 Alice Egerton
Alice Egerton Akarmi Bhachu1
Akarmi Bhachu1 Kate Merritt
Kate Merritt Grant McQueen
Grant McQueen Agata Szulc
Agata Szulc
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 28 April 2017
Sec. Neuroimaging
Volume 8 - 2017 | https://doi.org/10.3389/fpsyt.2017.00066
This article is part of the Research TopicMR Spectroscopy in NeuropsychiatryView all 10 articles
Schizophrenia is associated with brain glutamate dysfunction, but it is currently unclear whether antipsychotic administration can reduce the extent of glutamatergic abnormality. We conducted a systematic review of proton magnetic resonance spectroscopy (1H-MRS) studies examining the effects of antipsychotic treatment on brain glutamate levels in schizophrenia. The Medline database was searched to identify relevant articles published until December 2016. Inclusion required that studies examined longitudinal changes in brain glutamate metabolites in patients with schizophrenia before and after initiation of first antipsychotic treatment or a switch in antipsychotic treatment. The searches identified eight eligible articles, with baseline and follow-up measures in a total of 168 patients. The majority of articles reported a numerical reduction in brain glutamate metabolites with antipsychotic treatment, and the estimated overall mean reduction of 6.5% in Glx (the combined signal from glutamate and glutamine) across brain regions. Significant reductions in glutamate metabolites in at least one brain region were reported in four of the eight studies, and none of the studies reported a significant glutamatergic increase after antipsychotic administration. Relationships between the degree of change in glutamate and the degree of improvement in symptoms have been inconsistent but may provide limited evidence that antipsychotic response may be associated with lower glutamate levels before treatment and a greater extent of glutamatergic reduction during treatment. Further longitudinal, prospective studies of glutamate and antipsychotic response are required to confirm these findings.
Animal, postmortem, and genetic studies indicate that schizophrenia is associated with abnormalities in glutamatergic neurotransmission (1), but it is unclear whether antipsychotic treatment may impact on glutamate dysfunction in patients. Our recent meta-analysis suggests that schizophrenia is associated with a general elevation in glutamate metabolites, with some variation observed across brain regions and patient subgroups (2). Elevations in frontal cortical or hippocampal glutamate may lead to secondary elevations in striatal dopamine release, characteristic of schizophrenia (3–6). Observations of glutamate dysfunction prior to illness onset (7–9), and in patients who are antipsychotic naïve or who have received minimal antipsychotic treatment (10, 11), suggest that glutamatergic dysregulation is a pathological feature of schizophrenia, rather than an effect of antipsychotic exposure. However, it is unknown whether antipsychotic treatment can reduce, or indeed worsen, glutamatergic abnormalities. Potentially, modulation of glutamatergic transmission with antipsychotic treatment could occur via downstream effects of D2 antagonism and/or via interactions at other receptor subtypes. Alternatively, if current antipsychotics do not adequately address glutamatergic dysfunction, this would support the suggestion that adjunctive treatment with glutamatergic agents may have additional therapeutic benefit.
The idea that antipsychotics may modulate glutamatergic neurotransmission is supported by experimental animal studies showing a reduction in frontal cortical glutamate following administration of some, but not all antipsychotics (12–19). In rodents, decreases in frontal glutamate have been observed using ex vivo proton nuclear magnetic resonance (1H-NMR) following administration of clozapine and olanzapine, but not haloperidol, risperidone, or aripiprazole (12). In vivo microdialysis studies in rodents have demonstrated reductions in pharmacologically induced elevations in frontal glutamate by risperidone, paliperidone, clozapine, aripipirazole, olanzapine, and haloperidol (13–19). However, a lack of significant effects of haloperidol on resting or stimulated glutamate metabolites in the rat brain have also been reported (19–22), and when antipsychotics are administered in the absence of pharmacologically stimulated glutamate release, antipsychotic-induced glutamate elevations may also be observed (23, 24). Together, these studies may suggest that antipsychotic glutamate-modulatory effects may be dependent on the animal model or glutamatergic assay, as well as the level of basal glutamatergic tone. Differential effects of antipsychotics may also be mediated by differing receptor binding profiles, and it has been suggested that downregulation of 5HT2A receptors by antipsychotics with high 5HT2A affinity may be important in reducing glutamatergic signaling (15, 16).
In man, brain glutamate levels can be measured in vivo using proton magnetic resonance spectroscopy (1H-MRS), usually within a specific a priori brain region of interest. Depending on the achieved resolution, 1H-MRS can provide concentration estimates for glutamate and glutamine or, at lower field strengths, the combined glutamate plus glutamine signal, which is termed Glx. The idea that antipsychotics may reduce glutamate elevation in schizophrenia is supported by a cross-sectional study that detected an elevation in Glx in non-medicated, but not in medicated schizophrenia (25). In our recent meta-analysis, meta-regression showed no significant relationship between regional glutamate, glutamine, or Glx and mean chlorpromazine equivalent antipsychotic dose (2). Nonetheless, longitudinal studies examining glutamate metabolites before and after antipsychotic treatment are required to address this question directly. Several such studies have now been published and the purpose of this article is to provide systematic review of their findings.
As a second objective, we also review the relationships between glutamate and symptomatic response in longitudinal studies of antipsychotic treatment. Cross-sectional 1H-MRS studies show that glutamate metabolite levels differ between patients who have or have not responded well to antipsychotic treatment (26–29). This may suggest that glutamate level may predict the degree to which symptoms are likely to respond to antipsychotic administration, or that symptom reduction occurs in parallel with antipsychotic glutamatergic modulation. We, therefore, appraised the evidence from longitudinal studies for the value of glutamate levels in predicting or monitoring antipsychotic response.
The review was conducted in accordance with PRISMA guidelines (30). The Medline electronic database was searched to identify journal articles published until 19 December 2016, using the following freeform and MeSH search terms: (“GLUTAMATE” OR “GLX”) AND (“SPECTROSCOPY” OR “MRS”) AND (“ANTIPSYCHOTIC”) AND (“SCHIZOPHRENIA” OR “PSYCHOSIS”). Reference lists of the returned articles were hand searched for further relevant publications.
Inclusion required that articles were published in peer-reviewed journals in English or English translation. Inclusion also required that studies reported 1H-MRS glutamate, glutamine, or Glx before and after either first initiation of antipsychotic medication or initiation of a change in antipsychotic administration. Inclusion was limited to investigations performed in patients with first episode psychosis, schizophrenia, or schizoaffective disorder. Where separate articles reported overlapping datasets, the article reporting the largest dataset was included. Where articles reported glutamate measures over multiple time-points, the glutamate measure at longest time-point was included to provide maximal time for any antipsychotic effects to emerge.
Returned articles were initially screened for inclusion through reading of article titles and abstracts. Full text was then screened for articles potentially meeting the inclusion criteria. Two authors independently performed the searches and identified articles for inclusion (Akarmi Bhachu and Alice Egerton).
For qualitative comparison of antipsychotic-induced change in glutamate metabolites across studies, the percentage difference in glutamatergic metabolite level over the antipsychotic treatment period was extracted from each article. This was calculated as the percentage mean difference (PMD), where PMD = [(mean glutamate metabolite level after treatment − mean glutamate metabolite level before treatment)/mean glutamate metabolite level before treatment] × 100. Where these values were not reported (31, 32), they were extracted from figures using WebPlotDigitizer (http://arohatgi.info/WebPlotDigitizer). Extracted data also included the reported statistical significance of the change in glutamatergic metabolite over the antipsychotic treatment period, the demographic and clinical characteristics of the sample, the antipsychotic/s prescribed and the duration of treatment, and the brain region investigated.
The initial search identified 51 articles, of which 40 were excluded at the title and abstract screening stage. At the full-text screening, one study was excluded as it was not available in English (33). Analysis of the sample reported in Ref. (34) was subsequently extended (35). One article (36) was excluded as it reported a subsample of participants included in a larger cohort and longer term follow-up (31). This resulted in final inclusion of eight original articles (10, 31, 32, 34, 37–40).
The methodological characteristics of the included articles are provided in Table 1. Eight articles provided glutamate measures at baseline and after antipsychotic administration in a total of 168 patients. The sample sizes completing to 1H-MRS follow-up ranged from 7 to 42 patients. Three studies recruited patients with first episode psychosis who were antipsychotic naïve or had received minimal antipsychotic exposure (10, 31, 37). A further study in first episode psychosis did not describe antipsychotic exposure prior to baseline glutamatergic measurement (39). Two studies in chronic schizophrenia included washout periods of at least 7 days prior to baseline glutamate measurement and antipsychotic re-initiation (34, 40). One study in chronic schizophrenia investigated the change in glutamate measures following a switch from conventional antipsychotic treatment to olanzapine and did not include a washout period (38). The final study did not specify stage of illness, but included only patients who were antipsychotic naïve or antipsychotic-free for a minimum of 6 months (32).
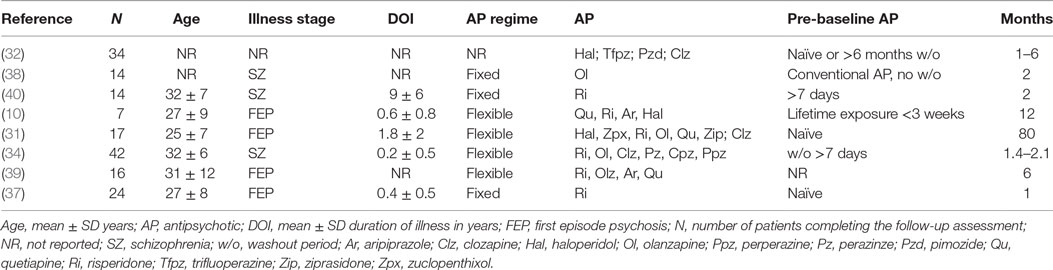
Table 1. Methodological characteristics of studies investigating the effects of antipsychotics on brain glutamate measures.
The majority of studies examined glutamatergic change in samples including patients on a variety of typical and atypical antipsychotic drugs, administered as standard clinical care (10, 31, 32, 34, 39) (Table 1). The remaining three studies investigated specific antipsychotic compounds, with one study investigating olanzapine (38) and two studies investigating risperidone (37, 40). The antipsychotic treatment period ranged from 1 to 80 months.
Four studies acquired glutamate measures at a field strength of 1.5-T and, therefore, reported glutamate primarily as the combined Glx signal (32, 34, 38, 40). Two studies were performed at 3-T, one of which reported Glx (39) and one of which reported both glutamate and Glx (37). The remaining two studies, performed at 4-T, reported glutamate, glutamine (10, 31), and Glx (31). Glutamatergic measures were reported as water-scaled values in ratio to voxel creatine (Cr) (32, 34, 38–40) or corrected for voxel cerebrospinal fluid (CSF) content (10, 31, 37). Brain regions investigated included the frontal cortex (seven studies), thalamus (four studies), temporal cortex (two studies), basal ganglia or striatum (two studies), parieto–occipital cortex (one study), and cerebellum (one study) (Table 2).
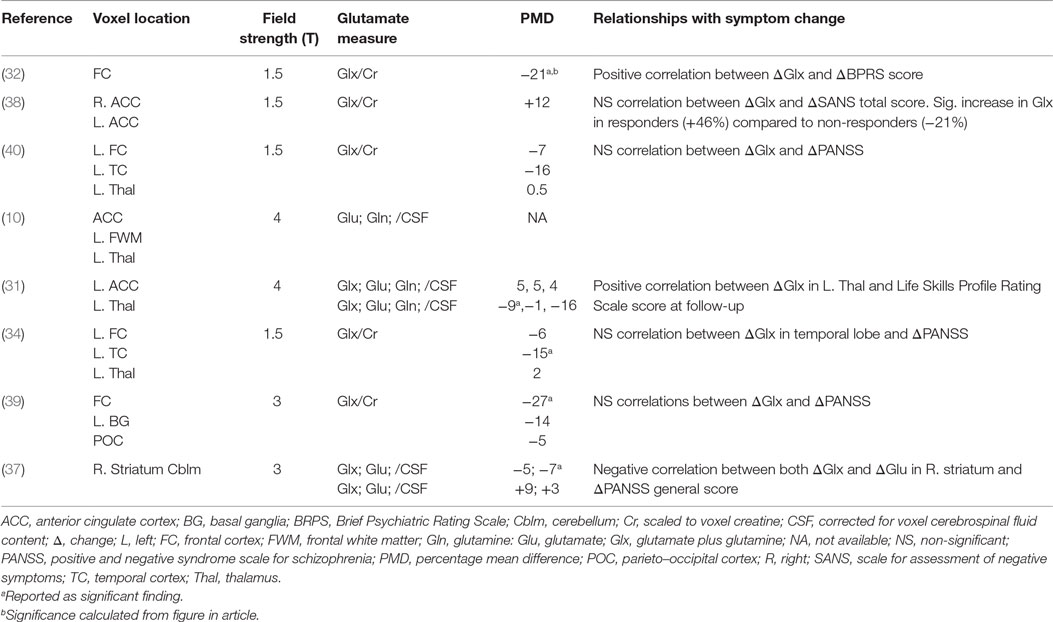
Table 2. Results of studies investigating the effects of antipsychotics on brain glutamate measures and relationships with symptoms.
Across brain regions, the PMD in Glx after antipsychotic treatment ranged from a 12.5% increase (38) to a 27% decrease (39). In the majority of observations (10 out of 15), there was a numerical reduction in Glx, with an overall mean decrease of 6.5% (Figure 1). The PMD in Glx could not be calculated from the information available in one article (10). A decrease in Glx of 6.5% (±11%) can be estimated to be associated with an effect size of approximately dz = 0.6. Accordingly, this translates to a within-subjects’ sample size of 24 patients to observe a significant change in Glx over antipsychotic treatment, at 80% power and α = 0.05, two-tailed.
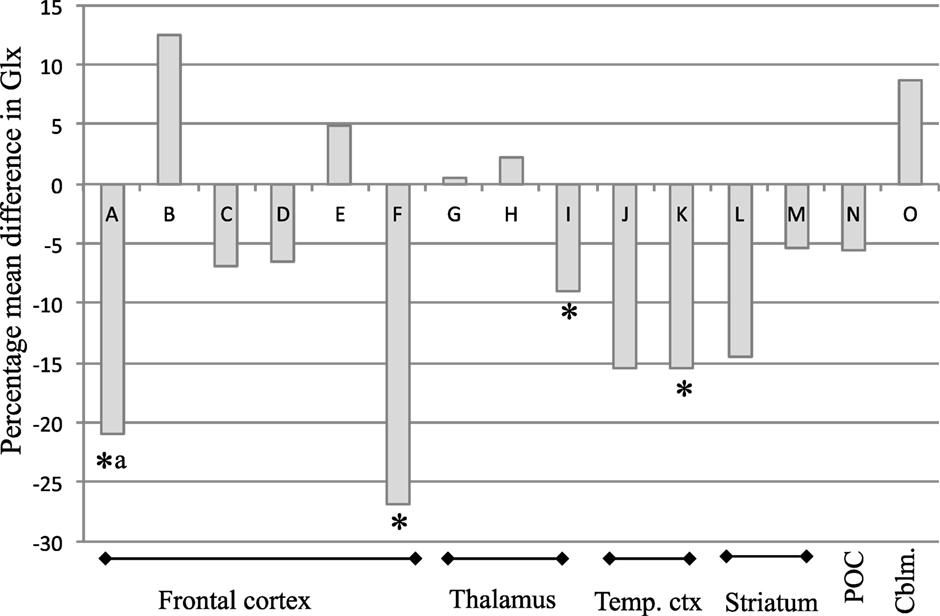
Figure 1. Percentage mean difference (PMD) in Glx in individual studies of Glx at baseline and after antipsychotic administration in schizophrenia. PMD was calculated as [(mean Glx level at after treatment−mean Glx level before treatment)/mean Glx level before treatment] × 100. Abbreviations: Glx, combined signal from glutamate and glutamine; Temp. ctx, temporal cortex; POC, parieto–occipital cortex; Cblm, cerebellum. Letters relate to articles as follows: A (32) B (38) C (40) D (34) E (31) F (39) G (40) H (34) I (31) J (40) K (34) L (39) M (37) N (39) O (37). *Reported as significant finding; aSignificance calculated from figure in article.
Four of the eight included studies reported statistically significant glutamatergic reductions over antipsychotic treatment in at least one brain region investigated (31, 34, 37, 39). Specifically, significant reductions in Glx occurred in the thalamus over 80 months of mixed antipsychotic administration (31), in the left temporal lobe over 2 months of mixed antipsychotic administration (34), and in the frontal lobe over 6 months of mixed antipsychotic administration (39), and in glutamate in right striatum over 1 month of risperidone administration (37). Additionally, Choe et al. (32) also reported a reduction in prefrontal cortical Glx over 1–6 months antipsychotic treatment in 29/34 patients but did not report the statistical outcome associated with this finding. Extraction and analysis of data from the figure provided in the article found a significant reduction in frontal cortical Glx [mean ± SD Glx at baseline: 0.95 ± 0.30; follow-up: 0.75 ± 0.31; paired samples t-test: t(29) = 3.06; P = 0.005]. There were no reports of significant increases in glutamate metabolites with antipsychotic treatment. Three of the eight studies did not report significant effects of antipsychotic treatment on glutamate measures in any brain area across the patient sample (10, 38, 40).
The included studies reported mixed findings regarding associations between the degree of change in glutamate and the degree of symptomatic reduction with antipsychotic treatment (Table 2). Significant positive associations were reported for change in frontal Glx and change in the total score on the Brief Psychiatric Rating Scale (BPRS) (32), and for change in striatal glutamate and Glx and change in Positive and Negative Syndrome Scale (PANSS) general symptom score (37). One study reported a positive correlation between the change in thalamic Glx over 80 months antipsychotic treatment and Life Skills Profile Rating Scale Score at 80 months (31). These observations suggest that a greater degree of glutamate reduction is associated with a greater degree of symptomatic improvement during antipsychotic treatment. However, four of the eight included studies reported no significant correlations between changes in glutamate metabolite levels and changes in symptom severity (34, 38–40). While one of these studies found no significant correlation between change in glutamate and change in the Scale for Assessment of Negative Symptoms (SANS) total score on olanzapine administration (38), a secondary analysis dividing the group into responders and non-responders based on the change in SANS total score found a 46% Glx increase in responders, compared to a 21% Glx decrease in non-responders, and a significant difference between these groups (38). This is broadly inconsistent with the reports of positive associations between glutamate reduction and symptom reduction (32, 37).
None of the studies included in the review examined whether glutamate measures before antipsychotic treatment predicted the degree of subsequent symptomatic response to antipsychotic administration. However, a separate article (35) presenting additional analysis of data presented in a previous article (34) found that frontal Glx at baseline was significantly lower in subsequent responders than non-responders.
The main finding of this review is that the majority of studies reported a numerical reduction in glutamate metabolites following antipsychotic treatment in schizophrenia, with half of the reviewed studies finding significant reductions in at least one brain region. In contrast, no significant increases in glutamate metabolites were reported. Schizophrenia is associated with a general increase in glutamate metabolites, which varies with region and with illness stage (2). This review provides some suggestion that antipsychotics may reduce glutamatergic elevations in schizophrenia but indicates that this effect may be relatively small or limited to subgroups of patients.
The mean change in Glx over antipsychotic treatment was estimated from the available data as a decrease in the range of 6.5% across regions, which would require a within-subjects’ sample size of 24 patients to achieve 80% power. Only three of the eight available studies had sample sizes of 24 or more patients (32, 34, 37) and the three studies with the smallest sample sizes (10, 38, 40) were the studies that did not find significant effects of antipsychotics on glutamatergic measures, suggesting that they may have been underpowered. Two of these smaller studies (38, 40) also differed in that they recruited patients with long antipsychotic medication histories, which may have also limited the ability to observe further glutamatergic reduction.
Our estimates of percentage reduction in glutamatergic metabolites following antipsychotic treatment in schizophrenia and the associated sample size calculations are limited by the availability of relatively few published studies. These studies have also investigated different brain regions, and glutamate dysfunction in schizophrenia may vary by region as well as by illness stage (2). In some cases, it was also necessary to extract values from published figures, which is less accurate than using reported values, and measures included both creatine-scaled or voxel CSF corrected data. As further studies become available, estimates of the percentage reduction can be updated and formal meta-analyses of effect sizes can be performed including inspection of potential influences such as regional specificity, patient subgroups, and duration of antipsychotic treatment. General limitations of 1H-MRS include the estimation of the total concentrations of glutamate metabolites in the voxel, rather than imaging of glutamate involved in neurotransmission specifically, and inability to resolve glutamate from glutamine at lower field strengths and thereby interpretation of the combined Glx signal. Advanced methodological approaches, such as 1H-MRS at high field strengths or 13C-MRS to inspect glutamate cycling may address some of these issues in future studies.
Reductions in glutamate metabolites following antipsychotic treatment would be consistent with the cross-sectional report of elevated prefrontal cortical Glx in non-antipsychotic medicated but not antipsychotic-medicated schizophrenia (25). While the mechanism by which antipsychotics might reduce glutamate is not yet clear, rodent studies also find reductions in basal or stimulated glutamate following administration of some antipsychotics (12–19), which may relate to the 5HT2A antagonist activity of atypical agents (16, 41, 42). Atypical antipsychotics may also indirectly modulate glutamate release by modifying glutamatergic receptor activity or density (43–45). The majority of patients included in the reviewed studies were prescribed atypical antipsychotics, which may, therefore, have increased the potential to observe glutamatergic reductions. Future studies could specifically compare the ability of D2-selective antipsychotics to antipsychotics with marked 5HT2A affinity to modulate glutamate in schizophrenia.
The timescales over which antipsychotic effects on glutamatergic measures have been evaluated range from 4 weeks (37) to 7 years (31). It might be predicted that longer durations of antipsychotic treatment may be associated with progressive glutamate reductions. Across the included studies, there was not suggestion of this, for example significant decreases in frontal Glx were observed after administration of antipsychotics for 1–6 months (32, 39), but not 12 or 80 months (10, 31). Individual studies that included repeated glutamatergic measurement also do not evidence progressive glutamatergic reduction (10, 31). This may suggest that antipsychotic effects on glutamatergic systems are most apparent within the initial stages of treatment, which could be explored in future studies. A further consideration for longitudinal assessment of glutamatergic measures in schizophrenia is the potential interacting effects of other confounds, such as disease progression, aging effects, or volumetric changes. The study of longest duration detected correlations between the reductions in thalamic glutamine and gray matter loss in the superior temporal gyrus over 30 months (36), and gray matter loss in frontal, parietal, temporal, and limbic regions over 7 years (31), possibly reflecting excitotoxic processes.
The second aim of this article was to review the potential relationships between glutamate and the degree of antipsychotic response in longitudinal studies. Our review only identified one study prospectively examining the relationship between baseline glutamate and subsequent response, which reported higher baseline Glx/Cr across voxels in the frontal and temporal lobes and thalamus in subsequent antipsychotic non-responders than responders (35). Two studies also reported correlations between the extent of glutamatergic reduction and the extent of symptomatic improvement over the antipsychotic treatment period (32, 37). These findings are consistent with observations of higher frontal glutamate levels in some (26, 27, 46), but not all (29), cross-sectional studies comparing treatment-responsive and non-responsive schizophrenia, and of higher striatal Glx/Cr in treatment resistant compared to treatment-responsive patients (29). The observation that glutamate levels may predict antipsychotic response (35) is also supported by recent pharmacogenomic findings that single nucleotide polymorphisms in genes encoding glutamatergic proteins associate with response to risperidone in first episode psychosis (47). In contrast, the conflicting result of an increase in frontal Glx/Cr in olanzapine responders compared to non-responders was reported in one study (38), and several studies reported no correlations between glutamatergic change and symptomatic improvement (34, 38–40). The idea that antipsychotic administration is less effective in reducing glutamate and improving symptoms in patients with the highest levels of glutamate before treatment could be further explored through re-analysis of existing longitudinal 1H-MRS studies, and investigated in future longitudinal studies potentially combining glutamate 1H-MRS with glutamate genetic approaches.
AE and AB performed the literature searches and data extraction. KM and GM checked data for accuracy. All authors contributed to interpretation of the data and the final draft of the manuscript.
The views expressed are those of the authors and do not necessarily represent those of the NHS, NIHR, or Department of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by a grant from the Medical Research Council, UK to Dr. Egerton (MR/L003988/1). This study presents independent research funded in part by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley National Health Service (NHS) Foundation Trust and King’s College London.
2. Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK. Nature of glutamate alterations in schizophrenia: a meta-analysis of proton magnetic resonance spectroscopy studies. JAMA Psychiatry (2016) 73:665–74. doi: 10.1001/jamapsychiatry.2016.0442
3. Laruelle M, Kegeles LS, Abi-Dargham A. Glutamate, dopamine, and schizophrenia: from pathophysiology to treatment. Ann N Y Acad Sci (2003) 1003:138–58. doi:10.1196/annals.1300.063
4. Carlsson A. Neurocircuitries and neurotransmitter interactions in schizophrenia. Int Clin Psychopharmacol (1995) 10(Suppl 3):21–8. doi:10.1097/00004850-199509003-00004
5. Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci (2008) 31:234–42. doi:10.1016/j.tins.2008.02.005
6. Stone JM, Morrison PD, Pilowsky LS. Glutamate and dopamine dysregulation in schizophrenia – a synthesis and selective review. J Psychopharmacol (2007) 21:440–52. doi:10.1177/0269881106073126
7. Stone JM, Day F, Tsagaraki H, Valli I, McLean MA, Lythgoe DJ, et al. Glutamate dysfunction in people with prodromal symptoms of psychosis: relationship to gray matter volume. Biol Psychiatry (2009) 66:533–9. doi:10.1016/j.biopsych.2009.05.006
8. Egerton A, Stone JM, Chaddock CA, Barker GJ, Bonoldi I, Howard RM, et al. Relationship between brain glutamate levels and clinical outcome in individuals at ultra high risk of psychosis. Neuropsychopharmacology (2014) 39:2891–9. doi:10.1038/npp.2014.143
9. de la Fuente-Sandoval C, Leon-Ortiz P, Favila R, Stephano S, Mamo D, Ramirez-Bermudez J, et al. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacology (2011) 36:1781–91. doi:10.1038/npp.2011.65
10. Bustillo JR, Rowland LM, Mullins P, Jung R, Chen H, Qualls C, et al. (1)H-MRS at 4 Tesla in minimally treated early schizophrenia. Mol Psychiatry (2010) 15:629–36. doi:10.1038/mp.2009.121
11. Theberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J, et al. Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry (2002) 159:1944–6. doi:10.1176/appi.ajp.159.11.1944
12. McLoughlin GA, Ma D, Tsang TM, Jones DN, Cilia J, Hill MD, et al. Analyzing the effects of psychotropic drugs on metabolite profiles in rat brain using 1H NMR spectroscopy. J Proteome Res (2009) 8:1943–52. doi:10.1021/pr800892u
13. Roenker NL, Gudelsky G, Ahlbrand R, Bronson SL, Kern JR, Waterman H, et al. Effect of paliperidone and risperidone on extracellular glutamate in the prefrontal cortex of rats exposed to prenatal immune activation or MK-801. Neurosci Lett (2011) 500:167–71. doi:10.1016/j.neulet.2011.06.011
14. Abekawa T, Ito K, Nakagawa S, Nakato Y, Koyama T. Effects of aripiprazole and haloperidol on progression to schizophrenia-like behavioural abnormalities and apoptosis in rodents. Schizophr Res (2011) 125:77–87. doi:10.1016/j.schres.2010.08.011
15. Abekawa T, Ito K, Koyama T. Different effects of a single and repeated administration of clozapine on phencyclidine-induced hyperlocomotion and glutamate releases in the rat medial prefrontal cortex at short- and long-term withdrawal from this antipsychotic. Naunyn Schmiedebergs Arch Pharmacol (2007) 375:261–71. doi:10.1007/s00210-007-0154-x
16. Lopez-Gil X, Artigas F, Adell A. Role of different monoamine receptors controlling MK-801-induced release of serotonin and glutamate in the medial prefrontal cortex: relevance for antipsychotic action. Int J Neuropsychopharmacol (2009) 12:487–99. doi:10.1017/S1461145708009267
17. Lopez-Gil X, Babot Z, Amargos-Bosch M, Sunol C, Artigas F, Adell A. Clozapine and haloperidol differently suppress the MK-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology (2007) 32:2087–97. doi:10.1038/sj.npp.1301356
18. Amitai N, Kuczenski R, Behrens MM, Markou A. Repeated phencyclidine administration alters glutamate release and decreases GABA markers in the prefrontal cortex of rats. Neuropharmacology (2012) 62:1422–31. doi:10.1016/j.neuropharm.2011.01.008
19. Carli M, Calcagno E, Mainolfi P, Mainini E, Invernizzi RW. Effects of aripiprazole, olanzapine, and haloperidol in a model of cognitive deficit of schizophrenia in rats: relationship with glutamate release in the medial prefrontal cortex. Psychopharmacology (Berl) (2011) 214:639–52. doi:10.1007/s00213-010-2065-7
20. Bustillo J, Barrow R, Paz R, Tang J, Seraji-Bozorgzad N, Moore GJ, et al. Long-term treatment of rats with haloperidol: lack of an effect on brain N-acetyl aspartate levels. Neuropsychopharmacology (2006) 31:751–6. doi:10.1038/sj.npp.1300874
21. Lindquist DM, Dunn RS, Cecil KM. Long term antipsychotic treatment does not alter metabolite concentrations in rat striatum: an in vivo magnetic resonance spectroscopy study. Schizophr Res (2011) 128:83–90. doi:10.1016/j.schres.2011.02.019
22. Huang M, Panos JJ, Kwon S, Oyamada Y, Rajagopal L, Meltzer HY. Comparative effect of lurasidone and blonanserin on cortical glutamate, dopamine, and acetylcholine efflux: role of relative serotonin (5-HT)2A and DA D2 antagonism and 5-HT1A partial agonism. J Neurochem (2014) 128:938–49. doi:10.1111/jnc.12512
23. Yamamura S, Ohoyama K, Hamaguchi T, Kashimoto K, Nakagawa M, Kanehara S, et al. Effects of quetiapine on monoamine, GABA, and glutamate release in rat prefrontal cortex. Psychopharmacology (Berl) (2009) 206:243–58. doi:10.1007/s00213-009-1601-9
24. Tanahashi S, Yamamura S, Nakagawa M, Motomura E, Okada M. Clozapine, but not haloperidol, enhances glial d-serine and l-glutamate release in rat frontal cortex and primary cultured astrocytes. Br J Pharmacol (2012) 165:1543–55. doi:10.1111/j.1476-5381.2011.01638.x
25. Kegeles LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X, et al. Elevated prefrontal cortex gamma-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry (2012) 69:449–59. doi:10.1001/archgenpsychiatry.2011.1519
26. Egerton A, Brugger S, Raffin M, Barker GJ, Lythgoe DJ, McGuire PK, et al. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology (2012) 37:2515–21. doi:10.1038/npp.2012.113
27. Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry (2013) 75:e11–3. doi:10.1016/j.biopsych.2013.06.011
28. Mouchlianitis E, Bloomfield MA, Law V, Beck K, Selvaraj S, Rasquinha N, et al. Treatment-resistant schizophrenia patients show elevated anterior cingulate cortex glutamate compared to treatment-responsive. Schizophr Bull (2016) 42:744–52. doi:10.1093/schbul/sbv151
29. Goldstein ME, Anderson VM, Pillai A, Kydd RR, Russell BR. Glutamatergic neurometabolites in clozapine-responsive and -resistant schizophrenia. Int J Neuropsychopharmacol (2015) 18:pyu117. doi:10.1093/ijnp/pyu117
30. Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol (2009) 62:1006–12. doi:10.1016/j.jclinepi.2009.06.005
31. Aoyama N, Theberge J, Drost DJ, Manchanda R, Northcott S, Neufeld RW, et al. Grey matter and social functioning correlates of glutamatergic metabolite loss in schizophrenia. Br J Psychiatry (2011) 198:448–56. doi:10.1192/bjp.bp.110.079608
32. Choe BY, Suh TS, Shinn KS, Lee CW, Lee C, Paik IH. Observation of metabolic changes in chronic schizophrenia after neuroleptic treatment by in vivo hydrogen magnetic resonance spectroscopy. Invest Radiol (1996) 31:345–52. doi:10.1097/00004424-199606000-00006
33. de la Fuente-Sandoval C, Favila R, Alvarado P, Leon-Ortiz P, Diaz-Galvis L, Amezcua C, et al. [Glutamate increase in the associative striatum in schizophrenia: a longitudinal magnetic resonance spectroscopy preliminary study]. Gac Med Mex (2009) 145:109–13.
34. Szulc A, Galinska B, Tarasow E, Waszkiewicz N, Konarzewska B, Poplawska R, et al. Proton magnetic resonance spectroscopy study of brain metabolite changes after antipsychotic treatment. Pharmacopsychiatry (2011) 44:148–57. doi:10.1055/s-0031-1279739
35. Szulc A, Konarzewska B, Galinska-Skok B, Lazarczyk J, Waszkiewicz N, Tarasow E, et al. Proton magnetic resonance spectroscopy measures related to short-term symptomatic outcome in chronic schizophrenia. Neurosci Lett (2013) 547:37–41. doi:10.1016/j.neulet.2013.04.051
36. Theberge J, Williamson KE, Aoyama N, Drost DJ, Manchanda R, Malla AK, et al. Longitudinal grey-matter and glutamatergic losses in first-episode schizophrenia. Br J Psychiatry (2007) 191:325–34. doi:10.1192/bjp.bp.106.033670
37. de la Fuente-Sandoval C, Leon-Ortiz P, Azcarraga M, Stephano S, Favila R, Diaz-Galvis L, et al. Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first-episode psychosis: a longitudinal proton magnetic resonance spectroscopy study. JAMA Psychiatry (2013) 70:1057–66. doi:10.1001/jamapsychiatry.2013.289
38. Goff DC, Hennen J, Lyoo IK, Tsai G, Wald LL, Evins AE, et al. Modulation of brain and serum glutamatergic concentrations following a switch from conventional neuroleptics to olanzapine. Biol Psychiatry (2002) 51:493–7. doi:10.1016/S0006-3223(01)01321-X
39. Goto N, Yoshimura R, Kakeda S, Nishimura J, Moriya J, Hayashi K, et al. Six-month treatment with atypical antipsychotic drugs decreased frontal-lobe levels of glutamate plus glutamine in early-stage first-episode schizophrenia. Neuropsychiatr Dis Treat (2012) 8:119–22. doi:10.2147/NDT.S25582
40. Szulc A, Galinska B, Tarasow E, Dzienis W, Kubas B, Konarzewska B, et al. The effect of risperidone on metabolite measures in the frontal lobe, temporal lobe, and thalamus in schizophrenic patients. A proton magnetic resonance spectroscopy (1H MRS). Pharmacopsychiatry (2005) 38:214–9. doi:10.1055/s-2005-873156
41. Ceglia I, Carli M, Baviera M, Renoldi G, Calcagno E, Invernizzi RW. The 5-HT receptor antagonist M100,907 prevents extracellular glutamate rising in response to NMDA receptor blockade in the mPFC. J Neurochem (2004) 91:189–99. doi:10.1111/j.1471-4159.2004.02704.x
42. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science (1998) 281:1349–52. doi:10.1126/science.281.5381.1349
43. Eastwood SL, Porter RH, Harrison PJ. The effect of chronic haloperidol treatment on glutamate receptor subunit (GluR1, GluR2, KA1, KA2, NR1) mRNAs and glutamate binding protein mRNA in rat forebrain. Neurosci Lett (1996) 212:163–6. doi:10.1016/0304-3940(96)12801-9
44. Laruelle M, Frankle WG, Narendran R, Kegeles LS, Abi-Dargham A. Mechanism of action of antipsychotic drugs: from dopamine D(2) receptor antagonism to glutamate NMDA facilitation. Clin Ther (2005) 27(Suppl A):S16–24. doi:10.1016/j.clinthera.2005.07.017
45. Molteni R, Calabrese F, Racagni G, Fumagalli F, Riva MA. Antipsychotic drug actions on gene modulation and signaling mechanisms. Pharmacol Ther (2009) 124:74–85. doi:10.1016/j.pharmthera.2009.06.001
46. Mouchliantis ED, Bloomfield MA, Law V, Beck K, Selvaraj S, Rasquinham R, et al. Treatment resistant schizophrenia patients show anterior cingulate cortex glutamate increase compared to treatment responsive patients. Schizophr Bull (2016) 42(3):744–52. doi:10.1093/schbul/sbv151
Keywords: schizophrenia, magnetic resonance spectroscopy, glutamates, antipsychotics, treatment response
Citation: Egerton A, Bhachu A, Merritt K, McQueen G, Szulc A and McGuire P (2017) Effects of Antipsychotic Administration on Brain Glutamate in Schizophrenia: A Systematic Review of Longitudinal 1H-MRS Studies. Front. Psychiatry 8:66. doi: 10.3389/fpsyt.2017.00066
Received: 31 January 2017; Accepted: 10 April 2017;
Published: 28 April 2017
Edited by:
Paul Croarkin, Mayo Clinic, USACopyright: © 2017 Egerton, Bhachu, Merritt, McQueen, Szulc and McGuire. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alice Egerton, YWxpY2UuZWdlcnRvbkBrY2wuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.