 Florence Noble1,2,3
Florence Noble1,2,3 Nicolas Marie1,2,3*
Nicolas Marie1,2,3*- 1CNRS ERL 3649, “Neuroplasticité et thérapies des addictions”, Paris, France
- 2INSERM UMR-S 1124, Paris, France
- 3Centre Universitaire des Saints Pères, Université Paris Descartes, Paris, France
With the opioid crisis in North America, opioid addiction has come in the spotlight and reveals the weakness of the current treatments. Two main opioid substitution therapies (OST) exist: buprenorphine and methadone. These two molecules are mu opioid receptor agonists but with different pharmacodynamic and pharmacokinetic properties. In this review, we will go through these properties and see how they could explain why these medications are recognized for their efficacy in treating opioid addiction but also if they could account for the side effects especially for a long-term use. From this critical analysis, we will try to delineate some guidelines for the design of future OST.
Introduction
When people talk about opioid problems or addiction to opioids, they think of opioids that some people get on the street, such as heroin, with the idea that only a minority of persons is concerned. But the truth is very different and anyone who uses an opioid can develop addictive behaviors. This is not a specific problem for heroin users as opioids are very useful molecules and powerful medications that are generally prescribed to relieve severe pain. Thus, problematic opioid use may also include the misuse of prescription opioid medications, such as oxycodone, morphine, or codeine, or the use of a drug for which no personal prescription has been received. As a result, the number of people over-using or dependent on opioids is increasing dramatically and is a public health problem. Over the past few years, both the U.S. and Canada have seen a spectacular increase in opioid overdose rates. From 1999 to 2016, more than 350,000 people died from opioid overdose in U.S. (https://www.cdc.gov/drugoverdose/epidemic/index.html). This so-called “opioid epidemic” or “opioid crisis” started in the 1990's with the conjunction of different factors including propaganda by pharmaceutical companies claiming that their opioids had a low liability to induce addiction mainly because of the extended release formula, and a better pain management which led to a widespread use of opioid drugs for the treatment of moderate pain (1, 2).
Behind these perfectly quantified data, there is another figure that is difficult to quantify, but which is most certainly very high, people with opioid addiction. This crisis shed light on the weakness of available treatments to manage opioid addiction. Two main medications—the opioid substitution treatments (OST) are used: buprenorphine and methadone. After a rapid review of neurobiology of opoid addiction, we will review some properties of these OST that could explain why they have a certain success [for an extensive review on methadone and/or buprenorphine, see (3)]. However, this success is only relative (relapses very often occur even when patients are under OST) and as banning opioids is not an option, it is therefore important to discuss the future of opioid research. A table with the opioids cited in the present review is included to facilitate the reading of the manuscript (Table 1).
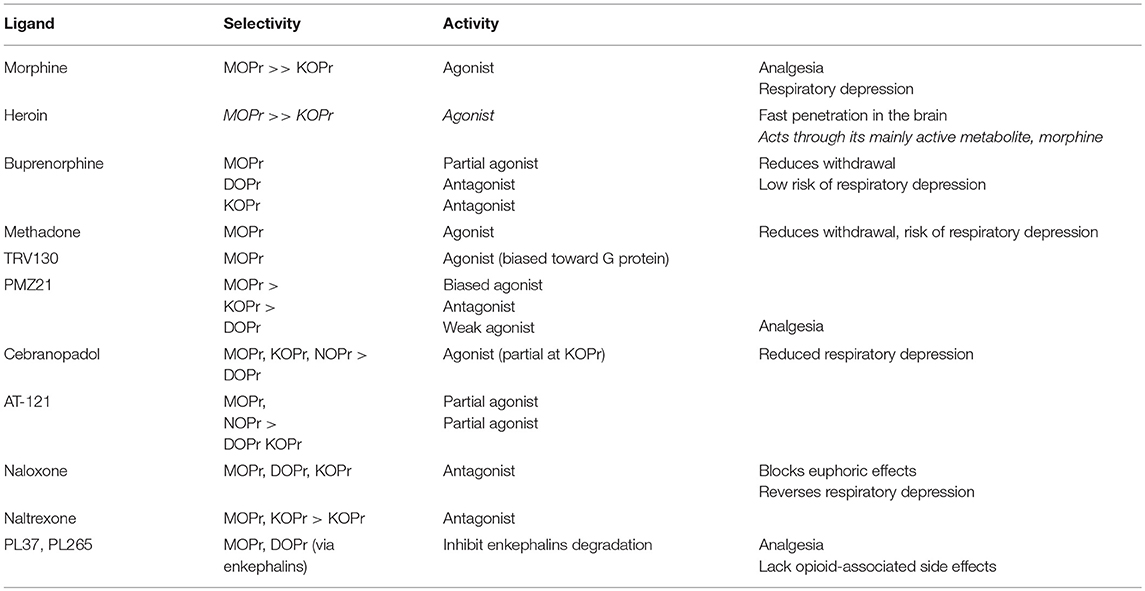
Table 1. Examples of preclinical and clinical opioid drugs.
The Neurobiological Basis of Opioid Addiction
It has been known for a long time that opioids such as morphine, heroin, and derivatives induce numerous pharmacological responses, including analgesia, dependence, respiratory depression or euphoria (4, 5). From these observations, evidence that different opioid drug effects could only be explained by the existence of stereospecific receptors has emerged. In the 1970s, the endogenous opioid receptors were discovered (6–8), followed by the characterization of the endogenous opioid peptides (9). Since these identifications numerous studies have been conducted in the opioid field.
Historically, three opioid receptors have been characterized, mu (MOPr), delta (DOPr), and kappa (KOPr). Additional receptor types have been identified, but are no longer considered as “classical” opioid receptors (e.g., sigma, nociceptin/orphanin receptor, NOPr) (10). The three opioid receptors were cloned in the early nineties (11–14). Since this period, several knockout mice lines, each harboring deletions of the genes encoding a particular opioid receptor, have been used to clarify the specific role of the different receptors in vivo and in many physiopathological conditions (15). In this review the focus will be on reward and addiction.
It is well-known that all drugs of abuse increase extracellular dopamine levels in the nucleus accumbens (Nac), either directly (e.g., cocaine and amphetamine directly target dopamine transporters), or indirectly (e.g., opioids decrease GABA release in the ventral tegmental area, leading to an activation of dopamine neurons). Several lines of evidence indicate that MOPr play a key role in mediating the rewarding effects of opioids, while the role of DOPr remains debatable, and KOPr are considered to have opposite functions to those of MOPr in the regulation of reward and addiction. KOPr agonists have dysphoric and aversive effects in humans and rodents (16, 17), in good agreement with decreases in dopamine release in the Nac observed following injection of selective agonists in this brain structure (18).
The pharmacological responses induced by opioids (e.g., conditioned place preference, intravenous self-administration, locomotor activity, analgesia) are abolished in MOPr knockout mice, demonstrating that MOPr represent the primary in vivo molecular target for these ligands (15). Morphine-induced conditioned place preference in an unbiased procedure is also reduced in DOPr knockout mice (19, 20), but these animals show normal motivation to obtain morphine in intravenous self-administration paradigm (20). These results, combined with other data obtained from other experimental approaches suggest that morphine reward and motivation to obtain opioids are intact in DOPr knockout mice, however drug-context association is more certainly impaired.
Both with most clinically useful (e.g., morphine, fentanyl, oxycodone) and most largely abused (heroin) opioids, opioid-use disorder is a public health problem. The number of opioid prescriptions sharply increased in the past two decades, increasing risks for addiction and overdoses. Addiction to prescribed opioids is associated with transition to illicit opioid use like heroin (21), and overdoses have strongly risen since the 1990s (22). As mentioned earlier the notion of “opioid crisis” or “opioid epidemic” has emerged in North America, and to a lesser extent in Australia (23). European countries appear to be less affected (24), but even if the risk in Europe appears relatively limited, vigilance is needed (25).
Opioid addiction is a brain disorder, involving alterations in neuronal circuits with complex neuroadaptative mechanisms that lead to dependence, craving, and relapse; thus contributing to the maintenance of drug use. Until now, no medication can reverse the drug-induced changes observed in the brain that are involved in the relapsing nature of opioid-use disorders, even after a protracted abstinence. Currently, the therapeutic approach using an agonist strategy with methadone and buprenorphine, has shown physical and psychosocial improvements in drug users, but these molecules possess MOPr agonist properties which limit their clinical usefulness, as described below.
Characteristics of the Opioid Substitution Treatments
The Way They Reach Their Target: Pharmacokinetic Properties
The therapeutic action of a compound strongly depends on its pharmacokinetic properties (26). The opioid users seek a rapid and intense euphoria which is obtained with heroin, which is a prodrug. Indeed, although it has a low affinity toward MOPr, its action is mainly mediated by its metabolites including morphine (27, 28). The intense and rapid euphoria following heroin administration is partly due to its high lipophilic nature, enabling the molecule to readily cross the blood-brain barrier (29). Another very important characteristic that determines the fast action of heroin is the route of administration: the intravenous route being the fastest (30). OST are both oral medications, methadone as a syrup or pills and buprenorphine as sublingual tablet or films. Methadone has a good oral bioavailability (between 40 and 95%) (31), conversely, buprenorphine has a poor oral bioavailability. In any case, both oral and sublingual routes allow the OST to diffuse slowly, thus avoiding peak effects which contribute to addiction. Therefore, after ingestion, the peak effects and peak plasma levels are reached between 1 and 6 h for methadone (average: at 4 h) (32), whereas the peak levels occur ~1 h after buprenorphine administration (33, 34). One of the mandatory features to be a good OST is that it needs to have slow metabolism and elimination profiles which avoid patients experiencing withdrawal. Methadone and buprenorphine fulfill these criteria, with an average half-life of 22 and 32 h, respectively (31, 33), therefore these medicines are taken once a day, which favors the observance. Opioid pharmacokinetics are influenced by their interaction with enzymes that metabolize xenobiotic, such as cytochromes P450 and efflux pumps. For instance, the two diphenylpropylamine opioids loperamide and methadone, which display similar structures, have different fates once administered. Whereas, methadone transport to the brain is partly restricted by the multidrug resistance protein 1 (MDR-1) (35), loperamide is unable to cross the brain blood-brain barrier due to the presence of the same efflux pump (36) showing that loperamide is a better substrate for MDR-1 than methadone. Many pharmacogenetic studies of cytochromes P450 such as CYP450 3A4 (one of the main cytochromes involved in OST metabolism) or efflux pumps have been conducted to explain the variability in OST dosing. Overall, it appears that although some variants of these genes are associated with OST plasma levels, their influence on dose requirement is very low (37). OST pharmacokinetics is more likely to be influenced by co-prescribed drugs, which interact with their metabolism. For instance, delavirdine, an antiretroviral medication used in HIV treatment, inhibits CYP450 3A4 and thus induces an elevation of methadone plasmatic concentration and drug delayed clearance (38).
The Way They Interact With the Target: Pharmacodynamic Properties
Methadone and buprenorphine bind MOPr with a higher affinity as compared to morphine. Therefore, when a patient under OST uses heroin, its effects will be reduced, as the morphine concentration in the brain will not be high enough to displace methadone or buprenorphine from the receptor. This highlights the issue of the optimum dose of OST, so each patient must have a sufficiently high brain concentration to avoid withdrawal symptoms. In addition, buprenorphine has a very low receptor dissociation rates (39–41) conferring a long duration of action (which contributes to its long half-life) and reinforcing its inability to be displaced by other opioids. Opioid overdoses cause death by respiratory depression: indeed, whereas tolerance to analgesia develops rapidly, tolerance to respiratory depression is far weaker and slower to appear (42). Methadone is a full agonist at the MOPr (43) and its potency and efficacy increase the risk of overdose, thus requiring this drug to be administered to treat opioid dependency only in designated medical units with trained staff. Buprenorphine has a particular pharmacological profile and is described as a MOPr partial agonist (44). In pioneering studies conducted in rodents, buprenorphine displayed a ceiling effect, exerting only partial analgesia compared to morphine or more effective agonists (45). Nevertheless, more recent studies have not shown this ceiling effect in other species such as humans where buprenorphine is quite powerful (46)—probably because there is a greater MOPr reserve [i.e., more spare receptors (47)]. The ceiling effect is probably rather more specific to the target system (e.g., respiration) than to the species (48) and may be explained by differences in the receptor reserve in the different pathways (pain, respiration…), probably explaining the lack of severe respiratory depression at analgesic doses with this drug (46). As a consequence, it was allowed to prescribe buprenorphine as an ambulatory medicine in many countries including UK, France, USA. Buprenorphine is also depicted as a KOPr antagonist, which might contribute to its antidepressant effect (49).
Why Searching for New Treatments for Opioid Addiction?
It is undeniable that the actual OST, methadone and buprenorphine, have brought a substantial benefit in the opioid addiction treatments. Indeed, when associated with a risk reduction policy they substantially reduced death by overdoses and the transmission of blood-borne diseases. They help addicts to follow their recovery program and contribute to their social reintegration. OST were also shown to preserve immune (50) and memory (51) functions, have positive effects on psychopathology (52, 53) and reduce polyabuse (54).
However, like any other medications, OST are not fully effective as many patients under OST might still relapse (55, 56), and because they are MOPr agonists they may be misused (57). The promised safety of buprenorphine was challenged as soon it arrived on the market and for example in France, several death cases were reported where buprenorphine was diverted (intravenous use). Whereas, several of these cases included the concomitant use of buprenorphine with other depressants of the respiratory system (ethanol and/or benzodiazepines), some of them reported only buprenorphine use (58). More recently, when gabapentin was used with opioids a substantial increase in the risk of opioid-related death was measured (59). Beyond the high risk of fatal respiratory depression (see above), methadone is associated with prolongation of the electrocardiographic QT interval (60, 61). However, the link to cardiac dysrhythmia and sudden cardiac death remains an open question. Indeed, recent studies did not confirm the role of methadone in sudden cardiac death (62) as it was previously suspected.
Many side effects have been reported with these OST such as a decrease of cognitive performance (63) or sexual dysfunction in men (64, 65). Finally, as they remain MOPr agonists, they will contribute to maintain—very likely to a lesser extent—the allostasis generated by previously abused opioids. In rodents, a short treatment (5 days) with buprenorphine or methadone is able to induce behavioral and neurochemical modifications until 35 days after withdrawal (66, 67). It therefore appears necessary to find new MOPr agonists, or new combinations of MOPr agonists and other ligands, that would not induce the neuroadaptations responsible for the harmful effects of opioids (e.g., addiction, respiratory depression), and would therefore gradually restore homeostasis, thus allowing for instance a complete escape from addiction.
On the other hand, to avoid buprenorphine diversion, different formulations of buprenorphine are currently evaluated and usually consist of transdermal patches, subcutaneous depot injections, or subdermal implants (68). An alternative strategy to limit diversion is to combine buprenorphine with an opioid antagonist, naloxone (suboxone). Naloxone has a poor oral bioavailability, but when injected intravenously (in the case of misuse), it will precipitate withdrawal. Human studies shown that it has a reduced abuse potential (69), however recent preclinical (70) and clinical (71, 72) data questioned the lower level of rewarding properties of intravenous suboxone.
Some Leads on the Future of Opioid Research
The “opioid crisis” dramatically exposes the need for more research in at least two main directions. One is to find better opioid analgesics with less and even virtually no addictive potential. The other direction is the discovery of new medications to treat opioid addiction. We will discuss these two directions focusing on opioid-based drugs.
Since the 1990's, studies have demonstrated that different ligands could induce (or select) different receptor conformations that could promote different signaling pathways. This concept is now known as biased agonism or functional selectivity (73). For opioid receptors, this notion combined with the pioneer work of Bohn and co-workers paved the way to design new opioids. It is now well-established that following ligand binding, MOPr activation could result in the activation of multiple downstream pathways through either G protein dependent processes (e.g., regulation of ion channels, adenylate cyclase inhibition) or G protein independent processes (e.g., beta-arrestin signaling). Beta-arrestin is a protein that binds the activated and phosphorylated receptor and is responsible for its desensitization and endocytosis (74). Bohn and co-workers found that in beta-arrestin-2 knockout mice, morphine analgesia was increased and prolonged (75, 76) with a decrease of respiratory depression and acute constipation (77). Therefore, it has been suggested that biased opioid agonists toward G protein pathway will retain analgesic effects with a reduction of side effects including tolerance mediated by beta-arrestin activation. This last point is of particular importance as tolerance, by increasing the dose required to induce the same effects, will contribute to dependence and overdose. So, recently few opioid biased agonists for the treatment of pain have been developed including TRV130, a compound recently entered in phase 3 to treat moderate and severe acute pain (78). This molecule is biased toward G proteins and shows less tolerance and respiratory depression as compared to morphine (79). Using the recent discovery of MOPr structure (80), Manglik and co-workers discovered PMZ21 a molecule that displays a protracted analgesia as compared to morphine and like the TRV130 has no rewarding effects in the conditioned place preference paradigm (81). However, this lack of rewarding effects has been recently challenged by Altarifi and colleagues who found that TRV130 reduced the threshold of intracranial self-stimulation (82). These results are not surprising as these molecules selectively target MOPr, so alternative strategies are currently considered such as targeting multiple opioid receptors to reduce some side effects and increase efficacy (83). For instance, cebranopadol a mixed MOPr/DOPr/KOPr/NOPr receptor agonist was found to be efficient in acute and chronic pain and development of tolerance was delayed as compared to equianalgesic doses of morphine (84). More recently, Ding et al. reported the discovery of AT-121, a MOPr/NOPr mixed agonist with analgesic effects in non-human primates and a lack of common opioid-associated side effects such as physical dependence, abuse potential, respiratory depression, and opioid-induced hyperalgesia (85). Finally, instead of activating opioid receptors with synthetic compounds that could results in unwanted effects (due to overstimulation in many target systems) the use of endogenous ligands has been proposed through the blockade of the catabolism of the endogenous peptides. This approach was developed by Roques and co-workers in the 1980's who published the first study showing that blocking enzymatic degradation of enkephalins enhances their physiological effects (86). It has the advantage to target only the structures where enkephalins are expressed, thus explaining why multiple preclinical studies demonstrated that these compounds are as effective as morphine to produce analgesia but without promotion of tolerance, physical dependence, constipation or respiration depression (87). Indeed, enkephalins are highly expressed in pain-control centers (88) whereas they are found in low amount in respiratory centers (89) or locus ceruleus (90), a structure involved in the expression of opioid physical dependence (91). At the moment, two of these molecules, PL37 and PL265, are in clinical development for treating acute and chronic pain.
Regarding the treatment of opioid addiction, no real progress has been made since the introduction of methadone and buprenorphine and most of the current research consists of work related to these compounds or other marketed opioids such as modifying the formulation to obtain slow-release compounds. For instance, it has been proposed to use slow release morphine for patients who cannot tolerate methadone (92).
Recently, some opioid antagonists (e.g., naltrexone, naloxone) have been approved for opioid addiction but only for abstinent patients because of the risk of withdrawal. They have multiple benefits: lack of reinforcing effects, blockade of the euphoric effects of opioids, relative safety (no respiratory depression) (93). Even so, the adherence to these medications is generally poor, thus limiting their efficacies for the prevention of relapse in patients with opioid-use disorder. To circumvent this low treatment observance, an injectable extended-release naltrexone was developed. The first meta-analysis on its efficacy mainly revealed that, unsurprisingly, the success of extended-release naltrexone was higher in opioid detoxified patients. However, when randomization occurred after detoxification, extended-release naltrexone showed similar efficacy to buprenorphine, whereas when randomization occurred prior to detoxification, buprenorphine efficacy was superior (94). The fact remains that opioid antagonists are very efficient in emergency medicine, by preventing opioid overdose fatalities (95). Naloxone is actually the only opioid antagonist approved for treating opioid overdose. Its efficacy is based on a rapid onset of action via intravenous route (2–3 min) (96), but its shorter half-life than that of most opioid agonists, requires multiple injections or continuous administration to reverse respiratory depression. A recent study showed that it was also able to reverse buprenorphine-induced respiratory depression (97). It is noteworthy that fast opioid detoxification in opioid-dependent patients might lead to acute opioid withdrawal syndrome accompanied by catecholamine releases, responsible for cardiac and respiratory functions impairment (98).
Conclusion
This review was focused on opioids, but knowing whether if they will remain the gold standard in pain management is an open question considering the opioid crisis. In addition, long-term treatment with OST, more than restoring the neurobiological equilibrium disturbed by the opioid misuse, will maintain drug-induced neuroplastic changes. So, besides the short and mid-term necessary research on the discovery of safer opioids, other pharmacological strategies have to be envisioned based either on different use of existing treatments or on other neurotransmitter systems with the objectives of having painkillers devoid of any activity on the reward system.
Author Contributions
NM and FN wrote the manuscript. All authors take responsibility for final content. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Van Zee A. The promotion and marketing of oxycontin: commercial triumph, public health tragedy. Am J Public Health (2009) 99:221–7. doi: 10.2105/AJPH.2007.131714
2. Morone NE, Weiner DK. Pain as the fifth vital sign: exposing the vital need for pain education. Clin Ther. (2013) 35:1728–32. doi: 10.1016/j.clinthera.2013.10.001
3. Maremmani I, Gerra G. Buprenorphine-based regimens and methadone for the medical management of opioid dependence: selecting the appropriate drug for treatment. Am J Addict. (2010) 19:557–68. doi: 10.1111/j.1521-0391.2010.00086.x
4. Dhawan BN, Cesselin F, Raghubir R, Reisine T, Bradley PB, Portoghese PS, et al. International Union of Pharmacology. XII. Classification of opioid receptors. Pharmacol Rev. (1996) 48:567–92.
5. Kieffer BL. Opioids: first lessons from knockout mice. Trends Pharmacol Sci. (1999) 20:19–26. doi: 10.1016/S0165-6147(98)01279-6
6. Terenius L. Stereospecific interaction between narcotic analgesics and a synaptic plasm a membrane fraction of rat cerebral cortex. Acta Pharmacol Toxicol. (1973) 32:317–20. doi: 10.1111/j.1600-0773.1973.tb01477.x
7. Simon EJ, Hiller JM, Edelman I. Stereospecific binding of the potent narcotic analgesic (3H) Etorphine to rat-brain homogenate. Proc Natl Acad Sci USA. (1973) 70:1947–9. doi: 10.1073/pnas.70.7.1947
8. Pert CB, Snyder SH. Opiate receptor: demonstration in nervous tissue. Science (1973) 179:1011–4. doi: 10.1126/science.179.4077.1011
9. Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature (1975) 258:577–80. doi: 10.1038/258577a0
10. Stein C. Opioid receptors. Annu Rev Med. (2016) 67:433–51. doi: 10.1146/annurev-med-062613-093100
11. Yasuda K, Raynor K, Kong H, Breder CD, Takeda J, Reisine T, et al. Cloning and functional comparison of kappa and delta opioid receptors from mouse brain. Proc Natl Acad Sci USA. (1993) 90:6736–40. doi: 10.1073/pnas.90.14.6736
12. Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol Pharmacol. (1993) 44:8–12.
13. Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci USA. (1992) 89:12048–52. doi: 10.1073/pnas.89.24.12048
14. Evans CJ, Keith DE, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science (1992) 258:1952–5. doi: 10.1126/science.1335167
15. Charbogne P, Kieffer BL, Befort K. 15 years of genetic approaches in vivo for addiction research: Opioid receptor and peptide gene knockout in mouse models of drug abuse. Neuropharmacology (2014) 76(Pt B):204–17. doi: 10.1016/j.neuropharm.2013.08.028
16. Mucha RF, Herz A. Motivational properties of kappa and mu opioid receptor agonists studied with place and taste preference conditioning. Psychopharmacology (1985) 86:274–80. doi: 10.1007/BF00432213
17. Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science (1986) 233:774–6. doi: 10.1126/science.3016896
18. Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci USA. (1992) 89:2046–50. doi: 10.1073/pnas.89.6.2046
19. Chefer VI, Shippenberg TS. Augmentation of morphine-induced sensitization but reduction in morphine tolerance and reward in delta-opioid receptor knockout mice. Neuropsychopharmacology (2009) 34:887–98. doi: 10.1038/npp.2008.128
20. Le Merrer J, Plaza-Zabala A, Del Boca C, Matifas A, Maldonado R, Kieffer BL. Deletion of the delta opioid receptor gene impairs place conditioning but preserves morphine reinforcement. Biol Psychiatry (2011) 69:700–3. doi: 10.1016/j.biopsych.2010.10.021
21. Jones CM. Heroin use and heroin use risk behaviors among nonmedical users of prescription opioid pain relievers - United States, 2002-2004 and 2008-2010. Drug Alcohol Depend. (2013) 132:95–100. doi: 10.1016/j.drugalcdep.2013.01.007
22. Schiller EY. Opioid, overdose. In: Mechanic OJ, editor. StatPearls. Treasure Island, FL: StatPearls Publishing LLC.
23. Shipton EA, Shipton EE, Shipton AJ. A review of the opioid epidemic: what do we do about it? Pain Ther. (2018) 7:23–36. doi: 10.1007/s40122-018-0096-7
24. Hauser W, Schug S, Furlan AD. The opioid epidemic and national guidelines for opioid therapy for chronic noncancer pain: a perspective from different continents. Pain Rep. (2017) 2:e599. doi: 10.1097/PR9.0000000000000599
25. van Amsterdam J, van den Brink W. The misuse of prescription opioids: a threat for Europe? Curr Drug Abuse Rev. (2015) 8:3–14. doi: 10.2174/187447370801150611184218
26. Marie N, Canestrelli C, Noble F. Role of pharmacokinetic and pharmacodynamic parameters in neuroadaptations induced by drugs of abuse, with a focus on opioids and psychostimulants. Neurosci Biobehav Rev. (2018). doi: 10.1016/j.neubiorev.2018.06.006. [Epub ahead of print].
27. Inturrisi CE, Schultz M, Shin S, Umans JG, Angel L, Simon EJ. Evidence from opiate binding studies that heroin acts through its metabolites. Life Sci. (1983) 33(Suppl. 1):773–6. doi: 10.1016/0024-3205(83)90616-1
28. Rook EJ, Huitema AD, van den Brink W, van Ree JM, Beijnen JH. Pharmacokinetics and pharmacokinetic variability of heroin and its metabolites: review of the literature. Curr Clin Pharmacol. (2006) 1:109–18. doi: 10.2174/157488406775268219
29. Oldendorf WH, Hyman S, Braun L, Oldendorf SZ. Blood-brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. Science (1972) 178:984–6. doi: 10.1126/science.178.4064.984
30. Comer SD, Collins ED, MacArthur RB, Fischman MW. Comparison of intravenous and intranasal heroin self-administration by morphine-maintained humans. Psychopharmacology (1999) 143:327–38. doi: 10.1007/s002130050956
31. Eap CB, Buclin T, Baumann P. Interindividual variability of the clinical pharmacokinetics of methadone: implications for the treatment of opioid dependence. Clin Pharmacokinet. (2002) 41:1153–93. doi: 10.2165/00003088-200241140-00003
32. Inturrisi CE, Verebely K. The levels of methadone in the plasma in methadone maintenance. Clin Pharmacol Ther. (1972) 13:633–7. doi: 10.1002/cpt1972135part1633
33. Chiang CN, Hawks RL. Pharmacokinetics of the combination tablet of buprenorphine and naloxone. Drug Alcohol Depend. (2003) 70:S39–47. doi: 10.1016/S0376-8716(03)00058-9
34. Strain EC, Moody DE, Stoller KB, Walsh SL, Bigelow GE. Relative bioavailability of different buprenorphine formulations under chronic dosing conditions. Drug Alcohol Depend. (2004) 74:37–43. doi: 10.1016/j.drugalcdep.2003.11.008
35. Wang J-S, Ruan Y, Taylor RM, Donovan JL, Markowitz JS, DeVane CL. Brain penetration of methadone (R)- and (S)-enantiomers is greatly increased by P-glycoprotein deficiency in the blood-brain barrier of Abcb1a gene knockout mice. Psychopharmacology (2004) 173:132–8. doi: 10.1007/s00213-003-1718-1
36. Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. (1996) 97:2517–24. doi: 10.1172/JCI118699
37. Fonseca F, Torrens M. Pharmacogenetics of methadone response. Mol Diagn Ther. (2018) 22:57–78. doi: 10.1007/s40291-017-0311-y
38. McCance-Katz EF, Rainey PM, Smith P, Morse GD, Friedland G, Boyarsky B, et al. Drug interactions between opioids and antiretroviral medications: interaction between methadone, LAAM, and delavirdine. Am J Addict. (2006) 15:23–34. doi: 10.1080/10550490500419029
39. Mégarbane B, Marie N, Pirnay S, Borron SW, Gueye PN, Risède P, et al. Buprenorphine is protective against the depressive effects of norbuprenorphine on ventilation. Toxicol Appl Pharmacol. (2006) 212:256–67. doi: 10.1016/j.taap.2005.08.002
40. Villiger JW. Binding of buprenorphine to opiate receptors. Regulation by guanyl nucleotides and metal ions. Neuropharmacology (1984) 23:373–5.
41. Yassen A, Olofsen E, Romberg R, Sarton E, Teppema L, Danhof M, et al. Mechanism-based PK/PD modeling of the respiratory depressant effect of buprenorphine and fentanyl in healthy volunteers. Clin Pharmacol Ther. (2007) 81:50–8. doi: 10.1038/sj.clpt.6100025
42. Mohammed W, Alhaddad H, Marie N, Tardy F, Lamballais F, Risede P, et al. Comparison of tolerance to morphine-induced respiratory and analgesic effects in mice. Toxicol Lett. (2013) 217:251–9. doi: 10.1016/j.toxlet.2012.12.021
43. Selley DE, Liu Q, Childers SR. Signal transduction correlates of Mu opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTPγS binding in mMOR-CHO cells and rat thalamus. J Pharmacol Exp Ther. (1998) 285:496–505.
44. Huang P, Kehner GB, Cowan A, Liu-Chen LY. Comparison of pharmacological activities of buprenorphine and norbuprenorphine: norbuprenorphine is a potent opioid agonist. J Pharmacol Exp Ther. (2001) 297:688–95.
45. Cowan A. Buprenorphine: the basic pharmacology revisited. J Addict Med. (2007) 1:68–72. doi: 10.1097/ADM.0b013e31806c9202
46. Dahan A, Yassen A, Romberg R, Sarton E, Teppema L, Olofsen E, et al. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br J Anaesth. (2006) 96:627–32. doi: 10.1093/bja/ael051
47. Kelly E. Efficacy and ligand bias at the μ-opioid receptor. Br J Pharmacol. (2013) 169:1430–46. doi: 10.1111/bph.12222
48. Raffa RB, Haidery M, Huang H-M, Kalladeen K, Lockstein DE, Ono H, et al. The clinical analgesic efficacy of buprenorphine. J Clin Pharm Ther. (2014) 39:577–83. doi: 10.1111/jcpt.12196
49. Falcon E, Browne CA, Leon RM, Fleites VC, Sweeney R, Kirby LG, et al. Antidepressant-like effects of buprenorphine are mediated by kappa opioid receptors. Neuropsychopharmacology (2016) 41:2344–51. doi: 10.1038/npp.2016.38
50. Sacerdote P, Franchi S, Gerra G, Leccese V, Panerai AE, Somaini L. Buprenorphine and methadone maintenance treatment of heroin addicts preserves immune function. Brain Behav Immun. (2008) 22:606–13. doi: 10.1016/j.bbi.2007.12.013
51. Elkana O, Adelson M, Doniger GM, Sason A, Peles E. Cognitive function is largely intact in methadone maintenance treatment patients. World J Biol Psychiatry (2017). doi: 10.1080/15622975.2017.1342047. [Epub ahead of print].
52. Maremmani AG, Rovai L, Pani PP, Pacini M, Lamanna F, Rugani F, et al. Do methadone and buprenorphine have the same impact on psychopathological symptoms of heroin addicts? Ann Gen Psychiatry (2011) 10:17. doi: 10.1186/1744-859X-10-17
53. Pani PP, Maremmani I, Pacini M, Lamanna F, Maremmani AG, Dell'osso L. Effect of psychiatric severity on the outcome of methadone maintenance treatment. Eur Addict Res. (2011) 17:80–9. doi: 10.1159/000321465
54. Bizzarri JV, Casetti V, Sanna L, Maremmani AGI, Rovai L, Bacciardi S, et al. The newer Opioid Agonist Treatment with lower substitutive opiate doses is associated with better toxicology outcome than the older Harm Reduction Treatment. Ann Gen Psychiatry (2016) 15:34. doi: 10.1186/s12991-016-0109-z
55. Kakko J, Svanborg KD, Kreek MJ, Heilig M. 1-year retention and social function after buprenorphine-assisted relapse prevention treatment for heroin dependence in Sweden: a randomised, placebo-controlled trial. Lancet (2003) 361:662–8. doi: 10.1016/S0140-6736(03)12600-1
56. Sees KL, Delucchi KL, Masson C, Rosen A, Clark HW, Robillard H, et al. Methadone maintenance vs 180-day psychosocially enriched detoxification for treatment of opioid dependence: a randomized controlled trial. JAMA (2000) 283:1303–10. doi: 10.1001/jama.283.10.1303
57. Wright N, D'Agnone O, Krajci P, Littlewood R, Alho H, Reimer J, et al. Addressing misuse and diversion of opioid substitution medication: guidance based on systematic evidence review and real-world experience. J Public Health (2016) 38:e368–74. doi: 10.1093/pubmed/fdv150
58. Mégarbane B, Hreiche R, Pirnay S, Marie N, Baud FJ. Does high-dose buprenorphine cause respiratory depression? Possible mechanisms and therapeutic consequences. Toxicol Rev. (2006) 25:79–85. doi: 10.2165/00139709-200625020-00002
59. Gomes T, Juurlink DN, Antoniou T, Mamdani MM, Paterson JM, van den Brink W. Gabapentin, opioids, and the risk of opioid-related death: A population-based nested case–control study. PLoS Med. (2017) 14:e1002396. doi: 10.1371/journal.pmed.1002396
60. Chou R, Weimer MB, Dana T. Methadone overdose and cardiac arrhythmia potential: findings from a review of the evidence for an American Pain Society and College on Problems of Drug Dependence clinical practice guideline. J Pain (2014) 15:338–65. doi: 10.1016/j.jpain.2014.01.495
61. Alinejad S, Kazemi T, Zamani N, Hoffman RS, Mehrpour O. A systematic review of the cardiotoxicity of methadone. EXCLI J. (2015) 14:577–600. doi: 10.17179/excli2015-553
62. Bart G, Wyman Z, Wang Q, Hodges JS, Karim R, Bart BA. Methadone and the QTc interval: paucity of clinically significant factors in a retrospective cohort. J Addict Med. (2017) 11:489–93. doi: 10.1097/ADM.0000000000000353
63. Pujol CN, Paasche C, Laprevote V, Trojak B, Vidailhet P, Bacon E, et al. Cognitive effects of labeled addictolytic medications. Prog Neuropsychopharmacol Biol Psychiatry (2018) 81:306–32. doi: 10.1016/j.pnpbp.2017.09.008
64. Gerra G, Manfredini M, Somaini L, Maremmani I, Leonardi C, Donnini C. Sexual dysfunction in men receiving methadone maintenance treatment: clinical history and psychobiological correlates. Eur Addict Res. (2016) 22:163–75. doi: 10.1159/000441470
65. Yee A, Loh HS, Hisham Hashim HMB, Ng CG. Clinical factors associated with sexual dysfunction among men in methadone maintenance treatment and buprenorphine maintenance treatment: a meta-analysis study. Int J Impot Res. (2014) 26:161–6. doi: 10.1038/ijir.2014.18
66. Allouche S, Le Marec T, Coquerel A, Noble F, Marie N. Striatal dopamine D1 and D2 receptors are differentially regulated following buprenorphine or methadone treatment. Psychopharmacology (2015) 232:1527–33. doi: 10.1007/s00213-014-3785-x
67. Allouche S, Le Marec T, Noble F, Marie N. Different patterns of administration modulate propensity of methadone and buprenorphine to promote locomotor sensitization in mice. Prog Neuropsychopharmacol Biol Psychiatry (2013) 40:286–91. doi: 10.1016/j.pnpbp.2012.10.013
68. Rosenthal RN, Goradia VV. Advances in the delivery of buprenorphine for opioid dependence. Drug Des Dev Ther. (2017) 11:2493–505. doi: 10.2147/DDDT.S72543
69. Mendelson J, Jones RT. Clinical and pharmacological evaluation of buprenorphine and naloxone combinations: why the 4:1 ratio for treatment? Drug Alcohol Depend. (2003) 70:S29–37. doi: 10.1016/S0376-8716(03)00057-7
70. Canestrelli C, Marie N, Noble F. Rewarding or aversive effects of buprenorphine/naloxone combination (Suboxone) depend on conditioning trial duration. Int J Neuropsychopharmacol. (2014) 17:1367–73. doi: 10.1017/S146114571400025X
71. Comer SD, Sullivan MA, Vosburg SK, Manubay J, Amass L, Cooper ZD, et al. Abuse liability of intravenous buprenorphine/naloxone and buprenorphine alone in buprenorphine-maintained intravenous heroin abusers. Addiction (2010) 105:709–18. doi: 10.1111/j.1360-0443.2009.02843.x
72. Monte AA, Mandell T, Wilford BB, Tennyson J, Boyer EW. Diversion of buprenorphine/naloxone coformulated tablets in a region with high prescribing prevalence. J Addict Dis. (2009) 28:226–31. doi: 10.1080/10550880903014767
73. Madariaga-Mazón A, Marmolejo-Valencia AF, Li Y, Toll L, Houghten RA, Martinez-Mayorga K. Mu-Opioid receptor biased ligands: a safer and painless discovery of analgesics? Drug Discov Today (2017) 22:1719–9. doi: 10.1016/j.drudis.2017.07.002
74. Allouche S, Noble F, Marie N. Opioid receptor desensitization: mechanisms and its link to tolerance. Front Pharmacol. (2014) 5:280. doi: 10.3389/fphar.2014.00280
75. Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science (1999) 286:2495–8. doi: 10.1126/science.286.5449.2495
76. Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature (2000) 408:720–3. doi: 10.1038/35047086
77. Raehal KM, Walker JKL, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther. (2005) 314:1195–201. doi: 10.1124/jpet.105.087254
78. Viscusi ER, Webster L, Kuss M, Daniels S, Bolognese JA, Zuckerman S, et al. A randomized, phase 2 study investigating TRV130, a biased ligand of the μ-opioid receptor, for the intravenous treatment of acute pain. Pain (2016) 157:264–72. doi: 10.1097/j.pain.0000000000000363
79. DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. (2013) 344:708–17. doi: 10.1124/jpet.112.201616
80. Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, et al. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature (2012) 485:321–6. doi: 10.1038/nature10954
81. Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature (2016) 537:185–90.
82. Altarifi AA, David B, Muchhala KH, Blough BE, Akbarali H, Negus SS. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J Psychopharmacol. (2017) 31:730–9. doi: 10.1177/0269881116689257
83. Günther T, Dasgupta P, Mann A, Miess E, Kliewer A, Fritzwanker S, et al. Targeting multiple opioid receptors - improved analgesics with reduced side effects? Br J Pharmacol. (2018) 175:2857–68. doi: 10.1111/bph.13809
84. Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M, et al. Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J Pharmacol Exp Ther. (2014) 349:535–48. doi: 10.1124/jpet.114.213694
85. Ding H, Kiguchi N, Yasuda D, Daga PR, Polgar WE, Lu JJ, et al. A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Sci Transl Med. (2018) 10: eaar3483. doi: 10.1126/scitranslmed.aar3483
86. Roques BP, Fournié-Zaluski MC, Soroca E, Lecomte JM, Malfroy B, Llorens C, et al. The enkephalinase inhibitor thiorphan shows antinociceptive activity in mice. Nature (1980) 288:286–8.
87. Roques BP, Fournié-Zaluski M-C, Wurm M. Inhibiting the breakdown of endogenous opioids and cannabinoids to alleviate pain. Nat Rev Drug Discov. (2012) 11:292–310. doi: 10.1038/nrd3673
88. McLean S, Skirboll LR, Pert CB. Comparison of substance P and enkephalin distribution in rat brain: an overview using radioimmunocytochemistry. Neuroscience (1985) 14:837–52. doi: 10.1016/0306-4522(85)90147-2
89. Boudinot E, Morin-Surun M, Foutz AS, Fournié-Zaluski M, Roques BP, Denavit-Saubié M. Effects of the potent analgesic enkephalin-catabolizing enzyme inhibitors RB101 and kelatorphan on respiration. Pain (2001) 90:7–13. doi: 10.1016/S0304-3959(00)00382-1
90. Williams JT, Christie MJ, North RA, Roques BP. Potentiation of enkephalin action by peptidase inhibitors in rat locus ceruleus in vitro. J Pharmacol Exp Ther. (1987) 243:397–401.
91. Maldonado R, Stinus L, Gold LH, Koob GF. Role of different brain structures in the expression of the physical morphine withdrawal syndrome. J Pharmacol Exp Ther. (1992) 261:669–77.
92. Hämmig R, Köhler W, Bonorden-Kleij K, Weber B, Lebentrau K, Berthel T, et al. Safety and tolerability of slow-release oral morphine versus methadone in the treatment of opioid dependence. J Subst Abuse Treat. (2014) 47:275–81. doi: 10.1016/j.jsat.2014.05.012
93. Ling W, Mooney L, Wu L-T. Advances in opioid antagonist treatment for opioid addiction. Psychiatr Clin North Am. (2012) 35:297–308. doi: 10.1016/j.psc.2012.03.002
94. Jarvis BP, Holtyn AF, Subramaniam S, Tompkins DA, Oga EA, Bigelow GE, et al. Extended-release injectable naltrexone for opioid use disorder: a systematic review. Addiction (2018) 113:1188–209. doi: 10.1111/add.14180
95. Kerensky T, Walley AY. Opioid overdose prevention and naloxone rescue kits: what we know and what we don't know. Addict Sci Clin Pract. (2017) 12:4. doi: 10.1186/s13722-016-0068-3
96. Johnstone RE, Jobes DR, Kennell EM, Behar MG, Smith TC. Reversal of Morphine Anesthesia with Naloxone. Anesthesiology (1974) 41:361–7. doi: 10.1097/00000542-197410000-00010
97. van Dorp E, Yassen A, Sarton E, Romberg R, Olofsen E, Teppema L, et al. Naloxone reversal of buprenorphine-induced respiratory depression. Anesthesiology (2006) 105:51–7. doi: 10.1097/00000542-200607000-00012
Keywords: addiction, morphine, buprenorphine, methadone, substitution treatment
Citation: Noble F and Marie N (2019) Management of Opioid Addiction With Opioid Substitution Treatments: Beyond Methadone and Buprenorphine. Front. Psychiatry 9:742. doi: 10.3389/fpsyt.2018.00742
Received: 09 July 2018; Accepted: 14 December 2018;
Published: 18 January 2019.
Edited by:
Dominique Massotte, UPR3212 Institut des Neurosciences Cellulaires et Intégratives (INCI), FranceReviewed by:
Brian Cox, Uniformed Services University, United StatesMariana Spetea, University of Innsbruck, Austria
Copyright © 2019 Noble and Marie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolas Marie, bmljb2xhcy5tYXJpZUBwYXJpc2Rlc2NhcnRlcy5mcg==