 Meridith T. Robins1,2
Meridith T. Robins1,2 Terrance Chiang1,2
Terrance Chiang1,2 Kendall L. Mores1
Kendall L. Mores1 Doungkamol Alongkronrusmee1,2
Doungkamol Alongkronrusmee1,2 Richard M. van Rijn1,2*
Richard M. van Rijn1,2*
- 1Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, IN, United States
- 2Purdue University Institute for Integrative Neuroscience, Purdue University, West Lafayette, IN, United States
The transition from non-dependent alcohol use to alcohol dependence involves increased activity of the dorsal striatum. Interestingly, the dorsal striatum expresses a large number of inhibitory G-protein-coupled receptors (GPCRs), which when activated may inhibit alcohol-induced increased activity and can decrease alcohol consumption. Here, we explore the hypothesis that dorsal striatal Gi/o-protein activation is sufficient to reduce voluntary alcohol intake. Using a voluntary, limited-access, two-bottle choice, drink-in-the-dark model of alcohol (10%) consumption, we validated the importance of Gi/o signaling in this region by locally expressing neuron-specific, adeno-associated-virus encoded Gi/o-coupled muscarinic M4 designer receptors exclusively activated by designer drugs (DREADD) in the dorsal striatum and observed a decrease in alcohol intake upon DREADD activation. We validated our findings by activating Gi/o-coupled delta-opioid receptors (DORs), which are natively expressed in the dorsal striatum, using either a G-protein biased agonist or a β-arrestin-biased agonist. Local infusion of TAN-67, an in vitro-determined Gi/o-protein biased DOR agonist, decreased voluntary alcohol intake in wild-type and β-arrestin-2 knockout (KO) mice. SNC80, a β-arrestin-2 biased DOR agonist, increased alcohol intake in wild-type mice; however, SNC80 decreased alcohol intake in β-arrestin-2 KO mice, thus resulting in a behavioral outcome generally observed for Gi/o-biased agonists and suggesting that β-arrestin recruitment is required for SNC80-increased alcohol intake. Overall, these results suggest that activation Gi/o-coupled GPCRs expressed in the dorsal striatum, such as the DOR, by G-protein biased agonists may be a potential strategy to decrease voluntary alcohol consumption and β-arrestin recruitment is to be avoided.
Introduction
Alcoholism and alcohol abuse is a widespread health issue, placing a large burden at both the individual and societal level. Yet, pharmacological treatment options are still limited. Currently, only three drugs have been approved by the Food and Drug Administration for the treatment of alcohol use disorders (AUD), and each come with their own limitations in therapeutic efficacy (1); therefore, it is imperative to identify novel targets for more effective drug development, with hopes of increasing the number of treatment options and compliance for AUD management.
One potential AUD treatment approach is to increase inhibition of the dorsal striatum, a brain region with observed increasing activation upon alcohol tasting in heavy alcohol drinking human subjects (2). In contrast to the ventral striatum, which is implicated in reward-associated learning and behavior, the dorsal striatum is heavily involved in the transition to compulsive drug or alcohol seeking and taking (2–5). In rats, habitual alcohol self-administration increases habit-like responding with decreased sensitivity to alcohol devaluation (6). This shift toward habit-like responding, as well as reports of increased hyperexcitability and altered glutamatergic and GABAergic transmission in the dorsomedial striatum upon alcohol exposure (7–9), suggests molecular alterations in this brain region lead to behavioral reinforcement of alcohol intake resulting in habitual, excessive alcohol intake (3, 7, 9). We hypothesized that one conceivable strategy to inhibit this alcohol-induced neuronal excitability is by activation of metabotropic, inhibitory Gi/o-protein signaling pathways via G-protein-coupled receptors (GPCRs) expressed on neurons in this region.
Interestingly, a large number of GPCRs expressed in the dorsal striatum couple to inhibitory G proteins (Gi/o) (10, 11), thereby providing an ideal target for inhibiting this hyperexcitability observed in the dorsal striatum following persistent alcohol use. To investigate our hypothesis, Gi/o-coupled designer receptors exclusively activated by designer drugs (DREADDs) can provide powerful tools (12, 13) to increase Gi/o signaling in a specific brain region, such as the dorsal striatum, on an experimenter’s predetermined time point to determine the role of the dorsal striatum in modulating alcohol consumption. In addition to artificially increasing Gi/o signaling using viral DREADD strategies, agonists have been developed to preferentially activate Gi/o-protein signal pathways over the competing β-arrestin recruitment and signaling pathways for receptors endogenously expressed in the dorsal striatum, with recent advances in opioid receptor pharmacology being a prime example (14–16). For this study, the delta-opioid receptor (DOR), a Gi/o-coupled GPCR with strong expression in the dorsal striatum (17), provided us with a powerful tool to investigate our hypothesis that Gi/o signaling in the dorsal striatum can reduce alcohol use. DORs are thought to play a protective role in AUD, as DOR knockout (KO) mice display increased alcohol consumption and preference compared with wild-type, kappa-, or mu-opioid receptor KO mice, suggesting that DOR expression prevents escalated alcohol intake compared with other opioid receptor subtypes (18). Moreover, DORs are heavily expressed in the dorsal striatum presynaptically on corticostriatal glutamatergic inputs (19), both pre- and postsynaptically on interneurons within this brain region, and enriched on D2 receptor-expressing MSNs (as compared with D1 receptor-expressing MSNs) (20–22). Furthermore, direct activation (23) or indirect activation of DORs via alcohol-induced release of endogenous enkephalins (24) in the dorsal striatum induces long-term depression (LTD).
The importance of the activation of dorsal striatal DORs in the modulation of alcohol intake was first evident in a report by Nielsen et al., where infusion of the DOR-selective agonist SNC80 into the dorsal striatum increased alcohol intake in rats while the DOR antagonist naltrindole reduced intake (25). This finding that DOR agonist SNC80 increased alcohol was somewhat surprising as DOR expression was previously mentioned to be protective against increased alcohol intake (18). Yet, our recent work investigating a panel of DOR agonists suggests that SNC80 prefers to recruit β-arrestin protein through a mechanism called biased signaling (also termed functional selectivity) (26, 27), where we have additionally correlated in vitro β-arrestin recruitment with in vivo increased alcohol intake (28). In that same study investigating the behavioral effects of DOR biased signaling, we also observed that DOR agonists that weakly recruit β-arrestin, particularly TAN-67 (and thus are G-protein-biased), decreased alcohol intake in mice in a limited-access, drinking-in-the-dark (DID) protocol to 10% alcohol (28).
Therefore, here we hypothesized that activation of Gi/o signaling in the dorsal striatum would be beneficial in reducing alcohol intake, whereas β-arrestin signaling will lead to enhanced alcohol use. To begin to investigate this hypothesis, we first utilized hM4Di DREADD technology (12) to identify the broad role of Gi/o-coupled receptor activation in the dorsal striatal on voluntary alcohol intake in C57Bl/6 male mice using a two-bottle choice, limited-access DID protocol. In addition, we selectively infused our previously identified differentially biased DOR agonists in wild-type and β-arrestin-2 KO mice to more specifically investigate the effect of increased dorsal striatal DOR Gi/o-protein signaling (versus β-arrestin) on voluntary alcohol intake.
Materials and Methods
Drugs and Chemicals
SNC80 and SB205607 (TAN-67) were purchased from Tocris, R&D systems (Minneapolis, MN, USA); naltrindole hydrochloride, forskolin, 200 proof ethyl alcohol, leu-enkephalin, sodium chloride, DMSO, cocaine hydrochloride, and clozapine-N-oxide (CNO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). For dorsal striatal infusion studies, TAN-67 and SNC80 were diluted in 0.9% saline to a concentration of 10 µM; for cellular assays, drugs were dissolved in water. Cocaine was dissolved in 0.9% saline for an administered dose of 15 mg/kg, and CNO was dissolved in 100% DMSO and diluted to a concentration of 0.2 mg/ml in saline (final DMSO concentration of 0.5% and administered dose of 2 mg/kg). Both cocaine and CNO were injected intraperitoneally (i.p.) during experimentation. Non-Cre-dependent AAV8-hSyn-hM4Di-mCherry (7.4 × 1012 vg/ml) virus and AAV8-hSyn-EGFP (3.9 × 1012 vg/ml) virus were obtained from the University of North Carolina Vector Core. Both viruses were chosen as they specifically express in neurons through use of the synapsin promoter. A 100 mg/kg ketamine (Henry Schein, Dublin, OH, USA):10 mg/kg xylazine (Sigma-Aldrich) cocktail was administered to induce anesthesia for cannulation surgeries and before transcardial perfusion. All systemic drugs were injected at a volume of 10 ml/kg.
Cell Culture and Biased Signaling Assays
Competition binding assays were performed using the Tag-lite assay according to the manufacturer’s protocol (Cis-Bio, Bedford, MA, USA). In short, Tb-labeled HEK293-SNAP-hDOR cells/well (4,000 cells/well) were plated in 10 µl Tag-lite medium into low-volume 384-well plates in the presence of 5 µl 8 nM fluorescent naltrexone (final concentration) and 5 µl of an increasing concentration of TAN-67, leu-enkephalin, or SNC80 and incubated at room temperature for 3 h. cAMP inhibition and β-arrestin-2 recruitment assays were performed as previously described (28). In brief, for cAMP inhibition assays HEK293 (Life Technologies, Grand Island, NY, USA) cells (15,000 cells/well, 7.5 µl) transiently expressing FLAG-mDOR (29, 30), SNAP-rDOR, or SNAP-hDOR (Cis-Bio), and pGloSensor22F-cAMP plasmids (Promega, Madison, WI, USA) were incubated with Glosensor reagent (Promega, 7.5 µl, 2% final concentration) for 90 min at 37°C/5% CO2. Cells were stimulated with 5 µl DOR agonist 20 min before 30 µM forskolin (5 µl) stimulation for an additional 15 min. For β-arrestin-2 recruitment assays, CHO-hDOR PathHunter β-arrestin-2 cells (DiscoverX, Fremont, CA, USA) were plated (2,500 cells/well, 10 µl) before stimulation with 2.5 µl DOR agonists for 90 min at 37°C/5% CO2, after which cells were incubated with 6 µl cell assay buffer for 60 min at room temperature as per the manufacturer’s protocol. Luminescence and fluorescence for each of the assays were measured using a FlexStation3 plate reader (Molecular Devices, Sunnyvale, CA, USA).
SNAP-rDOR Construction
Rat DOR cDNA was amplified from the pUC17-rDOR plasmid (Versaclone cDNA NP_036749, R&D systems) using the following forward (5′-CTTCGATATCTTGGAGCCGGTGCCTTCTG-3′) and a standard M13 reverse primer using the Pfu Ultra II Hotstart PCR Mastermix (Agilent, Santa Clara, CA, USA) according to the manufacturer’s protocol. The amplified rDOR PCR product and the pSNAP-hDOR plasmid (Cis-Bio) were restricted using EcoRV and XhoI restriction enzymes (New England BioLabs, Ipswich, MA, USA), and the rDOR construct was exchanged with the hDOR gene followed by ligation with T4 DNA Ligase (New England BioLabs) and transformation into NEB5α competent cells (New England BioLabs). The SNAP-rDOR was fully sequenced to ensure correct orientation and absence of point mutations introduced during amplification.
Animals
37 male C57BL/6 mice (age 6 weeks) were purchased from Harlan and habituated for to the facility 1 week before surgery. For β-arrestin-2 KO animals, animals were bred in house, and 16 animals were selected for surgery [for complete details on strain origin see Ref. (28)]. Throughout the experiment, animals were kept in at ambient temperature of 21°C in a room maintained on a reversed 12L:12D cycle (lights off at 10:00, lights on at 22:00) in Purdue University’s animal facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. This study was carried out in accordance with the recommendations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol (#1305000864) was approved by the Purdue University Institutional Animal Care and Use Committee.
Surgical Cannulation
Directly before surgery, mice were anesthetized with ketamine/xylazine (i.p.). A Kopf model 1900 stereotaxic alignment system (David Kopf Instruments, Tujunga, CA, USA) was used to drill two holes using Kyocera #69 drill bits at the following coordinates from bregma: AP = +1 mm, ML = ±1.5 mm, DV: −3.25 mm (31, 32). For experiments involving drug infusion, an additional two holes were drilled using Kyocera #60 drill bits at the following coordinates from bregma: AP = −2.4 mm, ML = ±1.6 mm, and 1 mm screws were positioned to ensure head-cap stability. A bilateral 22-gage guide cannula (cut 1.5 mm below pedestal, PlasticsOne, Roanoke, VA, USA) was attached to the skull using Geristore dental cement (DenMat, Lompoc, CA, USA). In total, two animals did not wake up from surgery, and eight animals were euthanized after their cannulas came off postoperation or throughout alcohol training and/or experimentation.
Viral Injection
After cannulation surgery, animals were single housed in double grommet cages to allow recovery and individual measurement of fluid intake. One-week postsurgery, mice were anesthetized as previously described and injected bilaterally with 450 nl of virus using a Harvard Apparatus infusion pump at a speed of 50 nl/min via internal cannula with 0.5 mm projection (PlasticsOne). The internal cannula was left in place for an additional 5 min to allow viral dispersion and prevent backflow of the viral solution into the injection syringe. All biohazard work was approved by the Institutional Biohazard Committee at Purdue University (#13-013-16).
Voluntary Alcohol Intake
One-week postsurgery and/or 1-week post-viral injection, mice were exposed to a limited-access (4 h/day), two-bottle choice (water versus 10% ethanol), DID protocol 3 h into their active cycle (dark phase) until the alcohol intake was stable as previously described (29). This model has previously shown that TAN-67 administration before the 4-h session decreases alcohol intake with a correlated decrease in blood ethanol concentration (with no TAN-67 effects on alcohol metabolism) (29). Mice were trained for 3 weeks during which the mice initially increased their alcohol intake before reaching steady state consumption. Bottle weights were measured directly before and after the 4-h access period to the second decimal point to determine fluid intake during this access period. Weights of bottles were corrected for any spillage with fluid bottles placed on empty cages.
Drug Infusion or Injection
After 3 weeks of exposure to the drinking model described earlier, alcohol and water intake on the day of infusion (Friday) was compared with the average alcohol intake over the preceding 3 days (Tuesday–Thursday) to determine if either drug injection or infusion altered voluntary alcohol intake in the following manipulations. For experiments involving viral expression, the AAV injected mice were injected with i.p. saline (with 0.5% DMSO) for vehicle measurements in week 4 and 2 mg/kg CNO (i.p.) the following week (week 5). The dose of CNO of 2 mg/kg was utilized as it has previously been shown to be effective in mice in activating expressed DREADDs (33, 34). Also, this relatively low dose limits high concentrations of clozapine caused by metabolism of CNO (35). For experiments involving direct drug infusion into the dorsal striatum, animals received a 150 nl bilateral infusion of saline into the dorsal striatum on Friday of the fourth week of alcohol exposure. In weeks 5 and 6, animals received either a 150 nl infusion of 10 µM TAN-67 or SNC80, respectively, thereby allowing for a within subjects’ analysis. The order of the drug infusions was chosen to mitigate potential DOR internalization and/or degradation as SNC80 is a high internalizing agonist in vitro and in vivo (36, 37). Doses of TAN-67 and SNC80 were determined based on previous studies of SNC80 infusions in rats (25) and in vivo specificity of TAN-67 and SNC80 for the DOR over MOR or KOR had been previously established using KO animals (29, 38). Importantly, no seizure behavior was observed up SNC80 infusion (39) following any dorsal striatal infusions.
Locomotor Activity
Square locomotor boxes from Med Associates (L 27.3 cm × W 27.3 cm × H 20.3 cm, St. Albans, VT, USA) were used to monitor locomotor activity during the active/dark phase as previously described (28). For AAV experiments, animals were placed in the locomotor box 15 min before CNO (2 mg/kg, i.p.) injection to allow baseline locomotor activity scoring. After 15 min, all animals were injected with CNO and then placed back into the box for an additional 60 min of testing to measure the total distance traveled in 60 min following drug injection. For intra-dorsal striatal infusion of SNC80, animals received either 10 µM SNC80 or vehicle (saline 0.9%) infusion and were placed immediately in the boxes for 90 min; locomotor data were analyzed 30 min after drug infusion as this is when drinking experiments began in the previously described alcohol intake studies.
Cannula Location and Immunohistochemical Analysis
For animals undergoing drug infusions, animals were sacrificed via transcardial perfusion within 1 week following their final drinking session. During analysis, it was determined that the cannula of one mouse from these experiments was not positioned properly and this animal was removed from analysis (placement was too ventral). Cannulation location and viral expression was verified with confocal microscopy (Nikon A1, Nikon, Melville, NY, USA) with an area of capture of 1.69 mm2. The experimenter performing analysis was blind to the experimental conditions; all images were evaluated in greyscale to prevent unintentional bias.
Cocaine-Induced c-Fos Activation in DREADD-Expressing Animals
For viral expression studies, both groups of mice were injected with 2 mg/kg CNO (i.p.) during the dark/active phase for each animal. Twenty minutes later, animals were injected with 15 mg/kg cocaine (i.p.) to induce expression of immediate-early gene c-Fos. Brains were collected 90 min following cocaine exposure via transcardial perfusion. Extracted brain samples embedded and frozen in Tissue-Tek® O.C.T. compound (VWR, Radnor, PA, USA) in tissue molds (VWR) and sliced into 50 µm coronal sections via cryostat (Leica Microsystems Inc., Buffalo Grove, IL, USA). Immunohistochemical staining was conducted using primary rabbit anti-c-Fos antibody (sc-52, Santa Cruz Biotechnology, Dallas, TX, USA), diluted 1:1,000. Control-GFP animal brains were applied Alexa-Fluor 594 goat anti-rabbit antibody (A-11012, Life Technologies, Grand Island, NY, USA) diluted 1:1,000. hM4Di-mCherry animal brains were applied Alexa-Fluor 488 goat anti-rabbit (A-11008, Life Technologies, Grand Island, NY, USA) diluted 1:1,000. Brain slices were mounted onto microscope slides (Fisher Scientific, Hampton, NH, USA) for confocal microscopy with an area of the capture of 0.40 mm2. Images were processed using ImageJ software (NIH, Bethesda, MD, USA) for the number of c-Fos positive cells in the dorsal striatum surrounding the viral injection site in infected cell populations. The experimenter performing analysis was blind to the experimental conditions; all images were evaluated in greyscale to prevent unintentional bias.
Statistical Analysis
All data are presented as means ± SEM and was performed using GraphPad Prism7 software (GraphPad Software, La Jolla, CA, USA). Differences between control-GFP and hM4Di-mCherry animals were analyzed by student two-tailed t-test for differences in baseline water intake, alcohol intake, alcohol preference, locomotion after CNO injection, and c-Fos expression in the dorsal striatum. Differences in alcohol intake after saline injection and CNO injection were evaluated by repeated measures, multiple comparisons (Bonferroni) two-way ANOVA. For in vitro assays, non-linear regression using a dose–response to either inhibition (binding, cAMP) or stimulation (β-arrestin-2 recruitment) was conducted to determine pIC50 or pEC50, respectively. In direct dorsal striatal drug infusion experiments, differences in voluntary alcohol intake, water intake, and alcohol preference were analyzed by repeated measures, multiple comparisons (Tukey) two-way ANOVA. The Grubb’s outlier test (alpha = 0.05) was used to identify potential outliers throughout the study. Statistical analysis was conducted in guidance with and approved by Purdue University’s Department of Statistics.
Results
Activation of a Gi/o-Coupled DREADD in the Dorsal Striatum Decreases Alcohol Intake
Cannula placement was verified postmortem (n = 10–11) through immunohistochemical analysis of viral expression (Figure 1A). Viral infusions of control-GFP (green fluorescent protein) or hM4Di-mCherry in the dorsal striatum did not alter baseline alcohol intake, water intake, or alcohol preference when comparing the two groups [Figure 1B, t(20) = 0.81, p = 0.32; Figure 1C, t(20) = 0.60, p = 0.42; Figure S1A in Supplementary Material, t(20) = 1.01, p = 0.55]. Vehicle injection (0.5% DMSO, i.p.) did not affect alcohol intake for control or hM4Di-expressing animals in alcohol intake (Figure 1D; see Table 1 for full statistical analysis for experimental group), water intake (Figure S1B in Supplementary Material), or alcohol preference in control-GFP or hM4Di-mCherry mice (Figure S1E in Supplementary Material). Unlike saline injection, CNO injection (2 mg/kg, i.p.) significantly reduced alcohol intake in hM4Di-expressing mice compared with GFP-control, as evaluated by two-way ANOVA (Figure 1E; Figure S1D in Supplementary Material, effect of drug × virus: p = 0.03), where Bonferroni posttest analysis revealed that CNO significantly reduced alcohol intake only in hM4Di-expressing animals and not control-GFP-expressing mice (p < 0.002). No significant change in water intake was observed after CNO injection in the testing period for in either group of animals (Figure S1C in Supplementary Material). CNO injection did not alter alcohol preference in control-GFP or hM4Di-mCherry mice (Figure S1F in Supplementary Material).
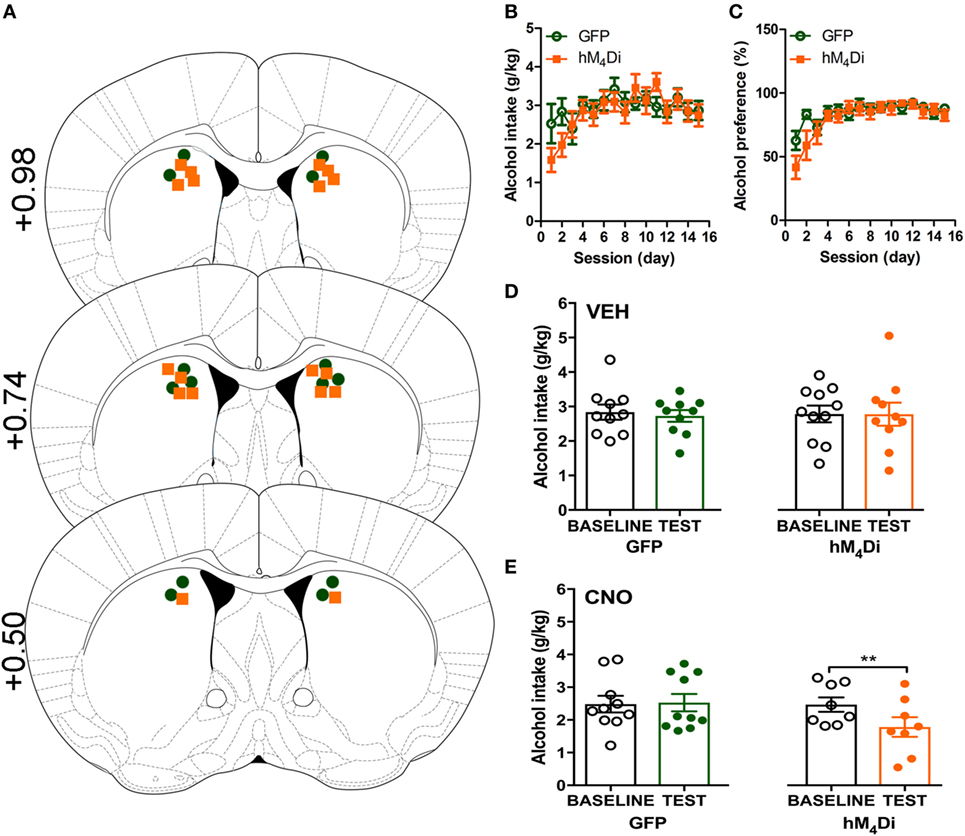
Figure 1. Activation of virally expressed hM4Di in the dorsal striatum decreases alcohol intake in mice. Cannula placement was verified for all animals included in behavioral analysis (A). C57BL/6 mice (n = 10–11) injected in the dorsal striatum with either AAV8-hSyn-EGFP (GFP) or AAV8-hSyn-hM4Di-mCherry (hM4Di) were trained to consume alcohol in a two-bottle, drinking-in-the-dark protocol. Both groups of animals displayed a similar increase in alcohol intake (B) and preference (C). Vehicle injection (saline 0.9%, i.p.) did not change alcohol intake (D). Systemic clozapine-N-oxide (CNO) injection (2 mg/kg i.p.) significantly decreased alcohol intake in mice expressing hM4Di, but not GFP, in the dorsal striatum (E). Significance by unpaired, Student’s t-test for AUC or two-way ANOVA with Bonferroni posttest for matching, **p < 0.01.

Table 1. Analysis of alcohol-related behaviors in control-GFP versus hM4Di-mCherry designer receptors exclusively activated by designer drugs (DREADD)-expressing mice.
Both viruses properly expressed in the dorsal striatum (Figure 2A). Differences in visualization of the control-GFP and hM4Di-mCherry expression may potentially result from differences in viral load and protein expression or inherent differences in quantum yield and extinction coefficients between GFP and mCherry (40). Considering that the striatum is part of the basal ganglia that controls movement (6, 41), we determined whether CNO activation of dorsal striatal hM4Di altered locomotor activity where we observed that CNO did not alter locomotor activity between control-GFP and hM4Di-mCherry expressing mice in a 60-min locomotor period after injection [Figure 2B, t(19) = 0.78, p = 0.45]. To confirm the inhibitory functionality of hM4Di expression, we determined if CNO activation of hM4Di could prevent cocaine-induced c-Fos expression (42, 43), an acceptable approach previously used in other studies to validate functionality of inhibitory DREADDs (42–45). We observed that activation of striatal hM4Di with CNO (2 mg/kg, i.p.) before a cocaine challenge (15 mg/kg, i.p.) significantly inhibited c-Fos activation in animals expressing hM4Di versus GFP controls (control were also administered CNO before cocaine challenge) [Figures 2C,D; t(13) = 2.78, p < 0.02], suggesting that activation of hM4Di via CNO before cocaine challenge inhibited cAMP pathway activity by Gi/o-coupled inhibition. The variability in c-Fos expression in control-GFP may be a result of intrinsic differences in response to psychostimulants between animals, which has been commonly observed in C57Bl/6 mice (46).
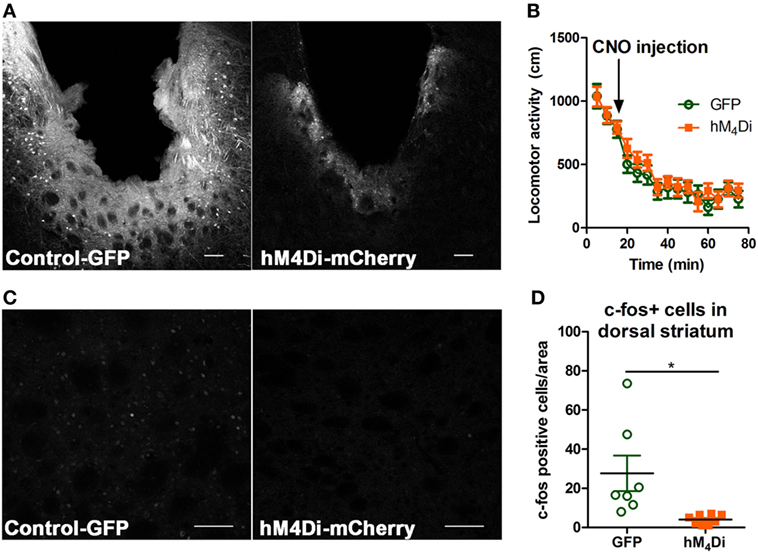
Figure 2. Verification of viral expression and functionality of control-GFP or hM4Di-mCherry in the dorsal striatum. Viral expression verification via confocal microscopy of control-GFP (left) and hM4Di-mCherry (right) in the dorsal striatum (scale bar = 100 µm) (A). C57BL/6 mice (n = 10–11 per group) expressing GFP or hM4Di in the dorsal striatum did not display significant clozapine-N-oxide (CNO) (2 mg/kg, i.p.) induced locomotor activity in the 60-min session after CNO injection (B). Immunohistochemical representation of c-Fos activation in the dorsal striatum in animals expressing control-GFP (left) and hM4Di-mCherry (right) in the dorsal striatum (scale bar = 100 µm) (C). Decreased c-Fos expression in dorsal striatum after cocaine challenge (15 mg/kg, i.p.) in C57BL/6 mice (n = 7–8) expressing hM4Di-mCherry versus control-GFP observed confocal microscopy (D). Significance by unpaired two-tailed t-test, *p < 0.05.
In Vitro Characterization of the β-Arrestin-2 Biased DOR Agonist, SNC80
We have previously established that systemic activation of the Gi/o-coupled DOR with TAN-67, an agonist that only weakly recruits β-arrestin-2 to hDOR (Figure 3A), reduces voluntary intake in mice, but that SNC80, an hDOR agonist that strongly recruits β-arrestin-2 (Figure 3A) increases alcohol intake (28). However, we previously had not determined if a difference in receptor binding was observed between TAN-67 and SNC80 at hDOR to potentially explain differences in ligand bias. Using a SNAP-tag HTRF® (Cis-Bio) approach we found that hDOR, TAN-67 exhibited a pKi = 7.7 ± 0.1 and SNC80 a pKi = 7.2 ± 0.2, with pKi = 5.8 ± 0.1 for leu-enkephalin (Figure 3B), suggesting that the only clear difference between TAN-67 and SNC80 is β-arrestin-2 recruitment efficacy. The surprisingly low affinity observed for leu-enkephalin may be an artifact of the fluorescent binding assay that relies on a large N-terminal SNAP-tag, which may potential interfere with the binding of relatively large peptide ligand, such as leu-enkephalin, but not small molecules.
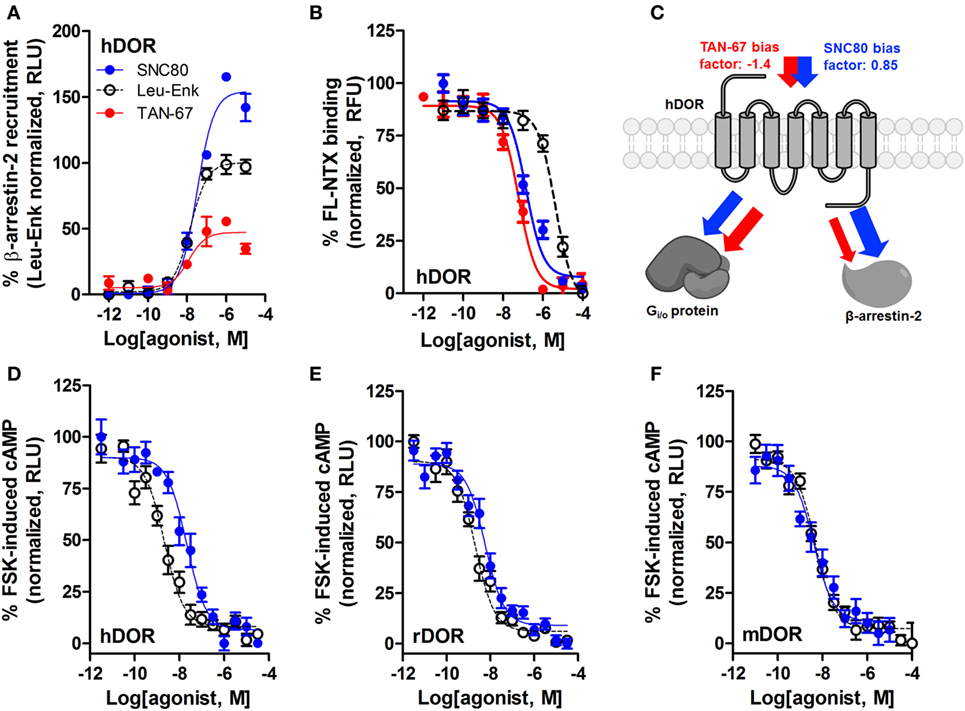
Figure 3. SNC80 is a β-arrestin-biased agonist with comparable potency across species in heterologous cell systems. At the hDOR, SNC80 acts as a β-arrestin-2 super-agonist compared with the endogenous agonist leu-enkephalin and the weak β-arrestin-2 recruiter TAN-67 (A). SNC80 and TAN-67 bind to hDOR with similar affinity (B). Schematic representation of the observed ligand bias of TAN-67 and SNC80 at hDOR, with calculated bias factor (C). SNC80 and Leu-enkephalin have similar potency to inhibit forskolin-induced cAMP production at hDOR (D), rDOR (E), and mDOR (F). A representative summation is shown (n ≥ 3).
Expanding from our previous study, we determined the equiactive bias factors for TAN-67 and SNC80 at hDOR using leu-enkephalin as a reference ligand (47) for β-arrestin-2 recruitment compared with Gi/o-stimulated cAMP inhibition (a more positive bias factor = indicative of bias toward β-arrestin-2, more negative bias factor = indicative of bias toward cAMP activity). TAN-67 displayed a bias factor of −1.4 (cAMP biased) versus a +0.85 bias factor for SNC80 (β-arrestin-2-biased) (Figure 3C). To estimate what concentration of SNC80 to infuse in vivo, we relied on the Nielsen et al. reported findings in rat (25). Our in vitro assays suggest minimal differences in cAMP inhibition between human hDOR (Figure 3D), rat rDOR (Figure 3E), and mDOR (Figure 3F) for SNC80 (pIC50 = 7.8 ± 0.3, n = 3, pIC50 = 8.4 ± 0.1, n = 5, pIC50 = 8.4 ± 0.4, n = 3, respectively) and leu-enkephalin (pIC50 = 8.7 ± 0.2, n = 5, pIC50 = 8.9 ± 0.2, n = 5, pIC50 = 8.3 ± 0.1, n = 6, respectively). Because the affinity and efficacy of TAN-67 is very comparably to SNC80 (Figures 3A,B), we decided to infuse 10 nM TAN-67 and SNC80 into the mouse dorsal striatum to investigate the role of Gi/o signaling versus β-arrestin-2 recruitment in the modulation of alcohol use.
Differential Modulation of Alcohol Intake Following Dorsal Striatal DOR Activation by Gi/o-Biased Versus β-Arrestin-2-Biased DOR Agonists
Cannula terminus location and patency were validated via trypan blue dye infusion into the dorsal striatum upon experimental completion (Figure 4A). Wild-type male animals (n = 9–10) were successfully trained to consume alcohol using a limited-access, two-bottle-choice (water versus 10% alcohol), DID protocol as shown by increased daily alcohol intake and preference (Figures 4B,C) compared with water intake (Figure S2A in Supplementary Material). For intra-striatal infusions, a significant drug (p = 0.03, see Table 2 for full statistical analysis for experimental group and Table S1 in Supplementary Material for Tukey comparisons between infusion weeks) and drug × test session effect (p < 0.0001) was observed, with no effect of test session alone, where Tukey multiple comparisons test revealed that 10 µM of TAN-67 significantly decreased voluntary alcohol intake (p = 0.04) while 10 µM SNC80 significantly increased alcohol intake (p = 0.0005). Importantly, vehicle (saline 0.9%) infusion did not affect alcohol intake (Figure 4D). No changes in water intake or alcohol preference were noted during these drug infusion testing sessions (Figures S2B,C in Supplementary Material).
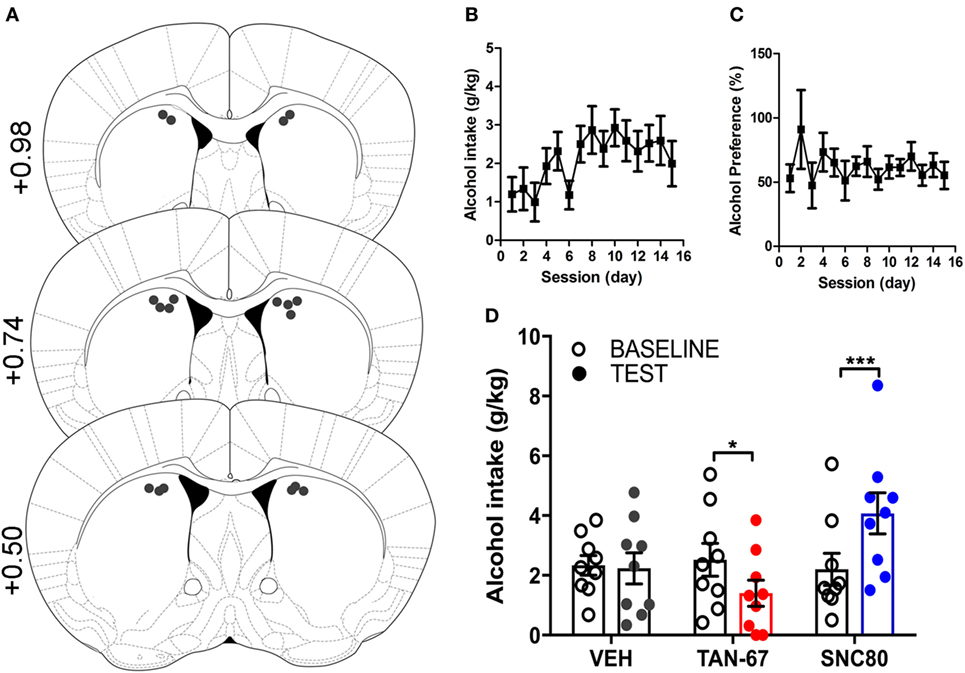
Figure 4. Dorsal striatal infusion of Gi/o-protein biased delta-opioid receptor (DOR) agonist TAN-67 decreases voluntary alcohol intake, while β-arrestin-2 biased DOR agonist SNC80 increases alcohol intake in wild-type mice. Cannula placement was verified for all animals included in behavioral analysis (A). C57BL/6 male, wild-type mice (n = 9–10) were trained to consume 10% alcohol over the course of 3 weeks, during which they increased their alcohol intake (B) and alcohol preference (C). Vehicle saline (0.9%) infusion did not change alcohol intake while TAN-67 (10 µM) significantly decreased alcohol intake and SNC80 (10 µM) significantly increased alcohol intake (D). Significance by repeated measures, multiple comparisons (Tukey) two-way ANOVA, *p < 0.05 and ***p < 0.001.
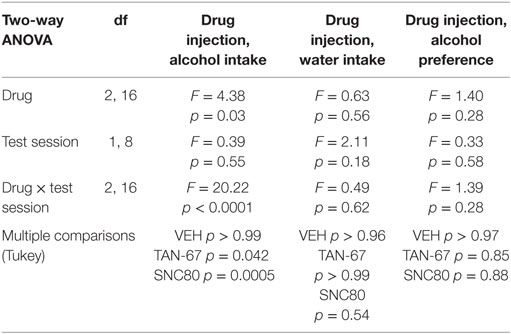
Table 2. Two-way, repeated measures ANOVA of alcohol-related behaviors in wild-type mice upon biased delta-opioid receptor agonist infusion in the dorsal striatum.
Genetic KO of β-Arrestin-2 Provides Additional Support for the Critical Role of DOR-Mediated Gi/o-Coupling in the Dorsal Striatum in Decreasing Alcohol Intake
β-Arrestin-2 KO male C57Bl/6 mice (n = 12) were surgically implanted with a bilateral cannula into the dorsal striatum before alcohol training, and cannula terminus location and patency were validated via trypan blue dye infusion upon experimental completion (Figure 5A). KO animals were successfully trained to consume alcohol using a limited-access, two-bottle-choice (water versus 10% alcohol), DID protocol (Figures 5B,C) compared with water intake (Figure S3A in Supplementary Material). A significant effect of drug (p = 0.003, see Table 3 for full statistical analysis for experimental group and Table S2 in Supplementary Material for Tukey comparisons between infusion weeks), test session (p = 0.002), and drug × test session (p = 0.0021) was identified for intra-dorsal striatal infusions, where multiple comparisons test found no effect of vehicle (saline 0.9%, p = 0.968) on alcohol intake. 10 µM of TAN-67 significantly decreased voluntary alcohol intake (p = 0.0113), and 10 µM of SNC80 also significantly decreased alcohol intake (p = 0.0021) (Figure 5D). This decrease was in contrast with that observed in wild-type animals and is the first report of SNC80’s ability to decrease voluntary alcohol intake, further suggesting that β-arrestin-2 functionality is key for SNC80-increased voluntary alcohol intake. No changes in water intake were noted during testing periods (Figure S3B in Supplementary Material), but a decrease in alcohol preference was noted for SNC80 infusion (p = 0.0018, Figure S3C in Supplementary Material). We have previously observed hyperlocomotion upon systemic SNC80 administration in both wild-type and β-arrestin-2 KO mice with increased alcohol intake or no change in alcohol intake, respectively (28, 48). Therefore, we questioned whether the decrease in alcohol intake upon dorsal striatal SNC80 infusion in the β-arrestin-2 KO was the result of changes in locomotion. However, SNC80 (10 µM) infusion into the dorsal striatum of β-arrestin-2 KO animals did not cause hyperlocomotion compared with vehicle infusion [Figures S4A,B in Supplementary Material, paired two-tailed Student’s t-test: t(6) = 1.68, p = 0.14], although the trend (albeit not significant) toward a decrease in locomotor activity suggests that there may be a potential influence of SNC80 on locomotor activity with respect to the decrease in alcohol intake observed upon SNC80 infusion in β-arrestin-2 KO animals.
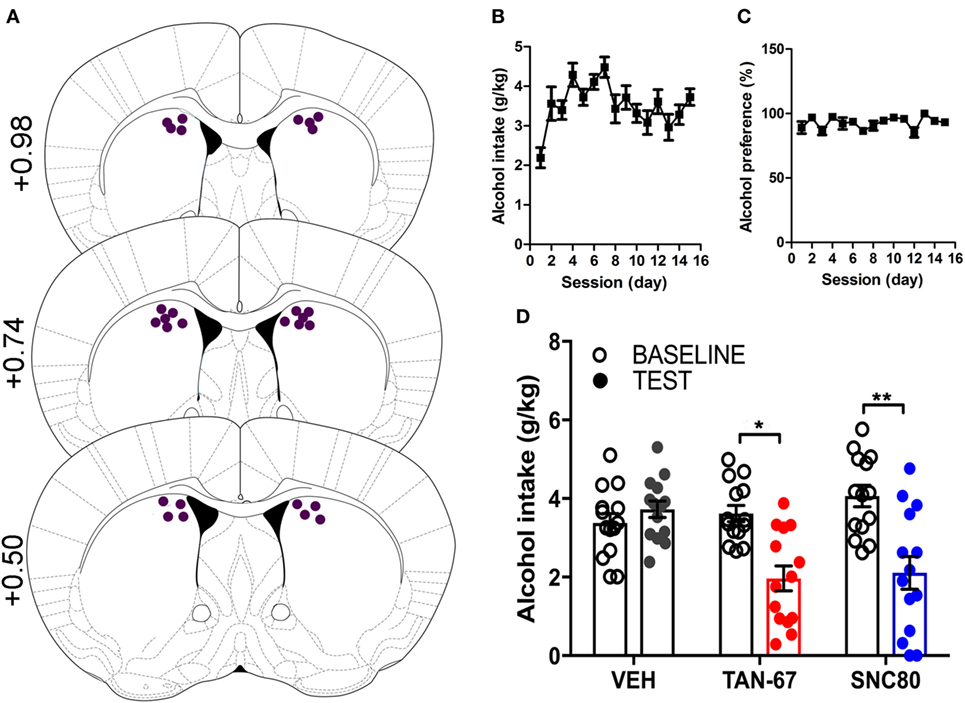
Figure 5. Genetic knockout (KO) of β-arrestin-2 reveals critical role of Gi/o signaling in reducing alcohol intake via dorsal striatal delta-opioid receptor activation. Cannula placement was verified for all animals included in behavioral analysis (A). C57BL/6 male, β-arrestin-2 KO mice (n = 12) were trained to consume 10% alcohol over the course of 3 weeks, during which they increased their alcohol intake (B) and alcohol preference (C). Vehicle saline (0.9%) infusion did not change alcohol intake, but both TAN-67 and SNC80 (10 µM) significantly decreased alcohol intake (D). Significance by repeated measures, multiple comparisons (Tukey) by two-way ANOVA, *p < 0.05 and **p < 0.01.
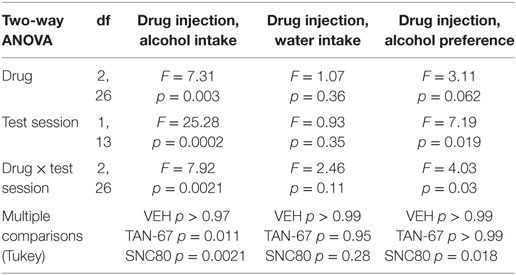
Table 3. Two-way, repeated measures ANOVA of alcohol-related behaviors in β-arrestin-2 knockout mice upon biased delta-opioid receptor agonist infusion in the dorsal striatum.
Discussion
Through both chemogenetic and pharmacologic activation of Gi/o-protein signaling, we observed that activation of Gi/o-protein-coupled receptors in the dorsal striatum significantly decreases alcohol intake in male C57BL/6 mice by either inhibitory DREADD activation or activation of endogenously expressed DORs using a G-protein biased agonist. We specifically targeted the dorsal striatum as it plays an important role in modulating habitual alcohol use (2, 3, 7, 9), has strong DOR expression (20), and, crucially, is a region where DOR agonist SNC80 has been shown to increase alcohol intake in rats (25). Here, activation of virally expressed Gi/o-coupled DREADDs in the dorsal striatum was capable of decreasing alcohol intake while no effect was observed in control-GFP animals upon CNO administration (Figure 1). For activation of endogenous dorsal striatal DORs, our findings that local dorsal striatal infusion of TAN-67 decreased alcohol intake and SNC80 increased alcohol intake (Figure 4; Figure S3 in Supplementary Material) agreed with our systemic findings (28) and also confirmed the previously observed alcohol intake increase following local dorsal striatal infusion of SNC80 in rats (25). Furthermore, through the use of β-arrestin-2 KO mice, we were able to shift the direction of alcohol intake modulation by SNC80 from significantly increasing intake to significantly decreasing consumption when β-arrestin-2 signaling pathways is not present (Figure 5; Figure S3 in Supplementary Material). This was expected as TAN-67 and SNC80 displayed similar binding and G-protein pathway efficacy at DOR in vitro, suggesting that the removal of potential β-arrestin-2 recruitment would allow the agonists to behave similarly (Figure 3). This shift is in agreement with our hypothesis that DOR-mediated Gi/o signaling is a potential strategy to reduce alcohol use, whereas DOR-mediated β-arrestin signaling is to be avoided.
While the dorsal striatum as a region in general is implicated in procedural learning (49–51), the dorsolateral striatum subregion is heavily associated with habitual behavior (behavioral actions that persist despite reward devaluation) (52) and the dorsomedial striatum with goal-directed learning (53). Chronic alcohol exposure may preferentially activate the dorsolateral striatum versus the dorsomedial striatum, as observed by increased glutamatergic transmission (54) and decreased GABAergic transmission (54, 55) in this subregion in animals exposed to chronic intermittent alcohol. Moreover, in rats, alcohol self-administration upregulates brain-derived neurotrophic factor (BDNF) in both the DLS and DMS, but with more robust increases in BDNF in the DLS (56, 57). Furthermore, infusion of BDNF in the DLS decreases alcohol self-administration (57). In rats, initial alcohol seeking was attenuated upon inactivation of the DMS (with no effect upon inactivation of the DLS). However, upon longer exposure to operant alcohol training, animals became insensitive to alcohol devaluation, and inactivation of the DLS re-sensitizes the animals to devaluation (6). Our results presented here did not differentiate between the DMS and the DLS, although future studies warrant investigation of Gi/o-protein activity in these dorsal striatal subregions for potential subregion-specific differences in alcohol intake upon Gi/o-protein activation.
To broadly validate the role of the dorsal striatum in alcohol consumption, we first virally expressed a Gi/o-coupled DREADD (hM4Di) to artificially activate Gi/o-protein signaling pathways in this region to determine how increased Gi/o-protein activity altered alcohol intake. In the present study, activation by the hM4Di DREADD ligand CNO decreased alcohol intake of animals expressing hM4Di in the dorsal striatum and had no effect on control-GFP animals (Figure 1). Despite recent concerns on the use of DREADD technology and CNO’s conversion to clozapine in vivo, the low dose of 2 mg/kg was specific in its behavioral effects on the hM4Di-expressing mice compared with control GFP-expressing animals in drinking behavior (35), thus ruling out the potential issue that decreased consumption resulted from CNO (or clozapine following CNO conversion) activating endogenous muscarinic M4 receptors, which are also highly expressed in the striatum (58). In addition, as previously mentioned, no differences in locomotor activity were observed upon CNO administration in either control or DREADD-expressing mice, suggesting that the observed decrease in consumption did not result from hypolocomotion (Figure 2). Our viral AAV8-DREADD construct was expressed under a human synapsin promoter which specifically targets neurons (59), and given that the majority of the dorsal striatum consists of MSNs and AAV8 has been shown to infect GABAergic neurons in the mouse striatum (60), activation of virally expressed striatal hM4Di receptors in our experimental design likely inhibited both the D1R-MSNs and D2R-MSNs of the direct and indirect pathways, respectively. This net inhibition may be responsible for the observed no net change in locomotor activity and a modest—albeit significant—decrease in alcohol intake (3, 6, 8, 35, 41). We did not verify the potential of preferential tropism of the AAV8-DREADD construct [although AAV8 transduction in the striatum suggests that serotype 8 successfully transduces GABAergic neurons in the mouse striatum (60, 61)], thus limiting our conclusions on the specificity of increased Gi/o-protein activity by DREADD activation on striatal GABAergic and/or cholinergic neurons. Furthermore, while our DREADD strategy was successful in confirming that inhibition of dorsal striatum by increased Gi/o-protein signaling can decrease alcohol consumption, CNO is known to be an unbiased ligand for DREADD receptors (62, 63). Therefore, we next continued with an approach where we could more selectively activate endogenous Gi/o-protein signaling over β-arrestin pathways.
Because of the limitations of potential tropism and possible β-arrestin-2 recruitment in our DREADD strategy, we next investigated changes in alcohol intake upon activation of Gi/o-protein activity by infusing DOR agonists into the dorsal striatum, where DORs are endogenously expressed presynaptically on corticostriatal glutamatergic inputs (19), pre- and postsynaptically on cholinergic interneurons, and on D2-MSNs (20–22). In designing our DOR drug infusion experiments, we infused known DOR agonists into the dorsal striatum of either wild-type or β-arrestin-2 KO mice once a week (following 3 weeks of alcohol drinking) to assess changes in voluntary alcohol intake in response to drug infusion. In the first infusion test week, we infused vehicle (saline 0.9%) to ensure that handling and infusion alone did not change voluntary alcohol intake (Figures 4 and 5). In the second infusion test week, TAN-67 was infused, followed by SNC80 infusion in the third infusion test week. This specific order of drug infusion was determined based upon the in vitro β-arrestin-2 recruitment profiles of TAN-67 and SNC80 (Figure 3) and previously published work on SNC80’s ability to cause rapid DOR internalization [and potential degradation (64)] in vitro and in vivo (in the striatum) (20, 37). Thus, we infused TAN-67 first to prevent potential SNC80-induced desensitization of the DOR system and we did not counterbalance our drug infusions, thus limiting our conclusions on how observed SNC80 responses may be confounded by potential inflammation upon repeated drug infusion into this brain region. Because we specifically observe different behavioral effects with SNC80, which was injected last in both wild-type and β-arrestin-2 KO mice, we would argue that the observed responses represent a true pharmacological effect and are not a negative or positive consequence of repeated infusions.
Our findings that activation of Gi/o signaling in the dorsal striatum reduces alcohol intake would suggest a role for adenylyl cyclase and cAMP in this behavior. Recently, reductions in cAMP levels in the dorsal striatum by adenylyl cyclase type 1 (AC1) inhibition and AC1-KO have been associated with decreased ethanol-induced locomotor sensitization (65). Furthermore, blockade of dorsal striatal Gs-coupled dopamine D1 receptors (but not blockade of Gi/o-coupled dopamine D2) attenuates alcohol consumption (8), suggesting indeed that inhibition of cAMP production in the dorsal striatum may contribute to reduced alcohol use. In the dorsal striatum, alcohol can induce LTD of fast spiking interneuron-medium spiny neuron synapses via a mechanism involving DORs, as this LTD was blocked by a DOR antagonist and the effect was mimicked when using the DOR agonist DPDPE (24). Moreover, the effects of DPDPE can also be blocked by activating adenylyl cyclases with forskolin (24). In our hands, we find that DPDPE is relatively unbiased and thus also efficiently recruits β-arrestin (28). This may be relevant as it has been shown that LTD may also rely on functional β-arrestin-2 expression: activation of hippocampal metabotropic glutamatergic receptors attenuated LTD in β-arrestin-2 KO animals (66, 67) and, upon metabotropic glutamate receptor activation, β-arrestin-2 scaffolding proteins increase the synaptic strength of hippocampal neurons (68). Currently, no studies have investigated the role of β-arrestin-2 in alcohol and DOR-mediated LTD in the dorsal striatum, nor have studies investigated if contributions of cAMP and β-arrestin to LTD change in alcohol-exposed or alcohol-dependent animals.
The observation that β-arrestin-2 activation in the dorsal striatum increases alcohol intake in mice is in agreement with reported elevated expression levels of β-arrestin-2 gene (Arrb2) and β-arrestin-2 protein levels in the striatum of ethanol-preferring alko alcohol rats in comparison with alko non-alcohol rat counterparts, as well as decreased voluntary alcohol intake in β-arrestin-2 KO (69). Despite these connections of β-arrestin expression and voluntary alcohol intake, conflicting results exist on how alcohol intake is altered in β-arrestin-2 KO animals. Li et al. (70) observed that their β-arrestin-2 KO mice displayed increased voluntary alcohol consumption compared with wild-type mice, in line with behavior by our β-arrestin-2 KO mice which also showed slightly higher alcohol intake than wild-type mice (Figure 5) (28). One potential explanation is that the Björk et al. study used alcohol solutions that contained saccharin (28, 69, 70). Importantly, as a number of these aforementioned studies (including ours presented here) utilize global β-arrestin-2 KO animal models, we are limited in our interpretation on how global β-arrestin-2 expression affects general alcohol behavior because of potential compensatory expression of the β-arrestin-1 isoform, particularly because isoform-selective differences in behavior have been observed (71, 72). The effect of β-arrestin expression on alcohol intake is noteworthy as altered levels of β-arrestin-2 have been observed as a result of acute and/or chronic morphine exposure in rats (73), elevated glucocorticoid activity in vitro (74), during inflammation in vivo in synoviocytes, and after cerebral hypoxia/ischemia (75). It is possible that alcohol intake and preference by subjects in these situations is enhanced, and that effectiveness of therapeutic drugs may be altered in these subjects, i.e., an unbiased drug may become β-arrestin-biased and increase alcohol use.
The dorsal striatum contains a large variety of Gi/o-coupled GPCRs besides DORs, including the muscarinic M4 and serotonin 5-HT1B receptors (10, 11). In line with our current findings, all three Gi/o-coupled receptors the respective KO animals (DOR KO, M4R KO, and 5-HT1B KO mice) consume more alcohol compared with wild-type littermates (18, 76, 77). Here, our findings indicate that activation of dorsal striatal Gi/o-coupled receptors, either via endogenous DORs or by virally expressed DREADDs, is sufficient to decrease voluntary alcohol intake in C57Bl/6 male mice. As β-arrestin-2 recruitment is associated with rapid internalization of DORs in vitro and in vivo [where DORs are degraded upon internalization (36, 37, 64)], we hypothesize that β-arrestin-2 recruitment to DORs by SNC80 can lead to rapid desensitization of endogenously expressed DORs, resulting in increased alcohol similar to that observed in DOR KO mice (18). In addition, SNC80-induced β-arrestin-2 recruitment may lead to β-arrestin-dependent signaling events (78), such as increased phosphorylation of ERK (79, 80). Previously, we discovered that agonists of the Gi/o-coupled DOR can either decrease or increase alcohol intake in mice (29, 81, 82), and closer examination of the pharmacology of the DOR agonists revealed that agonists that strongly recruit β-arrestin-2 increased alcohol intake, whereas agonists that were Gi/o-protein biased decreased alcohol intake in mice (28), suggesting that Gi/o-protein biased ligands may be a therapeutic option in treating AUD. Combined with our current results, these studies suggest a potentially broad role for striatal Gi/o-coupled signaling to decrease alcohol intake, which could be accomplished via G-protein biased ligands that activate Gi/o-coupled receptors robustly expressed in the dorsal striatum, such as the DOR. Therefore, the development of Gi/o-protein biased DOR agonists or agonists for other striatal Gi/o-coupled receptors, such as the M4, 5-HT1b, dopamine D2 (83), kappa-opioid (84), and/or GPR88 receptor (85), could present a novel strategy to treat AUD by decreasing excessive alcohol consumption.
Ethics Statement
This study was carried out in accordance with the recommendations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol (#1305000864) was approved by the Purdue University Institutional Animal Care and Use Committee.
Author Contributions
MR performed cannulation surgeries, alcohol intake studies, locomotor studies, perfusions, cocaine challenge, and immunohistochemistry, and wrote the main draft of the manuscript. TC performed alcohol intake studies and bred and genotyped β-arrestin-2 KO mice. KM cloned the rDOR construct and performed the in vitro assays. DA performed locomotor studies and also bred and genotyped β-arrestin-2 KO mice. RR designed the experiments, assisted with the in vitro assays, cannulation surgeries, and alcohol intake studies, and wrote the manuscript. All the authors analyzed and interpreted data and proofread the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was funded by an NARSAD Young Investigator Award from the Brain and Behavior Research Foundation (#23603 to RR) and the National Institute on Alcohol Abuse and Alcoholism (AA025368 to RR). The authors would like to thank Robert Cassell for assistance in running the binding and cAMP assays.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fpsyt.2018.00112/full#supplementary-material.
References
1. Zindel LR, Kranzler HR. Pharmacotherapy of alcohol use disorders: seventy-five years of progress. J Stud Alcohol Drugs Suppl (2014) 75(Suppl 17):79–88. doi:10.15288/jsads.2014.75.79
2. Filbey FM, Claus E, Audette AR, Niculescu M, Banich MT, Tanabe J, et al. Exposure to the taste of alcohol elicits activation of the mesocorticolimbic neurocircuitry. Neuropsychopharmacology (2008) 33(6):1391–401. doi:10.1038/sj.npp.1301513
3. Everitt BJ, Robbins TW. From the ventral to the dorsal striatum: devolving views of their roles in drug addiction. Neurosci Biobehav Rev (2013) 37(9 Pt A):1946–54. doi:10.1016/j.neubiorev.2013.02.010
4. DePoy L, Daut R, Brigman JL, MacPherson K, Crowley N, Gunduz-Cinar O, et al. Chronic alcohol produces neuroadaptations to prime dorsal striatal learning. Proc Natl Acad Sci U S A (2013) 110(36):14783–8. doi:10.1073/pnas.1308198110
5. Vollstädt-Klein S, Wichert S, Rabinstein J, Bühler M, Klein O, Ende G, et al. Initial, habitual and compulsive alcohol use is characterized by a shift of cue processing from ventral to dorsal striatum. Addiction (2010) 105(10):1741–9. doi:10.1111/j.1360-0443.2010.03022.x
6. Corbit LH, Nie H, Janak PH. Habitual alcohol seeking: time course and the contribution of subregions of the dorsal striatum. Biol Psychiatry (2012) 72(5):389–95. doi:10.1016/j.biopsych.2012.02.024
7. Wang J, Carnicella S, Phamluong K, Jeanblanc J, Ronesi JA, Chaudhri N, et al. Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. J Neurosci (2007) 27(13):3593–602. doi:10.1523/JNEUROSCI.4749-06.2007
8. Wang J, Cheng Y, Wang X, Roltsch Hellard E, Ma T, Gil H, et al. Alcohol elicits functional and structural plasticity selectively in dopamine D1 receptor-expressing neurons of the dorsomedial striatum. J Neurosci (2015) 35(33):11634–43. doi:10.1523/JNEUROSCI.0003-15.2015
9. Cheng Y, Huang CCY, Ma T, Wei X, Wang X, Lu J, et al. Distinct synaptic strengthening of the striatal direct and indirect pathways drives alcohol consumption. Biol Psychiatry (2017) 81(11):918–29. doi:10.1016/j.biopsych.2016.05.016
10. Xie K, Martemyanov KA. Control of striatal signaling by g protein regulators. Front Neuroanat (2011) 5:49. doi:10.3389/fnana.2011.00049
11. Komatsu H, Maruyama M, Yao S, Shinohara T, Sakuma K, Imaichi S, et al. Anatomical transcriptome of G protein-coupled receptors leads to the identification of a novel therapeutic candidate GPR52 for psychiatric disorders. PLoS One (2014) 9(2):e90134. doi:10.1371/journal.pone.0090134
12. Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A (2007) 104(12):5163–8. doi:10.1073/pnas.0700293104
13. Vardy E, Robinson JE, Li C, Olsen RHJ, DiBerto JF, Giguere PM, et al. A new DREADD facilitates the multiplexed chemogenetic interrogation of behavior. Neuron (2015) 86(4):936–46. doi:10.1016/j.neuron.2015.03.065
14. Singla N, Minkowitz H, Soergel D, Burt D, Skobieranda F. (432) Respiratory safety signal with oliceridine (TRV130), a novel µ receptor G protein pathway selective modulator (µ-GPS), vs morphine: a safety analysis of a phase 2b randomized clinical trial. J Pain (2016) 17(4S):S82. doi:10.1016/j.jpain.2016.01.409
15. Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, et al. Biased agonism of the µ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: a randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain (2014) 155(9):1829–35. doi:10.1016/j.pain.2014.06.011
16. Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature (2016) 537(7619):185–90. doi:10.1038/nature19112
17. Le Merrer J, Becker JA, Befort K, Kieffer BL. Reward processing by the opioid system in the brain. Physiol Rev (2009) 89(4):1379–412. doi:10.1152/physrev.00005.2009
18. Roberts AJ, Gold LH, Polis I, McDonald JS, Filliol D, Kieffer BL, et al. Increased ethanol self-administration in delta-opioid receptor knockout mice. Alcohol Clin Exp Res (2001) 25(9):1249–56. doi:10.1111/j.1530-0277.2001.tb02344.x
19. Jiang ZG, North RA. Pre- and postsynaptic inhibition by opioids in rat striatum. J Neurosci (1992) 12(1):356–61.
20. Scherrer G, Tryoen-Tóth P, Filliol D, Matifas A, Laustriat D, Cao YQ, et al. Knockin mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. Proc Natl Acad Sci U S A (2006) 103(25):9691–6. doi:10.1073/pnas.0603359103
21. Banghart MR, Neufeld SQ, Wong NC, Sabatini BL. Enkephalin disinhibits mu opioid receptor-rich striatal patches via delta opioid receptors. Neuron (2015) 88(6):1227–39. doi:10.1016/j.neuron.2015.11.010
22. Oude Ophuis RJ, Boender AJ, van Rozen AJ, Adan RA. Cannabinoid, melanocortin and opioid receptor expression on DRD1 and DRD2 subpopulations in rat striatum. Front Neuroanat (2014) 8:14. doi:10.3389/fnana.2014.00014
23. Atwood BK, Kupferschmidt DA, Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat Neurosci (2014) 17(4):540–8. doi:10.1038/nn.3652
24. Patton MH, Roberts BM, Lovinger DM, Mathur BN. Ethanol disinhibits dorsolateral striatal medium spiny neurons through activation of a presynaptic delta opioid receptor. Neuropsychopharmacology (2016) 41(7):1831–40. doi:10.1038/npp.2015.353
25. Nielsen CK, Simms JA, Li R, Mill D, Yi H, Feduccia AA, et al. Delta-opioid receptor function in the dorsal striatum plays a role in high levels of ethanol consumption in rats. J Neurosci (2012) 32(13):4540–52. doi:10.1523/JNEUROSCI.5345-11.2012
26. Zhou L, Bohn LM. Functional selectivity of GPCR signaling in animals. Curr Opin Cell Biol (2014) 27:102–8. doi:10.1016/j.ceb.2013.11.010
27. Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther (2011) 336(2):296–302. doi:10.1124/jpet.110.173948
28. Chiang T, Sansuk K, van Rijn RM. Beta-arrestin 2 dependence of delta opioid receptor agonists is correlated with alcohol intake. Br J Pharmacol (2016) 173(2):323–43. doi:10.1111/bph.13374
29. van Rijn RM, Whistler JL. The delta(1) opioid receptor is a heterodimer that opposes the actions of the delta(2) receptor on alcohol intake. Biol Psychiatry (2009) 66(8):777–84. doi:10.1016/j.biopsych.2009.05.019
30. Whistler JL, Tsao P, von Zastrow M. A phosphorylation-regulated brake mechanism controls the initial endocytosis of opioid receptors but is not required for post-endocytic sorting to lysosomes. J Biol Chem (2001) 276(36):34331–8. doi:10.1074/jbc.M104627200
31. Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. 3rd ed. San Diego: Academic (2008).
32. Pan WX, Mao T, Dudman JT. Inputs to the dorsal striatum of the mouse reflect the parallel circuit architecture of the forebrain. Front Neuroanat (2010) 4:147. doi:10.3389/fnana.2010.00147
33. Zhu H, Pleil KE, Urban DJ, Moy SS, Kash TL, Roth BL. Chemogenetic inactivation of ventral hippocampal glutamatergic neurons disrupts consolidation of contextual fear memory. Neuropsychopharmacology (2014) 39(8):1880–92. doi:10.1038/npp.2014.35
34. Parnaudeau S, Taylor K, Bolkan SS, Ward RD, Balsam PD, Kellendonk C. Mediodorsal thalamus hypofunction impairs flexible goal-directed behavior. Biol Psychiatry (2015) 77(5):445–53. doi:10.1016/j.biopsych.2014.03.020
35. Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science (2017) 357(6350):503–7. doi:10.1126/science.aan2475
36. Pradhan AA, Perroy J, Walwyn WM, Smith ML, Vicente-Sanchez A, Segura L, et al. Agonist-specific recruitment of arrestin isoforms differentially modify delta opioid receptor function. J Neurosci (2016) 36(12):3541–51. doi:10.1523/JNEUROSCI.4124-15.2016
37. Pradhan AA, Becker JA, Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, et al. In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoS One (2009) 4(5):e5425. doi:10.1371/journal.pone.0005425
38. van Rijn RM, Brissett DI, Whistler JL. Emergence of functional spinal delta opioid receptors after chronic ethanol exposure. Biol Psychiatry (2012) 71(3):232–8. doi:10.1016/j.biopsych.2011.07.015
39. Jutkiewicz EM, Rice KC, Traynor JR, Woods JH. Separation of the convulsions and antidepressant-like effects produced by the delta-opioid agonist SNC80 in rats. Psychopharmacology (Berl) (2005) 182(4):588–96. doi:10.1007/s00213-005-0138-9
40. Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods (2005) 2(12):905–9. doi:10.1038/nmeth819
41. Palmiter RD. Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann N Y Acad Sci (2008) 1129:35–46. doi:10.1196/annals.1417.003
42. Burnett CJ, Krashes MJ. Resolving behavioral output via chemogenetic designer receptors exclusively activated by designer drugs. J Neurosci (2016) 36(36):9268–82. doi:10.1523/JNEUROSCI.1333-16.2016
43. Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest (2011) 121(4):1424–8. doi:10.1172/JCI46229
44. Pina MM, Young EA, Ryabinin AE, Cunningham CL. The bed nucleus of the stria terminalis regulates ethanol-seeking behavior in mice. Neuropharmacology (2015) 99:627–38. doi:10.1016/j.neuropharm.2015.08.033
45. Cassataro D, Bergfeldt D, Malekian C, Van Snellenberg JX, Thanos PK, Fishell G, et al. Reverse pharmacogenetic modulation of the nucleus accumbens reduces ethanol consumption in a limited access paradigm. Neuropsychopharmacology (2014) 39(2):283–90. doi:10.1038/npp.2013.184
46. Merritt KE, Bachtell RK. Initial d2 dopamine receptor sensitivity predicts cocaine sensitivity and reward in rats. PLoS One (2013) 8(11):e78258. doi:10.1371/journal.pone.0078258
47. Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, Dewire SM, et al. Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol (2011) 80(3):367–77. doi:10.1124/mol.111.072801
48. Ito S, Mori T, Sawaguchi T. Dopamine-independent psychostimulant activity of a delta-agonist. Behav Pharmacol (2008) 19(2):113–9. doi:10.1097/FBP.0b013e3282f62c1b
49. Packard MG, McGaugh JL. Double dissociation of fornix and caudate nucleus lesions on acquisition of two water maze tasks: further evidence for multiple memory systems. Behav Neurosci (1992) 106(3):439–46. doi:10.1037/0735-7044.106.3.439
50. Packard MG, Hirsh R, White NM. Differential effects of fornix and caudate nucleus lesions on two radial maze tasks: evidence for multiple memory systems. J Neurosci (1989) 9(5):1465–72.
51. Dickinson A, Wood N, Smith JW. Alcohol seeking by rats: action or habit? Q J Exp Psychol B (2002) 55(4):331–48. doi:10.1080/0272499024400016
52. Packard MG, McGaugh JL. Inactivation of hippocampus or caudate nucleus with lidocaine differentially affects expression of place and response learning. Neurobiol Learn Mem (1996) 65(1):65–72. doi:10.1006/nlme.1996.0007
53. Quinn JJ, Pittenger C, Lee AS, Pierson JL, Taylor JR. Striatum-dependent habits are insensitive to both increases and decreases in reinforcer value in mice. Eur J Neurosci (2013) 37(6):1012–21. doi:10.1111/ejn.12106
54. Cuzon Carlson VC, Seabold GK, Helms CM, Garg N, Odagiri M, Rau AR, et al. Synaptic and morphological neuroadaptations in the putamen associated with long-term, relapsing alcohol drinking in primates. Neuropsychopharmacology (2011) 36(12):2513–28. doi:10.1038/npp.2011.140
55. Wilcox MV, Cuzon Carlson VC, Sherazee N, Sprow GM, Bock R, Thiele TE, et al. Repeated binge-like ethanol drinking alters ethanol drinking patterns and depresses striatal GABAergic transmission. Neuropsychopharmacology (2014) 39(3):579–94. doi:10.1038/npp.2013.230
56. Darcq E, Morisot N, Phamluong K, Warnault V, Jeanblanc J, Longo FM, et al. The neurotrophic factor receptor p75 in the rat dorsolateral striatum drives excessive alcohol drinking. J Neurosci (2016) 36(39):10116–27. doi:10.1523/JNEUROSCI.4597-14.2016
57. Jeanblanc J, He DY, Carnicella S, Kharazia V, Janak PH, Ron D. Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. J Neurosci (2009) 29(43):13494–502. doi:10.1523/JNEUROSCI.2243-09.2009
58. Zhang W, Basile AS, Gomeza J, Volpicelli LA, Levey AI, Wess J. Characterization of central inhibitory muscarinic autoreceptors by the use of muscarinic acetylcholine receptor knock-out mice. J Neurosci (2002) 22(5):1709–17.
59. Hioki H, Kameda H, Nakamura H, Okunomiya T, Ohira K, Nakamura K, et al. Efficient gene transduction of neurons by lentivirus with enhanced neuron-specific promoters. Gene Ther (2007) 14(11):872–82. doi:10.1038/sj.gt.3302924
60. Aschauer DF, Kreuz S, Rumpel S. Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS One (2013) 8(9):e76310. doi:10.1371/journal.pone.0076310
61. Low K, Aebischer P, Schneider BL. Direct and retrograde transduction of nigral neurons with AAV6, 8, and 9 and intraneuronal persistence of viral particles. Hum Gene Ther (2013) 24(6):613–29. doi:10.1089/hum.2012.174
62. Wess J, Nakajima K, Jain S. Novel designer receptors to probe GPCR signaling and physiology. Trends Pharmacol Sci (2013) 34(7):385–92. doi:10.1016/j.tips.2013.04.006
63. Alvarez-Curto E, Prihandoko R, Tautermann CS, Zwier JM, Pediani JD, Lohse MJ, et al. Developing chemical genetic approaches to explore G protein-coupled receptor function: validation of the use of a receptor activated solely by synthetic ligand (RASSL). Mol Pharmacol (2011) 80(6):1033–46. doi:10.1124/mol.111.074674
64. Milan-Lobo L, Enquist J, van Rijn RM, Whistler JL. Anti-analgesic effect of the mu/delta opioid receptor heteromer revealed by ligand-biased antagonism. PLoS One (2013) 8(3):e58362. doi:10.1371/journal.pone.0058362
65. Bosse KE, Oginsky MF, Susick LL, Ramalingam S, Ferrario CR, Conti AC. Adenylyl cyclase 1 is required for ethanol-induced locomotor sensitization and associated increases in NMDA receptor phosphorylation and function in the dorsal medial striatum. J Pharmacol Exp Ther (2017) 363(2):148–55. doi:10.1124/jpet.117.242321
66. Stoppel LJ, Auerbach BD, Senter RK, Preza AR, Lefkowitz RJ, Bear MF. β-Arrestin2 couples metabotropic glutamate receptor 5 to neuronal protein synthesis and is a potential target to treat fragile X. Cell Rep (2017) 18(12):2807–14. doi:10.1016/j.celrep.2017.02.075
67. Pontrello CG, Sun MY, Lin A, Fiacco TA, DeFea KA, Ethell IM. Cofilin under control of β-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci U S A (2012) 109(7):E442–51. doi:10.1073/pnas.1118803109
68. Eng AG, Kelver DA, Hedrick TP, Swanson GT. Transduction of group I mGluR-mediated synaptic plasticity by β-arrestin2 signalling. Nat Commun (2016) 7:13571. doi:10.1038/ncomms13571
69. Björk K, Rimondini R, Hansson AC, Terasmaa A, Hyytiä P, Heilig M, et al. Modulation of voluntary ethanol consumption by β-arrestin 2. FASEB J (2008) 22(7):2552–60. doi:10.1096/fj.07-102442
70. Li H, Tao Y, Ma L, Liu X, Ma L. β-Arrestin-2 inhibits preference for alcohol in mice and suppresses Akt signaling in the dorsal striatum. Neurosci Bull (2013) 29(5):531–40. doi:10.1007/s12264-013-1350-y
71. Zurkovsky L, Sedaghat K, Ahmed MR, Gurevich VV, Gurevich EV. Arrestin-2 and arrestin-3 differentially modulate locomotor responses and sensitization to amphetamine. Neuropharmacology (2017) 121:20–9. doi:10.1016/j.neuropharm.2017.04.021
72. Robins MT, Chiang T, Berry JN, Ko MJ, Ha JE, van Rijn RM. Behavioral characterization of β-arrestin 1 knockout mice in anxiety-like and alcohol behaviors. Front Behav Neurosci (2018) 12:54. doi:10.3389/fnbeh.2018.00054
73. Fan XL, Zhang JS, Zhang XQ, Yue W, Ma L. Differential regulation of beta-arrestin 1 and beta-arrestin 2 gene expression in rat brain by morphine. Neuroscience (2003) 117(2):383–9. doi:10.1016/S0306-4522(02)00930-2
74. Oakley RH, Revollo J, Cidlowski JA. Glucocorticoids regulate arrestin gene expression and redirect the signaling profile of G protein-coupled receptors. Proc Natl Acad Sci U S A (2012) 109(43):17591–6. doi:10.1073/pnas.1209411109
75. Lombardi MS, van den Tweel E, Kavelaars A, Groenendaal F, van Bel F, Heijnen CJ. Hypoxia/ischemia modulates G protein-coupled receptor kinase 2 and beta-arrestin-1 levels in the neonatal rat brain. Stroke (2004) 35(4):981–6. doi:10.1161/01.STR.0000121644.82596.7e
76. de la Cour C, Sørensen G, Wortwein G, Weikop P, Dencker D, Fink-Jensen A, et al. Enhanced self-administration of alcohol in muscarinic acetylcholine M4 receptor knockout mice. Eur J Pharmacol (2015) 746:1–5. doi:10.1016/j.ejphar.2014.10.050
77. Crabbe JC, Phillips TJ, Feller DJ, Hen R, Wenger CD, Lessov CN, et al. Elevated alcohol consumption in null mutant mice lacking 5-HT1B serotonin receptors. Nat Genet (1996) 14(1):98–101. doi:10.1038/ng0996-98
78. Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev (2010) 62(2):305–30. doi:10.1124/pr.109.002436
79. Ibba F, Vinci S, Spiga S, Peana AT, Assaretti AR, Spina L, et al. Ethanol-induced extracellular signal regulated kinase: role of dopamine D1 receptors. Alcohol Clin Exp Res (2009) 33(5):858–67. doi:10.1111/j.1530-0277.2009.00907.x
80. Thorsell A, Tapocik JD, Liu K, Zook M, Bell L, Flanigan M, et al. A novel brain penetrant NPS receptor antagonist, NCGC00185684, blocks alcohol-induced ERK-phosphorylation in the central amygdala and decreases operant alcohol self-administration in rats. J Neurosci (2013) 33(24):10132–42. doi:10.1523/JNEUROSCI.4742-12.2013
81. van Rijn RM, Brissett DI, Whistler JL. Distinctive modulation of ethanol place preference by delta opioid receptor-selective agonists. Drug Alcohol Depend (2012) 122(1–2):156–9. doi:10.1016/j.drugalcdep.2011.09.024
82. van Rijn RM, Brissett DI, Whistler JL. Dual efficacy of delta opioid receptor selective ligands for ethanol drinking and anxiety. J Pharmacol Exp Ther (2010) 335(1):133–9. doi:10.1124/jpet.110.170969
83. Dyr W, McBride WJ, Lumeng L, Li TK, Murphy JM. Effects of D1 and D2 dopamine receptor agents on ethanol consumption in the high-alcohol-drinking (HAD) line of rats. Alcohol (1993) 10(3):207–12. doi:10.1016/0741-8329(93)90037-O
84. Logrip ML, Janak PH, Ron D. Blockade of ethanol reward by the kappa opioid receptor agonist U50,488H. Alcohol (2009) 43(5):359–65. doi:10.1016/j.alcohol.2009.05.001
Keywords: dorsal striatum, alcohol intake, biased signaling, delta-opioid receptor, β-arrestin, designer receptors exclusively activated by designer drugs, C57BL/6 mice
Citation: Robins MT, Chiang T, Mores KL, Alongkronrusmee D and van Rijn RM (2018) Critical Role for Gi/o-Protein Activity in the Dorsal Striatum in the Reduction of Voluntary Alcohol Intake in C57Bl/6 Mice. Front. Psychiatry 9:112. doi: 10.3389/fpsyt.2018.00112
Received: 20 December 2017; Accepted: 19 March 2018;
Published: 05 April 2018
Edited by:
Dominique Massotte, UPR3212 Institut des Neurosciences Cellulaires et Intégratives (INCI), FranceReviewed by:
Candice Contet, The Scripps Research Institute, United StatesJay McLaughlin, University of Florida, United States
Copyright: © 2018 Robins, Chiang, Mores, Alongkronrusmee and van Rijn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard M. van Rijn, cnZhbnJpam5AcHVyZHVlLmVkdQ==