
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Psychiatry , 09 July 2019
Sec. Molecular Psychiatry
Volume 10 - 2019 | https://doi.org/10.3389/fpsyt.2019.00458
This article is part of the Research Topic The Role of Immune Components in Psychiatric Disorders View all 11 articles
 Gara Arteaga-Henríquez1,2,3†
Gara Arteaga-Henríquez1,2,3† Maria S. Simon1†
Maria S. Simon1† Bianka Burger4
Bianka Burger4 Elif Weidinger1
Elif Weidinger1 Annemarie Wijkhuijs5Volker Arolt6
Annemarie Wijkhuijs5Volker Arolt6 Tom K. Birkenhager7
Tom K. Birkenhager7 Richard Musil1
Richard Musil1 Norbert Müller1,4‡
Norbert Müller1,4‡ Hemmo A. Drexhage2‡*
Hemmo A. Drexhage2‡*Low-grade inflammation plays a role not only in the pathogenesis of major depressive disorder (MDD) but probably also in the poor responsiveness to regular antidepressants. There are also indications that anti-inflammatory agents improve the outcomes of antidepressants.
Aim: To study whether the presence of low-grade inflammation predicts the outcome of antidepressants, anti-inflammatory agents, or combinations thereof.
Methods: We carried out a systematic review of the literature on the prediction capability of the serum levels of inflammatory compounds and/or the inflammatory state of circulating leukocytes for the outcome of antidepressant/anti-inflammatory treatment in MDD. We compared outcomes of the review with original data (collected in two limited trials carried out in the EU project MOODINFLAME) on the prediction capability of the inflammatory state of monocytes (as measured by inflammatory gene expression) for the outcome of venlafaxine, imipramine, or sertraline treatment, the latter with and without celecoxib added.
Results: Collectively, the literature and original data showed that: 1) raised serum levels of pro-inflammatory compounds (in particular of CRP/IL-6) characterize an inflammatory form of MDD with poor responsiveness to predominately serotonergic agents, but a better responsiveness to antidepressant regimens with a) (add-on) noradrenergic, dopaminergic, or glutamatergic action or b) (add-on) anti-inflammatory agents such as infliximab, minocycline, or eicosapentaenoic acid, showing—next to anti-inflammatory—dopaminergic or lipid corrective action; 2) these successful anti-inflammatory (add-on) agents, when used in patients with low serum levels of CRP/IL-6, decreased response rates in comparison to placebo. Add-on aspirin, in contrast, improved responsiveness in such “non-inflammatory” patients; 3) patients with increased inflammatory gene expression in circulating leukocytes had a poor responsiveness to serotonergic/noradrenergic agents.
Conclusions: The presence of inflammation in patients with MDD heralds a poor outcome of first-line antidepressant therapies. Immediate step-ups to dopaminergic or glutamatergic regimens or to (add-on) anti-inflammatory agents are most likely indicated. However, at present, insufficient data exist to design protocols with reliable inflammation parameter cutoff points to guide such therapies, the more since detrimental outcomes are possible of anti-inflammatory agents in “non-inflamed” patients.
It is well accepted that immune dysregulation plays an important role in the pathogenesis of at least a proportion of patients with major depressive disorder (MDD) (1–16). Genetic defects and/or polymorphisms, childhood trauma, and chronic stress are all capable of eliciting such immune dysregulations (17–19). In the last decades, special interest has been raised for the role of low-grade inflammation in the immune system dysregulation of MDD. Low-grade inflammation is characterized by an increase in the level of circulating pro-inflammatory compounds, such as acute phase proteins [e.g., C-reactive protein (CRP)] and cytokines [e.g., interleukin (IL)-6], and/or by a pro-inflammatory activity of circulating or tissue resident immune cells (20–23).
A wide range of medications is currently available for the treatment of MDD. First-line agents are the well-known serotonin reuptake inhibitors (SSRIs; e.g., sertraline, escitalopram, or citalopram), which show a predominantly serotonergic action (24). First-line agents are also the serotonin-noradrenaline reuptake inhibitors (SNRIs), which show a predominantly serotonergic action at low doses and a combined serotonergic–noradrenergic action at moderate to high doses (25). Tricyclic antidepressants (TCAs) show a similar mechanism of action as SNRIs regarding the dual serotonergic-noradrenergic action, but because of more side effects, they are actually used as second-line agents. Third-line agents are drugs with a predominantly noradrenergic/dopaminergic action, such as mirtazapine or bupropion, or agents with other mechanisms of action, such as ketamine [i.e., an N-methyl-d-aspartate (NMDA) receptor antagonist, elevating glutamate levels]. Despite this wide range of medications, response rates to treatment are still insufficient, with about half of the patients not responding adequately to an installed treatment (26, 27).
Since most of the antidepressant drugs have—next to their neurotransmission modulatory effects—also immune modulating capacities (28, 29), it is thought that the inflammatory state of patients might play a role in non-responsiveness. To enforce the mood-regulating effects of antidepressants, and being aware of the notion that low-grade inflammation plays a role, various studies have been undertaken to use anti-inflammatory agents as add-ons to regular antidepressant therapies. In this way, acetylsalicylic acid (i.e., aspirin, a COX1 and COX2 inhibitor), selective COX-2 inhibitors (e.g., celecoxib), minocycline (a tetracyclin with anti-inflammatory effects), and anti-TNF monoclonal antibodies (e.g., infliximab) have been used experimentally (30–33). Besides these anti-inflammatory agents, agents such as cholesterol-lowering fish oil (eicosapentaenoic acid) and anti-oxidative n-acetylcysteine have also been used (33, 34). These agents also have anti-inflammatory actions, since both the cholesterol metabolism and the anti-oxidative machinery are linked to inflammation (35, 36). Though it seems that anti-inflammatory agents did show limited beneficial effects in most of the reported studies (30–34), there is still doubt on the real validity of such interventions, particularly due to the paucity and preliminary character of the studies, while there is also the feeling that such anti-inflammatory agents might only work in a proportion of patients.
Collectively, the abovementioned notions lead to the view that there is a need for a personalized medicine approach to select patients who, in particular, will respond to first-line agents and those needing immediate step-up therapies to drugs other than the first-line drugs and/or an add-on of a first-line agent with an anti-inflammatory agent. In such an approach, it is the question whether a pre-existent state of enhanced low-grade inflammation (present in around one-third of patients) (37) indeed plays a role in non-responsiveness to antidepressants and whether such a state is capable of predicting the outcome of the abovementioned antidepressant therapy regimens.
For this report, we have carried out a systematic review searching for the relevant literature on the prediction capability of soluble inflammatory compounds/cytokines in serum/plasma/CSF and/or the inflammatory state of circulating leukocytes for the outcome of antidepressant/anti-inflammatory treatments in MDD. We combined the outcomes of the systematic review with experimental data collected in the EU-MOODINFLAME consortium on the prediction capability of the inflammatory state of circulating monocytes (as measured by inflammatory gene expression). Two EU-MOODINFLAME trials could be evaluated, a trial carried out on patients with MDD collected at the Rotterdam site and treated in first line with venlafaxine or imipramine (38), and a small trial carried out on patients with MDD collected at the Munich site and treated with sertraline plus add-on celecoxib or placebo.
We conducted a systematic literature search in the PubMed/MEDLINE and Web of Science databases to identify immune-inflammatory predictors for treatment response to antidepressants, anti-inflammatory agents, and/or their combination with anti-inflammatory agents (or anti-inflammatory agents alone) in MDD from inception (for anti-inflammatory) and from 2008 (for antidepressant) until August 16, 2018. To find additional relevant studies, citation lists of included articles were tracked in Google Scholar (39) or citation lists of topic-related reviews and meta-analyses were checked. The last author of a significant paper concerning celecoxib and an expert in the field (NM) was also contacted and asked of awareness of any additional studies.
The following search terms were used: (mdd OR major depressive disorder OR depression) AND (inflammation) AND (therapy OR treatment OR antidepressant drugs OR sertraline OR venlafaxine OR escitalopram OR citalopram OR tricyclic OR ssri OR snri) AND (biomarker OR cytokines OR il-6 OR t cells OR nk cells OR th17 OR leukocytes OR macrophages OR crp OR genes) AND (response OR prediction), (mood disorder OR depression OR bipolar) AND (anti-inflammatory OR inflammation) AND (therapy OR treatment OR medication OR drugs OR add-on OR adjunct OR anti TNF OR infliximab OR CRP OR aspirin OR ASA OR acetyl salicylic acid OR minocycline OR omega 3 fatty acids OR NAC OR acetylcysteine OR cox 2 inhibitor OR celecoxib) AND (biomarker OR cytokines OR macrophages OR t cells OR NK cells OR leukocytes OR CRP OR genes).
The initial search of 7,047 studies resulted in 174 relevant studies selected by title. Inclusion criteria for further selection were:
1. publications written in the English language;
2. human clinical trials;
3. the diagnosis of MDD. Because of the paucity of studies in unipolar depression, both unipolar and bipolar depression were included for the studies on (add-on) anti-inflammatory agents. To make comparisons possible, we indicate in the result section (Table 1C, marked with B and C) which studies included bipolar depressed patients, and we discuss in the Discussion section putative differences stemming from this inclusion.
4. the absence of severe somatic diseases (especially inflammation-related);
5. the assessment of immune biomarkers;
6. the use of first-line or other antidepressant agents or the use of an anti-inflammatory agent added to antidepressant treatment or alone;
7. the assessment of symptom reduction with standardized measure [e.g., Hamilton Rating Scale for Depression (HAMD), Montgomery–Asberg Depression Rating Scale (MADRS), Beck’s depression inventory (BDI)] and
8. the analysis of responder and non-responder subgroups.
By reading the abstracts, methods, and results sections and applying the inclusion criteria and by removing duplicate records, 36 studies were selected. Further exclusion criteria were:
1. no predictive information provided;
2. use of parameters that are not inflammatory biomarkers in a narrower sense [e.g., serotonin and kynurenine metabolites, brain-derived neurotrophic factor (BDNF), calcium-binding protein B (S100B), macrophage-derived chemokine (MDC), platelet-derived growth factor (PDGF), and Eotaxin-1/CCL11];
3. genetic studies were excluded except for leukocyte gene expression level studies;
4. the use of agents whose anti-inflammatory mechanisms are not direct and even questionable (e.g., l-methylfolate, pioglitazone, modafinil).
By applying these exclusion criteria, we finally included 24 reports in the systematic review.
With the purpose of providing a comprehensive presentation, we decided to split the remaining studies into studies concerning circulating inflammatory compounds/cytokines (n = 19, see Tables 1A–C) and gene expression in circulating leukocytes (n = 5; see Table 2). For detailed information about the study selection, see Figure 1.
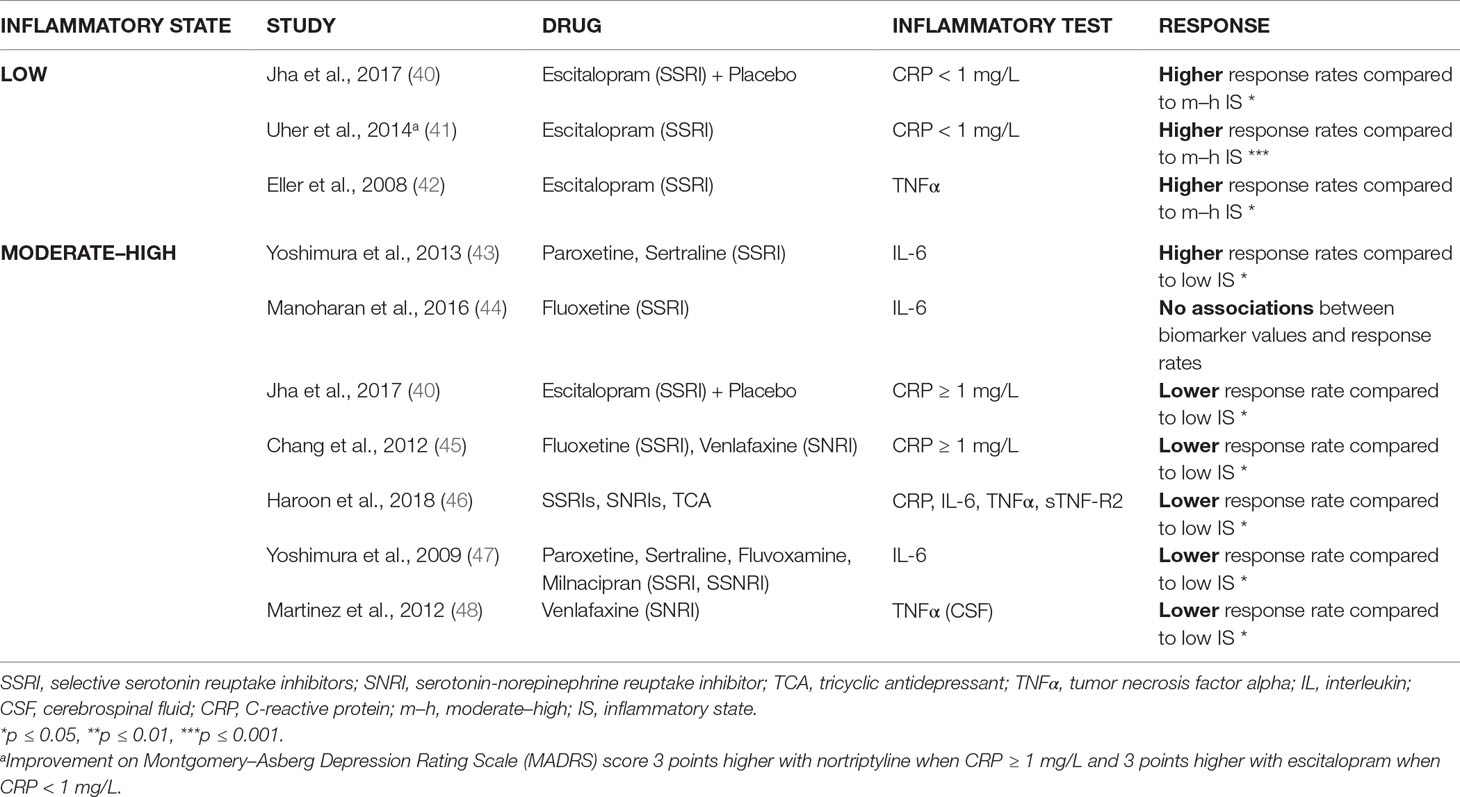
Table 1a Predominantly serotonergic action: higher response rates in low inflammatory state vs. moderate–high inflammatory state (prior to treatment).

Table 1b Predominantly noradrenergic, predominantly dopaminergic, and glutamatergic action: higher response rates in moderate–high inflammatory state vs. low inflammatory state (prior to treatment).
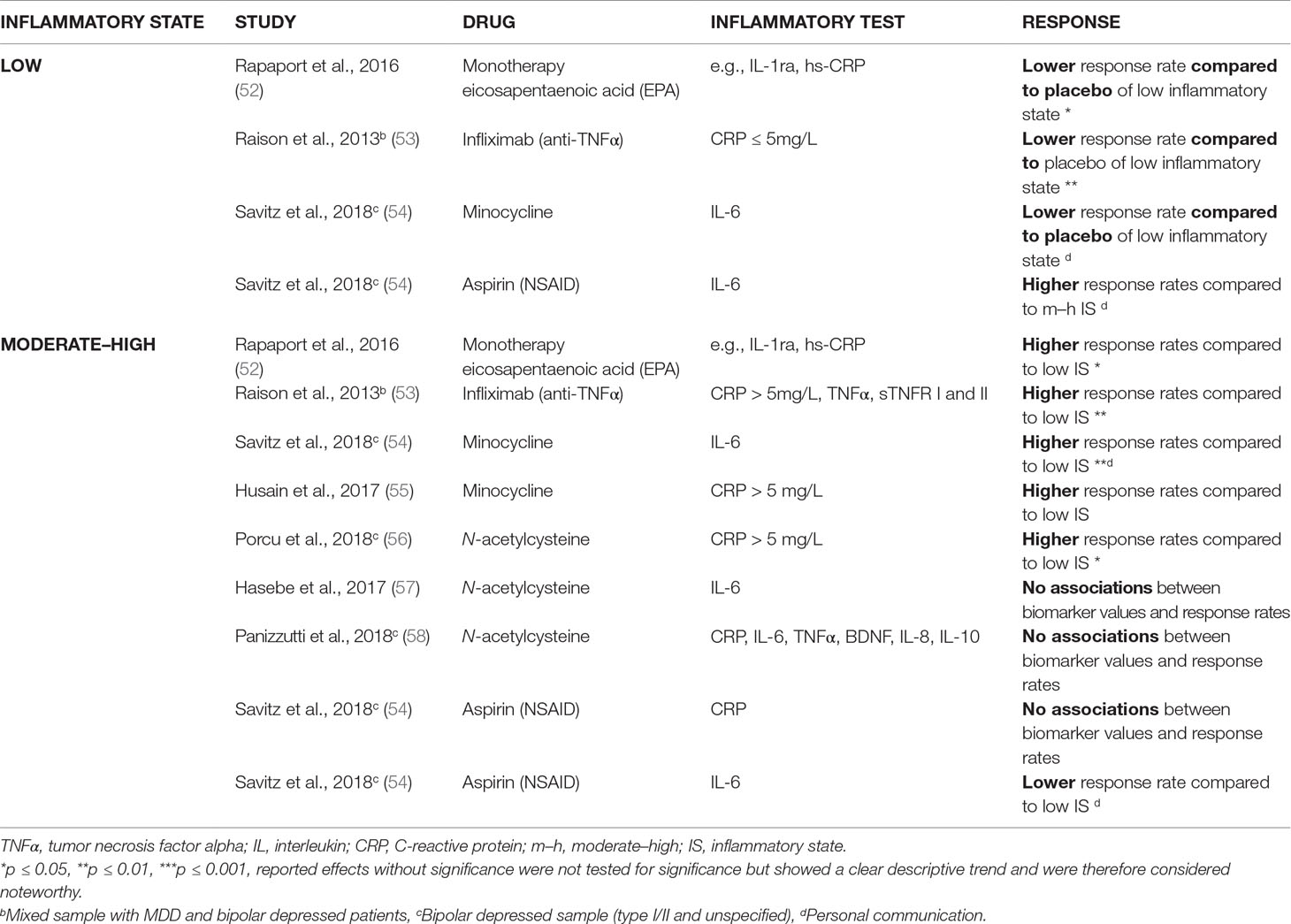
Table 1c Anti-inflammatory agents (added to an antidepressant regimen, except for one study): lower response rates in low inflammatory state (prior to treatment) versus placebo and higher response rates in moderate–high inflammatory state versus low inflammatory state (prior to treatment).
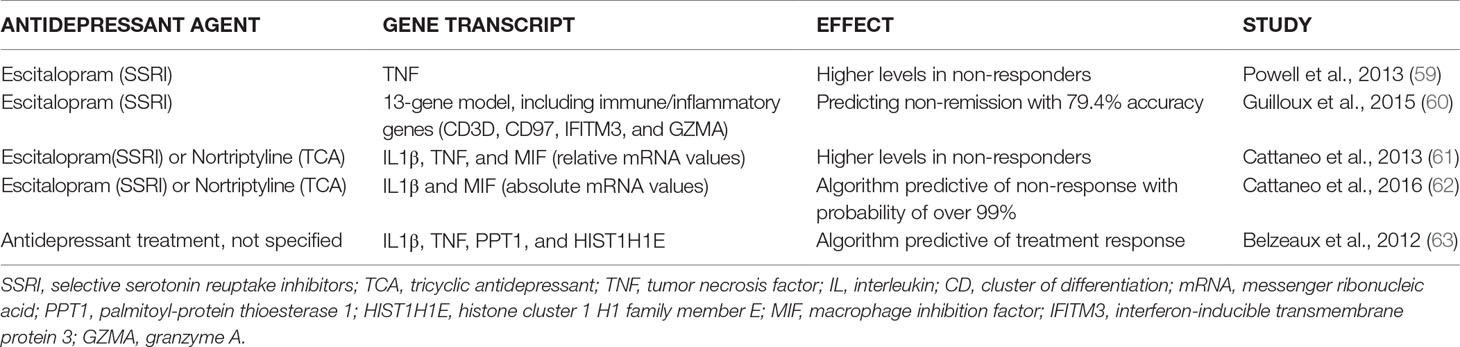
Table 2 The predictive capability of inflammatory state prior to therapy measured by circulating leukocyte gene expression for the response to various antidepressant regimens in MDD.
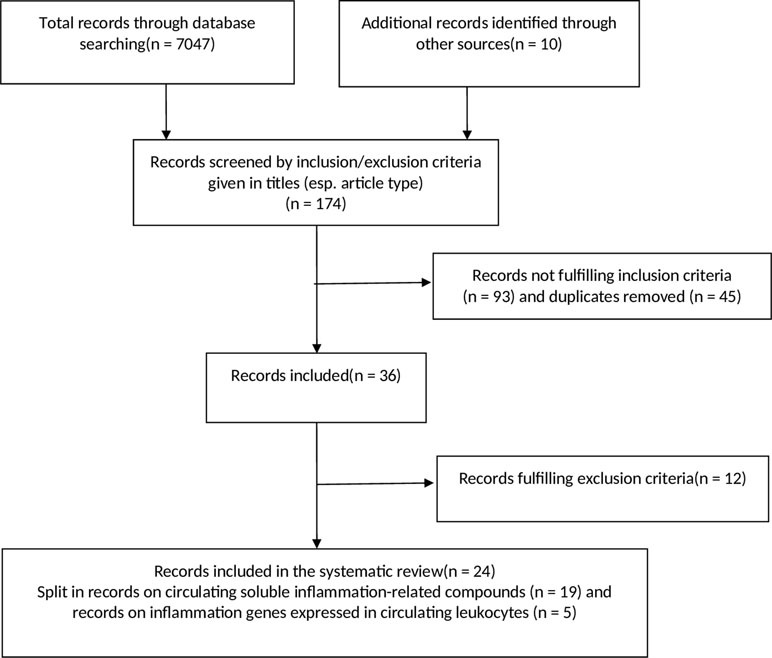
Figure 1 Flow diagram of the systematic research. See materials and methods section for further explanation.
Details on the inclusion and exclusion criteria, as well as on the clinical instruments and characteristics of patients, have been published before (38, 64). In short, in- and outpatients were recruited from the Departments of Psychiatry at the Erasmus Medical Centre (ErasmusMC) in Rotterdam (The Netherlands) and at the University Hospital of the Ludwig Maximilian University (LMU) in Munich (Germany). All patients were diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders, Text Revision (DSM-IV-TR) (65) and confirmed by using the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID-I) (66). Included were patients with a minimum score of 17 (Rotterdam) or 22 (Munich) on the Hamilton Rating Scale for Depression (HAMD, 17-item-version) (67).
Studies had been approved by the ethics committee of the medical faculty at the LMU, Munich (Germany), and the medical ethics committee of the ErasmusMC, Rotterdam (the Netherlands). The study was conducted in compliance with standards of Good Clinical Practice (CGP), assuring that the rights, safety, and well-being of patients were protected in accordance with the principles that have their origin in the Declaration of Helsinki (June 1964, last amendment Fortaleza 2013). Additionally, the relevant national and European regulations were adhered, too. After study procedures had been fully explained, all subjects provided written informed consent.
Healthy controls (HCs) were recruited from the same communities (Rotterdam and Munich). Details on the HC can be found in Refs. (64) and (68). In short, the inclusion criteria for HC were the absence of major DSM-IV-TR Axis I disorders including schizophrenia, psychotic disorders, mood disorders, anxiety disorders, or substance-related disorders according to DSM-IV criteria; the absence of usage of psychiatric drugs; and the absence of severe medical illness. HC had to be in self-proclaimed good health and free of any obvious medical illness for at least 2 weeks prior to the blood withdrawal, including acute infections and allergic reactions.
Being both double-blind studies, subjects, investigators, and study staff had been blinded to the treatment assignment for the duration of the study.
Prior to the start of antidepressants, patients with MDD underwent a wash-out period for at least 1 week. The use of benzodiazepines was allowed up to a maximum daily dose of 3 mg lorazepam or the corresponding equivalent. Subsequently, patients were randomly assigned to a 7-week monotherapy with either the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine (mean daily dose 371 mg, range dose of 300–375 mg/day) or with the TCA imipramine (mean dose 206 mg, range dose of 50–450 mg/day). The duration of the treatment trial was 7 weeks to ensure that patients treated with imipramine had adequate plasma levels for at least 4 weeks. Response to treatment was defined as ≥50% reduction of the initial HAM-D score.
Prior to the start of treatment, patients with MDD underwent a washout period for 3 days. The use of lorazepam or zopiclon was allowed in this period and also during the study, up to a maximum daily dose of 3 or 15 mg, respectively. Subsequently, patients were randomly assigned in a 1:1 ratio to a 6-week therapy with either the selective serotonin reuptake inhibitor (SSRI) sertraline plus placebo, or with sertraline plus the selective COX-2 inhibitor celecoxib. The dose of sertraline was flexible and ranged between 50 and 100 mg/day. A daily dose higher than 100 mg was not recommended, but in the expectation of more clinical benefit, a daily dose of 150 mg sertraline was allowed. The daily dose of celecoxib was 400 mg (200 mg in the morning and 200 mg in the evening). Patients from the placebo group received two identical capsules (morning and evening). As in the Rotterdam cohort, response to treatment was defined as ≥50% reduction of the initial HAM-D score.
Only patients and HC with full data regarding the expression levels of all key genes for monocyte inflammatory activation could be used for the present study. The Rotterdam sample therefore consisted of 34 MDD patients and 45 HC. Of the patient group, 14 patients were treated with venlafaxine and 20 patients were treated with imipramine. The Munich sample consisted therefore of 35 MDD patients and 42 HC. Of the patient group, 19 patients were treated with sertraline plus placebo, and 16 patients were treated with sertraline plus celecoxib.
Blood was collected in sodium-heparin tubes (36 ml) for immune cell preparation just prior to treatment. From the heparinized blood, peripheral blood mononuclear cell (PBMC) suspensions were prepared by low-density gradient centrifugation via Ficoll-Paque PLUS (GE Healthcare, Uppsala, Sweden) within 8 h to avoid erythrophagy-related activation of the monocytes. PBMCs were frozen in 10% dimethylsulfoxide and stored in liquid nitrogen. This enabled us to test immune cells of patients and controls together at a later stage. Tests were done at ErasmusMC.
CD14+ monocytes were isolated from aliquots of the frozen and thawed PBMCs by a magnetic cell sorting system (auto MACS Pro, Miltenyi Biotec, B.V., Bergisch Gladbach, Germany). The average viability was 86.3 ± 10.4 (Trypan blue staining) and the purity of monocytes was 95.1 ± 3.0% (flow cytometry). RNA was isolated from the purified monocytes using RNA easy mini kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). On average, monocytes cell yield after isolation was 2.0 ± 1.6 × 106/subject and the quantity of RNA in monocytes was 3.2 ± 1.8 μg. One microgram of RNA was reverse-transcribed using the cDNA high capacity reverse transcription kit (Applied Biosystems, Foster City, CA, USA). qPCR was performed using Taqman Arrays, format 48 (Applied Biosystems), according to the manufacturer’s protocol and validated against the single RT-qPCR method. Per fill port, 400 ng of cDNA (converted from total RNA) was loaded. PCR amplification was performed using an Applied Biosystems Prism 7900HT sequence detection system with TaqMan Array block. Thermal cycler conditions were 2 min at 50°C, 10 min at 94.5°C, 30 s at 97°C, and 1 min at 59.7°C for 40 cycles.
Based on several previous studies on mood disorders (21, 64, 69), we decided to include in our panel the most consistently abnormally expressed inflammatory genes in the studies. Therefore, relative to the housekeeping gene ABL1, the expression of a total of up to 49 genes was determined (also because of the maximum of fill ports in the Taqman assay) and expression values were calculated using the comparative threshold cycle (CT) method [see, for technical details, Refs. (21, 64, 69)]. The mentioned earlier studies also carried out a hierarchical clustering of these genes and found two main distinct clusters of gene expression. The first cluster is found consistently in virtually all of our monocyte inflammatory gene expression studies (also besides disease conditions such as mood disorders), and this cluster is composed of well-known pro-inflammatory cytokines and chemokines and important enzymes or transcription factors to produce these compounds. For the calculation of the “positivity of this inflammatory compound cluster”, we took the expression level of the top 10 genes [the most consistently overexpressed genes in all our studies thus far; see Ref. (64)] of this cluster into consideration, i.e., IL1β, CCL20, EREG, IL6, TNFAIP3, CXCL2, PDE4B, ATF3, PTX3, and IL1A. These genes accounted for 70–99% of the inflammatory cluster response. For each of the 10 genes, we determined a range of the HC gene expression (using the 2−ΔCt values). The range was defined by the mean of the values for that gene in HC monocytes ± 1 standard deviation (SD). Then, we used this range as a standard of comparison for the MDD patients’ gene expression. We decided to refer to a patient’s top gene as upregulated, if the patient’s gene expression was higher than HC’s mean plus 1×SD, or downregulated when it was lower than HC’s mean minus 1×SD. This was done for all 10 above given genes. Then, we declared the monocyte population of a given patient as “pro-inflammatory positive” if 6 of these 10 top inflammatory genes (or more) were upregulated. These data are given in Table 3 in the Results section. Similar calculations/algorithms for monocyte inflammatory positivity have been used by us before (21, 69–71). Further methodological details of the calculation can be found in these publications. Original Q-PCR data have been uploaded and can be retrieved via the GEO repository ref number GSE132315: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132315
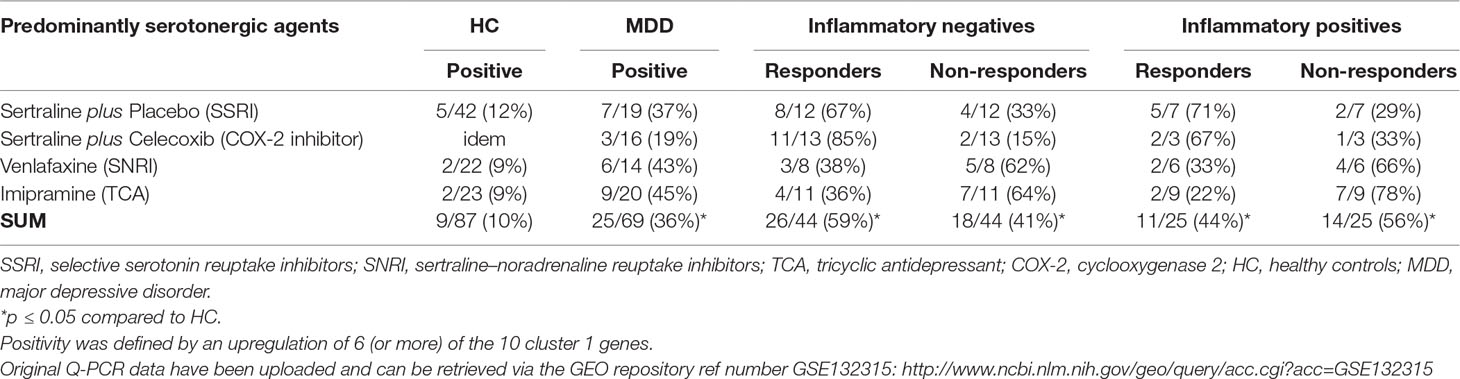
Table 3 Proportions of patients with a positive inflammatory gene signature of circulating monocytes prior to therapy measured in total and by response.
Statistical analyses were performed using IBM SPSS v.21 for Mac. Continuous sample characteristics are reported as mean (± standard deviation). Group comparisons (e.g., MDD vs. HC, responders vs. non-responders) were analyzed using analysis of variance (ANOVA) tests for continuous data (e.g., age) and using Pearson’s chi-square (χ²) tests for categorical data (e.g., gender). For group comparisons of positivity of monocyte gene expression (e.g., MDD vs. HC, responders/non-responders vs. HC, responders vs. non-responders), Pearson’s chi-square (χ²) tests were applied, too. All hypotheses were tested with α ≤ 0.05 (two-sided).
Tables 1A, 1B, and 1C show the data of the systematic review of the 19 selected articles (see the section Search Strategy for Systematic Review) regarding the predictive capability of inflammatory state [assessed by serum/plasma immune compounds (mainly CRP and cytokines) (only one study used CSF)] in patients with MDD for the response rates to various classes of antidepressant drugs and to anti-inflammatory agents added to an antidepressant regimen (except for one study, in which the anti-inflammatory agent was used as monotherapy) (52). For comprehensibility, we have grouped the outcomes in Table 1 according to the regimen used.
Table 1A shows that, in three out of three studies (40–42), antidepressants with a predominant serotonergic action [i.e., escitalopram (SSRI)] induced a better response in patients with low inflammatory markers as compared to patients with high inflammatory markers in the same study. On the contrary, when inflammatory markers were high, five out of seven of the studies (40, 45–48) showed that drugs with a predominant serotonergic action (i.e., SSRIs, SNRIs, and TCAs) induced reduced response rates as compared to patients with low inflammatory markers in the same study. Cutoff points for low and high levels were defined for CRP in the reviewed studies at 1 mg/L; for IL-6 and TNFα, values depended on the actual sensitivity of the assay used in the report. Two studies formed an exception. Manoharan et al. (44) did not find any effect of pre-selection of the inflammatory state. However, this study was special in that treatment duration was of only 6 weeks, and patients had a relatively low to moderate depression severity (HAMD score ≥ 13). The other study (43) showed the opposite message (i.e., an improved response to SSRIs in patients with a high inflammatory state as compared to a low inflammatory state). This study was special, in that many patients were treated with paroxetine (SSRIs), which—apart from its serotonergic action—also exerts a considerable dopaminergic action (72).
Taken together, predominantly serotonergic agents showed, in general, insufficient response rates in those patients with signs of moderate to high inflammation as measured by circulating inflammatory compounds.
The review also delivered that, in such conditions of moderate to high signs of inflammation, drugs with another mechanism of action than primarily serotonergic do show an effect. Using nortriptyline, mirtazapine, or ketamine alone, or combinations of an SSRI with nortriptyline or bupropion resulted, in 5 out of 5 studies, in improved responses rates (40, 41, 49–51) as compared to the patients with low inflammatory markers (Table 1B).
Similar beneficial effects existed for combinations of antidepressant drugs with anti-inflammatory agents. Table 1C shows that five out of seven studies (52–56) found a significant improvement of an (add-on) anti-inflammatory therapy, when patients with high signs were compared to patients with low signs of inflammation. The anti-inflammatory agents used in these studies were infliximab, minocycline, n-acetylcysteine, and fish oil (the latter as monotherapy, and compared to placebo).
It must be mentioned that the study of Savitz et al. (54) only noted such improving effect with minocycline; aspirin had no such effect in their study. Aspirin did work in their “non-inflamed” patients, yet had no effect or even a reduced effect in patients with high signs of inflammation, depending on the inflammatory serum marker used to determine the state of low-grade inflammation (CRP or IL-6, see Table 1C). The study of Savitz was also special in that both unipolar and bipolar depressed patients were included.
Table 1C additionally shows that there are also two out of three studies (57, 58) that showed that in the case of add-on n-acetyl cysteine, it was of no use to stratify the patients in low- or high-grade inflammation prior to therapy. Two of the studies of add-on n-acetyl cysteine (one showing and one not showing an effect of prior determination of the inflammatory state) involved both unipolar and bipolar depressed patients.
It was remarkable that when an add-on anti-inflammatory agent was given to patients with low signs of inflammation, reduced responses were obtained as compared to patients with high signs of inflammation and even to placebo (two out of three of such studies) (53, 54). Also, when fish oil (an agent with both lipid-correcting and anti-inflammatory properties) was given as a monotherapy to patients with a low inflammatory state, reduced responses were seen as compared to placebo ( (Table 1C). As mentioned above, add-on aspirin did induce an increased response in patients with low signs of inflammation in the study of Savitz et al. (54).
Taking these literature data together, it is difficult to draw a simple conclusion on the usefulness of a prior measurement of serum inflammatory markers for the determination of the effect of (add-on) anti-inflammatory agents. There is a clear trend that (add-on) anti-inflammatory agents, such as infliximab, minocycline, and fish oil are effective if inflammatory markers are clearly present, but this does not apply to aspirin and n-acetylcysteine. However, it is also safe to say that special caution must be given when there is an absence of circulating inflammatory markers in patients with MDD: the chances are high that the use of the effective anti-inflammatory agents (such as infliximab, minocycline, and fish oil) in states of moderate–high inflammation actually has an opposite effect than expected in such patients, namely, a reduced responsiveness.
Table 2 shows the studies we selected that dealt with the gene message for pro-inflammatory cytokine production in the circulating leukocyte pool prior to treatment and predictive for treatment outcome. We found five relevant articles.
In 2013, Powell et al. (59) described a significantly increased baseline expression of TNF in escitalopram non-responders (n = 21) compared to responders (n = 25) taken from the GENDEP study. In the same year, Cattaneo et al. (61) reported on data of the GENDEP study and found higher baseline mRNA levels for IL1β, macrophage inhibiting factor (MIF), and TNF in antidepressant (escitalopram or nortriptyline) non-responders compared to responders, the three cytokine expressions together explaining 46% of the variance of treatment response.
Belzeaux et al. (63) identified an algorithm of four mRNAs, including two cytokine genes (TNF and IL1β, together with PPT1 and HIST1H1E) to be predictive of the treatment response in MDD. However, the weakness of their study was that a whole scale of antidepressants was used, while numbers of patients and HC were limited (16 vs. 13). Guilloux et al. (60) predicted non-remission following escitalopram treatment in MDD with an accuracy of 79.4% using a 13-gene model including four genes associated with immune and inflammatory activation (however, TNF was not part of the 13 genes). Mediation of cell proliferation was another important function of the remaining genes, but not exclusively. In 2016, Cattaneo et al. (62) took the data of the GENDEP study further and reported that absolute values of the message for IL1β and MIF together could predict non-responsiveness to escitalopram or nortriptyline in over 99%. These outcomes were confirmed in an independent, naturalistic replication sample.
Taken together, it is clear that non-responsiveness to an SSRI or to a TCA (nortriptyline) can likely be predicted by determining the expression level of combinations of important immune genes (IL1β, MIF, TNF, and CD3) in preparations of circulating leukocytes of patients with MDD.
Prior to treatment, we could test 34 patients with MDD [mean age: 52.2 (±9.9) years, 59% females, collected at the ErasmusMC, Rotterdam] for inflammatory gene expression in their circulating monocytes. As a control group, we tested 45 HC of comparable age [mean age: 49.1 (±9.4) years] and gender (44% females). Of the 34 patients, 14 were treated with venlafaxine and 20 were treated with imipramine. An overall response rate of 11% was found in this trial, with 11/34 patients responding to treatment. The difference between the response rates for both treatment arms were not statistically significant, i.e., a response rate of 36% (5/14) for patients treated with venlafaxine and of 30% (6/20) for patients treated with imipramine. Vermeiden et al. (38) have reported extensively on this study and described that in the entire group of patients (n = 85), 45% of the patients responded to this first line of drug treatment (measured as 50% HAM-D reduction).
The other series of patients involved 35 patients with MDD [mean age: 41.4 (±10.8) years, 46.7% females, collected at the LMU, Munich] and 42 HC of comparable age [mean age: 37.9 (±11.9) years] and gender (61.9% females). Of the 35 patients, 19 were treated with sertraline plus placebo and 16 were treated with sertraline plus celecoxib. A high overall response rate was found in this trial, i.e., 26/35 (74.3%) of patients responded to treatment. The difference between the response rates for both treatment arms was not statistically significant, i.e., a response rate of 68.4% (13/19) for patients treated with sertraline plus placebo and 81.3% (13/16) for patients treated with sertraline plus celecoxib.
We determined with an already published algorithm [see the Section Monocyte Inflammatory Gene Expression and Ref. (45)] the inflammatory state of the monocytes using the top 10 cluster 1 inflammatory genes (IL1β, CCL20, EREG, IL6, TNFAIP3, CXCL2, PDE4B, ATF3, PTX3, and IL1A). We controlled the patient monocyte tests with the outcomes of the same tests carried out in HC. Table 3 shows that, in each study group, a significant larger proportion of patients had—prior to therapy—circulating monocytes with a positive inflammatory gene signature as compared to the respective HC. Taking all patients from the four study groups together, 25 of the 69 (36%) patients with MDD had circulating monocytes with a pro-inflammatory gene signature, while only 9 of 87 (10%) HC had such monocyte signature (p < 0.05). This observation is in accord with earlier observations that monocytes of part of the patients with MDD show signs of a high inflammatory state (21).
For the purpose of this study, we divided the total patient group in those with a negative monocyte inflammatory gene score and those with a positive score. The data in Table 3 show that in the response rates in three out of four patient groups, patients with a positive inflammatory gene score had a lower response rate than those without a positive score. This, however, did not apply to the sertraline plus placebo group, and also significant differences were not reached in any of the groups. The phenomenon of better responsiveness in “non-inflamed” MDD patients could also be seen in the total MDD patient group; patients with a positive inflammatory gene score had a lower response rate than patients without a positive inflammatory gene score (i.e., 44% vs. 59%); however, a statistical significance was not reached in the total group of patients.
The data of the systematic review and experimental monocyte data, as presented in this study, collectively point in the direction that the state of so-called low-grade inflammation does play a role in the outcome of antidepressant therapy of patients with MDD.
Low-grade inflammation is characterized by an increase in the serum level of pro-inflammatory compounds (e.g., CRP, IL-1 β, IL-6, and TNF-α) and/or an activation state of circulating or tissue resident immune cells, including the brain microglia. Both an increase in pro-inflammatory compounds in the blood of patients with MDD and a pro-inflammatory activation of microglia and/or of myeloid cells in the periphery (i.e., monocytes) have been documented in a considerable (≈30–40%) proportion of patients with MDD (20, 73, 74). Moreover, imaging and histological techniques have shown microglial activation in the hippocampus of depressed patients (75).
By producing an array of neurotrophic factors, pro- and anti-inflammatory cytokines (e.g., IL-6), as well as axon guidance molecules, non-inflammatory activated microglia has been implicated both in white matter integrity and in the adequate development and function of important stress-regulating systems in the healthy brain (76, 77). On the contrary, inflammatory activated microglia (and/or a transfer of peripheral pro-inflammatory cytokines to the brain) is thought to hamper the normal development, growth, and synaptic function of stress-regulating systems and brain connections important for mood regulation, such as the white matter tracts between the forebrain and the limbic system. To illustrate this, raised serum pro-inflammatory cytokine levels have been associated in mood disorder patients with increased activation of threat- and anxiety-related neuro-circuits (78), reduced neural responses to negative stimuli in frontal brain regions involved in cognitive and emotional functions (79), and compromised integrity of myelin sheaths in cortico-limbic networks involved in mood regulation (80).
Importantly, low-grade inflammation has been shown to influence not only brain development and function but also neurotransmission, with excellent reviews on the inhibitory effects of pro-inflammatory cytokines, such as IL-1β and TNF-α, on the synaptic availability of monoamines and BDNF, while the same cytokines have been shown to increase extracellular glutamate, all important molecular determinants in MDD pathogenesis and response to treatment (15).
The data from the here presented systematic review on circulating inflammatory compounds indicate that patients with MDD with an activated inflammatory state (as measured by, e.g., moderate to high levels of circulating CRP, IL-6, and/or TNF-α) show reduced response rates to antidepressant regimens with a primarily serotonergic action (e.g., escitalopram), while showing improved response rates to antidepressant regimens with a primarily noradrenergic (e.g., nortriptyline), dopaminergic (e.g., bupropion, mirtazapine), or glutamatergic action (i.e., ketamine).
The systematic review data on the inflammatory gene expression in circulating leukocytes confirmed this phenomenon, showing that patients with a high gene expression level of IL1β, TNF, and/or MIF did not respond well to interventions with an SSRI in comparison to MDD patients with a low expression of these genes. However, gene data in circulating leukocytes disagreed with the data reported for circulating inflammatory compounds regarding TCA. While the primarily noradrenergic TCA nortriptyline did not give a satisfactory response in patients with MDD with a high gene expression level in circulating leukocytes (see Table 2), nortriptyline did in patients with high levels of circulating inflammatory compounds (see Table 1B).
Apparently, inflammatory gene expression in leukocytes does not measure the same level of inflammation than the measurement of circulating inflammatory compounds in serum/plasma; a high inflammatory gene expression might typify a state of “stronger/other” inflammation in MDD needing a treatment with drugs beyond the serotonergic and noradrenergic drugs. In other studies, we have also noted that inflammatory gene expression in circulating cells does not correlate one to one with the circulating protein gene product in serum/plasma (81). We explained this phenomenon by assuming that resident cells, such as the endothelial cells and resident macrophages in the tissues, also contribute to the level of circulating inflammatory compounds.
Although the data of our experiments on inflammatory gene expression levels in circulating monocytes (a subset of the circulating leukocytes) did not deliver statistically significant results, they were, by and large, in agreement with the above-described findings for the gene expression in all circulating leukocytes and showed that patients with “inflammatory” monocytes showed reduced response rates to predominantly serotonergic drug interventions and patients with “non-inflammatory” monocytes showed higher response rates to these type of agents.
Collectively, we deduce from these data that MDD patients with an activated inflammatory state (as measured by moderate to high circulating levels of, e.g., CRP, TNF-α, and IL-6, or a high gene expression of, e.g., IL1β, TNF, and MIF in circulating leukocytes) need more than a monotherapy with a predominantly serotonergic agent to improve clinically in a satisfactory way. An option then seems to be an immediate step up to agents with also a strong dopaminergic or glutamatergic action.
The reason for a better response to dopaminergic or glutamatergic drugs in the case of signs of enhanced inflammation can only be speculated on. It is possible that these drugs are needed because they also have clear anti-inflammatory actions, counteracting the detrimental effects of the high inflammatory state on the signs and symptoms of depression. There is ample evidence that dopamine and ketamine can reduce the production of pro-inflammatory cytokines and enhance that of anti-inflammatory cytokines (82, 83). On the other hand, the pro-inflammatory state itself may lead to an altered neurotransmitter metabolism, necessitating more than a primarily serotonin reuptake inhibition, but also an intervention in the dopamine or glutamate metabolism. Pro-inflammatory cytokines have been reported to activate neuronal mitogen-activated protein kinase (MAPK) pathways, increasing monoamine transporter expression and activity in general, which leads to an increased pre-synaptic reuptake of not only serotonin but also other neuroactive amines (84, 85). Furthermore, the state of enhanced inflammation is thought to lead to an enhanced tryptophan breakdown via the kynurenine pathway, resulting in various neuroactive compounds, among which NMDA agonists and antagonists, aggravating glutamatergic neurotransmitter imbalances (86, 87). This might also necessitate more than only a serotonin reuptake inhibition to be effective.
A step up to dopaminergic and glutamatergic antidepressants was more effective in “inflammatory” MDD patients than in “non-inflammatory” patients, and a combination of an antidepressant with an anti-inflammatory agent increased the response rates in these “inflammatory” patients with MDD as compared to “non-inflammatory” MDD patients. Though the reviewed literature data are scarce, the best prediction results seem to be obtained for infliximab (anti-TNF-α agent), minocycline (tetracycline), and eicosapentaenoic acid (fish oil). For n-acetylcysteine, the inflammatory state did show conjectural prediction effects, while for aspirin (acetylsalicylic acid), a reduced response was actually seen in “inflammatory” patients as compared to “non-inflammatory” patients.
The strength or character of the anti-inflammatory agents may have played a role in this variation of predictability of the state of inflammation for the (add-on) anti-inflammatory agents. Both anti-TNF agents and minocycline are in clinical practice and are considered stronger anti-inflammatory drugs than n-acetylcysteine and aspirin. However, for fish oil, a high inflammatory state was also predictive for a better effect in MDD patients, while fish oil is considered a relatively weak anti-inflammatory agent. Interestingly, fish oil exerts its anti-inflammatory effects via changing the “bad” pro-inflammatory lipid state of individuals (88), and perhaps the high state of inflammation in MDD patients is primarily driven by a bad lipid profile, which is then best corrected by fish oil.
Also, direct or indirect neurotransmitter effects of the anti-inflammatory agents may have played a role in the success or failure to predict their improved responsiveness in “inflammatory” MDD patients. Interestingly, two of the three add-on anti-inflammatory agents (minocycline and fish oil) that worked better in “inflammatory” than in “non-inflammatory” patients possess dopaminergic activities (89, 90). N-acetylcysteine, of which it is conjectural whether it works better as add-on in “inflammatory” than in “non-inflammatory” MDD patients, influences both dopamine and glutamate levels in the brain (91). Add-on aspirin, in contrast, had fewer effects in “inflammatory” MDD patients as compared to “non-inflammatory” patients; interestingly, aspirin has anti-glutamatergic actions (92, 93). These varying neuro-modulating actions of anti-inflammatory drugs make complex interactions in the neuro-immune network possible, inducing varying outcomes of combinations of antidepressants and anti-inflammatory agents. Of note also is that three of the reviewed studies of add-on anti-inflammatory agents had included bipolar depressed patients (54, 56, 58). This applies in particular to the study on aspirin (54), in which a reducing effect was found in “inflammatory” versus “non-inflammatory” patients. Intrinsic differences between bipolar and unipolar depression, such as differences in the immune and the glutamate state (94–98), may have played a role here.
Despite the above-listed uncertainties, it is nevertheless tempting to postulate—based on the outcomes of the literature review—that when MDD patients are “inflammatory”, (add-on) anti-inflammatory drugs are also an option to improve responsiveness and then the best results are probably obtained when anti-inflammatory agents are potent, influence lipid metabolism, and/or influence primarily dopaminergic synaptic transmission.
Regarding the use of (add-on) anti-inflammatory agents, another important message emerges from our systematic review of the literature. Interestingly, three out of four reports (52–54) indicated that “non-inflammatory” MDD patients showed a reduced response rate as compared to even placebo to the effective (add-on) intervention with an anti-inflammatory agent. In other words, the addition of the anti-inflammatory drugs effective in “inflammatory” patients was detrimental, and the anti-inflammatory drugs inhibited the effect of the antidepressants or delayed natural recovery. Such an outcome of an anti-inflammatory regimen is counterintuitive, if one assumes that inflammation contributes to depressive symptomatology (see before). The authors of one of the papers describing this phenomenon (53) explain their finding, that perhaps a small activation of the inflammatory system is needed for mental well-being and that both an extreme low and an extreme high activity of the inflammatory response system is disadvantageous for mental health. In other words, there would be an optimal set point for the inflammatory state of an individual for mental health. Downregulating this optimal state with an effective anti-inflammatory agent would, in such a view, be counterproductive and would open the way for the development of depressive symptoms.
Another explanation is that there exists a form of MDD that is non-immune and characterized by absent serological markers of immune activation. As indicated, (add-on) aspirin has a beneficial effect in patients and it can be hypothesized that it is in particular the neuro-modulating effect (anti-glutamatergic) of aspirin that induces this effect.
Based on this literature review, what appears to be the best and easiest assay system to measure the inflammatory state of MDD patients? The systematic review data on the gene expression level of cytokines in circulating leukocytes showed that two of the leukocyte gene expression studies resulted in very good accuracy rates of prediction of non-responsiveness, and algorithms could be developed, which showed high accuracies from 75% to even a 100% to predict non-responsiveness to an SSRI/TCA drug intervention (60, 62). Apparently, high levels of inflammatory cytokine gene message in circulating leukocytes are a precise sign of poor (treatment) outcome, and perhaps even better than high levels of inflammatory compounds/cytokines in serum/plasma. Nevertheless, it is technically less demanding to measure inflammatory compounds/cytokines in serum/plasma than to perform a gene expression assay in circulating leukocytes. Regarding the inflammatory markers best to be measured in serum/plasma to determine a raised inflammatory state in patients with MDD, it is worthy to note that the most consistent effects were found in our literature analysis with circulating CRP and/or IL-6 levels. These inflammatory compounds were tested in a large proportion of the here reviewed studies on serum inflammatory compounds (15/19), and outcomes and conclusions were congruent between these studies regarding these two inflammation markers.
Circulating TNF-α was measured in only six studies; hence, sufficient information on the validity of this parameter is lacking. Importantly, one of the studies showed that circulating TNF-α levels were not in agreement with the general rule, finding that a high TNF-α level was not predictive of a decreased responsiveness to an SSRI/SNRI (while a high IL-6 level in the same study was) (43). Other circulating inflammatory compounds (e.g., IL-8, IL-10, and IL-1) have also been tested in the here reported studies, but in only very few studies, and therefore data cannot be reliably evaluated. They nevertheless showed the general trend for serum/plasma factors that, in a state of inflammation, more than a monotherapy with a predominantly serotonergic agent might be needed.
Collectively, it seems that for predicting responsiveness to regular antidepressants, the avenue exploring the usefulness of serum/plasma CRP and IL-6 determination is the easiest and clinically the most feasible and promising approach. High levels of CRP/IL-6 would indicate that treatment with a serotonergic drug is not effective enough. However, the data reviewed here also indicate that the gene expression in circulating leukocytes cannot be neglected as a predicting parameter due to the reported high levels of accuracy to predict non-responsiveness to SSRI/SNRI and TCA therapy.
In this article, we only focused on inflammation parameters as determinants for the outcome of treatment. The various other determinants important for treatment outcome have recently been reviewed by Perlman et al. (99). The authors described not only that inflammation-related determinants are important but also that a whole array of genetic, endocrine, neuroimaging, sociodemographic, and symptom-based predictors turn out to influence outcome. However, due to heterogeneous sample sizes, effect sizes, publication biases, and methodological disparities across reviews, Perlman et al. (99) concluded that they could not accurately assess the strength and directionality of the predictors, and the authors therefore highlighted the importance of large-scale research initiatives and the use of clinically easily accessible biomarkers, as well as the need for replication studies of current findings. Clearly, we support such view and underscore the notion that our review data are also affected by the heterogeneous sample sizes, effect sizes, publication biases, and methodological disparities and that the data do not yet give a clear-cut picture. Also, our own experimental data on monocyte gene expression were underpowered and too limited to obtain clear-cut results and significances. Thus, clearly more studies are needed using standardized add-on anti-inflammatory treatments to standardized single antidepressant medications to develop a clearer picture of the actual response rates in immune and otherwise stratified patients with MDD.
There are excellent recent reviews on the discovered signs of low-grade inflammation in psychiatric patients that have transformed our understanding of neuropsychiatric diseases and urge for new diagnostic and therapeutic criteria in the emerging field of immuno-psychiatry (100). There are, however, at present, insufficient data and reliable concepts on the inflammation pathogenesis of MDD to design clinically applicable treatment drug protocols with reliable cutoff points for inflammatory parameters to guide therapy regimens.
Despite this limitation, a few generalizations can nevertheless be made from our study regarding inflammation as a predictor. Of the inflammation parameters, the serum CRP and IL-6 seem to be the most promising parameters for further clinical development. They are relatively easy to determine and, thus, useful in clinical studies. Using these parameters, a state of raised inflammation (as evidenced by raised serum CRP and IL-6 levels) characterizes a form of MDD with a relatively poor outcome and a non-responsiveness to agents with a predominant serotonergic action. Such cases might need a faster step-up to drug regimens with agents with dopaminergic (e.g., mirtazapine and bupropion) or glutamatergic (e.g., ketamine) effects, or a combination of a first-line antidepressant with an anti-inflammatory agent such as infliximab, minocycline, or fish oil (but not aspirin), most of them showing dopaminergic action. Varying anti-inflammatory properties of antidepressants as well as varying neuro-modulatory effects of anti-inflammatory agents (and/or complex interactions thereof) may play a role in the therapeutic success or failure of the step-ups.
A word of caution is needed regarding the regimens using as add-on the successful anti-inflammatory agents infliximab, minocycline, and fish oil: There must indeed be laboratory signs of inflammation (i.e., raised serum levels of CRP or IL-6) for this addition to be effective. If not, even response rates lower than the non-add-on situation might be obtained.
These studies were carried in compliance with standards of CGP, assuring that the rights, safety, and well-being of patients were protected in accordance with the principles that have their origin in the Declaration of Helsinki (June 1964, last amendment Fortaleza 2013). Additionally, the relevant national and European regulations were adhered, too. After study procedures had been fully explained, all subjects provided written informed consent.
GA and MS designed the strategy of the present review. BB and EW collected part of the study cohort, GA, AW and HD evaluated the data. GAH and MS wrote the first draft of the paper, HD and NM contributed with supervision and expert advice and critically revised the draft. All other authors contributed to the manuscript revision and approved the submitted version.
This study was financially supported by the EU via the MOODINFLAME project (EU-FP7-HEALTH-F2-2008-222963), the PSYCHAID (EU-FP7-PEOPLE-2009-IAPP-MarieCurie-286334), and the MOODSTRATIFICATION project (H2020-EU.3.1.1., GA754740). NM and GA were additionally supported by the foundation “Immunität und Seele.” The funders had no role in study design, data collection, analysis and interpretation of data, the writing of the report, and the decision to submit the paper for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to acknowledge the excellent technical assistance of Harm de Wit, Thomas Hoogenboezem, and Angelique van Rijswijk. Mu Myint collected much of the material in Munich.
1. Müller N, Hofschuster E, Ackenheil M, Mempel W, Eckstein R. Investigations of the cellular immunity during depression and the free interval: evidence for an immune activation in affective psychosis. Prog Neuropsychopharmacol Biol Psychiatry (1993) 17(5):713–30. doi: 10.1016/0278-5846(93)90055-W
2. Connor TJ, Leonard BE. Depression, stress and immunological activation: the role of cytokines in depressive disorders. Life Sci (1998) 62(7):583–606. doi: 10.1016/S0024-3205(97)00990-9
3. Anisman H, Merali Z. Anhedonic and anxiogenic effects of cytokine exposure. Adv Exp Med Biol (1999) 461:199–233. doi: 10.1007/978-0-585-37970-8_12
4. Mikova O, Yakimova R, Bosmans E, Kenis G, Maes M. Increased serum tumor necrosis factor alpha concentrations in major depression and multiple sclerosis. Eur Neuropsychopharmacol (2001) 11(3):203–8. doi: 10.1016/S0924-977X(01)00081-5
5. Myint A-M, Leonard BE, Steinbusch HWM, Kim Y-K. Th1, Th2, and Th3 cytokine alterations in major depression. J Affect Disord (2005) 88(2):167–73. doi: 10.1016/j.jad.2005.07.008
6. Irwin MR, Miller AH. Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav Immun (2007) 21(4):374–83. doi: 10.1016/j.bbi.2007.01.010
7. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci (2008) 9(1):46–56. doi: 10.1038/nrn2297
8. Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med (2009) 71(2):171–86. doi: 10.1097/PSY.0b013e3181907c1b
9. Miller AH. Depression and immunity: a role for T cells? Brain Behav Immun (2010) 24(1):1–8. doi: 10.1016/j.bbi.2009.09.009
10. Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry (2010) 67(5):446–57. doi: 10.1016/j.biopsych.2009.09.033
11. Drexhage RC, Weigelt K, van Beveren N, Cohen D, Versnel MA, Nolen WA, et al. Immune and neuroimmune alterations in mood disorders and schizophrenia. Int Rev Neurobiol (2011) 101:169–201. doi: 10.1016/B978-0-12-387718-5.00007-9
12. Eyre HA, Stuart MJ, Baune BT. A phase-specific neuroimmune model of clinical depression. Prog Neuropsychopharmacol Biol Psychiatry (2014) 54:265–74. doi: 10.1016/j.pnpbp.2014.06.011
14. Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimäki M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav Immun (2015) 49:206–15. doi: 10.1016/j.bbi.2015.06.001
15. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol (2016) 16(1):22–34. doi: 10.1038/nri.2015.5
16. Grosse L, Hoogenboezem T, Ambrée O, Bellingrath S, Jörgens S, de Wit HJ, et al. Deficiencies of the T and natural killer cell system in major depressive disorder: T regulatory cell defects are associated with inflammatory monocyte activation. Brain Behav Immun (2016) 54:38–44. doi: 10.1016/j.bbi.2015.12.003
17. Gibney SM, Drexhage HA. Evidence for a dysregulated immune system in the etiology of psychiatric disorders. J Neuroimmune Pharmacol (2013) 8(4):900–20. doi: 10.1007/s11481-013-9462-8
18. Bergink V, Gibney SM, Drexhage HA. Autoimmunity, inflammation, and psychosis: a search for peripheral markers. Biol Psychiatry (2014) 75(4):324–31. doi: 10.1016/j.biopsych.2013.09.037
19. Poletti S, de Wit H, Mazza E, Wijkhuijs AJM, Locatelli C, Aggio V, et al. Th17 cells correlate positively to the structural and functional integrity of the brain in bipolar depression and healthy controls. Brain Behav Immun (2017) 61:317–25. doi: 10.1016/j.bbi.2016.12.020
20. Beumer W, Gibney SM, Drexhage RC, Pont-Lezica L, Doorduin J, Klein HC, et al. The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J Leukoc Biol (2012) 92(5):959–75. doi: 10.1189/jlb.0212100
21. Grosse L, Carvalho LA, Wijkhuijs AJM, Bellingrath S, Ruland T, Ambrée O, et al. Clinical characteristics of inflammation-associated depression: monocyte gene expression is age-related in major depressive disorder. Brain Behav Immun (2015) 44:48–56. doi: 10.1016/j.bbi.2014.08.004
22. Gałecki P, Talarowska M. Inflammatory theory of depression. Psychiatr Pol (2018) 52(3):437–47. doi: 10.12740/PP/76863
23. Medina-Rodriguez EM, Lowell JA, Worthen RJ, Syed SA, Beurel E. Involvement of innate and adaptive immune systems alterations in the pathophysiology and treatment of depression. Front Neurosci (2018) 12. doi: 10.3389/fnins.2018.00547
24. Pundiak TM, Case BG, Peselow ED, Mulcare L. Discontinuation of maintenance selective serotonin reuptake inhibitor monotherapy after 5 years of stable response: a naturalistic study. J Clin Psychiatry (2008) 69(11):1811–7. doi: 10.4088/JCP.v69n1117
25. Dell’Osso B, Palazzo MC, Oldani L, Altamura AC. The noradrenergic action in antidepressant treatments: pharmacological and clinical aspects. CNS Neurosci Ther (2011) 17(6):723–32. doi: 10.1111/j.1755-5949.2010.00217.x
26. Furukawa TA, Salanti G, Atkinson LZ, Leucht S, Ruhe HG, Turner EH, et al. Comparative efficacy and acceptability of first-generation and second-generation antidepressants in the acute treatment of major depression: protocol for a network meta-analysis. BMJ Open (2016) 6(7):e010919. doi: 10.1136/bmjopen-2015-010919
27. Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet (2009) 373(9665):746–58. doi: 10.1016/S0140-6736(09)60046-5
28. Frick LR, Rapanelli M. Antidepressants: influence on cancer and immunity? Life Sci (2013) 92(1):525–32. doi: 10.1016/j.lfs.2013.01.020
29. Gałecki P, Mossakowska-Wójcik J, Talarowska M. The anti-inflammatory mechanism of antidepressants—SSRIs, SNRIs. Prog Neuropsychopharmacol Biol Psychiatry (2018) 80(Pt C):291–94. doi: 10.1016/j.pnpbp.2017.03.016
30. Fond G, Hamdani N, Kapczinski F, Boukouaci W, Drancourt N, Dargel A, et al. Effectiveness and tolerance of anti-inflammatory drugs’ add-on therapy in major mental disorders: a systematic qualitative review. Acta Psychiatrica Scandinavica (2014) 123(3):163–79. doi: 10.1111/acps.12211
31. Müller N. Immunological aspects of the treatment of depression and schizophrenia. Dialogues Clin Neurosci (2017) 19:1.
32. Husain MI, Strawbridge R, Stokes PR, Young AH. Anti-inflammatory treatments for mood disorders: systematic review and meta-analysis. J Psychopharmacol (Oxford) (2017) 31(9):1137–48. doi: 10.1177/0269881117725711
33. Adzic M, Brkic Z, Mitic M, Francija E, Jovicic MJ, Radulovic J, et al. Therapeutic strategies for treatment of inflammation-related depression. Curr Neuropharmacol (2018) 16(2):176–209. doi: 10.2174/1570159X15666170828163048
34. Fernandes BS, Dean OM, Dodd S, Malhi GS, Berk M. Acetylcysteine in depressive symptoms and functionality: a systematic review and meta-analysis. J Clin Psychiatry (2016) 77(4):e457–66. doi: 10.4088/JCP.15r09984
35. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol (2015) 15(2):104–16. doi: 10.1038/nri3793
36. Palacio JR, Markert UR, Martínez P. Anti-inflammatory properties of N-acetylcysteine on lipopolysaccharide-activated macrophages. Inflamm Res (2011) 60(7):695–704. doi: 10.1007/s00011-011-0323-8
37. Raison CL, Miller AH. Is depression an inflammatory disorder? Curr Psychiatry Rep (2011) 13(6):467–75. doi: 10.1007/s11920-011-0232-0
38. Vermeiden M, Mulder PG, van den Broek WW, Bruijn JA, Birkenhäger TK. A double-blind randomized study comparing plasma level-targeted dose imipramine and high-dose venlafaxine in depressed inpatients. J Psychiatr Res (2013) 47(10):1337–42. doi: 10.1016/j.jpsychires.2013.05.029
39. Bakkalbasi N, Bauer K, Glover J, Wang L. Three options for citation tracking: Google Scholar, Scopus and Web of Science. Biomed Digit Libr (2006) 3:7. doi: 10.1186/1742-5581-3-7
40. Jha MK, Minhajuddin A, Gadad B, Greer T, Grannemann B, Soyombo A, et al. Can C-reactive protein inform antidepressant medication selection in depressed outpatients? Findings from the CO-MED trial. Psychoneuroendocrinology (2017) 78:105–13. doi: 10.1016/j.psyneuen.2017.01.023
41. Uher R, Tansey KE, Dew T, Maier W, Mors O, Hauser J, et al. An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am J Psychiatry (2014) 171(12):1278–86. doi: 10.1176/appi.ajp.2014.14010094
42. Eller T, Vasar V, Shlik J, Maron E. Pro-inflammatory cytokines and treatment response to escitalopram in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32(2):445–50. doi: 10.1016/j.pnpbp.2007.09.015
43. Yoshimura R, Hori H, Ikenouchi-Sugita A, Umene-Nakano W, Katsuki A, Atake K, et al. Plasma levels of interleukin-6 and selective serotonin reuptake inhibitor response in patients with major depressive disorder: IL-6 and SSRI response. Hum Psychopharmacol (2013) 28(5):466–70. doi: 10.1002/hup.2333
44. Manoharan A, Rajkumar RP, Shewade DG, Sundaram R, Muthuramalingam A, Paul A. Evaluation of interleukin-6 and serotonin as biomarkers to predict response to fluoxetine. Hum Psychopharmacol (2016) 31(3):178–84. doi: 10.1002/hup.2525
45. Chang HH, Lee IH, Gean PW, Lee S-Y, Chi MH, Yang YK, et al. Treatment response and cognitive impairment in major depression: association with C-reactive protein. Brain Behav Immun (2012) 26(1):90–5. doi: 10.1016/j.bbi.2011.07.239
46. Haroon E, Daguanno AW, Woolwine BJ, Goldsmith DR, Baer WM, Wommack EC, et al. Antidepressant treatment resistance is associated with increased inflammatory markers in patients with major depressive disorder. Psychoneuroendocrinology (2018) 95:43–9. doi: 10.1016/j.psyneuen.2018.05.026
47. Yoshimura R, Hori H, Ikenouchi-Sugita A, Umene-Nakano W, Ueda N, Nakamura J. Higher plasma interleukin-6 (IL-6) level is associated with SSRI- or SNRI-refractory depression. Prog Neuropsychopharmacol Biol Psychiatry (2009) 33(4):722–6. doi: 10.1016/j.pnpbp.2009.03.020
48. Martinez JM, Garakani A, Yehuda R, Gorman JM. Proinflammatory and “resiliency” proteins in the CSF of patients with major depression. Depress Anxiety (2012) 29(1):32–8. doi: 10.1002/da.20876
49. Harley J, Luty S, Carter J, Mulder R, Joyce P. Elevated C-reactive protein in depression: a predictor of good long-term outcome with antidepressants and poor outcome with psychotherapy. J Psychopharmacol (2010) 24(4):625–6. doi: 10.1177/0269881109102770
50. Yang J, Wang N, Yang C, Shi J, Yu H, Hashimoto K. Serum interleukin-6 is a predictive biomarker for ketamine’s antidepressant effect in treatment-resistant patients with major depression. Biol Psychiatry (2015) 77(3):e19–e20. doi: 10.1016/j.biopsych.2014.06.021
51. Gupta R, Gupta K, Tripathi AK, Bhatia MS, Gupta LK. Effect of mirtazapine treatment on serum levels of brain-derived neurotrophic factor and tumor necrosis factor-α in patients of major depressive disorder with severe depression. Pharmacology (2016) 97(3–4):184–8. doi: 10.1159/000444220
52. Rapaport MH, Nierenberg AA, Schettler PJ, Kinkead B, Cardoos A, Walker R, et al. Inflammation as a predictive biomarker for response to omega-3 fatty acids in major depressive disorder: a proof of concept study. Mol Psychiatry (2016) 21(1):71–9. doi: 10.1038/mp.2015.22
53. Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression. JAMA Psychiatry (2013) 70(1):31–41. doi: 10.1001/2013.jamapsychiatry.4
54. Savitz JB, Teague TK, Misaki M, Macaluso M, Wurfel BE, Meyer M, et al. Treatment of bipolar depression with minocycline and/or aspirin: an adaptive, 2×2 double-blind, randomized, placebo-controlled, phase IIA clinical trial. Transl Psychiatry (2018) 8(1):27. doi: 10.1038/s41398-017-0073-7
55. Husain MI, Chaudhry IB, Husain N, Khoso AB, Rahman RR, Hamirani MM, et al. Minocycline as an adjunct for treatment-resistant depressive symptoms: a pilot randomised placebo-controlled trial. J Psychopharmacol (2017) 31(9):1166–75. doi: 10.1177/0269881117724352
56. Porcu M, Urbano MR, Verri WA, Barbosa DS, Baracat M, Vargas HO, et al. Effects of adjunctive N-acetylcysteine on depressive symptoms: modulation by baseline high-sensitivity C-reactive protein. Psychiatry Res (2018) 263:268–74. doi: 10.1016/j.psychres.2018.02.056
57. Hasebe K, Gray L, Bortolasci C, Panizzutti B, Mohebbi M, Kidnapillai S, et al. Adjunctive N-acetylcysteine in depression: exploration of interleukin-6, C-reactive protein and brain-derived neurotrophic factor. Acta Neuropsychiatrica (2017) 29(6):337–46. doi: 10.1017/neu.2017.2
58. Panizzutti B, Bortolasci C, Hasebe K, Kidnapillai S, Gray L, Walder K, et al. Mediator effects of parameters of inflammation and neurogenesis from a N-acetyl cysteine clinical-trial for bipolar depression. Acta Neuropsychiatrica (2018) 30(6):334–41. doi: 10.1017/neu.2018.13
59. Powell TR, Schalkwyk LC, Heffernan AL, Breen G, Lawrence T, Price T, et al. Tumor necrosis factor and its targets in the inflammatory cytokine pathway are identified as putative transcriptomic biomarkers for escitalopram response. Eur Neuropsychopharmacol (2013) 23(9):1105–14. doi: 10.1016/j.euroneuro.2012.09.009
60. Guilloux J-P, Bassi S, Ding Y, Walsh C, Turecki G, Tseng G, et al. Testing the predictive value of peripheral gene expression for nonremission following citalopram treatment for major depression. Neuropsychopharmacology (2015) 40(3):701–10. doi: 10.1038/npp.2014.226
61. Cattaneo A, Gennarelli M, Uher R, Breen G, Farmer A, Aitchison KJ, et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline “predictors” and longitudinal “targets”. Neuropsychopharmacology (2013) 38(3):377–85. doi: 10.1038/npp.2012.191
62. Cattaneo A, Ferrari C, Uher R, Bocchio-Chiavetto L, Riva MA, Pariante CM. Absolute measurements of macrophage migration inhibitory factor and interleukin-1-β mRNA levels accurately predict treatment response in depressed patients. Int J Neuropsychopharmacol (2016) 19(10):pyw045. doi: 10.1093/ijnp/pyw045
63. Belzeaux R, Bergon A, Jeanjean V, Loriod B, Formisano-Tréziny C, Verrier L, et al. Responder and nonresponder patients exhibit different peripheral transcriptional signatures during major depressive episode. Transl Psychiatry (2012) 2(11):e185. doi: 10.1038/tp.2012.112
64. Carvalho LA, Bergink V, Sumaski L, Wijkhuijs J, Hoogendijk WJ, Birkenhager TK, et al. Inflammatory activation is associated with a reduced glucocorticoid receptor alpha/beta expression ratio in monocytes of inpatients with melancholic major depressive disorder. Transl Psychiatry (2014) 4(1):e344. doi: 10.1038/tp.2013.118
65. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed., text rev). Washington, DC: American Psychiatric Association (2000).
66. First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I disorders (SCID I). New York: Biometric Research Department (1997).
67. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry (1960) 23:56–62. doi: 10.1037/t04100-000
68. Müller N, Krause D, Barth R, Myint AM, Weidinger E, Zill P, et al. Childhood adversity and current stress are related to pro- and anti-inflammatory cytokines in major depression. J Affect Disord (2019) 253:270–76. doi: 10.1016/j.jad.2019.04.088
69. Vogels RJ, Koenders MA, van Rossum EF, Spijker AT, Drexhage HA. T Cell Deficits and overexpression of hepatocyte growth factor in anti-inflammatory circulating monocytes of middle-aged patients with bipolar disorder characterized by a high prevalence of the metabolic syndrome. Front Psychiatry (2017) 8(34):1–11. doi: 10.3389/fpsyt.2017.00034
70. Mesman E, Hillegers MH, Ambree O, Arolt V, Nolen WA, Drexhage HA. Monocyte activation, brain-derived neurotrophic factor (BDNF), and S100B in bipolar offspring: a follow-up study from adolescence into adulthood. Bipolar Disord (2015) 17(1):39–49. doi: 10.1111/bdi.12231
71. Snijders G, Schiweck C, Mesman E, Grosse L, De Wit H, Nolen WA, et al. A dynamic course of T cell defects in individuals at risk for mood disorders. Brain Behav Immun (2016) 58:11–7. doi: 10.1016/j.bbi.2016.05.007
72. Nakayama K. Effect of paroxetine on extracellular serotonin and dopamine levels in the prefrontal cortex. Naunyn Schmiedebergs Arch Pharmacol (2002) 365(2):102–5. doi: 10.1007/s00210-001-0497-7
73. Köhler CA, Freitas TH, Maes M, de Andrade NQ, Liu CS, Fernandes BS, et al. Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. Acta Psychiatr Scand (2017) 135(5):373–87. doi: 10.1111/acps.12698
74. Hanisch UW. Microglia as a source and target of cytokines. Glia (2002) 40(2):140–55. doi: 10.1002/glia.10161
75. Czéh B, Nagy SA. Clinical findings documenting cellular and molecular abnormalities of glia in depressive disorders. Front Mol Neurosci (2018) 11(56):1–16. doi: 10.3389/fnmol.2018.00056
76. Sato K. Effects of microglia on neurogenesis. Glia (2015) 63(8):1394–1405. doi: 10.1002/glia.22858
77. Hökfelt T, Stanic D, Sanford SD, Gatlin JC, Nilsson I, Paratcha G, et al. NPY and its involvement in axon guidance, neurogenesis, and feeding. Nutrition (2008) 24(9):860–68. doi: 10.1016/j.nut.2008.06.010
78. Slavich GM, Way BM, Eisenberger NI, Taylor SE. Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc Natl Acad Sci USA (2010) 107(33):14817–22. doi: 10.1073/pnas.1009164107
79. Poletti S, Leone G, Hoogenboezem TA, Ghiglino D, Vai B, de Wit H, et al. Markers of neuroinflammation influence measures of cortical thickness in bipolar depression. Psychiatry Res Neuroimaging (2019) 285:64–6. doi: 10.1016/j.pscychresns.2019.01.009
80. Benedetti F, Poletti S, Hoogenboezem TA, Mazza E, Ambree O, de Wit H, et al. Inflammatory cytokines influence measures of white matter integrity in bipolar disorder. J Affect Disord (2016) 202:1–9. doi: 10.1016/j.jad.2016.05.047
81. Baldeón Rojas L, Weigelt K, de Wit H, Ozcan B, van Oudenaren A, Sempértegui F, et al. Study on inflammation-related genes and microRNAs, with special emphasis on the vascular repair factor HGF and miR-574-3p, in monocytes and serum of patients with T2D. Diabetol Metab Syndr (2016) 8(6):1–12. doi: 10.1186/s13098-015-0113-5
82. Beck G, Brinkkoetter P, Hanusch C, Schulte J, van Ackern K, van der Woude FJ, et al. Clinical review: immunomodulatory effects of dopamine in general inflammation. Crit Care (2004) 8(6):485–91. doi: 10.1186/cc2879
83. Loix S, KocK MD, Henin P. The anti-inflammatory effects of ketamine: state of the art. Acta Anaesthesiol Belg (2011) 62(1):47–58..
84. Zhu C-B, Blakely RD, Hewlett WA. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology (2006) 31(10):2121–31. doi: 10.1038/sj.npp.1301029
85. Zhu C-B, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology (2010) 35(13):2510–20. doi: 10.1038/npp.2010.116
86. Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry (2007) 12(11):988–1000. doi: 10.1038/sj.mp.4002006
87. Walker FR. A critical review of the mechanism of action for the selective serotonin reuptake inhibitors: do these drugs possess anti-inflammatory properties and how relevant is this in the treatment of depression? Neuropharmacology (2013) 67:304–17. doi: 10.1016/j.neuropharm.2012.10.002
88. Brooks JD, Norris PC, Dennis EA. Effects of omega-3 fatty acids on lipid metabolism and signaling using lipidomic analyses. FASEB J (2010) 24:475.1-475.1. doi: 10.1096/fasebj.24.1_supplement.475.1
89. Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci (2001) 98(25):14669–74. doi: 10.1073/pnas.251341998
90. Shin SS, Dixon CE. Oral fish oil restores striatal dopamine release after traumatic brain injury. Neurosci Lett (2011) 496(3):168–71. doi: 10.1016/j.neulet.2011.04.009
91. Berk M, Malhi GS, Gray LJ, Dean OM. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Sci (2013) 34(3):167–77. doi: 10.1016/j.tips.2013.01.001
92. De Cristóbal J, Moro MA, Dávalos A, Castillo J, Leza JC, Camarero J, et al. Neuroprotective effect of aspirin by inhibition of glutamate release after permanent focal cerebral ischaemia in rats. J Neurochem (2001) 79(2):456–9. doi: 10.1046/j.1471-4159.2001.00600.x
93. De Cristóbal J, Cárdenas A, Lizasoain I, Leza JC, Fernández-Tomé P, Lorenzo P, et al. Inhibition of glutamate release via recovery of ATP levels accounts for a neuroprotective effect of aspirin in rat cortical neurons exposed to oxygen-glucose deprivation. Stroke (2002) 33(1):261–7. doi: 10.1161/hs0102.101299
94. Becking K, Haarman BCM, Grosse L, Nolen WA, Claes S, Arolt V, et al. The circulating levels of CD4+ t helper cells are higher in bipolar disorder as compared to major depressive disorder. J Neuroimmunol (2018) 319:28–36. doi: 10.1016/j.jneuroim.2018.03.004
95. Dager SR, Friedman SD, Parow A, Demopulos C, Stoll AL, Lyoo IK, et al. Brain metabolic alterations in medication-free patients with bipolar disorder. Arch Gen Psychiatry (2004) 61(5):450–8. doi: 10.1001/archpsyc.61.5.450
96. Yüksel C, Öngür D. Magnetic resonance spectroscopy studies of glutamate-related abnormalities in mood disorders. Biol Psychiatry (2010) 68(9):785–94. doi: 10.1016/j.biopsych.2010.06.016
97. Gigante AD, Bond DJ, Lafer B, Lam RW, Young LT, Yatam LN. Brain glutamate levels measured by magnetic resonance spectroscopy in patients with bipolar disorder: a meta-analysis. Bipolar Disord (2012) 14(5):478–87. doi: 10.1111/j.1399-5618.2012.01033.x
98. Jun C, Choi Y, Lim SM, Bae S, Hong YS, Kim JE, et al. Disturbance of the glutamatergic system in mood disorders. Exp Neurobiol (2014) 23(1):28–35. doi: 10.5607/en.2014.23.1.28
99. Perlman K, Benrimoh D, Israel S, Rollins C, Brown E, Tunteng J-F, et al. A systematic meta-review of predictors of antidepressant treatment outcome in major depressive disorder. J Affect Disord (2019) 243:503–15. doi: 10.1016/j.jad.2018.09.067
Keywords: major depression, inflammation, antidepressant therapy, anti-inflammatory therapy, therapy prediction
Citation: Arteaga-Henríquez G, Simon MS, Burger B, Weidinger E, Wijkhuijs A, Arolt V, Birkenhager TK, Musil R, Müller N and Drexhage HA (2019) Low-Grade Inflammation as a Predictor of Antidepressant and Anti-Inflammatory Therapy Response in MDD Patients: A Systematic Review of the Literature in Combination With an Analysis of Experimental Data Collected in the EU-MOODINFLAME Consortium. Front. Psychiatry 10:458. doi: 10.3389/fpsyt.2019.00458
Received: 03 December 2018; Accepted: 11 June 2019;
Published: 09 July 2019.
Edited by:
Iris E. Sommer, University Medical Center Graniger, NetherlandsReviewed by:
Eva E. Redei, Northwestern University, United StatesCopyright © 2019 Arteaga-Henríquez, Simon, Burger, Weidinger, Wijkhuijs, Arolt, Birkenhager, Musil, Müller and Drexhage. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hemmo A. Drexhage, aC5kcmV4aGFnZUBlcmFzbXVzbWMubmw=
†These authors share first authorship.
‡These authors share last authorship.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.