
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 27 March 2019
Sec. Psychopharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fpsyt.2019.00157
This article is part of the Research Topic Current Topics in Opioid Research View all 26 articles
 Jordan K. Zjawiony1*
Jordan K. Zjawiony1* Antônio S. Machado2
Antônio S. Machado2 Ricardo Menegatti2
Ricardo Menegatti2 Paulo C. Ghedini3Elson A. Costa3
Paulo C. Ghedini3Elson A. Costa3 Gustavo R. Pedrino4Scott E. Lukas5
Gustavo R. Pedrino4Scott E. Lukas5 Octávio L. Franco6,7,8
Octávio L. Franco6,7,8 Osmar N. Silva6
Osmar N. Silva6 James O. Fajemiroye4,9*
James O. Fajemiroye4,9*Over the years, pain has contributed to low life quality, poor health, and economic loss. Opioids are very effective analgesic drugs for treating mild, moderate, or severe pain. Therapeutic application of opioids has been limited by short and long-term side effects. These side effects and opioid-overuse crisis has intensified interest in the search for new molecular targets and drugs. The present review focuses on salvinorin A and its analogs with the aim of exploring their structural and pharmacological profiles as clues for the development of safer analgesics. Ethnopharmacological reports and growing preclinical data have demonstrated the antinociceptive effect of salvinorin A and some of its analogs. The pharmacology of analogs modified at C-2 dominates the literature when compared to the ones from other positions. The distinctive binding affinity of these analogs seems to correlate with their chemical structure and in vivo antinociceptive effects. The high susceptibility of salvinorin A to chemical modification makes it an important pharmacological tool for cellular probing and developing analogs with promising analgesic effects. Additional research is still needed to draw reliable conclusions on the therapeutic potential of salvinorin A and its analogs.
Pain management is a challenging medical issue that requires a wide range of expertise and innovative ideas (1). Medicinal chemistry as well as extensive analysis of opioid receptors have increased the possibility of developing novel analgesics that are devoid of detrimental actions (2–6). Pain as an unpleasant sensory and emotional experience has been managed by different classes of drugs such as non-steroidal anti-inflammatory drugs, glucocorticoids, sodium channels inhibitors (local anesthetics), anti-epileptic drugs, tricyclic antidepressants, and opiates (2).
Opioid medications that mimic endogenous opioid peptides (dynorphins, endorphins, and enkephalins) typically bind to subtypes of opioid receptors (kappa-KOP, mu-MOP, and/or delta-DOP) to suppress pain (7). In addition, the activation of nociceptin/orphanin FQ peptide receptor (the fourth members of the opioid family of G protein-coupled receptors) by its endogenous peptide nociceptin/orphanin FQ (N/OFQ) modulates stress, reward and pain circuitry in several brain areas (8–11). A schematic representation of the pain and opioid sites of action, as shown in Figure 1, identifies important structures and pain modulatory circuits (12). The detailed account of signal transduction through opioid receptors, as illustrated by Figure 2, has been widely reported (13–17).
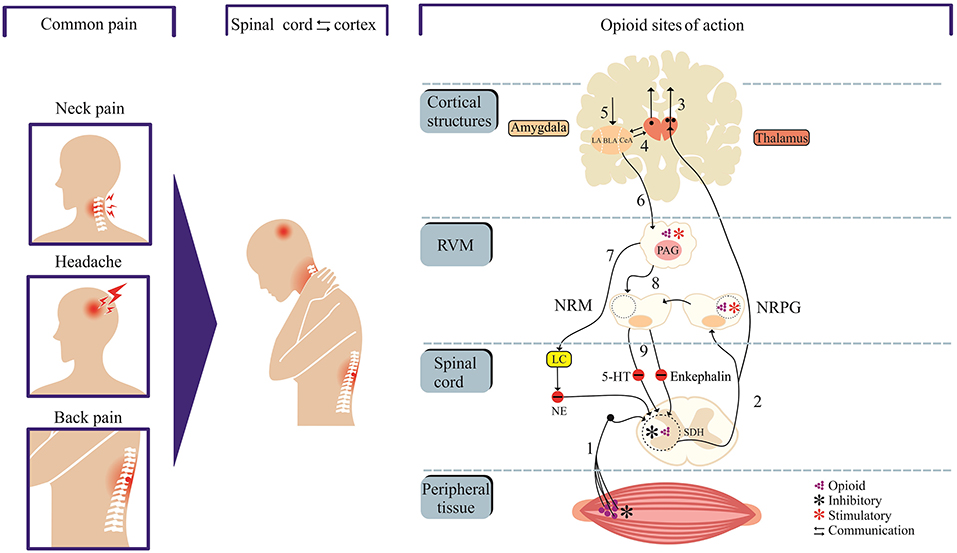
Figure 1. Neural circuits of pain and opioid site of action. Cortex and spinal cord communication modulate pain perception and offer targets for opioid drugs. Neural projections from peripheral tissue transmit nociceptive inputs through primary afferent fibers (1) to the spinal dorsal horn (SDH) before reaching the thalamus (2). Neural projections from thalamus target cortical sites (centers of pain processing, cognition, perceptions and integration) and amygdala (“emotional site”). The amygdala receives nociceptive inputs from the thalamus and cortex. The descending pain control system is mediated through projections from structures such as amygdala and hypothalamus to the periaqueductal gray matter (PAG) which in turn communicates with the rostral ventromedial medulla (RVM). The neural components within RVM [the nucleus raphe magnus (NRM) and nucleus reticularis paragigantocellularis (NRPG)] project to the spinal or medullary dorsal horns to directly or indirectly enhance or attenuate nociceptive transmission. The 5-hydroxytryptamine (5-HT) and enkephalin-containing neurons in the NRM project to the substantia gelatinosa of the dorsal horn and exert an inhibitory influence on transmission. Opioids sites of action include dorsal horn and peripheral terminals of nociceptive afferent neurons where opioids inhibit transmission. Opioids stimulate PAG and NRPG (blue asterisk), which in turn project to the rostroventral medulla. The locus coeruleus (LC) which receives inputs from the PAG releases noradrenalin to the dorsal horn, which in turn inhibits nociceptive transmission. Areas labeled 2–4 in red color and 5–8 in green color represent ascending and descending tracts, respectively.
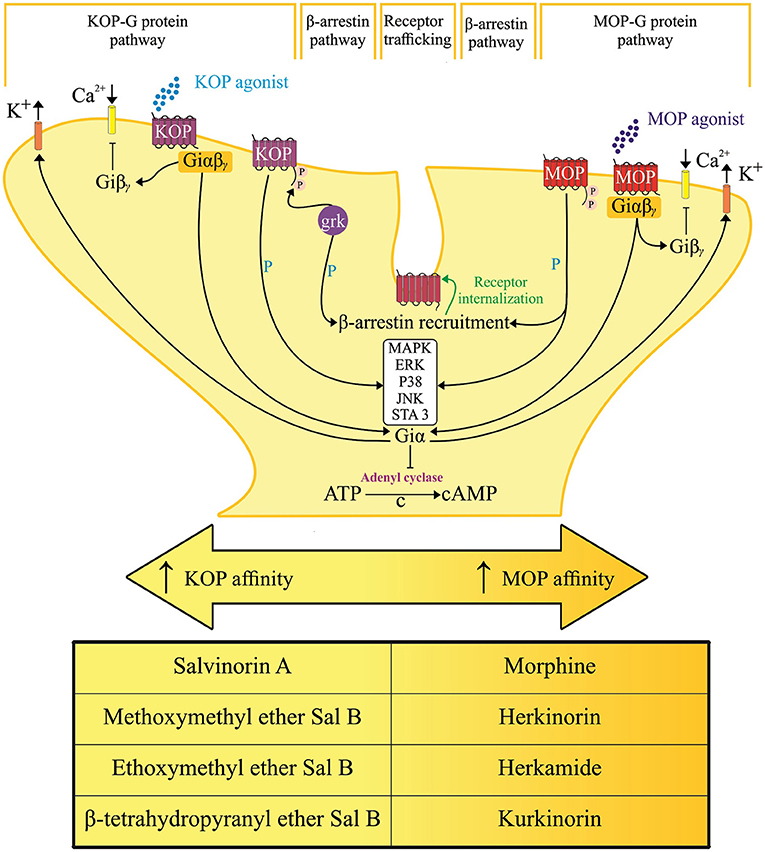
Figure 2. Hypothetical representation of signal transduction and trafficking of mu [μ] and kappa [κ] opioid receptor. Converging downstream pathways are activated by salvinorin A and its analogs with selective action and varying affinity on their respective opioid receptor subtypes. Arrows, activation; T lines, blockade of function; βγ, G protein β-γ subunit; cAMP, cyclic adenosine monophosphate; ERK, extracellular signal-regulated kinase; JNK, c-jun N-terminal kinase; MAPK, mitogen-activated protein kinases; GRK-3, G protein-receptor kinase 3; P, phosphorylation; C → , cyclization of adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP) through the cleavage of pyrophosphate.
The KOP ligands are important research tools and promising molecules for safer treatment of pain (18). The antagonists or partial agonists of KOP could prevent relapse to drug dependence (19–23). The blockade of KOP on dopamine terminals could disinhibit dopamine release in the nucleus accumbens and prevent drug withdrawal-induced dysphoria (24). This receptor remains an important cellular mediator of stress, reward, abuse, emotion, perception (25, 26), sedation (27), hypothermia (28), depression (29, 30), hallucination (31), conditioned place aversion, and locomotion impairment (32). Despite the possibility of undesirable KOP-mediated effects (33, 34), evidence has shown that this receptor subtype is an alternative molecular target for the development of safer analgesics (35).
Recently, Che et al. (3) conducted research on the active-state crystal structure of the KOP complex with a high-affinity agonist to provide molecular details of KOP and overcome the therapeutic limitations of its agonist. In this study, the authors identified residues that are critical for KOP activation and illuminate key molecular determinants of subtype selectivity and signaling bias. The affinity and specificity of drugs to KOP are fundamental to the array of inducible-biological effects (Table 1). The development of drugs that clearly separate pain relief from unwanted side effects has remained challenging and elusive.
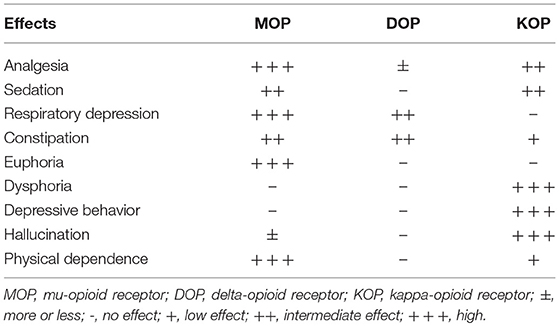
Table 1. Varying degree of opioid receptor involvement in some pharmacological effect.
Natural products are important sources of new drugs (36). Several active principles from medicinal plants have been used for pain relief (37). The main active principle of Salvia divinorum (38), salvinorin A, had been suggested as a useful research tool toward the development of analgesic drugs (39). Salvinorin A has a distinctive mode of action and pharmacology. Unlike psilocybin and lysergic acid diethylamide (alkaloidal hallucinogens which interact with specific serotonin receptor subtypes), the hallucinogenic effect of salvinorin A has been associated with its potent and selective KOP agonism. Salvinorin A shows no significant binding to over 50 other pharmacologically important receptors, transporter proteins and ion channels (40). As a non-nitrogenous KOP agonist (40), this compound differs from typical alkaloid opioid agonists. Recently, the potent antinociceptive effect of salvinorin A was reported in the neuropathic pain model (41). Over the years, medicinal chemists have synthesized several hundred analogs of this compound including herkinorin, kurkinorin, P-37, PR-38, methoxymethyl- and ethoxymethyl ether of salvinorin B, and β-tetrahydropyranyl ether of salvinorin B (4, 6, 42). Previous preclinical reports on salvinorin A and its analogs have revealed their promising antinociceptive effects (41, 43, 44). Hence, this review explored the structural and pharmacological profiles of salvinorin A and its analogs toward the development of new analgesic drugs.
The unique biological effects of salvinorin A (Figure 3A) have motivated many scientists to seek correlations between its chemical structure and pharmacological activity (4, 6, 42).
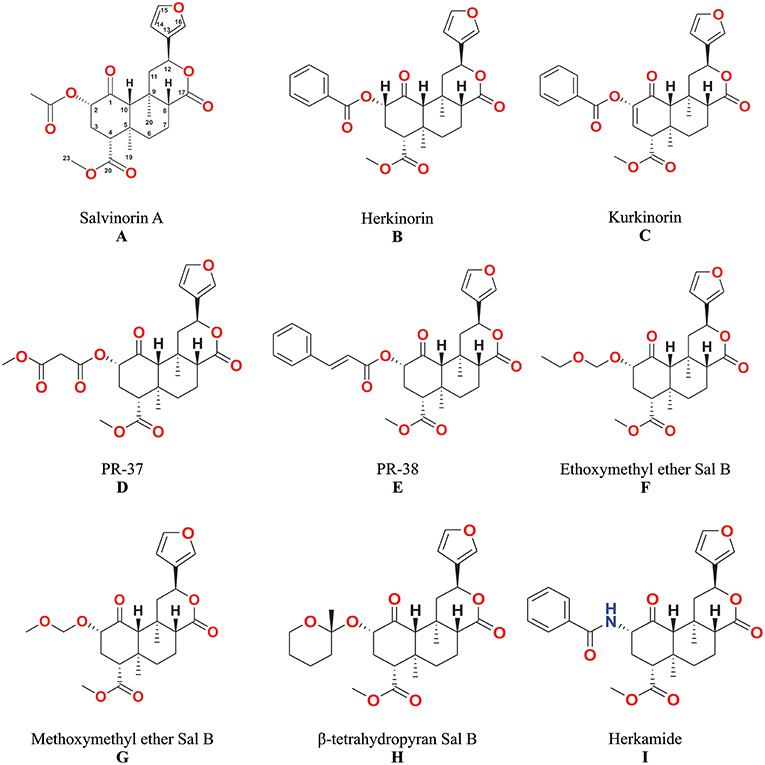
Figure 3. The structures of salvinorin A (A) and its analogs (B–I).
Following the determination of the salvinorin A structure through A single-crystal x-ray analysis [(38, 45), molecular modeling studies were performed to determine the interaction of this compound with KOP (40). Initially, the salvinorin A crystal structure (45) was docked by superimposition of its aromatic centroids and the carbonyl atoms with those of bound U69593 (known KOP agonist which shares structural similarity such as an aromatic ring and ester carbonyl groups separated by a short bond with salvinorin A). As a hydrogen bond acceptor, the carbonyl functionality supports the proposed role of Y139 and its interaction with the lactone carbonyl of salvinorin A (40, 46).
Previous study showed the list of residues that could form the salvinorin A-binding site of the KOP (40). The KOP models could accommodate the furan oxygen and 4-methoxycarbonyl functionality but not the 2-acetoxy group (40). The key residues in KOP that are responsible for the high binding affinity and efficacy of salvinorin A as well as important contacts between this compound and KOP have been identified through mutagenesis studies (6, 47). Potent and efficacious interactions of this compound with KOP are due to novel binding modes within a common three-dimensional space for binding and activating KOP (47).
Additional studies correlated the structure and activities of salvinorin A with the potential binding site on KOP. For instance, a change to the furan ring resulted in analogs that are more sterically demanding than a one-for-one aryl ring replacement (6). Sterically hindered environment of C-1 carbonyl of salvinorin A is not essential for activity as it is incapable of forming specific donor/acceptor contacts with residues in the receptor model, (48). In contrary, the 2-acetoxy group of salvinorin A which makes specific donor/acceptor contacts in the model is required for activity (45). Meanwhile, the lack of consensus binding model makes generalization of structure-activity relationships a challenge (6).
According to Yan (47), salvinorin A uses its flexible functional groups at C-2, C-4 and C-12 to optimize KOP interactions and stabilize itself in the binding site. Moreso, this compound also takes advantage of the conformational changes induced by G protein-coupling to facilitate active state stabilization and activation of downstream signaling events.
Salvinorin A, a neo-clerodane diterpenoid with seven stereogenic centers and three different types of ester functionalities, is a challenging substrate for chemical modifications because minor modifications can result in a complete loss or increase in pharmacological activity. For example, a product of hydrolysis of the C2 acetoxy side chain (salvinorin B) is totally devoid of activity, but other changes to this position actually demonstrate the highest KOP binding affinities (4, 6). Structural modifications of salvinorin A at the C1, C4, C12, and the C17-positions have been mostly associated with a reduction in KOP binding affinity (4, 6, 49). Over the years, the carbonate, carbamate, ester, ether, amine, amide, sulfonic ester, sulfonamide, thioester, halide, and other groups have been introduced to the salvinorin A molecule with a wide variety of outcomes (6, 50).
Furthermore, potential interactions of salvinorin A with other receptors have been either hypothesized or demonstrated preclinically by some researchers. Previous study showed that salvinorin A allosterically modulates MOP binding (51). The in vitro testing which showed the binding affinity of salvinorin A (EC50 values of 89 nM) against the D2 High receptor and blockade by 10 μM S-sulpiride (an antagonist of DRD2) has resulted into the discussion of partial agonism of salvinorin A at D2 receptor (52). Moreso, computational studies have predicted CB1, CB2, or DRD2 as a potential targets of salvinorin A (53). In an in vivo test, the attenuation of neuropathic pain by Salvinorin A was blocked by CB1 and KOP antagonists (41). The inhibition of the effects of salvinorin A on colonic motility by antagonists of OPRK, CB1 and CB2 in vitro and largely by antagonists of OPRK in vivo (54) suggests mechanistic complexity in the activity of salvinorin A as against widely acclaimed KOP selectivity.
Analogs with bulky alkyl esters at C-2 resulted in a loss in affinity for KOP (55), but the replacement of the alkyl with aryl esters at C-2 results in a lower affinity and potency for KOP; and an increase in affinity for MOP (56). Herkinorin (Figure 3B) is the first example of salvinorin A derivative with Ki at MOP (12 ± 1.0 nM) while still retaining lower affinity at KOP (90 ± 2.0 nM).
These compounds have an alkoxyalkyl ether bond, which replaced acetoxy group at C-2. The alkoxy methyl ether substituents improved KOP affinity and potency. The methoxymethyl ether of salvinorin B (Figure 3G) has higher binding affinity to KOP (Ki = 0.60 ± 0.1 nM) and potency (EC50 = 0.40 ± 0.04 nM) than salvinorin A. The putative synergistic binding interactions of the additional oxygen in the substituent have been associated with the higher affinity and potency (57, 58). The ethoxymethyl ether of salvinorin B (Figure 3F) also displayed a higher KOP binding affinity (Ki = 0.32 nM) and potency (EC50 = 0.14 nM) than other salvinorin A analogs (4, 59).
Previous studies have shown the synthesis and biological activities of Michael acceptor-type of salvinorin A analogs, such as methyl salvinorin B-2-O-malonate (PR-37) and 2-O-cinnamoylsalvinorin B (PR-38) (42, 54, 60). The addition of a second H-binding acceptor leads to the development of a malonate analog (PR-37) (Figure 3D) that displayed a 3-fold improvement in KOP affinity (Ki = 2.0 ± 0.9 nM) (42). However, other malonic ester substitutions with different carbonyl spacings reduced biological activity (6). The replacement of the acetate substituent with the spirolactone group caused a restriction in bond rotation and a decrease in potency (61). The analog with cinnamic ester functionality (PR-38) (Figure 3E) displayed not only KOP affinity (Ki = 9.6 ± 2.0 nM) but also MOP (Ki = 52 ± 9.0 nM) with 5.4 MOP/KOP selectivity (42).
Tidgewell (62) showed a lower KOP affinity as a result of bioisosteric exchange of the 2-acetoxy subunit of salvinorin A (Ki = 1.9 ± 0.2) with acetamide (Ki = 30 ± 2.0 nM). Although the introduction of a phenyl ring in the herkamide analog (Figure 3I) decreased KOP affinity, an increase in affinity for MOP - Ki = 3.1 ± 0.4 as compared to herkinorin Ki = 12 ± 1.0 nM was reported. In addition, herkamide MOP selectivity (KOP/MOP = 0.0004) was shown to be higher than that of herkinorin (KOP/MOP = 0.13).
The introduction of a double bond between C-2 and C-3 in herkinorin resulted in the new analog kurkinorin (Figure 3C). In vitro functional assay revealed that kurkinorin was more selective for MOP (>8,000-fold selectivity over KOP) than morphine (66-fold selectivity over KOP) and herkinorin (4.25-fold selectivity over KOP). Moreover, kurkinorin has similar potency when compared to MOP agonist such as DAMGO in forskolin-induced cAMP accumulation assays (63).
The relative flexibility of the acetoxy (C-2) subunit and potential adoption of different conformations when interacting with KOP has been hypothesized and studied in β-tetrahydropyranyl ether of salvinorin B (Figure 3H). Prevatt-Smith et al. (59) applied the concept of conformational restriction toward the development of ligands as tools to elucidate KOP affinity and potency. The new analog β-tetrahydropyranyl ether of salvinorin B showed slightly higher KOP affinity (Ki = 6.21 ± 0.4) than salvinorin A (Ki = 7.40 ± 0.4). This result showed that the rotational restriction strategy as proposed by Prevatt-Smith et al. (59) only led to small changes in binding values.
Unlike the modification of salvinorin A at C-2, the structural modifications at the C-4 present challenges because selective hydrolysis of methyl ester requires a more drastic condition that often leads to C-8 epimerization (6). Hence, additional efforts are often needed toward the separation of diastereoisomers. This seems to be part of the reason why analogs from C-4 position are fewer than that of C-2. A total loss of KOP binding affinity (Ki > 1000 nM) was reported for methyl, propyl and methoxymethyl esters at C-4 (57, 64).
The loss of KOP binding affinity in the long-chain ester has been associated with the fact that the pocket where the methyl ester fits is small and delimited by Trp287 and Tyr320 amino acids residues (65). Reduction of ester to alcohol has also led to an 87-fold decrease in KOP binding affinity as compared to salvinorin A (58). Consistent with these data, other studies have shown 33-fold and 385-fold losses in KOP affinity (64, 66). Some esters with modified regiochemistry with the exception of cyclopropyl ester showed a 17-fold loss of affinity (64).
The replacement of methyl ester at C-4 with amides or amines resulted in a 535-fold loss or a total loss (Ki > 10,000 nM) of KOP binding affinity, respectively (66). With the exception of alanine, the introduction of amino acid at C-4 resulted in a total loss of affinity (57, 64). Some substitutions with functional groups, such as carboxylic acid and aldehyde, have also resulted in total loss of KOP binding affinity (64).
The study of the analogs with substitutions at C-12 (the furan ring of salvinorin A) has attracted attention as a result of their metabolic stability (67). Although the interaction between furan and KOP is not well-established, the removal of this ring in salvinorin A resulted in total loss of activity while its hydrogenation resulted in a 7-fold loss (Ki = 14 ± 1.0 vs. 1.9 ± 0.2) in KOP binding affinity (68). Perhaps the possibility of hydrogen bonding, hydrophobic interactions or even π-π stacking type is essential for a receptor's recognition. (69), the regiochemical modification of the furan resulted in a 2-fold decrease in potency (EC50 = 12.2 ± 4.4 nM) without significant change in efficacy (Emax = 97 ± 2%) when compared to salvinorin A (vs. EC50 = 6.11 ± 0.04 nM and vs. Emax = 97 ± 8%).
Medicinal chemists have consistently modified salvinorin A structures to produce a wide range of analogs (4, 6). These efforts have helped to further understanding of salvinorin A chemistry and pharmacology as well as developing new compounds with potential therapeutic values (70). Since the identification of salvinorin A by Bücheler et al. (71), scientists have shown interests in its analgesic potential. Recently, the effectiveness of salvinorin A in a rodent model of pain showed that this compound could be beneficial for neuropathic pain relief (41). Salvinorin A is lipophilic, and it is mainly absorbed through the respiratory tract and to a lesser extent by the oral mucosa (72). Following the isolation and characterization of salvinorin A in vitro (receptor binding and functional assays) and in vivo, chemical modification of this compound led to changes in pharmacological parameters including stability, bioavailability, binding affinity, potency, functional activities, and selectivity (6, 73, 74). Hence, the activities of salvinorin A and its analogs offer clues toward the development of safer analgesics.
The pharmacological characterization of salvinorin A has been widely published (75). Salvinorin A was reported as a selective agonist of KOP through a competitive radioligand binding affinity assay (40). This pharmacological profile was subsequently replicated and confirmed by the findings of Chavkin and co-collaborators (76). As an important pharmacological target, the KOP has been implicated in the antinociceptive effect of salvinorin A (28, 77–79). Consistent with these reports, the potent antineuropathic pain of salvinorin A was blocked by the administration of a KOP antagonist (41). In addition to its selectivity to KOP, salvinorin A has a very high KOP potency. For instance, doses as low as 200 micrograms of this compound produce hallucination (31).
The pharmacology of salvinorin A is considered unique as a result of its structure and binding to the KOP (5). Despite being a potent activator of KOP-mediated G protein signaling, receptor internalization by salvinorin A is still poorly known (79). The internalization of receptor and β-arrestin recruitment are two cellular events that often accompany G protein activation (5). These cellular events have been linked to the underlying mechanism of unwanted side effects (80, 81). Hence, specific functional groups or structural features of salvinorin A that are critical to KOP interaction could be explored to repurpose analogs with only antinociceptive effect.
As highlighted above, binding affinity parameter has consistently been used for preclinical screening and as a basis for structural activity relationship studies. However, varying values of binding affinity data of salvinorin A and its analogs from different laboratories have raised questions about their reliability. Inconsistent data or lack of replicability of binding data could have resulted from the use of different radioligands to measure binding constants for the same analog (6).
Nature provides important chemical and pharmacological clues through the hydrolysis of salvinorin A at C-2 position that leads to salvinorin B and eliminates KOP activity (4, 48, 76). In this manner, the C-2 position represents a critical pharmacophore for salvinorin A and KOP interaction (57, 66, 76). In addition, the therapeutic potential of salvinorin A is limited as a result of its fast hydrolysis at the C-2 position by esterases (76, 82). Previous study which showed loss of the antinociceptive activity of salvinorin A after 20 min of intrathecal injection confirmed its short duration of action (77).
Medicinal chemists have advanced understanding of salvinorin A through its analogs (4, 6, 42). As mentioned earlier in this review, the aromatic substitution through the introduction of a phenyl group at C-2 as in herkinorin reduced KOP and increased MOP binding affinity (56). The structural change and selective activation of opioid receptors seem to be important clues to the antinociceptive effect of this compound. However, there is no data to exclude the possible hallucinations and physical dependence that are often associated with KOP and MOP agonists, respectively. Selective activation of opioid receptor-mediated beneficial pathways over deleterious signaling pathways offers an alternative therapeutic opportunity (3, 83, 84). According to some authors, the selective activation of Gi/o protein-mediated pathways over arrestin-mediated signaling could be a clue to designing safer drugs (85–87).
Some experimental data on salvinorin A analogs have shown preferential activation of G protein, β-arrestin recruitment among other molecular targets. Previous data showed that herkinorin promoted phosphorylation of MAP kinases ERK1/2 independent from β-arrestin-2 signaling and without promoting MOP recruitment of β-arrestin-2 (88). The β-arrestin-2 knockout mice with opioid treatment exhibited reduced opioid tolerance, improved the antinociceptive effect devoid of respiratory depression and constipation (89–91). Some authors have associated opioid dependence with the internalization of G protein—coupled receptors (80, 81, 92). The fact that herkinorin did not promote MOP internalization makes its potential application as an analgesic far more interesting.
Kurkinorin, which is considered to be extremely selective to MOP, showed a complete pharmacological change from salvinorin A which is known for a very high KOP binding affinity (63, 76). Moreover, kurkinorin also has greater selectivity for MOP than herkinorin. However, kurkinorin was found to recruit β-arrestin 2 (EC50 > 140 nM) with an efficacy of 96% and a bias factor of 0.57 when compared to DAMGO (63). Although these data suggest that kurkinorin may produce a morphine-like antinociceptive effect, chemical changes in the structure of these compounds provide important information on the molecular features that are necessary for molecular recognition of a ligand by opioid receptors. Hence, additional modification could be sufficient to prevent potential undesirable activity of kurkinorin without compromising antinociceptive property.
Animal models of abdominal pain and pruritus have also been explored to further the study on some salvinorin A analogs and their potential antinociceptive effect (43, 93). The aromatic analogs such as PR-37 and PR-38, which displayed lower affinity for KOP, blocked nociceptive responses. The intraperitoneal administration of PR-38 (10 mg/kg) and salvinorin A (3 mg/kg) elicited a significant decrease in pain-related behaviors. The higher dose of this analog suggests that salvinorin A is more potent than PR-38. In 2015, Salaga et al. showed attenuation of compound 48/80-induced itch responses in mice by PR-37 and PR-38 (93). The antiscratch activity of PR-37 was blocked by the selective nor-binaltorphimine (KOP antagonist), and that of PR-38 by β-funaltrexamine (selective MOP antagonist). In this study, both PR-37 and PR-38 induced antiscratch activity at the same doses of 10 and 20 mg/kg.
Pharmacological evaluation of β-tetrahydropyranyl ether of salvinorin B has provided effective insight into the antinociceptive activity of this analog and salvinorin A. The non-linear regression analysis of hot water tail-withdrawal latency revealed β-tetrahydropyranyl ether of salvinorin B to be more potent (ED50 1.4 mg/kg) than salvinorin A (ED50 2.1 mg/kg) (44). In addition, salvinorin A and β-tetrahydropyranyl ether of salvinorin B reduced both phase 1 nociceptive pain and phase 2 inflammatory pain in formalin test. The β-tetrahydropyranyl ether of salvinorin B produced a longer duration of action in the tail-withdrawal assay when compared to the salvinorin A. An increased duration of action has been attributed to the substitutions of tetrahydropyran group at C-2 position (44).
Currently, except for the ethnopharmacological reports, there is a dearth of clinical data to support the analgesic property of S. divinorum and salvinorin A (94). Salvinorin A has a long history of use as an entheogen by the shamans/healers of the Mazatec people (95). The ingestion of this plant species induces a short-lived inebriant state with intense, bizarre feelings of depersonalization (71, 96). At low infusion doses, the plant leaves have been used to treat headache, rheumatism, anemia, constipation, anuria, and diarrhea (97, 98). These pharmacological effects among others have been attributed to salvinorin A (99).
Headache is a daily painful experience that affects individuals of all ages (100, 101). Medicinal plant application to ameliorate unpleasant sensory and emotional experience that is associated with a headache is a common practice. The analgesic effect of S. divinorum leaf through infusion, chewing and swallowing could be attributed to its salvinorin A content. One kilogram of dried leaves or eight kilograms of fresh leaves of S. divinorum delivers about 1.5 g of salvinorin A when smoked, vaporized and inhaled (31, 99).
In addition to the antiheadache property of salvinorin A, its potential role in the antirheumatoid activity of S. divinorum leaf preparation further supports the antinociceptive property. The origins of rheumatoid arthritis (RA) remains controversial, and its origins in the New or Old World are subjects of several scientific works (102). However, several studies have shown higher prevalence of RA among the natives and women between the ages of 35 and 50 (103, 104). The treatment of RA among the Mazatec peoples is expected given its high incidence. The reports of the use of S. divinorum for RA treatment are plausible, in view of the potent antinociceptive effect of salvinorin A against chronic and neuropathic pain (41).
The effect of S. divinorum infusion against anemia, constipation, anuria, and diarrhea could provide additional therapeutic benefits not only from its main isolate but also from some of salvinorin A analogs (105, 106). Gastrointestinal tract discomfort, constipation and diarrhea are among the therapeutic limitations of some analgesic medications including NSAIDs and opioids (107).
Salvinorin A as a selective KOP agonist does not elicit an addictive effect. This property has stimulated research into its semi-synthetic analogs as therapeutic agents (108). The activation of KOP produces anti-addictive effects by regulating dopamine levels in the brain (106). Unlike salvinorin A, there is dearth of scientific data on the addictive or anti-addictive tendency of its analogs. This may be connected with the fact that most of these compounds were not studied in vivo. However, based on the neurobiology of addiction, analogs with high affinity for MOP including kurkinorin, herkamide, and herkinorin need to be evaluated for addictive property and compared with the available drugs being used to manage pain. In addition, the potential addictive property of analogs with high KOP affinity including methoxymethyl ether of salvinorin B, β-tetrahydropyranyl ether of salvinorin B, and ethoxymethyl ether of salvinorin B needs to be evaluated since KOP often promote aversion, withdrawal and abstinence (109). There are possibilities of analog such as 2-O-cinnamoylsalvinorin B with moderate dual MOP/KOP agonism to retain analgesic effect without addiction. However, biased activation of different signaling pathways that are associated with KOP is key to non-addictive, addictive, or anti-addictive effect (106). According to (109), both MOP and KOP contribute to specific aspects of addiction by triggering its onset and progression.
The reports on the side effects of salvinorin A such as locomotor decreases aversion, anhedonia, memory impairment, depressant-like behaviors, hallucinations among others (73, 106, 110) may have negatively reduced its therapeutic values. In the previous report, the intraperitoneal administration of salvinorin A significantly lowered dopamine levels in the caudate putamen to elicit conditioned place aversion in rodents (32). Salvinorin A-induced potentiation of dopamine re-uptake transporter function has been reported as a plausible mechanism of the decreases in dopamine levels (106). The neurobiology of salvinorin A induced memory impairment and other side effects is still unclear. Although there are no established structural activity relationships in respect of these side effects, the analogs of salvinorin A still hold promise for the future development of analgesic drugs without addictive and other side effects. Hence, robust preclinical studies and clinical trials will ultimately reveal the therapeutic potential of these analogs.
Salvinorin A was the first non-nitrogenous opioid receptor agonist. Non-nitrogenous nature of this compound can be attributed to its unique biological activities. According to Cunningham et al. (4), non-nitrogenous or non-alkaloids are promising scaffolds for new drug development. Despite the reports on the opioid receptor mediated antinociceptive effect of salvinorin A, its instability, short duration of action and side effects remains sources of concern. Systematical modification has increased understanding of the important role of substitutions at different positions of the salvinorin A scaffold and increased the possibility of developing safer analgesic drugs. Currently, the data on the binding affinity of salvinorin A analogs are yet to be correlated with possible low side effects and therapeutic advantage over existing drugs. As ligands can bind well without stabilizing the receptor's active conformation, lower or higher binding affinity is not synonymous with efficacy and potency.
Renewed focus on molecular targets seems to be promising because the activation of KOP or MOP could selectively affect β-arrestin or G-protein signaling. As the arrestin signaling pathway is responsible for many adverse effects of opioids, biased agonism for the G-protein pathway could retain analgesic effects with a reduced side effect. Hence, additional research efforts are still needed toward: (i) the modification of salvinorin A, (ii) comprehensive study of opioid receptors and associated molecular targets, (iii) extensive in vivo assays of salvinorin A analogs, iv. optimization of structural and pharmacological clues to develop safer analgesics.
On a final note, it is clear that FDA approval of salvinorin A as an analgesic constitutes an uphill task, however, the body of work reviewed here shows that some analogs of salvinorin A could translate to valuable drugs for the management of pain.
JF and JZ conceived the presented idea. JF, AM, PG, EC, GP, and OS wrote the manuscript with support from JZ, SL, RM, and OF. All authors discussed the content and contributed to the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Brazilian funding agencies CNPq, CAPES, FAPEG, FADP-DF, FINEP, and FUNDECT. OS holds a postdoctoral scholarship from CNPq and FUNDECT - Brazil [300583/2016-8].
1. Goesling J, Lin LA, Clauw DJ. Psychiatry and pain management: at the intersection of chronic pain and mental health. Curr Psychiatry Rep. (2018) 20:12. doi: 10.1007/s11920-018-0872-4
2. Chan HCS, McCarthy D, Li J, Palczewski K, Yuan S. Designing safer analgesics via μ-opioid receptor pathways. Trends Pharmacol Sci. (2017) 38:1016–37. doi: 10.1016/j.tips.2017.08.004
3. Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, et al. Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell. (2018) 172:55–67. doi: 10.1016/j.cell.2017.12.011
4. Cunningham CW, Rothman RB, Prisinzano TE. Neuropharmacology of the naturally occurring -opioid hallucinogen salvinorin A. Pharmacol Rev. (2011) 63:316–47. doi: 10.1124/pr.110.003244
5. Riley AP, Groer CE, Young D, Ewald AW, Kivell BM, Prisinzano TE. Synthesis and κ-opioid receptor activity of furan-substituted salvinorin A analogues. J Med Chem. (2014) 57:10464–75. doi: 10.1021/jm501521d
6. Roach JJ, Shenvi RA. A review of salvinorin analogs and their kappa-opioid receptor activity. Bioorganic Med Chem Lett. (2018) 28:1436–45. doi: 10.1016/j.bmcl.2018.03.029
7. Pathan H, Williams J. Basic opioid pharmacology: an update. Br J Pain. (2012) 6:11–6. doi: 10.1177/2049463712438493
8. Zaveri NT, Marquez PV, Meyer ME, Hamid A, Lutfy K. The nociceptin receptor (NOP) Agonist AT-312 blocks acquisition of morphine- and cocaine-induced conditioned place preference in mice. Front Psychiatry. (2018) 9:638. doi: 10.3389/fpsyt.2018.00638
9. Neal CR, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Watson SJ. Localization of orphanin FQ (nociceptin) peptide and messenger RNA in the central nervous system of the rat. J Comp Neurol. (1999) 406:503–47.
10. Anand P, Yiangou Y, Anand U, Mukerji G, Sinisi M, Fox M, et al. Nociceptin/orphanin FQ receptor expression in clinical pain disorders and functional effects in cultured neurons. Pain. (2016) 157:1960–9. doi: 10.1097/j.pain.0000000000000597
11. Whiteside GT, Kyle DJ. A review of the NOP (ORL-1)-Nociceptin/Orphanin FQ System covering receptor structure, distribution, role in analgesia and reward and interactions with other receptors. ACS Symp Series. (2013) 1131:327–68. doi: 10.1021/bk-2013-1131.ch016
12. Ossipov MH, Dussor GO, Porreca F. Central modulation of pain. J Clin Invest. (2010) 120:3779–87. doi: 10.1172/JCI43766
13. Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. (2011) 115:1363–81. doi: 10.1097/ALN.0b013e318238bba6
14. Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, et al. Structural insights into the dynamic process of beta2-adrenergic receptor signaling. Cell. (2015) 161:1101–11. doi: 10.1016/j.cell.2015.04.043
15. Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. (2011) 477:549–55. doi: 10.1038/nature10361
16. Siuda ER, Carr R, Rominger DH, Violin JD. Biased mu-opioid receptor ligands: a promising new generation of pain therapeutics. Curr Opin Pharmacol. (2017) 32:77–84. doi: 10.1016/j.coph.2016.11.007
18. Le Naour M, Lunzer MM, Powers MD, Kalyuzhny AE, Benneyworth MA, Thomas MJ, et al. Putative kappa opioid heteromers as targets for developing analgesics free of adverse effects. J Med Chem. (2014) 57:6383–92. doi: 10.1021/jm500159d
19. Al-Hasani R, McCall JG, Bruchas MR. Exposure to chronic mild stress prevents kappa opioid-mediated reinstatement of cocaine and nicotine place preference. Front Pharmacol. (2013) 4:96. doi: 10.3389/fphar.2013.00096
20. Butelman ER, Yuferov V, Kreek MJ. κ-opioid receptor/dynorphin system: Genetic and pharmacotherapeutic implications for addiction. Trends Neurosci. (2012) 35:587–96. doi: 10.1016/j.tins.2012.05.005
21. Grosshans M, Mutschler J, Kiefer F. Treatment of cocaine craving with as-needed nalmefene, a partial κ opioid receptor agonist. Int Clin Psychopharmacol. (2015) 30:237–8. doi: 10.1097/YIC.0000000000000069
22. Schlosburg JE, Whitfield TW, Park PE, Crawford EF, George O, Vendruscolo LF, et al. Long-term antagonism of opioid receptors prevents escalation of and increased motivation for heroin intake. J Neurosci. (2013) 33:19384–92. doi: 10.1523/JNEUROSCI.1979-13.2013
23. Smith MA, Cole KT, Iordanou JC, Kerns DC, Newsom PC, Peitz GW, et al. The mu/kappa agonist nalbuphine attenuates sensitization to the behavioral effects of cocaine. Pharmacol Biochem Behav. (2013) 104:40–6. doi: 10.1016/j.pbb.2012.12.026
24. Volkow ND, Morales M. The brain on drugs: from reward to addiction. Cell. (2015) 162:712–25. doi: 10.1016/j.cell.2015.07.046
25. Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. (2010) 1314:44–55. doi: 10.1016/j.brainres.2009.08.062
26. Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. (1982) 215:413–5. doi: 10.1126/science.6120570
27. Fantegrossi WE, Kugle KM, Valdes LJ, Koreeda M, Woods JH. Kappa-opioid receptor-mediated effects of the plant-derived hallucinogen, salvinorin A, on inverted screen performance in the mouse. Behav Pharmacol. (2005) 16:627–33. doi: 10.1097/00008877-200512000-00005
28. Ansonoff MA, Zhang J, Czyzyk T, Rothman RB, Stewart J, Xu H, et al. Antinociceptive and hypothermic effects of Salvinorin A are abolished in a novel strain of kappa-opioid receptor-1 knockout mice. J Pharmacol Exp Ther. (2006) 318:641–8. doi: 10.1124/jpet.106.101998
29. Taylor GT, Manzella F. Kappa opioids, salvinorin A and major depressive disorder. Curr Neuropharmacol. (2016) 14:165–76. doi: 10.2174/1570159X13666150727220944
30. Harden MT, Smith SE, Niehoff JA, McCurdy CR, Taylor GT. Antidepressive effects of the κ-opioid receptor agonist salvinorin A in a rat model of anhedonia. Behav Pharmacol. (2012) 23:710–5. doi: 10.1097/FBP.0b013e3283586189
31. Siebert DJ. Salvia divinorum and salvinorin A: new pharmacologic findings. J Ethnopharmacol. (1994) 43:53–6. doi: 10.1016/0378-8741(94)90116-3
32. Zhang Y, Butelman ER, Schlussman SD, Ho A, Kreek MJ. Effects of the plant-derived hallucinogen salvinorin A on basal dopamine levels in the caudate putamen and in a conditioned place aversion assay in mice: Agonist actions at kappa opioid receptors. Psychopharmacology. (2005) 179:551–8. doi: 10.1007/s00213-004-2087-0
33. Laurence L, Gulebru A, Brigitte LK, Lutz PE. The kappa opioid receptor: From addiction to depression, and back. Front Psychiatry. (2014) 5:170. doi: 10.3389/fpsyt.2014.00170
34. Maillet EL, Milon N, Heghinian MD, Fishback J, Schürer SC, Garamszegi N, et al. Noribogaine is a G-protein biased κ-opioid receptor agonist. Neuropharmacology. (2015) 99:675–88. doi: 10.1016/j.neuropharm.2015.08.032
35. Bruchas MR, Roth BL. New technologies for elucidating opioid receptor function. Trends Pharmacol Sci. (2016) 37:279–89. doi: 10.1016/j.tips.2016.01.001
36. Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981-2014. J Nat Prod. (2016) 79:629–61. doi: 10.1021/np200906s
37. Veeresham C. Natural products derived from plants as a source of drugs. J Adv Pharm Technol Res. (2012) 3:200. doi: 10.4103/2231-4040.104709
38. Ortega A, Blount JF, Manchand PS. Salvinorin, a new trans-neoclerodane diterpene from Salvia divinorum(Labiatae). J Chem Soc Perkin Trans. (1982) 1:2505. doi: 10.1039/p19820002505
39. Peet MM, Baker LE. Salvinorin B derivatives, EOM-Sal B and MOM-Sal B, produce stimulus generalization in male Sprague-Dawley rats trained to discriminate salvinorin A. Behav Pharmacol. (2011) 22:450–7. doi: 10.1097/FBP.0b013e328349fc1b
40. Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, et al. Salvinorin A: A potent naturally occurring nonnitrogenous opioid selective agonist. Proc Natl Acad Sci USA. (2002) 99:11934–9. doi: 10.1073/pnas.182234399
41. Coffeen U, Canseco-Alba A, Simón-Arceo K, Almanza A, Mercado F, León-Olea M, et al. Salvinorin A reduces neuropathic nociception in the insular cortex of the rat. Eur J Pain. (2018) 22:311–8. doi: 10.1002/ejp.1120
42. Polepally PR, Huben K, Vardy E, Setola V, Mosier PD, Roth BL, et al. Michael acceptor approach to the design of new salvinorin A-based high affinity ligands for the kappa-opioid receptor. Eur J Med Chem. (2014) 85:818–29. doi: 10.1016/j.ejmech.2014.07.077
43. Sałaga M, Polepally PR, Zakrzewski PK, Cygankiewicz A, Sobczak M, Kordek R, et al. Novel orally available salvinorin a analog PR-38 protects against experimental colitis and reduces abdominal pain in mice by interaction with opioid and cannabinoid receptors. Biochem Pharmacol. (2014) 92:618–26. doi: 10.1016/j.bcp.2014.09.018
44. Paton KF, Kumar N, Crowley RS, Harper JL, Prisinzano TE, Kivell BM. The analgesic and anti-inflammatory effects of Salvinorin A analogue β-tetrahydropyran Salvinorin B in mice. Eur J Pain. (2017) 21:1039–50. doi: 10.1002/ejp.1002
45. Valdes LJ, Butler WM, Hatfield GM, Paul AG, Koreeda M. Divinorin A, a psychotropic terpenoid, and divinorin B from the hallucinogenic Mexican mint, Salvia divinorum. J Org Chem. (1984) 49:4716–20. doi: 10.1021/jo00198a026
46. Lavecchia A, Greco G, Novellino E, Vittorio F, Ronsisvalle G. Modeling of kappa-opioid receptor/agonists interactions using pharmacophore-based and docking simulations. J Med Chem. (2000) 43:2124–34. doi: 10.1021/jm991161k
47. Yan F. Molecular Mechanisms by Which Salvinorin A Binds to and Activates the Kappa-Opioid Receptor. Case Western Reserve University, ProQuest Dissertations Publishing. 3301865 (2018).
48. Valdés LJ, Chang HM, Visger DC, Koreeda M. Salvinorin C, a new neoclerodane diterpene from a bioactive fraction of the hallucinogenic Mexican mint Salvia divinorum. Org Lett. (2001) 3:3935–7. doi: 10.1021/ol016820d
49. Lozama A, Cunningham CW, Caspers MJ, Douglas JT, Dersch CM, Rothman RB, et al. Opioid receptor probes derived from cycloaddition of the hallucinogen natural product salvinorin A. J Nat Prod. (2011) 74:718–26. doi: 10.1021/np1007872
50. Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, et al. Synthesis of salvinorin A analogues as opioid receptor probes. J Nat Prod. (2006) 69:914–8. doi: 10.1021/np060094b
51. Rothman RB, Murphy DL, Xu H, Godin JA, Dersch CM, Partilla JS, et al. Salvinorin A: allosteric interactions at the mu-opioid receptor. J Pharmacol Exp Ther. (2007) 320:801–10. doi: 10.1124/jpet.106.113167
52. Seeman P, Guan HC, Hirbec H. Dopamine D2High receptors stimulated by phencyclidines, lysergic acid diethylamide, salvinorin A, and modafinil. Synapse. (2009) 63:698–704. doi: 10.1002/syn.20647
53. Xu X, Ma S, Feng Z, Hu G, Wang L, Xie XQ. Chemogenomics knowledgebase and systems pharmacology for hallucinogen target identification-Salvinorin A as a case study. J Mol Graph Model. (2016) 70:284–95. doi: 10.1016/j.jmgm.2016.08.001
54. Fichna J, Schicho R, Andrews C, Bashashati M, Klompus M, McKay D, et al. Salvinorin A inhibits colonic transit and neurogenic ion transport in mice by activating κ-opioid and cannabinoid receptors. Neurogastroenterol Motil. (2009) 21:1326–e128. doi: 10.1111/j.1365-2982.2009.01369.x
55. Béguin C, Duncan KK, Munro TA, Ho DM, Xu W, Liu-Chen LY, et al. Modification of the furan ring of salvinorin A: identification of a selective partial agonist at the kappa opioid receptor. Bioorg Med Chem. (2009) 17:1370–80. doi: 10.1016/j.bmc.2008.12.012
56. Harding W W, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, et al. Neoclerodane diterpenes as a novel scaffold for μ opioid receptor ligands. J. Med. Chem. (2005) 48:4765–71. doi: 10.1021/jm048963m
57. Lee DYW, Karnati VVR, He M, Liu-Chen LY, Kondaveti L, Ma Z, et al. Synthesis and in vitro pharmacological studies of new C(2) modified salvinorin A analogues. Bioorganic Med Chem Lett. (2005) 15:3744–7. doi: 10.1016/j.bmcl.2005.05.048
58. Munro TA, Duncan KK, Xu W, Wang Y, Liu-Chen LY, Carlezon WA, et al. Standard protecting groups create potent and selective κ opioids: salvinorin B alkoxymethyl ethers. Bioorganic Med Chem. (2008) 16:1279–86. doi: 10.1016/j.bmc.2007.10.067
59. Prevatt-Smith KM, Lovell KM, Simpson DS, Day VW, Douglas JT, Bosch P, et al. Potential drug abuse therapeutics derived from the hallucinogenic natural product salvinorin A. Medchemcomm. (2011) 2:1217–22. doi: 10.1039/c1md00192b
60. Polepally PR, White K, Vardy E, Roth BL, Ferreira D, Zjawiony JK. Kappa-opioid receptor-selective dicarboxylic ester-derived salvinorin A ligands. Bioorg Med Chem Lett. (2013) 23:2860–2. doi: 10.1016/j.bmcl.2013.03.111
61. Sherwood AM, Crowley RS, Paton KF, Biggerstaff A, Neuenswander B, Day VW, et al. Addressing structural flexibility at the A-ring on salvinorin A: discovery of a potent kappa-opioid agonist with enhanced metabolic stability. J Med Chem. (2017) 60:3866–78. doi: 10.1021/acs.jmedchem.7b00148
62. Tidgewell K, Groer CK, Harding WW, Lozama A, Schmidt M, Marquam A, et al. Herkinorin analogues with differential β-arrestin-2 interactions. J Med Chem. (2008) 51:2421–31. doi: 10.1021/jm701162g
63. Crowley RS, Riley AP, Sherwood AM, Groer CE, Shivaperumal N, Biscaia M, et al. Synthetic studies of neoclerodane diterpenes from salvia divinorum: identification of a potent and centrally acting μ opioid analgesic with reduced abuse liability. J Med Chem. (2016) 59:11027–38. doi: 10.1021/acs.jmedchem.6b01235
64. Lee DYW, He M, Liu-Chen LY, Wang Y, Li JG, Xu W, et al. Synthesis and in vitro pharmacological studies of new C(4)-modified salvinorin A analogues. Bioorganic Med Chem Lett. (2006) 16:5498–502. doi: 10.1016/j.bmcl.2006.08.051
65. Vardy E, Mosier PD, Frankowski KJ, Wu H, Katritch V, Westkaemper RB, et al. Chemotype-selective modes of action of k-opioid receptor agonists. J Biol Chem. (2013) 288:34470–83. doi: 10.1074/jbc.M113.515668
66. Béguin C, Richards MR, Li JG, Wang Y, Xu W, Liu-Chen LY, et al. Synthesis and in vitro evaluation of salvinorin A analogues: Effect of configuration at C(2) and substitution at C(18). Bioorg Med Chem Lett. (2006) 16:4679–85. doi: 10.1016/j.bmcl.2006.05.093
67. St Jean DJ, Fotsch C. Mitigating heterocycle metabolism in drug discovery. J. Med. Chem. (2012) 55:6002–6020. doi: 10.1021/jm300343m
68. Simpson DS, Katavic PL, Lozama A, Harding WW, Parrish D, Deschamps JR, et al. Synthetic studies of neoclerodane diterpenes from Salvia divinorum: Preparation and opioid receptor activity of salvinicin analogues. J Med Chem. (2007) 50:3596–603. doi: 10.1021/jm070393d
69. Lovell KM, Vasiljevik T, Araya JJ, Lozama A, Prevatt-Smith KM, Day VW, et al. Semisynthetic neoclerodanes as kappa opioid receptor probes. Bioorg Med Chem. (2012) 20:3100–10. doi: 10.1016/j.bmc.2012.02.040
70. Sherwood AM, Williamson SE, Crowley RS, Abbott LM, Day VW, Prisinzano TE. Modular approach to pseudo-neoclerodanes as designer κ-opioid ligands. Org Lett. (2017b) 19:5414–7. doi: 10.1021/acs.orglett.7b02684
71. Bücheler R, Gleiter CH, Schwoerer P, Gaertner I. Use of nonprohibited hallucinogenic plants: Increasing relevance for public health? A case report and literature review on the consumption of Salvia divinorum (Diviner's Sage). Pharmacopsychiatry. (2005) 38:1–5. doi: 10.1055/s-2005-837763
72. Orton E, Liu R. Salvinorin A: a mini review of physical and chemical properties affecting its translation from research to clinical applications in humans. Transl Perioper Pain Med. (2014) 1:9–11. doi: 10.31480/2330-4871/004
73. Butelman ER, Kreek MJ. Salvinorin A, a kappa-opioid receptor agonist hallucinogen: Pharmacology and potential template for novel pharmacotherapeutic agents in neuropsychiatric disorders. Front Pharmacol. (2015) 6:190. doi: 10.3389/fphar.2015.00190
74. Hooker JM, Munro TA, Béguin C, Alexoff D, Shea C, Xu Y, et al. Salvinorin A and derivatives: protection from metabolism does not prolong short-term, whole-brain residence. Neuropharmacology. (2009) 57:386–91. doi: 10.1016/j.neuropharm.2009.06.044
75. Grundmann O, Phipps SM, Zadezensky I, Butterweck V. Salvia divinorum and salvinorin A: an update on pharmacology and analytical methodology. Planta Med. (2007) 73:1039–46. doi: 10.1055/s-2007-981566
76. Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, et al. Salvinorin A, an Active component of the hallucinogenic sage salvia divinorum is a highly efficacious {{}kappa{}}-opioid receptor agonist: structural and functional considerations. J Pharmacol Exp Ther. (2004) 308:1197–203. doi: 10.1124/jpet.103.059394
77. John TF, French LG, Erlichman JS. The antinociceptive effect of Salvinorin A in mice. Eur J Pharmacol. (2006) 545:129–33. doi: 10.1016/j.ejphar.2006.06.077
78. McCurdy CR, Sufka KJ, Smith GH, Warnick JE, Nieto MJ. Antinociceptive profile of salvinorin A, a structurally unique kappa opioid receptor agonist. Pharmacol Biochem Behav. (2006) 83:109–13. doi: 10.1016/j.pbb.2005.12.011
79. Wang Y. Comparison of pharmacological activities of three distinct ligands (Salvinorin A, TRK-820 and 3FLB) on opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J Pharmacol Exp Ther. (2004) 312:220–30. doi: 10.1124/jpet.104.073668
80. Connor M, Osborne PB, Christie MJ. μ-Opioid receptor desensitization: is morphine different? Br J Pharmacol. (2004) 143:685–96. doi: 10.1038/sj.bjp.0705938
81. Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein–coupled receptors and neuronal functions. Annu Rev Neurosci. (2004) 27:107–44. doi: 10.1146/annurev.neuro.27.070203.144206
82. Teksin ZS, Lee IJ, Nemieboka NN, Othman AA, Upreti VV, Hassan HE, et al. Evaluation of the transport, in vitro metabolism and pharmacokinetics of Salvinorin A, a potent hallucinogen. Eur J Pharm Biopharm. (2009) 72:471–7. doi: 10.1016/j.ejpb.2009.01.002
83. Kenakin T, Christopoulos A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat Rev Drug Discov. (2013) 12:205–16. doi: 10.1038/nrd3954
84. Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. (2006) 320:1–13. doi: 10.1124/jpet.106.104463
85. Brust TF, Morgenweck J, Kim SA, Rose JH, Locke JL, Schmid CL, et al. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci Signal. (2016) 9:ra117. doi: 10.1126/scisignal.aai8441
86. Spetea M, Eans SO, Ganno ML, Lantero A, Mairegger M, Toll L, et al. Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br. J. Pharmacol. (2017) 174:2444–56. doi: 10.1111/bph.13854
87. White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, et al. The G protein-biased -opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther. (2014) 352:98–109. doi: 10.1124/jpet.114.216820
88. Groer C, Tidgewell K, Moyer R. An opioid agonist that does not induce μ-opioid receptor — arrestin interactions or receptor internalization. Mol Pharmacol. (2007) 71:549–57. doi: 10.1124/mol.106.028258.of
89. Bohn LM, Lefkowitz RJ, Caron MG. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J Neurosci. (2002) 22:10494–500. doi: 10.1523/JNEUROSCI.22-23-10494.2002
90. Przewlocka B, Sieja A, Starowicz K, Maj M, Bilecki W, Przewlocki R. Knockdown of spinal opioid receptors by antisense targeting β-arrestin reduces morphine tolerance and allodynia in rat. Neurosci Lett. (2002) 325:107–10. doi: 10.1016/S0304-3940(02)00246-X
91. Raehal KM. Morphine side effects in -arrestin 2 knockout mice. J Pharmacol Exp Ther. (2005) 314:1195–201. doi: 10.1124/jpet.105.087254
92. Bohn LM, Gainetdinov RR, Caron MG. G protein-coupled receptor kinase/beta-arrestin systems and drugs of abuse: psychostimulant and opiate studies in knockout mice. Neuromolecular Med. (2004) 5:41–50. doi: 10.1385/nmm:5:1:041
93. Salaga M, Polepally PR, Zielinska M, Marynowski M, Fabisiak A, Murawska N, et al. Salvinorin A analogues PR-37 and PR-38 attenuate compound 48/80-induced itch responses in mice. Br J Pharmacol. (2015) 172:4331–41. doi: 10.1111/bph.13212
94. Wu L-T, Woody G, Yang C, Li J-H, Blazer DG. Recent national trends in Salvia divinorum use and substance-use disorders among recent and former Salvia divinorum users compared with nonusers. Subst Abuse Rehabil. (2011) 2011:53–68. doi: 10.2147/SAR.S17192
95. Valdés LJ, Díaz J, Paul AG. Ethnopharmacology of ska María Pastora (Salvia divinorum, Epling AND Játiva-M.). J. Ethnopharmacol. (1983) 7:287–312. doi: 10.1016/0378-8741(83)90004-1
96. Prisinzano TE. Psychopharmacology of the hallucinogenic sage Salvia divinorum. Life Sci. (2005) 78:527–31. doi: 10.1016/j.lfs.2005.09.008
97. Jenks AA, Walker JB, Kim SC. Evolution and origins of the Mazatec hallucinogenic sage, Salvia divinorum (Lamiaceae): A molecular phylogenetic approach. J Plant Res. (2011) 124:593–600. doi: 10.1007/s10265-010-0394-6
98. Valdes LJ, Hatheld GM, Koreeda M, Paul AG. Studies of salvia divinorum (Lamiaceae), an hallucinogenic mint from the Sierra Mazateca in Oaxaca, Central Mexico. Econ Bot. (1987) 41:283–91. doi: 10.1007/BF02858975
99. Valdés LJ. Salvia divinorum and the unique diterpene hallucinogen, salvinorin (divinorin) A. J Psychoactive Drugs. (1994) 26:277–83. doi: 10.1080/02791072.1994.10472441
100. Rolan PE. Understanding the pharmacology of headache. Curr Opin Pharmacol. (2014) 14:30–3. doi: 10.1016/j.coph.2013.10.005
101. Solomon GD. Pharmacology and use of headache medications. Cleve Clin J Med. (1990) 57:627–35. doi: 10.3949/ccjm.57.7.627
102. Entezami P, Fox DA, Clapham PJ, Chung KC. Historical perspective on the etiology of rheumatoid arthritis. Hand Clin. (2011) 27:1–10. doi: 10.1016/j.hcl.2010.09.006
103. Beasley J. Osteoarthritis and rheumatoid arthritis: conservative therapeutic management. J Hand Ther. (2012) 25:163–72. doi: 10.1016/j.jht.2011.11.001
104. Rothschild BM, Woods RJ, Rothschild C, Sebes JI. Geographic distribution of rheumatoid arthritis in ancient North America: implications for pathogenesis. Semin Arthritis Rheum. (1992) 22:181–7. doi: 10.1016/0049-0172(92)90018-9
105. Capasso R, Borrelli F, Cascio MG, Aviello G, Huben K, Zjawiony JK, et al. Inhibitory effect of salvinorin A, from Salvia divinorum, on ileitis-induced hypermotility: cross-talk between κ-opioid and cannabinoid CB 1 receptors. Br J Pharmacol. (2008) 155:681–9. doi: 10.1038/bjp.2008.294
106. Kivell BM, Ewald AWM, Prisinzano TE. Salvinorin A analogs and other kappa-opioid receptor compounds as treatments for cocaine abuse. Adv Pharmacol. (2014) 69:482–511. doi: 10.1016/B978-0-12-420118-7.00012-3
107. Raehal KM, Schmid CL, Groer CE, Bohn LM. Functional selectivity at the -opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev. (2011) 63:1001–19. doi: 10.1124/pr.111.004598
108. Cruz A, Domingos S, Gallardo E, Martinho A. A unique natural selective kappa-opioid receptor agonist, salvinorin A, and its roles in human therapeutics. Phytochemistry. (2017) 137:9–14. doi: 10.1016/j.phytochem.2017.02.001
109. Darcq E, Kieffer BL. Opioid receptors: drivers to addiction? Nat Rev Neurosci. (2018) 19:499–514. doi: 10.1038/s41583-018-0028-x
Keywords: analgesic, opioid receptors, salvinorin A, side effects, analogs
Citation: Zjawiony JK, Machado AS, Menegatti R, Ghedini PC, Costa EA, Pedrino GR, Lukas SE, Franco OL, Silva ON and Fajemiroye JO (2019) Cutting-Edge Search for Safer Opioid Pain Relief: Retrospective Review of Salvinorin A and Its Analogs. Front. Psychiatry 10:157. doi: 10.3389/fpsyt.2019.00157
Received: 22 July 2018; Accepted: 04 March 2019;
Published: 27 March 2019.
Edited by:
Nazzareno Cannella, University of Camerino, ItalyReviewed by:
Jinhua Wu, Florida Atlantic University, United StatesCopyright © 2019 Zjawiony, Machado, Menegatti, Ghedini, Costa, Pedrino, Lukas, Franco, Silva and Fajemiroye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordan K. Zjawiony, am9yZGFuQG9sZW1pc3MuZWR1
James O. Fajemiroye, b2x1bG9sb0B5YWhvby5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.