
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry , 25 March 2015
Sec. Neuropharmacology
Volume 6 - 2015 | https://doi.org/10.3389/fpsyt.2015.00041
This article is part of the Research Topic The Role of Cannabinoid Signaling in Emotional Processing: Implications for Neuropsychiatric Disorders View all 7 articles
 Islam Hany Gamaleddin1,2†
Islam Hany Gamaleddin1,2† Jose M. Trigo1†
Jose M. Trigo1† Aliou B. Gueye1
Aliou B. Gueye1 Alexander Zvonok3
Alexander Zvonok3 Alexandros Makriyannis3Steven R. Goldberg4
Alexandros Makriyannis3Steven R. Goldberg4 Bernard Le Foll1,5,6,7,8*
Bernard Le Foll1,5,6,7,8*Several lines of evidence have shown that the endogenous cannabinoids are implicated in several neuropsychiatric diseases. Notably, preclinical and human clinical studies have shown a pivotal role of the cannabinoid system in nicotine addiction. The CB1 receptor inverse agonist/antagonist rimonabant (also known as SR141716) was effective to decrease nicotine-taking and nicotine-seeking in rodents, as well as the elevation of dopamine induced by nicotine in brain reward area. Rimonabant has been shown to improve the ability of smokers to quit smoking in randomized clinical trials. However, rimonabant was removed from the market due to increased risk of psychiatric side-effects observed in humans. Recently, other components of the endogenous cannabinoid system have been explored. Here, we present the recent advances on the understanding of the role of the different components of the cannabinoid system on nicotine’s effects. Those recent findings suggest possible alternative ways of modulating the cannabinoid system that could have implication for nicotine dependence treatment.
Among addictive substances, nicotine use is one of the most prevalent worldwide. The World Health Organization (WHO) estimates that nearly six million tobacco smokers will die every year as a consequence of their tobacco use (1). Therefore, tobacco smoking represents the largest preventable cause of death in the world. Nicotine exerts its effects on the main neurotransmitter systems, such as acetylcholine, dopamine, noradrenaline, serotonin, opioid, glutamate, and gamma-aminobutyric acid (GABA) systems (2–7). There is also mounting evidence supporting the existence of a significant role of the endocannabinoid system in mediating the reinforcing and other addiction-related effects of nicotine. The close overlap of cannabinoid and nicotinic acetylcholine receptors (nAChRs) in certain brain areas such as the midbrain, known to mediate the reinforcing properties of nicotine, but also the hippocampus and the amygdala that are involved in nicotine-associated memory certainly facilitates the interaction between both systems (8–10). There is also evidence of the existence of modulatory interactions between endocannabinoid and cholinergic signaling systems (11–13). Behavioral experiments have shown specific functional interactions between nicotine and the endocannabinoid system that could be mediated by brain structures involved in motivation (14).
The cannabinoid system includes the cannabinoid CB1 and CB2 receptors, endogenous cannabinoids, and the processes responsible for their biosynthesis, cellular uptake, and metabolism (15–18). Endocannabinoids are synthesized on-demand and can activate cannabinoid CB1 and/or CB2 receptors (19, 20). CB1 receptors are believed to be the main mediators of the psychoactive properties of delta-9-tetrahydrocannabinol (THC), which is the main psychoactive component of cannabis (21). CB1 receptors are among the most abundant G-protein-coupled receptors in the central nervous system (CNS) (22). Cannabinoids modify the synaptic efficacy of central neuronal circuits involved in reward and other processes by acting at CB1 receptors located pre-synaptically (23). Although CB2 receptor protein can be detected in the brainstem neurons using western blotting and immunohistochemistry (24), yet, levels of expression of brain CB2 receptors are much lower than those of CB1 receptors (25, 26). CB2 receptor mRNAs were detected in certain regions of the rat brain such as, the cerebellum, cortex, and brainstem using reverse transcription polymerase chain reaction (RT-PCR) (24). In contrast to the predominant pre-synaptic localization of CB1 receptors in the brain, immunoreactivity studies suggest a more likely post-synaptic localization of CB2 receptors (25, 26).
There are several endocannabinoids, of which the most studied have been arachidonoyl ethanolamide (anandamide; AEA) and 2-arachidonoylglycerol (2-AG). Anandamide synthesis is regulated through the conversion of a minor phosphoglyceride, N-arachidonyl phosphatidylethanolamine (N-arachPE), through two possible pathways. The first pathway involves phospholipase D (NAPE-PLD) (27) and the second involves two enzymes, alpha beta hydrolase (ABH4) and glycerophosphodiesterase (GDE1) (28). The exact processes regulating the involvement of these enzymes in the synthesis of anandamide have not been fully elucidated. Cessation of endocannabinoid signaling is hypothesized to happen through the transport inside the cell and the later degradation by specific enzymes. It is further hypothesized that anandamide and 2-AG actually share same the intracellular transport mechanism (17, 23). Anandamide and 2-AG seem to diffuse passively through lipid membranes due their lipophilic nature. Nevertheless, the diffusion process might be facilitated by a selective carrier system (29, 30). After anandamide’s uptake inside the cell, the enzyme fatty acid amide hydrolase (FAAH) degrades it into arachidonic acid and ethanolamine (31, 32). FAAH and CB1 receptors are widely distributed in the CNS and show partial overlap. However, FAAH is mainly available at the post-synaptic neurons whereas CB1 receptors are located at the pre-synaptic neurons (33, 34). On the other hand, 2-AG has its own distinct structure and different biosynthesis and degradation pathways. Moreover, 2-AG appears to be formed under conditions different from those required for the synthesis of anandamide and is modulated by different pharmacological mechanisms. 2-AG is synthesized in response to cellular activation from arachidonic acid-containing membrane phospholipids. The most important pathway for 2-AG synthesis is the phosphatidylinositol (PI)-phospholipase C (PLC)/DAG lipase [diglyceride lipase (DAGL)] pathway, which involves the hydrolysis of inositol phospholipids by PLC. The second pathway for producing 2-AG is through the sequential hydrolysis of PI (35). Although a large number of enzymes are involved in the hydrolysis of monoacyl glycerols, evidence has shown that MAG lipase [monoglyceride lipase (MAGL)] might play a fundamental role on 2-AG degradation. The remaining 2-AG seems to be hydrolyzed by ABHD6 and ABHD12 enzymes (36), although the information on this regard is limited. Interestingly, it has been demonstrated that anandamide enhances the metabolism and in turn attenuates 2-AG effects in the striatum (37). These findings suggest that anandamide and 2-AG might have different actions according to the different physiological or pathophysiological conditions under which they are synthesized (17). Endocannabinoids function as non-conventional neuromodulators whose functions include retrograde signaling and they mediate several types of synaptic plasticity (38). Once released by post-synaptic neurons, endocannabinoids will inhibit neurotransmitter release by pre-synaptic neurons. The respective inhibition of GABA or glutamate release by endocannabinoids mediate depolarization-induced suppression of “inhibition” (DSI) (39, 40) or the depolarization-induced suppression of “excitation” (DSE) (41), respectively. The occurrence of DSE and DSI in the mesocorticolimbic system (42–44) is relevant because of the importance of this system in addiction.
Emerging evidence has shown that the endogenous cannabinoid ligands are implicated in drug addiction processes (10, 45, 46). We will review the literature on nicotine only. Initial preclinical and human clinical studies suggested that the use of CB1 receptor inverse agonists/antagonists such as rimonabant (also known as SR141716) and AM251 might be effective for the treatment of nicotine addiction (47). In fact, rimonabant was able to improve smoking cessation rates in controlled trials (47). However, the use of rimonabant was associated with higher rates of anxiety and depression (48, 49), and consequently this medication was withdrawn from the market in 2008 (50). In this article, we will review the recent advances that have occurred in the last few years regarding our understanding of the different components of the cannabinoid system and how it possibly modulates nicotine addiction.
To better interpret how endocannabinoids modulate the reinforcing effects of nicotine, it is important to understand how nicotine interacts with the different brain reward pathways, particularly the mesolimbic dopaminergic pathway. The psychoactive effects of nicotine are believed to occur through its activation of the nAChRs. These receptors are located in a variety of brain areas and are not limited to the central cholinergic pathways. The nAchRs have been detected in high densities in the thalamus and caudate nucleus, moderate densities in the frontal and temporal parietal cortices, and in the cerebellum with low levels in white matter tracts (51). Exposure to nicotine in a chronic manner leads to desensitization (52) and up regulation (53) of α4β2* subtype of high-affinity nAChRs (54). This receptor subtype has been shown to play a major role in mediating the reinforcing and antinociceptive effects of nicotine. In the ventral tegmental area (VTA), nicotine binds to nAChRs located on nerve terminals of GABAergic and glutamate neurons projecting on the dopaminergic neurons, but also on nAChRs located directly on dopamine neurons (55). The dopamine neurons project to several brain regions implicated in reward including the nucleus accumbens (NAc). Nicotine administration ultimately stimulates the release of dopamine in the dorsal and ventral striatal terminals, notably the NAc (52). These findings have been validated using positron emission tomography (PET) approach (56, 57). Notably, a recent PET study using [11C]-(+)-PHNO PET tracer have shown that tobacco smoking produced elevation of dopamine in the limbic striatum and in extra-striatal area (the ventral pallidum) (57). Interestingly, this study identified that in smokers, dopamine release in the limbic striatum was associated with motivation to smoke, anticipation of pleasure from cigarettes, and relief of withdrawal symptoms. Furthermore, studies have shown that the lesion of the mesolimbic dopamine system (58) or administration of selective dopamine antagonists results in a significant decrease of nicotine self-administration in rats (59). On the other hand, several neurotransmitter systems have shown to play a vital role in nicotine dependence. Studies have shown that nicotine-induced dopamine release can be reduced significantly by atropine (muscarinic receptor antagonist), eticlopride (dopamine D1/2 receptor antagonist), and MK801 [N-methyl-d-aspartate (NMDA) antagonist] (60). It has been shown that smoking cigarettes enhances plasma levels of endogenous opioids (61, 62) and that nicotine stimulates the release of β-endorphins in neuronal cell cultures (63). Furthermore, nicotine-conditioned place preference (CPP) and nicotine-induced antinociception were significantly attenuated in μ-opioid knockout mice compared to wild-type mice (64). Additionally, naloxone has been shown to block nicotine CPP in mice (65).
The dopaminergic system has long been hypothesized to play an essential role in the formulation of goal-directed behaviors of natural rewards and drugs of abuse including nicotine (58, 66, 67). Furthermore, the conditioned-reinforcing properties of drugs of abuse and their associated stimuli are also mediated through the dopaminergic system (68, 69). Dopamine is further involved in the development of behavioral sensitization that follows the repeated administration of drugs of abuse, as well as non-drug stimuli (70).
Several lines of evidence have demonstrated the significance of the dopaminergic system in cue associations. Using a discriminative stimulus and a conditioned stimulus as conditioning tasks associated with a food reward, Miller and colleagues, demonstrated an increase in neuronal firing in the VTA and substantia nigra (71). Similarly, in monkeys, phasic neuronal responses were recorded in response to conditioned stimuli in dopaminergic neurons (72).
The modulatory role of the endocannabinoid system on signaling in the mesolimbic dopamine reward system (73, 74), is believed to be substantiated by its abundant presence within the VTA (75, 76). The ability of endocannabinoids to act as retrograde neurotransmitters (44) allows them to attenuate the activity of external afferents (pre-synaptic neurons) (42) and allows dopamine neurons (post-synaptic neurons) to regulate their own function (43). This topic has been recently reviewed by Wang and Lupica (77) and will be not developed here. It appears that the main endocannabinoid regulating dopamine firing is 2-AG and it has been proposed that the burst firing activity pattern of dopamine neurons as well as the long-term plasticity effects induced by drugs of abuse are regulated by 2-AG (77, 78). The critical role of endocannabinoids mediating the ability of drugs of abuse, including nicotine, to stimulate reward pathway is shown by multiple pharmacological studies (14, 79, 80).
In rats, the selective CB1 receptor inverse agonist/antagonist rimonabant decreases intravenous nicotine self-administration behavior and also nicotine-induced elevations in extracellular dopamine in the NAc (14). We and others subsequently reported that rimonabant decreases the motivation to self-administer nicotine, as measured using progressive-ratio schedules of reinforcement (81) (see Figure 1), blocks the development of nicotine-induced CPP (82–84), and the reinstatement of previously extinguished nicotine-seeking behavior in rats (81, 85, 86) (see Figure 2). Several studies have shown that genetic deletion of cannabinoid CB1 receptors reduces nicotine-induced CPP (87, 88). Cannabinoid CB1 receptor stimulation, in contrast, increased the motivation to self-administer nicotine as measured using a progressive-ratio schedule of reinforcement (89) (see Figure 3), enhanced cue-induced reinstatement of nicotine-seeking behavior and the discriminative stimulus effects of low doses of nicotine in rats (89) (see Figure 4). Several of these responses in rats were blocked by the CB1 inverse agonist/antagonist rimonabant. However, the CB2 antagonist AM630, was not able to block CB1 stimulation effects, supporting the critical role of CB1 receptors in mediating nicotine-dependent processes (89, 90). Interestingly, blockade of the CB1 receptors in the shell of the NAc, the basolateral amygdala, and the prelimbic cortex (91), but also in the bed nucleus of the stria terminalis (92), is able to reduce nicotine-seeking behavior. Nicotine-taking appears to be controlled by CB1 receptors located in the VTA, but not in the NAc (93). Taken together, these findings indicate that CB1 receptors have a bi-directional role on both nicotine reward/reinforcement and on relapse to nicotine-seeking behavior in abstinent subjects.
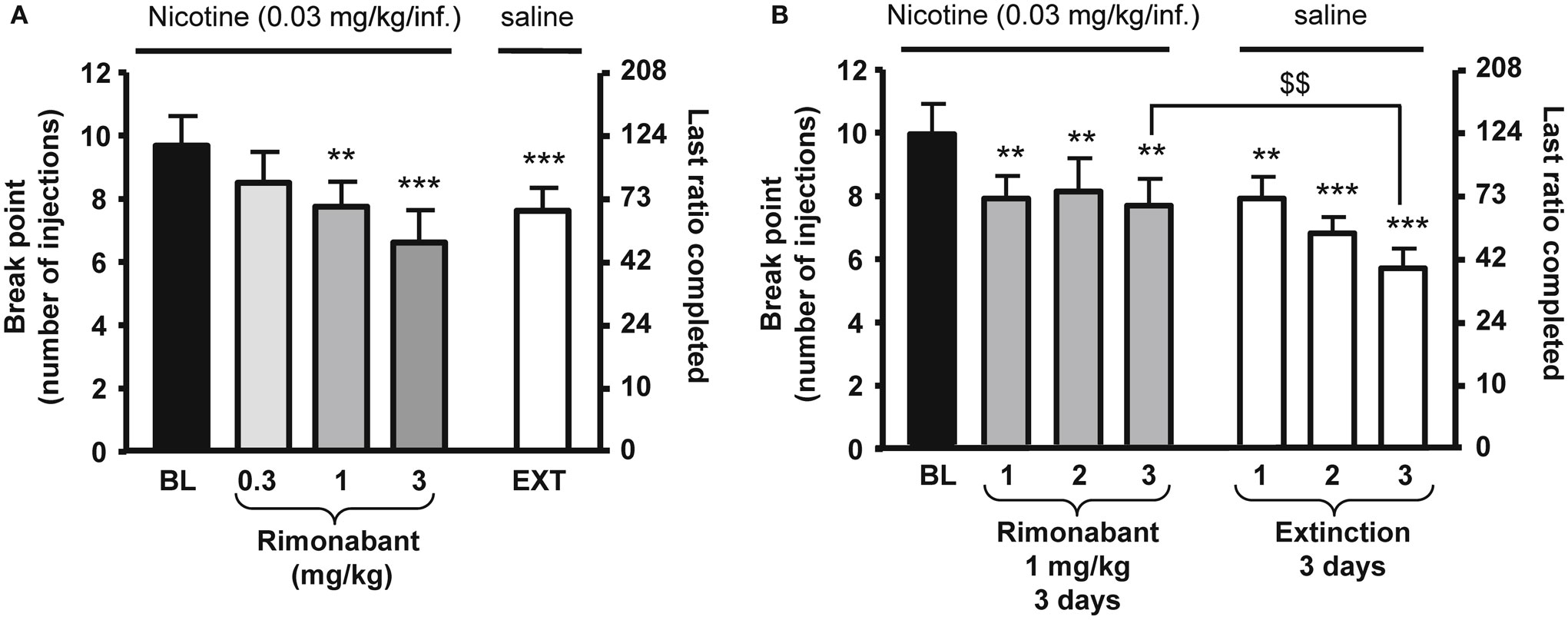
Figure 1. Effects of rimonabant on motivation for nicotine in rats. In (A), rimonabant [0.3–3 mg/kg, IP 60 min pre-treatment time (PTT)] dose-dependently reduced nicotine (0.03 mg/kg/injection) self-administration under a progressive-ratio schedule. Data are expressed as means (±SEM) of the number of injections (break-point, left y-axis) and of the last ratio completed (in number of lever presses, right y-axis) during baseline (BL) conditions, rimonabant pre-treatment, and vehicle pre-treatment and substitution of nicotine with saline (EXT). N = 8. **p < 0.01; ***p < 0.001 vs. baseline (BL), Dunnett’s test after significant ANOVA for repeated measures. In (B), effects of rimonabant (1 mg/kg, IP 60 min PTT) on nicotine self-administration under a progressive-ratio schedule during three consecutive sessions. Data are expressed as means (±SEM) of the number of injections (break-point, left y-axis) and of the last ratio completed (in number of lever presses, right y-axis) during baseline (BL) conditions, during three consecutive sessions with rimonabant pre-treatment (1 mg/kg) and during three consecutive sessions with vehicle pre-treatment and substitution of nicotine with saline. N = 9. **p < 0.01; ***p < 0.001 vs. baseline; $$p < 0.01 vs. vehicle extinction group, Student Newman–Keuls multiple comparison test after significant ANOVA for repeated measures. The figure and its caption have been reproduced with permission from Ref. (81).
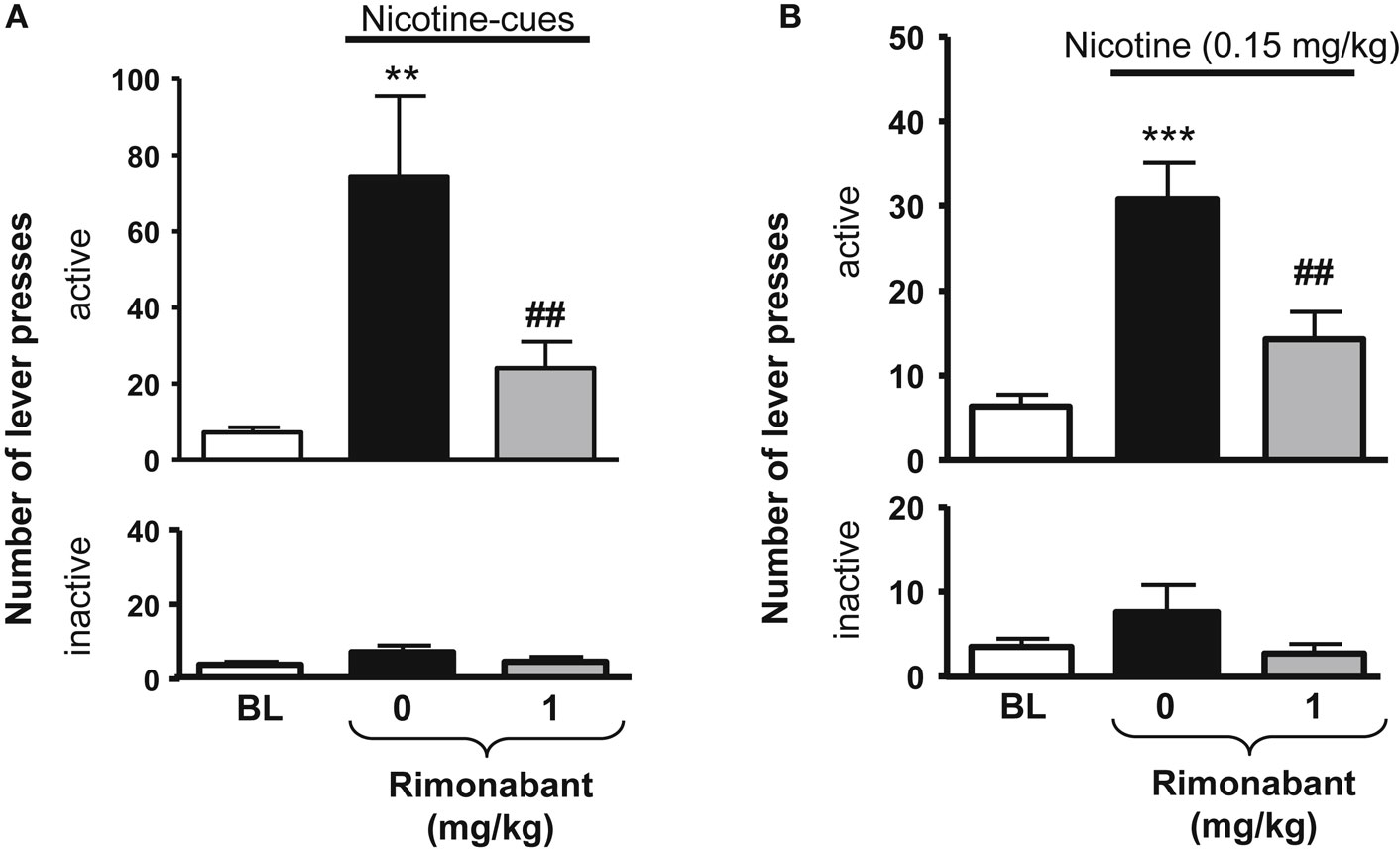
Figure 2. Effects of rimonabant on nicotine-seeking in rats. In (A), effects of rimonabant [0.1 mg/kg, IP 60 min pre-treatment time (PTT)] on the active (top) and the inactive (below) levers responses during cue-induced reinstatement of nicotine-seeking. **p < 0.01 vs. baseline; ##p < 0.01 vs. vehicle pre-treatment. In (B), effects of rimonabant (1 mg/kg, IP 70 min, PTT) on the active (top) and the inactive (below) levers responses during a nicotine-induced (0.15 mg/kg, SC, 10 min) reinstatement of nicotine-seeking. ***p < 0.001 vs. baseline; ##p < 0.001 vs. vehicle pre-treatment. The figure and its caption have been reproduced with permission from Ref. (81).
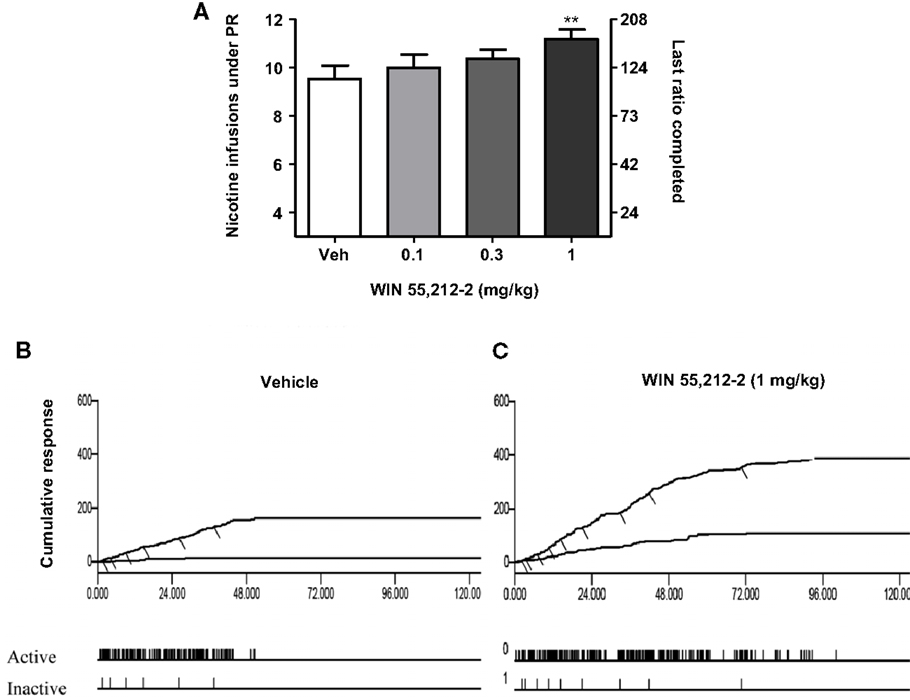
Figure 3. Effects of the stimulation of cannabinoid receptors on motivation for nicotine. In (A), effects of pre-treatment with WIN 55,212-2 [0.1–1 mg/kg, IP 15 min pre-treatment time (PTT)] on nicotine (0.03 mg/kg/infusion) self-administration under a progressive-ratio schedule. Data are expressed as means (±SEM) of the number of infusions obtained during the 4-h sessions. **p < 0.01 vs. vehicle pre-treatment (Dunnett’s test after significant ANOVA for repeated measures N = 9). In (B,C), individual representative cumulative responses on the active and inactive levers during nicotine self-administration under progressive-ratio schedule in rats pre-treated with vehicle (B) or 1 mg/kg WIN 55,212-2 (C). Each short upward mark on the cumulative lever-press records indicates one nicotine infusion. Break-point values are indicated and the pattern of response across time on active and inactive levers is provided below. The figure and its caption have been reproduced with permission from Ref. (89).
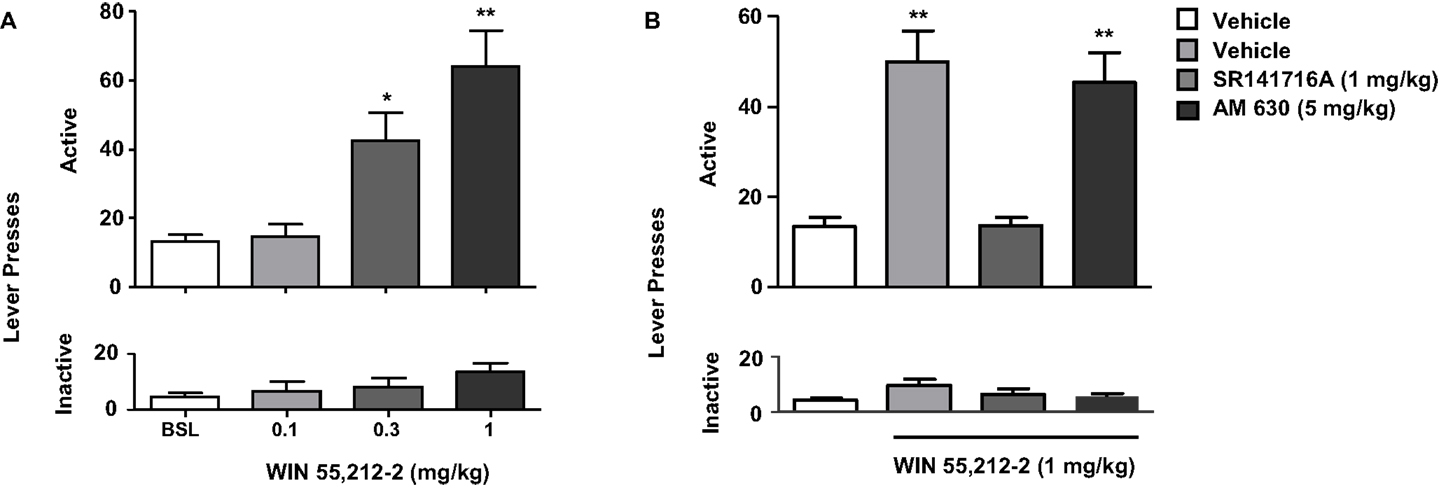
Figure 4. Effects of the stimulation of cannabinoid receptors on nicotine-seeking. In (A), rats trained to self-administer nicotine underwent an extinction phase after which they were pre-treated with WIN 55,212-2 [0.1–1 mg/kg, IP 15 min pre-treatment time (PTT)]. Figure shows responses on the active lever (top) and inactive lever (bottom). WIN 55,212-2 (0.3 and 1 mg/kg) significantly reinstated nicotine-seeking, assessed by the number of responses on the active lever (*p < 0.05 and *p < 0.001). No significant changes in responding on the inactive lever were observed. In (B), pre-treatment with the CB1 antagonist rimonabant (SR141716A) (1 mg/kg, IP), but not with the CB2 antagonist AM630 (5 mg/kg, IP) reversed reinstatement of nicotine-seeking induced by WIN 55,212-2. The figure and its caption have been reproduced with permission from Ref. (89).
Several lines of evidence indicate that central CB2 receptors could be implicated in modulating several neuropsychiatric disorders, including drug addiction (94–96). In fact, the effects of CB1 and CB2 receptor activation (at high doses) can produce similar effects such as antinociception and catalepsy (97, 98). However, the activation of CB2 receptors by the selective CB2 agonist AM1241 did not have effects on motivation to obtain nicotine or nicotine intake in rats (90). Similarly, the selective CB2 antagonist AM630 did not modify nicotine-taking or motivation to obtain nicotine under the progressive-ratio schedule of reinforcement. Moreover, the CB2 agonist and antagonist were not able to affect cue or nicotine-induced reinstatement of nicotine-seeking behavior (90). Together, these results further support the current literature on the distinct behavioral, neurochemical, and immunological profiles, of CB1 and CB2 receptors. On the other hand, it should be noted that in mice, CB2 receptors have been shown to modulate some addictive properties of drugs of abuse using both genetic and pharmacological models (99, 100). Another recent study by Zhang and colleagues, reported that activation of CB2 receptors in the VTA can attenuate cocaine self-administration in WT and CB1 knockout but not in CB2 knockout mice (101). Moreover, recent studies have documented the relevance of CB2 receptors on the rewarding/reinforcing properties of nicotine in mice (102, 103). Altogether, these results suggest that there may be important species differences that mediate these effects. Further studies performed in non-human primates or human subjects would allow better exploration of these discrepancies.
A recent study have shown that the volitional intake of nicotine (i.e., nicotine self-administration in rats) was able to modify anandamide and oleoylethanolamide (OEA) levels in the VTA (104). This is an interesting finding considering that several studies have described the existence of modulatory effects in signaling between the endocannabinoid and cholinergic systems (11–13). The understanding of effects of anandamide and 2-AG in modulating nicotine-reinforcing properties has been facilitated by the discovery of drugs able to interfere with the different processes involved in the synthesis, reuptake, and inactivation of these endocannabinoids. Among those drugs, N-(4-hydroxyphenyl)-arachidonamide (AM404) and cyclohexyl carbamic acid 3′-carbamoyl-3-yl ester (URB597) produce elevation of anandamide levels by blocking anandamide reuptake or by inhibiting FAAH, respectively (105, 106).
Based on observations of the effects of cannabinoid receptor agonists and antagonists on nicotine’s rewarding properties, one could speculate that increasing brain anandamide levels might enhance nicotine’s rewarding/reinforcing effects. Consistent with this prediction, Merritt et al. (88) observed enhanced nicotine CPP in FAAH knockout mice. Similarly, the pre-treatment with URB597 enhanced nicotine CPP in mice (88). Administration of a sub-threshold dose of nicotine that did not produce CPP in wild-type mice effectively produced CPP in FAAH knockout mice, and this effect was mediated by CB1 receptors (88). The enhancement of nicotine’s rewarding effects in FAAH knockout mice further supports previous studies where co-administration of sub-threshold doses of nicotine and THC produced nicotine CPP in mice (107). On the other hand, no differences on nicotine CPP were observed between FAAH knockout and wild-type mice when higher doses of nicotine were tested (88).
In a marked contrast to the results of Merritt et al. (88) in mice, FAAH inhibition by URB597 has been shown to reverse some addiction-related behavioral and neurochemical effects of nicotine in rats (81, 108). The inhibition of FAAH by URB597 was able to prevent the development of nicotine-induced CPP, reduced acquisition of nicotine self-administration behavior, and inhibited reinstatement of nicotine-seeking behavior induced by nicotine priming and cue-induced reinstatement in abstinent rats, while demonstrating no rewarding effects per se (108). However, there was no impact on nicotine-taking assessed using a fixed-ratio or a progressive-ratio schedule of reinforcement (81). A possible explanation for the differences observed between those studies is the fact that different species were used in these studies: rats (81, 108) vs. mice (88). However, URB597-induced increases in anandamide brain levels do not differ between mice and rats (109).
There is a surprising similarity between the effects of FAAH inhibition by URB597, which is expected to enhance anandamide levels and, thus, enhance cannabinoid CB1 receptor signaling, and those of rimonabant, described earlier, which blocks cannabinoid CB1 receptor signaling. In addition to a similarity in behavioral effects, it has been shown that both compounds, URB597 and rimonabant, were able to block nicotine-induced increases of dopamine levels in the NAc (14, 108). The similar effects of URB597 and rimonabant described above could be explained by the involvement of non-cannabinoid peroxisome proliferator-activated nuclear receptor (PPAR-α) systems (110, 111). Therefore, PPAR-α receptors seems to mediate the effects of FAAH inhibition on nicotine’s abuse-related behavioral and neurochemical effects in both rats and monkeys while CB1 receptors may play the major role in mediating the effects of FAAH inhibition on nicotine’s abuse-related behavioral and neurochemical effects in mice. This is supported by findings by Fegley et al. (109), showing that 2 h after treatment with 0.3 mg/kg URB597, there was only a twofold increase in brain levels of the endogenous PPAR-α ligand OEA and palmitoylethanolamide (PEA) in mice compared to a four to fivefold increase in OEA and PEA levels in rats (109). In contrast, as commented above, URB597-induced increases in anandamide brain levels do not differ between mice and rats (109). Moreover, the PPAR-α receptor antagonist MK-886 blocked URB597-induced reductions in nicotine’s effects on dopaminergic neuronal activity in rats (112). Similar to URB597, a variety of natural and synthetic PPAR-α receptor agonists were shown to decrease nicotine-reinforcing properties and reinstatement nicotine-seeking in different species (111). Another mechanism by which PPAR-α ligands are proposed to modulate the reinforcing effects of nicotine is downstream that activation of α7-nAChR subtype. During low activity, acetylcholine preferentially binds to high-affinity β2-nAChRs. This binding does not trigger nAChRs-mediated modulation of PPAR-α ligands. However, upon activation of cholinergic receptors, low affinity α7-nAChRs located in the extra dendritic regions of dopaminergic neurons are activated. This activation leads to an increase in intracellular Ca2+ which stimulates the synthesis of the PPAR-α ligands OEA and PEA as well as, anandamide. These ligands in turn activate PPAR-α which exerts negative modulation of β2-nAChRs through tyrosine kinase-mediated phosphorylation of β2-nAChRs. This mechanism demonstrates how dopaminergic neurons in the VTA have an ability to self-regulate their firing through selectively increasing OEA and PEA levels (110, 113). Thus, PPAR-α receptors could also be mediating their inhibitory effect on nicotine’s addiction-related behavioral and neurochemical effects through a non-FAAH pathway (111).
Anandamide might also modulate nicotine effects by targeting other receptors such as transient potential receptor of vanilloid type 1 (TRPV1) or even nicotine receptors. Indeed, anandamide has been shown to inhibit α4β2-nAChRs function in a CB1 receptor-independent manner (114, 115). Therefore, the effect of endocannabinoids on nicotine-reinforcing properties seems to be complex and suitable of being affected by different variables including the species and nicotine doses being tested. Other non-cannabinoid systems as vanilloid and PPAR-α might also be involved.
In support of the results obtained with the FAAH inhibitor URB 597, the anandamide uptake inhibitor VDM11 (5Z, 8Z, 11Z, 14Z)-N-(4-hydroxy-2-methylphenyl)-5, 8, 11, 14-eicosatetraenamide, was able to reduce both cue- and nicotine-induced reinstatement of nicotine-seeking behavior. However, VDM11 did not affect responding for nicotine under fixed-ratio or progressive-ratio schedules of reinforcement (116) (see Figure 5). Findings with VDM11 were further confirmed using AM404, another anandamide uptake inhibitor, which also attenuated cue and nicotine priming reinstatement of nicotine-seeking behavior (117). Additionally, AM404 decreased nicotine CPP and its reinstatement. AM404 has been also shown to attenuate dopamine increase on the NAc shell following nicotine injection (118). The fact that two different ligands developed to elevate anandamide levels produce similar effects, strongly suggest that those effects are mediated by anandamide elevation.
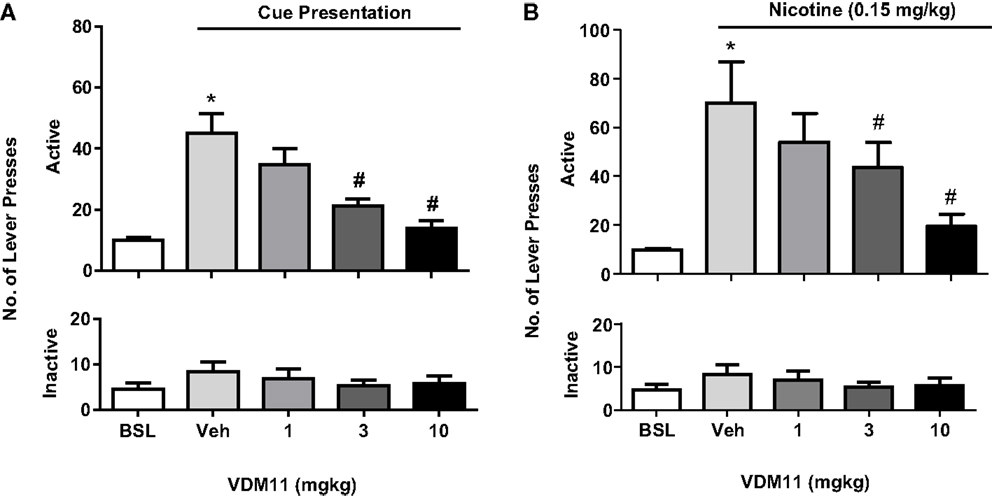
Figure 5. Effects of VDM11 on cue and nicotine priming reinstatement of nicotine-seeking behavior. In (A), a significant reinstatement of nicotine- seeking behavior was produced by presentation of nicotine-associated cues in non-treated animals (*p < 0.01). Pre-treatment with VDM11 [3 and 10 mg/kg, IP 30 min pre-treatment time (PTT)] significantly reduced cue-induced reinstatement of nicotine-seeking behavior (#p < 0.05). In (B), a significant reinstatement of nicotine-seeking was also produced by pre-treatment with nicotine (0.15 mg/kg) (*p < 0.01). VDM11 (3 and 10 mg/kg, IP 30 min PTT) significantly reduced the reinstatement of nicotine-seeking behavior induced by a priming injection of 0.15 mg/kg nicotine administered 10 min before the session (#p < 0.05). Data are expressed as means (±SEM) of the number of active lever presses during extinction (BSL) vehicle pre-treatment (visual cues). The figure and its caption have been reproduced with permission from Ref. (116).
The effects observed with VDM11 and AM404 on nicotine reward/reinforcement, were further supported by Oleson and Cheer (119) using the intracranial brain stimulation paradigm. Elevating levels of anandamide using the anandamide uptake inhibitor VDM11 reduced NAc neural encoding of reward-predictive cues and attenuated reward seeking, defined as the time occurring between cue presentation and a reward-directed behavioral response (120). A possible explanation for these findings is that VDM11 increases anandamide to a greater extent than 2-AG in vivo (121). Since anandamide functions as a partial agonist, the elevation of its levels in the brain induced by VDM11 might allow it to compete with 2-AG, a full CB1 receptor agonist (122), thereby anandamide might block 2-AG effects on reward seeking. In fact, comparing different behavioral studies suggest that anandamide and 2-AG may have opposite effects on reward-seeking behavior (81, 116, 119, 120). Interestingly, anandamide and 2-AG are effective reinforcers as evaluated using the intravenous drug self-administration paradigm in squirrel monkeys (123, 124). However, while 2-AG increases dopamine neurotransmission (125) and facilitates reward-directed behavior (125, 126), the elevation of anandamide levels attenuates the ability of cues to motivate reward-seeking behavior (81, 116, 119, 120).
It has been suggested that VDM11 might be actually a substrate for FAAH. Thus, it has been shown that VDM11 reduces FAAH hydrolysis of anandamide in vitro [(127, 128), also see Ref. (129)]. Therefore, the above described effects of VDM11 attenuating reward seeking might be due FAAH inhibition as well as reduced anandamide uptake. In that case, other fatty acid amides affected by FAAH catabolic effects, such as PEA and OEA, might also potentiate the effects of anandamide at TRPV1 receptors (130). As the specific pharmacological mechanisms of action of endocannabinoids remain unclear, it would be interesting to investigate the brain regions involved in mediating the effects observed with increasing levels of anandamide (e.g., intraregional injection of anandamide uptake inhibitors in areas of the brain that may be involved in mediating reward/reinforcement such as the basolateral amygdala and the NAc shell) (131). The ubiquitous nature of the endocannabinoid system might provide another interpretation regarding the pharmacological mechanism of action by which anandamide uptake inhibitors modulate neural mechanisms of reward seeking (132–134).
An alternative explanation of how VDM11 might attenuate reward seeking comes from the pharmacological inhibition of the membrane transporter. The bi-directional role of the membrane transporter in transporting anandamide and 2-AG (29, 30, 135) might explain the effects of VDM11 in reward seeking. In fact, recent studies using VDM11 have reported effects that are in close resemblance to those observed following CB1 receptor blockade (116, 119).
Endocannabinoids seem to be implicated in the response to withdrawal from nicotine. The concomitant treatment with nicotine and THC in mice induced an increased withdrawal syndrome when these mice were challenged with rimonabant (107). Additionally, the administration of THC seems to attenuate the magnitude of nicotine withdrawal (136). On the other hand, rimonabant failed to induce withdrawal in nicotine-dependent animals (136), suggesting that the cannabinoid system is not involved in the expression of nicotine physical dependence. However, more recent findings have reported modification in the number of nicotine receptors following chronic exposure to cannabinoids (137). Additionally, Cippitteli and colleagues reported that withdrawal from nicotine is associated with fluctuations in anandamide but not in 2-AG (138). Interestingly, the same study showed that administration of URB597, decreased nicotine withdrawal associated anxiety, but did not alter somatic signs of nicotine withdrawal (138).
It appears, from the experiments conducted in preclinical models (and partly validated by the testing of rimonabant in human clinical trials) that the CB1 receptors are critically involved in mediating nicotine reward/reinforcement. CB1 receptors also seem to be involved in mediating cue and nicotine priming reinstatement of nicotine-seeking behavior. Further work is needed to explore the role of CB1 receptors in nicotine-seeking induced by stress. It appears that many of these findings hold true in non-human primate studies as well as rodents (46, 47, 139). Thus, the CB1 receptor appears to be a logical target for drug development. As inverse agonist/antagonists have been found to have negative side-effects in humans (i.e., anxiety/depression), novel approaches, such as neutral CB1 receptor antagonists that might be effective against drug addiction without inverse agonist/antagonists side-effects, could represent a better therapeutic option (140–142). Limited experiments conducted in rats, suggest that the CB2 receptor has no involvement in mediating nicotine self-administration and relapse to nicotine-seeking. However, since differences have been reported in the role of CB2 receptors in mice and rats, it would be interesting to further explore the role of CB2 receptors before concluding that this receptor is not an interesting target. Recent experiments suggest that elevating brain anandamide levels would be an effective strategy to reduce relapse to nicotine-seeking behavior, but not to reduce ongoing nicotine-taking behavior. This is of great interest as such a strategy has been shown to decrease anxiety and depression in some animal models and may therefore be better tolerated than CB1 receptor inverse agonist/antagonists. However, we have no validation in humans of these interesting findings. Finally, exploring the role of the endogenous cannabinoid receptor ligand 2-AG on nicotine-taking and -seeking behavior will improve our understanding of the respective behavioral role of different endocannabinoids. Overall, those recent studies suggest that the endocannabinoid system still presents many opportunities for development of novel therapeutic strategies for nicotine addiction.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. Wilkie GI, Hutson PH, Stephens MW, Whiting P, Wonnacott S. Hippocampal nicotinic autoreceptors modulate acetylcholine release. Biochem Soc Trans (1993) 21:429–31.
3. Clarke PB, Reuben M. Release of [3H]-noradrenaline from rat hippocampal synaptosomes by nicotine: mediation by different nicotinic receptor subtypes from striatal [3H]-dopamine release. Br J Pharmacol (1996) 117:595–606. doi: 10.1111/j.1476-5381.1996.tb15232.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Pontieri FE, Tanda G, Orzi F, Di Chiara G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature (1996) 382:255–7. doi:10.1038/382255a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Yang X, Criswell HE, Breese GR. Nicotine-induced inhibition in medial septum involves activation of presynaptic nicotinic cholinergic receptors on gamma-aminobutyric acid-containing neurons. J Pharmacol Exp Ther (1996) 276:482–9.
6. Kenny PJ, File SE, Neal MJ. Evidence for a complex influence of nicotinic acetylcholine receptors on hippocampal serotonin release. J Neurochem (2000) 75:2409–14. doi:10.1046/j.1471-4159.2000.0752409.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Berrendero F, Robledo P, Trigo JM, Martin-Garcia E, Maldonado R. Neurobiological mechanisms involved in nicotine dependence and reward: participation of the endogenous opioid system. Neurosci Biobehav Rev (2010) 35:220–31. doi:10.1016/j.neubiorev.2010.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Picciotto MR, Caldarone BJ, King SL, Zachariou V. Nicotinic receptors in the brain. Links between molecular biology and behavior. Neuropsychopharmacology (2000) 22:451–65. doi:10.1016/S0893-133X(99)00146-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci (2001) 22:565–72. doi:10.1016/S0165-6147(00)01805-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Le Foll B, Goldberg SR. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J Pharmacol Exp Ther (2005) 312:875–83. doi:10.1124/jpet.104.077974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Narushima M, Uchigashima M, Fukaya M, Matsui M, Manabe T, Hashimoto K, et al. Tonic enhancement of endocannabinoid-mediated retrograde suppression of inhibition by cholinergic interneuron activity in the striatum. J Neurosci (2007) 27:496–506. doi:10.1523/JNEUROSCI.4644-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Adermark L. Modulation of endocannabinoid-mediated long-lasting disinhibition of striatal output by cholinergic interneurons. Neuropharmacology (2011) 61:1314–20. doi:10.1016/j.neuropharm.2011.07.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Oldenburg IA, Ding JB. Cholinergic modulation of synaptic integration and dendritic excitability in the striatum. Curr Opin Neurobiol (2011) 21:425–32. doi:10.1016/j.conb.2011.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Cohen C, Perrault G, Voltz C, Steinberg R, Soubrie P. SR141716, a central cannabinoid (CB(1)) receptor antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats. Behav Pharmacol (2002) 13:451–63. doi:10.1097/00008877-200209000-00018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Di Marzo V, De Petrocellis L, Bisogno T. Endocannabinoids Part I: molecular basis of endocannabinoid formation, action and inactivation and development of selective inhibitors. Expert Opin Ther Targets (2001) 5:241–65. doi:10.1517/14728222.5.2.241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Sugiura T, Waku K. Cannabinoid receptors and their endogenous ligands. J Biochem (2002) 132:7–12. doi:10.1093/oxfordjournals.jbchem.a003200
17. Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci (2003) 4:873–84. doi:10.1038/nrn1247
18. Di Marzo V. A brief history of cannabinoid and endocannabinoid pharmacology as inspired by the work of British scientists. Trends Pharmacol Sci (2006) 27:134–40. doi:10.1016/j.tips.2006.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther (1997) 74:129–80. doi:10.1016/S0163-7258(97)82001-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci (1998) 21:521–8. doi:10.1016/S0166-2236(98)01283-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Psychol (2013) 64:21–47. doi:10.1146/annurev-psych-113011-143739
22. Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, De Costa BR, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A (1990) 87:1932–6. doi:10.1073/pnas.87.5.1932
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev (2003) 83:1017–66. doi:10.1152/physrev.00004.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science (2005) 310:329–32. doi:10.1126/science.1115740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res (2006) 1071:10–23. doi:10.1016/j.brainres.2005.11.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Onaivi ES, Ishiguro H, Gong JP, Patel S, Perchuk A, Meozzi PA, et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci (2006) 1074:514–36. doi:10.1196/annals.1369.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Okamoto Y, Tsuboi K, Ueda N. Enzymatic formation of anandamide. Vitam Horm (2009) 81:1–24. doi:10.1016/S0083-6729(09)81001-7
28. Simon GM, Cravatt BF. Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J Biol Chem (2008) 283:9341–9. doi:10.1074/jbc.M707807200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Ligresti A, Morera E, Van Der Stelt M, Monory K, Lutz B, Ortar G, et al. Further evidence for the existence of a specific process for the membrane transport of anandamide. Biochem J (2004) 380:265–72. doi:10.1042/BJ20031812
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci (2004) 24:1673–9. doi:10.1523/JNEUROSCI.5214-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Desarnaud F, Cadas H, Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem (1995) 270:6030–5. doi:10.1074/jbc.270.11.6030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Hillard CJ, Edgemond WS, Campbell WB. Characterization of ligand binding to the cannabinoid receptor of rat brain membranes using a novel method: application to anandamide. J Neurochem (1995) 64:677–83. doi:10.1046/j.1471-4159.1995.64020677.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Tsou K, Nogueron MI, Muthian S, Sanudo-Pena MC, Hillard CJ, Deutsch DG, et al. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett (1998) 254:137–40. doi:10.1016/S0304-3940(98)00700-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Egertova M, Cravatt BF, Elphick MR. Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience (2003) 119:481–96. doi:10.1016/S0306-4522(03)00145-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Ueda H, Kobayashi T, Kishimoto M, Tsutsumi T, Okuyama H. A possible pathway of phosphoinositide metabolism through EDTA-insensitive phospholipase A1 followed by lysophosphoinositide-specific phospholipase C in rat brain. J Neurochem (1993) 61:1874–81. doi:10.1111/j.1471-4159.1993.tb09829.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol (2007) 14:1347–56. doi:10.1016/j.chembiol.2007.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, et al. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci (2008) 11:152–9. doi:10.1038/nn2042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev (2009) 89:309–80. doi:10.1152/physrev.00019.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron (2001) 29:729–38. doi:10.1016/S0896-6273(01)00247-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature (2001) 410:588–92. doi:10.1038/35069076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron (2001) 29:717–27. doi:10.1016/S0896-6273(01)00246-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, et al. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci (2001) 21:9506–18.
43. Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci (2001) 21:109–16.
44. Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci (2004) 24:53–62. doi:10.1523/JNEUROSCI.4503-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. De Vries TJ, Shaham Y, Homberg JR, Crombag H, Schuurman K, Dieben J, et al. A cannabinoid mechanism in relapse to cocaine seeking. Nat Med (2001) 7:1151–4. doi:10.1038/nm1001-1151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Maldonado R, Valverde O, Berrendero F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci (2006) 29:225–32. doi:10.1016/j.tins.2006.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Le Foll B, Forget B, Aubin HJ, Goldberg SR. Blocking cannabinoid CB1 receptors for the treatment of nicotine dependence: insights from pre-clinical and clinical studies. Addict Biol (2008) 13:239–52. doi:10.1111/j.1369-1600.2008.00113.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Moreira FA, Grieb M, Lutz B. Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: focus on anxiety and depression. Best Pract Res Clin Endocrinol Metab (2009) 23:133–44. doi:10.1016/j.beem.2008.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Nathan PJ, O’neill BV, Napolitano A, Bullmore ET. Neuropsychiatric adverse effects of centrally acting antiobesity drugs. CNS Neurosci Ther (2011) 17:490–505. doi:10.1111/j.1755-5949.2010.00172.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Le Foll B, Gorelick DA, Goldberg SR. The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology (Berl) (2009) 205:171–4. doi:10.1007/s00213-009-1506-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol (2000) 61:75–111. doi:10.1016/S0301-0082(99)00045-3
52. Pidoplichko VI, Debiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature (1997) 390:401–4. doi:10.1038/37120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol (1992) 41:31–7.
54. Whiting PJ, Liu R, Morley BJ, Lindstrom JM. Structurally different neuronal nicotinic acetylcholine receptor subtypes purified and characterized using monoclonal antibodies. J Neurosci (1987) 7:4005–16.
55. Balfour DJ. The role of mesoaccumbens dopamine in nicotine dependence. Curr Top Behav Neurosci (2015) 24:55–98. doi:10.1007/978-3-319-13482-6_3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Brody AL, Olmstead RE, London ED, Farahi J, Meyer JH, Grossman P, et al. Smoking-induced ventral striatum dopamine release. Am J Psychiatry (2004) 161:1211–8. doi:10.1176/appi.ajp.161.7.1211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Le Foll B, Guranda M, Wilson AA, Houle S, Rusjan PM, Wing VC, et al. Elevation of dopamine induced by cigarette smoking: novel insights from a [11C]-+-PHNO PET study in humans. Neuropsychopharmacology (2014) 39:415–24. doi:10.1038/npp.2013.209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Corrigall WA, Franklin KB, Coen KM, Clarke PB. The mesolimbic dopaminergic system is implicated in the reinforcing effects of nicotine. Psychopharmacology (Berl) (1992) 107:285–9. doi:10.1007/BF02245149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Corrigall WA, Coen KM. Selective dopamine antagonists reduce nicotine self-administration. Psychopharmacology (Berl) (1991) 104:171–6. doi:10.1007/BF02244174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Sziraki I, Sershen H, Hashim A, Lajtha A. Receptors in the ventral tegmental area mediating nicotine-induced dopamine release in the nucleus accumbens. Neurochem Res (2002) 27:253–61. doi:10.1023/A:1014844823534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Pomerleau OF, Fertig JB, Seyler LE, Jaffe J. Neuroendocrine reactivity to nicotine in smokers. Psychopharmacology (Berl) (1983) 81:61–7. doi:10.1007/BF00439275
62. del Arbol JL, Munoz JR, Ojeda L, Cascales AL, Irles JR, Miranda MT, et al. Plasma concentrations of beta-endorphin in smokers who consume different numbers of cigarettes per day. Pharmacol Biochem Behav (2000) 67:25–8. doi:10.1016/S0091-3057(00)00291-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Boyadjieva NI, Sarkar DK. Role of microglia in ethanol’s apoptotic action on hypothalamic neuronal cells in primary cultures. Alcohol Clin Exp Res (2010) 34:1835–42. doi:10.1111/j.1530-0277.2010.01271.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Berrendero F, Kieffer BL, Maldonado R. Attenuation of nicotine-induced antinociception, rewarding effects, and dependence in mu-opioid receptor knock-out mice. J Neurosci (2002) 22:10935–40.
65. Walters CL, Cleck JN, Kuo YC, Blendy JA. Mu-opioid receptor and CREB activation are required for nicotine reward. Neuron (2005) 46:933–43. doi:10.1016/j.neuron.2005.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Carboni E, Imperato A, Perezzani L, Di Chiara G. Amphetamine, cocaine, phencyclidine and nomifensine increase extracellular dopamine concentrations preferentially in the nucleus accumbens of freely moving rats. Neuroscience (1989) 28:653–61. doi:10.1016/0306-4522(89)90012-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Di Ciano P, Everitt BJ. The GABA(B) receptor agonist baclofen attenuates cocaine- and heroin-seeking behavior by rats. Neuropsychopharmacology (2003) 28:510–8. doi:10.1038/sj.npp.1300088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Schultz W, Tremblay L, Hollerman JR. Reward prediction in primate basal ganglia and frontal cortex. Neuropharmacology (1998) 37:421–9. doi:10.1016/S0028-3908(98)00071-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Stuber GD, Klanker M, De Ridder B, Bowers MS, Joosten RN, Feenstra MG, et al. Reward-predictive cues enhance excitatory synaptic strength onto midbrain dopamine neurons. Science (2008) 321:1690–2. doi:10.1126/science.1160873
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Bourdelais A, Kalivas PW. High sensitivity HPLC assay for GABA in brain dialysis studies. J Neurosci Methods (1991) 39:115–21. doi:10.1016/0165-0270(91)90077-D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Miller JD, Sanghera MK, German DC. Mesencephalic dopaminergic unit activity in the behaviorally conditioned rat. Life Sci (1981) 29:1255–63. doi:10.1016/0024-3205(81)90231-9
72. Schultz W, Apicella P, Ljungberg T. Responses of monkey dopamine neurons to reward and conditioned stimuli during successive steps of learning a delayed response task. J Neurosci (1993) 13:900–13.
73. Fernandez-Ruiz J, Hernandez M, Ramos JA. Cannabinoid-dopamine interaction in the pathophysiology and treatment of CNS disorders. CNS Neurosci Ther (2010) 16:e72–91. doi:10.1111/j.1755-5949.2010.00144.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Melis M, Muntoni AL, Pistis M. Endocannabinoids and the processing of value-related signals. Front Pharmacol (2012) 3:7. doi:10.3389/fphar.2012.00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Matyas F, Urban GM, Watanabe M, Mackie K, Zimmer A, Freund TF, et al. Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology (2008) 54:95–107. doi:10.1016/j.neuropharm.2007.05.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Buczynski MW, Parsons LH. Quantification of brain endocannabinoid levels: methods, interpretations and pitfalls. Br J Pharmacol (2010) 160:423–42. doi:10.1111/j.1476-5381.2010.00787.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Wang H, Lupica CR. Release of endogenous cannabinoids from ventral tegmental area dopamine neurons and the modulation of synaptic processes. Prog Neuropsychopharmacol Biol Psychiatry (2014) 52:24–7. doi:10.1016/j.pnpbp.2014.01.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Melis M, Perra S, Muntoni AL, Pillolla G, Lutz B, Marsicano G, et al. Prefrontal cortex stimulation induces 2-arachidonoyl-glycerol-mediated suppression of excitation in dopamine neurons. J Neurosci (2004) 24:10707–15. doi:10.1523/JNEUROSCI.3502-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Cheer JF, Wassum KM, Sombers LA, Heien ML, Ariansen JL, Aragona BJ, et al. Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci (2007) 27:791–5. doi:10.1523/JNEUROSCI.4152-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Grieder TE, George O, Tan H, George SR, Le Foll B, Laviolette SR, et al. Phasic D1 and tonic D2 dopamine receptor signaling double dissociate the motivational effects of acute nicotine and chronic nicotine withdrawal. Proc Natl Acad Sci U S A (2012) 109:3101–6. doi:10.1073/pnas.1114422109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Forget B, Coen KM, Le Foll B. Inhibition of fatty acid amide hydrolase reduces reinstatement of nicotine seeking but not break point for nicotine self-administration – comparison with CB(1) receptor blockade. Psychopharmacology (Berl) (2009) 205:613–24. doi:10.1007/s00213-009-1569-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Le Foll B, Goldberg SR. Rimonabant, a CB1 antagonist, blocks nicotine-conditioned place preferences. Neuroreport (2004) 15:2139–43. doi:10.1097/00001756-200409150-00028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Forget B, Hamon M, Thiebot MH. Cannabinoid CB1 receptors are involved in motivational effects of nicotine in rats. Psychopharmacology (Berl) (2005) 181:722–34. doi:10.1007/s00213-005-0015-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Hashemizadeh S, Sardari M, Rezayof A. Basolateral amygdala CB1 cannabinoid receptors mediate nicotine-induced place preference. Prog Neuropsychopharmacol Biol Psychiatry (2014) 51:65–71. doi:10.1016/j.pnpbp.2014.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Cohen C, Perrault G, Griebel G, Soubrie P. Nicotine-associated cues maintain nicotine-seeking behavior in rats several weeks after nicotine withdrawal: reversal by the cannabinoid (CB(1)) receptor antagonist, rimonabant (SR141716). Neuropsychopharmacology (2005) 30:145–55. doi:10.1038/sj.npp.1300541
86. Diergaarde L, De Vries W, Raaso H, Schoffelmeer AN, De Vries TJ. Contextual renewal of nicotine seeking in rats and its suppression by the cannabinoid-1 receptor antagonist rimonabant (SR141716A). Neuropharmacology (2008) 55:712–6. doi:10.1016/j.neuropharm.2008.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Castane A, Valjent E, Ledent C, Parmentier M, Maldonado R, Valverde O. Lack of CB1 cannabinoid receptors modifies nicotine behavioural responses, but not nicotine abstinence. Neuropharmacology (2002) 43:857–67. doi:10.1016/S0028-3908(02)00118-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Merritt LL, Martin BR, Walters C, Lichtman AH, Damaj MI. The endogenous cannabinoid system modulates nicotine reward and dependence. J Pharmacol Exp Ther (2008) 326:483–92. doi:10.1124/jpet.108.138321
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Gamaleddin I, Wertheim C, Zhu AZ, Coen KM, Vemuri K, Makryannis A, et al. Cannabinoid receptor stimulation increases motivation for nicotine and nicotine seeking. Addict Biol (2012) 17:47–61. doi:10.1111/j.1369-1600.2011.00314.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Gamaleddin I, Zvonok A, Makriyannis A, Goldberg SR, Le Foll B. Effects of a selective cannabinoid CB2 agonist and antagonist on intravenous nicotine self administration and reinstatement of nicotine seeking. PLoS One (2012) 7:e29900. doi:10.1371/journal.pone.0029900
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Kodas E, Cohen C, Louis C, Griebel G. Cortico-limbic circuitry for conditioned nicotine-seeking behavior in rats involves endocannabinoid signaling. Psychopharmacology (Berl) (2007) 194:161–71. doi:10.1007/s00213-007-0813-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Reisiger AR, Kaufling J, Manzoni O, Cador M, Georges F, Caille S. Nicotine self-administration induces CB1-dependent LTP in the bed nucleus of the stria terminalis. J Neurosci (2014) 34:4285–92. doi:10.1523/JNEUROSCI.3149-13.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Simonnet A, Cador M, Caille S. Nicotine reinforcement is reduced by cannabinoid CB1 receptor blockade in the ventral tegmental area. Addict Biol (2013) 18:930–6. doi:10.1111/j.1369-1600.2012.00476.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Ishiguro H, Iwasaki S, Teasenfitz L, Higuchi S, Horiuchi Y, Saito T, et al. Involvement of cannabinoid CB2 receptor in alcohol preference in mice and alcoholism in humans. Pharmacogenomics J (2007) 7:380–5. doi:10.1038/sj.tpj.6500431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Onaivi ES, Ishiguro H, Gong JP, Patel S, Meozzi PA, Myers L, et al. Brain neuronal CB2 cannabinoid receptors in drug abuse and depression: from mice to human subjects. PLoS One (2008) 3:e1640. doi:10.1371/journal.pone.0001640
96. Ishiguro H, Horiuchi Y, Ishikawa M, Koga M, Imai K, Suzuki Y, et al. Brain cannabinoid CB2 receptor in schizophrenia. Biol Psychiatry (2010) 67:974–82. doi:10.1016/j.biopsych.2009.09.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology (2005) 48:658–72. doi:10.1016/j.neuropharm.2004.12.008
98. Rahn EJ, Makriyannis A, Hohmann AG. Activation of cannabinoid CB1 and CB2 receptors suppresses neuropathic nociception evoked by the chemotherapeutic agent vincristine in rats. Br J Pharmacol (2007) 152:765–77. doi:10.1038/sj.bjp.0707333
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Xi ZX, Peng XQ, Li X, Song R, Zhang HY, Liu QR, et al. Brain cannabinoid CB(2) receptors modulate cocaine’s actions in mice. Nat Neurosci (2011) 14:1160–6. doi:10.1038/nn.2874
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Aracil-Fernandez A, Trigo JM, Garcia-Gutierrez MS, Ortega-Alvaro A, Ternianov A, Navarro D, et al. Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB(2) receptors. Neuropsychopharmacology (2012) 37:1749–63. doi:10.1038/npp.2012.22
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Zhang HY, Gao M, Liu QR, Bi GH, Li X, Yang HJ, et al. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci U S A (2014) 111:E5007–15. doi:10.1073/pnas.1413210111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Ignatowska-Jankowska BM, Muldoon PP, Lichtman AH, Damaj MI. The cannabinoid CB2 receptor is necessary for nicotine-conditioned place preference, but not other behavioral effects of nicotine in mice. Psychopharmacology (Berl) (2013) 229:591–601. doi:10.1007/s00213-013-3117-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Navarrete F, Rodriguez-Arias M, Martin-Garcia E, Navarro D, Garcia-Gutierrez MS, Aguilar MA, et al. Role of CB2 cannabinoid receptors in the rewarding, reinforcing, and physical effects of nicotine. Neuropsychopharmacology (2013) 38:2515–24. doi:10.1038/npp.2013.157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Buczynski MW, Polis IY, Parsons LH. The volitional nature of nicotine exposure alters anandamide and oleoylethanolamide levels in the ventral tegmental area. Neuropsychopharmacology (2013) 38:574–84. doi:10.1038/npp.2012.210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science (1997) 277:1094–7. doi:10.1126/science.277.5329.1094
106. Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med (2003) 9:76–81. doi:10.1038/nm803
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Valjent E, Mitchell JM, Besson MJ, Caboche J, Maldonado R. Behavioural and biochemical evidence for interactions between Delta 9-tetrahydrocannabinol and nicotine. Br J Pharmacol (2002) 135:564–78. doi:10.1038/sj.bjp.0704479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Scherma M, Panlilio LV, Fadda P, Fattore L, Gamaleddin I, Le Foll B, et al. Inhibition of anandamide hydrolysis by URB597 reverses abuse-related behavioral and neurochemical effects of nicotine in rats. J Pharmacol Exp Ther (2008) 327:482–90. doi:10.1124/jpet.108.142224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, et al. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3’-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther (2005) 313:352–8. doi:10.1124/jpet.104.078980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Melis M, Carta S, Fattore L, Tolu S, Yasar S, Goldberg SR, et al. Peroxisome proliferator-activated receptors-alpha modulate dopamine cell activity through nicotinic receptors. Biol Psychiatry (2010) 68:256–64. doi:10.1016/j.biopsych.2010.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Mascia P, Pistis M, Justinova Z, Panlilio LV, Luchicchi A, Lecca S, et al. Blockade of nicotine reward and reinstatement by activation of alpha-type peroxisome proliferator-activated receptors. Biol Psychiatry (2011) 69:633–41. doi:10.1016/j.biopsych.2010.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Luchicchi A, Lecca S, Carta S, Pillolla G, Muntoni AL, Yasar S, et al. Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: involvement of PPAR-alpha nuclear receptors. Addict Biol (2010) 15:277–88. doi:10.1111/j.1369-1600.2010.00222.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Melis M, Carta G, Pistis M, Banni S. Physiological role of peroxisome proliferator-activated receptors type alpha on dopamine systems. CNS Neurol Disord Drug Targets (2013) 12:70–7. doi:10.2174/1871527311312010012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Spivak CE, Lupica CR, Oz M. The endocannabinoid anandamide inhibits the function of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol (2007) 72:1024–32. doi:10.1124/mol.107.036939
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Butt C, Alptekin A, Shippenberg T, Oz M. Endogenous cannabinoid anandamide inhibits nicotinic acetylcholine receptor function in mouse thalamic synaptosomes. J Neurochem (2008) 105:1235–43. doi:10.1111/j.1471-4159.2008.05225.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Gamaleddin I, Guranda M, Goldberg SR, Le Foll B. The selective anandamide transport inhibitor VDM11 attenuates reinstatement of nicotine seeking behaviour, but does not affect nicotine intake. Br J Pharmacol (2011) 164:1652–60. doi:10.1111/j.1476-5381.2011.01440.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Gamaleddin I, Guranda M, Scherma M, Fratta W, Makriyannis A, Vadivel SK, et al. AM404 attenuates reinstatement of nicotine seeking induced by nicotine-associated cues and nicotine priming but does not affect nicotine- and food-taking. J Psychopharmacol (2013) 27:564–71. doi:10.1177/0269881113477710
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Scherma M, Justinova Z, Zanettini C, Panlilio LV, Mascia P, Fadda P, et al. The anandamide transport inhibitor AM404 reduces the rewarding effects of nicotine and nicotine-induced dopamine elevations in the nucleus accumbens shell in rats. Br J Pharmacol (2012) 165:2539–48. doi:10.1111/j.1476-5381.2011.01467.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Oleson EB, Cheer JF. Paradoxical effects of the endocannabinoid uptake inhibitor VDM11 on accumbal neural encoding of reward predictive cues. Synapse (2012) 66:984–8. doi:10.1002/syn.21587
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Oleson EB, Cheer JF. A brain on cannabinoids: the role of dopamine release in reward seeking. Cold Spring Harb Perspect Med (2012) 2:a012229. doi:10.1101/cshperspect.a012229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. van der Stelt M, Mazzola C, Esposito G, Matias I, Petrosino S, De Filippis D, et al. Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci (2006) 63:1410–24. doi:10.1007/s00018-006-6037-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Savinainen JR, Jarvinen T, Laine K, Laitinen JT. Despite substantial degradation, 2-arachidonoylglycerol is a potent full efficacy agonist mediating CB(1) receptor-dependent G-protein activation in rat cerebellar membranes. Br J Pharmacol (2001) 134:664–72. doi:10.1038/sj.bjp.0704297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Justinova Z, Solinas M, Tanda G, Redhi GH, Goldberg SR. The endogenous cannabinoid anandamide and its synthetic analog R(+)-methanandamide are intravenously self-administered by squirrel monkeys. J Neurosci (2005) 25:5645–50. doi:10.1523/JNEUROSCI.0951-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Justinova Z, Yasar S, Redhi GH, Goldberg SR. The endogenous cannabinoid 2-arachidonoylglycerol is intravenously self-administered by squirrel monkeys. J Neurosci (2011) 31:7043–8. doi:10.1523/JNEUROSCI.6058-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Oleson EB, Beckert MV, Morra JT, Lansink CS, Cachope R, Abdullah RA, et al. Endocannabinoids shape accumbal encoding of cue-motivated behavior via CB1 receptor activation in the ventral tegmentum. Neuron (2012) 73:360–73. doi:10.1016/j.neuron.2011.11.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Gutierrez-Lopez MD, Llopis N, Feng S, Barrett DA, O’shea E, Colado MI. Involvement of 2-arachidonoyl glycerol in the increased consumption of and preference for ethanol of mice treated with neurotoxic doses of methamphetamine. Br J Pharmacol (2010) 160:772–83. doi:10.1111/j.1476-5381.2010.00720.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Vandevoorde S, Fowler CJ. Inhibition of fatty acid amide hydrolase and monoacylglycerol lipase by the anandamide uptake inhibitor VDM11: evidence that VDM11 acts as an FAAH substrate. Br J Pharmacol (2005) 145:885–93. doi:10.1038/sj.bjp.0706253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Hillard CJ, Shi L, Tuniki VR, Falck JR, Campbell WB. Studies of anandamide accumulation inhibitors in cerebellar granule neurons: comparison to inhibition of fatty acid amide hydrolase. J Mol Neurosci (2007) 33:18–24. doi:10.1007/s12031-007-0045-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Fegley D, Kathuria S, Mercier R, Li C, Goutopoulos A, Makriyannis A, et al. Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci U S A (2004) 101:8756–61. doi:10.1073/pnas.0400997101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Di Marzo V. Anandamide serves two masters in the brain. Nat Neurosci (2010) 13:1446–8. doi:10.1038/nn1210-1446
131. Soria-Gomez E, Matias I, Rueda-Orozco PE, Cisneros M, Petrosino S, Navarro L, et al. Pharmacological enhancement of the endocannabinoid system in the nucleus accumbens shell stimulates food intake and increases c-Fos expression in the hypothalamus. Br J Pharmacol (2007) 151:1109–16. doi:10.1038/sj.bjp.0707313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Bisogno T, Maccarrone M, De Petrocellis L, Jarrahian A, Finazzi-Agro A, Hillard C, et al. The uptake by cells of 2-arachidonoylglycerol, an endogenous agonist of cannabinoid receptors. Eur J Biochem (2001) 268:1982–9. doi:10.1046/j.1432-1327.2001.02072.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Hajos N, Kathuria S, Dinh T, Piomelli D, Freund TF. Endocannabinoid transport tightly controls 2-arachidonoyl glycerol actions in the hippocampus: effects of low temperature and the transport inhibitor AM404. Eur J Neurosci (2004) 19:2991–6. doi:10.1111/j.0953-816X.2004.03433.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, et al. Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol (2005) 569:751–60. doi:10.1113/jphysiol.2005.097477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Chicca A, Marazzi J, Nicolussi S, Gertsch J. Evidence for bidirectional endocannabinoid transport across cell membranes. J Biol Chem (2012) 287:34660–82. doi:10.1074/jbc.M112.373241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Balerio GN, Aso E, Berrendero F, Murtra P, Maldonado R. Delta9-tetrahydrocannabinol decreases somatic and motivational manifestations of nicotine withdrawal in mice. Eur J Neurosci (2004) 20:2737–48. doi:10.1111/j.1460-9568.2004.03714.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Mateos B, Borcel E, Loriga R, Luesu W, Bini V, Llorente R, et al. Adolescent exposure to nicotine and/or the cannabinoid agonist CP 55,940 induces gender-dependent long-lasting memory impairments and changes in brain nicotinic and CB(1) cannabinoid receptors. J Psychopharmacol (2011) 25:1676–90. doi:10.1177/0269881110370503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Cippitelli A, Astarita G, Duranti A, Caprioli G, Ubaldi M, Stopponi S, et al. Endocannabinoid regulation of acute and protracted nicotine withdrawal: effect of FAAH inhibition. PLoS One (2011) 6:e28142. doi:10.1371/journal.pone.0028142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Scherma M, Fadda P, Le Foll B, Forget B, Fratta W, Goldberg SR, et al. The endocannabinoid system: a new molecular target for the treatment of tobacco addiction. CNS Neurol Disord Drug Targets (2008) 7:468–81. doi:10.2174/187152708786927859
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Janero DR, Makriyannis A. Cannabinoid receptor antagonists: pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin Emerg Drugs (2009) 14:43–65. doi:10.1517/14728210902736568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. McLaughlin PJ. Reports of the death of CB1 antagonists have been greatly exaggerated: recent preclinical findings predict improved safety in the treatment of obesity. Behav Pharmacol (2012) 23:537–50. doi:10.1097/FBP.0b013e3283566a8c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Kangas BD, Delatte MS, Vemuri VK, Thakur GA, Nikas SP, Subramanian KV, et al. Cannabinoid discrimination and antagonism by CB(1) neutral and inverse agonist antagonists. J Pharmacol Exp Ther (2013) 344:561–7. doi:10.1124/jpet.112.201962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: cannabinoid system, nicotine, addiction, endogenous cannabinoids
Citation: Gamaleddin IH, Trigo JM, Gueye AB, Zvonok A, Makriyannis A, Goldberg SR and Le Foll B (2015) Role of the endogenous cannabinoid system in nicotine addiction: novel insights. Front. Psychiatry 6:41. doi: 10.3389/fpsyt.2015.00041
Received: 15 September 2014; Accepted: 06 March 2015;
Published online: 25 March 2015.
Edited by:
Steven R. Laviolette, University of Western Ontario, CanadaReviewed by:
Marco Pistis, University of Cagliari, ItalyCopyright: © 2015 Gamaleddin, Trigo, Gueye, Zvonok, Makriyannis, Goldberg and Le Foll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernard Le Foll, Translational Addiction Research Laboratory, Centre for Addiction and Mental Health (CAMH), University of Toronto, 33 Russell Street, Toronto, ON M5S 2S1, Canada e-mail:YmVybmFyZC5sZWZvbGxAY2FtaC5jYQ==
†Islam Hany Gamaleddin and Jose M. Trigo have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.