 Mami Noda
Mami Noda Masataka Ifuku2†
Masataka Ifuku2† Md. Shamim Hossain
Md. Shamim Hossain Toshihiko Katafuchi
Toshihiko Katafuchi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Psychiatry, 16 November 2018
Sec. Molecular Psychiatry
Volume 9 - 2018 | https://doi.org/10.3389/fpsyt.2018.00589
Fatigue is commonly reported in a variety of illnesses and has major impact on quality of life. Chronic fatigue syndrome (CFS) is a debilitating syndrome of unknown etiology. The clinical symptoms include problems in neuroendocrine, autonomic, and immune systems. It is becoming clear that the brain is the central regulator of CFS. For example, neuroinflammation, especially induced by activation of microglia and astrocytes, may play a prominent role in the development of CFS, though little is known about molecular mechanisms. Many possible causes of CFS have been proposed. However, in this mini-review, we summarize evidence for a role for microglia and astrocytes in the onset and the maintenance of immunologically induced CFS. In a model using virus mimicking synthetic double-stranded RNA, infection causes sequential signaling such as increased blood brain barrier (BBB) permeability, microglia/macrophage activation through Toll-like receptor 3 (TLR3) signaling, secretion of IL-1β, upregulation of the serotonin transporter (5-HTT) in astrocytes, reducing extracellular serotonin (5-HT) levels and hence reduced activation of 5-HT1A receptor subtype. Hopefully, drug discovery targeting these pathways may be effective for CFS therapy.
CFS, also often described as myalgic encephalomyelitis, is characterized not only by severe and prolonged fatigue drastically impairing life quality, and much more attention has been paid in the past year than ever (1–6). CFS is observed even in children and adolescents (7). CFS is induced by impairment of neuronal–endocrine–immune interactions with various symptoms including circadian rhythm abnormalities (8, 9). Though systematic reviews were carried out recently, remarkable ways to reduce fatigue severity are still missing (1, 10–17). In the central nervous system (CNS), activation or dysfunction of glial cells are involved in various neurodegenerative disorders and psychiatric diseases or symptoms. It has been reported that neuroinflammation is present in widespread brain areas in CFS patients in a positron emission tomography (PET) study utilizing 11C-(R)-PK11195, a ligand for a translocator protein expressed in activated microglia or astrocytes (18). This finding suggests that activation of glial cells play an important role also in CFS. Though the brain regions involved in CFS still remain mysterious, a clinical study with CFS patients showed involvement of the prefrontal cortex (PFC) and frontal networks (19, 20). In addition, single-photon emission-captured tomography (SPECT) studies in CFS patients revealed that production of glutamate in the PFC was low (21). These findings suggest that fatigue sensation and dysfunction of the PFC are correlated, although the molecular/cellular mechanism is only partially known. Here, based on the recent reviews mentioned above, possible causes of CFS proposed during the last decade are summarized. Then, infection-related/immunological CFS is focused and how glial cells are involved in the development of fatigue sensation is described, giving a new insight as an alternative strategy for the treatment of CFS.
CFS may be explained by both central and peripheral pathophysiological mechanisms (22), together with anatomical changes such as a decrease in gray matter volume in the brain (23). The etiology of CFS is still little advanced over the last decade (24). There are many potential causes, consisting of several categories. One is metabolic abnormalities or changes in neural activity, which were suggested by the studies with magnetic resonance spectroscopy (MRS) or magnetoencephalography (MEG) of muscles and brain in some CFS patients (25–28). The cellular metabolic abnormalities contain, for example, a decrease in nuclear factor erythroid-2-related factor 2 (Nrf2), antioxidant enzymes (superoxide dismutase 1&2, glutathione peroxidase, heme oxygenase-1, and catalase), and glycogen, and adenosine triphosphate (ATP). Though, it has been claimed that metabolic biomarkers for CFS were still not available (29), oxidative stress index was proposed as a potential biomarker for fatigue (30) and a diagnostic biomarker was proposed by comprehensive metabolomic analyses of blood samples obtained from patients (31, 32), for example, decreased plasma levels of carnitine-choline and phosphatidylcholine, while increased plasma levels of ceramide, triglyceride, and phosphatidylethanolamine. Dysfunction of the tricarboxylic acid (TCA) and urea cycles in CFS (33) and increase in lactic acid and blood urea nitrogen could also be the cause of CFS (34), which could become metabolite markers of CFS. Consequently, reduced cellular-energy availability and non-adaptive energy expenditure in CFS has been reported (35). Nevertheless, the reader should be aware that we do not know if these metabolic changes are a cause or a consequence of CFS. Recently, the question has been raised whether CFS is a metabolic disease or the result of disturbed homeostasis, and a multiplicity of physiological/pathophysiological factors related to CFS have been listed (36). On the other hand, a new concept called circadian metabolomics, the relationship between circadian rhythms and energy metabolism, has been proposed to explain circadian rhythm abnormalities in CFS patients (8).
The second possible mechanism is functional and structural abnormalities. For example, lymphatic dysfunction was postulated because cerebrospinal fluid drainage was beneficial in some CFS patients (37, 38). Other functional changes such as immune dysregulation, mitochondrial dysfunction (39), AMP-activated protein kinase, skeletal muscle cell acidosis are summarized in a recent review (29). In addition, recent genome-wide epigenetic analyses revealed different patterns of DNA methylation, which are related to cell signaling in immune system and could become possible diagnostic markers (40). Furthermore, changes in the amount and the size of circulating extracellular vesicles were also reported as potential biomarkers in CFS (41).
Finally, classical but still relevant studies revealed that CFS can occur after infection with viral and non-viral pathogens (42, 43), though virus infection as a cause of CFS has been questioned (44). Several lines of evidence indicate that pathogens entering the body induces expression of cytokines in the CNS (45, 46). Alternatively, CFS might be an autoimmune disease. Indeed, autoantibodies against various antigens, such as nuclear and membrane structures and neurotransmitters, have been identified in some CFS patients (47). The possible causes mentioned above are summarized in Table 1.
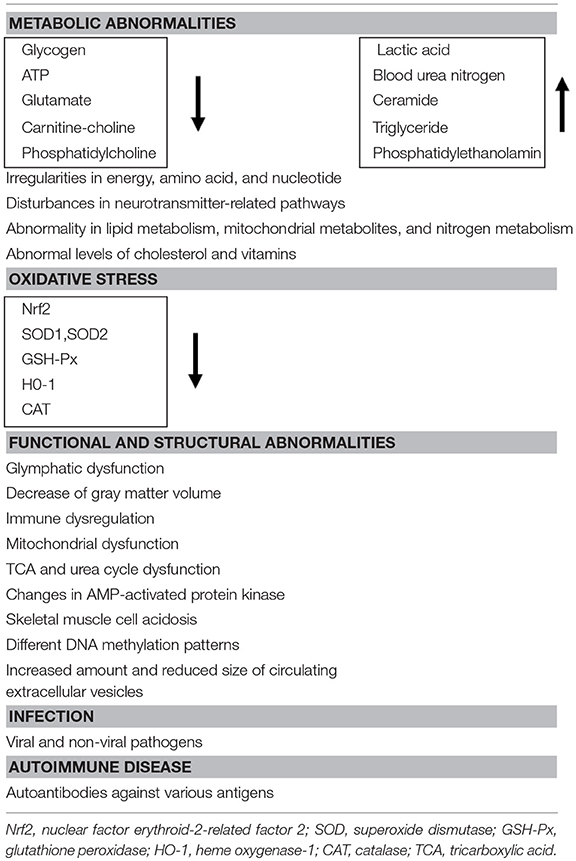
Table 1. Possible causes of CFS.
In addition to immune dysfunction, activation of the immune system occurs in both infection- or autoantigens-related CFS. In the CNS, glial cells, especially microglial cells, play an important role the immune system. Therefore, inflammation-induced functional changes in neuron-glial interaction in the CNS, affecting peripheral organs, could be fundamental cause of CFS.
In the case of infection-related CFS, it is well-known that inflammatory cytokines in the brain can affect the sympathetic nervous system, the hypothalamic–pituitary–adrenal (HPA) axis, peripheral cellular immunity, learning and memory, and behavioral activity (46, 48, 49). It is most likely that cytokines might play a role in the development of CFS initiated/triggered by viral/non-viral pathogens. To investigate this, systemic injection of poly-I:C, virus-mimicking synthetic double-stranded RNA, is a useful approach. Interestingly, poly-I:C injection causes a sustained decrease in spontaneous running wheel activity that is accompanied by enhanced expression of interferon-α (IFN-α), IL-1β, and 5-HTT in the rat brain (48, 50). It must be noted that in this experimental model the reduced activity is not attributable to sickness behavior or depression, as the activity decrease is still observed after the poly-I:C-induced acute-phase responses, such as fever and adrenal responses, were abolished (51). Curiously, however, open field tests showed no differences from controls (52).
A question arises whether or not intraperitoneal (i.p.) injection of poly-I:C causes breakdown of BBB. There is evidence that poly-I:C increases BBB permeability in a rat model of CFS (48). The breakdown of the BBB can be assessed by quantification of extravasated Evans Blue in the brain (53–56). With this method, extravasated Evans Blue content in the brain indeed increases at 12 and 24 h after poly-I:C injection. Interestingly, the relative expression levels of microglial activation marker mRNAs began to increase 12 h after poly-I:C administration, which showed a similar time course in the change of BBB permeability (48). This may partly, if not entirely, explain how i.p. injection of poly I:C induces morphological activation of microglia in the brain.
TLR3 recognizes viral double-stranded (ds)RNA, mimicked by poly-I:C. As a byproduct of viral replication, dsRNA serves as the signature molecule for viral infection via TLR3 (57). Poly I:C, as a TLR3 agonist, induces suppression of spontaneous activity in rats (51). The involvement of TLR3 on West Nile virus infection, causing lethal encephalitis, has also been reported (58). Viral infections emerge as important events in the etiology of CNS damage, and it was reported that dsRNA/TLR3-activated astrocytes initiate a battery of rapid innate immune responses (57). However, activation of astrocytes turned out to be secondary to TLR3 signaling in the CNS (48).
IL-1β expression is deeply associated with poly-I:C-induced fatigue in rats (48, 51). IL-1β is the typical pro-inflammatory cytokines produced by activated microglia. It was indeed shown that direct application of poly-I:C to primary cultured rat microglia enhances IL-1β expression (48). Therefore, it is likely that i.p. injection of poly-I:C induces IL-1β expression in microglia/macrophages at least partly via direct action of poly-I:C. TLR agonists also upregulate other inflammatory cytokines such as IL-4, IL-5, IL-10, IL-13, and TNF-α (59). However, these cytokines are not often analyzed in CFS animal models. The importance of IL-1β in CFS was proved using a neutralizing antibody against IL-1β (48) or IL-1 receptor antagonist (51) injected into the lateral ventricle of rats.
Instead of blocking IL-1β, prevention of microglial activation may also be useful as well. Pretreatment with minocycline, an inhibitor of microglia/macrophage activation, prevented the increased expression of microglial IL-1β and induction of CFS (48). Since activation of microglia is the key event of other neurological disorders, many beneficial effects of minocycline have recently been reported. The inhibitory effects of minocycline on the neurological diseases, however, seem to be only effective by pretreatment. For example, once microglial cells in spinal cord were activated by sciatic nerve injury, minocycline could not inhibit the neuropathic pain (Akimoto et al., unpublished data). In addition, microglial activation at the onset and the chronic phase of neurological diseases seems quite different. For example, after traumatic brain injury, minocycline reduced chronic microglial activation, while a marker of neurodegeneration increased (60). These findings suggest that microglial activation may have a reparative effect in the chronic phase of neurological disorders, presumably including CFS. The function of activated microglia/macrophages may depend on the time and microenvironment of the brain pathology.
Glial interaction between microglia and astrocytes is crucial for neuronal function in the brain (61, 62). The poly-I:C-induced upregulation of 5-HTT in astrocytes, not in microglia, in the brain of CFS is also the case. The direct effect of poly-I:C is actually on microglia, which then affect astrocytes, because the upregulated expression of 5-HTT in the brain of poly-I:C-injected rats was abolished by pretreatment of minocycline, an inhibitor of microglial activation (48). Therefore, activation of microglia is the trigger for upregulation of 5-HTT in astrocytes.
The messenger from microglia to astrocytes is IL-1β. Among pro-inflammatory cytokines or other factors released from activated microglia (61), IL-1β plays a pivotal role in CFS. The interaction between microglial IL-1β and astrocytic 5-HTT was proved by using IL-1 receptor antagonists. Upregulation of 5-HTT in astrocytes from poly-I:C-injected rat brain was completely blocked by IL-1 receptor antagonists. Furthermore, it was shown that IL-1β directly upregulates the expression of 5-HTT in primary cultured rat astrocytes (48). The function of 5-HTT in astrocytes has been already demonstrated in the rat brain (63).
On the other hand, treatment with IFN-α, one of the factors upregulated by TLR agonists (50), did not affect 5-HTT expression in primary cultured astrocytes (48). Though other factors have not yet been fully investigated, IL-1β is probably the most important factor among poly-I:C-related factors.
5-HTT expressed in astrocytes is sensitive to antidepressants and contributes to control extracellular 5-HT concentration, i.e., 5-HT taken up by astrocytes is inactivated through deamination by monoamine oxidase A, then scavenged into the cerebrospinal fluid. On the other hand, 5-HT taken up by neurons is re-utilized as a neurotransmitter (64, 65). The microdialysis experiment demonstrated that administration of poly-I:C decreased the extracellular levels of 5-HT, which was abolished by imipramine, a non-selective 5-HT reuptake inhibitor (50). Therefore, it is possible that functional 5-HTT overexpressed on astrocytes scavenges 5-HT, thereby leading to the reduction in the 5-HT levels following poly-I:C injection. Additionally, the importance of the 5-HT1A receptor, but not 5-HT2, 5-HT3 or dopamine D3 receptor, has been suggested (50, 66). Considering these information, a specific agonist for the 5-HT1A receptor, as well as 5-HTT reuptake inhibitor, would be promising therapeutic drugs for CFS.
Neuronal function is altered by glial cells in many ways. In a model of immunologically induced fatigue, poly-I:C increases BBB permeability and activates microglia/macrophages through TLR3 signaling. Activated microglia/macrophages secrete IL-1β, which induces 5-HTT expression in astrocytes, reducing extracellular 5-HT levels, and finally less activation of 5-HT1A receptor. Consequently, these cascades possibly produce fatigue sensation (Figure 1). Therefore, inhibition of microglial IL-1β and astrocytic 5-HTT, or specific activation of 5-HT1A receptor could be promising ways to ameliorate the various symptoms of CFS, at least in infection-related cases.
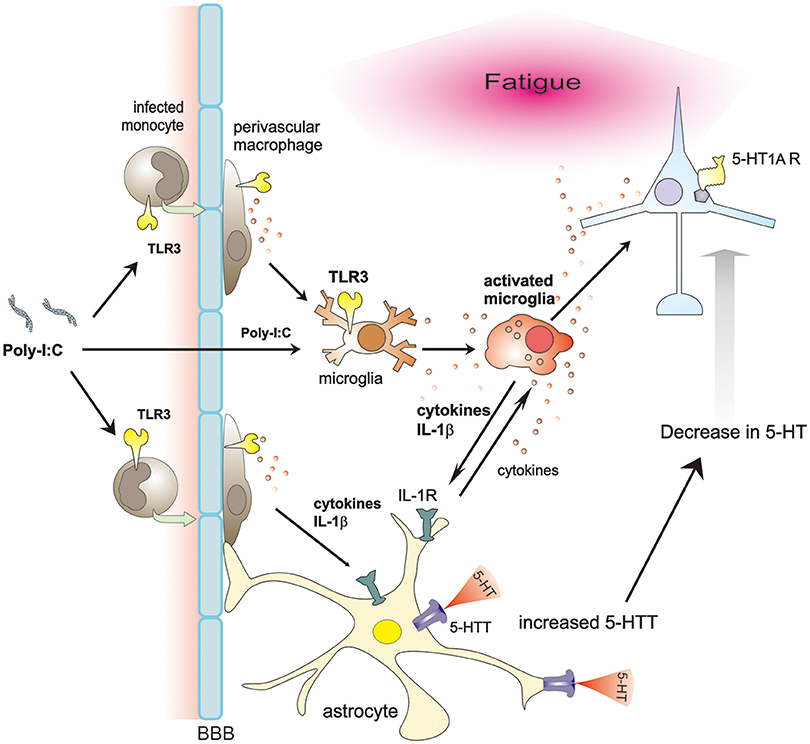
Figure 1. Proposed schema on glia-neuron interaction in a model of immunologically induced fatigue. Poly-I:C, a synthetic double-stranded RNAs, increases BBB permeability, entering the CNS and activating microglia/macrophages through TLR3 signaling. Activated microglia/macrophages secrete various cytokines. Among them, IL-1β upregulates expression of 5-HTT in astrocytes. Due to increased expression of 5-HTT, extracellular 5-HT decreases, impairing the 5-HT signaling, especially via 5-HT1A receptors, thereby possibly inducing fatigue sensation.
MN wrote the manuscript. MI performed experiments and contributed writing manuscript. MSH helped experimenting and editing manuscript. TK used to manage the experiments and wrote the abstract draft.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work is dedicated to the late TK. We thank Prof. David A. Brown (UCL, UK) for helpful comments and correcting the manuscript.
1. Geraghty KJ, Blease C. Myalgic encephalomyelitis/chronic fatigue syndrome and the biopsychosocial model: a review of patient harm and distress in the medical encounter. Disabil Rehabil. (2018) 21:1–10. doi: 10.1080/09638288.2018.1481149
2. Komaroff AL, Takahashi R, Yamamura T, Sawamura M. Neurologic abnormalities in Myalgic Encephalomyelitis/chronic fatigue syndrome: a review. Brain Nerve (2018) 70:41–54. doi: 10.11477/mf.1416200948
3. Monro JA, Puri BK. A molecular neurobiological approach to understanding the aetiology of chronic fatigue syndrome (Myalgic Encephalomyelitis or systemic exertion intolerance disease) with treatment implications. Mol Neurobiol. (2018) 55:7377–88. doi: 10.1007/s12035-018-0928-9
4. Roberts D. Chronic fatigue syndrome and quality of life. Patient Relat Outcome Meas. (2018) 9:253–62. doi: 10.2147/PROM.S155642
5. Rusin A, Seymour C, Mothersill C. Chronic fatigue and immune deficiency syndrome (CFIDS), cellular metabolism, and ionizing radiation: a review of contemporary scientific literature and suggested directions for future research. Int J Radiat Biol. (2018) 94:212–28. doi: 10.1080/09553002.2018.1422871
6. Sharif K, Watad A, Bragazzi NL, Lichtbroun M, Martini M, Perricone C, et al. On chronic fatigue syndrome and nosological categories. Clin Rheumatol. (2018) 37:1161–70. doi: 10.1007/s10067-018-4009-2
7. Crawley E. Pediatric chronic fatigue syndrome: current perspectives. Pediatric Health Med Ther. (2017) 9:27–33. doi: 10.2147/PHMT.S126253
8. Cambras T, Castro-Marrero J, Zaragoza MC, Diez-Noguera A, Alegre J. Circadian rhythm abnormalities and autonomic dysfunction in patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. PLoS ONE (2018) 13:e0198106. doi: 10.1371/journal.pone.0198106
9. Morris G, Stubbs B, Kohler CA, Walder K, Slyepchenko A, Berk M, et al. The putative role of oxidative stress and inflammation in the pathophysiology of sleep dysfunction across neuropsychiatric disorders: focus on chronic fatigue syndrome, bipolar disorder and multiple sclerosis. Sleep Med Rev. (2018) 41:255–65. doi: 10.1016/j.smrv.2018.03.007
10. Corbitt M, Campagnolo N, Staines D, Marshall-Gradisnik S. A systematic review of probiotic interventions for gastrointestinal symptoms and irritable bowel syndrome in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis (CFS/ME). Probiotics Antimicrob Proteins (2018) 10:466–77. doi: 10.1007/s12602-018-9397-8
11. Estevez-Lopez F, Castro-Marrero J, Wang X, Bakken IJ, Ivanovs A, Nacul L, et al. Prevalence and incidence of myalgic encephalomyelitis/chronic fatigue syndrome in Europe-the Euro-epiME study from the European network EUROMENE: a protocol for a systematic review. BMJ Open (2018) 8:e020817. doi: 10.1136/bmjopen-2017-020817
12. Fragkos KC, Barragry J, Fernando CS, Novelli M, Begent J, Zarate-Lopez N. Severe eosinophilic colitis caused by neuropathic agents in a patient with chronic fatigue syndrome and functional abdominal pain: case report and review of the literature. Z Gastroenterol. (2018) 56:573–7. doi: 10.1055/a-0596-7981
13. Galeoto G, Sansoni J, Valenti D, Mollica R, Valente D, Parente M, et al. The effect of physiotherapy on fatigue and physical functioning in chronic fatigue syndrome patients: a systematic review. Clin Ter. (2018) 169:e184–8. doi: 10.7417/T.2018.2076
14. Roman P, Carrillo-Trabalon F, Sanchez-Labraca N, Canadas F, Estevez AF, Cardona D. Are probiotic treatments useful on fibromyalgia syndrome or chronic fatigue syndrome patients? A systematic review. Benef Microbes (2018) 9:603–11. doi: 10.3920/BM2017.0125
15. Teodoro T, Edwards MJ, Isaacs JD. A unifying theory for cognitive abnormalities in functional neurological disorders, fibromyalgia and chronic fatigue syndrome: systematic review. J Neurol Neurosurg Psychiatry (2018). doi: 10.1136/jnnp-2017-317823. [Epub ahead of print].
16. Tollit M, Politis J, Knight S. Measuring school functioning in students with chronic fatigue syndrome: a systematic review. J Sch Health (2018) 88:74–89. doi: 10.1111/josh.12580
17. Vink M, Vink-Niese A. Multidisciplinary rehabilitation treatment is not effective for myalgic encephalomyelitis/chronic fatigue syndrome: a review of the FatiGo trial. Health Psychol Open (2018) 5:2055102918792648. doi: 10.1177/2055102918792648
18. Nakatomi Y, Mizuno K, Ishii A, Wada Y, Tanaka M, Tazawa S, et al. Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis: an (1)(1)C-(R)-PK11195 PET study. J Nucl Med. (2014) 55:945–50. doi: 10.2967/jnumed.113.131045
19. Pardini M, Bonzano L, Mancardi GL, Roccatagliata L. Frontal networks play a role in fatigue perception in multiple sclerosis. Behav Neurosci. (2010) 124:329–36. doi: 10.1037/a0019585
20. Pardini M, Krueger F, Raymont V, Grafman J. Ventromedial prefrontal cortex modulates fatigue after penetrating traumatic brain injury. Neurology (2010) 74:749–54. doi: 10.1212/WNL.0b013e3181d25b6b
21. Kuratsune H, Yamaguti K, Lindh G, Evengard B, Hagberg G, Matsumura K, et al. Brain regions involved in fatigue sensation: reduced acetylcarnitine uptake into the brain. Neuroimage (2002) 17:1256–65. doi: 10.1006/nimg.2002.1260
22. Rutherford G, Manning P, Newton JL. Understanding muscle dysfunction in chronic fatigue syndrome. J Aging Res. (2016) 2016:2497348. doi: 10.1155/2016/2497348
23. Kokubun K, Nemoto K, Oka H, Fukuda H, Yamakawa Y, Watanabe Y. Association of fatigue and stress with gray matter volume. Front Behav Neurosci. (2018) 12:154. doi: 10.3389/fnbeh.2018.00154
24. Afari N, Buchwald D. Chronic fatigue syndrome: a review. Am J Psychiatry (2003) 160:221–36. doi: 10.1176/appi.ajp.160.2.221
25. Chaudhuri A, Behan PO. In vivo magnetic resonance spectroscopy in chronic fatigue syndrome. Prostaglandins Leukot Essent Fatty Acids (2004) 71:181–3. doi: 10.1016/j.plefa.2004.03.009
26. Ishii A, Tanaka M, Watanabe Y. The neural mechanisms of re-experiencing physical fatigue sensation: a magnetoencephalography study. Exp Brain Res. (2016) 234:2433–46. doi: 10.1007/s00221-016-4648-y
27. Ishii A, Tanaka M, Watanabe Y. Neural mechanisms to predict subjective level of fatigue in the future: a magnetoencephalography study. Sci Rep. (2016) 6:25097. doi: 10.1038/srep25097
28. Tanaka M, Ishii A, Watanabe Y. Physical fatigue increases neural activation during eyes-closed state: a magnetoencephalography study. Behav Brain Funct. (2015) 11:35. doi: 10.1186/s12993-015-0079-3
29. Tomas C, Newton J. Metabolic abnormalities in chronic fatigue syndrome/myalgic encephalomyelitis: a mini-review. Biochem Soc Trans. (2018) 46:547–53. doi: 10.1042/BST20170503
30. Fukuda S, Nojima J, Motoki Y, Yamaguti K, Nakatomi Y, Okawa N, et al. A potential biomarker for fatigue: oxidative stress and anti-oxidative activity. Biol Psychol. (2016) 118:88–93. doi: 10.1016/j.biopsycho.2016.05.005
31. Nagy-Szakal D, Barupal DK, Lee B, Che X, Williams BL, Kahn EJR, et al. Insights into myalgic encephalomyelitis/chronic fatigue syndrome phenotypes through comprehensive metabolomics. Sci Rep. (2018) 8:10056. doi: 10.1038/s41598-018-28477-9
32. Yamano E, Kataoka Y. New diagnostic biomarkers for chronic fatigue syndrome. Brain Nerve (2018) 70:27–34. doi: 10.11477/mf.1416200946
33. Yamano E, Sugimoto M, Hirayama A, Kume S, Yamato M, Jin G, et al. Index markers of chronic fatigue syndrome with dysfunction of TCA and urea cycles. Sci Rep. (2016) 6:34990. doi: 10.1038/srep34990
34. Wang X, Qu Y, Zhang Y, Li S, Sun Y, Chen Z, et al. Antifatigue potential activity of sarcodon imbricatus in acute excise-treated and chronic fatigue syndrome in mice via regulation of Nrf2-mediated oxidative stress. Oxid Med Cell Longev. (2018) 2018:9140896. doi: 10.1155/2018/9140896
35. Lacourt TE, Vichaya EG, Chiu GS, Dantzer R, Heijnen CJ. The high costs of low-grade inflammation: persistent fatigue as a consequence of reduced cellular-energy availability and non-adaptive energy expenditure. Front Behav Neurosci. (2018) 12:78. doi: 10.3389/fnbeh.2018.00078
36. Hatziagelaki E, Adamaki M, Tsilioni I, Dimitriadis G, Theoharides TC. Myalgic encephalomyelitis/chronic fatigue syndrome-metabolic disease or disturbed homeostasis? J Pharmacol Exp Ther. (2018) 367:155–67. doi: 10.1124/jpet.118.250845
37. Wostyn P, De Deyn PP. Can cerebrospinal fluid diversion be beneficial in the treatment of chronic fatigue syndrome? Med Hypotheses (2018) 118:174. doi: 10.1016/j.mehy.2018.04.020
38. Wostyn P, De Deyn PP. The putative glymphatic signature of chronic fatigue syndrome: a new view on the disease pathogenesis and therapy. Med Hypotheses (2018) 118:142–5. doi: 10.1016/j.mehy.2018.07.007
39. Torrell H, Alonso Y, Garrabou G, Mulet D, Catalan M, Valiente-Palleja A, et al. Mitochondrial dysfunction in a family with psychosis and chronic fatigue syndrome. Mitochondrion (2017) 34:1–8. doi: 10.1016/j.mito.2016.10.007
40. Trivedi MS, Oltra E, Sarria L, Rose N, Beljanski V, Fletcher MA, et al. Identification of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome-associated DNA methylation patterns. PLoS ONE (2018) 13:e0201066. doi: 10.1371/journal.pone.0201066
41. Castro-Marrero J, Serrano-Pertierra E, Oliveira-Rodriguez M, Zaragoza MC, Martinez-Martinez A, Blanco-Lopez MDC, et al. Circulating extracellular vesicles as potential biomarkers in chronic fatigue syndrome/myalgic encephalomyelitis: an exploratory pilot study. J Extracell Vesicles (2018) 7:1453730. doi: 10.1080/20013078.2018.1453730
42. Glaser R, Kiecolt-Glaser JK. Stress-associated immune modulation: relevance to viral infections and chronic fatigue syndrome. Am J Med. (1998) 105:35S−42S.
43. Hickie I, Davenport T, Wakefield D, Vollmer-Conna U, Cameron B, Vernon SD, et al. Post-infective and chronic fatigue syndromes precipitated by viral and non-viral pathogens: prospective cohort study. BMJ (2006) 333:575. doi: 10.1136/bmj.38933.585764.AE
44. Newberry F, Hsieh SY, Wileman T, Carding SR. Does the microbiome and virome contribute to myalgic encephalomyelitis/chronic fatigue syndrome? Clin Sci (Lond). (2018) 132:523–42. doi: 10.1042/CS20171330
45. Oka T, Tanahashi T, Sudo N, Lkhagvasuren B, Yamada Y. Changes in fatigue, autonomic functions, and blood biomarkers due to sitting isometric yoga in patients with chronic fatigue syndrome. Biopsychosoc Med. (2018) 12:3. doi: 10.1186/s13030-018-0123-2
46. Ousman SS, Kubes P. Immune surveillance in the central nervous system. Nat Neurosci. (2012) 15:1096–101. doi: 10.1038/nn.3161
47. Sotzny F, Blanco J, Capelli E, Castro-Marrero J, Steiner S, Murovska M, et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome - evidence for an autoimmune disease. Autoimmun Rev. (2018) 17:601–9. doi: 10.1016/j.autrev.2018.01.009
48. Ifuku M, Hossain SM, Noda M, Katafuchi T. Induction of interleukin-1beta by activated microglia is a prerequisite for immunologically induced fatigue. Eur J Neurosci. (2014) 40:3253–63. doi: 10.1111/ejn.12668
49. Rothwell NJ, Hopkins SJ. Cytokines and the nervous system II: actions and mechanisms of action. Trends Neurosci. (1995) 18:130–6.
50. Katafuchi T, Kondo T, Take S, Yoshimura M. Enhanced expression of brain interferon-alpha and serotonin transporter in immunologically induced fatigue in rats. Eur J Neurosci. (2005) 22:2817–26. doi: 10.1111/j.1460-9568.2005.04478.x
51. Yamato M, Tamura Y, Eguchi A, Kume S, Miyashige Y, Nakano M, et al. Brain interleukin-1β and the intrinsic receptor antagonist control peripheral Toll-like receptor 3-mediated suppression of spontaneous activity in rats. PLoS ONE (2014) 9:e90950. doi: 10.1371/journal.pone.0090950
52. Katafuchi T, Kondo T, Yasaka T, Kubo K, Take S, Yoshimura M. Prolonged effects of polyriboinosinic:polyribocytidylic acid on spontaneous running wheel activity and brain interferon-alpha mRNA in rats: a model for immunologically induced fatigue. Neuroscience (2003) 120:837–45. doi: 10.1016/S0306-4522(03)00365-8
53. Chan PH, Yang GY, Chen SF, Carlson E, Epstein CJ. Cold-induced brain edema and infarction are reduced in transgenic mice overexpressing CuZn-superoxide dismutase. Ann Neurol. (1991) 29:482–6. doi: 10.1002/ana.410290506
54. Murakami K, Kondo T, Chan PH. Blood-brain barrier disruption, edema formation, and apoptotic neuronal death following cold injury. Acta Neurochir Suppl. (1997) 70:234–6.
55. Murakami K, Kondo T, Sato S, Li Y, Chan PH. Occurrence of apoptosis following cold injury-induced brain edema in mice. Neuroscience (1997) 81:231–7.
56. Sato S, Gobbel GT, Li Y, Kondo T, Murakami K, Sato M, et al. Blood-brain barrier disruption, HSP70 expression and apoptosis due to 3-nitropropionic acid, a mitochondrial toxin. Acta Neurochir Suppl. (1997) 70:237–9.
57. Scumpia PO, Kelly KM, Reeves WH, Stevens BR. Double-stranded RNA signals antiviral and inflammatory programs and dysfunctional glutamate transport in TLR3-expressing astrocytes. Glia (2005) 52:153–62. doi: 10.1002/glia.20234
58. Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. (2004) 10:1366–73. doi: 10.1038/nm1140
59. Kopecka J, Rozkova D, Sediva A. Plasmacytoid DCs, exposed to TSLP in synergy with TLR ligands, acquire significant potential towards Th2 polarization. Med Sci Monit Basic Res. (2013) 19:291–9. doi: 10.12659/MSMBR.889791
60. Scott G, Zetterberg H, Jolly A, Cole JH, De Simoni S, Jenkins PO, et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain (2018) 141:459–71. doi: 10.1093/brain/awx339
61. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. (2011) 91:461–553. doi: 10.1152/physrev.00011.2010
62. Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, et al. Role of microglia in central nervous system infections. Clin Microbiol Rev. (2004) 17:942–64, table of contents doi: 10.1128/CMR.17.4.942-964.2004
63. Hirst WD, Price GW, Rattray M, Wilkin GP. Serotonin transporters in adult rat brain astrocytes revealed by [3H]5-HT uptake into glial plasmalemmal vesicles. Neurochem Int. (1998) 33:11–22.
64. Bal N, Figueras G, Vilaro MT, Sunol C, Artigas F. Antidepressant drugs inhibit a glial 5-hydroxytryptamine transporter in rat brain. Eur J Neurosci. (1997) 9:1728–38.
65. Fitzgerald LW, Kaplinsky L, Kimelberg HK. Serotonin metabolism by monoamine oxidase in rat primary astrocyte cultures. J Neurochem. (1990) 55:2008–14.
66. Kajitani K, Yamaguchi H, Dan Y, Furuichi M, Kang D, Nakabeppu Y. MTH1, an oxidized purine nucleoside triphosphatase, suppresses the accumulation of oxidative damage of nucleic acids in the hippocampal microglia during kainate-induced excitotoxicity. J Neurosci. (2006) 26:1688–98. doi: 10.1523/JNEUROSCI.4948-05.2006
Keywords: chronic fatigue syndrome, poly I:C, TLR3, IL-1beta, serotonin transporter
Citation: Noda M, Ifuku M, Hossain MS and Katafuchi T (2018) Glial Activation and Expression of the Serotonin Transporter in Chronic Fatigue Syndrome. Front. Psychiatry 9:589. doi: 10.3389/fpsyt.2018.00589
Received: 19 May 2018; Accepted: 25 October 2018;
Published: 16 November 2018.
Edited by:
Margit Burmeister, University of Michigan, United StatesReviewed by:
Mikhail Pletnikov, Johns Hopkins University, United StatesCopyright © 2018 Noda, Ifuku, Hossain and Katafuchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mami Noda, bm9kYUBwaGFyLmt5dXNodS11LmFjLmpw
†Present Address: Masataka Ifuku, Pharmaceutical Research Division, Takeda Pharmaceutical Company Limited, Fujisawa, Japan
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.