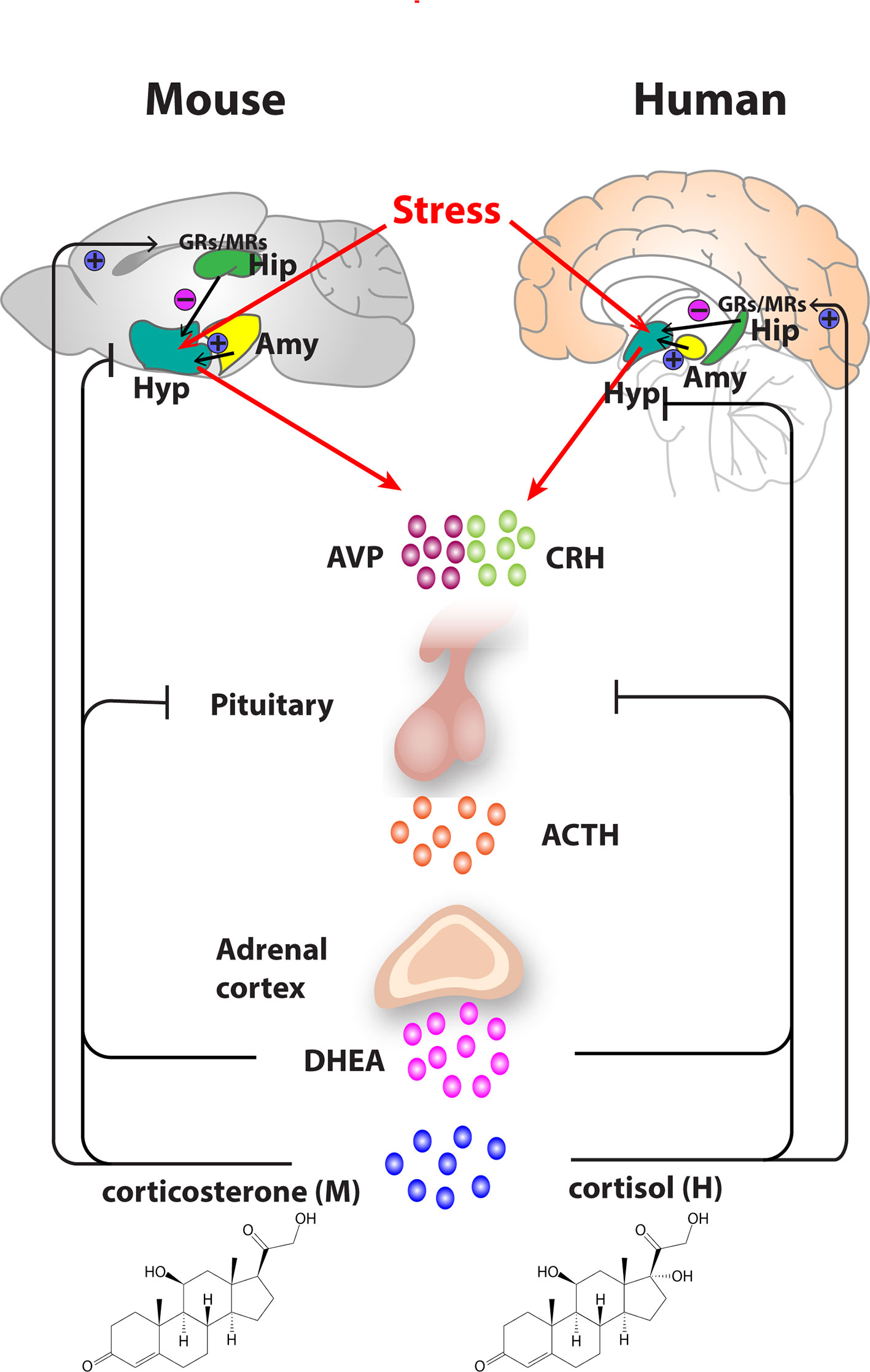
Figure 1 Organization of the hypothalamic–pituitary–adrenal (HPA) axis in mouse and human. In both mouse and humans, stress leads to secretion of corticotropin releasing hormone (CRH) and arginine vasopressin (AVP) from the hypothalamus, which subsequently stimulates the secretion of adrenocorticotropic hormone (ACTH) from the pituitary gland. ACTH transported via blood to the adrenal cortex of the adrenal gland stimulates the synthesis and secretion of glucocorticoids—primarily corticosterone, in mice and cortisol in humans as well as secretion of other major steroid dehydroepiandrosterone (DHEA). Glucocorticoids regulate their own secretion via a negative feedback control mechanism via mineralocorticoid (MR) and glucocorticoid receptors (GR) at the level of the hippocampus, hypothalamus, and pituitary gland. MRs, which have a high affinity for endogenous glucocorticoids, are found primarily in the hippocampus and determine the basal activity of the HPA axis. In contrast, the activation GRs upon binding of glucocorticoids in the hippocampus, hypothalamus, and pituitary leads to an inhibitory feedback onto the hypothalamus to control the stress response. A facilitatory input from amygdala to the hypothalamus also plays a role in the activation of the HPA axis response. Arrows represent stimulation, and T-shaped lines represent inhibition. Amy, Amygdala; Hip, Hippocampus; Hyp, Hypothalamus.
With respect to the severity of MDD, a number of studies have shown that the severity of depressive symptoms is proportionate to cortisol level (16). More severe MDD subtypes, such as psychotic (17–20) and melancholic MDD (21, 22), consistently show higher baseline and challenge salivary and serum cortisol levels than observed in atypical MDD, a less severe MDD variant. Atypical MDD sufferers have been shown to have cortisol responses that are closer to those of healthy controls, especially when contrasted against the levels seen in melancholic MDD (21, 22). It has also been suggested that elevated cortisol may be responsible for the emergence of psychotic symptoms in severe MDD (23, 24). There are also marked HPA abnormalities in bipolar depression, which has a more severe presentation than milder forms of MDD (25). Elevated cortisol levels are also associated with the cognitive impairment seen in psychotic MDD (24).
Taken together, the evidence supports the notion that cortisol dysfunction is proportionate to, and consistent with, current nosological classifications of MDD severity. It appears that cortisol dysregulation, specifically in response to stress, is reliably associated with severe and acute presentations of MDD. Chronic and less severe subtypes of MDD, such as atypical depression, do not exhibit this robust association, which is consistent with data suggesting that personality and social factors may be major drivers of these subtypes (26). The association of HPA dysregulation with MDD severity might be mechanistically explained by parallel findings from receptor, immunological, and imaging studies. Melancholic and psychotic, but not atypical, depression show increased cortisol response to laboratory stress challenge (27). This may come about through altered functioning of GR expression, which then affects the homeostatic regulation of cortisol (28). The resultant hypercortisolism drives changes to serotonin receptors (29, 30), and this has been proposed as the mechanism underlying the specific symptoms of severe MDD (31). In atypical MDD, such symptoms are absent and hence serotonin receptor function is likely preserved (32). Elevated cortisol levels have also been proposed to contribute to the psychosis seen in psychotic MDD, via increased dopaminergic activity (33). Supporting this theory, mifepristone, a GR antagonist, has been shown to specifically reduce the psychotic symptoms of psychotic MDD (34, 35). Atypical MDD shows elevated inflammatory markers compared to melancholic MDD (22). Individuals with psychotic (36) and melancholic (37) MDD have been found to have smaller hippocampal volumes than those with less severe forms of depression. Frontal lobe volume is reduced in psychotic MDD (38), and the sylvian fissure is enlarged in melancholic MDD (39). In contrast to atypical depression, melancholic depression is characterized by reduced blood flow to the right frontal lobe, perhaps supporting the importance of social and personality in the former (40).
Risk Factors for MDD and Cortisol Elevation
Early Life Stress, Cortisol, and MDD
Early life stress (ELS) programs for abnormal adult cortisol responses to stress challenge (41), potentially via glucocorticoid resistance, increased central CRH activity (42) and changes to GR and mineralocorticoid receptor (MR) binding (43). Abnormal cortisol responses persist into adulthood (44) and likely underpin the impaired HPA axis function seen in several trauma associated psychiatric disorders, including MDD. Nonetheless, the precise way that ELS affects adult HPA function appears complex. In an instructive study, for adolescents with a history of stressful life events, those who experienced recurrent depression exhibited a persistently elevated cortisol response to a laboratory stress task (32). In the same cohort, those who were currently experiencing their first episode of MDD exhibited a blunted cortisol response to the same task (32). Despite all the adolescents having had a prior history of ELS, there were no differences in baseline cortisol levels, and correlation with MDD was only demonstrated by variance in cortisol reactivity, rather than absolute level, to the task (32). This range of state and task dependent cortisol changes in a single cohort parallels the varied findings in the cortisol and ELS literature (45). Several studies report increased cortisol reactivity to stress in ELS populations (44, 46–49), whereas, contrary data showing reduced cortisol responses have also been reported (50–53). Variance in HPA axis outcome to ELS have been shown to be dependent on the timing of the stress (54). ELS from birth to age 5 years does not appear to affect adult HPA function, whereas ELS from age 6 to11 years results in exaggerated adult cortisol response to stress, and ELS from age 12 to 16 years is associated with blunting (54). These differing outcomes might be due to age-dependent variance in neural plasticity (42) and the impact of puberty (55). In any case, adults who have experienced ELS all appear to have smaller hippocampal volumes (56, 57), regardless of age of trauma and differing effects on HPA function. Stress in adulthood is also a risk factor for developing MDD, with up to 75% of MDD episodes following stress exposure (58, 59).
Gender
Female gender is an established risk factor for MDD (60), which has generated interest into probing whether females show higher magnitude cortisol responses to stress than males. A simple relationship, however, has not been established. When exposed to a stressful public speaking task, females in luteal phase showed similarly elevated salivary cortisol levels to men, whereas women in follicular phase, or on oral contraceptives, actually had lower cortisol levels than men and luteal phase women (61). The total serum cortisol level did not differ between groups (61). In contrast, higher cortisol levels following dexamethasone challenge were found in females, but not males, in a depressed cohort (62). In this study, higher cortisol was associated with greater burden of neurovegetative symptoms, providing additional evidence that cortisol abnormalities appear to be positively correlated with depressive severity (62). However, the findings that males with MDD can have higher baseline cortisol levels than females with MDD (63, 64) argue against there being a simple relationship between overall cortisol level and the increased incidence of MDD in women.
A clearer picture emerges from the finding that males with MDD and matched male controls show similar baseline cortisol levels, whereas females with MDD have significantly higher baseline cortisol than their matched controls (65). This suggests that the increased incidence of MDD in females might be better explained by intra-gender cortisol level variability (65). Women show differences in HPA regulation based on menopausal status, with menopausal depressed women being more likely to be corticotrophin non-suppressors than pre-menopausal women (66). Women in the luteal phase also show decreased cortisol suppression to DST compared to results obtained in the follicular phase (60). This may be due to a decrease in GR density during the luteal phase changing negative feedback sensitivity to dexamethasone, resulting in phase dependent intra-individual cortisol response variability (60). Young et al. hypothesize that it is this variable, rather than simply higher, female HPA responsiveness to stress across menstrual phase, and then across the lifetime, which explains the greater incidence of MDD in women (60). In a similar manner to the relationship between stress tasks, cortisol, and major depression, it again appears that the variability, rather than magnitude, of the cortisol response that correlates with MDD.
Relationship Between Cortisol and MDD Treatment
Antidepressants
For monoamine reuptake inhibitor antidepressants, acute dosing is consistently associated with an increase in serum cortisol levels (see Supplementary Table 1). For example, in healthy volunteers, single infusions of the Selective serotonin reuptake inhibitor (SSRI) citalopram (20 mg), fluvoxamine (100 mg), and fluoxetine (80 mg) all produced increases in serum cortisol (67–69). Likewise, acute administration of reboxetine (4 mg), a noradrenaline reuptake inhibitor (NRI), and venlafaxine, a serotonin and noradrenaline reuptake inhibitor antidepressant (SNRI), also increased the level of cortisol in healthy subjects (70, 71). The ability of various monoamine reuptake inhibitor antidepressants to rapidly increase cortisol levels after acute administration might be due to increased monoamine concentrations directly affecting anterior pituitary hormone secretion (72).
In contrast to the monoamine reuptake inhibitors, a single dose of mirtazapine (15 mg) decreased serum cortisol, ACTH, and urinary free cortisol in healthy male volunteers (73). Mirtazapine is a presynaptic alpha2 and postsynaptic serotonin 5-HT2, 5-HT3 receptors and histamine H1 receptor antagonist that does not use reuptake inhibition to increase serotonin and noradrenaline levels (74). Modulation of these receptors in the rodent hypothalamus can inhibit CRH release, which may explain mirtazapine's ability to rapidly decrease cortisol levels in humans (8).
In studies of MDD patients, chronic antidepressant treatment has also been shown to normalize cortisol levels, irrespective of their differing pharmacological profiles (see Supplementary Table 1) (8). It has been suggested that normalization of GR function is the common antidepressant mechanism that allows improved negative feedback to the HPA and restoration of normal neuroendocrine function (75). For example, in a study where MDD and controls had similar baseline urinary cortisol levels, the clinical response after treatment with fluoxetine over 4 months was associated with a reduction in urinary cortisol (76).
Reductions in cortisol level are not, however, consistently associated with the clinical response to antidepressants (see Supplementary Table 1). For example, in a comparative study of 94 MDD patients treated with either amitriptyline (a tricyclic antidepressant; 150 mg) or paroxetine (a SSRI antidepressant; 40 mg), only the amitriptyline responders showed a decrease in salivary cortisol over 5 weeks, despite the similar clinical efficacy of these drugs (77). The consistency of the antidepressant effect on cortisol can also vary over time (see Supplementary Table 1). For example, after 7 days of mirtazapine treatment, the serum cortisol level decreased in 12 MDD responders for 5 weeks but then increased to higher levels than baseline, and was ultimately unable to distinguish them from non-responders (78).
Antipsychotics and Lithium
There is increasing evidence that atypical antipsychotics are efficacious in the treatment of MDD (8). The older generation “typical” antipsychotics was mostly limited to antagonizing dopaminergic D2 receptors, whereas newer “atypical” antipsychotics have additional antagonist activity at 5-HT2A, potentially allowing direct modulation of the hypothalamus to influence cortisol levels (8). For example, in healthy controls, acute dosing with the atypical antipsychotics olanzapine, quetiapine, and ziprasidone has been reported to reduce cortisol levels, whereas the typical antipsychotic haloperidol had no effect (79, 80). With respect to chronic dosing, after 1 week of treatment with quetiapine (300 mg), MDD patients had a significant reduction in cortisol in response to dexamethasone challenge; however, after 5 weeks cortisol secretion had increased to near pre-treatment levels (81). Lithium treatment in mood disorders has also been associated with increased cortisol levels in response to dexamethasone and DST challenge without reliably mapping to clinical response (82, 83).
GR Antagonists and Cortisol Synthesis Inhibitors
Probing the relationship between cortisol and mood disorder, the GR antagonist mifepristone has gathered considerable interest as a potential treatment for MDD. While initially showing promise for the treatment of psychotic depression (35, 84), further work has shown that its ameliorative effect is limited only to psychotic, rather than depressive, symptoms (85, 86). This result may be due to the wide variation in serum mifepristone concentrations across patients despite standard dosing (87), with higher serum concentrations associated with reducing psychotic symptoms only (34). Somewhat paradoxically, mifepristone, which appears to increase cortisol levels (85), reached Phase 3 trials, despite elevated cortisol being associated with worsening psychosis via increased dopaminergic tone (33). Mifepristone's modest antipsychotic mechanism remains poorly understood and interest in further antidepressant trials has waned after three failed Phase 3 studies (88).
Agents that block cortisol synthesis, rather than acting directly at the GR, have also been pursued. Metyrapone is an aromatic ketone that inhibits the synthesis of cortisol and has long been of interest for the treatment of MDD (89). More recent trials have confirmed that metyrapone is associated with an improvement in mood in MDD patients (90, 91) but beneficial effects do not appear to be related to cortisol levels and its underlying mechanism(s) remain unclear (92). Another cortisol synthesis inhibitor, ketoconazole, has only produced inconclusive results in MDD (93, 94). Notably, several HPA modulating agents showing promise in preclinical studies remain in the early stages of investigation and are yet to reach Phase 3 clinical trials (88, 95).
Electroconvulsive Therapy
Electroconvulsive therapy (ECT) is a somatic treatment for MDD that has been shown to be particularly efficacious in psychotic and melancholic depression (96). In a study of nine patients with psychotic MDD, ECT produced a reduction in cerebrospinal fluid CRH (97). Thirty minutes after treatment, ECT produced an increase in serum cortisol in 13 patients with melancholic MDD (98). In two studies of unspecified MDD, ECT led to a normalization of serum cortisol with resolution of depressive symptoms (99, 100).
Confounders to Interpreting Cortisol in MDD
Cortisol response to stress can be measured across three phases: (a) basal, or unstressed, phase; (b) reactivity phase, where cortisol rises in response to a stressor; and (c) recovery phase; where cortisol returns to baseline following stress. These phases are regulated by different receptors, with GR regulating cortisol during reactivity phase, and MR regulating cortisol at periods of baseline activity. In a meta-analysis, Burke et al. reviewed these varying cortisol phases in depressed patients versus controls (101). Depressed patients tended to show a relatively flat unresponsive pattern of cortisol secretion with both blunted stress reactivity and impaired recovery phases. This association also appears to hold true for prospective MDD risk. In patients who had undergone coronary artery bypass grafting (a stressor), those whose salivary cortisol returned to normal more slowly during the recovery phase were most likely to have MDD a year later (102). Similarly, in adolescents with a history of stressful life events, those who also had a history of recurrent depression also had a persistently elevated reactivity phase to stress (32). The meta-analysis showed that blunted response curves are most strongly associated with severe MDD. In contrast, non-depressed controls showed greater reactivity variability and faster recovery phase following stressors. Conclusions from the meta-analysis were limited, however, as it included studies focusing on total salivary, rather than free, cortisol fraction.
Salivary cortisol has frequently been used to assay stress in MDD studies. While being relatively inexpensive and pain-free to test in subjects, it is limited by only being able to measure unbound free cortisol (103). In contrast, serum cortisol assays are able to measure unbound and protein bound cortisol fractions but have the disadvantage of being more difficult to collect and the added confounder of stress associated with venepuncture (104). There is a high correlation between salivary cortisol and free serum cortisol levels, which is maintained during DST and ACTH challenge (105). The usefulness of this correlation, however, relies on the presumption that only free serum cortisol is biologically active (103). This notion has been challenged by the finding that cortisol bound to cortisol binding globulin (CBG) can have physiological effects on tissue (106). Furthermore, the distinction between free and bound cortisol is not very clear, with hepatic uptake of bound cortisol being threefold faster than free cortisol followed by rapid dissociation, suggesting that CBG may be acting as an active delivery mechanism, rather than passive storage, in serum (106). In contrast to its relationship with free serum cortisol, salivary cortisol does not have a linear relationship with total cortisol. Instead, the ratio of salivary cortisol to total cortisol is concentration dependent, with 1–2% at lower concentrations and rising to 8–9% at higher concentrations (105). Salivary cortisol levels also rise dramatically once CBG is saturated via sex steroids such as during the follicular phase, pregnancy, and usage of the oral contraceptive pill (61, 106–107). Confounders such as these have likely contributed to the marked heterogeneity of results in a meta-analysis of salivary cortisol levels in 1,354 MDD patients and 1,052 controls, which concluded that salivary cortisol alone was unable to distinguish MDD (108). Recent work has also shown how differences in dexamethasone metabolism, rather than MDD symptoms, can better explain variance in cortisol levels (109), though studies have attempted to control for this by directly sampling serum dexamethasone (14). To counter difficulties with salivary cortisol, it is possible to measure free and bound fractions of serum cortisol directly via liquid chromatography-tandem mass spectrometry; however, reference ranges using this technique for suppression tests are yet to be established (110). Furthermore, the DST only probes GR, whereas the less frequently used, but more physiologically relevant, prednisolone suppression test examines both GR and MR function (111), both of which interact abnormally in MDD pathophysiology.
Understanding the Neurobiological Mechanisms of Depression: Modelling MDD in Preclinical Studies
Despite progress in the development of non-invasive techniques to study MDD in humans, detailed examination of the molecular and cellular mechanisms and neural circuitry underpinning the disorder remains limited. Some limitations can be addressed via animal models that critically confer the ability to study cause and effect relationships under controlled conditions. For example, although human genome studies have revealed genes associated with depression (112), additional human studies cannot prospectively address how such genes might cause depression. In contrast, being able to manipulate these genes in transgenic animals provides an opportunity to correlate depression-associated behaviors with candidate mechanisms (113).
Different behavioral responses are scored and quantified in preclinical models as surrogates of mood-related changes. For example, in rodent models, the amount of time spent immobile in the forced swim test and the tail suspension test are used to measure behavioral despair, which is thought to be analogous to the hopelessness seen in MDD (114). The sucrose preference test has been used to determine anhedonic behavior in rodents, potentially corresponding to the diminished interest in pleasurable experiences seen in depressed patients (114). In rodent colonies, the social interaction test has been used to model the social withdrawal seen in MDD (115). Due to the high comorbidity of depression and anxiety (116), behavioral tests that examine rodent anxiety, such as the elevated plus maze, novelty-suppressed feeding, open field, and dark-light box tests, are also used to study depression (117, 118). Utilizing multiple behavioral assays thus provides an experimental system that features analogues of human MDD and is open to detailed experimental manipulation.
Given that both susceptible genes and environmental risk factors are implicated in the development of depression, most animal models employ a combination of genetic manipulation and environmental stressors to produce animals that exhibit depression-like behaviors. The evolutionary conservation of the stress response brain circuitry between humans and rodents remains fundamental to translational studies (Figure 1). In rodents, stress induces secretion of corticosterone via the HPA axis, with rodent corticosterone and human cortisol having functionally equivalent roles (119).
Corticosterone Levels in Genetic Models of Depression
Most genetic models of MDD in rodents are generated via selective breeding. Animals with the desired features are bred over several generations, yielding inbred strains that reliably exhibit specific physiological or behavioral abnormalities (120). For example, Flinders sensitive line (FSL) rats display both depression-like behavior (121) and vulnerability to stress-induced anhedonia-like behavior (122). These behavioral traits are also associated with elevated corticosterone levels (123–125). Wistar-Kyoto (WKY) rats are characterized by depression-like and anxiety-like behaviors (126), and exhibit an upregulated basal serum corticosterone level at the start of the dark cycle, which is reminiscent of the circadian rhythm abnormalities seen in human MDD (127, 128). Importantly, prolonged corticosterone stress responses and DST non-suppressibility have been observed in WKY rats (128, 129). Congenitally learned helpless (cLH) rats show pronounced helplessness behavior without prior exposure to uncontrollable shock (130). Baseline corticosterone levels are similar between cLH rats and controls; however, cLH rats experiencing acute stress have a decreased cortisol response compared with adult controls (131, 132), reminiscent of the differences in the stress cortisol response between traumatized and non-traumatized MDD patients.
Corticosterone Levels in Environmental Models of Depression
Chronic mild stress (CMS) paradigms are often used to model MDD in rodents due to their ability to induce behavioral despair, and anxiety-like and anhedonia-like behaviors (133). In CMS models, there is a consistent increase in serum corticosterone following stressor exposure (134–138). Notably, reducing corticosterone levels via pharmacological inhibition of corticosterone synthesis or adrenalectomy has been shown to block the development of depressive behaviors in CMS models (139, 140). Compared to physical stressors, such as restraint, damp bedding, and food deprivation, social stressors, such as social isolation (141) and social defeat (142), tend to more reliably produce depression-like behaviors but are associated with variable basal corticosterone levels while leading to increase in corticosterone levels in response to acute stress (141–150).
Models of ELS, induced by maternal separation or limited nesting, have also been used to recapitulate the association between adverse childhood events and MDD (151, 152). In rodents, the effects of ELS on corticosterone levels are highly variable, being stressor-specific as well as dependent on the developmental period during which the stressors are administered and the age at which animals are assessed (152–159). This is reminiscent of the weak association between early life trauma and dysregulated HPA response to acute stress in adults.
Collectively, the findings from rodent models display some similarities to those observed in humans, where it is the variable responsiveness to repeated stress, rather than the basal level of corticosterone, which is associated with depression-like behavior.
Modelling Depression Using Exogeneous Corticosterone Administration
To simulate the HPA dysregulation seen in the genetic and stress-based models of depression, researchers have used exogeneous corticosterone administration in rodents (160). Robust and highly reproducible anxiety- and depression-like behaviors have been reported following chronic oral administration of corticosterone, which may or may not lead to an elevation in the level of serum corticosterone (133, 161–169). In addition to the behavioral changes, chronic exogeneous corticosterone is associated with several neurobiological changes seen in MDD models including the disruption of adult hippocampal neurogenesis (170), and decreased hippocampal brain derived neurotrophic factor (BDNF) (171). Chronic treatment with classical antidepressants such as fluoxetine and imipramine (160, 172–174), as well as a single dose of faster-acting antidepressant ketamine (164, 175), have been shown to reverse corticosterone-induced depression-like behaviors. In a subset of these studies, beneficial behavioral effects of antidepressant treatment was accompanied by normalization of serum corticosterone levels (172–174). Notably, sub-chronic treatment with GR selective antagonist, RU-43044, has also been shown to reverse corticosterone-induced depression-like behavior (176) suggesting GR as a potential molecular target to combat HPA axis dysregulation.
Classical Antidepressants and Corticosterone
Mirroring findings in humans, acute and sub-chronic treatment with classical antidepressants have been reported to increase corticosterone levels in rodents (see Supplementary Table 2). For example, in non-stressed animals, a single injection of fluoxetine (10 mg/kg) or imipramine (30 mg/kg) increased serum corticosterone levels (177, 178). It has been suggested that following antidepressant administration, hippocampal GR and mineralocorticoid (MR) receptors are downregulated, which reduces HPA negative feedback, thereby leading to an increase in corticosterone (179). This hypothesis is supported by the finding that 9 days of fluoxetine (10 mg/kg) and venlafaxine (10 mg/kg) treatment results in downregulation of hippocampal GRs and MRs (180). In contrast to these effects, chronic antidepressant treatment had no effect on basal corticosterone levels in non-stressed rodents. For example, 24 days of either paroxetine, (7.5 mg/kg) (181), venlafaxine (10 mg/kg) (181), or desipramine (7.5 mg/kg) (181, 182) did not affect basal serum corticosterone levels.
Paralleling findings in human MDD described above (summarized in Supplementary Table 1), multiple antidepressants have been shown to normalize corticosterone levels in stressed animals (see Supplementary Table 2). For example, treatment with fluoxetine for more than 2 weeks normalized basal corticosterone levels in chronic unpredictable stress and social defeat stress rodent models (183–185). Similarly, chronic treatment with venlafaxine or imipramine normalized basal corticosterone in chronic unpredictable stress and prenatal stress exposed rodents (186–190).
Atypical Antidepressants, Lithium, and Corticosterone
In rodents, the effect of atypical antidepressants on corticosterone levels is similar to those seen with classical antidepressants (see Supplementary Table 2). For example, a single injection of mirtazapine increased basal corticosterone levels (177), whereas a longer treatment period had no effect on either basal or post-stress levels (177, 191). In chronic unpredictable stress and prenatal stress, chronic mirtazapine treatment normalized basal and post-stress corticosterone (188, 190). Chronic treatment with tianeptine (10 mg/kg), a tricyclic antidepressant that locks the serotonin transporter into the open position, also normalized basal corticosterone in a prenatal stress model (190). Single (1.4 meq Li+/kg) and repeated (45 meq Li+ mixed with 1 kg food, 6 days) lithium treatment of non-stressed rats increased corticosterone levels (192). However, in socially isolated mice, a single high dose (10 mg/kg) injection of lithium decreased post-stress, but not basal, corticosterone levels (193). Similarly, repeated lithium treatment at a moderate dose (2.5 mg/kg) decreased basal corticosterone levels in a chronic unpredictable stress model in rats (194).
Conclusions and Future Perspectives
In summary, clinical studies show that MDD is most consistently associated with stress-induced variability in HPA axis response rather than absolute levels of cortisol. Elevated baseline cortisol levels are only seen in more severe subtypes of MDD and are conspicuously absent in those with chronic or atypical presentations. Furthermore, a reduction in cortisol is not reliably associated with successful treatment with monoaminergic antidepressants, regardless of type. Accordingly, baseline cortisol, or corticosterone levels, cannot be used as a simple proxy for modelling MDD in general, or evaluating treatment response in either humans or animals.
Recent work has resulted in a rapid expansion of the potential mechanisms that could underlie the increasingly complex relationship between HPA axis dysfunction and MDD. Arginine vasopressin (AVP), a peptide hormone secreted from the posterior pituitary, acts synergistically with CRH to increase the release of ACTH and cortisol (Figure 1) (196). AVP is increased in MDD (195, 196), possibly contributing to the variable cortisol response to stress challenge in these populations. Interestingly, recent work has also shown that melancholic MDD is specifically associated increased AVP activity (197), which is linked to increased vasopressin V3 receptor expression (198). It remains unknown whether such changes are also present in milder forms of MDD. MDD also features HPA axis changes at the receptor level. GR and MR act in a complementary fashion during the stress response but are thought to be in imbalance in MDD (10, 199), which may lead to variable cortisol expression during stress via competition between these pathways. In addition to this abnormal relationship between MR and GR, negative feedback through GR is unpredictably impaired in MDD, further exacerbating cortisol variability (200, 201). In addition to these emerging receptor and endocrine mechanisms, genetics and epigenetic are also likely contributing to the varied results in extant literature.
Evidence from clinical and animal studies suggests that genetic and epigenetic mechanisms link chronic stress, HPA axis dysregulation, and alterations to glucocorticoid levels and signaling. For example, polymorphisms in promoter region of the GR gene, NR3C1, have been associated with either increased sensitivity (202, 203) or resistance to cortisol (204). There is also increased DNA methylation of hippocampal NRC31 associated with depression, including in the brains of suicide victims with a history of ELS (205, 206). Expression of GR mRNA is decreased in those with a history of childhood sexual abuse (205) and GR gene methylation is increased in children who have experienced trauma (207). In addition to genetic and epigenetic regulation of the GR gene, polymorphism in FK506 binding protein (FKBP5), an important regulator of GR function, has been associated with DST hyperresponsiveness (208). The importance of this gene to stress-related disorders has been further supported by studies of FKBP5 knockout mice that show an increased sensitivity of GRs (209, 210). Similarly, polymorphism in the CRH receptor (CRHR1) gene has also been linked to MDD (211) and is associated with HPA axis dysregulation.
The finding that a diverse range of antidepressants rapidly stabilize cortisol levels following acute administration could potentially explain why their beneficial effects are observed within hours of commencement compared to placebo (212). We speculate that antidepressants might be overriding ELS-induced adult cortisol variability in response to stress, which allows recovery to commence. However, at present, the inconsistent relationship between chronic antidepressant administration, clinical response, and cortisol remains difficult for any mechanistic interrogation. Fortunately, the data showing that multiple antidepressants have similar effects on stress hormones in humans and rodents increases confidence that there is sufficient evolutionary conservation between species to carefully model HPA axis dysregulation in rodents to further probe MDD neurobiology. While rodent models have proved extremely valuable in gaining mechanistic insights into the pathophysiology, emerging studies using the non-human primates have begun to offer additional dimensions to the endocrine and behavioral correlates of chronic stress induced MDD (213, 214). Exploring the effects of HPA axis modulation in these animal models could provide a bridge to improve our mechanistic understanding of how changes in cortisol cascade to abnormalities in metabolism, gene regulation, and immune function, all of which have been independently implicated in MDD. This combined with high-throughput longitudinal monitoring of group-housed animals, as reported recently for mice (215), could yield a variety of behavioral readouts of individual animals and help stratify vulnerable versus resilient subpopulations in response to various stress such as early life or social stress.
An ideal rodent or non-human primate model should therefore reliably produce exaggerated or blunted HPA axis responses to acute or chronic stress a priori, which may, or may not, alter baseline cortisol levels. Such a model would provide an evidence-based neurochemical environment that can inform subsequent behaviors as targets for reversal. The value of such a model would be to correlate the established biomarker of HPA dysregulation with multiple emerging biomarkers using modern neuroscience tools. For example, recent advances in small animal imaging using magnetic resonance imaging (MRI) confers the ability to directly measure structural and functional changes in the rodent brain, thereby providing a bridge to translate findings from animals to humans. Although structural MRI has been used to report robust subcortical brain alterations in MDD (216), a recent approach (217) exploiting differences in the resting state connectivity between brain regions among MDD patients to define novel subtypes of depression has excited clinical and preclinical researchers alike. We therefore envisage that structural and functional MRI-based changes, together with HPA tissue biomarkers, will correlate with clinical subtypes of MDD and guide new treatment approaches.
Recent advances in identifying genetic risks (112), together with access to brain-wide transcriptome data from post-mortem clinical samples (218), will also advance our understanding of brain region-specific roles for select genes and molecular networks in MDD. Defining the roles of these genes, as well as selective manipulation of the underlying brain circuits, using tools such as optogenetics, in HPA informed animal models will likely drive further insight into MDD.
Thus, a research pathway that encompasses multi-modal investigations, including molecular, cellular, neurocircuitry, and behavioral studies in preclinical models, and which is informed and refined by clinical findings will accelerate understanding of MDD and provide multiple strategies to effectively diagnose and treat this heterogeneous disorder. It is vital that we persevere and strengthen such interactions between preclinical and clinical research to drive multiple iterations of this pathway.
Author Contributions
LSN and DJ conceptualized the review. LSN, MB, MZ, and DJ conducted critical evaluation of literature and contributed to the draft manuscript. LSN and DJ wrote the paper.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Acknowledgments
This work was supported by Mater Foundation Fellowship to DJ. We thank Rowan Tweedale for editorial assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2019.00974/full#supplementary-material
References
1. Kessler RC, Bromet EJ. The epidemiology of depression across cultures. Annu Rev Public Health (2013) 34:119–38. doi: 10.1146/annurev-publhealth-031912-114409
2. Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry (1965) 122:509–22. doi: 10.1176/ajp.122.5.509
3. Czéh B, Fuchs E, Wiborg O, Simon M. Animal models of major depression and their clinical implications. Prog Neuropsychopharmacol Biol Psychiatry (2016) 64:293–310. doi: 10.1016/J.PNPBP.2015.04.004
4. Rosenblat JD, Cha DS, Mansur RB, McIntyre RS. Inflamed moods: a review of the interactions between inflammation and mood disorders. Prog Neuropsychopharmacol Biol Psychiatry (2014) 53:23–34. doi: 10.1016/J.PNPBP.2014.01.013
5. Hyman SE. Revitalizing psychiatric therapeutics. Neuropsychopharmacology (2014) 39:220–9. doi: 10.1038/npp.2013.181
6. Board F, Wadeson R, Persky H. Depressive affect and endocrine functions: blood levels of adrenal cortex and thyroid hormones in patients suffering from depressive reactions. Arch Neurol Psychiatry (1957) 78:612–20. doi: 10.1001/archneurpsyc.1957.02330420072015
7. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology (2000) 23:477–501. doi: 10.1016/S0893-133X(00)00159-7
8. Horstmann S, Dose T, Lucae S, Kloiber S, Menke A, Hennings J, et al. Suppressive effect of mirtazapine on the HPA system in acutely depressed women seems to be transient and not related to antidepressant action. Psychoneuroendocrinology (2009) 34:238–48. doi: 10.1016/j.psyneuen.2008.09.004
9. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry (2003) 160:1554–65. doi: 10.1176/appi.ajp.160.9.1554
10. Young EA, Lopez JF, Murphy-Weinberg V, Watson SJ, Akil H. Mineralocorticoid receptor function in major depression. Arch Gen Psychiatry (2003) 60:24–8. doi: 10.1001/archpsyc.60.1.24
11. Carroll BJ, Curtis GC, Mendels J. Neuroendocrine regulation in depression. II. Discrimination of depressed from nondepressed patients. Arch Gen Psychiatry (1976) 33:1051–8. doi: 10.1001/archpsyc.1976.01770090041003
12. Ising M, Künzel HE, Binder EB, Nickel T, Modell S, Holsboer F. The combined dexamethasone/CRH test as a potential surrogate marker in depression. Prog Neuropsychopharmacol Biol Psychiatry (2005) 29:1085–93. doi: 10.1016/j.pnpbp.2005.03.014
13. Dam H, Mellerup ET, Rafaelsen OJ. The dexamethasone suppression test in depression. J Affect Disord (1985) 8:95–103.
14. Watson S, Gallagher P, Del-Estal D, Hearn A, Ferrier IN, Young AH. Hypothalamic-pituitary-adrenal axis function in patients with chronic depression. Psychol Med (2002) 32:1021–8. doi: 10.1017/S0033291702005998
15. Holsen LM, Lancaster K, Klibanski A, Whitfield-Gabrieli S, Cherkerzian S, Buka S, et al. HPA-axis hormone modulation of stress response circuitry activity in women with remitted major depression. Neuroscience (2013) 250:732–42. doi: 10.1016/j.neuroscience.2013.07.042
16. Zobel AW, Nickel T, Sonntag A, Uhr M, Holsboer F, Ising M. Cortisol response in the combined dexamethasone/CRH test as predictor of relapse in patients with remitted depression. a prospective study. J Psychiatr Res (2001) 35:83–94. doi: 10.1016/s0022-3956(01)00013-9
17. Posener JA, DeBattista C, Williams GH, Kraemer HC, Kalehzan BM, Schatzberg AF. 24-Hour monitoring of cortisol and corticotropin secretion in psychotic and nonpsychotic major depression. Arch Gen Psychiatry (2000) 57:755–60. doi: 10.1001/archpsyc.57.8.755
18. Schatzberg AF, Keller J, Tennakoon L, Lembke A, Williams G, Kraemer FB, et al. HPA axis genetic variation, cortisol and psychosis in major depression. Mol Psychiatry (2014) 19:220–7. doi: 10.1038/mp.2013.129
19. Sachar EJ, Hellman L, Roffwarg HP, Halpern FS, Fukush DK, Gallagher TF. Disrupted 24 hour patterns of cortisol secretion in psychotic depressives. Arch Gen Psychiatry (1973) 28:19–24. doi: 10.1001/archpsyc.1973.01750310011002
20. Keller J, Flores B, Gomez RG, Solvason HB, Kenna H, Williams GH, et al. Cortisol circadian rhythm alterations in psychotic major depression. Biol Psychiatry (2006) 60:275–81. doi: 10.1016/j.biopsych.2005.10.014
21. Karlović D, Serretti A, Jevtović S, Vrkić N, Serić V, Peleš AM. Diagnostic accuracy of serum brain derived neurotrophic factor concentration in antidepressant naïve patients with first major depression episode. J Psychiatr Res (2013) 47:162–7. doi: 10.1016/j.jpsychires.2012.09.017
22. Lamers F, Vogelzangs N, Merikangas KR, De Jonge P, Beekman ATF, Penninx BWJH. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression. Mol Psychiatry (2013) 18:692–9. doi: 10.1038/mp.2012.144
23. Holsboer F, Ising M. Central CRH system in depression and anxiety — Evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol (2008) 583:350–7. doi: 10.1016/j.ejphar.2007.12.032
24. Keller J, Gomez R, Williams G, Lembke A, Lazzeroni L, Murphy GM, et al. HPA axis in major depression: cortisol, clinical symptomatology and genetic variation predict cognition. Mol Psychiatry (2017) 22:527–36. doi: 10.1038/mp.2016.120
25. Rush AJ, Giles DE, Schlesser MA, Orsulak PJ, Weissenburger JE, Fulton CL, et al. Dexamethasone response, thyrotropin-releasing hormone stimulation, rapid eye movement latency, and subtypes of depression. Biol Psychiatry (1997) 41:915–28. doi: 10.1016/S0006-3223(97)00148-0
26. Angst J, Gamma A, Benazzi F, Ajdacic V, Rössler W. Melancholia and atypical depression in the Zurich study: epidemiology, clinical characteristics, course, comorbidity and personality. Acta Psychiatr Scand (2007) 115:72–84. doi: 10.1210/edrv-17-2-187
27. Stetler C, Miller GE. Depression and hypothalamic-pituitary-adrenal activation: a quantitative summary of four decades of research. Psychosom Med (2011) 73:114–26. doi: 10.1097/PSY.0b013e31820ad12b
28. Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry (2001) 49:391–404. doi: 10.1016/S0006-3223(00)01088-X
29. Holsboer F, Barden N. Antidepressants and hypothalamic-pituitary adrenocortical regulation. Endocr Rev (1996) 17:187–205. doi: 10.1210/edrv-17-2-187
30. de Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease 1. Endocr Rev (1998) 19:269–301. doi: 10.1210/edrv.19.3.0331
31. Leonard BE. The HPA and immune axes in stress: the involvement of the serotonergic system. Eur Psychiatry (2005) 20(Suppl 3):S302–6. doi: 10.1016/s0924-9338(05)80180-4
32. Mazurka R, Wynne-Edwards KE, Harkness KL. Stressful life events prior to depression onset and the cortisol response to stress in youth with first onset versus recurrent depression. J Abnorm Child Psychol (2016) 44:1173–84. doi: 10.1007/s10802-015-0103-y
33. Schatzberg AF, Rothschild AJ, Langlais PJ, Bird ED, Cole JO. A corticosteroid/dopamine hypothesis for psychotic depression and related states. J Psychiatr Res (1985) 19:57–64. doi: 10.1016/0022-3956(85)90068-8
34. Block TS, Kushner H, Kalin N, Nelson C, Belanoff J, Schatzberg A. Combined analysis of mifepristone for psychotic depression: plasma levels associated with clinical response. Biol Psychiatry (2018) 84:46–54. doi: 10.1016/j.biopsych.2018.01.008
35. Belanoff JK, Flores BH, Kalezhan M, Sund B, Schatzberg AF. Rapid reversal of psychotic depression using mifepristone. J Clin Psychopharmacol (2001) 21:516–21. doi: 10.1097/00004714-200110000-00009
36. Bijanki KR, Hodis B, Brumm MC, Harlynn EL, McCormick LM. Hippocampal and left subcallosal anterior cingulate atrophy in psychotic depression. PloS One (2014) 9:e110770. doi: 10.1371/journal.pone.0110770
37. Hickie I, Naismith S, Ward PB, Turner K, Scott E, Mitchell P, et al. Reduced hippocampal volumes and memory loss in patients with early- and late-onset depression. Br J Psychiatry (2005) 186:197–202. doi: 10.1192/bjp.186.3.197
38. Simpson S, Baldwin RC, Jackson A, Burns A. The differentiation of DSM-III-R psychotic depression in later life from nonpsychotic depression: comparisons of brain changes measured by multispectral analysis of magnetic resonance brain images, neuropsychological findings, and clinical features. Biol Psychiatry (1999) 45:193–204. doi: 10.1016/S0006-3223(98)00006-7
39. Pujol J, Cardoner N, Benlloch L, Urretavizcaya M, Deus J, Losilla JM, et al. CSF spaces of the sylvian fissure region in severe melancholic depression. Neuroimage (2002) 15:103–6. doi: 10.1006/nimg.2001.0928
40. Fountoulakis KN, Iacovides A, Gerasimou G, Fotiou F, Ioannidou C, Bascialla F, et al. The relationship of regional cerebral blood flow with subtypes of major depression. Prog Neuropsychopharmacol Biol Psychiatry (2004) 28:537–46. doi: 10.1016/j.pnpbp.2004.01.006
41. Hunter AL, Minnis H, Wilson P. Altered stress responses in children exposed to early adversity: a systematic review of salivary cortisol studies. Stress (2011) 14:614–26. doi: 10.3109/10253890.2011.577848
42. Heim C, Binder EB. Current research trends in early life stress and depression: review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp Neurol (2012) 233:102–11. doi: 10.1016/j.expneurol.2011.10.032
43. Juruena MF. Early-life stress and HPA axis trigger recurrent adulthood depression. Epilepsy Behav (2014) 38:148–59. doi: 10.1016/j.yebeh.2013.10.020
44. Heim C, Newport DJ, Bonsall R, Miller AH, Nemeroff CB. Altered pituitary-adrenal axis responses to provocative challenge tests in adult survivors of childhood abuse. Am J Psychiatry (2001) 158:575–81. doi: 10.1176/appi.ajp.158.4.575
45. Agorastos A, Pervanidou P, Chrousos GP, Baker DG. Developmental trajectories of early life stress and trauma: a narrative review on neurobiological aspects beyond stress system dysregulation. Front Psychiatry (2019) 10:118. doi: 10.3389/fpsyt.2019.00118
46. Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA (2000) 284:592–7. doi: 10.1001/jama.284.5.592
47. Heim C, Mletzko T, Purselle D, Musselman DL, Nemeroff CB. The dexamethasone/corticotropin-releasing factor test in men with major depression: role of childhood trauma. Biol Psychiatry (2008) 63:398–405. doi: 10.1016/j.biopsych.2007.07.002
48. Tyrka AR, Wier L, Price LH, Ross N, Anderson GM, Wilkinson CW, et al. Childhood parental loss and adult hypothalamic-pituitary-adrenal function. Biol Psychiatry (2008) 63:1147–54. doi: 10.1016/j.biopsych.2008.01.011
49. Pesonen A-K, Räikkönen K, Feldt K, Heinonen K, Osmond C, Phillips DIW, et al. Childhood separation experience predicts HPA axis hormonal responses in late adulthood: a natural experiment of World War II. Psychoneuroendocrinology (2010) 35:758–67. doi: 10.1016/j.psyneuen.2009.10.017
50. De Bellis MD, Chrousos GP, Dorn LD, Burke L, Helmers K, Kling MA, et al. Hypothalamic-pituitary-adrenal axis dysregulation in sexually abused girls. J Clin Endocrinol Metab (1994) 78:249–55. doi: 10.1210/jcem.78.2.8106608
51. Carpenter LL, Carvalho JP, Tyrka AR, Wier LM, Mello AF, Mello MF, et al. Decreased adrenocorticotropic hormone and cortisol responses to stress in healthy adults reporting significant childhood maltreatment. Biol Psychiatry (2007) 62:1080–7. doi: 10.1016/j.biopsych.2007.05.002
52. Carpenter LL, Shattuck TT, Tyrka AR, Geracioti TD, Price LH. Effect of childhood physical abuse on cortisol stress response. Psychopharmacol (Berl) (2011) 214:367–75. doi: 10.1007/s00213-010-2007-4
53. Voellmin A, Winzeler K, Hug E, Wilhelm FH, Schaefer V, Gaab J, et al. Blunted endocrine and cardiovascular reactivity in young healthy women reporting a history of childhood adversity. Psychoneuroendocrinology (2015) 51:58–67. doi: 10.1016/j.psyneuen.2014.09.008
54. Bosch NM, Riese H, Reijneveld SA, Bakker MP, Verhulst FC, Ormel J, et al. Timing matters: long term effects of adversities from prenatal period up to adolescence on adolescents' cortisol stress response. The TRAILS study. Psychoneuroendocrinology (2012) 37:1439–47. doi: 10.1016/j.psyneuen.2012.01.013
55. McEwen BS. Invited review: Estrogens effects on the brain: multiple sites and molecular mechanisms. J Appl Physiol (2001) 91:2785–801. doi: 10.1152/jappl.2001.91.6.2785
56. Vythilingam M, Heim C, Newport J, Miller AH, Anderson E, Bronen R, et al. Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry (2002) 159:2072–80. doi: 10.1176/appi.ajp.159.12.2072
57. Vaiserman AM. Epigenetic programming by early-life stress: evidence from human populations. Dev Dyn (2015) 244:254–65. doi: 10.1002/dvdy.24211
58. Mazure CM. Life stressors as risk factors in depression. Clin Psychol Sci Pract (1998) 5:291–313. doi: 10.1111/j.1468-2850.1998.tb00151.x
59. Brown GW, Harris T. Social origins of depression: A study of psychiatric disorder in women. Routledge (1978).
60. Young E, Korszun A. Sex, trauma, stress hormones and depression. Mol Psychiatry (2010) 15:23–8. doi: 10.1038/mp.2009.94
61. Kirschbaum C, Kudielka BM, Gaab J, Schommer NC, Hellhammer DH. Impact of gender, menstrual cycle phase, and oral contraceptives on the activity of the hypothalamus-pituitary-adrenal axis. Psychosom Med (1999) 61:154–62. doi: 10.1097/00006842-199903000-00006
62. Künzel HE, Binder EB, Nickel T, Ising M, Fuchs B, Majer M, et al. Pharmacological and nonpharmacological factors influencing hypothalamic–pituitary–adrenocortical axis reactivity in acutely depressed psychiatric in-patients, Measured by the Dex-CRH Test. Neuropsychopharmacology (2003) 28:2169–78. doi: 10.1038/sj.npp.1300280
63. Seeman TE, Singer B, Wilkinson CW. Bruce McEwen. Gender differences in age-related changes in HPA axis reactivity. Psychoneuroendocrinology (2001) 26:225–40. doi: 10.1016/S0306-4530(00)00043-3
64. Kudielka BM, Kirschbaum C. Sex differences in HPA axis responses to stress: a review. Biol Psychol (2005) 69:113–32. doi: 10.1016/j.biopsycho.2004.11.009
65. Young EA, Haskett RF, Murphy Weinberg V, Watson SJ, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry (1991) 48:693–9. doi: 10.1001/archpsyc.1991.01810320017003
66. Young EA, Kotun J, Haskett RF, Grunhaus L, Greden JF, Watson SJ, et al. Dissociation between pituitary and adrenal suppression to dexamethasone in depression. Arch Gen Psychiatry (1993) 50:395–403. doi: 10.1001/archpsyc.1993.01820170073010
67. Seifritz E, Baumann P, Müller MJ, Annen O, Amey M, Hemmeter U, et al. Neuroendocrine effects of a 20-mg citalopram infusion in healthy males a placebo-controlled evaluation of citalopram as 5-HT function probe. Neuropsychopharmacology (1996) 14:253–63. doi: 10.1016/0893-133X(95)00117-V
68. Skene D, Bojkowski C, Arendt J. Comparison of the effects of acute fluvoxamine and desipramine administration on melatonin and cortisol production in humans. Br J Clin Pharmacol (1994) 37:181–6. doi: 10.1111/j.1365-2125.1994.tb04258.x
69. von Bardeleben U, Steiger A, Gerken A, Holsboer F. Effects of fluoxetine upon pharmacoendocrine and sleep-EEG parameters in normal controls. Int Clin Psychopharmacol (1989) 4(Suppl 1):1–5.
70. Schüle C, Baghai T, Laakmann G. Mirtazapine decreases stimulatory effects of reboxetine on cortisol, adrenocorticotropin and prolactin secretion in healthy male subjects. Neuroendocrinology (2004) 79:54–62. doi: 10.1159/000076046
71. Daffner-Bugía C, Laakmann G, Voderholzer U, Haag C, Baghai T, Kolmsee S, et al. The neuroendocrine effects of venlafaxine in healthy subjects. Hum Psychopharmacol (1996) 11:1–9. doi: 10.1002/(SICI)1099-1077(199601)11:1<1::AID-HUP732>3.3.CO;2-Q
72. Laakmann G, Wittmann M, Gugath M, Mueller OA, Treuschl J, Wahlster U, et al. Effects of psychotropic drugs (desimipramine, chlorimipramine, sulpiride and diazepam) on the human HPA axis. Psychopharmacol (Berl) (1984) 84:66–70. doi: 10.1007/BF00432027
73. Schüle C, Baghai T, Goy J, Bidlingmaier M, Strasburger C, Laakmann G. The influence of mirtazapine on anterior pituitary hormone secretion in healthy male subjects. Psychopharmacol (Berl) (2002) 163:95–101. doi: 10.1007/s00213-002-1148-5
74. de Boer T. The pharmacologic profile of mirtazapine. J Clin Psychiatry (1996) 57(Suppl 4):19–25.
75. Reul JM, Stec I, Söder M, Holsboer F. Chronic treatment of rats with the antidepressant amitriptyline attenuates the activity of the hypothalamic-pituitary-adrenocortical system. Endocrinology (1993) 133:312–20. doi: 10.1210/endo.133.1.8391426
76. Vythilingam M, Vermetten E, Anderson GM, Luckenbaugh D, Anderson ER, Snow J, et al. Hippocampal volume, memory, and cortisol status in major depressive disorder: Effects of treatment. Biol Psychiatry (2004) 56:101–12. doi: 10.1016/j.biopsych.2004.04.002
77. Deuschle M, Hamann B, Meichel C, Krumm B, Lederbogen F, Kniest A, et al. Antidepressive treatment with amitriptyline and paroxetine: effects on saliva cortisol concentrations. J Clin Psychopharmacol (2003) 23:201–5. doi: 10.1097/00004714-200304000-00014
78. Schüle C, Baghai T, Zwanzger P, Ella R, Eser D, Padberg F, et al. Attenuation of hypothalamic-pituitary-adrenocortical hyperactivity in depressed patients by mirtazapine. Psychopharmacol (Berl) (2003) 166:271–5. doi: 10.1007/s00213-002-1356-z
79. Cohrs S, Röher C, Jordan W, Meier A, Huether G, Wuttke W, et al. The atypical antipsychotics olanzapine and quetiapine, but not haloperidol, reduce ACTH and cortisol secretion in healthy subjects. Psychopharmacol (Berl) (2006) 185:11–8. doi: 10.1007/s00213-005-0279-x
80. Meier A, Neumann AC, Jordan W, Huether G, Rodenbeck A, Rüther E, et al. Ziprasidone decreases cortisol excretion in healthy subjects. Br J Clin Pharmacol (2005) 60:330–6. doi: 10.1111/j.1365-2125.2005.02431.x
81. Sarubin N, Nothdurfter C, Schmotz C, Wimmer AM, Trummer J, Lieb M, et al. Impact on cortisol and antidepressant efficacy of quetiapine and escitalopram in depression. Psychoneuroendocrinology (2014) 39:141–51. doi: 10.1016/j.psyneuen.2013.10.008
82. Bschor T, Adli M, Baethge C, Eichmann U, Ising M, Uhr M, et al. Lithium augmentation increases the ACTH and cortisol response in the combined DEX/CRH test in unipolar major depression. Neuropsychopharmacology (2002) 27:470–8. doi: 10.1016/S0893-133X(02)00323-8
83. Bschor T, Ritter D, Winkelmann P, Erbe S, Uhr M, Ising M, et al. Lithium monotherapy increases ACTH and cortisol response in the DEX/CRH test in unipolar depressed subjects. A study with 30 treatment-naive patients. PloS One (2011) 6:e27613. doi: 10.1371/journal.pone.0027613
84. Belanoff JK, Rothschild AJ, Cassidy F, DeBattista C, Baulieu E-E, Schold C, et al. An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatry (2002) 52:386–92. doi: 10.1016/s0006-3223(02)01432-4
85. Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology (2006) 31:628–36. doi: 10.1038/sj.npp.1300884
86. DeBattista C, Belanoff J, Glass S, Khan A, Horne RL, Blasey C, et al. Mifepristone versus placebo in the treatment of psychosis in patients with psychotic major depression. Biol Psychiatry (2006) 60:1343–9. doi: 10.1016/j.biopsych.2006.05.034
87. Brogden RN, Goa KL, Faulds D. Mifepristone: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs (1993) 45:384–409. doi: 10.2165/00003495-199345030-00007
88. Rizvi SJ, Kennedy SH. Emerging drugs for major depressive disorder: An update. Expert Opin Emerg Drugs (2012) 17:285–94. doi: 10.1517/14728214.2012.681301
89. Jeffcoate WJ, Silverstone JT, Edwards CRW, Besser GM. Psychiatric manifestations of cushing's syndrome: response to lowering of plasma cortisol. QJM (1979) 48:465–72. doi: 10.1093/oxfordjournals.qjmed.a067587
90. O'Dwyer AM, Lightman SL, Marks MN, Checkley SA. Treatment of major depression with metyrapone and hydrocortisone. J Affect Disord (1995) 33:123–8. doi: 10.1016/0165-0327(94)00082-k
91. Jahn H, Schick M, Kiefer F, Kellner M, Yassouridis A, Wiedemann K. Metyrapone as additive treatment in major depression: a double-blind and placebo-controlled trial. Arch Gen Psychiatry (2004) 61:1235–44. doi: 10.1001/archpsyc.61.12.1235
92. Raven PW, O'Dwyer AM, Taylor NF, Checkley SA. The relationship between the effects of metyrapone treatment on depressed mood and urinary steroid profiles. Psychoneuroendocrinology (1996) 21:277–86. doi: 10.1016/0306-4530(95)00057-7
93. Wolkowitz OM, Reus VI, Chan T, Manfredi F, Raum W, Johnson R, et al. Antiglucocorticoid treatment of depression: double-blind ketoconazole. Biol Psychiatry (1999) 45:1070–4. doi: 10.1016/S0006-3223(98)00267-4
94. Malison RT, Anand A, Pelton GH, Kirwin P, Carpenter L, McDougle CJ, et al. Limited efficacy of ketoconazole in treatment-refractory major depression. J Clin Psychopharmacol (1999) 19:466–70. doi: 10.1097/00004714-199910000-00011
95. Menke A. Is the HPA axis as target for depression outdated, or is there a new hope? Front Psychiatry (2019) 10:101. doi: 10.3389/fpsyt.2019.00101
96. Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med (2007) 357:1939–45. doi: 10.1056/NEJMct075234
97. Nemeroff CB, Bissette G, Akil H, Fink M. Neuropeptide concentrations in the cerebrospinal fluid of depressed patients treated with electroconvulsive therapy: corticotropin-releasing factor, b-endorphin and somatostatin. Br J Psychiat (1991) 158:59–63. doi: 10.1192/bjp.158.1.59
98. Apéria B, Bergman H, Engelbrektson K, Thorén M, Wetterberg L. Effects of electroconvulsive therapy on neuropsychological function and circulating levels of ACTH, cortisol, prolactin, and TSH in patients with major depressive illness. Acta Psychiatr Scand (1985) 72:536–41. doi: 10.1111/j.1600-0447.1985.tb02651.x
99. Burgese DF, Bassitt DP. Variation of plasma cortisol levels in patients with depression after treatment with bilateral electroconvulsive therapy. Trends Psychiatry Psychother (2015) 37:27–36. doi: 10.1590/2237-6089-2014-0031
100. Linkowski P, Mendlewicz J, Kerkhofs M, Leclercq R, Golstein J, Brasseur M, et al. Cauter E Van. 24-Hour profiles of adrenocorticotropin, cortisol, and growth hormone in major depressive illness: effect of antidepressant treatment. J Clin Endocrinol Metab (1987) 65:141–52. doi: 10.1210/jcem-65-1-141
101. Burke HM, Davis MC, Otte C, Mohr DC. Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendocrinology (2005) 30:846–56. doi: 10.1016/j.psyneuen.2005.02.010
102. Poole L, Kidd T, Ronaldson A, Leigh E, Jahangiri M, Steptoe A. Depression 12-months after coronary artery bypass graft is predicted by cortisol slope over the day. Psychoneuroendocrinology (2016) 71:155–8. doi: 10.1016/J.PSYNEUEN.2016.05.025
103. Vining RF, McGinley RA, Maksvytis J, Ho K. Salivary cortisol: a better measure of adrenal cortical function than serum cortisol. Ann Clin Biochem (1983) 20:329–35. doi: 10.1177/000456328302000601
104. Meeran K, Hattersley A, Mould G, Bloom SR. Venepuncture causes rapid rise in plasma ACTH. Br J Clin Pract (1993) 47:246–7.
105. Hellhammer DH, Wüst S, Kudielka BM. Salivary cortisol as a biomarker in stress research. Psychoneuroendocrinology (2009) 34:163–71. doi: 10.1016/j.psyneuen.2008.10.026
106. Levine A, Zagoory-Sharon O, Feldman R, Lewis JG, Weller A. Measuring cortisol in human psychobiological studies. Physiol Behav (2007) 90:43–53. doi: 10.1016/j.physbeh.2006.08.025
107. Meulenberg PMM, Ross HA, Swinkels LMJW, Benraad TJ. The effect of oral contraceptives on plasma-free and salivary cortisol and cortisone. Clin Chim Acta (1987) 165:379–85. doi: 10.1016/0009-8981(87)90183-5
108. Knorr U, Vinberg M, Kessing LV, Wetterslev J. Salivary cortisol in depressed patients versus control persons: a systematic review and meta-analysis. Psychoneuroendocrinology (2010) 35:1275–86. doi: 10.1016/j.psyneuen.2010.04.001
109. Menke A, Arloth J, Best J, Namendorf C, Gerlach T, Czamara D, et al. Time-dependent effects of dexamethasone plasma concentrations on glucocorticoid receptor challenge tests. Psychoneuroendocrinology (2016) 69:161–71. doi: 10.1016/J.PSYNEUEN.2016.04.003
110. El-Farhan N, Rees DA, Evans C. Measuring cortisol in serum, urine and saliva – are our assays good enough? Ann Clin Biochem (2017) 54:308–22. doi: 10.1177/0004563216687335
111. Juruena MF, Cleare AJ, Papadopoulos AS, Poon L, Lightman S, Pariante CM. Different responses to dexamethasone and prednisolone in the same depressed patients. Psychopharmacol (Berl) (2006) 189:225–35. doi: 10.1007/s00213-006-0555-4
112. Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet (2018) 50:668–81. doi: 10.1038/s41588-018-0090-3
113. Neumann ID, Wegener G, Homberg JR, Cohen H, Slattery DA, Zohar J, et al. Animal models of depression and anxiety: What do they tell us about human condition? Prog Neuropsychopharmacol Biol Psychiatry (2011) 35:1357–75. doi: 10.1016/j.pnpbp.2010.11.028
114. Powell TR, Fernandes C, Schalkwyk LC. Depression-related behavioral tests. Curr Protoc Mouse Biol (2012) 2:119–27. doi: 10.1002/9780470942390.mo110176
115. Kenny DA. The design and analysis of social-interaction research. Annu Rev Psychol (1996) 47:59–86. doi: 10.1146/annurev.psych.47.1.59
116. Cummings CM, Caporino NE, Kendall PC. Comorbidity of anxiety and depression in children and adolescents: 20 years after. Psychol Bull (2014) 140:816–45. doi: 10.1037/a0034733
117. Ramaker MJ, Dulawa SC. Identifying fast-onset antidepressants using rodent models. Mol Psychiatry (2017) 22:656–65. doi: 10.1038/mp.2017.36
118. Chiba S, Numakawa T, Ninomiya M, Richards MC, Wakabayashi C, Kunugi H. Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry (2012) 39:112–9. doi: 10.1016/j.pnpbp.2012.05.018
119. Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci (2006) 8:383–95. doi: 10.1038/nrendo.2011.222
120. El Yacoubi M, Vaugeois J-M. Genetic rodent models of depression. Curr Opin Pharmacol (2007) 7:3–7. doi: 10.1016/J.COPH.2006.11.002
121. Overstreet DH, Pucilowski O, Rezvani AH, Janowsky DS. Administration of antidepressants, diazepam and psychomotor stimulants further confirms the utility of flinders sensitive line rats as an animal model of depression. Psychopharmacol (Berl) (1995) 121:27–37. doi: 10.1007/BF02245589
122. Pucilowski O, Overstreet DH, Rezvani AH, Janowsky DS. Chronic mild stress-induced anhedonia: greater effect in a genetic rat model of depression. Physiol Behav (1993) 54:1215–20. doi: 10.1016/0031-9384(93)90351-F
123. Malkesman O, Maayan R, Weizman A, Weller A. Aggressive behavior and HPA axis hormones after social isolation in adult rats of two different genetic animal models for depression. Behav Brain Res (2006) 175:408–14. doi: 10.1016/J.BBR.2006.09.017
124. Thiele S, Spehl TS, Frings L, Braun F, Ferch M, Rezvani AH, et al. SsLong-term characterization of the flinders sensitive line rodent model of human depression: Behavioral and PET evidence of a dysfunctional entorhinal cortex. Behav Brain Res (2016) 300:11–24. doi: 10.1016/j.bbr.2015.11.026
125. Overstreet DH, Friedman E, Mathé AA, Yadid G. The flinders sensitive line rat: a selectively bred putative animal model of depression. Neurosci Biobehav Rev (2005) 29:739–59. doi: 10.1016/J.NEUBIOREV.2005.03.015
126. Nam H, Clinton SM, Jackson NL, Kerman IA. Learned helplessness and social avoidance in the Wistar-Kyoto rat. Front Behav Neurosci (2014) 8:109. doi: 10.3389/fnbeh.2014.00109
127. Solberg LC, Olson SL, Turek FW, Redei E. Altered hormone levels and circadian rhythm of activity in the WKY rat, a putative animal model of depression. Am J Physiol Integr Comp Physiol (2001) 281:R786–94. doi: 10.1152/ajpregu.2001.281.3.R786
128. Rittenhouse PA, López-Rubalcava C, Stanwood GD, Lucki I. Amplified behavioral and endocrine responses to forced swim stress in the Wistar–Kyoto rat. Psychoneuroendocrinology (2002) 27:303–18. doi: 10.1016/S0306-4530(01)00052-X
129. De La Garza R, Mahoney JJ. A distinct neurochemical profile in WKY rats at baseline and in response to acute stress: implications for animal models of anxiety and depression. Brain Res (2004) 1021:209–18. doi: 10.1016/J.BRAINRES.2004.06.052
130. Henn FA, Vollmayr B. Stress models of depression: forming genetically vulnerable strains. Neurosci Biobehav Rev (2005) 29:799–804. doi: 10.1016/J.NEUBIOREV.2005.03.019
131. Edwards E, King J, Fray JC. Increased basal activity of the HPA axis and renin-angiotensin system in congenital learned helpless rats exposed to stress early in development. Int J Dev Neurosci (1999) 17:805–12. doi: 10.1016/S0736-5748(99)00062-3
132. King JA, Edwards E. Early stress and genetic influences on hypothalamic–pituitary–adrenal axis functioning in adulthood. Horm Behav (1999) 36:79–85. doi: 10.1006/HBEH.1999.1525
133. Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology (2005) 52:90–110. doi: 10.1159/000087097
134. Toth E, Gersner R, Wilf-Yarkoni A, Raizel H, Dar DE, Richter-Levin G, et al. Age-dependent effects of chronic stress on brain plasticity and depressive behavior. J Neurochem (2008) 107:522–32. doi: 10.1111/j.1471-4159.2008.05642.x
135. Ayuob NN, El Wahab MGA, Ali SS, Abdel-Tawab HS. Ocimum basilicum improve chronic stress-induced neurodegenerative changes in mice hippocampus. Metab Brain Dis (2018), 33:795–804. doi: 10.1007/s11011-017-0173-3
136. Bielajew C, Konkle ATM, Merali Z. The effects of chronic mild stress on male Sprague-Dawley and Long Evans rats: I. Biochemical and physiological analyses. Behav Brain Res (2002) 136:583–92. doi: 10.1016/S0166-4328(02)00222-X
137. Garcia LSB, Comim CM, Valvassori SS, Réus GZ, Stertz L, Kapczinski F, et al. Ketamine treatment reverses behavioral and physiological alterations induced by chronic mild stress in rats. Prog Neuropsychopharmacol Biol Psychiatry (2009) 33:450–5. doi: 10.1016/j.pnpbp.2009.01.004
138. de Andrade JS, Céspedes IC, Abrão RO, dos Santos TB, Diniz L, Britto LRG, et al. Chronic unpredictable mild stress alters an anxiety-related defensive response, Fos immunoreactivity and hippocampal adult neurogenesis. Behav Brain Res (2013) 250:81–90. doi: 10.1016/j.bbr.2013.04.031
139. Kvarta MD, Bradbrook KE, Dantrassy HM, Bailey AM, Thompson SM. Corticosterone mediates the synaptic and behavioral effects of chronic stress at rat hippocampal temporoammonic synapses. J Neurophysiol (2015) 114:1713–24. doi: 10.1152/jn.00359.2015
140. Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry (2008) 13:717–28. doi: 10.1038/sj.mp.4002055
141. Weiss IC, Pryce CR, Jongen-Rêlo AL, Nanz-Bahr NI, Feldon J. Effect of social isolation on stress-related behavioural and neuroendocrine state in the rat. Behav Brain Res (2004) 152:279–95. doi: 10.1016/j.bbr.2003.10.015
142. Hollis F, Kabbaj M. Social defeat as an animal model for depression. ILAR J (2014) 55:221–32. doi: 10.1093/ilar/ilu002
143. Perelló M, Chacon F, Cardinali DP, Esquifino AI, Spinedi E. Effect of social isolation on 24-h pattern of stress hormones and leptin in rats. Life Sci (2006) 78:1857–62. doi: 10.1016/j.lfs.2005.08.029
144. Dronjak S, Gavrilović L, Filipović D, Radojčić MB. Immobilization and cold stress affect sympatho–adrenomedullary system and pituitary–adrenocortical axis of rats exposed to long-term isolation and crowding. Physiol Behav (2004) 81:409–15. doi: 10.1016/J.PHYSBEH.2004.01.011
145. Hermes GL. Social isolation and the inflammatory response: sex differences in the enduring effects of a prior stressor. AJP Regul Integr Comp Physiol (2005) 290:R273–82. doi: 10.1152/ajpregu.00368.2005
146. Pisu MG, Garau A, Boero G, Biggio F, Pibiri V, Dore R, et al. Sex differences in the outcome of juvenile social isolation on HPA axis function in rats. Neuroscience (2016) 320:172–82. doi: 10.1016/j.neuroscience.2016.02.009
147. Mouri A, Ukai M, Uchida M, Hasegawa S, Taniguchi M, Ito T, et al. Juvenile social defeat stress exposure persistently impairs social behaviors and neurogenesis. Neuropharmacology (2018) 133:23–37. doi: 10.1016/j.neuropharm.2018.01.016
148. Macedo GC, Morita GM, Domingues LP, Favoretto CA, Suchecki D, Quadros IMH. Consequences of continuous social defeat stress on anxiety- and depressive-like behaviors and ethanol reward in mice. Horm Behav (2018) 97:154–61. doi: 10.1016/j.yhbeh.2017.10.007
149. Lin YE, Chou ST, Lin SH, Lu KH, Panyod S, Lai YS, et al. Antidepressant-like effects of water extract of Gastrodia elata Blume on neurotrophic regulation in a chronic social defeat stress model. J Ethnopharmacol (2018) 215:132–9. doi: 10.1016/j.jep.2017.12.044
150. Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell (2007) 131:391–404. doi: 10.1016/j.cell.2007.09.018
151. Schmidt MV, Wang XD, Meijer OC. Early life stress paradigms in rodents: Potential animal models of depression? Psychopharmacol (Berl) (2011) 214:131–40. doi: 10.1007/s00213-010-2096-0
152. van Bodegom M, Homberg JR, Henckens MJAG. Modulation of the hypothalamic-pituitary-adrenal axis by early life stress exposure. Front Cell Neurosci (2017) 11:87. doi: 10.3389/fncel.2017.00087
153. Vargas J, Junco M, Gomez C, Lajud N. Early life stress increases metabolic risk, HPA axis reactivity, and depressive-like behavior when combined with postweaning social isolation in rats. PloS One (2016) 11:e0162665. doi: 10.1371/journal.pone.0162665
154. Biggio F, Talani G, Locci V, Pisu MG, Boero G, Ciarlo B, et al. Low doses of prenatal ethanol exposure and maternal separation alter HPA axis function and ethanol consumption in adult male rats. Neuropharmacology (2018) 131:271–81. doi: 10.1016/j.neuropharm.2017.12.005
155. Fuentes IM, Pierce AN, Di Silvestro ER, Maloney MO, Christianson JA. Differential influence of early life and adult stress on urogenital sensitivity and function in male mice. Front Syst Neurosci (2018) 11:97. doi: 10.3389/fnsys.2017.00097
156. Odeon MM, Yamauchi L, Grosman M, Acosta GB. Long-term effects of repeated maternal separation and ethanol intake on HPA axis responsiveness in adult rats. Brain Res (2017) 1657:193–201. doi: 10.1016/j.brainres.2016.11.034
157. De Palma G, Blennerhassett P, Lu J, Deng Y, Park AJ, Green W, et al. Microbiota and host determinants of behavioural phenotype in maternally separated mice. Nat Commun (2015) 6:7735. doi: 10.1038/ncomms8735
158. Koe AS, Salzberg MR, Morris MJ, O'Brien TJ, Jones NC. Early life maternal separation stress augmentation of limbic epileptogenesis: The role of corticosterone and HPA axis programming. Psychoneuroendocrinology (2014) 42:124–33. doi: 10.1016/j.psyneuen.2014.01.009
159. Feldcamp L, Doucet JS, Pawling J, Fadel MP, Fletcher PJ, Maunder R, et al. Mgat5 modulates the effect of early life stress on adult behavior and physical health in mice. Behav Brain Res (2016) 312:253–64. doi: 10.1016/j.bbr.2016.06.033
160. David DJ, Samuels BA, Rainer Q, Wang J-W, Marsteller D, Mendez I, et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron (2009) 62:479–93. doi: 10.1016/j.neuron.2009.04.017
161. Mendez-David I, Boursier C, Domergue V, Colle R, Falissard B, Corruble E, et al. Differential peripheral proteomic biosignature of fluoxetine response in a mouse model of anxiety/depression. Front Cell Neurosci (2017) 11:237. doi: 10.3389/fncel.2017.00237
162. Gourley SL, Kiraly DD, Howell JL, Olausson P, Taylor JR. Acute hippocampal brain-derived neurotrophic factor restores motivational and forced swim performance after corticosterone. Biol Psychiatry (2008) 64:884–90. doi: 10.1016/j.biopsych.2008.06.016
163. Li YC, Wang LL, Pei YY, Shen JD, Li HB, Wang BY, et al. Baicalin decreases SGK1 expression in the hippocampus and reverses depressive-like behaviors induced by corticosterone. Neuroscience (2015) 311:130–7. doi: 10.1016/j.neuroscience.2015.10.023
164. Pazini FL, Cunha MP, Rosa JM, Colla ARS, Lieberknecht V, Oliveira Á, et al. Creatine, similar to ketamine, counteracts depressive-like behavior induced by corticosterone via PI3K/Akt/mTOR pathway. Mol Neurobiol (2016) 53:6818–34. doi: 10.1007/s12035-015-9580-9
165. Xie X, Shen Q, Ma L, Chen Y, Zhao B, Fu Z. Chronic corticosterone-induced depression mediates premature aging in rats. J Affect Disord (2018) 229:254–61. doi: 10.1016/j.jad.2017.12.073
166. Johnson SA, Fournier NM, Kalynchuk LE. Effect of different doses of corticosterone on depression-like behavior and HPA axis responses to a novel stressor. Behav Brain Res (2006) 168:280–8. doi: 10.1016/j.bbr.2005.11.019
167. Forbes NF, Stewart CA, Matthews K, Reid IC. Chronic mild stress and sucrose consumption: validity as a model of depression. Physiol Behav (1996) 60:1481–4. doi: 10.1016/S0031-9384(96)00305-8
168. Vollmayr B, Henn FA. Learned helplessness in the rat: improvements in validity and reliability. Brain Res Protoc (2001) 8:1–7. doi: 10.1016/S1385-299X(01)00067-8
169. Nestler EJ, Gould E, Manji H. Preclinical models: status of basic research in depression. Biol Psychiatry (2002) 52:503–28. doi: 10.1016/S0006-3223(02)01405-1
170. Miller BR, Hen R. The current state of the neurogenic theory of depression and anxiety. Curr Opin Neurobiol (2015) 30:51–8. doi: 10.1016/j.conb.2014.08.012
171. Demuyser T, Bentea E, Deneyer L, Albertini G, Massie A, Smolders I. Disruption of the HPA-axis through corticosterone-release pellets induces robust depressive-like behavior and reduced BDNF levels in mice. Neurosci Lett (2016) 626:119–25. doi: 10.1016/j.neulet.2016.05.026
172. Weng L, Guo X, Li Y, Yang X, Han Y. Apigenin reverses depression-like behavior induced by chronic corticosterone treatment in mice. Eur J Pharmacol (2016) 774:50–4. doi: 10.1016/J.EJPHAR.2016.01.015
173. Ali SH, Madhana RM, Athira KV, Kasala ER, Bodduluru LN, Pitta S, et al. Resveratrol ameliorates depressive-like behavior in repeated corticosterone-induced depression in mice. Steroids (2015) 101:37–42. doi: 10.1016/j.steroids.2015.05.010
174. Yan T, Xu M, Wan S, Wang M, Wu B, Xiao F, et al. Schisandra chinensis produces the antidepressant-like effects in repeated corticosterone-induced mice via the BDNF/TrkB/CREB signaling pathway. Psychiatry Res (2016) 243:135–42. doi: 10.1016/J.PSYCHRES.2016.06.037
175. Fukumoto K, Toki H, Iijima M, Hashihayata T, Yamaguchi J-I, Hashimoto K, et al. Antidepressant potential of (R)-Ketamine in rodent models: comparison with (S)-Ketamine. J Pharmacol Exp Ther (2017) 361:9–16. doi: 10.1124/jpet.116.239228
176. Ago Y, Arikawa S, Yata M, Yano K, Abe M, Takuma K, et al. Antidepressant-like effects of the glucocorticoid receptor antagonist RU-43044 are associated with changes in prefrontal dopamine in mouse models of depression. Neuropharmacology (2008) 55:1355–63. doi: 10.1016/J.NEUROPHARM.2008.08.026
177. Weber C-C, Eckert GP, Müller WE. Effects of antidepressants on the brain/plasma distribution of corticosterone. Neuropsychopharmacology (2006) 31:2443–8. doi: 10.1038/sj.npp.1301076
178. Li B, Zhao J, Lv J, Tang F, liu L, Sun Z, et al. Additive antidepressant-like effects of fasting with imipramine via modulation of 5-HT2 receptors in the mice. Prog Neuropsychopharmacol Biol Psychiatry (2014) 48:199–206. doi: 10.1016/J.PNPBP.2013.08.015
179. Pariante CM, Thomas SA, Lovestone S, Makoff A, Kerwin RW. Do antidepressants regulate how cortisol affects the brain? Psychoneuroendocrinology (2004) 29:423–47. doi: 10.1016/j.psyneuen.2003.10.009
180. Yau JL, Noble J, Hibberd C, Seckl JR. Short-term administration of fluoxetine and venlafaxine decreases corticosteroid receptor mRNA expression in the rat hippocampus. Neurosci Lett (2001) 306:161–4. doi: 10.1016/S0304-3940(01)01890-0
181. Connor TJ, Kelliher P, Shen Y, Harkin A, Kelly JP, Leonard BE. Effect of subchronic antidepressant treatments on behavioral, neurochemical, and endocrine changes in the forced-swim test. Pharmacol Biochem Behav (2000) 65:591–7. doi: 10.1016/S0091-3057(99)00192-6
182. Conti AC. Inducible cAMP early repressor regulates corticosterone suppression after tricyclic antidepressant treatment. J Neurosci (2004) 24:1967–75. doi: 10.1523/JNEUROSCI.4804-03.2004
183. Wu J, Du J, Xu C, Le J, Xu Y, Liu B, et al. Icariin attenuates social defeat-induced down-regulation of glucocorticoid receptor in mice. Pharmacol Biochem Behav (2011) 98:273–8. doi: 10.1016/j.pbb.2011.01.008
184. Cai L, Li R, Tang W, Meng G, Hu X, Wu T. Antidepressant-like effect of geniposide on chronic unpredictable mild stress-induced depressive rats by regulating the hypothalamus–pituitary–adrenal axis. Eur Neuropsychopharmacol (2015) 25:1332–41. doi: 10.1016/J.EURONEURO.2015.04.009
185. Xing H, Zhang K, Zhang R, Shi H, Bi K, Chen X. Antidepressant-like effect of the water extract of the fixed combination of Gardenia jasminoides, Citrus aurantium and Magnolia officinalis in a rat model of chronic unpredictable mild stress. Phytomedicine (2015) 22:1178–85. doi: 10.1016/J.PHYMED.2015.09.004
186. Xing Y, He J, Hou J, Lin F, Tian J, Kurihara H. Gender differences in CMS and the effects of antidepressant venlafaxine in rats. Neurochem Int (2013) 63:570–5. doi: 10.1016/j.neuint.2013.09.019
187. Zhai XJ, Chen F, Chen C, Zhu CR, Lu YN. LC-MS/MS based studies on the anti-depressant effect of hypericin in the chronic unpredictable mild stress rat model. J Ethnopharmacol (2015) 169:363–9. doi: 10.1016/j.jep.2015.04.053
188. Zhang Y, Gu F, Chen J, Dong W. Chronic antidepressant administration alleviates frontal and hippocampal BDNF deficits in CUMS rat. Brain Res (2010) 1366:141–8. doi: 10.1016/j.brainres.2010.09.095
189. Mizuki D, Matsumoto K, Tanaka K, Thi Le X, Fujiwara H, Ishikawa T, et al. Antidepressant-like effect of Butea superba in mice exposed to chronic mild stress and its possible mechanism of action. J Ethnopharmacol (2014) 156:16–25. doi: 10.1016/J.JEP.2014.08.014
190. Szymańska M, Budziszewska B, Jaworska-Feil L, Basta-Kaim A, Kubera M, Leśkiewicz M, et al. The effect of antidepressant drugs on the HPA axis activity, glucocorticoid receptor level and FKBP51 concentration in prenatally stressed rats. Psychoneuroendocrinology (2009) 34:822–32. doi: 10.1016/J.PSYNEUEN.2008.12.012
191. Dazzi L, Serra M, Spiga F, Pisu MG, Jentsch JD, Biggio G. Prevention of the stress-induced increase in frontal cortical dopamine efflux of freely moving rats by long-term treatment with antidepressant drugs. Eur Neuropsychopharmacol (2001) 11:343–9. doi: 10.1016/S0924-977X(01)00105-5
192. Vatal M, Aiyar AS. Some aspects of corticosterone metabolism in lithium treated rats. Chem Biol Interact (1983) 45:277–82. doi: 10.1016/0009-2797(83)90074-1
193. Haj-Mirzaian A, Amiri S, Kordjazy N, Momeny M, Razmi A, Rahimi-Balaei M, et al. Lithium attenuated the depressant and anxiogenic effect of juvenile social stress through mitigating the negative impact of interlukin-1β and nitric oxide on hypothalamic-pituitary-adrenal axis function. Neuroscience (2016) 315:271–85. doi: 10.1016/j.neuroscience.2015.12.024
194. Silva R, Mesquita AR, Bessa J, Sousa JC, Sotiropoulos I, Leão P, et al. Lithium blocks stress-induced changes in depressive-like behavior and hippocampal cell fate: the role of glycogen-synthase-kinase-3β. Neuroscience (2008) 152:656–69. doi: 10.1016/j.neuroscience.2007.12.026
195. Bao A-M, Meynen G, Swaab DF. The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res Rev (2008) 57:531–53. doi: 10.1016/j.brainresrev.2007.04.005
196. Purba JS, Hoogendijk WJ, Hofman MA, Swaab DF. Increased Number of Vasopressin- and Oxytocin-Expressing Neurons in the Paraventricular Nucleus of the Hypothalamus in Depression. Arch Gen Psychiatry (1996) 53:137. doi: 10.1001/archpsyc.1996.01830020055007
197. Dinan TG, O'Brien S, Lavelle E, Scott LV. Further neuroendocrine evidence of enhanced vasopressin V3 receptor responses in melancholic depression. Psychol Med (2004) 34:169–72. doi: 10.1017/s0033291703001004
198. Dinan TG, Scott LV. Anatomy of melancholia: focus on hypothalamic-pituitary-adrenal axis overactivity and the role of vasopressin. J Anat (2005) 207:259–64. doi: 10.1111/j.1469-7580.2005.00443.x
199. De Kloet ER, DeRijk RH, Meijer OC. Therapy insight: is there an imbalanced response of mineralocorticoid and glucocorticoid receptors in depression? Nat Clin Pract Endocrinol Metab (2007) 3:168–79. doi: 10.1038/ncpendmet0403
200. Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci (2008) 31:464–8. doi: 10.1016/j.tins.2008.06.006
201. Modell S, Yassouridis A, Huber J, Holsboer F. Corticosteroid receptor function is decreased in depressed patients. Neuroendocrinology (1997) 65:216–22. doi: 10.1159/000127275
202. DeRijk RH, van Leeuwen N, Klok MD, Zitman FG. Corticosteroid receptor-gene variants: modulators of the stress-response and implications for mental health. Eur J Pharmacol (2008) 585:492–501. doi: 10.1016/j.ejphar.2008.03.012
203. Manenschijn L, Van Den Akker ELT, Lamberts SWJ, Van Rossum EFC. Clinical features associated with glucocorticoid receptor polymorphisms. Ann N Y Acad Sci (2009) 1179:179–98. doi: 10.1111/j.1749-6632.2009.05013.x
204. van Rossum EFC, Koper JW, Huizenga NATM, Uitterlinden AG, Janssen JAMJL, Brinkmann AO, et al. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes (2002) 51:3128–34. doi: 10.2337/diabetes.51.10.3128
205. McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci (2009) 12:342–8. doi: 10.1038/nn.2270
206. Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci (2004) 7:847–54. doi: 10.1038/nn1276
207. Romens SE, McDonald J, Svaren J, Pollak SD. Associations between early life stress and gene methylation in children. Child Dev (2015) 86:303–9. doi: 10.1111/cdev.12270
208. Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA (2008) 299:1291. doi: 10.1001/jama.299.11.1291
209. Hartmann J, Wagner KV, Liebl C, Scharf SH, Wang X-D, Wolf M, et al. The involvement of FK506-binding protein 51 (FKBP5) in the behavioral and neuroendocrine effects of chronic social defeat stress. Neuropharmacology (2012) 62:332–9. doi: 10.1016/j.neuropharm.2011.07.041
210. Touma C, Gassen NC, Herrmann L, Cheung-Flynn J, Büll DR, Ionescu IA, et al. FK506 binding protein 5 shapes stress responsiveness: modulation of neuroendocrine reactivity and coping behavior. Biol Psychiatry (2011) 70:928–36. doi: 10.1016/j.biopsych.2011.07.023
211. Liu Z, Zhu F, Wang G, Xiao Z, Wang H, Tang J, et al. Association of corticotropin-releasing hormone receptor1 gene SNP and haplotype with major depression. Neurosci Lett (2006) 404:358–62. doi: 10.1016/j.neulet.2006.06.016
212. Brühl AB, Kaffenberger T, Herwig U. Serotonergic and noradrenergic modulation of emotion processing by single dose antidepressants. Neuropsychopharmacology (2010) 35:521–33. doi: 10.1038/npp.2009.159
213. Qin D, Li Z, Li Z, Wang L, Hu Z, Lü L, et al. Chronic glucocorticoid exposure induces depression-like phenotype in rhesus macaque (Macaca Mulatta). Front Neurosci (2019) 13:188. doi: 10.3389/fnins.2019.00188
214. Qin DD, Rizak J, Feng XL, Yang SC, Lü LB, Pan L, et al. Prolonged secretion of cortisol as a possible mechanism underlying stress and depressive behaviour. Sci Rep (2016) 6:1–9. doi: 10.1038/srep30187
215. Forkosh O, Karamihalev S, Roeh S, Alon U, Anpilov S, Touma C, et al. Identity domains capture individual differences from across the behavioral repertoire. Nat Neurosci (2019) 22:2023–2028. doi: 10.1038/s41593-019-0516-y
216. Schmaal L, Veltman DJ, van Erp TGM, Sämann PG, Frodl T, Jahanshad N, et al. Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry (2016) 21:806–12. doi: 10.1038/mp.2015.69
217. Drysdale AT, Grosenick L, Downar J, Dunlop K, Mansouri F, Meng Y, et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat Med (2017) 23:28–38. doi: 10.1038/nm.4246
Keywords: major depressive disorder, cortisol, stress, antidepressants, preclinical models, behavior
Citation: Nandam LS, Brazel M, Zhou M and Jhaveri DJ (2020) Cortisol and Major Depressive Disorder—Translating Findings From Humans to Animal Models and Back. Front. Psychiatry 10:974. doi: 10.3389/fpsyt.2019.00974
Received: 05 June 2019; Accepted: 09 December 2019;
Published: 22 January 2020.
Edited by:
Julian Macoveanu, Copenhagen University Hospital, DenmarkReviewed by:
Hiroaki Hori, National Center of Neurology and Psychiatry, JapanHideki Tsumura, Tokushima University, Japan
Copyright © 2020 Nandam, Brazel, Zhou and Jhaveri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: L. Sanjay Nandam, lawrence.nandam@health.qld.gov.au; Dhanisha J. Jhaveri, dhanisha@uq.edu.au
†These authors have contributed equally to this work.