 Shui Jiang
Shui Jiang Lynne Postovit
Lynne Postovit Annamaria Cattaneo
Annamaria Cattaneo Elisabeth B. Binder
Elisabeth B. Binder Katherine J. Aitchison
Katherine J. Aitchison
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 08 November 2019
Sec. Molecular Psychiatry
Volume 10 - 2019 | https://doi.org/10.3389/fpsyt.2019.00808
This article is part of the Research TopicEarly Life Adversity’s Impact on Brain and Body: Understanding Disrupted Circuits to Identify Preventive StrategiesView all 15 articles
Adverse childhood experiences (ACEs) may be referred to by other terms (e.g., early life adversity or stress and childhood trauma) and have a lifelong impact on mental and physical health. For example, childhood trauma has been associated with posttraumatic stress disorder (PTSD), anxiety, depression, bipolar disorder, diabetes, and cardiovascular disease. The heritability of ACE-related phenotypes such as PTSD, depression, and resilience is low to moderate, and, moreover, is very variable for a given phenotype, which implies that gene by environment interactions (such as through epigenetic modifications) may be involved in the onset of these phenotypes. Currently, there is increasing interest in the investigation of epigenetic contributions to ACE-induced differential health outcomes. Although there are a number of studies in this field, there are still research gaps. In this review, the basic concepts of epigenetic modifications (such as methylation) and the function of the hypothalamic-pituitary-adrenal (HPA) axis in the stress response are outlined. Examples of specific genes undergoing methylation in association with ACE-induced differential health outcomes are provided. Limitations in this field, e.g., uncertain clinical diagnosis, conceptual inconsistencies, and technical drawbacks, are reviewed, with suggestions for advances using new technologies and novel research directions. We thereby provide a platform on which the field of ACE-induced phenotypes in mental health may build.
Stressful or traumatic events experienced in childhood or adolescence can be driven by a broad range of life events, including but not limited to physical injury, natural disaster, bullying, and childhood maltreatment (1). They are referred to by many terms, including early life adversity, early life stress, early life trauma, and adverse childhood experiences (ACEs) (2). It is reported that the worldwide average trauma exposure rate is 69.7% for children and adults (3). In the United States, around 60% of adults reported that they had experienced at least one type of ACE (2).
ACEs/childhood trauma are associated with negative health outcomes, both mentally and physically (4). Individuals exposed to multiple types of childhood trauma show an increased risk of early mortality, which decreases their lifespan up to 20 years (5). Physically, childhood trauma has been associated with increased risk of cardiovascular disease (6), autoimmune disease (7), gastrointestinal symptoms (8), poor dental health (9), obesity, and type 2 diabetes (10). Psychologically, childhood trauma is regarded as one of the major risk factors for psychopathology. Childhood trauma has been associated with many mental disorders (11). Specifically, childhood trauma has been linked to posttraumatic stress disorder (PTSD) (12), insomnia (13), anxiety (14), depression (15, 16), bipolar disorder (17, 18), maladaptive daydreaming (MD) (19), hallucinations (20), borderline personality disorder (21), disruptive behavior (22), risky behaviors (23, 24), substance abuse (25, 26), antisocial behavior (27), and eating disorders (28, 29).
Childhood trauma impacts children to different extents. Some people are more vulnerable, whereas, others show the characteristic of “resilience,” with the ability to “bounce back” even after adversity (30). Multiple factors, e.g., genetic, epigenetic, and environmental factors, and their interactions contribute to the differential health outcomes induced by childhood trauma. According to a neural diathesis-stress model, genetic predisposition and environmental factors contribute synergistically to the development of mental disorders. The magnitude of the heritability of a phenotype is one way of estimating the relative magnitude of the genetic contribution. In the case of ACE-associated psychiatric disorders such as PTSD, the heritability is in fact low to moderate (31). Similarly, the heritability of resilience is low to moderate, varying in research reports from 25% to 60% (32–34). These heritability values suggest that there may be other mechanisms contributing to these phenotypes, such as gene by gene interaction and gene by environment interactions, and epigenetic mechanisms. Consequently, it might well be productive to explore genetic, epigenetic, and environmental interactions in resilience and ACE-associated health outcomes.
The human genome is made up of DNA, which stands for deoxyribonucleic acid, the genetic code which is a continuous sequence of four “letters” or “bases,” A, G, C, T (A = adenine, C = cytosine, G = guanine, T = thymine). This encodes heritable information from parents to offspring. Part of this sequence is “read” during a process known as “transcription.” Transcriptional machines, which have a complicated structure and are made up of several protein subunits, are needed to start this process. Following transcription of the locus, the noncoding DNA areas (known as “introns”) are spliced out and the regions that are coding proteins/peptides, known as “exons,” are converted into mRNA sequences. These mRNAs are then used to build different protein structures from “building blocks” known as amino acids. In the next, “posttranslational,” stage, further modifications may occur and influence the function of the protein. In general, gene expression is a complicated dynamic process and controlled by diverse regulators at different levels, such as transcriptional regulation (cis: e.g., promoters, trans: e.g., DNA-binding proteins), RNA processing (RNA splicing, noncoding RNA, miRNAs, etc.), translational regulation, and posttranslational regulation (acetylation, phosphorylation, and glycation, etc.) (35).
Epigenetic modifications regulate this dynamic process of DNA to protein. Epigenetics, which means “outside conventional genetics” (36), focuses on the regulation of “turning on or off” genes without changing the DNA sequence, but rather the accessibility of regulatory transcription factors to the gene. Epigenetic modifications impact on multiple nuclear processes, such as DNA packaging and chromatin structure, and thus on gene expression, with various directions of effect (which may be conceptualized as “epigenetic readers, writers, and erasers”) (37). Such modifications include changes in the spatial positioning of chromosomal territories (38). There are three main types of epigenetic modifications: DNA methylation, histone modifications, and various RNA-mediated processes (39, 40). Epigenetic modifications may be cell-type-specific.
Cytosine methylation (5-methylcytosine, 5-mC) is very common in both eukaryotes and prokaryotes (41). It largely happens at cytosine followed by guanine residues (CpG) sites; less commonly, cytosine may be methylated at CpA, CpT, or CpC sites. A family of enzymes named DNA methyltransferases (DNMTs) regulates DNA methylation through transferring a methyl group to the DNA base cytosine (42). Methylation, which is similar to a protective cover on the DNA, generally suppresses gene expression by physically preventing transcription factor binding (43). It also suppresses gene expression by interacting with other mechanisms, e.g., histone deacetylase (HDACs) complex recruitment. For example, methyl-CpG-binding proteins (MeCP) 2 binds tightly to chromatin in a methylation-dependent way, which induces the formation of the histone deacetylase complex. This complex induces transcriptional suppression by changing chromatin structures (44). However, DNA methylation also enhances gene expression through more complicated mechanisms such as the methylation of CCCTC-binding factor (CTCF) binding sites and/or intronic regions (45–49). Hydroxymethylcytosine (5-hmC) is another form of DNA methylation. It is in fact converted from5-mC through an oxidative reaction, by the ten-eleven translocation methylcytosine dioxygenase (TET) 1 enzyme. Similarly, 5-hmC is able to both activate and suppress gene expression in a bidirectional manner (50). The expression of 5-hmC is highly concentrated in the brain and is dynamic during fetal development (51). It has been reported to play important roles in neuronal function, learning and memory, and stress-mediated responses (52).
As for histone modification, it impacts chromatin structure and DNA packaging (37) [e.g., the acetylation of histones may render DNA more or less accessible to transcription factors, leading to enhanced or reduced transcriptional activity (53)]. It has been estimated that the full length of DNA is more than 500 billion kilometres in the human body (54). Consequently, DNA needs to be packed tightly into cells. Histones are directly involved in the packaging process. Basically, a core of DNA (around 146 base pairs) wraps around each histone to form a structure known as a “nucleosome.” Subsequently, an octamer comprising four different histones (H3, H4, H2A, and H2B) is formed. This is further packed into chromatin fibres and then into chromosomes, a unit now visible under a light microscopes. There are several types of histone modifications, including acetylation, methylation, phosphorylation and ubiquitylation. The specific modification patterns of histones, which are called histone codes, work with the other chromatin associated proteins, change the structure of the local chromatin, and thus impact the process of gene expression, such as transcription, replication and DNA repair. The proper topological structure of chromatin is essential in gene expression and genome maintenance (55).
Lastly, noncoding small RNAs (e.g., miRNAs) are also able to mediate sequence-specific modulation of gene expression by different mechanisms (56). For example, miRNAs bind to their target mRNAs via complementary sequences, which induces the cleavage and degradation of the corresponding mRNA (57). More recently, additional epigenetic modifications have been discovered, including for example, RNA methylation (58).
Each cell in the living organism, under normal conditions, essentially shares the same copy of DNA, but eventually develops and differentiates to different cell types under regulatory mechanisms. Epigenetic modifications such as genetic imprinting (59) are necessary for embryogenesis and gametogenesis (60, 61), differentiation, and development. In fact, epigenetic regulation occurs throughout the lifespan and can be induced by random changes (62) or by multiple different environmental factors (63). For example, changes in human epigenome have been associated with processes related to adaptation and evolution (64, 65), and have also been linked to several diseases, such as cancer (66), type 2 diabetes (67), and autoimmune rheumatic disorders, such as systemic lupus erythematosus (SLE) (68). Epigenomic alterations are also associated with pathologies characterized by behavioral or/and cognitive problems, e.g., Alzheimer’s disease (69), Rett syndrome (70), Cushing’s syndrome (71), depression (72), addiction (73), aggression and antisocial behavior (74), and also with illnesses characterized by childhood trauma exposures, such as mental disorders (75).
Early life is a special period characterized by a high level of plasticity and fast development (76). Thus, the impact of childhood trauma is particularly deleterious, since the developmental trajectory of the brain is affected, with resultant alteration of the circuitry for threat detection, emotional regulation, and the reward system (77).
In this paper, we will focus on the epigenetic modification of DNA methylation, as this has the most data relevant to childhood trauma.
Why does childhood trauma impact health outcomes? One mechanism is by the induction of toxic stress. In fact, stress can be classified into “good stress,” “bearable stress,” and “toxic stress” (78), and has acute, delayed and long-term effects on the body (79). “Good stress” can be coped with by physiological mechanisms, encouraging healthy growth; “bearable stress” states may eventually be turned into homeostasis through successful interventions; whereas, “toxic stress,” which is characterized by prolonged or frequent activation and dysregulation of the stress response pathway, induces long-term changes and damage not only to the brain but also to the rest of body (2, 80).
The central biological pathway involved in the response to stress is the hypothalamic-pituitary-adrenal (HPA) axis (Figure 1). In 1914, Walter B. Cannon put forward the “fight or flight” model, which described the body’s response toward stress (81). Around the 1950s, Selye’s general adaptation syndrome was put forward: that chronic stress could induce a nonspecific response in the body, such as increased heart rate and blood pressure (82). More recently, more in-depth research has illustrated that alterations in the HPA axis have been associated with the process of dealing with stress, especially toxic stress-induced negative health outcomes (83).
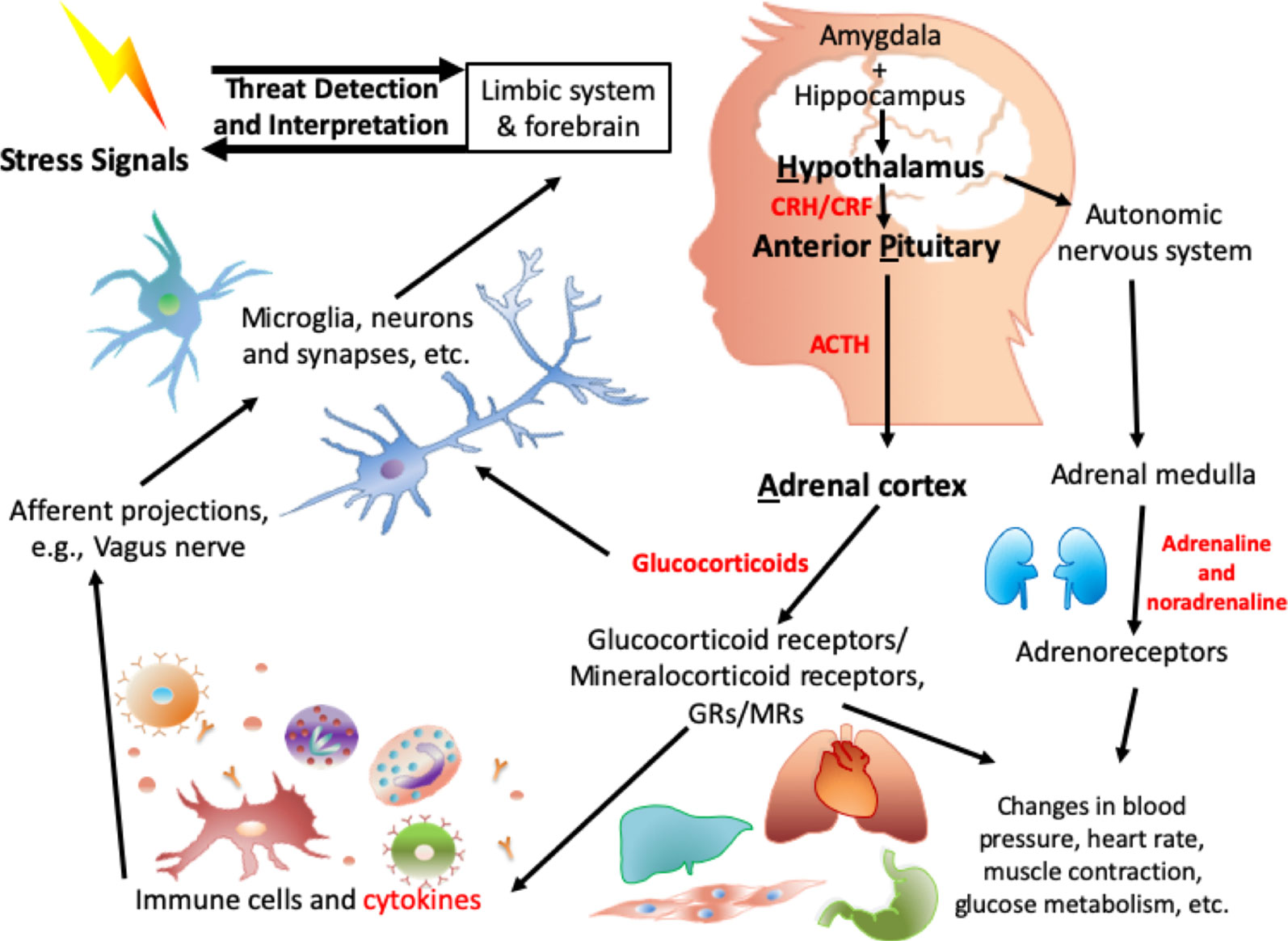
Figure 1 Biological mechanisms involved in HPA axis induced dysregulation in response to toxic stress.
When a threat signal is recognized, the central nervous system (CNS): amygdala (84), hypothalamus (85), and parts of brainstem such as the locus coeruleus, (86–88), which are regarded as the central components of the stress response, will be activated. Neurotransmitters such as glutamate, serotonin (89), and γ-aminobutyric acid (GABA) are involved in this signal transmission. On receipt of the neuronal signal from the amygdala and locus coeruleus, numerous neuropeptides are released from the hypothalamus, including arginine vasopressin (AVP) and stress-induced corticotropin-releasing factor/hormone (CRF/H) (90). The CRF Receptor 1 (CRFR1) on the anterior pituitary is activated, which results in the secretion of adrenocorticotropic hormone (ACTH). AVP works together with CRH to contribute to the ACTH response (91). ACTH acts on receptors in the adrenal cortex, leading to the release of stress-related hormones: glucocorticoids (cortisol) and mineralocorticoids (aldosterone). These stress-related hormones mediate the stress response (92) to induce changes in heart rate, blood pressure, metabolism (93), and immune function (94). Other neuropeptides/neurotrophic factors, such as neuropeptide Y (95), dynorphin (96), and oxytocin as well as brain-derived neurotrophic factor (BDNF), are also involved in the HPA axis and in the orchestration of the response to stress.
On the other hand, in the sympathetic adrenal medullary (SAM) axis, a signal from the hypothalamus activates the adrenal medulla, and then induces the secretion of the catecholamines adrenaline and noradrenaline. Peripheral organs (e.g., heart, liver), glands, and vessels have receptors for these hormones and are in addition regulated by the sympathetic autonomic neurons. Together with the HPA axis as mentioned above, the downstream effects, e.g., increased heart rate and blood pressure, are intended to be biologically adaptive, to enhance the individual’s ability to respond to the stressor.
Importantly, cortisol provides negative feedback are the level of the hypothalamus (97) to stop the HPA axis from being excessively activated with consequent deleterious health effects. In addition, within the autonomic nervous system, parasympathetic neurons balance the activation of the sympathetic system, inducing a “rest and digest” body state. Childhood stress and trauma alter the HPA axis (98) and the long-term dysregulation of the HPA axis induced by childhood stress/trauma has been associated with increased risk of adverse health outcomes. For some of these, the effects of adversity appears to be dose-dependent (99–101).
There is increasing interest in the investigation of epigenetic and environmental interactions in ACE-induced differential health outcomes. In humans, studies have mainly focused on peripheral tissues, such as peripheral blood, buccal cells, or saliva. There are also studies with human postmortem brain tissue. For example, Labonte and colleagues reported that in hippocampal tissues derived from those who had died by suicide, comparing those with and without a history of childhood abuse, there were 362 differentially methylated promoter sites. Among these, 248 sites were hypermethylated and 114 were hypomethylated (102). Similarly, there was a bidirectional regulation of methylation in the cingulate cortex of those with/without childhood trauma who has had depression and died by suicide, with the highest differential methylation occurring in genes that related to myelin (103). In a 2017 systematic review of epigenetic associations with childhood trauma in first episode psychosis patients and healthy individuals, childhood trauma was associated with global hypomethylation in peripheral blood samples (104, 105).
A key limitation of such epigenetic research as described above is nonetheless the tissue specificity of effects, which means that for only very limited sites scan congruent changes across tissues be expected (106, 107). In fact, even with the same sample, e.g., saliva taken at different times from the same individual, the cellular composition (proportion of different cells) may vary, which brings challenges to the analysis of methylation results (108).
Relevant biological systems relevant to the HPA axis are summarized in Figure 1 with highlights provided below.
The FKBP5 gene encodes a heat shock protein 90 (HSP90) cochaperone that modifies the sensitivity of steroid receptor hormones, interacting with the glucocorticoid receptor (GR), the progesterone receptor (PR), and the androgen receptor (AR). Together with other chaperone proteins such as Hsp90, FKBP5 inhibits GR function by slowing ligand-receptor complex translocation to the nucleus (109). It has been reported that in the HPA axis, the activation of GR inhibits the expression of CRH and ACTH, thus restraining overactivation of the HPA axis (110). Although GR activation increases the expression of FKBP5, the increased binding of FKBP5 to the GR suppresses GR activity in a negative-feedback way. Thus, alterations in FKBP5 hinders this negative feedback loop and induces “glucocorticoid resistance” (111).
Genetic variants of FKBP5 impact gene expression. For example, the SNP rs1360780, which has been associated with a change in the three-dimensional structure of the genetic locus, influences the physical contact between RNA polymerase and the transcription start site, as well as the hormone responsive elements (HREs) located in intron 2 (112, 113). Consequently, FKBP5 genetic variants may induce differential health stress-related outcomes via their influence on GR sensitivity (114), the HPA axis, and subsequent regulation of neuronal function and synaptic plasticity (115). Exposure to childhood trauma has been shown to interact with the rs1360780 risk allele (T) to increase risk for a number of psychiatric disorders (115). It has been proposed that rs1360780 risk allele carriers show an increased FKBP5 mRNA response on exposure to stress (through enhanced binding of the promoter and the intron 2 glucocorticoid response element GRE), along with decreased negative feedback signal to the GR, which induces prolonged cortisol secretion. Enhanced cortisol secretion induces decreased DNA methylation in the intron 7 GRE, which in turn further enhances the activation of FKBP5 (116). This dual genetic and epigenetic disinhibition may increase FKBP5 levels and induce downstream changes in cellular and circuit function to a level that promotes risk for psychiatric disorders (116). For example, in major depressive disorder (MDD) patients who have been exposed to childhood trauma, the risk allele (T) of rs1360780 has been associated with a lower methylation level of FKBP5 in peripheral blood cells, and lower methylation of FKBP5 has been linked to functional as well as to structural alterations in the inferior frontal orbital gyrus (117). This region of the brain belongs to the anterior limbic and paralimbic structures and plays an important role in response inhibition and cognitive function (118). Also, alterations of this area have been associated with symptoms of PTSD induced by childhood sexual abuse (119). Interestingly, it has been found that the FKBP5 rs3800373 minor allele alters the secondary structure of FKBP5 mRNA, decreasing the binding of a stress- and pain-associated microRNA, miR-320a. This results in relatively greater FKBP5 translation, unchecked by miR-320a, increasing glucocorticoid resistance and increasing vulnerability to stress such as posttraumatic pain (120).
Other genes that have been associated with the effects of childhood trauma are the monoamine oxidase (MAO) gene (located on the X chromosome), encoding the mitochondrial bound isoenzyme MAO A and B, which break down monoamine neurotransmitters such as dopamine, serotonin and noradrenaline (121). This degrading function of MAOA and MAOB is essential for the maintenance of synaptic transmission and thus proper motor control, emotional regulation, and cognitive function (122). There are more data relevant to this review on MAOA than on MAOB.
In 1993, it was reported that a point mutation in exon eight of the MAOA gene (leading to a premature stop codon) contributes to Brunner Syndrome, with prominent aggressive behaviors (123). Polymorphisms in MAOA have in fact been associated with antisocial behavior in general (27, 124), as well as with panic disorder (125), restless legs syndrome (126) and attention control (127).
The variable number tandem repeats (VNTRs) in the MAOA gene have been associated with differential health outcomes after stressful life events. VNTR may be variously defined, generally referring to short tandem repeats of 20–100 nucleotides. They regulate gene expression and have been associated with human diseases (128) such as spinocerebellar and Friedreich ataxia, fragile X syndrome, Huntington’s disease (128), and other psychiatric disorders.
There is a VNTR comprising CCCCTCCCCG (known as the “A repeat”) and CTCCTCCCCG (known as the “B repeat”) of a 10-base pair unit near the transcription start site (TSS) of MAOA that contributes to antisocial behavior after exposure to childhood trauma in females (129). The first six repeats are the same within different individuals, ABABAB; variants occur in at or after the seventh repeat. For example, seven repeats (7R) is ABABABA, and 10 repeats (10R) is ABABABAAAA. The risk allele (comprising 10 repeats) is associated with lower MAOA activity. Lower MAOA activity, which is associated with higher levels of the relevant neurotransmitters, has been associated with increased risk of conduct disorder and antisocial behavior (130, 131). Another well-studied VNTR in the MAOA gene is the 30 base pair (bp) upstream VNTR (u-VNTR) with a repeat sequence of 5’-ACCGGCACCGGCACCAGTACCCGCACCAGT-3’ (132). The risk allele is three repeats, which has been associated with a significantly decreased level of MAOA expression (133). Similarly, maltreated children with the risk MAOA u-VNTR genotype develop conduct disorder, antisocial personality, and violent criminality in adulthood (131).
Moreover, different genetic variants have been associated with differential methylation status of MAOA and corresponding phenotypes. For example, nine-repeat (9R) carriers (the lower risk allele) of the 10 bp MAOA VNTR show a lower methylation level in the MAOA promoter in females (129). In regard to the 30 bp u-VNTR, carriers of the lower-MAOA-activity variants (i.e., the higher risk alleles such as 3.5) had a higher risk of depression with histories of childhood trauma in females compared to those who without trauma histories, and the overall methylation of MAOA was reduced in depressed patients (130). Interestingly, the lower-activity MAOA-allele (3.5 repeats) of the MAOA u-VNTR has been associated with other epigenetic modifications, such as NR3C1 hypermethylation after childhood trauma (130).
In monozygotic twin studies, hypomethylation of all MAOA CpG sites has been negatively associated with depressive symptoms, but not with childhood trauma; whereas, hypermethylation of the MAOB gene shows a nominally significant association with childhood sexual abuse (134).
Another well-studied candidate in the HPA axis is the GR gene: nuclear receptor subfamily 3 group C member 1 (NR3C1). NR3C1 encodes the GR. The binding of glucocorticoid to the GRs plays important roles in glucose homeostasis (135) and regulates the stress response through both genetic (136) and epigenetic mechanisms (137). Childhood trauma and early stress alter the methylation status of this gene and its expression.
Research has shown that altered methylation status in this gene has been associated with childhood trauma, especially the CpG sites located in the noncoding first exons of NR3C1 (138). In a rat model, pups who received less early nurturing behaviors (low licking and grooming (LG) and arched-back nursing (ABN) from the mother rat) presented significantly higher levels of methylation in the exon 17 GR promoter nerve growth factor-inducible protein A (NGFI)-A binding site (139). Since NGFI binding decreases GR expression, this alteration is thought to be associated with impaired regulation of the HPA axis (140, 141).
In humans, it has been shown that experiencing childhood trauma increases methylation of NR3C1 (142). In Melas and colleagues’ study, one specific type of childhood trauma (parental death) was associated with hypermethylation of the NR3C1 CpGs close to the NGFI-A binding site, at, in association with the L-allele (3.5 repeats) of the MAOA u-VNTR in salivary DNA samples (130). In postmortem brain tissue (hippocampus) from those who have died by suicide, there was decreased expression of GRs, along with enhanced cytosine methylation of the NR3C1 promoter in those with a history of childhood trauma. Also, in the same group there was decreased NGF1-A transcription factor binding and NGF1-induced gene transcription (137). Labonte and colleagues also investigated methylation and NR3C1 expression in postmortem (suicide) brains. In the hippocampus, total GR expression, and the 1B,1C, 1H GR RNA variant levels decreased with history of childhood trauma. Site-specific methylation showed that the methylation of variants 1B and 1C was negatively associated not only with their expression but also with total GR mRNA level. Variant 1H was associated with effects in the opposite direction (143). Other tissues, such as cord blood, peripheral blood, buccal epithelial cells and placental tissues were also tested, and the majority of them showed similar results in regard to enhanced methylation of NR3C1 in association with early life adversity (138, 144).
Stressful life events occurring slightly later in life, such as in adolescence, are associated with a further independent increase in methylation of NR3C1 (142). Interestingly, the effects of methylation within the NR3C1 promoter can be sex-specific. Vukojevic et al. showed that enhanced methylation of NR3C1 promoter at the NGFI-A binding site has been associated with less intrusive memory, and thus decreased risk of PTSD among survivors of the Rwandan genocide, but only in males (145).
In a recent study in mice, hemizygosity of a deletion of nr3c1 (nr3c1-/+ heterozygote) has been associated with changes in DNA methylation in fetal placenta, and these changes have been associated with methylation changes in the adult prefrontal cortex, as well as with increased anxiety-like behaviors in the same animals (146). In addition, hydroxymethylation modifications of nr3c1 have been suggested to be involved in the stress response pathway. Li and colleagues reported that acute stress induces accumulation of 5-hmC in the nr3c1 3’ untranslated region (UTR) in the mouse hippocampus (147). Further investigation of molecular mechanisms involving 5-hmC and childhood trauma in not only NR3C1 but also in other genes could be productive.
Serotonin or 5-hydroxytryptamine (5-HT) is a monoamine neurotransmitter. It can be found in the gastrointestinal (GI) tract, blood platelets, and the CNS (148). In the CNS, serotonergic neurons are widely distributed in the brain, especially the limbic system (149). Serotonin contributes to brain development (149) and to maintenance of normal brain function. It promotes neural and glial cell growth, differentiation, survival and synapse formation (150). Alterations in the serotonin system have been associated with structural and functional changes in the brain (149). Stress induces brain serotonin turnover (151, 152). Excessively raised serotonin is associated with neurotoxicity (153). Stress-induced serotonin turnover has been associated with conditions such as impulsive violence (154), depressive symptoms (155), and substance dependence (156).
The HTR genes encode serotonin receptors, which are widely distributed in the CNSs including the prefrontal, parietal, and somatosensory cortices as well as the olfactory tubercle. Variants in these gene have been associated with differential treatment responses and with various psychiatric disorders, such as panic disorder (157), impulsive disorder (158), PTSD (159), and eating disorder (160). In children, HTR2A variants are related to differential risk of being hyperactive (161) with harsh parenting styles (162, 163) or after experiencing childhood abuse (164). It has been reported that early life adversity alters HTR2A methylation, and the effects were allele-specific. Contextual stress experienced in childhood induces enhanced HTR2A methylation at site-1420, in those of A/A genotype at rs6311- (-1438 A/G). Moreover, enhanced methylation of HTR2A at site-1420 was negatively associated with PTSD and depression; whereas, those of G/G genotype presented decreased methylation (165). Notably, hypermethylation of site-1420 has also been found in schizophrenia and bipolar patients (166). In the serotonin 3A receptor (HTR3A) gene, the mother’s life-time exposure to interpersonal violence is associated with altered methylation in the HTR3A gene, which has been associated with their children’s secure base distortion (167). In addition to the HTR2A and HTR3A, there are several other serotoninergic genes that undergo epigenetic modifications and have been associated with childhood trauma, such as that encoding the serotonin transporter.
These serotonin transporter (responsible for serotonin reuptake) encoded by SLC6A4 (also known as the 5-HTT gene) is in fact a frequently studied candidate in psychiatric genetics and epigenetics. Higher methylation of SLC6A4 has been associated with childhood trauma, and with worse clinical presentation in MDD (168). In women, there is a significant association between sexual abuse and SLC6A4 hypermethylation, which has been linked to antisocial behavior (74). In newborn babies whose mothers have depressive symptoms in the second trimester, methylation at SLC6A4 promoter CpG sites is lower in both the newborns and in the mothers compared to controls (169). Methylation also mediates allele-specific cortisol response patterns of the 5-HTT linked polymorphic region (5-HTTLPR) (rs25531) (170). The 5-HTTLPR, consisting of a 20 to 23 bp GC-rich VNTR repeated 14 times in the short allele (S) and 16 times in the long allele (L), is located 1 kb upstream of the SLC6A4 gene. The short variant is associated with reduced 5-HTT expression (171). The S/S HTTLPR genotype has been associated with increased risk of depression and suicide attempts after stressful events (172), as well as with adult depression after childhood trauma (173). In Alexander and colleagues’ study, it was showed that those of S/S genotype with lower methylation level exhibited higher cortisol response; while the association of the 5-HTTLPR short allele with enhanced cortisol response disappeared at a higher SLC6A4 methylation level (170).
BDNF has been investigated not only for association with childhood trauma, but also for association with mental health outcomes such as psychotic or depressive symptoms (174–176). BDNF encodes a neurotrophin, which promotes the growth, differentiation and survival of neurons. BDNF is also involved in neuroplasticity. Structural brain changes are seen after trauma and BDNF is hypothesized to be involved in these (177). For example, early isolation (one type of ACEs) causes decreased BDNF levels in the amygdala and infralimbic cortex; however, the combination of resocializing and the antidepressant fluoxetine reverses the reduction of bdnf in rodents (178). In a rat model, early stress (being abused by caretakers) induces enhanced BDNF methylation and decreased bdnf gene expression in the prefrontal cortex in exon 9 and 14, which includes the transcription start site (TSS) and cyclic adenosine monophosphate (cAMP) response element and the enhanced methylation persists until adulthood (179). In rodents, the bdnf gene contains 9 noncoding exons, each of which can be linked to the protein coding exon IX (9) after splicing, and transcription can be initiated before the protein coding exon in the intronic area. Exon-specific transcription is tissue-specific. Importantly, methylation-induced altered gene expression of BDNF is also cell-type specific (180, 181).
In humans, childhood trauma has been associated with decreased serum levels of BDNF (182). Also, childhood maltreatment induces alterations in BDNF promoter methylation (75). It has been shown that a lower maternal care condition is associated with higher BDNF DNA methylation levels (183). Furthermore, differential BDNF methylation has been associated with structural brain changes. For example, socioeconomic disadvantage has been negatively associated with BDNF DNA methylation, specifically at the exon IV promoter site, and this lower level of BDNF methylation has been linked to greater thickness of the lateral orbitofrontal cortex (IOFC), medial frontal cortex and decreased thickness of the bilateral IOFC in adolescence (age 12–13) (184). These brain areas are relevant to decision-making, emotion, and memory processing.
In addition, BDNF works synergistically with other genes after childhood trauma, such as the 5-HTTLPR (182), noradrenaline/norepinephrine transporter (NET) and corticotropin releasing hormone receptor 1 (CRHR1) genes (185), as well as tryptophan hydroxylase (TPH) 2 (186). In fact, the BDNF receptor TrkB and GRs, as well as mineralocorticoid receptors, are coexpressed in hippocampal neurons. Additionally, as mentioned above, BDNF directly regulates the HPA axis. The administration of BDNF in vivo induces increased CRH level and reduction of BDNF or its receptor normalizes the CRH level and thus, the HPA axis. This cross-talk between BDNF and CRH may be at least partly mediated by the CREB and MAPK pathway and is involved in the enhancement of fear memory under stress (187).
There are other candidate genes with at least some data in childhood trauma and epigenetic alterations, such as COMT, IL-6 (188), and OXTR (189).
The catechol-O-methyltransferaseenzyme encoded by the COMT gene on chromosome 22q11.2 (190), is involved in the metabolism of catecholamines including the neurotransmitters dopamine, adrenaline, and noradrenaline, in reactions that involve the transfer of a methyl group from S-adenosyl-methionine (SAM) to a hydroxyl group (191). There appear to be epistatic effects between COMT and NR3C1 on working memory (192). In addition, methylation of the COMT promoter has been associated with a change in prefrontal cortical connectivity in schizophrenia (193), as well as in depression (194).
Interleukin 6 (IL-6) encodes the IL-6 protein, which is a proinflammatory cytokine. Alterations in IL-6 have been associated with psychiatric disorders, such as depression (195). In addition, patients with schizophrenia and a history of childhood trauma have a pro-inflammatory phenotype (196). Inflammatory factors can in fact be regarded as mediators that connect the environmental stimulus of childhood trauma with clinical symptoms. Changes in the IL-6 methylation has been associated with childhood trauma related phenotypes. In African American men, there was an association with decreased methylation of IL-6 and enhanced IL-6 protein level after childhood trauma (197). Importantly, altered expression of IL-6 can be associated with other genetic variants that are involved in neural pathways. For example, women who carry two short alleles of the 5-HTTLPR present a higher IL-6/IL-10 ratio when dealing with stress (198).
Oxytocin is a neuropeptide hormone facilitating labor and breastfeeding in mammals. In the brain, oxytocin receptors (OXTRs) are expressed mainly in the central nucleus of the amygdala (cAmyg) and the ventromedial nucleus of the hypothalamus (VMH) (199). The cAmyg regulates the fear response (200) while the VMH regulates a range of behaviors including female sex behaviors (201). Oxytocin and its receptor are involved in the regulation of attachment, social behavior and the stress response (202). In a recent study, there was hypermethylation at OXTR CpG sites in children who had experienced childhood trauma, and hypermethylation has been associated with decreased grey matter volumes in the orbitofrontal cortex (OFC), which may be related to insecure attachment styles (189).
Research has shown that altered methylation has been associated with childhood trauma-induced phenotypes. Several candidate genes (FKBP5, MAOA, NR3C1, HTR and SLC6A4, BDNF) have been discussed in this review. However, the actual regulatory network and mechanisms are more complicated.
Firstly, multiple functional pathways or circuits are involved in processes relevant to stress, including both the reward and the fear circuits, emotional regulation and executive cognitive function. Secondly, in the HPA axis, molecules and their receptors interact and cross talk with each other. Thirdly, there are potential gene by gene, gene by environment, gene by epigenetic modification, and even epigenetic by epigenetic modifications interactions. All these components influence stress-related phenotypes.
For example, the reward pathway/circuit in the brain has been associated with trait optimism, which has been associated with resilience after stress (203).There are two main reward pathways in the brain: the mesocortical dopamine pathway and the mesolimibic dopamine pathway. Glutamatergic and GABAergic connections are also involved in the reward circuit (204). Similarly, glutamatergic and noradrenergic neuronal signalling (203) and dopaminergic connections participate in neuronal regulation in the fear circuit. In addition, adrenergic receptors (205) and GRs (206) are also involved in fear conditioning. The serotonergic and noradrenergic systems have an established role in mood regulation, while the former is involved in motivation as well, with both anxiogenic and anxiolytic effects (207). Dopamine is relevant to mood regulation too. Enzymes regulating these pathways, such as COMT, MAOA and MAOB, regulate these phenotypes.
At the molecular level, there are different levels of cross-talk. For example, the dopamine D1 receptor interacts with glutamate-mediated excitatory neurotransmission through protein-protein interactions (208). In addition, serotonin signalling, has been reported to interact with cannabinoid receptors (209). Acting as retrograde synaptic messengers (210), the endogenous cannabinoids, such as anandamide, sleep-inducing substance oleamide (211) and palmitoylethanolamide (212), regulate numerous biological processes such as neuronal migration (213), learning, memory (214), pain processing (215), motility (216), and emotional-and reward-related processing (217–219).Further, both serotonin and endocannabinoids are involved in stress-related phenotypes, such as anxiety (212). In addition, serotonin is also involved in the regulation of GRs, such as in primary hippocampal cell cultures (220) and in the rat brain (221). At the genetic level, it has been reported that different genotypes of the 5-HTT gene has been associated with the altered GRs’ mRNA level under conditions of childhood adversity (222). A variant in MAOA gene is associated with differential NR3C1 methylation (130). For BDNF, its expression level responds to stress-related HPA axis activation. Moreover, there is a feedback loop whereby directly regulates CRH, and thus, the HPA axis (187). Besides, as mentioned above, multiple other genes, act in concert with BDNF (185). These genes further interact with other genetic/epigenetic variants to form a sophisticated molecular and functional network, which has not yet been fully characterized. For example, TPH2 also interacts with the adenosine deaminase, RNA specific B1 (ADARB1) gene, which affects pre-mRNA splicing. The interaction of these two genes predicts risk of suicide attempts after childhood trauma (223). A given neurotransmitter/neuronal pathway may conduct more than one function, e.g., glutamate signaling has been associated with both activation and inhibition of the HPA axis through inotropic and kainite/group I metabotropic receptors respectively (224). Interestingly, cognitive therapy and cognitive reappraisal decreases amygdala and HPA activation in response to stress (225), suggesting that there is some “flexibility” in stress-related psychiatric phenotypic presentations. Hence, molecular mechanisms of the HPA axis and the stress response pathway more widely are not only highly complex and orchestrated but also require further illumination.
Limitations exist in this field. Even though numerous studies have been done, evidence of associations between epigenetic/epigenomic alterations and differential health outcomes induced by childhood trauma are limited (226). Additionally, there are inconsistencies in the field. For example, the association between childhood trauma and NR3C1 methylation has not been consistently replicated (138) and likewise the differences in SLC6A4 methylation between trauma- and nontrauma-impacted groups (104).
The full complements of molecular mechanisms involved in childhood trauma related health outcomes remain to be elucidated (31). As mentioned above, a further complication is the possibility of coordinate regulation of epigenetic processes in more than one gene/pathway. In addition, there may be pleiotropic or polygenic effects. Pleiotropy means that a gene is associated with more than one phenotype (e.g., the association between disrupted in schizophrenia 1 (DISC)1 mutations and various psychiatric disorders) (227), and polygenic means that one phenotype may be influenced by several genes (e.g., AOB blood type systems). Moreover, metastable epialleles, differential expression of alleles induced by epigenetic modification during early embryonic development have been identified in genetically identical individuals, and these may also induce phenotypic changes (228). Additionally, study heterogeneities may have limited the conclusions possible in this data synthesis.
Between study heterogeneity includes the investigation of different types of childhood trauma. Research has shown that different types of trauma stimulate different brain areas (77). Although psychological trauma might induce similar biological responses to physical trauma (229), the affected brain areas are different: physical stressors mainly impact the brainstem and hypothalamus (230); whereas, psychological stressors mainly impact regions that regulate emotion, learning, memory and decision making, e.g., the hippocampus, the amygdala and the prefrontal cortex (231, 232). Moreover, long-term stress and acute stress have different effects on the brain. Trauma timing, and frequency also impacts differential health outcomes owing to neurodevelopmental stages (233). However, the exact timing as well as the frequency are difficult to reliably record, since the most common type assessment for childhood maltreatment is retrospective self-report, which may map relatively poorly on to prospective assessments (234).
In addition, phenotypic measurement and diagnosis for children who experience childhood trauma may be ambiguous. In diagnosis, children exposed to childhood trauma may develop PTSD, but the potential outcomes are not limited to PTSD (235). Even with PTSD, there are arguments about the diagnostic criteria in DSM-5 (e.g., lack of connection between exposure to stressor and some specific symptoms, some very-well-documented symptoms failing to be captured in DSM-5, and lack of extensive field trial data (236)). Consequently, the term ‘complex PTSD’ has been put forward to describe complicated traumatic outcomes not captured by standard PTSD (237). Importantly, in behavioral measurement, it is necessary to develop appropriate mathematical models and measurements to correctly quantify within- and between-individual variability (238).In behavioral studies, it is hard to define associations between single genetic/epigenetic variants, as behavioral traits are usually controlled by multiple genes (239). In the definition of childhood trauma induced phenotypes, cultural and ethnic differences may bring additional between study heterogeneity (240). There are other factors such as sex/gender differences in response to stress (241, 242), and the use of different tissues (saliva, cord blood, whole blood and peripheral blood) by different researchers (243). The latter brings complexity to data comparisons since the epigenetic signature differs between and within tissues (244).
Crucially, more than one trait contributes to health outcomes after experiencing trauma. The same genetic/epigenetic modification may impact differently on different traits in one individual. For example, 7 repeat (7R) carriers of the DRD4 exon III VNTR exhibit the highest sensitivity toward parental-induced stress (245); however, they also show a higher level of emotional resilience due to the association between the 7R and specific personality types (246).
Although epigenetics is not a novel concept [the first scientific hypothesis of epigenetics was put forward by Malpighi (247) in 1673, with a key milestone of epigenetic development in 1975 by Riggis (248), Holliday and Pugh (249)], and may simply mean inherited altered gene expression states, it may also refer to inter- versus trans-generational effects, where the former refers to transmission across one generation, and the latter to transmission across multiple generations (250). Historically, these terms have been ambiguously defined (247, 251, 252). This has led to misunderstandings as well as to bias in methodologies and interpretations, especially in interdisciplinary research (253). Indeed, inherited epigenetic patterns (254–256) and environmental factors (257, 258) other than childhood trauma (such as heavy metals (259), parenting style, and early trauma such as maternal separation (260)) may all impact the epigenetic pattern and hence childhood trauma-induced differential health outcomes. However, how much these changes are due to these factors, and to what extent, remains, unclear (261).
In regard to methylation, except for CpG methylation, there are some non-CpG methylations, such as CpA, CpT, and CpC. These are expressed in cell types such as pluripotent stem cells, oocytes, neurons, and glial cells. Importantly, these non-CpG methylations are critical in maintaining neuronal function and are thus involved in neurological disorders (262). Kigar and colleagues posited that adenosine methylation could be regarded as an epigenetic marker of mammalian early life stress (263). However, more research is needed in regard to the above non-CpG methylations, and also that of 5-hmC. As for non-coding RNA, and histone acetylation, there are to date few investigations of associations between these and childhood trauma. Furthermore, the various epigenetic mechanisms, such as methylation, histone modification and noncoding RNA, while often studied one by one, may cooccur and act in concert.
Research has shown that the effects of trauma can be intergenerationally passed on through epigenetic mechanisms, such as methylation (264). Specifically, childhood trauma has been associated with alteration in methylation patterns in human sperm, which may induce intergenerational effects. Further such analyses in larger samples are required (265). Importantly, in addition to epigenetic modifications, other factors, such as epimutations (an mutation occur at the epigenetic level), fetal reprogramming (266), and even gut microbiome transfer (267) may induce intergenerational phenotypic changes. It is challenging and costly to investigate/exclude all of these factors in one human study.
Sex/gender differences exist in this research field. In stress-related psychiatric disorders, there are sex-associated differences in incidence, symptoms and treatment response (268). For example, in PTSD, the life time prevalence in females (10-12%) is 2-3 times higher than that in males (5-6%) (269). Similarly, depression is more common in females than males (268). Interestingly, both sex- and gender-related concepts contribute to these differences (270).
There are multiple reasons that may explain these phenomena, such as differential traumatic exposures, cognitive factors, coping strategies and biological factors between different sexes. There are also fundamental sex-dependent brain differences between males and females, e.g., the size of vasopressin (AVP) neurons (271). Moreover, when dealing with stress, males and females present different sex-specific cortico-striatal and limbic patterns. In the work of Cahill and colleagues (272), men showed greater activation of the right amygdala; whereas, women showed greater activation of the left amygdala when facing stress (272). In addition, brain connectivity in response to stress also differs by sex: e.g., there was greater connectivity between the anterior and dorsal anterior insula, as well as between the anterior and dorsal anterior mid-cingulate in males than females after stress (273, 274).Similarly, Helpman and colleagues showed that males present overactivation and increased connectivity of salience hubs (including anterior insula (AI) and dorsal anterior cingulate cortex (dACC)); whereas, females show an overactive and possibly enlarged amygdala. In addition, males lose more grey matter after stress in limbic system structures (prefrontal cortex, amygdala and the hippocampus (275). These differences contribute to differential fear processing, emotional regulation and decision-making. Moreover, males and females cope with stress differently. For example, when facing traumatic stress, females tend to be more emotion-focused and to use more palliative coping skills than males. Also, females tend to seek social support more and benefit more from psychotherapies (269). Differential stress-related phenotypes between males and females are also related to the gonadal hormones, which play important roles in the establishment, activation and regulation of the HPA axis (276). In animal models, both female rats and mice exhibit more robust responses of the HPA axis (such as a higher level of ACTH), owing to circulating estradiol. In rats, progesterone and estrogen have been shown to directly impact the stress response in females (277). Epigenetic modifications are also involved in gonadal hormone setting up and maintenance of sex differences in the brain, even before puberty (278). In rodents, it has been shown that females have significantly higher level of methylation in the estrogen receptor-alpha (ER-α) promoter than estradiol treated females or males (279). Note that, early exposure to estradiol induces masculinization/defeminisation (280, 281). Interestingly, these sex-dependent epigenetic changes are dynamic across the lifespan (279).
Current studies in regard to epigenetics and sex-dependent phenotypes mainly focus on steroid hormones and targets related to the HPA axis, such as NR3C1, and majority of them are association studies, e.g.,the enhanced methylation of NR3C1and PTSD risk (145). There are also neurotransmitter specific effects in sex differences. For example, in a study by Oswald and colleagues, the availability of the dopamine D2 receptor (D2R) has been associated with childhood trauma and pleasant drug (amphetamine) effects. In males, there was a positive association between childhood trauma and pleasant drug effects but not in females (282), which suggests that there may be by sex differences in the reward pathway after childhood trauma (283). Autonomic systems are also different between males and females (284), which may also contribute to sex differences in stress-induced phenotypes. Groleau and colleagues reported higher methylation of the DRD2 promoter in women with an eating disorder and a history of childhood trauma versus those without such a history (285). Comparison studies between both males and females are limited, probably owing to the different prevalence within different sexes; in some studies with both females and males, the sample sizes were too small to have enough power; the comparison study between the differences of self-identified gender and biological sex, which may provide us the biological and psychological effects about sex-dependent stress responses, are limited; in addition, current studies are mainly focused on the candidate genes that are related to steroid hormones, and they are mainly association studies, which can’t provide the information about the causality. Research about more in-depth molecular mechanisms between different sexes, and their interactions with other genetic, epigenetic, as well as environmental factors is limited. Thus, the epigenetic contribution to sex-dependent stress-related phenotypes is still filed for research exploration.
By sex and gender differences are still relatively new areas of research, and hence replications are required and interactions between the above components remain to be explored (285–288).
Interestingly, it has been reported that epigenetic patterns and phenotypic changes can be induced by a single genetic variant, combined with random epimutation (289). Hence, it has been recommended that when investigating epimutations and phenotypic changes, the DNA sequence, replication, GC%, and the topological structure of chromosomal bands, especially in unstable genomic areas, should be first analyzed (290) - in an integrated combined “omic” approach. Chromosomal banding was first used with light microscopy and divides chromosomes into regions visible at that level of magnification. These regions include G bands, which have a lower number of genes and lower gene expression level, which replicate late in the cell cycle, and R bands, which have a higher gene number, GC content and expression levels (291). Alterations in the topological structure of chromosomal bands have been associated with changes in gene expression and thus with phenotypes (292–294).
In epigenome-wide association studies (EWAS), although these provide the opportunity to investigate epigenetic variants (methylation, noncoding RNA and histone modification) on a genome-wide level, which could assist with identification of disorder-related markers in different populations (295), the individual CpG sites detected by array methods are limited (296). Genome-wide sequencing approaches can be helpful, but DNA methylation sequencing at a depth to reliably detect the small changes often observed in mixed tissues in human studies is very costly. Targeted assays with high sensitivity covering functionally relevant regions could be an interesting complement here (297). Nonetheless, issues such as cost, speed of delivery, errors of variant annotation, logical and methodological issues (e.g., the appropriate selection of the cohort, population stratification and statistical approaches) remain in human genomic and epigenomic studies (298, 299). Consequently, multiple validations via more than one method might bring more reliability.
New technologies and strategies have emerged in this field. For example, the nanopore sequencing framework, able to distinguish five types of methylation variants with high-throughput (300). The usage of this technology reduces sample preparation processes and increases the detection speed (300). In addition, nanopore sequencing is able to detect 5-hmC (301), which is not adequately covered by traditional array/bisulfite sequencing methods. We suggest a more in-depth investigation of molecular mechanisms including 5-hmC in relation to childhood trauma related effects.
In living cells, fluorescence recovery after photobleaching (FRAP) has been reported to be able to detect histone mobility (302), which permits real-time investigation of dynamic histone modification. In regard to chromatin structure, Stevens and colleagues reported that the combination of chromosome conformation capture (3C) and tagged fluorescent imaging was able to detect the folding of a genomic sequence <100bp in a single cell (303). This provides the opportunity to investigate how epigenetic modifications dynamically and spatially mold chromosomes and thus, cellular function and related phenotypes in animal models in vivo.
In addition, the CRISPR-CAS9 system can be used to study targeted genetic/epigenetic variant-induced phenotypic changes in animal models. In fact, usage of a modified CRISPR-cas9 system has been expanded beyond genome editing, to RNA targeting, chromatin topology, chromatin imaging, and developmental trajectories as well as to lineage tracing (188, 304). Since the effects of childhood trauma are neurodevelopmental stage-sensitive, a tracing-based technique may provide us with information about when sensitive periods toward different stress are, and how stress impacts on neuronal differentiation (305). The CRISPR-cas9 system can also be used as an effective tool to edit the epigenome (306). Liao and colleagues reported that the endogenous gene was activated via trans-epigenetic remodelling by using a CRISPR-cas9 system, and phenotypic changes were observed in acute kidney injury, type 1 diabetes and Duchenne muscular dystrophy rodent models (307). Thus, epigenome editing may help us to better understand the molecular mechanisms in diverse stress-related phenotypes with known targeted sequences. More in-depth molecular insight may also be helpful for improving the definitions and diagnoses of different psychiatric phenotypes.
Given the cell-type specificity of epigenetic changes, achieving single cell-, or at least single cell type-resolution is also an important goal. Single cell sequencing is able distinguish methylated changes in different cell types, and thereby reduce in errors/bias. Using such techniques in combination with sex-dependent stratification, different network mechanisms in males and females may be distinguished. So far, a number of single cell sequencing techniques have in fact been developed to facilitate investigation of methylation (308). For example, single-cell nucleosome, methylation and transcription sequencing (scNMT sequencing), combining epigenome and transcriptome data, are able to detect several “layers” of epigenomic and molecular dynamic coupling processes (309). Psychiatric disorders are more regarded as network dysfunctions (310). As mentioned above, focusing on only one cell type, brain area or neuronal pathway may not be sufficient. Thus, a combination of single cell sequencing and a pathway approach to the analysis of methylation patterns similar to network analysis in genomics (as exemplified by weighted gene coexpression network analysis or WGCNA) could be fruitful in this field.
Furthermore, the assay for transposase-accessible chromatin by sequencing (ATAC-seq) is able to get access to DNA sequences in open chromatin and to produce high quality data with a low background in a high-through output way (311). When being used at the single cell level, ATAC-seq detects DNA regulatory variations, e.g., trans-factors, cis-elements, which have been associated with induction or suppression of cell-to-cell variability. Such DNA variation data can be combined with chromatin accessibility and thus form a three-dimensional informative “regulome” in the genome (312). The concept of “connectomics” put forward by Fornito and colleagues, may also benefit this field of research (313). “Connectomics” was originally characterized as brain-network topological regulation of neural activities after injury (313). The combination of the different “omic,” such as genomic, epigenomic, transcriptomic, and even connectomics studies, may form interesting perspectives about how genetic/epigenetic and their molecular and topological mechanisms impact different cells and brain areas, and thus, stress-related phenotypes. So far, combined “omic” studies such as the combination of GWAS data with enhancer enrichment profiles, RNA sequencing data (RNA-seq) and chromatin status have been utilized (314). The integration of in vitro cell culture and multi “omic” analysis in the investigation of human germline epigenome reprogramming has been reported, producing some hints about the origin of neuropsychiatric disorders and transgenerational inheritance (315, 316).
In summary, by using new technologies, “omic” analysis and “big data”-integration of data from different platforms in a system biology approach-bias will be reduced and understanding of molecular mechanisms will be deepened (317). In the future, integration of genomics, epigenomics, transcriptomics, proteomics, metabolomics, regulomics, and connectomics could shed light on both basic biological processes in response to childhood trauma and disorder-related mechanisms, and thereby produce innovations in mental health and addiction health service provision.
SJ conducted the literature review and drafted the paper for a course (MDGEN605). LP, EB and AC reviewed the manuscript and provided some text and suggested edits. KA reviewed the manuscript, discussed with SJ, provided some text and suggested edits.
An Alberta Innovates Health Solutions Team Grant (Collaborative Research & Innovation Opportunities (CRIO) Population Resiliency Grant Prediction and Understanding of Resilience in Albertan Families: Longitudinal Study of Disaster Responses (PURLS)) contributed to the training of the first author.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
SJ would like to thank Dr Alan Underhill for his helpful steering and discussion of reading material by way of background material for this review, and sincerely appreciate the support from Margaret Pike during the submission process.
1. Hornor G. Childhood trauma exposure and toxic stress: what the pnp needs to know. J Pediatr Health Car (2015) 29(2):191–8. doi: 10.1016/j.pedhc.2014.09.006
2. Bucci M, Marques SS, Oh D, Harris NB. Toxic stress in children and adolescents. Adv Pediatr (2016) 63(1):403–28. doi: 10.1016/j.yapd.2016.04.002
3. Koenen KC, Ratanatharathorn A, Ng L, McLaughlin KA, Bromet EJ, Stein DJ, et al. Posttraumatic stress disorder in the world mental health Surveys. Psychol Med (2017) 47(13):2260–74. doi: 10.1017/S0033291717000708
4. Danese A, Moffitt TE, Harrington H, Milne BJ, Polanczyk G, Pariante CM, et al. Adverse childhood experiences and adult risk factors for age-related disease: depression, inflammation, and clustering of metabolic risk markers. Arch Pediatr Adolesc Med (2009) 163(12):1135–43. doi: 10.1001/archpediatrics.2009.214
5. Brown DW, Anda RF, Tiemeier H, Felitti VJ, Edwards VJ, Croft JB, et al. Adverse childhood experiences and the risk of premature mortality. Am J Prev Med (2009) 37(5):389–96. doi: 10.1016/j.amepre.2009.06.021
6. Murphy MO, Cohn DM, Loria AS. Developmental origins of cardiovascular disease: impact of early life stress in humans and rodents. Neurosci Biobehav Rev (2017) 74(Pt B):453–65. doi: 10.1016/j.neubiorev.2016.07.018
7. Dube SR, Fairweather D, Pearson WS, Felitti VJ, Anda RF, Croft JB. Cumulative childhood stress and autoimmune diseases in adults. Psychosom Med (2009) 71(2):243–50. doi: 10.1097/PSY.0b013e3181907888
8. Park SH, Videlock EJ, Shih W, Presson AP, Mayer EA, Chang L. Adverse childhood experiences are associated with irritable bowel syndrome and gastrointestinal symptom severity. Neurogastroenterology motility: official J Eur Gastrointestinal Motility Soc (2016) 28(8):1252–60. doi: 10.1111/nmo.12826
9. Bright MA, Alford SM, Hinojosa MS, Knapp C, Fernandez-Baca DE. Adverse childhood experiences and dental health in children and adolescents. Community Dent Oral Epidemiol (2015) 43(3):193–9. doi: 10.1111/cdoe.12137
10. Thomas C, Hypponen E, Power C. Obesity and type 2 diabetes risk in midadult life: the role of childhood adversity. Pediatrics (2008) 121(5):e1240–9. doi: 10.1542/peds.2007-2403
11. Green JG, McLaughlin KA, Berglund PA, Gruber MJ, Sampson NA, Zaslavsky AM, et al. Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication I: associations with first onset of DSM-IV disorders. Arch Gen Psychiatry (2010) 67(2):113–23. doi: 10.1001/archgenpsychiatry.2009.186
12. Alisic E, Zalta AK, van Wesel F, Larsen SE, Hafstad GS, Hassanpour K, et al. Rates of post-traumatic stress disorder in trauma-exposed children and adolescents: meta-analysis. Br J Psychiatry (2014) 204(5):335–40. doi: 10.1192/bjp.bp.113.131227
13. Bader K, Schafer V, Schenkel M, Nissen L, Schwander J. Adverse childhood experiences associated with sleep in primary insomnia. J sleep Res (2007) 16(3):285–96. doi: 10.1111/j.1365-2869.2007.00608.x
14. Mayer SE, Abelson JL, Lopez-Duran NL, Briggs H, Young EA. The roles of trauma exposure and timing and anxiety comorbidity in shaping HPA axis patterns in depression. Psychoneuroendocrino (2016) 71:68–. doi: 10.1016/j.psyneuen.2016.07.176
15. Williams LM, Debattista C, Duchemin AM, Schatzberg AF, Nemeroff CB. Childhood trauma predicts antidepressant response in adults with major depression: data from the randomized international study to predict optimized treatment for depression. Transl Psychiatry (2016) 6(5):e799. doi: 10.1038/tp.2016.61
16. Heim C, Newport DJ, Mletzko T, Miller AH, Hemeroff CB. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrino (2008) 33(6):693–710. doi: 10.1016/j.psyneuen.2008.03.008
17. Aas M, Henry C, Bellivier F, Lajnef M, Gard S, Kahn JP, et al. Affective lability mediates the association between childhood trauma and suicide attempts, mixed episodes and co-morbid anxiety disorders in bipolar disorders. psychol Med (2017) 47(5):902–12. doi: 10.1017/S0033291716003081 PubMed PMID: WOS:000396305100009
18. Daruy-Filho L, Brietzke E, Lafer B, Grassi-Oliveira R. Childhood maltreatment and clinical outcomes of bipolar disorder. Acta Psychiatr Scand (2011) 124(6):427–34. doi: 10.1111/j.1600-0447.2011.01756.x
19. Somer E, Herscu O. Childhood trauma, social anxiety, absorption and fantasy dependence: two potential mediated pathways to maladaptive daydreaming. J Addictive Behaviors Ther Rehabil (2018) 6(4):1–5. doi: 10.4172/2324-9005.1000170
20. Whitfield CL, Dube SR, Felitti VJ, Anda RF. Adverse childhood experiences and hallucinations. Child Abuse Negl (2005) 29(7):797–810. doi: 10.1016/j.chiabu.2005.01.004
21. Cattane N, Rossi R, Lanfredi M, Cattaneo A. Borderline personality disorder and childhood trauma: exploring the affected biological systems and mechanisms. BMC Psychiatry (2017) 17(1):221. doi: 10.1186/s12888-017-1383-2
22. van der Kolk BA, Perry JC, Herman JL. Childhood origins of self-destructive behavior. Am J Psychiatry (1991) 148(12):1665–71. doi: 10.1176/ajp.148.12.1665
23. Kendall-Tackett K. The health effects of childhood abuse: four pathways by which abuse can influence health. Child Abuse Neglect (2002) 26(6-7):715–29. doi: 10.1016/s0145-2134(02)00343-5
24. London S, Quinn K, Scheidell JD, Frueh BC, Khan MR. Adverse experiences in childhood and sexually transmitted infection risk from adolescence into adulthood. Sexually transmitted Dis (2017) 44(9):524–32. doi: 10.1097/olq.0000000000000640
25. Wu NS, Schairer LC, Dellor E, Grella C. Childhood trauma and health outcomes in adults with comorbid substance abuse and mental health disorders. Addict Behav (2010) 35(1):68–71. doi: 10.1016/j.addbeh.2009.09.003
26. Dube SR, Miller JW, Brown DW, Giles WH, Felitti VJ, Dong M, et al. Adverse childhood experiences and the association with ever using alcohol and initiating alcohol use during adolescence. J Adolesc Health (2006) 38(4):444. e1–. e10. doi: 10.1016/j.jadohealth.2005.06.006
27. Ducci F, Enoch MA, Hodgkinson C, Xu K, Catena M, Robin RW, et al. Interaction between a functional MAOA locus and childhood sexual abuse predicts alcoholism and antisocial personality disorder in adult women. Mol Psychiatr (2008) 13(3):334–47. doi: 10.1038/sj.mp.4002034
28. Monteleone AM, Monteleone P, Serino I, Scognamiglio P, Di Genio M, Maj M. Childhood trauma and cortisol awakening response in symptomatic patients with anorexia nervosa and bulimia nervosa. Int J Eat Disord (2015) 48(6):615–21. doi: 10.1002/eat.22375
29. Monteleone AM, Monteleone P, Esposito F, Prinster A, Ruzzi V, Canna A, et al. The effects of childhood maltreatment on brain structure in adults with eating disorders. World J Biol Psychiatry (2017), 20(4):1–10. doi: 10.1080/15622975.2017.1395071
30. Henderson J, Denny K. The resilient child, human development and the “postdemocracy”. BioSocieties (2015) 10(3):352–78. doi: 10.1057/biosoc.2015.24
31. Sheerin CM, Lind MJ, Bountress KE, Nugent NR, Amstadter AB. The genetics and epigenetics of PTSD: overview, recent advances, and future directions. Curr Opin Psychol (2017) 14:5–11. doi: 10.1016/j.copsyc.2016.09.003
32. Wolf EJ, Miller MW, Sullivan DR, Amstadter AB, Mitchell KS, Goldberg J, et al. A classical twin study of PTSD symptoms and resilience: evidence for a single spectrum of vulnerability to traumatic stress. Depression Anxiety (2018) 35(2):132–9. doi: 10.1002/da.22712
33. Amstadter AB, Maes HH, Sheerin CM, Myers JM, Kendler KS. The relationship between genetic and environmental influences on resilience and on common internalizing and externalizing psychiatric disorders. Soc Psych Psych Epid (2016) 51(5):669–78. doi: 10.1007/s00127-015-1163-6
34. Amstadter AB, Moscati A, Oxon MA, Maes HH, Myers JM, Kendler KS. Personality, cognitive/psychological traits and psychiatric resilience: a multivariate twin study. Pers Individ Dif (2016) 91:74–9. doi: 10.1016/j.paid.2015.11.041
35. Corella D, Ordovas JM. Basic concepts in molecular biology related to genetics and epigenetics. Rev Esp Cardiol (Engl Ed) (2017) 70(9):744–53. doi: 10.1016/j.rec.2017.05.011
36. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet (2003) 33:245–54. doi: 10.1038/ng1089
37. McKittrick E, Gaften PR, Ahmad K, Henikoff S. Histone H3.3 is enriched in covalent modifications associated with active chromatin. P Natl Acad Sci USA (2004) 101(6):1525–30. doi: 10.1073/pnas.0308092100
38. Cremer T, Cremer M, Dietzel S, Muller S, Solovei I, Fakan S. Chromosome territories - a functional nuclear landscape. Curr Opin Cell Biol (2006) 18(3):307–16. doi: 10.1016/j.ceb.2006.04.007
39. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature (2004) 429(6990):457–63. doi: 10.1038/nature02625
40. Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity (Edinb) (2010) 105(1):4–13. doi: 10.1038/hdy.2010.54
41. Bird A. The essentials of DNA methylation. Cell (1992) 70(1):5–8. doi: 10.1016/0092-8674(92)90526-i
42. Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature (2000) 406(6796):593–9. doi: 10.1038/35020506
43. Wade PA. Methyl CpG-binding proteins and transcriptional repression. Bioessays (2001) 23(12):1131–7. doi: 10.1002/bies.10008
44. Nan X, Ng H-H, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature (1998) 393(6683):386. doi: 10.1038/30764
45. Eden S, Constancia M, Hashimshony T, Dean W, Goldstein B, Johnson AC, et al. An upstream repressor element plays a role in Igf2 imprinting. EMBO J (2001) 20(13):3518–25. doi: 10.1093/emboj/20.13.3518
46. Sharma S, De Carvalho DD, Jeong S, Jones PA, Liang G. Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PloS Genet (2011) 7(2):e1001286. doi: 10.1371/journal.pgen.1001286
47. Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet (2001) 17(9):520–7. doi: 10.1016/S0168-9525(01)02366-6
48. Strong E, Butcher DT, Singhania R, Mervis CB, Morris CA, De Carvalho D, et al. Symmetrical dose-dependent dna-methylation profiles in children with deletion or duplication of 7q11.23. Am J Hum Genet (2015) 97(2):216–27. doi: 10.1016/j.ajhg.2015.05.019
49. Rizzardi LF, Hickey PF, Rodriguez DiBlasi V, Tryggvadottir R, Callahan CM, Idrizi A, et al. Neuronal brain-region-specific DNA methylation and chromatin accessibility are associated with neuropsychiatric trait heritability. Nat Neurosci (2019) 22(2):307–16. doi: 10.1038/s41593-018-0297-8
50. Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet (2012) 13(1):7. doi: 10.1038/nrg3080
51. Spiers H, Hannon E, Schalkwyk LC, Bray NJ, Mill J. 5-hydroxymethylcytosine is highly dynamic across human fetal brain development. BMC Genomics (2017) 18(1):738. doi: 10.1186/s12864-017-4091-x
52. Hack LM, Dick AL, Provençal N. Epigenetic mechanisms involved in the effects of stress exposure: focus on 5-hydroxymethylcytosine. Environ epigenetics (2016) 2(3):1–7. doi: 10.1093/eep/dvw016
53. Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev (1998) 12(5):599–606. doi: 10.1101/gad.12.5.599
55. Cavalli G, Misteli T. Functional implications of genome topology. Nat Struct Mol Biol (2013) 20(3):290. doi: 10.1038/nsmb.2474
56. Kaikkonen MU, Lam MTY, Glass CK. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res (2011) 90(3):430–40. doi: 10.1093/cvr/cvr097
57. Kundu P, Fabian MR, Sonenberg N, Bhattacharyya SN, Filipowicz W. HuR protein attenuates miRNA-mediated repression by promoting miRISC dissociation from the target RNA. Nucleic Acids Res (2012) 40(11):5088–100. doi: 10.1093/nar/gks148
58. Du KZ, Zhang LB, Lee T, Sun T. m(6)A RNA methylation controls neural development and is involved in human diseases. Mol Neurobiol (2019) 56(3):1596–606. doi: 10.1007/s12035-018-1138-1
59. Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature (2007) 447(7143):425–32. doi: 10.1038/nature05918
60. Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet (2002) 3(9):662–73. doi: 10.1038/nrg887
61. Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet (2001) 27(1):31–9. doi: 10.1038/83730
62. Massicotte R, Whitelaw E, Angers B. DNA methylation: a source of random variation in natural populations. Epigenetics (2011) 6(4):421–7. doi: 10.4161/epi.6.4.14532
63. Guerrero-Bosagna CM, Skinner MK. Environmental epigenetics and phytoestrogen/phytochemical exposures. J Steroid Biochem Mol Biol (2014) 139:270–6. doi: 10.1016/j.jsbmb.2012.12.011
64. Goossens L, van Roekel E, Verhagen M, Cacioppo JT, Cacioppo S, Maes M, et al. The genetics of loneliness: linking evolutionary theory to genome-wide genetics, epigenetics, and social science. Perspect Psychol Sci (2015) 10(2):213–26. doi: 10.1177/1745691614564878
65. Skinner MK. Environmental epigenetics and a unified theory of the molecular aspects of evolution: a neo-lamarckian concept that facilitates neo-darwinian evolution. Genome Biol Evol (2015) 7(5):1296–302. doi: 10.1093/gbe/evv073
66. Rius M, Lyko F. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene (2012) 31(39):4257. doi: 10.1038/onc.2011.601
67. Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes (2009) 58(12):2718–25. doi: 10.2337/db09-1003
68. Ballestar E. Epigenetic alterations in autoimmune rheumatic diseases. Nat Rev Rheumatology (2011) 7(5):263. doi: 10.1038/nrrheum.2011.16
69. Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY) (2015) 7(12):1198. doi: 10.18632/aging.100864
70. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet (1999) 23(2):185. doi: 10.1038/13810
71. Resmini E, Santos A, Aulinas A, Webb SM, Vives-Gilabert Y, Cox O, et al. Reduced DNA methylation of FKBP5 in Cushing’s syndrome. Endocrine (2016) 54(3):768–77. doi: 10.1007/s12020-016-1083-6
72. Höhne N, Poidinger M, Merz F, Pfister H, Brückl T, Zimmermann P, et al. FKBP5 genotype-dependent DNA methylation and mRNA regulation after psychosocial stress in remitted depression and healthy controls. Int J Neuropsychopharmacol (2015) 18(4):pyu087. doi: 10.1093/ijnp/pyu087
73. Lodhi RJ, Rossolatos D, Aitchison KJ. Genetics and genomics in addiction research. SAGE Handbook Drug Alcohol Studies: Biol Approaches (2016) 1:3–36. doi: 10.4135/9781473922143
74. Beach SRH, Brody GH, Todorov AA, Gunter TD, Philibert RA. Methylation at 5htt mediates the impact of child sex abuse on women’s antisocial behavior: an examination of the iowa adoptee sample. Psychosomatic Med (2011) 73(1):83–7. doi: 10.1097/PSY.0b013e3181fdd074
75. Weder N, Zhang H, Jensen K, Yang BZ, Simen A, Jackowski A, et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J Am Acad Child Adolesc Psychiatry (2014) 53(4):417–24. doi: 10.1016/j.jaac.2013.12.025
76. Dunn EC, Soare TW, Zhu Y, Simpkin AJ, Suderman MJ, Klengel T, et al. Sensitive periods for the effect of childhood adversity on DNA methylation: results from a prospective, longitudinal study. Biol Psychiatry (2019) 85(10):838–49. doi: 10.1016/j.biopsych.2018.12.023
77. Teicher MH, Samson JA, Anderson CM, Ohashi K. The effects of childhood maltreatment on brain structure, function and connectivity. Nat Rev Neurosci (2016) 17:652. doi: 10.1038/nrn.2016.111
78. Shonkoff JP, Boyce WT, McEwen BS. Neuroscience, molecular biology, and the childhood roots of health disparities: building a new framework for health promotion and disease prevention. JAMA (2009) 301(21):2252–9. doi: 10.1001/jama.2009.754
79. Joels M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci (2009) 10(6):459–66. doi: 10.1038/nrn2632
80. Franke HA. Toxic stress: effects, prevention and treatment. Children (Basel) (2014) 1(3):390–402. doi: 10.3390/children1030390
81. Cannon WB. The interrelations of emotions as suggested by recent physiological researches. Am J Psychol (1914) 25(2):256–82. doi: 10.2307/1413414
82. Selye H. Stress and the general adaptation syndrome. Br Med J (1950) 1(4667):1383–92. doi: 10.1136/bmj.1.4667.1383
83. Janak PH, Tye KM. From circuits to behaviour in the amygdala. Nature (2015) 517(7534):284–92. doi: 10.1038/nature14188
84. Roozendaal B, McEwen BS, Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci (2009) 10(6):423–33. doi: 10.1038/nrn2651
85. DiMicco JA, Samuels BC, Zaretskaia MV, Zaretsky DV. The dorsomedial hypothalamus and the response to stress: part renaissance, part revolution. Pharmacol Biochem Behav (2002) 71(3):469–80. doi: 10.1016/s0091-3057(01)00689-x
86. Buller KM. Neuroimmune stress responses: reciprocal connections between the hypothalamus and the brainstem. Stress (Amsterdam Netherlands) (2003) 6(1):11–7. doi: 10.1080/1025389031000092313
87. Korf J, Aghajanian GK, Roth RH. Increased turnover of norepinephrine in the rat cerebral cortex during stress: role of the locus coeruleus. Neuropharmacology (1973) 12(10):933–8. doi: 10.1016/0028-3908(73)90024-5
88. Danese A, McEwen BS. Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiol Behav (2012) 106(1):29–39. doi: 10.1016/j.physbeh.2011.08.019
89. Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci (2012) 13(1):22–37. doi: 10.1038/nrn3138
90. DeBold CR, Sheldon WR, DeCherney GS, Jackson RV, Alexander AN, Vale W, et al. Arginine vasopressin potentiates adrenocorticotropin release induced by ovine corticotropin-releasing factor. J Clin Invest (1984) 73(2):533–8. doi: 10.1172/JCI111240
91. Rotondo F, Butz H, Syro LV, Yousef GM, Di Ieva A, Restrepo LM, et al. Arginine vasopressin (AVP): a review of its historical perspectives, current research and multifunctional role in the hypothalamo-hypophysial system. Pituitary (2016) 19(4):345–55. doi: 10.1007/s11102-015-0703-0
92. Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci (2009) 10(6):459. doi: 10.1038/nrn2632
93. Suglia SF, Koenen KC, Boynton-Jarrett R, Chan PS, Clark CJ, Danese A, et al. Childhood and adolescent adversity and cardiometabolic outcomes: a scientific statement from the American Heart Association. Circulation (2018) 137(5):e15–28. doi: 10.1161/CIR.0000000000000536
94. Danese A, Baldwin JR. Hidden wounds? inflammatory links between childhood trauma and psychopathology. Annu Rev Psychol (2017) 68(1):517–44. doi: 10.1146/annurev-psych-010416-044208
95. Witt SH, Buchmann AF, Blomeyer D, Nieratschker V, Treutlein J, Esser G, et al. An interaction between a neuropeptide Y gene polymorphism and early adversity modulates endocrine stress responses. Psychoneuroendocrino (2011) 36(7):1010–20. doi: 10.1016/j.psyneuen.2010.12.015
96. Bailey CR, Cordell E, Sobin SM, Neumeister A. Recent progress in understanding the pathophysiology of post-traumatic stress disorder: implications for targeted pharmacological treatment. CNS Drugs (2013) 27(3):221–32. doi: 10.1007/s40263-013-0051-4
97. Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol (2004) 44:525–57. doi: 10.1146/annurev.pharmtox.44.101802.121410
98. Kuhlman KR, Geiss EG, Vargas I, Lopez-Duran N. HPA-axis activation as a key moderator of childhood trauma exposure and adolescent mental health. J Abnormal Child Psychol (2018) 46(1):149–57. doi: 10.1007/s10802-017-0282-9
99. Roper LJ, Purdon SE, Aitchison KJ. Childhood and later life stressors and psychosis. Clin Neuropsychiatr (2015) 12(6):148–56.
100. Monteleone AM, Monteleone P, Volpe U, De Riso F, Fico G, Giugliano R, et al. Impaired cortisol awakening response in eating disorder women with childhood trauma exposure: evidence for a dose-dependent effect of the traumatic load. Psychol Med (2018) 48(6):952–60. doi: 10.1017/s0033291717002409
101. Roper LJ. Delineating factors associated with vulnerability to psychosis in young people. [master’s thesis]. [Edmonton(AB)]: University of Alberta (2015) doi: 10.7939/R3GB1XT99
102. Labonté B, Suderman M, Maussion G, Navaro L, Yerko V, Mahar I, et al. Genome-wide epigenetic regulation by early-life trauma. Arch Gen Psychiatry (2012) 69(7):722–31. doi: 10.1001/archgenpsychiatry.2011.2287
103. Lutz PE, Tanti A, Gasecka A, Barnett-Burns S, Kim JJ, Zhou Y, et al. Association of a history of child abuse with impaired myelination in the anterior cingulate cortex: convergent epigenetic, transcriptional, and morphological evidence. Am J Psychiatry (2017) 174(12):1185–94. doi: 10.1176/appi.ajp.2017.16111286
104. Tomassi S, Tosato S. Epigenetics and gene expression profile in first-episode psychosis: the role of childhood trauma. Neurosci Biobehav Rev (2017) 83:226–37. doi: 10.1016/j.neubiorev.2017.10.018
105. Misiak B, Szmida E, Karpiński P, Loska O, Sąsiadek MM, Frydecka D. Lower LINE-1 methylation in first-episode schizophrenia patients with the history of childhood trauma. Epigenomics (2015) 7(8):1275–85. doi: 10.2217/epi.15.68
106. Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics (2015) 10(11):1024–32. doi: 10.1080/15592294.2015.1100786
107. Edgar RD, Jones MJ, Meaney MJ, Turecki G, Kobor MS. BECon: a tool for interpreting DNA methylation findings from blood in the context of brain. Transl Psychiatry (2017) 7(8):e1187. doi: 10.1038/tp.2017.171
108. Bearer EL, Mulligan BS. Epigenetic changes associated with early life experiences: saliva, a biospecimen for dna methylation signatures. Curr Genomics (2018) 19(8):676–98. doi: 10.2174/1389202919666180307150508
109. Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci (2013) 16(1):33–41. doi: 10.1038/nn.3275
110. Deng Q, Riquelme D, Trinh L, Low MJ, Tomić M, Stojilkovic S, et al. Rapid glucocorticoid feedback inhibition of ACTH secretion involves ligand-dependent membrane association of glucocorticoid receptors. Endocrinology (2015) 156(9):3215–27. doi: 10.1210/EN.2015-1265
111. Merkulov VM, Merkulova TI, Bondar NP. Mechanisms of brain glucocorticoid resistance in stress-induced psychopathologies. Biochem Biokhimiia (2017) 82(3):351–65. doi: 10.1134/s0006297917030142
112. Klengel T, Binder EB. Allele-specific epigenetic modification: a molecular mechanism for gene-environment interactions in stress-related psychiatric disorders? Epigenomics (2013) 5(2):109–12. doi: 10.2217/epi.13.11
113. Hubler TR, Scammell JG. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones (2004) 9(3):243–52. doi: 10.1379/csc-32r.1
114. Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrino (2009) 34 Suppl 1:S186–95. doi: 10.1016/j.psyneuen.2009.05.021
115. Matosin N, Halldorsdottir T, Binder EB. Understanding the molecular mechanisms underpinning gene by environment interactions in psychiatric disorders: the fkbp5 model. Biol Psychiatry (2018) 83(10):821–30. doi: 10.1016/j.biopsych.2018.01.021
116. Klengel T, Binder EB. FKBP5 allele-specific epigenetic modification in gene by environment interaction. Neuropsychopharmacology (2015) 40(1):244–6. doi: 10.1038/npp.2014.208
117. Tozzi L, Farrell C, Booij L, Doolin K, Nemoda Z, Szyf M, et al. Epigenetic changes of fkbp5 as a link connecting genetic and environmental risk factors with structural and functional brain changes in major depression. Neuropsychopharmacology (2018) 43(5):1138–45. doi: 10.1038/npp.2017.290
118. Petrides M. Lateral prefrontal cortex: architectonic and functional organization. Philos T Roy Soc B (2005) 360(1456):781–95. doi: 10.1098/rstb.2005.1631
119. Shin LM, McNally RJ, Kosslyn SM, Thompson WL, Rauch SL, Alpert NM, et al. Regional cerebral blood flow during script-driven imagery in childhood sexual abuse-related PTSD: A PET investigation. Am J Psychiatry (1999) 156(4):575–84. doi: 10.1176/ajp.156.4.575
120. Linnstaedt SD, Riker KD, Rueckeis CA, Kutchko KM, Lackey L, McCarthy KR, et al. A Functional riboSNitch in the 3’ untranslated region of fkbp5 alters microrna-320a binding efficiency and mediates vulnerability to chronic post-traumatic pain. J Neurosci: official J Soc Neurosci (2018) 38(39):8407–20. doi: 10.1523/JNEUROSCI.3458-17.2018
121. Hotamisligil GS, Breakefield XO. Human monoamine oxidase A gene determines levels of enzyme activity. Am J Hum Genet (1991) 49(2):383–92.
122. Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev (2008) 60(13-14):1527–33. doi: 10.1016/j.addr.2008.06.002
123. Brunner HG, Nelen M, Breakefield XO, Ropers HH, van Oost BA. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science (5133) (1993) 262:578–80. doi: 10.1126/science.8211186
124. McDermott R, Tingley D, Cowden J, Frazzetto G, Johnson DD. Monoamine oxidase A gene (MAOA) predicts behavioral aggression following provocation. Proc Natl Acad Sci (2009) 106(7):2118–23. doi: 10.1073/pnas.0808376106
125. Deckert J, Catalano M, Syagailo YV, Bosi M, Okladnova O, Di Bella D, et al. Excess of high activity monoamine oxidase A gene promoter alleles in female patients with panic disorder. Hum Mol Genet (1999) 8(4):621–4. doi: 10.1093/hmg/8.4.621
126. Desautels A, Turecki G, Montplaisir J, Brisebois K, Sequeira A, Adam B, et al. Evidence for a genetic association between monoamine oxidase A and restless legs syndrome. Neurology (2002) 59(2):215–9. doi: 10.1212/wnl.59.2.215
127. Das M, Das Bhowmik A, Sinha S, Chattopadhyay A, Chaudhuri K, Singh M, et al. MAOA promoter polymorphism and attention deficit hyperactivity disorder (ADHD) in Indian children. Am J Med Genet Part B: Neuropsychiatr Genet (2006) 141(6):637–42. doi: 10.1002/ajmg.b.30385
128. Hannan AJ. Tandem repeats mediating genetic plasticity in health and disease. Nat Rev Genet (2018) 19:286. doi: 10.1038/nrg.2017.115
129. Philibert RA, Wernett P, Plume J, Packer H, Brody GH, Beach SR. Gene environment interactions with a novel variable Monoamine Oxidase A transcriptional enhancer are associated with antisocial personality disorder. Biol Psychol (2011) 87(3):366–71. doi: 10.1016/j.biopsycho.2011.04.007
130. Melas PA, Wei Y, Wong CC, Sjoholm LK, Aberg E, Mill J, et al. Genetic and epigenetic associations of MAOA and NR3C1 with depression and childhood adversities. Int J Neuropsychopharmacol (2013) 16(7):1513–28. doi: 10.1017/S1461145713000102
131. Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science (2002) 297(5582):851–4. doi: 10.1126/science.1072290
132. Sabol SZ, Hu S, Hamer D. A functional polymorphism in the monoamine oxidase A gene promoter. Hum Genet (1998) 103(3):273–9. doi: 10.1007/s004390050816
133. Reif A, Rösler M, Freitag CM, Schneider M, Eujen A, Kissling C, et al. Nature and nurture predispose to violent behavior: serotonergic genes and adverse childhood environment. Neuropsychopharmacology (2007) 32(11):2375. doi: 10.1038/sj.npp.1301359
134. Peng H, Zhu Y, Strachan E, Fowler E, Bacus T, Roy-Byrne P, et al. Childhood trauma, DNA methylation of stress-related genes, and depression: findings from two monozygotic twin studies. Psychosom Med (2018) 80(7):599–608. doi: 10.1097/psy.0000000000000604
135. Majer-Łobodzińska A, Adamiec-Mroczek J. Glucocorticoid receptor polymorphism in obesity and glucose homeostasis. Adv Clin Exp Med: official Organ Wroclaw Med University (2017) 26(1):143–8. doi: 10.17219/acem/41231
136. DeRijk RH, van Leeuwen N, Klok MD, Zitman FG. Corticosteroid receptor-gene variants: modulators of the stress-response and implications for mental health. Eur J Pharmacol (2008) 585(2-3):492–501. doi: 10.1016/j.ejphar.2008.03.012
137. McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci (2009) 12(3):342–8. doi: 10.1038/nn.2270
138. Palma-Gudiel H, Cordova-Palomera A, Leza JC, Fananas L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: a critical review. Neurosci Biobehav R (2015) 55:520–35. doi: 10.1016/j.neubiorev.2015.05.016
139. Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci (2004) 7(8):847–54. doi: 10.1038/nn1276
140. Weaver IC, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ, et al. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci: official J Soc Neurosci (2005) 25(47):11045–54. doi: 10.1523/JNEUROSCI.3652-05.2005
141. Lutz P-E, Almeida D, Fiori LM, Turecki G. Childhood maltreatment and stress-related psychopathology: the epigenetic memory hypothesis. Curr Pharm Des (2015) 21(11):1413–7. doi: 10.2174/1381612821666150105124928
142. Van Der Knaap L, Riese H, Hudziak J, Verbiest M, Verhulst F, Oldehinkel A, et al. Glucocorticoid receptor gene (NR3C1) methylation following stressful events between birth and adolescence. The TRAILS study. Trans Psychiatry (2014) 4(4):e381. doi: 10.1038/tp.2014.22
143. Labonte B, Yerko V, Gross J, Mechawar N, Meaney MJ, Szyf M, et al. Differential glucocorticoid receptor exon 1(B), 1(C), and 1(H) expression and methylation in suicide completers with a history of childhood abuse. Biol Psychiatry (2012) 72(1):41–8. doi: 10.1016/j.biopsych.2012.01.034
144. Turecki G, Meaney MJ. Effects of the social environment and stress on glucocorticoid receptor gene methylation: a systematic review. Biol Psychiatry (2016) 79(2):87–96. doi: 10.1016/j.biopsych.2014.11.022
145. Vukojevic V, Kolassa I-T, Fastenrath M, Gschwind L, Spalek K, Milnik A, et al. Epigenetic modification of the glucocorticoid receptor gene is linked to traumatic memory and post-traumatic stress disorder risk in genocide survivors. J Neurosci (2014) 34(31):10274–84.
146. Schmidt M, Lax E, Zhou R, Cheishvili D, Ruder AM, Ludiro A, et al. Fetal glucocorticoid receptor (Nr3c1) deficiency alters the landscape of DNA methylation of murine placenta in a sex-dependent manner and is associated to anxiety-like behavior in adulthood. Trans Psychiatry (2019) 9(1):23. doi: 10.1038/s41398-018-0348-7
147. Li S, Papale LA, Kintner DB, Sabat G, Barrett-Wilt GA, Cengiz P, et al. Hippocampal increase of 5-hmC in the glucocorticoid receptor gene following acute stress. Behavioural Brain Res (2015) 286:236–40. doi: 10.1016/j.bbr.2015.03.002
148. Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med (2009) 60:355–66. doi: 10.1146/annurev.med.60.042307.110802
149. Nordquist N, Oreland L. Serotonin, genetic variability, behaviour, and psychiatric disorders–a review. Ups J Med Sci (2010) 115(1):2–10. doi: 10.3109/03009730903573246
150. Jacobs BL, Azmitia EC. Structure and function of the brain serotonin system. Physiol Rev (1992) 72(1):165–229. doi: 10.1152/physrev.1992.72.1.165
151. Van Loon GR, Shum A, Sole MJ. Decreased brain serotonin turnover after short term (two-hour) adrenalectomy in rats: a comparison of four turnover methods. Endocrinology (1981) 108(4):1392–402. doi: 10.1210/endo-108-4-1392
152. Browne CA, Clarke G, Dinan TG, Cryan JF. Differential stress-induced alterations in tryptophan hydroxylase activity and serotonin turnover in two inbred mouse strains. Neuropharmacology (2011) 60(4):683–91. doi: 10.1016/j.neuropharm.2010.11.020
153. Oates JA, Sjoerdsma A. Neurologic effects of tryptophan in patients receiving a monoamine oxidase inhibitor. Neurology (1960) 10(12):1076–. doi: 10.1212/wnl.10.12.1076
154. Virkkunen M, Goldman D, Nielsen DA, Linnoila M. Low brain serotonin turnover rate (low CSF 5-HIAA) and impulsive violence. J Psychiatry Neurosci (1995) 20(4):271.
155. Barton DA, Esler MD, Dawood T, Lambert EA, Haikerwal D, Brenchley C, et al. Elevated brain serotonin turnover in patients with depression: effect of genotype and therapy. Arch Gen Psychiatry (2008) 65(1):38–46. doi: 10.1001/archgenpsychiatry.2007.11
156. Heiander A, Beck O, Boysen L. 5-Hydroxytryptophol conjugation in man: influence of alcohol consumption and altered serotonin turnover. Life Sci (1995) 56(18):1529–34. doi: 10.1016/0024-3205(95)00115-m
157. Albert PR, Lemonde S. 5-HT1A receptors, gene repression, and depression: guilt by association. Neuroscientist (2004) 10(6):575–93. doi: 10.1177/1073858404267382
158. Stoltenberg SF, Christ CC, Highland KB. Serotonin system gene polymorphisms are associated with impulsivity in a context dependent manner. Prog Neuropsychopharmacol Biol Psychiatry (2012) 39(1):182–91. doi: 10.1016/j.pnpbp.2012.06.012
159. Cornelis MC, Nugent NR, Amstadter AB, Koenen KC. Genetics of post-traumatic stress disorder: review and recommendations for genome-wide association studies. Curr Psychiatry Rep (2010) 12(4):313–26. doi: 10.1007/s11920-010-0126-6
160. Sigurdh J, Allard P, Spigset O, Hagglof B. Platelet serotonin transporter and 5-HT2A receptor binding in adolescents with eating disorders. Int J Neurosci (2013) 123(5):333–8. doi: 10.3109/00207454.2012.761215
161. Gouin J-P, Zhou Q, Booij L, Boivin M, Côté S, Hébert M, et al. Associations among oxytocin receptor gene (OXTR) DNA methylation in adulthood, exposure to early life adversity, and childhood trajectories of anxiousness. Sci Rep (2017) 7(1):7446. doi: 10.1038/s41598-017-07950-x
162. Blaya C, Salum GA, Moorjani P, Seganfredo AC, Heldt E, Leistner-Segal S, et al. Panic disorder and serotonergic genes (SLC6A4, HTR1A and HTR2A): association and interaction with childhood trauma and parenting. Neurosci Lett (2010) 485(1):11–5. doi: 10.1016/j.neulet.2010.08.042
163. Leve LD, Harold GT, Ge X, Neiderhiser JM, Shaw D, Scaramella LV, et al. Structured parenting of toddlers at high versus low genetic risk: Two pathways to child problems. J Am Acad Child Adolesc Psychiatry (2009) 48(11):1102–9. doi: 10.1097/CHI.0b013e3181b8bfc0
164. Shinozaki G, Romanowicz M, Mrazek DA, Kung S. HTR2A gene–child abuse interaction and association with a history of suicide attempt among Caucasian depressed psychiatric inpatients. J Affect Disord (2013) 150(3):1200–3. doi: 10.1016/j.jad.2013.05.028
165. Parade SH, Novick AM, Parent J, Seifer R, Klaver SJ, Marsit CJ, et al. Stress exposure and psychopathology alter methylation of the serotonin receptor 2A (HTR2A) gene in preschoolers. Dev Psychopathol (2017) 29(5):1619–26. doi: 10.1017/S0954579417001274
166. Abdolmaleky HM, Yaqubi S, Papageorgis P, Lambert AW, Ozturk S, Sivaraman V, et al. Epigenetic dysregulation of HTR2A in the brain of patients with schizophrenia and bipolar disorder. Schizophrenia Res (2011) 129(2):183–90. doi: 10.1016/j.schres.2011.04.007
167. Schechter DS, Moser DA, Pointet VC, Aue T, Stenz L, Paoloni-Giacobino A, et al. The association of serotonin receptor 3A methylation with maternal violence exposure, neural activity, and child aggression. Behavioural Brain Res (2017) 325(Pt B):268–77. doi: 10.1016/j.bbr.2016.10.009
168. Kang H-J, Kim J-M, Stewart R, Kim S-Y, Bae K-Y, Kim S-W, et al. Association of SLC6A4 methylation with early adversity, characteristics and outcomes in depression. Prog Neuropsychopharmacol Biol Psychiatry (2013) 44:23–8. doi: 10.1016/j.pnpbp.2013.01.006
169. Devlin AM, Brain U, Austin J, Oberlander TF. Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth. PloS One (2010) 5(8):e12201. doi: ARTN e12201 10.1371/journal.pone.0012201
170. Alexander N, Wankerl M, Hennig J, Miller R, Zänkert S, Steudte-Schmiedgen S, et al. DNA methylation profiles within the serotonin transporter gene moderate the association of 5-HTTLPR and cortisol stress reactivity. Trans Psychiatry (2014) 4:e443. doi: 10.1038/tp.2014.88 https://www.nature.com/articles/tp201488#supplementary-information
171. Lesch K-P, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science (1996) 274(5292):1527–31. doi: 10.1126/science.274.5292.1527
172. Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science (2003) 301(5631):386–9. doi: 10.1126/science.1083968
173. Zalsman G, Huang Y-y, Oquendo MA, Burke AK, Hu X-z, Brent DA, et al. Association of a triallelic serotonin transporter gene promoter region (5-HTTLPR) polymorphism with stressful life events and severity of depression. Am J Psychiatry (2006) 163(9):1588–93. doi: 10.1176/ajp.2006.163.9.1588
174. Birkenhäger TK, Geldermans S, Van den Broek WW, van Beveren N, Fekkes D. Serum brain-derived neurotrophic factor level in relation to illness severity and episode duration in patients with major depression. J. Psy Res. (2012) 46(3):285–9. doi: 10.1016/j.jpsychires.2011.12.006
175. Han YX, Tao C, Gao XR, Wang LL, Jiang FH, Wang C, et al. BDNF-related imbalance of copine 6 and synaptic plasticity markers couples with depression-like behavior and immune activation in CUMS rats. Front Neurosci (2018) 12:731. doi: 10.3389/fnins.2018.00731
176. Wei Y, Melas PA, Wegener G, Mathe AA, Lavebratt C. Antidepressant-like effect of sodium butyrate is associated with an increase in TET1 and in 5-hydroxymethylation levels in the Bdnf gene. Int J Neuropsychopharmacol (2014) 18(2):pyu032. doi: 10.1093/ijnp/pyu032
177. van Velzen LS, Schmaal L, Jansen R, Milaneschi Y, Opmeer EM, Elzinga BM, et al. Effect of childhood maltreatment and brain-derived neurotrophic factor on brain morphology. Soc Cogn Affect Neurosci (2016) 11(11):1841–52. doi: 10.1093/scan/nsw086
178. Mikics E, Guirado R, Umemori J, Toth M, Biro L, Miskolczi C, et al. Social learning requires plasticity enhanced by fluoxetine through prefrontal Bdnf-TrkB Signaling to limit aggression induced by post-weaning social isolation. Neuropsychopharmacology (2018) 43(2):235–45. doi: 10.1038/npp.2017.142
179. Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry (2009) 65(9):760–9. doi: 10.1016/j.biopsych.2008.11.028
180. Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res (2006) 1067(1):1–12. doi: 10.1016/j.brainres.2005.10.004
181. Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res (2007) 85(3):525–35. doi: 10.1002/jnr.21139
182. Benedetti F, Ambree O, Locatelli C, Lorenzi C, Poletti S, Colombo C, et al. The effect of childhood trauma on serum BDNF in bipolar depression is modulated by the serotonin promoter genotype. Neurosci Lett (2017) 656:177–81. doi: 10.1016/j.neulet.2017.07.043
183. Unternaehrer E, Meyer AH, Burkhardt SC, Dempster E, Staehli S, Theill N, et al. Childhood maternal care is associated with DNA methylation of the genes for brain-derived neurotrophic factor (BDNF) and oxytocin receptor (OXTR) in peripheral blood cells in adult men and women. Stress (Amsterdam Netherlands) (2015) 18(4):451–61. doi: 10.3109/10253890.2015.1038992
184. Wrigglesworth J, Ryan J, Vijayakumar N, Whittle S. Brain-derived neurotrophic factor DNA methylation mediates the association between neighborhood disadvantage and adolescent brain structure. Psychiatry Res Neuroimaging (2019) 285:51–7. doi: 10.1016/j.pscychresns.2018.12.012
185. Cicchetti D, Rogosch FA. Genetic moderation of child maltreatment effects on depression and internalizing symptoms by serotonin transporter linked polymorphic region (5-HTTLPR), brain-derived neurotrophic factor (BDNF), norepinephrine transporter (NET), and corticotropin releasing hormone receptor 1 (CRHR1) genes in African American children. Dev Psychopathol (2014) 26(4 Pt 2):1219–39. doi: 10.1017/s0954579414000984
186. Nobile M, Rusconi M, Bellina M, Marino C, Giorda R, Carlet O, et al. The influence of family structure, the TPH2 G-703T and the 5-HTTLPR serotonergic genes upon affective problems in children aged 10-14 years. J Child Psychol Psychiatry Allied disciplines (2009) 50(3):317–25. doi: 10.1111/j.1469-7610.2008.01958.x
187. Jeanneteau FD, Lambert WM, Ismaili N, Bath KG, Lee FS, Garabedian MJ, et al. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. Proc Natl Acad Sci (2012) 109(4):1305–10. doi: 10.1073/pnas.1114122109
188. Nöthling J, Malan-Müller S, Abrahams N, Hemmings SMJ, Seedat S. Epigenetic alterations associated with childhood trauma and adult mental health outcomes: a systematic review. World J Biol Psychiatry (2019), 1–58. (just-accepted). doi: 10.1080/15622975.2019.1583369
189. Fujisawa TX, Nishitani S, Takiguchi S, Shimada K, Smith AK, Tomoda A. Oxytocin receptor DNA methylation and alterations of brain volumes in maltreated children. Neuropsychopharmacology (2019) 1:2045–53. doi: 10.1038/s41386-019-0414-8
190. van Rooij SJ, Stevens JS, Ely TD, Fani N, Smith AK, Kerley KA, et al. Childhood trauma and COMT genotype interact to increase hippocampal activation in resilient individuals. Front Psychiatry (2016) 7:156. doi: 10.3389/fpsyt.2016.00156
191. Retz W, Rösler M, Kissling C, Wiemann S, Hünnerkopf R, Coogan A, et al. Norepinephrine transporter and catecholamine-O-methyltransferase gene variants and attention-deficit/hyperactivity disorder symptoms in adults. J Neural transmission (2008) 115(2):323–9. doi: 10.1007/s00702-007-0822-5
192. El-Hage W, Phillips ML, Radua J, Gohier B, Zelaya F, Collier D, et al. Genetic modulation of neural response during working memory in healthy individuals: interaction of glucocorticoid receptor and dopaminergic genes. Mol Psychiatr (2013) 18(2):174. doi: 10.1038/mp.2011.145
193. Gao S, Cheng J, Li G, Sun T, Xu Y, Wang Y, et al. Catechol-O-methyltransferase gene promoter methylation as a peripheral biomarker in male schizophrenia. Eur Psychiatry (2017) 44:39–46. doi: 10.1016/j.eurpsy.2017.03.002
194. Na K-S, Won E, Kang J, Kim A, Choi S, Tae W-S, et al. Differential effect of COMT gene methylation on the prefrontal connectivity in subjects with depression versus healthy subjects. Neuropharmacology (2018) 137:59–70. doi: 10.1016/j.neuropharm.2018.04.030
195. Munjiza A, Kostic M, Pesic D, Gajic M, Markovic I, Tosevski DL. Higher concentration of interleukin 6 - A possible link between major depressive disorder and childhood abuse. Psychiatry Res (2018) 264:26–30. doi: 10.1016/j.psychres.2018.03.072
196. Dennison U, McKernan D, Cryan J, Dinan T. Schizophrenia patients with a history of childhood trauma have a pro-inflammatory phenotype. psychol Med (2012) 42(9):1865–71. doi: 10.1017/S0033291712000074
197. Janusek LW, Tell D, Gaylord-Harden N, Mathews HL. Relationship of childhood adversity and neighborhood violence to a proinflammatory phenotype in emerging adult African American men: an epigenetic link. Brain Behav Immun (2017) 60:126–35. doi: 10.1016/j.bbi.2016.10.006
198. Fredericks CA, Drabant EM, Edge MD, Tillie JM, Hallmayer J, Ramel W, et al. Healthy young women with serotonin transporter SS polymorphism show a pro-inflammatory bias under resting and stress conditions. Brain Behav Immunity (2010) 24(3):350–7. doi: 10.1016/j.bbi.2009.10.014
199. Bale TL, Dorsa DM, Johnston CA. Oxytocin receptor mRNA expression in the ventromedial hypothalamus during the estrous cycle. J Neurosci (1995) 15(7):5058–64.
200. Davis M. Neurobiology of fear responses: the role of the amygdala. J neuropsychiatry Clin Neurosci (1997) 9(3):382–402. doi: 10.1176/jnp.9.3.382
201. Pedersen CA, Boccia ML. Oxytocin maintains as well as initiates female sexual behavior: effects of a highly selective oxytocin antagonist. Hormones Behav (2002) 41(2):170–7. doi: 10.1006/hbeh.2001.1736
202. Heim C, Young LJ, Newport DJ, Mletzko T, Miller AH, Nemeroff CB. Lower CSF oxytocin concentrations in women with a history of childhood abuse. Mol Psychiatry (2008) 14:954. doi: 10.1038/mp.2008.112
203. Feder A, Nestler EJ, Charney DS. Psychobiology and molecular genetics of resilience. Nat Rev Neurosci (2009) 10(6):446. doi: 10.1038/nrn2649
204. Rao P, Bell RL, Engleman EA, Sari Y. Targeting glutamate uptake to treat alcohol use disorders. Front Neurosci (2015) 9:144. doi: 10.3389/fnins.2015.00144
205. Goode TD, Leong K-C, Goodman J, Maren S, Packard MG. Enhancement of striatum-dependent memory by conditioned fear is mediated by beta-adrenergic receptors in the basolateral amygdala. Neurobiol stress (2016) 3:74–82. doi: 10.1016/j.ynstr.2016.02.004
206. Ding J, da Silva MS, Lingeman J, Chen X, Shi Y, Han F, et al. Late glucocorticoid receptor antagonism changes the outcome of adult life stress. Psychoneuroendocrinology (2019) 107:169–78. doi: 10.1016/j.psyneuen.2019.05.014
207. Charney DS. Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am J Psychiatry (2004) 161(2):195–216. doi: 10.1176/appi.ajp.161.2.195
208. Lee FJS, Xue S, Pei L, Vukusic B, Chéry N, Wang Y, et al. Dual Regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine d1 receptor. Cell (2002) 111(2):219–30. doi: 10.1016/S0092-8674(02)00962-5
209. Hermann H, Marsicano G, Lutz B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience (2002) 109(3):451–60. doi: 10.1016/S0306-4522(01)00509-7
210. Lu H-C, Mackie K. An introduction to the endogenous cannabinoid system. Biol Psychiatry (2016) 79(7):516–25. doi: 10.1016/j.biopsych.2015.07.028
211. McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem (2005) 74:411–32. doi: 10.1146/annurev.biochem.74.082803.133450
212. Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med (2003) 9(1):76. doi: 10.1038/nm803
213. Zhou Y, Falenta K, Lalli G. Endocannabinoid signalling in neuronal migration. Int J Biochem Cell Biol (2014) 47:104–8. doi: 10.1016/j.biocel.2013.12.007
214. Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature (2002) 418(6897):530. doi: 10.1038/nature00839
215. Walker JM, Huang SM, Strangman NM, Tsou K, Sañudo-Peña MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci (1999) 96(21):12198–203. doi: 10.1073/pnas.96.21.12198
216. Calignano A, La GR, Makriyannis A, Lin SY, Beltramo M, Piomelli D. Inhibition of intestinal motility by anandamide, an endogenous cannabinoid. Eur J Pharmacol (1997) 340(2-3):R7–8.
217. Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature (2001) 410(6828):588. doi: 10.1038/35069076
218. Dincheva I, Drysdale AT, Hartley CA, Johnson DC, Jing D, King EC, et al. FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat Commun (2015) 6:6395. doi: 10.1038/ncomms7395
219. Mahler SV, Smith KS, Berridge KC. Endocannabinoid hedonic hotspot for sensory pleasure: anandamide in nucleus accumbens shell enhances ‘liking’ of a sweet reward. Neuropsychopharmacology (2007) 32(11):2267. doi: 10.1038/sj.npp.1301376
220. Laplante P, Diorio J, Meaney MJ. Serotonin regulates hippocampal glucocorticoid receptor expression via a 5-HT7 receptor. Dev Brain Res (2002) 139(2):199–203. doi: 10.1016/S0165-3806(02)00550-3
221. Smythe JW, Rowe WB, Meaney MJ. Neonatal handling alters serotonin (5-HT) turnover and 5-HT2 receptor binding in selected brain regions: relationship to the handling effect on glucocorticoid receptor expression. Dev Brain Res (1994) 80(1):183–9. doi: 10.1016/0165-3806(94)90103-1
222. Belay H, Burton CL, Lovic V, Meaney MJ, Sokolowski M, Fleming AS. Early adversity and serotonin transporter genotype interact with hippocampal glucocorticoid receptor mRNA expression, corticosterone, and behavior in adult male rats. Behav Neurosci (2011) 125(2):150. doi: 10.1037/a0022891
223. Karanović J, Ivković M, Jovanović VM, Šviković S, Pantović-Stefanović M, Brkušanin M, et al. Effect of childhood general traumas on suicide attempt depends on TPH2 and ADARB1 variants in psychiatric patients. J Neural Transmission (2017) 124(5):621–9. doi: 10.1007/s00702-017-1677-z
224. Evanson NK, Herman JP. Role of paraventricular nucleus glutamate signaling in regulation of HPA axis stress responses. Interdisciplinary Inf Sci (2015) 21(3):253–60. doi: 10.4036/iis.2015.B.10
225. Lelli L, Castellini G, Cassioli E, Monteleone AM, Ricca V. Cortisol levels before and after cognitive behavioural therapy in patients with eating disorders reporting childhood abuse: a follow-up study. Psychiatry Res (2019) 275:269–75. doi: 10.1016/j.psychres.2019.03.046
226. Marzi SJ, Sugden K, Arseneault L, Belsky DW, Burrage J, Corcoran DL, et al. Analysis of DNA methylation in young people: limited evidence for an association between victimization stress and epigenetic variation in blood. Am J Psychiatry (2018) 175(6):517–29. doi: 10.1176/appi.ajp.2017.17060693
227. Niwa M, Cash-Padgett T, Kubo K-I, Saito A, Ishii K, Sumitomo A, et al. DISC1 a key molecular lead in psychiatry and neurodevelopment: No-More Disrupted-in-Schizophrenia 1. Mol Psychiatry (2016) 21(11):1488–9. doi: 10.1038/mp.2016.154
228. Kazachenka A, Bertozzi TM, Sjoberg-Herrera MK, Walker N, Gardner J, Gunning R, et al. Identification, characterization, and heritability of murine metastable epialleles: implications for non-genetic inheritance. Cell (2018) 175(5):1259–71. e13. doi: 10.1016/j.cell.2018.09.043
229. Molina PE. Neurobiology of the stress response: contribution of the sympathetic nervous system to the neuroimmune axis in traumatic injury. Shock (2005) 24(1):3–10. doi: 10.1097/01.shk.0000167112.18871.5c
230. Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci (2009) 10(6):397–409. doi: 10.1038/nrn2647
231. McEwen BS. In pursuit of resilience: stress, epigenetics, and brain plasticity. Ann New York Acad Sci (2016) 1373(1):56–64. doi: 10.1111/nyas.13020
232. Mika A, Mazur GJ, Hoffman AN, Talboom JS, Bimonte-Nelson HA, Sanabria F, et al. Chronic stress impairs prefrontal cortex-dependent response inhibition and spatial working memory. Behav Neurosci (2012) 126(5):605–19. doi: 10.1037/a0029642
233. Zannas AS, Wiechmann T, Gassen NC, Binder EB. Gene–Stress–Epigenetic regulation of FKBP5: clinical and translational implications. Neuropsychopharmacology (2015) 41:261. doi: 10.1038/npp.2015.235
234. Baldwin JR, Reuben A, Newbury JB, Danese A. Agreement between prospective and retrospective measures of childhood maltreatment: a systematic review and meta-analysis. JAMA Psychiatry (2019) 76(6):584–93. doi: 10.1001/jamapsychiatry.2019.0097
235. van der Kolk BA, Roth S, Pelcovitz D, Sunday S, Spinazzola J. Disorders of extreme stress: The empirical foundation of a complex adaptation to trauma. J Trauma Stress (2005) 18(5):389–99. doi: 10.1002/jts.20047
236. Friedman MJ, Kilpatrick DG, Schnurr PP, Weathers FW. Correcting misconceptions about the diagnostic criteria for posttraumatic stress disorder in DSM-5. JAMA Psychiatry (2016) 73(7):753–4. doi: 10.1001/jamapsychiatry.2016.0745
237. van der Kolk BA, Courtois CA. Editorial comments: complex developmental trauma. J Trauma Stress (2005) 18(5):385–8. doi: 10.1002/jts.20046
238. Cleasby IR, Nakagawa S, Schielzeth H. Quantifying the predictability of behaviour: statistical approaches for the study of between-individual variation in the within-individual variance. Methods Ecol Evolution (2015) 6(1):27–37. doi: 10.1111/2041-210X.12281
239. Chesler EJ, Lu L, Shou SM, Qu YH, Gu J, Wang JT, et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet (2005) 37(3):233–42. doi: 10.1038/ng1518
240. Hjemdal O, Roazzi A, Maria da Graça B, Friborg O. The cross-cultural validity of the resilience scale for adults: a comparison between norway and brazil. BMC Psychol (2015) 3(1):18. doi: 10.1186/s40359-015-0076-1
241. Edelman S, Shalev I, Uzefovsky F, Israel S, Knafo A, Kremer I, et al. Epigenetic and genetic factors predict women’s salivary cortisol following a threat to the social self. PloS One (2012) 7(11):e48597. doi: 10.1371/journal.pone.0048597
242. Young E, Korszun A. Sex, trauma, stress hormones and depression. Mol Psychiatry (2010) 15(1):23–8. doi: 10.1038/mp.2009.94
243. Provenzi L, Giorda R, Beri S, Montirosso R. SLC6A4 methylation as an epigenetic marker of life adversity exposures in humans: A systematic review of literature. Neurosci Biobehav Rev (2016) 71:7–20. doi: 10.1016/j.neubiorev.2016.08.021
244. Skuse DH. Imprinting, the X-chromosome, and the male brain: explaining sex differences in the liability to autism. Pediatr Res (2000) 47(1):9–16. doi: 10.1203/00006450-200001000-00006
245. Bakermans-Kranenburg MJ, van IMH, Caspers K, Philibert R. DRD4 genotype moderates the impact of parental problems on unresolved loss or trauma. Attachment Hum Dev (2011) 13(3):253–69. doi: 10.1080/14616734.2011.562415
246. Das D, Cherbuin N, Tan X, Anstey KJ, Easteal S. DRD4-exonIII-VNTR moderates the effect of childhood adversities on emotional resilience in young-adults. PloS One (2011) 6(5):e20177. doi: 10.1371/journal.pone.0020177
247. Burggren WW, Crews D. Epigenetics in comparative biology: why we should pay attention. Integr Comp Biol (2014) 54(1):7–20. doi: 10.1093/icb/icu013
248. Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet (1975) 14(1):9–25. doi: 10.1159/000130315
249. Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science (1975) 187(4173):226–32.
250. Grabowski M. Neuroscience and media: new understandings and representations. New York, NY: Routledge (2014). doi: 10.4324/9781315749235
251. Greally JM. A user’s guide to the ambiguous word ‘epigenetics’. Nat Rev Mol Cell Bio (2018) 19(4):207–8. doi: 10.1038/nrm.2017.135
252. Horsthemke B. A critical view on transgenerational epigenetic inheritance in humans. Nat Commun (2018) 9(1):2973. doi: 10.1038/s41467-018-05445-5
253. Deans C, Maggert KA. What do you mean, “epigenetic”? Genetics (2015) 199(4):887–96. doi: 10.1534/genetics.114.173492
254. Trerotola M, Relli V, Simeone P, Alberti S. Epigenetic inheritance and the missing heritability. Hum Genomics (2015) 9(1):17. doi: 10.1186/s40246-015-0041-3
255. Zhu B, Reinberg D. Epigenetic inheritance: uncontested? Cell Res (2011) 21(3):435–41. doi: 10.1038/cr.2011.26
256. Audergon PN, Catania S, Kagansky A, Tong P, Shukla M, Pidoux AL, et al. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science (2015) 348(6230):132–5. doi: 10.1126/science.1260638
257. Peedicayil J. The importance of cultural inheritance in psychiatric genetics. Med Hypotheses (2002) 58(2):164–6. doi: 10.1054/mehy.2001.1503
258. Chasiotis A. An epigenetic view on culture: what evolutionary developmental psychology has to offer for cross-cultural psychology. Fundamental questions cross-cultural Psychol (2011), 376–404. doi: 10.1017/CBO9780511974090.016
259. Mishra S, Dwivedi SP., Singh R. A review on epigenetic effect of heavy metal carcinogens on human health. Open Nutraceuticals J (2010) 3:188–93. doi: 10.2174/1876396001003010188
260. Mulder RH, Rijlaarsdam J, Van Ijzendoorn MH. DNA Methylation: a mediator between parenting stress and adverse child development? In: deater-deckard K, Panneton R, editors. Parental Stress and Early Child Development. Cham, Switzerland: Springer International Publishing. (2017). p. 157–80. doi: 10.1007/978-3-319-55376-4_7
261. Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell (2014) 157(1):95–109. doi: 10.1016/j.cell.2014.02.045
262. Jang H, Shin W, Lee J, Do J. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes (2017) 8(6):148. doi: 10.3390/genes8060148
263. Kigar SL, Chang L, Guerrero CR, Sehring JR, Cuarenta A, Parker LL, et al. N(6)-methyladenine is an epigenetic marker of mammalian early life stress. Sci Rep (2017) 7(1):18078. doi: 10.1038/s41598-017-18414-7
264. Stenz L, Schechter DS, Serpa SR, Paoloni-Giacobino A. Intergenerational transmission of DNA methylation signatures associated with early life stress. Curr Genomics (2018) 19(8):665–75. doi: 10.2174/1389202919666171229145656
265. Roberts AL, Gladish N, Gatev E, Jones MJ, Chen Y, MacIsaac JL, et al. Exposure to childhood abuse is associated with human sperm DNA methylation. Transl Psychiatry (2018) 8(1):194. doi: 10.1038/s41398-018-0252-1
266. Conradt E, Adkins DE, Crowell SE, Raby KL, Diamond LM, Ellis B. Incorporating epigenetic mechanisms to advance fetal programming theories. Dev Psychopathol (2018) 30(3):807–24. doi: 10.1017/S0954579418000469
267. Hantsoo L, Jašarević E, Criniti S, McGeehan B, Tanes C, Sammel MD, et al. Childhood adversity impact on gut microbiota and inflammatory response to stress during pregnancy. Brain Behav immunity (2019) 75:240–50.
268. Labonté B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. Sex-specific transcriptional signatures in human depression. Nat Med (2017) 23(9):1102. doi: 10.1038/nm.4386
269. Olff M. Sex and gender differences in post-traumatic stress disorder: an update. Eur J Psychotraumatology (2017) 8(sup4):1351204. d
270. Christiansen DM, Hansen M. Accounting for sex differences in PTSD: A multi-variable mediation model. Eur J Psychotraumatology (2015) 6(1):26068. doi: 10.3402/ejpt.v6.26068
271. Ishunina TA, Swaab DF. Vasopressin and oxytocin neurons of the human supraoptic and paraventricular nucleus; size changes in relation to age and sex. J Clin Endocrinol metabolism (1999) 84(12):4637–44. doi: 10.1210/jcem.84.12.6187
272. Cahill L. Sex-and hemisphere-related influences on the neurobiology of emotionally influenced memory. Prog Neuropsychopharmacol Biol Psychiatry (2003) 27(8):1235–41. doi: 10.1016/j.pnpbp.2003.09.019
273. Moriguchi Y, Touroutoglou A, Dickerson BC, Barrett LF. Sex differences in the neural correlates of affective experience. Soc Cogn Affect Neurosci (2013) 9(5):591–600. doi: 10.1093/scan/nst030
274. Lungu O, Potvin S, Tikàsz A, Mendrek A. Sex differences in effective fronto-limbic connectivity during negative emotion processing. Psychoneuroendocrino (2015) 62:180–8. doi: 10.1016/j.psyneuen.2015.08.012
275. Helpman L, Zhu X, Suarez-Jimenez B, Lazarov A, Monk C, Neria Y. Sex Differences in trauma-related psychopathology: a critical review of neuroimaging literature (2014–2017). Curr Psychiatry Rep (2017) 19(12):104. doi: 10.1007/s11920-017-0854-y
276. Oyola MG, Handa RJ. Hypothalamic-pituitary-adrenal and hypothalamic-pituitary-gonadal axes: sex differences in regulation of stress responsivity. Stress (Amsterdam Netherlands) (2017) 20(5):476–94. doi: 10.1080/10253890.2017.1369523
277. Viau V, Meaney MJ. Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology (1991) 129(5):2503–11. doi: 10.1210/endo-129-5-2503
278. McCarthy MM, Nugent BM. At the frontier of epigenetics of brain sex differences. Front Behav Neurosci (2015) 9:221–. doi: 10.3389/fnbeh.2015.00221
279. Schwarz JM, Nugent BM, McCarthy MM. Developmental and hormone-induced epigenetic changes to estrogen and progesterone receptor genes in brain are dynamic across the life span. Endocrinology (2010) 151(10):4871–81. doi: 10.1210/en.2010-0142
280. Amateau SK, McCarthy MM. Induction of PGE 2 by estradiol mediates developmental masculinization of sex behavior. Nat Neurosci (2004) 7(6):643. doi: 10.1038/nn1254
281. Wu MV, Manoli DS, Fraser EJ, Coats JK, Tollkuhn J, Honda S-I, et al. Estrogen masculinizes neural pathways and sex-specific behaviors. Cell (2009) 139(1):61–72. doi: 10.1016/j.cell.2009.07.036
282. Oswald LM, Wand GS, Kuwabara H, Wong DF, Zhu S, Brasic JR. History of childhood adversity is positively associated with ventral striatal dopamine responses to amphetamine. Psychopharmacology (2014) 231(12):2417–33. doi: 10.1007/s00213-013-3407-z
283. Blum K, Chen TJ, Chen AL, Madigan M, Downs BW, Waite RL, et al. Do dopaminergic gene polymorphisms affect mesolimbic reward activation of music listening response? Therapeutic impact on reward Deficiency Syndrome (RDS). Med hypotheses (2010) 74(3):513–20. doi: 10.1016/j.mehy.2009.10.008
284. Nugent AC, Bain EE, Thayer JF, Sollers JJ, Drevets WC. Sex differences in the neural correlates of autonomic arousal: a pilot PET study. Int J Psychophysiology (2011) 80(3):182–91. doi: 10.1016/j.ijpsycho.2011.03.001
285. Groleau P, Joober R, Israel M, Zeramdini N, DeGuzman R, Steiger H. Methylation of the dopamine D2 receptor (DRD2) gene promoter in women with a bulimia-spectrum disorder: Associations with borderline personality disorder and exposure to childhood abuse. J Psychiatr Res (2014) 48(1):121–7. doi: 10.1016/j.jpsychires.2013.10.003
286. Talmi D, Anderson AK, Riggs L, Caplan JB, Moscovitch M. Immediate memory consequences of the effect of emotion on attention to pictures. Learn Memory (2008) 15(3):172–82. doi: 10.1101/lm.722908
287. Tiwari A, Gonzalez A. Biological alterations affecting risk of adult psychopathology following childhood trauma: a review of sex differences. Clin Psychol Rev (2018) 66:69–79. doi: 10.1016/j.cpr.2018.01.006
288. Thaler L, Gauvin L, Joober R, Groleau P, de Guzman R, Ambalavanan A, et al. Methylation of BDNF in women with bulimic eating syndromes: associations with childhood abuse and borderline personality disorder. Prog Neuropsychopharmacol Biol Psychiatry (2014) 54:43–9. doi: 10.1016/j.pnpbp.2014.04.010
289. Gueant JL, Chery C, Oussalah A, Nadaf J, Coelho D, Josse T, et al. APRDX1 mutant allele causes a MMACHC secondary epimutation in cblC patients. Nat Commun (2018) 9(1):67. doi: 10.1038/s41467-017-02306-5
290. Watanabe Y, Maekawa M. Methods and Strategies to determine epigenetic variation in human disease. In: Tollefsbol TO, editor. Epigenetics in Human Disease (Second Edition)., vol. 6. Cambridge, MA: Academic Press (2018). p. 13–37. doi: 10.1016/B978-0-12-388415-2.00002-0
291. Quante T, Bird A. Do short, frequent DNA sequence motifs mould the epigenome? Nat Rev Mol Cell Bio (2016) 17(4):257. doi: 10.1038/nrm.2015.31
292. Loviglio MN, Leleu M, Männik K, Passeggeri M, Giannuzzi G, van der Werf I, et al. Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes. Mol Psychiatr (2016) 22:836. doi: 10.1038/mp.2016.84
293. de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature (2013) 502(7472):499–506. doi: 10.1038/nature12753
294. Ibn-Salem J, Köhler S, Love MI, Chung H-R, Huang N, Hurles ME, et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol (2014) 15(9):423–. doi: 10.1186/s13059-014-0423-1
295. Verma M. Epigenome-Wide Association Studies (EWAS) in Cancer. Curr Genomics (2012) 13(4):308–13. doi: 10.2174/138920212800793294
296. Saffari A, Silver MJ, Zavattari P, Moi L, Columbano A, Meaburn EL, et al. Estimation of a significance threshold for epigenome-wide association studies. Genet Epidemiol (2018) 42(1):20–33. doi: 10.1002/gepi.22086
297. Roeh S, Wiechmann T, Sauer S, Kodel M, Binder EB, Provencal N. HAM-TBS: high-accuracy methylation measurements via targeted bisulfite sequencing. Epigenetics Chromatin (2018) 11(1):39. doi: 10.1186/s13072-018-0209-x
298. Latendresse SJ, Musci R, Maher BS. Critical issues in the inclusion of genetic and epigenetic information in prevention and intervention trials. Prev Sci (2018) 19(1):58–67. doi: 10.1007/s11121-017-0785-1
299. Lappalainen T, Greally JM. Associating cellular epigenetic models with human phenotypes. Nat Rev Genet (2017) 18(7):441–51. doi: 10.1038/nrg.2017.32
300. Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, et al. Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods (2017) 14(4):411–3. doi: 10.1038/nmeth.4189
301. Laszlo AH, Derrington IM, Brinkerhoff H, Langford KW, Nova IC, Samson JM, et al. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc Natl Acad Sci (2013) 110(47):18904–9. doi: 10.1073/pnas.1310240110
302. Ooga M, Wakayama T. FRAP analysis of chromatin looseness in mouse zygotes that allows full-term development. PloS One (2017) 12(5):e0178255. doi: ARTNe0178255
303. Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature (2017) 544(7648):59–64. doi: 10.1038/nature21429
304. Hwang B, Lee W, Yum S-Y, Jeon Y, Cho N, Jang G, et al. Lineage tracing using a Cas9-deaminase barcoding system targeting endogenous L1 elements. Nat Commun (2019) 10(1):1234. doi: 10.1038/s41467-019-09203-z
305. Raj B, Wagner DE, McKenna A, Pandey S, Klein AM, Shendure J, et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat Biotechnol (2018) 36:442. doi: 10.1038/nbt.4103
306. Pulecio J, Verma N, Mejía-Ramírez E, Huangfu D, Raya A. CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell (2017) 21(4):431–47. doi: 10.1016/j.stem.2017.09.006
307. Liao H-K, Hatanaka F, Araoka T, Reddy P, Wu M-Z, Sui Y, et al. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell (2017) 171(7):1495–507. e15. doi: 10.1016/j.cell.2017.10.025
308. Karemaker ID, Vermeulen M. Single-cell DNA methylation profiling: technologies and biological applications. Trends Biotechnol (2018) 36(9):952–65. doi: 10.1016/j.tibtech.2018.04.002
309. Clark SJ, Argelaguet R, Kapourani CA, Stubbs TM, Lee HJ, Alda-Catalinas C, et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun (2018) 9(1):781. doi: 10.1038/s41467-018-03149-4
310. Braun U, Schaefer A, Betzel RF, Tost H, Meyer-Lindenberg A, Bassett DS. From maps to multi-dimensional network mechanisms of mental disorders. Neuron (2018) 97(1):14–31. doi: 10.1016/j.neuron.2017.11.007
311. Montefiori L, Hernandez L, Zhang Z, Gilad Y, Ober C, Crawford G, et al. Reducing mitochondrial reads in ATAC-seq using CRISPR/Cas9. Sci Rep (2017) 7(1):2451. doi: 10.1038/s41598-017-02547-w
312. Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature (2015) 523(7561):486–90. doi: 10.1038/nature14590
313. Fornito A, Zalesky A, Breakspear M. The connectomics of brain disorders. Nat Rev Neurosci (2015) 16(3):159. doi: 10.1038/nrn3901
314. Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci USA (2013) 110(44):17921–6. doi: 10.1073/pnas.1317023110
315. Irie N, Weinberger L, Tang WW, Kobayashi T, Viukov S, Manor YS, et al. SOX17 is a critical specifier of human primordial germ cell fate. Cell (2015) 160(1-2):253–68. doi: 10.1016/j.cell.2014.12.013
316. Tang WW, Dietmann S, Irie N, Leitch HG, Floros VI, Bradshaw CR, et al. A unique gene regulatory network resets the human germline epigenome for development. Cell (2015) 161(6):1453–67. doi: 10.1016/j.cell.2015.04.053
Keywords: childhood trauma, stress disorders, mental health, the hypothalamic-pituitary-adrenal axis (HPA), epigenetic association studies
Citation: Jiang S, Postovit L, Cattaneo A, Binder EB and Aitchison KJ (2019) Epigenetic Modifications in Stress Response Genes Associated With Childhood Trauma. Front. Psychiatry 10:808. doi: 10.3389/fpsyt.2019.00808
Received: 07 June 2019; Accepted: 11 October 2019;
Published: 08 November 2019.
Edited by:
Naguib Mechawar, McGill University, CanadaReviewed by:
Benoit Labonte, Laval University, CanadaCopyright © 2019 Jiang, Postovit, Cattaneo, Binder and Aitchison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine J. Aitchison, a2FpdGNoaXNAdWFsYmVydGEuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.