 Cheryl Cheuk-Yan Leung1,2*
Cheryl Cheuk-Yan Leung1,2* Romayne Gadelrab2
Romayne Gadelrab2 Chukwuma Uchenna Ntephe2
Chukwuma Uchenna Ntephe2 Philip K. McGuire3,4
Philip K. McGuire3,4 Arsime Demjaha3,4*
Arsime Demjaha3,4*- 1Department of Old Age Psychiatry, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, United Kingdom
- 2South London and Maudsley NHS Foundation Trust, London, United Kingdom
- 3Department of Psychosis Studies, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, United Kingdom
- 4National Institute for Health Research (NIHR) Biomedical Research Centre (BRC), South London and Maudsley NHS Foundation Trust, London, United Kingdom
Despite considerable psychotherapeutic advancement since the discovery of chlorpromazine, almost one third of patients with schizophrenia remain resistant to dopamine-blocking antipsychotics, and continue to be exposed to unwanted and often disabling side effects, but little if any clinical benefit. Even clozapine, the superior antipsychotic treatment, is ineffective in approximately half of these patients. Thus treatment resistant schizophrenia (TRS), continues to present a major therapeutic challenge to psychiatry. The main impediment to finding novel treatments is the lack of understanding of precise molecular mechanisms leading to TRS. Not only has the neurobiology been enigmatic for decades, but accurate and early detection of patients who are at risk of not responding to dopaminergic blockade remains elusive. Fortunately, recent work has started to unravel some of the neurobiological mechanisms underlying treatment resistance, providing long awaited answers, at least to some extent. Here we focus on the scientific advances in the field, from the clinical course of TRS to neurobiology and available treatment options. We specifically emphasize emerging evidence from TRS imaging and genetic literature that implicates dysregulation in several neurotransmitters, particularly dopamine and glutamate, and in addition genetic and neural alterations that concertedly may lead to the formation of TRS. Finally, we integrate available findings into a putative model of TRS, which may provide a platform for future studies in a bid to open the avenues for subsequent development of effective therapeutics.
Introduction
Almost one third of patients with schizophrenia do not respond to dopamine (DA) blocking antipsychotic medication and are described as being treatment-resistant (1). Although clozapine can be effective in these patients, there is usually a long delay before it is used, and what is more around half of treatment-resistant patients do not respond to clozapine (2, 3). Treatment-resistant schizophrenia (TRS), is thus associated with particularly poor clinical outcomes (4), and presents a major therapeutic challenge to psychiatry. One of the main impediments to finding novel treatments for TRS patients is the lack of understanding of the molecular basis of TRS, despite over 50 years of scientific work in this field. Moreover, biomarkers that can identify patients who are unlikely to respond to conventional treatment remain elusive. Fortunately, recent work has started to unravel some of the mechanisms underlying treatment resistance. Here we describe these scientific advances and propose an integrated model of TRS that may facilitate the identification of biomarkers for TRS and provide a rationale for the development of novel therapeutic approaches.
Defining Treatment-Resistant Schizophrenia
Prior to embarking on finding reliable biomarkers and conduct promising clinical trials, it is of crucial importance to precisely stratify patients according to their response to treatment. The literature, however, has been limited by inconsistent TRS definitions. In the absence of a universally accepted definition, studies have opted for different TRS criteria according to their aims and population studied. This has resulted in marked heterogeneity in results and disparity in response rates. For instance, in Suzuki and colleagues’ systematic review, 33 studies reported treatment response rates ranging from 0% to 76% (5). Studies recruiting patients for novel antipsychotic drug trials may use more stringent criteria than those testing psychosocial interventions, thus reporting lower prevalence of TRS (5, 6).
Furthermore, the lack of precise and universal operational definitions of TRS may have important clinical and scientific implications. For instance, it hinders early detection of treatment resistance and, subsequently, may delay initiation of clozapine, and in research settings, it complicates comparisons and interpretation of results. To address these issues, International Treatment Response and Resistance in Psychosis (TRRIP) group has developed operationalized TRS definition criteria and reached consensus on “minimum requirements.” The group emphasizes that any definition of treatment resistance should indicate that the patient has received an adequate trial of antipsychotic medication in terms of dosage (equivalent to or greater than 600 mg of chlorpromazine per day), trial of two different antipsychotics for a duration of 6 weeks each at a therapeutic dose, strong advocation for acquiring treatment adherence measures (≥80% of prescribed doses), as well as the use of structured clinical assessments to ascertain symptom presence and severity (7). However, there are limitations to these criteria, such as the use of dichotomous classification, which does not account for the continuum of treatment response. As authors acknowledge, future revisions incorporating novel neurobiological findings are required prior to criteria being fully standardized and more applicable across research and clinical settings.
Heterogeneity of Clinical Course of TRS
For decades, researchers in the field of TRS debated whether treatment resistance is a stable phenotype, or whether it is a consequence of neurodegenerative process, evolving over time in the context of multiple episodes and repeated exposure to antipsychotic treatment.
In favor of the first notion were reports that emerged even prior to the existence of psychopharmacology, and indirectly suggested that unfavorable clinical outcomes may be related to more severe and enduring subtype of schizophrenic illness. Kraepelin, referring in his textbook to Albrecht’s observations that one third of his cases with hebephrenia reached terminal state within a year of onset, concluded: “Often enough the unmistakable symptoms of dementia appear already within the first year” (8). Decades later, Kolakowska and colleagues demonstrated that the majority of “poor responders” were unresponsive throughout their illness and remarked that treatment resistance was related to the “type,” rather than the “stage” of schizophrenia. (9). Analogously, two prospective studies observed that resistance to treatment was apparent in early stages of illness (10, 11). Furthermore, longitudinal first episode psychosis (FEP) studies (12) observed that between 5% and 25% of patients were unresponsive and had persistent positive symptoms during the initial phase of illness (12, 13). Such variability again, might be a consequence of the different TRS criteria employed in these studies.
Other authors, however, considered neurodegenerative hypothesis, attributing treatment resistance to chronicity of illness. Wyatt (1991) reviewed the evidence derived from 22 studies of predominantly FEP patients and concluded that early psychopharmacological intervention could improve the outcomes and prognosis of the disorder (14). It was proposed that a neurodegenerative process might be inherent to psychosis and thus adversely affect the clinical course in those who were non-compliant with treatment and subjected to multiple relapses.
More recently, the largest to date, a 10-year follow-up FEP study, designed to address these inconsistencies in literature, found that over 80% of treatment-resistant patients were persistently resistant from the very early stage of their illness (15).
This work, however, identified a small proportion of patients (16%), who although initially responded to medication, ultimately developed treatment resistance. These patients showed higher number of relapses associated with more inpatient admissions. The reasons for this remain elusive and warrant further exploration. As suggested by animal studies, it can be that chronic treatment with DA blocking agents may induce D2 receptor up-regulation leading to breakthrough DA supersensitivity, which may predispose some patients to treatment resistance (16, 17). Accordingly, it has been shown that in a proportion of patients not only the time to remission is longer in subsequent episodes, but less, if at all, achievable (18, 19). Furthermore, most recently, a study by Takeuchi et al. (20) has implicated that treatment response is unfavorably affected by symptomatic relapse following initial response. This finding could be particularly relevant to this subgroup of patients (20).
On the other hand, some treatment-resistant patients may achieve spontaneous remission or begin responding to treatment later in life (21), which is in line with previous observations that older patients with schizophrenia require much less intensive maintenance antipsychotic treatment than those who are younger in age (22–24). This can perhaps be explained by the fact that DA system is age-dependent, with significant reductions in dopaminergic transmission in older patients being observed (24, 25). This notion is intriguing and contradicts the recent findings of unaltered DA levels in TRS, but it can be that this sub-group of patients have different neurobiology altogether, which remains to be determined in larger and more stratified studies. Finally, up to 50% of treatment-resistant patients are resistant to clozapine recently termed as “ultra-treatment resistance” (26). Such non-response to clozapine, a last treatment resort for those who do not respond to first-line antipsychotics, is the major unmet clinical need in schizophrenia (Figure 1).
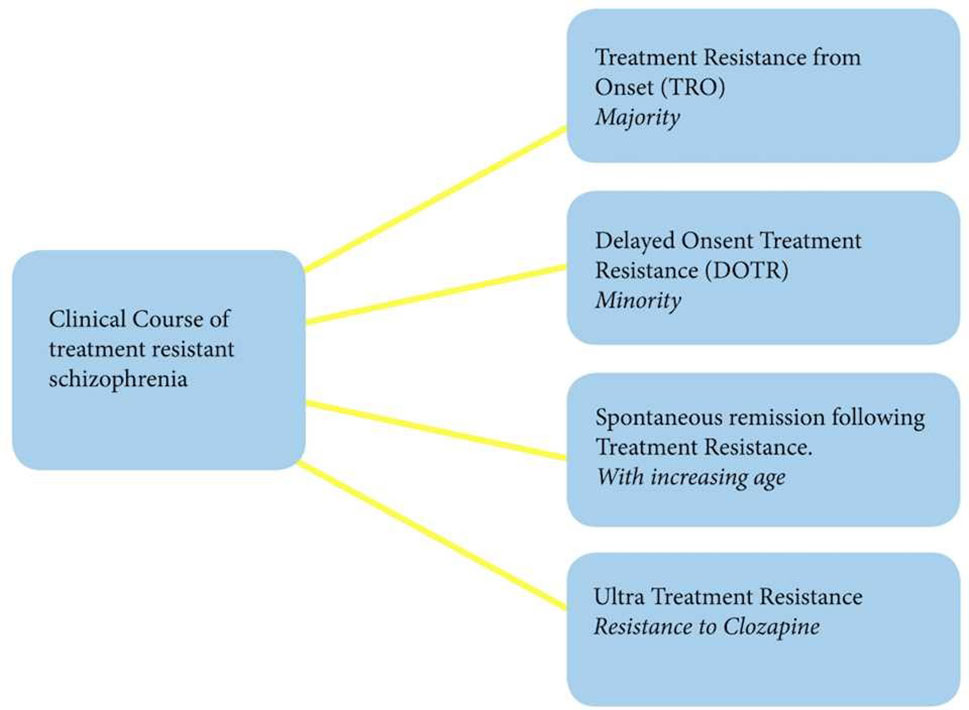
Figure 1 Clinical course of treatment-resistant schizophrenia.
Putative Predictors of TRS
Several studies have identified younger age at onset, longer duration of untreated psychosis, and negative symptomatology to be associated with treatment resistance (12, 15, 27, 28). Furthermore, severe cognitive impairment, poorer premorbid functioning (21, 29), obstetric complications (30), as well as neurological soft signs (31), have, in addition, shown significant associations with treatment resistance. Additionally, family history and, thus, increased genetic burden (32, 33) have been linked to poor prognosis of illness, and finally, a study comparing first-degree relatives of patients with and without TRS showed higher morbidity risk of schizophrenia in relatives of TRS patients (34).
The findings and observations, to date, indicate that treatment resistance in schizophrenia is heterogenous, as a disorder itself, assuming at least four different trajectories. However, a significant majority of patients with TRS appear to be resistant at the time of their first presentation. This form of treatment resistance may represent an enduring phenotype of schizophrenic illness, which is particularly associated with younger age at onset and negative symptoms (15). Such high prevalence at the early phase of illness should alert clinicians to commence clozapine as soon as possible. However, larger FEP studies are needed to delineate reliable predictors to facilitate early and accurate detection of patients who are not likely to respond to first-line antipsychotic treatment.
Neurobiology of Treatment-Resistant Schizophrenia
Until recently, the underlying neurobiology of treatment-resistant schizophrenia remained elusive. Emerging evidence from TRS imaging and genetic literature implicates dysregulation in several neurotransmitters, particularly DA and glutamate, and in addition genetic and neural alterations that concertedly may lead to the formation of treatment resistance in schizophrenia. The presented literature here is not exhaustive. Instead, we predominantly focus on the most robust and high-impact evidence and neurobiological aspects that may predispose to treatment resistance.
Neurotransmitters in TRS
The DA hypothesis remains an important part of our understanding of psychosis. DA blocking antipsychotics are effective in a majority of patients with schizophrenia, and illicit drugs that induce acute psychotic symptoms increase DA levels. Although this hypothesis does hold true for the patients who are responsive to treatment, it fails to provide explanations for patients with TRS.
To understand the dopaminergic mechanisms underlying treatment resistance, scientists have first focused on striatal DA D2 receptors. Positron emission tomography (PET) studies revealed significant associations between striatal DA D2 occupancy and prediction of short-term clinical response to antipsychotic treatment (35) and suggested that at least 60% of DA D2 receptor occupancy is necessary to reach adequate therapeutic response. This is true for both typical and atypical antipsychotics (31, 35, 36), excluding clozapine (37). Hypothesizing that the lack of response may result from inadequate DA D2 receptor blockade, Wolkin et al. (38), using PET, examined DA D2 receptor occupancy in patients with TRS schizophrenia and intriguingly demonstrated almost identical striatal DA D2 receptor occupancies in both treatment-responsive and treatment-resistant patients (38). Correspondingly, a [123I] IBZM Single Photon Emission Tomography (SPET) study reported a similar degree of DA D2 occupancy in both groups (39). Moreover, Kane et al. (40) in their seminal trial included the most severely ill and treatment-resistant patients with schizophrenia who failed a prospective trial of haloperidol at doses of at least 60 mg/day, which indirectly suggests that DA receptor occupancy was sufficiently achieved (40).
It became apparent that although DA D2 blockade may be necessary, it does not guarantee a response. Thus, the focus shifted to investigating presynaptic DA synthesis capacity (DSC). Increased DSC in patients with schizophrenia is considered one of the most replicated finding in dopaminergic studies of schizophrenia (41–43), and therefore, DSC anomalies are considered critical in the formation of positive psychotic symptoms. The biochemistry of DA synthesis is presented in a schematic diagram (Figure 2).
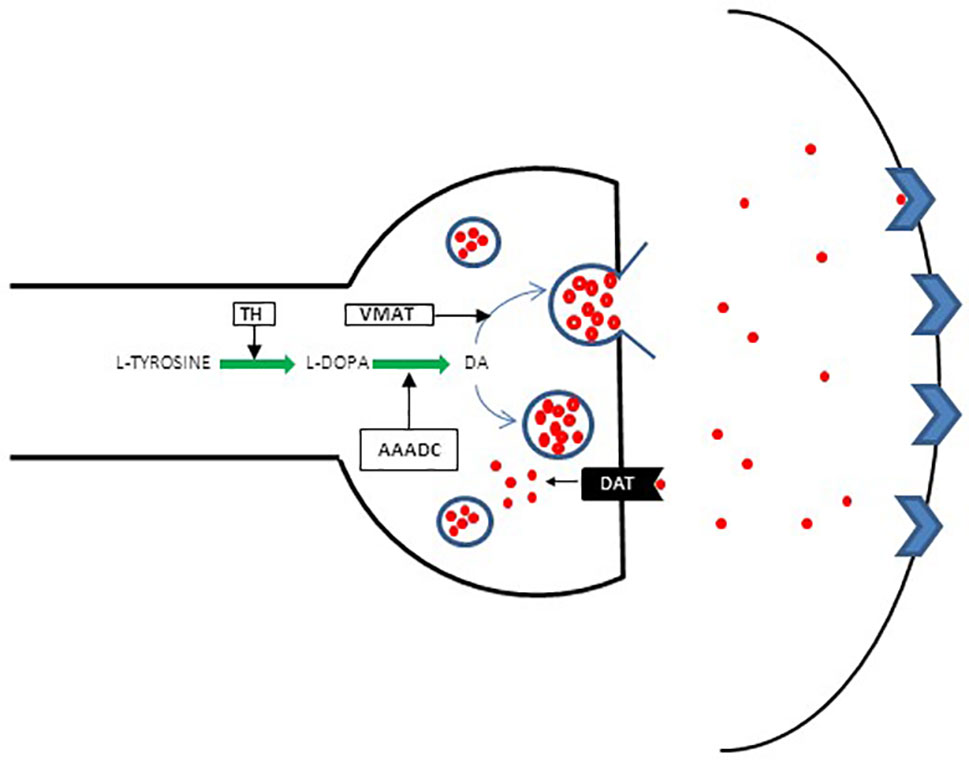
Figure 2 Schematic presentation of presynaptic DA regulation. Conversion of l-tyrosine (4-hydroxyphenylalanin) to l-3, 4 dihydroxyphenylalanine [l-DOPA] constitutes the first step in a complex pathway of DA synthesis. l-tyrosine is derived mainly from dietary sources, although a small quantity originates from l-phenylalanine converted to l-tyrosine by phenylalanine hydroxylase (PHA). l-tyrosine is converted to l-DOPA by tyrosine hydroxylase (TH). Aromatic L-amino acid decarboxylase (AAADC) then acts on l-DOPA to convert it to DA. The DA uptake transporter (DAT) plays an additional role in increasing cytoplasmic DA levels via the reuptake of extracellular DA and thus maintains extracellular DA homeostasis. From the cytoplasm, the majority of DA is stored in specialized synaptic vesicles by the vesicular monoamine transporter (VMAT) and is ready for release upon arrival of the action potential.
The first study to directly examine DSC in specifically treatment-resistant patients with schizophrenia, using PET and stringent criteria for TRS, demonstrated unaltered DSC in treatment-resistant patients. In a subgroup of these patients, authors measured glutamate levels using proton magnetic resonance spectroscopy (1H-MRS) and documented increased glutamatergic levels in anterior cingulate cortex (ACC) in TRS patients (44). Analogously, two subsequent studies reported increased glutamate levels in the ACC in treatment-resistant patients, but decreased levels in treatment responders (45, 46). To address the potential effects of medication or chronicity on these findings, the same group prospectively investigated DSC and ACC glutamate levels in initially medication-naïve FEP patients and confirmed that although striatal DSC is unaltered, ACC glutamate levels are increased in patients who subsequently do not respond to treatment (47). Most recently, in a multicenter longitudinal study of either minimally treated or medication naïve patients, higher levels of glutamate in the ACC were associated with treatment non-response to amisulpiride (48).
However, not all studies have observed glutamatergic alterations in relation to treatment response as discussed in recent systematic reviews (49, 50). The discrepancy in results may be related to differing methodology and, in particular, different TRS criteria studies. Thus, some studies may have misclassified TRS patients as responders or vice versa, which may lead to different outcomes and complicate comparisons as we discussed in previous sections.
Taken together, the neurochemical evidence to date supports the hypothesis that distinct neurochemical abnormalities, such as normal striatal DSC and increased ACC glutamate function, may underlie TRS. What is more, the demonstrated lack of DA abnormality in this subgroup of patients raises the possibility that other neurotransmitters, such as GABAaergic, glutamatergic, and endocannabinoid systems may be a promising target for novel antipsychotics.
However, the neurobiological underpinning of schizophrenia in general as well as that of TRS may involve complex interactions of these neurotransmitters. Carlsson and colleagues (2000, 2001) proposed that alterations in cortical glutamate levels, either acting directly as an “accelerator” or via GABA interneuron projections as a “brake,” modulates the firing of dopaminergic neurons that can in turn lead to either decrease or increase in dopaminergic activity (51, 52). Thus, for instance, the reduced glutamate activity enhances DA release in dopaminergic pathways, which then via negative feedback, mediated, at least in part, via the striatum and the thalamus, regulates glutamate release that would then act as “a brake” on cortical DA production (51, 52). How this mechanism operates in TRS remains to be determined in precise future pre-clinical models. At this stage, and based on available neurochemical imaging evidence, we could only speculate that in TRS, this mechanism involves the indirect pathway that involves GABA interneurons that exerts a “brake” effect on DA production, which may explain the absence of DSC increase in TRS. In turn, the absence of feedback from normal striatal DA status may lead to cortical hyperglutamatergia. In line with this, studies have reported an inverse correlation between cortical glutamate and striatal DSC (53, 54).
Genetic data also support to some degree the distinct neurobiology of TRS by suggesting a specific heritable vulnerability in TRS sub-group of patients. It has been suggested that TRS may be related to increased genetic burden (32). For instance, family history of psychosis has been shown to be associated with TRS (33). Studies that investigated several candidate genes, such as ABCB1, ABCC1, and ABCB11, demonstrated associations with response to antipsychotics as summarized by Vita et al. (50). Subsequent studies have examined polygenic risk scores (PRS) representing aggregate score of risk loci, that have been identified from genome-wide association studies (GWAS) in schizophrenia patients, to determine whether this approach can detect treatment non-response, but both chronic and medication-naive FEP studies have been negative (55, 56).
Functional and Structural Neuroimaging
Evidence from structural magnetic resonance imaging (MRI) studies indicates that patients with limited response to treatment have increased cortical atrophy in comparison with responders (57, 58). Reduced gyrification was observed across multiple brain regions at illness onset in FEP patients who subsequently do not respond to treatment (59). In addition, cortical thinning generally, but particularly in dorsolateral prefrontal cortex (DLPFC) was reported in TRS (60). Recent review has revealed that patients with TRS have larger number of regions with decreased GM when compared with responders (49).
Functional MRI studies have similarly been able to distinguish between responders and non-responders. Most recently, global functional connectivity decrease, particularly in frontotemporal and occipital regions, was reported to be associated with treatment resistance in several studies (61, 62). Two comprehensive reviews have demonstrated decreased metabolism in the prefrontal and frontotemporal regions and hypermetabolism in the basal ganglia in TRS patients (63, 64).
Therapeutic Approaches of Treatment-Resistant Schizophrenia
Clozapine—A Gold Standard
The discovery of chlorpromazine has stimulated the discovery of numerous DA-blocking antipsychotics that in most patients are effective. However, first-line antipsychotic treatment in considerable proportion of patients does not alleviate symptoms, but instead exposes these patients to unwanted and often disabling side effects. The only antipsychotic, to date, that has an adequate therapeutic effect in this subgroup of patients is clozapine, and as such remains superior to other antipsychotics for TRS patients (65–67).
Clozapine has been actualized by Kane and colleagues in their seminal work (40). They have shown clozapine to be more effective than chlorpromazine at symptomatic reduction (30% vs. 4%, respectively) in participants who failed a trial of haloperidol treatment (40). It is, however, underutilized (68) with documented delay of its initiation approximating 5 years (69). This delay has important clinical implications associated with reduced effectiveness, increased number of hospital admissions, and more frequent use of concurrent electroconvulsive therapy (ECT) (68, 70–72). Recent scientific reports advocate its use at much earlier stages of illness (15, 73). This is compounded by the fact that a great majority of TRS patients seem to be destined to non-response to medication at the earliest stages of their illness necessitating much earlier use of clozapine (15). A meta-analysis by Okhuijsen-Pfeifer and colleagues (2018) comparing clozapine with a number of conventional antipsychotics found significant benefit for early clozapine use (Hedges’ g = 0.220; P = 0.026; 95% CI = 0.026–0.414) (74), whereas a large three-phase switching clinical trial conducted by the OPTiMiSe study group found that following a failed initial response to amisulpiride switching to olanzapine resulted in no additional benefit, whereas switching to clozapine did improve clinical outcomes (73).
The precise psychopharmacology of clozapine is yet to be unraveled. Its efficacy in TRS may be related to the fact that clozapine is a weak DA blocker and that its action may be mediated via glutamatergic and serotonergic pathway as indicated by recent neurochemical imaging literature (45, 47, 75–79).
Treatment Strategies in Ultra-Treatment Resistance
Clozapine Augmentation With Other Psychotropic Agents
Almost half of TRS patients do not respond to clozapine (2, 3, 40, 80) and are termed ultra-treatment resistant. When faced with such treatment challenge, clinicians tend to resort to augmentation with other psychotropic agents, although there is limited evidence to support this therapeutic approach (32, 81). Antipsychotics are the most frequently utilized and studied agent, and of these, risperidone is the most frequently researched (82). A meta-analysis has shown no increased benefit to augmentation with risperidone (83) and another of 14-placebo controlled RCTs showed that augmentation with antipsychotic medication is of little benefit (effect size, −0.239; 95% CI, −0.45 to −0.026; P = 0.028) (84). Furthermore, augmentation with antipsychotics seems to be associated with a worsening of side effects (83). Similarly, the augmentation with mood stabilizers and SSRIs has yielded limited evidence for efficacy (85). Lamotrigine has garnered conflicting evidence (83, 86). While topiramate has some evidence supporting its effect at curtailing weight gain in patients taking clozapine, there is limited evidence for its reduction in psychotic symptoms (85). Augmentation, in theory, may be a useful approach to adopt in managing TRS as it utilizes already existing medication, whose mechanisms of action and side effect profiles have been well studied. Unfortunately, the evidence does not currently support their effectiveness.
Other Treatment Strategies
Evidence investigating the effectiveness of ECT in combination with clozapine has shown positive results (87). A recent meta-analysis by Wang and colleagues (2018) who analyzed data from 18 randomized control trials (n = 1769) found that adjunctive ECT was more beneficial for short-term recovery, compared with clozapine alone (standard mean difference = −0.54; 95% CI, −0.88 to −0.20; I2 = 77%, P = 0.002) (88).
Repetitive trans-magnetic stimulation (89) and transcranial direct stimulation (90) may be effective at reducing auditory hallucinations, though some of their effects may be short-lived (91). Their low-cost and mild side effect profile (92, 93) make them attractive options to treat schizophrenia and, more specifically, TRS, though with a scarcity of large clinical trials, more research is needed to delineate their effectiveness as sole or adjunct agents (81).
In summary, clozapine remains a gold standard treatment for patients with TRS (74). Research has shown that delay in clozapine initiation leads to a poorer response to treatment (72) and, worse outcomes (70, 71). However, there are significant issues with its tolerability, and there is still a significant subgroup of non-responders to clozapine who see, a modest, if any improvement with pharmacological (32, 81) and non-pharmacological augmentation (87, 89, 90). This strongly supports a need for new therapeutic targets. Recent meta-analytic work has demonstrated significant effects of glutamatergic agents, such as glycine/d-serine site antagonists, on negative symptoms (94) that are generally resistant to DA-blocking antipsychotics. In view of complex interplay of neurotransmitters governing schizophrenia and particularly treatment resistance, other promising therapeutic approaches include the stimulation of GABA receptors to overcome glutamatergic deficits, which is yet to be tested in clinical trials (95), as well as the use of cannabinoids, which have shown promising therapeutic effect in recent drug trials (96–98).
Conclusion
Taken together, the findings to date suggest that TRS is a distinct, more severe, and enduring subtype of schizophrenic illness, marked by greater neuroanatomical abnormalities and different molecular mechanisms. Such complex and intractable condition requires a more fine-grained conceptualization of underlying neurobiology, which may consequently lead to much-needed novel biologically determined treatments. It is crucial to develop clinical tools that will enable clinicians to predict whether a patient will or will not respond to DA blockade, so that clozapine or other novel alternatives can be commenced as early as possible. Here, we integrate the available findings into a putative predictive model of TRS (Figure 3), which may provide a platform for impending scientific developments. Carefully designed studies that address rigorously the heterogeneity of the disorder and that of the antipsychotic treatment response (97, 99) are urgently needed so that patients may be stratified accurately according to their likely therapeutic responses.
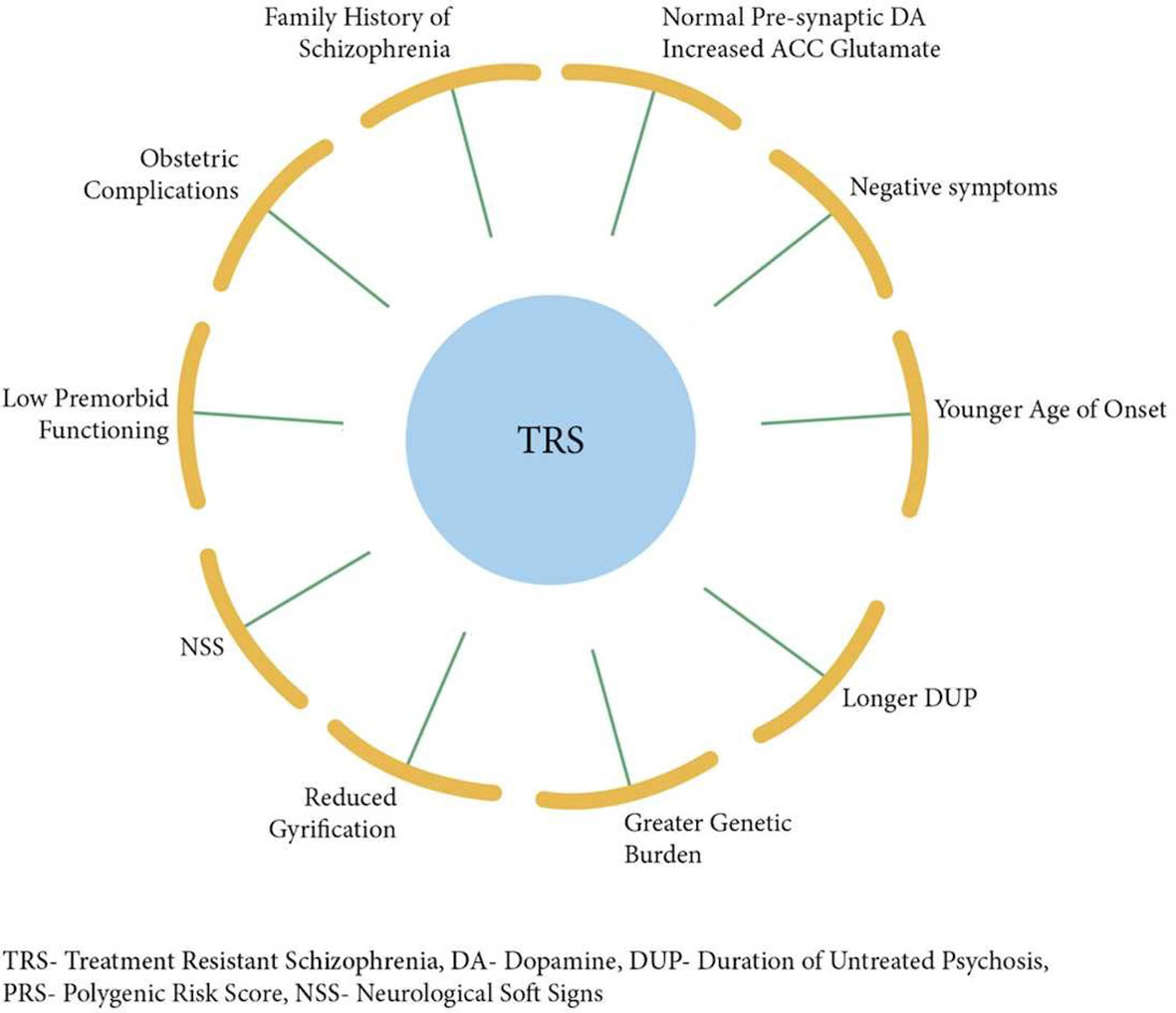
Figure 3 Putative model integrating factors that are associated with treatment resistance in schizophrenia.
Author Contributions
All authors contributed significantly to the conception and drafting of the manuscript. AD critically reviewed the manuscript and all authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS Med (2005) 2:e141. doi: 10.1371/journal.pmed.0020141
2. Chakos M, Lieberman J, Hoffman E, Bradford D, Sheitman B. Effectiveness of second-generation antipsychotics in patients with treatment-resistant schizophrenia: a review and meta-analysis of randomized trials. Am J Psychiatry (2001) 158:518–26. doi: 10.1176/appi.ajp.158.4.518
3. Lieberman JA, Safferman AZ, Pollack S, Szymanski S, Johns C, Howard A, et al. Clinical effects of clozapine in chronic schizophrenia: response to treatment and predictors of outcome. Am J Psychiatry (1994) 151:1744–52. doi: 10.1176/ajp.151.12.1744
4. Lasevoli F, Giordano S, Balletta R, Latte G, Formato MV, Prinzivalli E, et al. Treatment resistant schizophrenia is associated with the worst community functioning among severely-ill highly-disabling psychiatric conditions and is the most relevant predictor of poorer achievements in functional milestones. Prog Neuropsychopharmacol Biol Psychiatry (2016) 65:34–48. doi: 10.1016/j.pnpbp.2015.08.010
5. Suzuki T, Remington G, Mulsant BH, Rajji TK, Uchida H, Graff-Guerrero A, et al. Treatment resistant schizophrenia and response to antipsychotics: a review. Schizophr Res (2011) 133:54–62. doi: 10.1016/j.schres.2011.09.016
7. Leifker FR, Patterson TL, Heaton RK, Harvey PD. Validating measures of real-world outcome: the results of the VALERO expert survey and RAND panel. Schizophr Bull (2011) 37:334–43. doi: 10.1093/schbul/sbp044
8. Kraepelin E, Barclay RM, Robertson GM. Dementia praecox and paraphrenia. Edinburgh: E S Livingston (1919).
9. Kolakowska T, Williams AO, Ardern M, Reveley MA, Jambor K, Gelder MG, et al. Schizophrenia with good and poor outcome. I: Early clinical features, response to neuroleptics and signs of organic dysfunction. Br J Psychiatry (1985) 146:229–39. doi: 10.1192/bjp.146.3.229
10. Bleuler M. The schizophrenic disorders: long-term patient and family studies. New Haven: Yale University Press (1978).
11. Huber G, Gross G, Schuttler R. A long-term follow-up study of schizophrenia: psychiatric course of illness and prognosis. Acta Psychiatr Scand (1975) 52:49–57. doi: 10.1111/j.1600-0447.1975.tb00022.x
12. Robinson DG, Woerner MG, Alvir JM, Geisler S, Koreen A, Sheitman B, et al. Predictors of treatment response from a first episode of schizophrenia or schizoaffective disorder. Am J Psychiatry (1999) 156:544–9. doi: 10.1176/ajp.156.4.544
13. MacMillan JF, Crow TJ, Johnson AL, Johnstone EC. Short-term outcome in trial entrants and trial eligible patients. Br J Psychiatry (1986) 148:128–33. doi: 10.1192/bjp.148.2.128
14. Wyatt RJ. Neuroleptics and the natural course of schizophrenia. Schizophr Bull (1991) 17:325–51. doi: 10.1093/schbul/17.2.325
15. Demjaha A, Lappin JM, Stahl D, Patel MX, MacCabe JH, Howes OD, et al. Antipsychotic treatment resistance in first-episode psychosis: prevalence, subtypes and predictors. Psychol Med (2017) 47:1981–9. doi: 10.1017/S0033291717000435
16. Ginovart N, Wilson AA, Hussey D, Houle S, Kapur S. D2-receptor upregulation is dependent upon temporal course of D2-occupancy: a longitudinal [11C]-raclopride PET study in cats. Neuropsychopharmacology (2009) 34:662–71. doi: 10.1038/npp.2008.116
17. Samaha AN, Seeman P, Stewart J, Rajabi H, Kapur S. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci (2007) 27:2979–86. doi: 10.1523/JNEUROSCI.5416-06.2007
18. Lieberman JA. Prediction of outcome in first-episode schizophrenia. J Clin Psychiatry (1993) 54(suppl):13–7.
19. Sheitman BB, Lee H, Strous R, Lieberman JA. The evaluation and treatment of first-episode psychosis. Schizophr Bull (1997) 23:653–61. doi: 10.1093/schbul/23.4.653
20. Takeuchi H, Siu C, Remington G, Fervaha G, Zipursky RB, Foussias G, et al. Does relapse contribute to treatment resistance? Antipsychotic response in first- vs. second-episode schizophrenia. Neuropsychopharmacology (2019) 44:1036–42. doi: 10.1038/s41386-018-0278-3
21. Meltzer HY. Treatment-resistant schizophrenia: the role of clozapine. Curr Med Res Opin (1997) 14:1–20. doi: 10.1185/03007999709113338
22. Fenton WS, McGlashan TH. Sustained remission in drug-free schizophrenic patients. Am J Psychiatry (1987) 144:1306–9. doi: 10.1176/ajp.144.10.1306
23. Cohen CI, Freeman K, Ghoneim D, Vengassery A, Ghezelaiagh B, Reinhardt MM. Advances in the conceptualisation and study of schizophrenia in later life. Psychiatr Clin North Am (2018) 41:39–53. doi: 10.1016/j.psc.2017.10.004
24. Dreher JC, Meyer-Lindenberg A, Kohn P, Berman KF. Age-related changes in midbrain dopaminergic regulation of the human reward system. Proc Natl Acad Sci U S A (2008) 105:15106–11. doi: 10.1073/pnas.0802127105
25. Reeves S, Bench C, Howard R. Ageing and the nigrostriatal dopaminergic system. Int J Geriatr Psychiatry (2002) 17:359–70. doi: 10.1002/gps.606
26. Remington G, Agid O, Takeuchi H, Lee J, Chintoh A. Ultra-treatment resistance. In: Howes O, editor. Treatment Response and Resistance in Schizophrenia. Oxford University Press (2018). p. 144–62. doi: 10.1093/med/9780198828761.003.0012
27. McCreadie RG, Wiles D, Grant S, Crockett GT, Mahmood Z, Livingston MG, et al. The Scottish first episode schizophrenia study. VII. Two-year follow-up. Scottish Schizophrenia Research Group. Acta Psychiatr Scand (1989) 80:597–602. doi: 10.1111/j.1600-0447.1989.tb03032.x
28. Malaspina D, Goetz RR, Yale S, Berman A, Friedman JH, Tremeau F, et al. Relation of familial schizophrenia to negative symptoms but not to the deficit syndrome. Am J Psychiatry (2000) 157:994–1003. doi: 10.1176/appi.ajp.157.6.994
29. Lindenmayer JP. Treatment refractory schizophrenia. Psychiatry Q (2000) 71:373–84. doi: 10.1023/A:1004640408501
30. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry (2002) 159:1080–92. doi: 10.1176/appi.ajp.159.7.1080
31. Smith RC, Kadewari RP, Rosenberger JR, Bhattacharyya A. Nonresponding schizophrenia: differentiation by neurological soft signs and neuropsychological tests. Schizophr Bull (1999) 25:813–25. doi: 10.1093/oxfordjournals.schbul.a033421
32. Nucifora FC, Woznica E, Lee BJ, Cascella N, Sawa A. Treatment resistant schizophrenia: clinical, biological, and therapeutic perspectives. Neurobiol Dis (2018) 18:30494–7. doi: 10.1016/j.nbd.2018.08.016
33. Crespo-Facorro B, de la Foz VO, Ayesa-Arriola R, Perez-Iglesias R, Mata I, Suarez-Pinilla P, et al. Prediction of acute clinical response following a first episode of non affective psychosis: results of a cohort of 375 patients from the Spanish PAFIP study. Prog Neuropsychopharmacol Biol Psychiatry (2013) 44:162–7. doi: 10.1016/j.pnpbp.2013.02.009
34. Joober R, Rouleau GA, Lal S, Bloom DM, Lalonde P, Labelle A, et al. Increased prevalence of schizophrenia spectrum disorders in relatives of neuroleptic-nonresponsive schizophrenic patients. Schizophr Res (2005) 77:35–41. doi: 10.1016/j.schres.2005.01.008
35. Kapur S, Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first episode schizophrenia. Am J Psychiatry (2000) 157:514–20. doi: 10.1176/appi.ajp.157.4.514
36. Zipursky RB, Christensen BK, Daskalakis Z, Epstein I, Roy P, Furimsky I, et al. Treatment response to olanzapine and haloperidol and its association with dopamine D receptor occupancy in first-episode psychosis. Can J Psychiatry (2005) 50:462–9. doi: 10.1177/070674370505000806
37. Nordstrom AL, Farde L, Nyberg S, Karlsson P, Halldin C, Sedvall G. D2, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry (1995) 152:1444–9. doi: 10.1176/ajp.152.10.1444
38. Wolkin A, Barouche F, Wolf AP, Rotrosen J, Fowler JS, Shiue CY, et al. Dopamine blockade and clinical response: evidence for two biological subgroups of schizophrenia. Am J Psychiatry (1989) 146:905–8. doi: 10.1176/ajp.146.7.905
39. Pilowsky LS, Costa DC, Ell PJ, Murray RM, Verhoeff NP, Kerwin RW. Antipsychotic medication, D2 dopamine receptor blockade and clinical response: a 123I IBZM SPECT (single photon emission tomography) study. Psychol Med (1993) 23:791–7. doi: 10.1017/S0033291700025575
40. Kane K, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry (1988) 45:789–96. doi: 10.1001/archpsyc.1988.01800330013001
41. Demjaha A. Biological and clinical determinants of treatment resistant schizophrenia. [dissertation]. London (UK): King’s College London (2014).
42. Fusar-Poli P, Meyer-Lindenberg A. Striatal presynaptic dopamine in schizophrenia, Part II: Meta-analysis of [18F/11C]-DOPA PET studies. Schizophr Bull (2012) 39:33–42. doi: 10.1093/schbul/sbr180
43. Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry (2012) 69:776–86. doi: 10.1001/archgenpsychiatry.2012.169
44. Demjaha A, Murray RM, McGuire PK, Kapur S, Howes OD. Dopamine synthesis capacity in patients with treatment-resistant schizophrenia. Am J Psychiatry (2012) 169:1203–10. doi: 10.1176/appi.ajp.2012.12010144
45. Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry (2014) 75:e11–13. doi: 10.1016/j.biopsych.2013.06.011
46. Mouchlianitis E, Bloomfield MA, Law V, Beck K, Selvaraj S, Rasquinha N, et al. Treatment-resistant schizophrenia patients show elevated anterior cingulate cortex glutamate compared to treatment-responsive. Schizophr Bull (2015) 42:744–52. doi: 10.1093/schbul/sbv151
47. Jauhar S, Veronese M, Nour MM, Rogdaki M, Hathway P, Turkheimer FE, et al. Determinants of treatment response in first-episode psychosis: an 18F-DOPA PET study. Mol Psychiatry (2018) doi: 10.1038/s41380-018-0042-4
48. Egerton A, Broberg BV, Van Haren N, Merritt K, Barker GJ, Lythgoe DJ, et al. Response to initial antipsychotic treatment in first episode psychosis is related to anterior cingulate glutamate levels: a multicentre 1H-MRS study (OPTiMiSE). Mol Psychiatry (2018) 23(11):2145–55. doi: 10.1038/s41380-018-0082-9
49. Gillespie AL, Samanaite R, Mill J, Egerton A, MacCabe JH. Is treatment-resistant schizophrenia categorically distinct from treatment-responsive schizophrenia? A systematic review. BMC Psychiatry (2017) 17(1):12. doi: 10.1186/s12888-016-1177-y
50. Vita A, Minelli A, Barlati S, Deste G, Giacopuzzi E, Valsecchi P, et al. Treatment-resistant schizophrenia: genetic and neuroimaging correlates. Front Pharmacol (2019) 10:402. doi: 10.3389/fphar.2019.00402
51. Carlsson A, Waters N, Hom-Waters S, Tedroff K, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol (2001) 41:237– 60. doi: 10.1146/annurev.pharmtox.41.1.237
52. Carlsson A, Waters N, Waters S, Carlsson ML. Network interactions in schizophrenia: therapeutic implications. Brain Res Rev (2000) 31:342–9. doi: 10.1016/S0165-0173(99)00050-8
53. Gleich T, Deserno L, Lorenz RC, Boehme R, Pankow A, Buchert R, et al. Prefrontal and striatal glutamate differently relate to striatal dopamine: potential regulatory mechanisms of striatal presynaptic dopamine function? J Neurosci (2015) 35(26):9615–21. doi: 10.1523/JNEUROSCI.0329-15.2015
54. Jauhar S, McCutcheon R, Borgan F, Veronese M, Nour M, Pepper F, et al. The relationship between cortical glutamate and striatal dopamine in first-episode psychosis: a cross-sectional multimodal PET and magnetic resonance spectroscopy imaging study. Lancet Psychiatry (2018) 5(10):816–23. doi: 10.1016/S2215-0366(18)30268-2
55. Frank J, Lang M, Witt SH, Strohmaier J, Rujescu D, Cichon S, et al. Identification of increased genetic risk scores for schizophrenia in treatment-resistant patients. Mol Psychiatry (2015) 20:150–1. doi: 10.1038/mp.2014.56
56. Santoro ML, Ota V, de Jong S, Noto C, Spindola LM, Talarico F, et al. Polygenic risk score analyses of symptoms and treatment response in an antipsychotic-naïve first episode of psychosis cohort. Transl Psychiatry (2018) 8:174. doi: 10.1038/s41398-018-0230-7
57. Bilder RM, Wu H, Chakos MH, Bogerts B, Pollack S, Aronowitz J, et al. Cerebral morphometry and clozapine treatment in schizophrenia. J Clin Psychiatry (1994) 55(suppl B):53–6.
58. Stern RG, Kahn RS, Davidson M. Predictors of response to neuroleptic treatment in schizophrenia. Psychiatr Clin North Am (1993) 16:313–38. doi: 10.1016/S0193-953X(18)30176-X
59. Palaniyappan L, Marques TR, Taylor H, Handley R, Mondelli V, Bonaccorso S, et al. Cortical folding defects as markers of poor treatment response in first-episode psychosis. JAMA Psychiatry (2013) 70:1031–40. doi: 10.1001/jamapsychiatry.2013.203
60. Zugman A, Gadelha A, Assuncao I, Sato J, Ota VK, Rocha DL, et al. Reduced dorso-lateral prefrontal cortex in treatment resistant schizophrenia. Schizophr Res (2013) 148:81–6. doi: 10.1016/j.schres.2013.05.002
61. Ganella EP, Bartholomeusz CF, Seguin C, Whittle S, Bousman C, Phassouliotis C, et al. Functional brain networks in treatment-resistant schizophrenia. Schizophr Res (2017) 184:73–81. doi: 10.1016/j.schres.2016.12.008
62. Vanes LD, Mouchlianitis E, Collier T, Averbeck BB, Shergill SS. Differential neural reward mechanisms in treatment-responsive and treatment-resistant schizophrenia. Psychol Med (2018) 48(14):2418–27. doi: 10.1017/S0033291718000041
63. Nakajima S, Takeuchi H, Plitman E, Fervaha G, Gerretsen P, Caravaggio F, et al. Neuroimaging findings in treatment-resistant schizophrenia: a systematic review. Schizophr Res (2015) 164:164–75. doi: 10.1016/j.schres.2015.01.043
64. Mouchlianitis E, McCutcheon R, Howes OD. Brain-imaging studies of treatment-resistant schizophrenia: a systematic review. Lancet Psychiatry (2016) 3(5):451–63. doi: 10.1016/S2215-0366(15)00540-4
65. Taylor DM, Duncan-McConnell D. Refractory schizophrenia and atypical antipsychotics. J Psychopharmacol (2000) 14:409–18. doi: 10.1177/026988110001400411
66. McEvoy JP, Lieberman JA, Stroup TS, Davis SM, Meltzer HY, Rosenheck RA, et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry (2006) 163:600–10. doi: 10.1176/appi.ajp.163.4.600
67. Lewis SW, Barnes TR, Davies L, Murray RM, Dunn G, Hayhurst KP, et al. Randomized controlled trial of effect of prescription of clozapine versus other second-generation antipsychotic drugs in resistant schizophrenia. Schizophr Bull (2006) 32:715–23. doi: 10.1093/schbul/sbj067
68. Shah P, Iwata Y, Plitman E, Brown EE, Caravaggio F, Kim J, et al. The impact of delay in clozapine initiation on treatment outcomes in patients with treatment-resistant schizophrenia: a systematic review. Psychiatry Res (2018) 268:114–22. doi: 10.1016/j.psychres.2018.06.070
69. Patel MX, Bishara D, Jayakumar S, Zalewska K, Shiers D, Crawford MJ, et al. Quality of prescribing for schizophrenia: evidence from a national audit in England and Wales. Eur Neuropsychopharmacol (2014) 24:499–509. doi: 10.1016/j.euroneuro.2014.01.014
70. Harrison J, Janlov M, Wheeler AJ. Patterns of clozapine prescribing in a mental health service in New Zealand. Pharm World Sci (2010) 32:503–11. doi: 10.1007/s11096-010-9398-5
71. Yoshimura B, Yada Y, So R, Takaki M, Yamada N. The critical treatment window of clozapine in treatment-resistant schizophrenia: secondary analysis of an observational study. Psychiatry Res (2017) 250:65–70. doi: 10.1016/j.psychres.2017.01.064
72. Ucok A, Cikrikcili U, Karabulut S, Salaj A, Ozturk M, Tabak O, et al. Delayed initiation of clozapine may be related to poor response in treatment-resistant schizophrenia. Int Clin Psychopharmacol (2015) 30:290–5. doi: 10.1097/YIC.0000000000000086
73. Kahn RS, Winter van Rossum I, Leucht S, McGuire P, Lewis SW, Leboyer M, et al. Amisulpride and olanzapine followed by open-label treatment with clozapine in first-episode schizophrenia and schizophreniform disorder (OPTiMiSE): a three-phase switching study. Lancet Psychiatry (2018) 5(10):797–807. doi: 10.1016/S2215-0366(18)30252-9
74. Okhuijsen-Pfeifer C, Huijsman EAH, Hasan A, Sommer IEC, Leucht S, Kahn RS, et al. Clozapine as a first- or second-line treatment in schizophrenia: a systematic review and meta-analysis. Acta Psychiatr Scand (2018) 138:281–8. doi: 10.1111/acps.12954
75. Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A (2000) 97:8104–9. doi: 10.1073/pnas.97.14.8104
76. Wulff S, Pinborg LH, Svarer C, Jensen LT, Nielsen MO, Allerup P, et al. Striatal D(2/3) binding potential values in drug-naïve first-episode schizophrenia patients correlate with treatment outcome. Schizophr Bull (2015) 41:1143–52. doi: 10.1093/schbul/sbu220
77. Gellman RL, Aghajanian GK. Distribution of the serotonin 5-HT2 receptor family mRNAs: comparison between 5-HT2A and 5-HT2C receptors antagonism by atypical antipsychotic drugs. Neuroscience (1994) 58:515–25. doi: 10.1016/0306-4522(94)90077-9
78. Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry (2004) 9:984–97. doi: 10.1038/sj.mp.4001551
79. Tanahashi S, Yamamura S, Nakagawa M, Motomura E, Okada M. Clozapine, but not haloperidol, enhances glial d-serine and l-glutamate release in rat frontal cortex and primary cultured astrocytes. Br J Pharmacol (2012) 165:1543–55. doi: 10.1111/j.1476-5381.2011.01638.x
80. Meltzer HY. Antidepressive drug treatment of the schizophrenic patient. Schizophr Bull (1992) 18:515–42. doi: 10.1093/schbul/18.3.515
81. Miyamoto S, Jarskog LF, Fleischhacker WW. New therapeutic approaches for treatment-resistant schizophrenia: a look to the future. J Psychiatr Res (2014) 58:1–6. doi: 10.1016/j.jpsychires.2014.07.001
82. Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry (1999) 156:286–93. doi: 10.1176/ajp.156.2.286
83. Porcelli S, Balzarro B, Serretti A. Clozapine resistance: augmentation strategies. Eur Neuropsychopharmacol (2012) 22:165–82. doi: 10.1016/j.euroneuro.2011.08.005
84. Taylor DM, Smith L, Gee SH, Nielsen J. Augmentation of clozapine with a second antipsychotic—a meta-analysis. Acta Psychiatr Scand (2012) 125:15–24. doi: 10.1111/j.1600-0447.2011.01792.x
85. Sommer IE, Begemann MJ, Temmerman A, Leucht S. Pharmacological augmentation strategies for schizophrenia patients with insufficient response to clozapine: a quantitative literature review. Schizophr Bull (2012) 38:1003–11. doi: 10.1093/schbul/sbr004
86. Tiihonen K, Wahlbeck K, Kiviniemi V. The efficacy of lamotrigine in clozapine-resistant schizophrenia: a systematic review and meta-analysis. Schizophr Res (2009) 109:10–4. doi: 10.1016/j.schres.2009.01.002
87. Petrides G, Malur C, Braga RJ, Bailine SH, Schooler NR, Malhotra AK, et al. Electroconvulsive therapy augmentation in clozapine-resistant schizophrenia: a prospective, randomized study. Am J Psychiatry (2015) 172:52–8. doi: 10.1176/appi.ajp.2014.13060787
88. Wang G, Zheng W, Li XB, Wang SB, Cai DB, Yang XH, et al. ECT augmentation of clozapine for clozapine-resistant schizophrenia: a meta-analysis of randomized controlled trials. J Psychiatr Res (2018) 105:23–32. doi: 10.1016/j.jpsychires.2018.08.002
89. De Jesus DR, Gil A, Barbosa L, Lobato MI, Magalhaes PV, Favalli GP, et al. A pilot double-blind sham-controlled trial of repetitive transcranial magnetic stimulation for patients with refractory schizophrenia treated with clozapine. Psychiatry Res (2011) 188:203–7. doi: 10.1016/j.psychres.2010.11.022
90. Brunelin J, Mondino M, Gassab L, Haesebaert F, Gaha L, Suaud-Chagny MF, et al. Examining transcranial direct-current stimulation (tDCS) as a treatment for hallucinations in schizophrenia. Am J Psychiatry (2012) 169:719–24. doi: 10.1176/appi.ajp.2012.11071091
91. Slotema CW, Aleman A, Daskalakis ZJ, Sommer IE. Meta-analysis of repetitive transcranial magnetic stimulation in the treatment of auditory verbal hallucinations: update and effects after one month. Schizophr Res (2012) 142:40–5. doi: 10.1016/j.schres.2012.08.025
92. Andrade C. Transcranial direct current stimulation for refractory auditory hallucinations in schizophrenia. J Clin Psychiatry (2013) 74:e1054–1058. doi: 10.4088/JCP.13f08826
93. Slotema CW, Blom JD, Hoek HW, Sommer IE. Should we expand the toolbox of psychiatric treatment methods to include Repetitive Transcranial Magnetic Stimulation (rTMS)? A meta-analysis of the efficacy of rTMS in psychiatric disorders. J Clin Psychiatry (2010) 71:873–84. doi: 10.4088/JCP.08m04872gre
94. Tsai GE, Lin PY. Strategies to enhance N-methyl-d-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des (2010) 16:522–37. doi: 10.2174/138161210790361452
95. Kantrowitz JT, Javitt DC. Research on glutamatergic dysfunction may lead to therapies targeting negative and cognitive symptoms. Curr Psychiatry (2010) 10:68–74.
96. Leweke FM, Mueller JK, Lange B, Fritze S, Topor CE, Koethe D, et al. Role of the endocannabinoid system in the pathophysiology of schizophrenia: implications for pharmacological intervention. CNS Drugs (2018) 32:605–19. doi: 10.1007/s40263-018-0539-z
97. Demjaha A, Morgan K, Morgan C, Landau S, Dean K, Reichenberg A, et al. Combining dimensional and categorical representation of psychosis: the way forward for DSM-V and ICD-11? Psychol Med (2009) 39:1943–55. doi: 10.1017/S0033291709990651
98. McGuire P, Robson P, Cubala WJ, Vasile D, Morrison PD, Barron R, et al. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: a multicentre randomised controlled trial. Am J Psychiatry (2018) 175(3):225–31. doi: 10.1176/appi.ajp.2017.17030325
99. Demjaha A. On the brink of precision medicine for psychosis: treating the patient, not the disease: a commentary on: association between serum levels of glutamate and neurotrophic factors and response to clozapine treatment by Krivoy et al. 2017. Schizophr Res (2018) 193:487–8. doi: 10.1016/j.schres.2017.08.011
Keywords: schizophrenia, treatment-resistant, neurobiology, neuroimaging, clozapine
Citation: Leung CC-Y, Gadelrab R, Ntephe CU, McGuire PK and Demjaha A (2019) Clinical Course, Neurobiology and Therapeutic Approaches to Treatment Resistant Schizophrenia. Toward an Integrated View. Front. Psychiatry 10:601. doi: 10.3389/fpsyt.2019.00601
Received: 15 October 2018; Accepted: 29 July 2019;
Published: 03 September 2019.
Edited by:
Vincenzo De Luca, University of Toronto, CanadaReviewed by:
Mette Ødegaard Nielsen, Center for Neuropsychiatric Schizophrenia Research (CNSR), DenmarkHiroyoshi Takeuchi, Keio University, Japan
Copyright © 2019 Leung, Gadelrab, Ntephe, McGuire and Demjaha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheryl Cheuk-Yan Leung, Y2hlcnlsLmxldW5nQGtjbC5hYy51aw==; Arsime Demjaha, YXJzaW1lLmRlbWphaGFAa2NsLmFjLnVr