 Rajiv Radhakrishnan
Rajiv Radhakrishnan Samuel T. Wilkinson
Samuel T. Wilkinson Deepak Cyril D’Souza
Deepak Cyril D’Souza
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychiatry, 22 May 2014
Sec. Addictive Disorders
Volume 5 - 2014 | https://doi.org/10.3389/fpsyt.2014.00054
This article is part of the Research TopicClearing the smokescreen: The current evidence on cannabis useView all 20 articles
Cannabis is the most commonly used illicit drug worldwide, with ~5 million daily users worldwide. Emerging evidence supports a number of associations between cannabis and psychosis/psychotic disorders, including schizophrenia. These associations-based on case-studies, surveys, epidemiological studies, and experimental studies indicate that cannabinoids can produce acute, transient effects; acute, persistent effects; and delayed, persistent effects that recapitulate the psychopathology and psychophysiology seen in schizophrenia. Acute exposure to both cannabis and synthetic cannabinoids (Spice/K2) can produce a full range of transient psychotomimetic symptoms, cognitive deficits, and psychophysiological abnormalities that bear a striking resemblance to symptoms of schizophrenia. In individuals with an established psychotic disorder, cannabinoids can exacerbate symptoms, trigger relapse, and have negative consequences on the course of the illness. Several factors appear to moderate these associations, including family history, genetic factors, history of childhood abuse, and the age at onset of cannabis use. Exposure to cannabinoids in adolescence confers a higher risk for psychosis outcomes in later life and the risk is dose-related. Individuals with polymorphisms of COMT and AKT1 genes may be at increased risk for psychotic disorders in association with cannabinoids, as are individuals with a family history of psychotic disorders or a history of childhood trauma. The relationship between cannabis and schizophrenia fulfills many but not all of the standard criteria for causality, including temporality, biological gradient, biological plausibility, experimental evidence, consistency, and coherence. At the present time, the evidence indicates that cannabis may be a component cause in the emergence of psychosis, and this warrants serious consideration from the point of view of public health policy.
Psychotic disorders are arguably the most serious of mental illnesses, the best known being schizophrenia. As yet, the etiology of schizophrenia and other psychotic disorders remains unclear. There is emerging evidence to support a number of associations between cannabis and psychosis, but the precise nature of these associations remains unclear.
Cannabis is the most commonly used illicit drug by adults, with 18.1 million current users in the U.S. in 2011 (up from 14.5 million in 2007) and ~5 million daily cannabis users (1–3). In the U.S., it was also the most commonly used illicit drug by children 12–17 years (7.9%) in 2011. The age at onset of regular cannabis use appears to be occurring earlier. About 1.3% of eighth graders endorsed daily use of cannabis in 2011 (3). Additionally, the average delta-9-tetrahydrocannabinol (THC) content of cannabis has increased from 3.4% in 1993 to 8.8% in 2008, with concentrations in high potency varieties such as sinsemilla increasing to as high as 11.1% (4). “Medical” marijuana (cannabis) is being legalized increasingly across the U.S. (5, 6). Some states have legalized recreational cannabis use and others are projected to follow suit (7). As a result, individuals, including those with a higher risk for psychosis, who would not have risked the consequences of procuring an illegal drug previously, may now consider exposing themselves to cannabis.
In parallel, there is the emerging phenomenon of the recreational use of Spice, a mixture of synthetic cannabinoids, by young people (8). Among high school seniors, 11.4% reported using Spice in the past year (9). In contrast to THC, the synthetic cannabinoids present in Spice are highly potent full cannabinoid 1 receptor (CB1R) agonists (10, 11). There are a number of reports of acute and persistent psychosis immediately following the use of Spice, sometimes with catastrophic outcomes (12–14). In the U.S., emergency department visits related to cannabinoids (149 ED visits per 100,000 population) were second only to cocaine (157.8 ED visits per 100,000 population) (15).
Various lines of evidence point to associations between cannabinoids and psychosis [reviewed in Ref. (16–18)]. These associations may be categorized according to temporal proximity of the onset of psychosis to exposure, duration, and clinical significance of psychosis. Converging lines of evidence suggest that early and heavy exposure to cannabis is associated with a higher risk for psychotic outcomes, including schizophrenia in later life (18–28). In addition, cannabinoids can induce immediate-onset psychotomimetic symptoms that do not persist beyond the period of intoxication (~1 h), as reviewed by us (18). Finally, less well-characterized but perhaps clinically important, cannabinoids are also associated with acute episodes of psychosis that: (1) manifest immediately following exposure, (2) last beyond the period of intoxication, and (3) require clinical intervention (29, 30).
Furthermore, although the associations between cannabinoids and psychosis have gained increasing recognition, the moderators (i.e., variables that affect the direction and/or strength of the relation between an independent, predictor variable – such as cannabis use – and a dependent, outcome variable – such as psychosis) and mediators (i.e., variables that directly account for the relationship between cannabis use and psychosis) are less well-understood. Emerging evidence suggests the crucial role of age of exposure to cannabis (with the period of adolescence being identified as a period of exquisite vulnerability), familial risk, degree of schizotypy, childhood trauma, and the role of genetic factors in moderating this association.
As a preface to this review of the literature, several important issues should be considered. Firstly, cannabis contains more than 70 different cannabinoids (31) of which THC is thought to be the main psychoactive ingredient, while another cannabinoid, cannabidiol (CBD), is thought to have antipsychotic properties (32). THC is hence not the same as cannabis, although most of the experimental studies are conducted using THC. Secondly, cannabis grown in different conditions and different parts of the world has varying potencies based on the content of THC and CBD. The type or potency of cannabis has rarely been accounted for in epidemiological studies. Thirdly, it is important to make a distinction between psychosis as a syndrome and psychosis-like experiences (psychotomimetic effects). While psychosis refers to a heterogeneous group of disorders defined as consisting of positive symptoms (delusions, hallucinations, and thought-alienation phenomena), negative symptoms (alogia, avolition, anhedonia, asociality, and affective flattening), and disorganization/cognitive symptoms (deficits in attention, working memory, problem-solving, and executive function); psychosis-like experiences are characterized by a loss of reality-testing and include derealization, depersonalization, dissociation, hallucination, paranoia, impairment in concentration, and perceptual alterations, which are transient and self-limited. The fact that schizophrenia is a syndrome that is much more than positive symptoms needs to be considered. Negative (e.g., amotivation, asociality, and anhedonia) and cognitive symptoms (e.g., deficits in attention, memory, and executive function) contribute to the disease burden of schizophrenia just as positive symptoms do.
Below herewith, we review existing literature on the association between cannabinoids and psychosis with special focus on the recent critical literature. We categorized major findings into the following categories: immediate psychotic symptoms, psychosis outlasting intoxication, delayed and persistent effects, moderators, and mediators of the association (age of exposure, family history, history of childhood abuse, and genetics), and evidence for causality.
The evidence from anecdotal reports suggests that cannabis may induce acute psychotomimetic effects and precipitate the syndrome of psychosis. One of the earliest systematic studies of the psychotomimetic effects of cannabis was that by the French psychiatrist Jacques-Joseph Moreau (de Tours) in his 1845 book, Hashish and Mental Illness (33). He reported that hashish (cannabis resin) could precipitate “acute psychotic reactions, generally lasting but a few hours, but occasionally as long as a week; the reaction seemed dose-related and its main features included paranoid ideation, illusions, hallucinations, delusions, depersonalization, confusion, restlessness, and excitement. There can be delirium, disorientation, and marked clouding of consciousness” (33). Numerous case reports have since then documented the acute psychotomimetic symptoms of cannabis intoxication, including depersonalization, derealization, paranoia, ideas of reference, flight of ideas, pressured thought, disorganized thinking, persecutory delusions, grandiose delusions, auditory/visual hallucinations, and impairments in attention and memory (30, 33–42) in about 20–50% of individuals (43, 44).
In a survey of ultra-high-risk and recent-onset patients with psychosis (45), 37% of subjects reported that their first psychotic symptoms appeared during cannabis intoxication. The subjects also reported feeling more anxiety, depression, and suspiciousness immediately after cannabis use than cannabis-using controls. Another recent study of first-episode psychosis (FEP) patients (n = 109) found that daily cannabis users were significantly more likely to have an acute onset of psychosis than non-daily users (46). Evidence from case reports and surveys is limited, however, by confounds such as observer bias, effects of other illicit drugs, and failure to exclude negative and cognitive symptoms prior to onset of positive symptoms.
With the pioneering work of Mechoulam in 1964, the individual constituents of cannabis were characterized (47). The identification of THC as the main psychoactive agent led to the synthesis of dronabinol (synthetic THC) and other non-psychotropic synthetic cannabinoids such as levonantradol and nabilone (9-trans-ketocannabinoid), which were thought to have specific antiemetic, analgesic, and antispastic effects. The use of these agents for the treatment of pain syndromes, chemotherapy-induced nausea, and spasticity in multiple sclerosis was followed by reports of transient psychotomimetic effects among patients. The psychotomimetic effects reported were similar to that with cannabis including “loss of control,” thought disturbances, feelings of unreality, apprehension, fear and paranoia, anxiety and panic, dissociation, depersonalization, dysphoria, difficulty concentrating, hallucinations, perceptual alterations, amnesia, and anxiety (48–62). These effects were dose-related and proportional to the affinity of the compound for the CB1R. The high incidence of intolerable behavioral side effects in fact, led to the discontinuation of drug development of levonantradol as an analgesic. In a systematic review of 30 studies that examined the efficacy of dranabinol, nabilone, or levonantradol for chemotherapy-induced nausea and vomiting Machado Rocha et al. (63) found that synthetic cannabinoids was responsible for 30% of dropouts; with 6% patients developing hallucinations and 5% developing paranoia. In another systematic review, Tramer et al. (64) found that patients receiving synthetic cannabinoids had a higher relative risk of developing dysphoria or depression [RR 8.06 (95% CI 3.38–19.2)], hallucinations [RR 6.10 (95% CI 2.41–15.4)], and paranoia [RR 8.58 (95% CI 6.38–11.5)] than those receiving non-cannabinoid antiemetics. Importantly, hallucinations and paranoia were seen exclusively with cannabinoids, and not with other antiemetic agents; and these effects appeared to be related to dose, potency, and frequency of administration.
The emergence of potent synthetic cannabinoids as drugs of abuse in the last decade provide another source of evidence pointing to the link between cannabinoids and psychosis (8). These compounds, collectively referred to as Spice or K2, comprise a mixture of synthetic cannabinoids such as CP-47,497, CP-47,497-C8, JWH-018, JWH-073, JWH-081, JWH-122, JWH-210, JWH-250, HU-211, and RCS-4 (65–72). It should be noted that, unlike THC, which is a weak partial agonist of brain CB1Rs, the synthetic cannabinoids are highly potent, full agonists of CB1R, which would predict more robust effects. Spice has gained popularity as a drug of abuse since it is more psychoactive than cannabis, is readily available over the Internet (advertised as “natural herbs” or “harmless incense” under brand names such as Spice, K2, Yucatan Fire, Skunk, Moon Rocks), and is non-detectable in standard urine toxicological tests. In some countries, including much of the United States and Canada, synthetic cannabinoids are available at gas stations and head-shops as natural herbs and incense; this contributes to its perception as safe and legal among users.
There are no controlled-studies on the psychotomimetic effects of synthetic cannabinoids (73); available information about their effects in humans consists of retrospective case reports from emergency room (ER) visits (69, 70, 74), surveys (12–14), reports from the American Association of Poison Control Centers (AAPCC) (75), and from media and law-enforcement agencies on catastrophic events related to their use (76–81). There has been a substantial increase in ER visits resulting from acute behavioral effects following use of these synthetic cannabinoids. The psychotomimetic effects reported include anxiety, agitation, disorientation, hallucinations, and paranoia (69, 70, 82–84). In an Internet survey, Spice/K2 users most commonly endorsed feeling paranoid (11%), hallucinating (3%), and feeling as if in a dream-like state (26%) “most of the time” or “every time” they used “Spice” (14). The AAPCC reported an exponential increase in call volume related to the use of Spice/K2 from 53 calls in 2009 to over 6000 in 2011 with callers reporting symptoms of agitation, drowsiness, and hallucinations (62% of calls) (75).
Case reports document the ability of these compounds to precipitate a psychotic relapse in patients with pre-existing psychotic disorders and psychotic symptoms in those with no prior history of psychosis (12, 74, 85). Müller et al. (86) reported on a 25-year-old man with a history of psychotic episodes precipitated by cannabis use and a family history of schizophrenia who had been stable for 2 years and had a psychotic relapse comprising anxiety, paranoid delusions, and hallucinations after smoking Spice on three occasions in 1 month. Every-Palmer described sudden agitation, disorganization, and delusions in five forensic patient who had consumed Spice containing JWH-018 and/or CP-47,497 (85). Of the five patients, only one retained insight into the possible psychotogenic nature of “Spice” (85). In a follow up survey of 15 inpatients with serious mental illness in a forensic psychiatric facility, Every-Palmer reported that patients commonly experienced anxiety and psychotomimetic effects, few developed tolerance, and none reported withdrawal symptoms (12).
Psychotic symptoms are also reported in patients with no previous history of psychosis. The adverse clinical events documented in case reports include altered consciousness, confusion, anxiety, irritability, agitation, paranoia, hallucinations, and psychosis (70, 82, 85–87). However, the majority of case reports to date discuss people 25 years or younger (84, 88). Therefore, it is possible that “Spice” exacerbates a pre-existing prodromal syndrome. Case reports and cross-sectional surveys are only able to show an association and cannot elucidate causation.
The sparse literature on Spice/K2 effects reviewed above has a number of limitations, including selection bias, reliance on the accuracy of written record or subject recall, uncontrolled nature of the evidence, the inadequate characterization of cases, lack of standardized assessments, confounding effects of concomitant drug use, different doses and routes of administration, and variable individual expectancy, set, and setting. Cases reported by the media and law-enforcement may represent extremes that might not be generalizable. The temporal profile, range, and intensity of Spice/K2 effects, and whether the effects are dose-related or biphasic, are not known. Furthermore, the relationship between dose, effects, and blood/urine levels of the parent compound and metabolites is not known.
Experimental studies provide an opportunity to control variables such as dose, route of administration, and setting, while employing a randomized-control paradigm. Studies have variously examined the effects of smoked cannabis, cannabis extract, oral, and intravenous THC and CBD on positive psychotomimetic symptoms, negative symptoms, cognitive, and psychophysiological measures. Although, early semi-experimental studies of cannabis in the early 1900s using oral cannabis or cannabis extract [reviewed in Ref. (18)] demonstrated cognitive and perceptual effects of cannabis, D’Souza et al. (89) were the first to characterize the profile of positive psychotomimetic symptoms, negative symptoms, and cognitive effects of intravenous THC in healthy individuals. Despite varying routes of administration, experimental studies have yielded some consistent results regarding the effects of cannabis, THC, and CBD. There have, however, not been any controlled experimental studies of the synthetic cannabinoids in humans to date.
In the following sections, we provide a brief summary of the consistent effects noted with cannabis, THC, and CBD. Interestingly, cannabis and THC produce the full range of positive psychotomimetic symptoms, negative symptoms, and cognitive deficits seen in schizophrenia, while CBD has been shown to have anxiolytic properties and even inhibit the psychotomimetic effects of THC (90–92).
Cannabis extract containing predetermined quantities of THC (93, 94) and THC alone (32, 73, 89, 92, 94–99) have been shown to produce a range of transient, positive symptoms, that are qualitatively similar to the positive symptoms of schizophrenia. These symptoms include suspiciousness, paranoid and grandiose delusions, conceptual disorganization, fragmented thinking, and perceptual alterations. Additionally, cannabis and THC also result in depersonalization, derealization, alterations in sensory perception, and feelings of unreality. These effects have consistently been demonstrated with smoked cannabis, oral cannabis extract/THC (dose range 5–20 mg), intravenous THC (dose range 0.015–0.03 mg/kg), and intrapulmonary administration via a vaporizer (32, 73, 89, 92, 94–99). In the first study of its kind in a carefully controlled laboratory setting, D’Souza et al. (89), administered intravenous THC in two doses (2.5 and 5 mg), in a double-blind, randomized, placebo-controlled study in healthy adults (n = 22). Subjects were screened to rule out significant psychiatric disorder or family history of Axis I disorders (89). The study found that THC produced transient positive psychotic symptoms (Figure 1) including perceptual alterations, negative symptoms, mood symptoms such as euphoria and anxiety, and also cognitive deficits, especially in attention, working memory, and verbal recall (Figure 2). In a similar study in healthy individuals, using almost identical methods except for a lower dose of THC, Morrison et al. (95) showed that intravenous THC (2.5 mg) produced similar effects on positive psychotic symptoms, mood, and cognition.
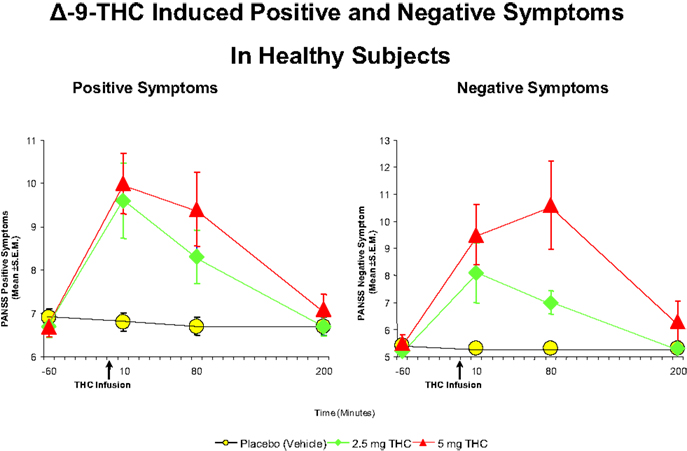
Figure 1. Effects of THC on the seven-item positive symptom and negative symptoms subscales of the Positive and Negative Syndrome Scale (PANSS). THC at both a low dose (2.5 mg) (green) and moderate dose (5 mg) (100) induce an increase in positive and negative symptoms, compared to placebo (yellow). Adapted from Ref. (89).
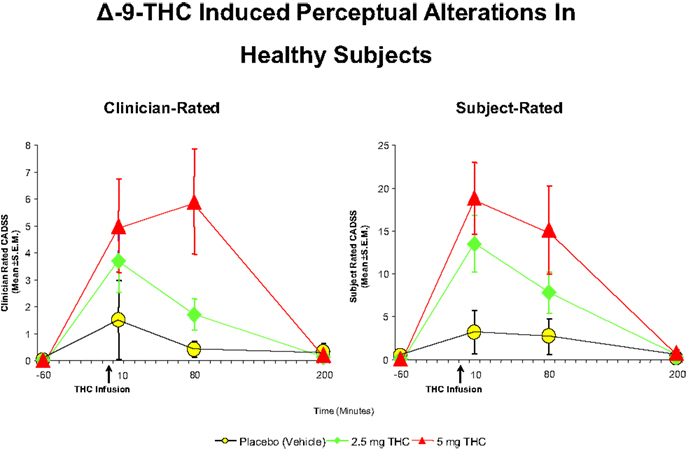
Figure 2. Effects of THC on the clinician- and subject-rated subscales of the Clinician Administered Dissociative Symptoms Scale (CADSS), a measure of perceptual alterations. THC at both a low dose (2.5 mg) (green) and moderate dose (5 mg) (100) induce an increase in perceptual alterations as rated by the clinician and the subject, compared to placebo (yellow). Adapted from Ref. (89).
The effects of dopamine D2-receptor antagonists on the psychotomimetic effects of THC are not clear. For example, in some studies, olanzapine (101) and haloperidol (102) were shown to attenuate the psychotomimetic effects of THC. However, D’Souza et al. showed that acute treatment with haloperidol did not attenuate the psychotomimetic effects of THC in healthy subjects (103) and chronic antipsychotic treatment failed to protect schizophrenia patients from the symptom exacerbating effects of THC (104). The potential antipsychotic and anxiolytic effects of CBD have drawn increasing attention. In a functional magnetic resonance (fMRI) study of brain responses to emotional expression of faces, Fusar-Poli et al. (90) found that while THC resulted in increased psychotic symptoms and increased skin conductance responses during processing of fearful faces; CBD, on the other hand led to a reduction in anxiety and a decrease in skin conductance response. A separate fMRI study showed that THC and CBD had opposite effects on blood oxygen-level dependent (BOLD) responses in tasks of verbal recall, response-inhibition, processing fearful facial expressions, auditory processing, and visual processing (91). Some limitations notwithstanding, this study provided some important leads into the differential effects of CBD and THC.
Time perception abnormalities are known to occur in schizophrenia, but have received little attention (105–108). Cannabinoids have been shown to alter time perception in both preclinical (109–112) and clinical studies (113–117). In the largest double-blind, randomized, cross-over, placebo-controlled study to date, Sewell et al., showed that THC at different doses induced time overestimation and underproduction compared with placebo (118). Cannabinoids have also been found to disrupt performance on visual information processing in the binocular depth inversion task, a potential surrogate marker for psychosis seen in patients with acute paranoid schizophrenic or schizophreniform psychosis (119). This effect has been observed with cannabis resin (120), nabilone (a synthetic analog of THC) (121), dronabinol (a synthetic isomer of THC) (119), and in chronic cannabis users (122).
Delta-9-tetrahydrocannabinol also produces a range of effects similar to the negative symptoms of schizophrenia, including blunted affect, emotional withdrawal, psychomotor retardation, lack of spontaneity, and reduced rapport (89, 97). It is difficult to determine whether these “negative symptoms” were primary or were a consequence of the sedating and cataleptic effects of cannabinoids observed in animal studies. Morrison et al. (97) however, showed that the effect of THC on negative symptoms was independent of effects on sedation. It is also unclear if the negative symptoms were a manifestation of internal preoccupation with positive psychotic experiences. Furthermore, acute pharmacological studies may be limited in their capacity to model negative symptoms.
Cannabis, THC and other synthetic cannabinoids also produce transient, dose-related cognitive impairments, especially in the domains of verbal learning, short-term memory, working memory, executive function, abstract ability, decision-making, and attention (123–129). These effects are not limited to humans but are also seen in rodents and non-human primates [reviewed in Ref. (130, 131)]. Interestingly, the profile of impairment observed in different cognitive domains is similar to that observed in schizophrenia (132).
The cognitive impairment produced by THC is most pronounced in the domain of verbal learning and memory (129), which is also one of the domains of significant impairment in schizophrenia (132). Figure 3 illustrates the effects of THC on the Hopkins Verbal Learning Test (HVLT) in healthy subjects (104). THC has been shown to produce robust dose-dependent impairments on both immediate and delayed (30 mins) verbal recall. THC also increased the number of “false positives” and “intrusions” on the HVLT. Similar findings have been recently reported by Henquet et al. (133) and Morrison et al. (95).
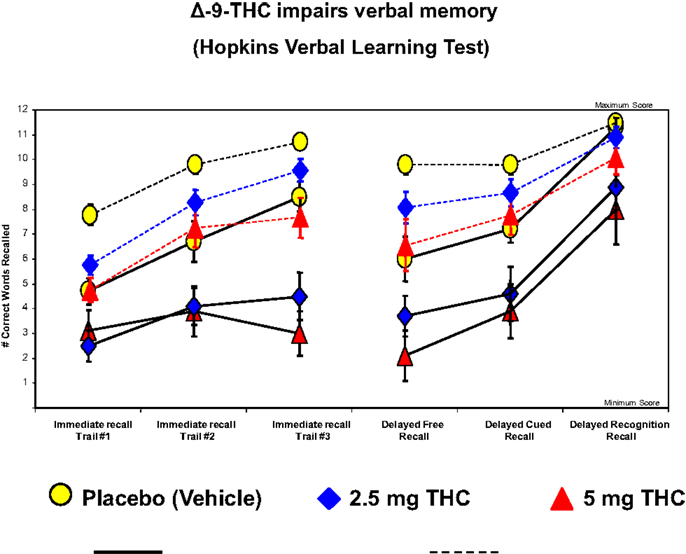
Figure 3. Effects of THC on the immediate free recall, delayed free recall, delayed cued, and recognition recall measured by a 12-word learning task (Hopkins Verbal Learning Test), a measure of verbal memory. THC at both a low dose (2.5 mg) (blue) and moderate dose (5 mg) (100) induce an immediate free recall, delayed free recall, delayed cued, and recognition in patients with schizophrenia (solid line) and healthy individuals (dotted line), compared to placebo (yellow). Adapted from Ref. (89).
The acute effects of cannabinoids are likely modulated by genetic and personality factors. This would explain why only a small minority of people experience the psychotomimetic effects of cannabinoids. Henquet and colleagues examined the effects of the interaction of Catechol-O-methyl transferase (COMT) polymorphism and a trait index of psychosis liability on smoked THC (0.3 mg/kg) on cognitive performance and psychosis in 30 healthy individuals (133). They found that individuals with the Val/Val polymorphism and high scores on psychosis liability had higher THC-induced psychotic symptoms.
Psychophysiological effects refer to measures that attempt to examine the physiological basis of psychological processes. In the study of cannabinoids, these effects have primarily been demonstrated using electroencephalography (EEG). EEG measures of information processing, such as event-related potentials (ERPs) and neural oscillations, offer a more proximal index of neural events in humans with exquisite temporal precision (134). ERPs are averaged EEG responses time-locked to particular stimuli or events. ERPs relevant to psychosis include: (1) P50 – a measure of auditory sensory gating, (2) P300b – a measure of directed attention, contextual updating of working memory, and the attribution of salience to deviant or novel stimuli (135), (3) P300a – a measure of novelty detection, and (4) mismatch negativity (MMN) – a measure of processing and memory of deviant stimuli. These ERP measures have been reported to be abnormal in schizophrenia and have been considered biomarkers of the disorder. Abnormalities in neural oscillations have also been noted in schizophrenia and in chronic cannabis users.
Deficits in auditory sensory gating, as demonstrated by a disruption in P50 response, have been shown in patients with schizophrenia (136–140). The cannabinoid agonists CP-55940 and WIN 55,212-2 have been shown to disrupt sensory gating in rats (141, 142). However, there are no studies that have examined the acute effects of cannabinoids on sensory gating (P50) in humans. In contrast, there are cross-sectional studies comparing heavy, chronic cannabis users to healthy controls that have shown that chronic cannabis users show disruptions in P50 suppression (143, 144), which was evident despite subjects abstaining for 24 h. These findings suggest that chronic cannabis use is associated with disruption in sensory gating. Furthermore, the degree of disruption in sensory gating was found to correlate positively with the magnitude of cannabis exposure (138, 145), suggesting a dose-response relationship.
The P300 is a late positive, post-attentional ERP component thought to be related to directed attention, contextual updating of working memory, and the attribution of salience to deviant or novel stimuli (135). Deficits in P300 amplitude and latency have been demonstrated in patients with schizophrenia (136, 139, 146–152). THC has been shown to cause a reduction in the amplitude of the P300 response in several paradigms such as a visuospatial N-back working memory task (153), and auditory choice reaction task (154, 155). D’Souza et al. examined the effect of several doses of intravenous THC on the P300 response in healthy individuals and showed that THC decreased the amplitude of both the novelty P300a and target P300b (155), while also producing concomitant psychotomimetic effects. There was no impairment in the latency of the P300 response or in the N100 response, indicating that THC disrupted cortical processes responsible for context updating (P300b) and the automatic orientation of attention (P300a), without affecting early sensory registration (N100) or processing speed. Studies of chronic cannabis users have however been equivocal, with studies variably showing decreased P300 amplitudes (156), increased P300 latency (157), increased P300 amplitude (157, 158), or shorter P300 latency (159). Although the reasons behind these discrepant results are unclear, it is possible that chronic cannabis users are impaired during more cognitively challenging selective attention tasks (156, 157, 160), but retain normal ERP responses during simple dual-stimulus discrimination tasks (158, 159).
Mismatch negativity is an automatic, pre-attentive, and negative-voltage ERP component that occurs ~100–200 ms after a deviant auditory stimulus that differs in frequency or duration from a sequence of standard auditory stimuli. It is thought to represent basic auditory information processing, and sensory memory generated primarily in the superior temporal and prefrontal cortex (PFC), while being relatively independent of attention (161, 162). Deficits in MMN have been shown in patients with schizophrenia, early psychosis, and high-risk subjects (163, 164). While oral THC did not produce any acute changes in MMN amplitude (93), studies in chronic cannabis users have demonstrated decreased MMN amplitudes in the frequency deviance condition (154, 165–167). There also appears to be a dose–response effect in the MMN response with long-term and heavier users of cannabis demonstrating significantly lower MMN amplitudes compared to short-term or light users and duration of cannabis exposure showing a negative correlation with MMN amplitudes (154, 165).
Cannabinoids have been shown to disrupt theta band (4–8 Hz) neural oscillations in rats (168). Similar disruption in theta band power was demonstrated following smoked cannabis (169). The degree of disruption in theta band power correlated with deficits in working memory performance in this study. Studies of neural oscillations in chronic cannabis users have demonstrated attenuation of high frequency activity in the beta range (13–29 Hz) (145, 170) and in the gamma range (30–50 Hz) (145, 171). These findings are very interesting in light of accumulating evidence that schizophrenia may be primarily a disorder of abnormal neural oscillations and synchrony [reviewed in Ref. (172)] and that neural oscillations may also be important in the organization of the networks in the brain (173).
The use of cannabinoids are also associated with acute episodes of psychosis that: (1) manifest immediately following exposure, (2) last beyond the period of intoxication, and (3) sometimes require clinical intervention. This is distinct from the effects previously described, which do not outlast the period of intoxication. Most of the literature about this phenomenon comes from small case series and case reports. The phenomenology, duration, and course of such cases – which we refer to as cannabis-induced acute and persistent psychosis (CIAPP) – have not been systematically characterized.
In the 1890s, the Indian Hemp Drugs Commission undertook a study to examine the effects of cannabis use. The commission reported that “excessive” cannabis use was responsible for psychotic reactions in 9.5% (222/2344) of cases in asylums in India. Chopra et al. reported a series of patients admitted to a psychiatric hospital in India for cannabis related psychosis (29, 30). The psychosis was typically preceded by ingestion of large doses of cannabis and was characterized by hallucinations, delusions, paranoia, depersonalization, amnesia, emotional lability, confusion, and disorientation. Similar case series have been reported from other geographical areas including Sweden, Denmark, the Caribbean, Scotland, UK, USA, and South Africa (37, 174–181). These case reports suggest that when cannabis use is stopped, the acute psychotic episodes resolve (quicker in comparison with “endogenous” psychoses) (37, 39, 177, 178, 180, 182–186), and do not recur unless cannabis use resumes [reviewed in Ref. (187)]. However, since follow up was only for a few months, the long-term course and outcome, the clinical implications, and prognostic significance of these cases remains unclear. Several recent large (n = ~20,000) studies suggest that, over long-term (~8 year) follow up, ~50% of patients without any pre-existing psychiatric disorder who were hospitalized for cannabis-induced psychosis, were later re-diagnosed with a schizophrenia-spectrum disorder (181, 188); that number increased to ~75% when the diagnosis included any psychotic outcome (181). These observations suggest that hospitalization for CIAPP may be a harbinger of a recurrent psychotic disorder that we currently classify as schizophrenia. More recent case reports and retrospective studies continue to demonstrate the close temporal relationship between use of cannabis and the onset of a psychotic disorder, sometimes quite indistinguishable from schizophrenia (189, 190). In fact, the International Classification of Diseases-10 (ICD-10) allows for the psychotic effects of cannabis to be coded as both an acute polymorphic psychotic disorder and a protracted substance-induced psychotic disorder. It is conceivable that, as suggested by Rounsaville (191), these cases may actually represent a distinct persistent psychotic disorder.
The evidence for persistent effects of cannabinoids in humans comes from large-scale epidemiological studies and from studies in chronic cannabis users. In the following section, we examine the evidence linking cannabis use and persistent psychotic disorder, including negative and cognitive symptoms.
The evidence for the association between cannabis use and persistent psychosis comes from both cross-sectional studies (192–196) and longitudinal epidemiological studies, including the Swedish military conscript cohort (197–199), the Netherlands Mental Health Survey and Incidence Study (NEMESIS) (20), the German prospective Early Developmental Stages of Psychopathology Study (EDSP) (24), the Dunedin cohort (19, 200), and the Christchurch Health and Development Study (CHDS) birth cohort (23).
The first study to draw attention to the association between cannabis use and psychosis was the Swedish conscript study (197), in which Andreasson et al. followed a cohort of 45,570 Swedish military conscripts (97% of all Swedish males aged 18–20 years) from 1969 to 1970. The investigators observed a dose–response relationship between cannabis use (via self report) at initiation of military service and hospitalization for a psychotic disorder over the ensuing 15 years, with maximal risk among those who had smoked cannabis more than 50 times. Conscripts who reported having used cannabis at least once in their lifetime had a 2.4-fold (95% confidence interval 1.8–3.3) increased risk of developing schizophrenia over the course of 15 years. This relative risk rose to sixfold (95% CI 4–8.9) in those who had used cannabis more than 50 times in their lifetime. The risk remained significantly high despite adjusting for other factors such as psychiatric illness at the time of conscription, solvent abuse, and parental separation. In a 27-year follow up study of the same cohort and a re-analysis of the data, Zammit et al. replicated the findings of Andreasson et al., showing that cannabis use was associated with a linear increase in the risk of developing schizophrenia; the relative risk increasing from 2.2 (95% CI 1.7–2.8) in those who had used cannabis at least once, to 6.7 (95% CI 4.5–10) in those who had used cannabis more than 50 times in their lifetime (198). When potential confounders such as IQ sore, disturbed behavior in childhood, psychiatric diagnosis at conscription, cigarette smoking, degree of social integration, and place of upbringing were included in the regression analysis, the adjusted relative risk was 1.5 (95% CI 1.1–2.0) in those who had used cannabis at least once and 3.1 (95% CI 1.7–5.5) in those who had used cannabis more than 50 times in their lifetime. The relative risk for schizophrenia was significantly higher in those who developed schizophrenia within 5 years of conscription, which raises questions about the direction of causality. In other words, this preliminary analysis could not distinguish whether cannabis use led to schizophrenia or whether subjects used cannabis in an attempt to self-medicate incipient symptoms of schizophrenia. In a secondary analysis that excluded those who developed a diagnosis of schizophrenia within 5 years of conscription, the adjusted relative risk remained significant only for those who had used cannabis more than 50 times (adjusted relative risk = 2.5, 95% CI 1.2–5.1). The study needs to be interpreted with caution: while 24.3% of the sample had used any drug, a very small percent (3.4%) had used only cannabis. While the analysis controlled for cigarette smoking, it failed to control for the use of stimulants and other drugs. Also, the fact that presumably weak confounders (such as “place of upbringing” and “cigarette smoking”) contributed substantially, along with other variables in reducing the adjusted relative risk by ~50% in the regression analysis highlights the difficulties inherent in interpreting epidemiological data and raises the issue of other unknown confounders. Similar criticisms of the studies from the Swedish conscript cohort have been raised by other authors (201–203), including the facts that: (a) the use of other drugs was more common in the cannabis-using group, (b) the association between cannabis use and schizophrenia may be mediated by a third, as yet unknown factor, and (c) the follow up study, a quarter century later, failed to address the issue of confounding due to use of other drugs, many of which are also known to precipitate psychosis.
Using the NEMESIS cohort, van Os et al. reported that cannabis use at baseline was associated with an increased risk of psychosis (20). The study assessed 7076 subjects at baseline (1996), 5618 subjects at a first time-point (1997), and 4848 subjects at a second time-point (1999) via telephonic interviews, and found 10 subjects who developed psychosis, while 38 subjects endorsed individual items on the Brief Psychiatric Rating Scale (BPRS). The findings of the study are limited by the small numbers in the outcome of interest (25) despite the large sample size.
The EDSP study, which used in-person interviews in the assessment of 923 individuals from the general population (aged 14–24 years), showed that cannabis use was associated with an increased risk of psychotic symptoms and persistent use increased this risk further (28). Importantly, this study yields evidence for a unidirectional relationship between cannabis use and psychosis. This is in contrast with another recent study (22), which showed the relationship to be bi-directional, alluding to the possibility of a phenomenon of “self-medication,” a topic that is further discussed below.
The Dunedin cohort study (19) examined data from 759 subjects of the population birth cohort comprising 1037 individuals born in Dunedin, New Zealand, in 1972–1973. The study collected information on psychotic symptoms at age 11, drug use at ages 15 and 18 years, and assessed psychiatric symptoms at age 26. Cannabis use by age 15 and 18 years was found to be associated with more schizophrenia symptoms at age 26 years; and the association remained significant despite controlling for the presence of psychotic symptoms at age 11 years. The association was also found to be stronger with earlier use. Those who used cannabis by age 15 years were also four times more likely to have a diagnosis of schizophreniform disorder; the risk was reduced by 31% and no longer significant after controlling for psychotic symptoms at age 11 years, pointing to the possibility of reverse-causality.
Fergusson et al. attempted to validate a possible causal link between cannabis use and psychosis in a dataset of a 25-year longitudinal study in New Zealand (the CHDS birth cohort comprising 1265 children) (23). The study showed that daily use of cannabis was associated with 2.3- to 3.3-fold higher risk of psychosis than among non-users. One of the limitations of the study is that the data was derived from 10 items of the Symptom Checklist-90, the items on which overlap with personality traits such as schizotypy and paranoia and that the study did not attempt to delineate psychotic symptoms due to the acute effects of cannabis use from persistent effects (204).
This finding of increased psychosis risk has been reported in several other prospective studies (19–21, 24). The cumulative evidence for the association between cannabis and psychosis have been examined in five systematic reviews (25, 205–208), four of which (25, 205, 207, 208) found a consistent association between cannabis use and psychosis. The review by Macleod et al. (206) did not find a consistent association, but has been critiqued for failure to perform a meta-analysis. The inconsistent results of the systematic reviews are also likely due to different inclusion/exclusion criteria, different methodology, and different outcome measures (209). In the latest systematic review by Moore et al., any cannabis use (pooled adjusted OR = 1.41, 95% CI 1.20–1.65) was associated with a 40% increased risk of psychotic disorder, and the risk increased in a dose-dependent fashion with greater cannabis exposure (OR = 2.09, 95% CI 1.54–2.84) (25).
While the evidence supporting an association between cannabis exposure in adolescence and later psychosis is largely consistent, the evidence has been challenged on many counts (210), including sampling bias; under-powered sample sizes; presence of unknown confounders; difficulty distinguishing psychotic symptoms from psychotic disorder in longitudinal studies; direction of causality; lifetime exposure to multiple drugs; and period-, time-, and cohort-effects.
Chronic and heavy cannabis use has been associated with a syndrome characterized by a predominance of negative symptoms, referred to as an “amotivational syndrome” (175, 187, 211–213). The features of this syndrome include apathy, amotivation, social withdrawal, narrowing of one’s personal repertoire of interests, lethargy, impairment in memory and concentration, impaired judgment and decision-making, and poor socio-occupational functioning. All these symptoms share similarities with the negative symptoms of schizophrenia. The nosological status of the syndrome is, however, debated. Further, the confounding effects of concomitant poly-substance abuse, poverty, low socio-economic status, or pre-existing psychiatric disorders may explain the association (214, 215).
This literature is in contrast with the finding that healthy, cannabis users have lower scores on negative schizotypy compared to healthy, drug-free individuals (158, 216), and that patients with schizophrenia who use cannabis have less negative symptoms compared to those who do not use cannabis (217, 218). The cross-sectional nature of these studies and lack of information regarding scores at baseline makes it difficult to conclude if cannabis does not indeed cause a worsening of negative symptoms compared to baseline.
Several studies suggest that chronic, heavy cannabis use leads to impairments in memory, attention, working memory, executive function and IQ (219–227). Solowij and Mitchie suggested that cognitive dysfunction associated with long-term or heavy cannabis use is a cognitive endophenotype of schizophrenia (139). In a comprehensive review, Solowij and Battisti concluded that chronic heavy cannabis use was associated with impairments in memory (224) that persisted beyond the period of acute intoxication and was related to the frequency, duration, dose, and age of onset of cannabis use. Fontes et al. evaluated the neuropsychological performance of 104 chronic, heavy cannabis users and found that, compared to controls, chronic cannabis users had significant impairment on the cognitive domains of sustained attention, impulse control, and executive functioning (226). Additionally, similar to the literature on the risk of psychosis, individuals who used cannabis in adolescence (before the age of 15 years) had greater deficits. The authors however, did not assess whether subjects were in withdrawal or had residual effects from their last use of cannabis at the time of assessment.
While chronic, heavy cannabis users have deficits in cognitive processes, especially memory and attention in the context of ongoing cannabis use, the question of whether these impairments are persistent or a result of withdrawal and residual effects is unclear. While one study demonstrated an absence of persistent neuropsychological deficits in frequent long-term cannabis users after 28 days of abstinence (228), other studies have shown variable durations to full recovery, ranging from a week (229), to 28 days (221), to 3 months of abstinence (230), with some studies showing recovery only after an average of 2 years of abstinence (187, 231). A recent review provides a summary of the literature to date (225). Among studies in which neuropsychological assessments were performed 3 weeks or later after last use of cannabis, five out of seven studies showed no impairment in attention (221, 228, 232–236), while two showed persisting impairment (222, 231). One study revealed a trend toward impairment in decision-making/risk-taking (237). There was no impairment on response-inhibition measured by the Stroop test (221, 222, 233–235), and on working memory (236) while all (221, 222, 233, 234) but one (235) found an impairment on the Wisconsin Card Sorting Test, a test of set shifting. There was no impairment in verbal memory in two (228, 233) of the three studies that used the Buschke’s Selective Reminding Test (BSRT), a test of memory of word lists. When the data from the third study (234) was stratified based on age at onset of cannabis use, significantly greater impairment was noticed in those who had first use cannabis before the age of 17 years, suggesting that, as for positive symptoms, earlier age of onset of cannabis use may be associated with greater persistent cognitive deficits. It is important to note that none of these studies were designed to determine whether the cognitive impairments predated cannabis use.
Previous cross-sectional experiments have reported inconsistent results with some suggesting that chronic cannabis use impairs performance on tests of intelligence (238, 239), while others finding no impairment (240, 241). A recent longitudinal study examined 1037 subjects followed from birth to age 38 years (242). Cannabis use was evaluated at ages 18, 21, 26, 32, and 38 years while neuropsychological testing was conducted at ages 13 and 38 years. The experiment determined that those who persistently use cannabis are more likely than non-users to experience a significant decline in IQ. The findings persisted even after controlling for level of education and impaired IQ was found to be particularly true for the subjects who began to use cannabis during adolescence as opposed to during adulthood. Those who began to use cannabis during adolescence exhibited an eight-point decrease in IQ between childhood and adulthood. Another important finding of the study was that the decline in IQ did not appear to reverse after cannabis use ceased (242).
Some studies that have examined cognitive performance among patients with schizophrenia have made a case that patients with schizophrenia and comorbid cannabis abuse have better cognitive performance than patients without comorbid cannabis abuse (243–246). Emerging evidence, however, suggests that patients with cannabis use have higher premorbid IQ (247). The findings are not inconsistent with the experimental data; it is likely that persons who smoke cannabis have higher premorbid IQ, as evidenced by their ability to procure an illegal substance while evading the law. Therefore, although continued cannabis use results in a decline in their individual cognitive performance (242, 248, 249), when compared to non-users they appear to have relatively better cognitive performance. Furthermore, abstinence from cannabis may be associated with better cognitive performance among male patients with schizophrenia (248).
Epidemiological evidence suggest that the earlier the age of exposure to cannabis, the greater the risk of a psychosis outcome (19). Dragt et al. showed that younger age of onset of cannabis use is associated with earlier symptoms of anxiety, social withdrawal, derealization, memory impairment, and difficulties in concentration, with effects being more pronounced in patients with heavier cannabis use (250). Another recent study found that early onset cannabis use was only associated with earlier onset of psychosis when cannabis use began by age 14 (251). A large meta-analysis of 83 studies found that the age of onset of psychosis in cannabis users was 2.7 years younger than in non-users (252). Animal studies have shown that exposure to cannabinoids in adolescence has more deleterious effects than exposure in adulthood (253–257).
It is being increasingly recognized that adolescence may be a particularly critical period of increased vulnerability to the effects of cannabis. Additionally, factors such as schizotypy, other trait measures of liability to psychosis, and childhood abuse may moderate the risk of schizophrenia with prolonged and persistent cannabis use. As discussed above, the 26-year longitudinal study of the Dunedin cohort showed that earlier cannabis use is associated with a greater risk of psychotic disorder. However, when adjusted for psychotic symptoms at age 11, the association between cannabis use and subsequent psychotic disorder was no longer significant but remained elevated (OR = 3.1) (19). The small sample size may limit the interpretation of these results.
These studies suggest a “window of vulnerability” hypothesis: a critical period during early adolescence where the brain is particularly susceptible to the psychosis-inducing effects of cannabis (19, 250, 251, 253–258). One possible explanation for the “window of vulnerability” theory is that cannabis may affect the brain during a critical period of development and maturation. Brain development and maturation processes – including neuronal migration and differentiation, synaptogenesis, axon formation, and dendritic proliferation, myelination, pruning, apoptosis, and activity-dependent changes – begin in utero but continue into the early 20s or even later (259–264). Cannabis may disrupt one or more of these processes.
A retrospective study of 997 subjects by Stefanis and colleagues showed that, after adjusting for family history, there was a consistent relationship between the age of cannabis initiation and FEP, with an average time of 7–8 years (265). This finding does not support the “window of vulnerability” hypothesis, but rather indicates that the brain (at least in years 12–19) is continually sensitive to cannabis.
The association between age of onset of cannabis use and worse outcomes could simply reflect that earlier use is more likely to become longstanding, thus increasing the overall exposure to cannabis. An alternate explanation for the association between age of exposure to cannabis and psychosis is that those prone to early psychosis may “self-medicate” with cannabis to relieve symptoms (22, 266). However, this has not been supported by recent literature (28, 250, 251, 267). These studies are limited in that they have relied on measuring only positive psychotic symptoms as an indication of psychosis onset, although it is known that negative symptoms and cognitive deficits predate the onset of positive symptoms (268) and even predict conversion to psychosis in high-risk individuals (269). The interpretation of the data is also limited by the fact that cannabis use at an early age may be part of a broader pattern of externalizing behavior in response to difficult family circumstances (270, 271). Children and adolescents who begin cannabis use at an earlier age may represent a distinct sub-population that differs in ways that have not been accounted for (such as history of abuse or family socio-economic level) in the aforementioned studies.
Early studies have indicated that a positive family history of schizophrenia may increase risk for cannabis-induced psychotic disorders. One such study found that among patients admitted for acute psychosis, those who tested positive for cannabinoids in urine toxicology screens were 10 times more likely (7.1 vs. 0.7%) to have a positive family history for schizophrenia than patients without a positive urine toxicology screen (272). This finding implicated a familial predisposition to persistent psychotic disorders precipitated by cannabis use. Thus, in a genetically predisposed sub-population, cannabis confers a marked risk for psychosis. Most studies since have confirmed an association between a family history of psychotic disorder and an increased risk of cannabis-induced psychosis, though the association is more modest than the original study. Bersani et al. found that among schizophrenia patients, 24% of cannabis users had a positive family history of psychotic disorder vs. 10% (217). The largest study to investigate this association (n = 2,276,309) found a 2.5-fold increased risk of developing cannabis-induced psychosis in children of mothers with schizophrenia but no increased risk of conversion to schizophrenia (273). Further studies that have followed patients over time have shown that among patients who are admitted with an initial diagnosis of cannabis-induced psychosis, almost 50% convert to schizophrenia or some other psychotic disorder (181, 188). Boydell et al., found, in a retrospective study of 757 first-episode schizophrenia patients (24% who used cannabis in the year prior to presentation), that among patients with schizophrenia, cannabis users did not differ significantly from those not using cannabis in terms of a positive family history of schizophrenia (15 vs. 12%) (274). More recently, investigators from the Genetic Risk and Outcome of Psychosis (GROUP) collaboration studied a large sample of patients with a psychotic disorder (n = 1120), their siblings (n = 1057), and community controls (n = 590). In this prospective, ongoing study, the investigators found that the effect size of the relationship between current cannabis use and both positive and negative schizotypy symptoms was significantly greater in siblings of patients with a psychotic disorder when compared to healthy, un-related control. Further, there was a significant association between cannabis-using siblings and their psychotic patient relatives (in terms of positive symptoms), whereas this association did not emerge among non-exposed siblings and their psychotic relatives. The authors proposed that the familial liability to psychosis is expressed partially in terms of psychotomimetic experiences with cannabis (GROUP).
More recently, the interactive effects of childhood maltreatment and cannabis abuse have been examined. In a cross-sectional study, Houston and colleagues found an odds ratio of 11.96 (95% CI 2.10–68.22) for having experienced psychosis among children with a history of abuse who used cannabis prior to age 16 (275). Another cross-sectional study by Harley et al. found a significant interactive effect of childhood trauma and cannabis use in moderating the risk of psychotic symptoms; the odds ratio of experiencing psychosis in adolescents with a history of exposure to trauma and cannabis was 20.9 (95% CI 2.3–173.5) (276). A longitudinal study has similarly shown a significant interaction between cannabis use and childhood maltreatment in the development of psychotic symptoms (277). Notably, there was no evidence in this study that baseline history of childhood abuse affected subsequent cannabis use. These findings, however, were not replicated in the EDSP dataset (278). It is important to interpret the above findings with caution. Some investigators (279) have shown a link between childhood abuse and subsequent cannabis use; others demonstrate a link between abuse and subsequent psychosis (280). Future studies, which examine the interaction between genetic liability, trait measures of psychosis liability, cannabis use, and other environmental factors may provide greater insights into the complex mechanisms that cause psychosis.
Genetic factors may confer vulnerability to psychosis outcomes following exposure to cannabis, i.e., a gene-environment interaction. In specific, Catechol-O-methyltransferase (COMT) and AKT1, have been implicated in conferring such vulnerability (see Table 1). Preliminary evidence suggests that other genes might also moderate the cannabis–psychosis interaction.
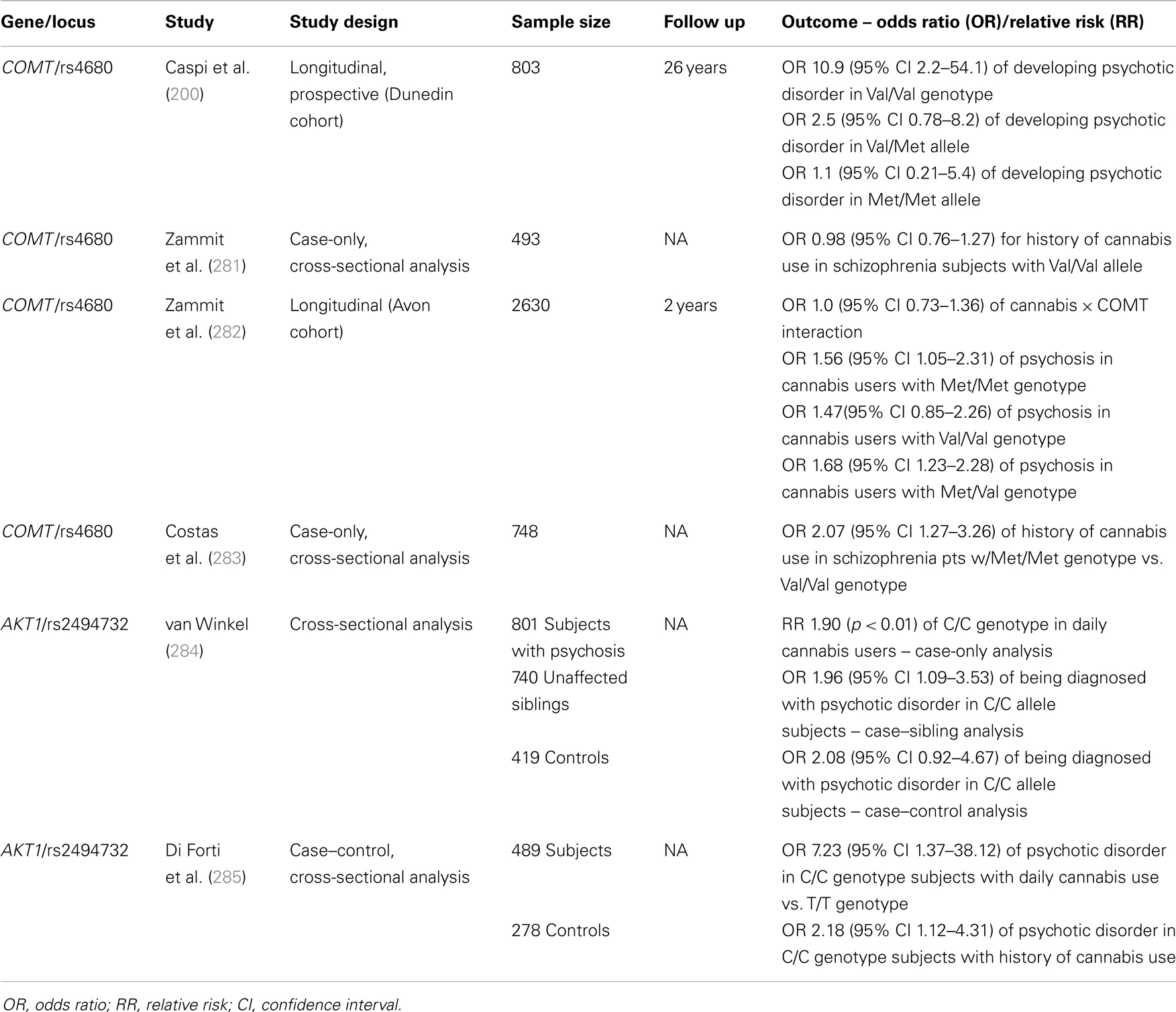
Table 1. Gene × cannabis interactions in moderating risk of psychosis.
In one of the first studies that drew attention to gene × environment interactions, Caspi et al. reported that the COMT gene moderated the risk of psychotic disorder with adolescent cannabis exposure. The enzyme COMT plays a critical role in the breakdown of dopamine in the PFC (286), in contrast to the striatum where DA is cleared by a transporter. The COMT gene has a common polymorphism in humans, which results in 40% higher enzymatic activity and thus more rapid degradation of dopamine when Valine (107) is substituted for Methionine (Met) at the 158/108 locus. Val/Val homozygotes have the lowest levels of dopamine; Met/Met homozygotes have the highest levels; and heterozygotes have intermediate levels. Lower cortical dopamine levels in individuals homozygous for the Val(158) polymorphism are associated with, among other things, poorer cognitive performance, and inefficient precortical functioning (287).
In a longitudinal prospective study (Dunedin cohort) of 803 individuals followed over 25 years, Caspi et al. showed that the risk of developing of psychotic disorder in association with cannabis exposure increased by 10-fold in those patients with the Val/Val allele (200). There were subsequent attempts to validate these findings with experimental evidence: a double-blind, placebo-controlled cross-over study showed that individuals with the Val polymorphism of the COMT gene have a higher chance of developing acute psychosis in response to THC exposure (133). These findings have been confirmed in a similar experiment (288).
Recent studies have failed to confirm the findings of the original 2005 study from Caspi and colleagues. A case-only analysis of 1438 individuals found no interaction between COMT polymorphism and cannabis use with regard to schizophrenia (281). Further, a 2-year longitudinal study of 2630 genotyped patients showed no interaction between COMT and cumulative cannabis use on the development of psychosis (282). A more recent case–control study also showed no COMT-mediated increased cannabis risk in the development of psychosis (284). Kantrowitz et al. were unable to find an interaction between COMT polymorphisms and cannabis-induced psychotic disorder in a population of 92 individuals with psychotic disorder, though this study was under-powered. Sub-analyses based on race (African American and Caucasian) did not yield significant findings (289). In contrast to the original Caspi et al. study (200), a case-only study from Spain (155 out of 748 total schizophrenia subjects who used cannabis) actually found an association between the low-activity Met allele of COMT and cannabis use in psychotic disorder (283). Estrada et al. (290) showed a dose-effect of COMT polymorphism on the age of onset of psychosis among cannabis users: individuals who were homozygous for the Val allele of COMT had the earliest age of onset of psychotic disorders at 15.4 years; homozygotes for the Met allele had the latest age of onset at 18.8 years; heterozygotes with intermediate enzymatic activity, had an age of onset of 17.1 years. Notwithstanding, there was no overall greater risk for psychotic disorder found among any of the polymorphism groups. A similar trend regarding the interaction of COMT polymorphism and cannabis use in association with the age of onset of psychosis has been shown, though not all results achieved statistical significance (291).
Other studies have examined the interactive effects of COMT polymorphisms and other environmental factors. A cross-sectional analysis of 918 individuals in Europe found a significant three-way interaction between the COMT Val allele, cannabis use, and childhood abuse in moderating psychosis. Individuals homozygous for the Val polymorphism were more likely to experience psychosis in association with cannabis use in the context of a history of childhood abuse than individuals homozygous or heterozygous for the Met allele. A replicative sample as part of the same study showed the same trend but did not achieve statistical significance (292). Confirming these findings, Alemany et al. found that the three-way interaction of COMT polymorphism (Val allele), cannabis use, and a positive history of child abuse significantly increased the risk of both positive and negative psychotic symptoms (293).
AKT1 is another gene thought to play a role in moderating the association between cannabis and psychotic disorders. The enzyme AKT1 functions to inactivate glycogen synthase kinase (GSK-3) by phosphorylation (294). The interaction between AKT1 and GSK-3 has been implicated to play a role in a number of important cellular processes, such as cell proliferation, apoptosis, and transcription (295). In vitro studies have shown that cannabinoids are capable of stimulating the AKT1 pathway via CB1 and CB2 receptors (296) and in vivo studies in mice have also confirmed this (297). Further, the gene product has been implicated in schizophrenia: postmortem studies have shown decreased AKT1 levels in lymphoblasts in the PFC of patients with schizophrenia (298, 299).
In a sample comprised of 801 patients with psychosis, 740 of their unaffected siblings, and 419 controls, van Winkel showed that cannabis users with the C/C genotype of a specific polymorphism (rs2494732) of the AKT1 gene had a twofold increase in risk of being diagnosed with psychotic disorder (284). Additionally, among psychotic patients, those homozygous for the C allele were twice as likely to have a history of daily cannabis use compared with T/T genotypes. The significance of the AKT1 × cannabis interaction held among case-only (p = 0.007) and case–sibling (p = 0.04) sub-analyses; in the case–control sub-analysis, the AKT1 × cannabis interaction approached statistical significance (p = 0.057). A more recent study has replicated these findings and found an even stronger interaction. Di Forti and colleagues studied 489 patients with FEP and 278 control subjects in a case–control design; among daily cannabis users, those who carried the C/C allele had, on average, a sevenfold increase in the risk of psychosis compared to T/T carriers (285). Notably, carriers of this genotype (C/C at SNP rs2494732) also have been shown to have increased cognitive side effects from cannabis use as evidenced by lower scores on tests of sustained attention (300). Preliminary experimental evidence has also implicated a different polymorphism of the AKT1 gene (the GG genotype of the SNP rs1130233) as a moderator of sensitivity to the acute psychosis-inducing effect of THC (301).
Another gene implicated in moderating the effects of cannabis on the development of psychosis is DAT1, which codes for the dopamine transporter, which is critical in removing DA from the synapse in striatal regions. A polymorphism involving a variable number of tandem repeats (VNTR) has been described in the 3′ untranslated region of the DAT1 gene (SLC6A3). One of the common alleles of this polymorphism (the nine-repeat allele) is associated with lower enzymatic activity and thus higher dopamine levels in the striatum. DAT1 has previously been associated with schizophrenia (independent of cannabis use) in gene association studies (302). Bhattacharyya et al., reported that individuals with the nine-repeat allele showed increased sensitivity to THC-induced psychotomimetic effects in a small laboratory based study (n = 35) (301). There was also a trend toward greater THC-induced psychotomimetic effects in individuals with the G/G genotype of the rs1130233 polymorphism of the AKT1 gene in the same sample. Furthermore, there was a synergistic interaction between these DAT and AKT1 genotypes on the psychotomimetic effects of THC. In addition to studying behavioral effects of THC, this study showed interactive effects of DAT1 genotype, AKT1 genotype, and THC on striatal and midbrain activation during encoding and recall of verbal information, respectively. Individuals with the GG allele at AKT1 and carriers of the nine-repeat allele of DAT1 also showed increased activation in the striatum in response to THC in comparison to the rest.
Neuregulin 1 (NRG1), a leading schizophrenia susceptibility gene, is relevant to several schizophrenia-related neurodevelopmental processes (303, 304). Heterozygous deletion of NRG1 results in increased sensitivity of mice to schizophrenia-like symptoms induced by THC especially under stressful conditions (305). These mice also showed greater increases in prepulse inhibition (PPI), a marker for sensorimotor gating known to be impaired in schizophrenia, following THC administration (305). However, to our knowledge, this work has not yet been extended to humans. Decoster et al. reported significant interactions between brain-derived neurotrophic factor (BDNF) genotype, cannabis exposure, and gender in a cohort of schizophrenia patients: in female patients only, cannabis use was associated with earlier age of onset of psychosis in BDNF Met-carriers relative to Val/Val-genotypes (306). Additionally, cannabis use may interact with specific genotypes of the cannabinoid receptor 1 (CNR1) gene to moderate cognitive impairment in schizophrenia patients (307), but thus far no significant interaction between CNR1 polymorphisms and cannabis exposure on the risk for the development of psychotic disorders has been reported (281).
The association between cannabis and psychosis fulfills many but not all of the standard criteria for causality (308), namely temporal relationship, biological gradient, biological plausibility, coherence, consistency, and experimental evidence.
As discussed above, evidence from experimental studies shows a clear temporal relationship between exposure to cannabinoids and symptoms of psychosis. Despite a number of limitations (discussed previously), several epidemiological studies have concluded that cannabis use generally precedes the development of psychotic disorder. In one of the earliest such studies, Allebeck and colleagues found that cannabis use preceded the onset of schizophrenia by at least 1 year in 69% of cases; in only 11% of cases did cannabis succeed psychosis (309). In a prospective cohort study, Linszen et al. found that in all but 1 patient from a sample of 24 cannabis-abusing patients, cannabis abuse preceded FEP by at least 1 year (310).
Studies from recent years suggest that in the majority of cases, cannabis use precedes the onset of psychosis, rather than vice versa. In a study of 28 FEP patients, cannabis use preceded psychosis in all patients (267). Another study of 45 psychotic disorder patients with a history of cannabis use showed that the onset of cannabis use preceded hallucinations in 74% of cases and preceded persecutory ideas in 90% of cases by at least on year (250). Schimmelmann and associates (251) reported that in 88% of cases (n = 201 FEP patients with cannabis use), drug exposure preceded psychotic symptoms by a mean of 5 years.
As discussed above, numerous additional studies have shown that cannabis users have a younger age of onset of psychotic disorders compared to non-users (197, 250, 258, 309–312). A recent meta-analysis of over 22,000 subjects found the onset of psychosis was 2.7 years younger in cannabis users than in non-users (252). These studies lend further evidence to the finding that cannabis usually precedes the onset of psychotic symptoms and argue against the “self-medication” hypothesis.
The findings from epidemiological studies regarding the temporal relationship between cannabis and psychosis must be qualified. Epidemiological studies have traditionally examined the relationship of cannabis use and psychosis as defined by positive psychotic symptoms. It is unclear whether the same temporal relationship holds for cognitive deficits or negative symptoms of psychosis, which usually predate the onset of positive psychotic symptoms. Furthermore, the data fails to explain why patients with schizophrenia continue to abuse cannabis. Cannabis continues to be among the most common illicit drug used by patients with schizophrenia. In the Australian Study of High Impact Psychoses (313), 49% of patients with schizophrenia reported exposure to cannabis in the past year (314). In a study among patients with schizophrenia using experience-sampling, Henquet et al. (315) found that compared to healthy control, patient with schizophrenia reported a reduction in negative affect after cannabis use, while the increase in positive affect that they experienced was comparable to controls. Schizophrenia is a disease of gradual onset and diagnosis usually occurs only when a patient’s symptoms are severe enough to cause disruptions in psychosocial functioning. If, as has been hypothesized, schizophrenia is a neurodevelopmental disorder in which neurobiological changes occur years before the onset of symptoms, then these studies have been unable to examine the true temporal relationship between psychosis and cannabis. On the other hand, if cannabis induces schizophrenia in individuals who are genetically vulnerable (see discussion below) and thus exhibit “prodromal” symptoms at baseline, then the exact temporal nature of this relationship is extremely relevant.
There are a number of limitations to assessing the dose of exposure of cannabis and its effect on psychotic outcome. Whereas cigarettes and alcoholic beverages have standardized and well-known quantities of nicotine and alcohol, the THC content of cannabis varies considerably. Further, when people smoke cannabis, they may smoke the same joint over several sessions or share a joint with others. Therefore, the number of times a person has smoked is a crude proxy of the “dose” of cannabis exposure. Finally, as discussed above, CBD is thought to have antipsychotic effects in opposition to the pro-psychotic effects of THC. The variable content of CBD in marijuana further complicates the interpretation of studies investigating a dose–response effect.
Despite these limitations, a consistent dose–response effect has been shown in numerous studies. One of the earliest studies showing a biological gradient in the association between cannabis use and psychotic symptoms was done by Andreasson and colleagues. Using the Swedish military conscript (n > 45,000) followed over a 15-year period (described in detail previously), the investigators found that individuals with heavier cannabis use (>50 occasions of consumption) had a greater chance of developing schizophrenia (relative risk 6.0); intermediate users (11–50 occasions of consumption) had a relative risk of 3.0 for developing schizophrenia. After adjusting for various potential confounders (school adjustment, socio-economic status, solvent abuse, psychiatric diagnosis or medications at baseline, and others, but not including childhood abuse/trauma), the relative risk remained elevated and statistically significant (197). A follow up of this same cohort at 27 years found that this dose–response relationship between cannabis consumption and risk of developing schizophrenia persisted over time (198). Other studies previously described, including the NEMESIS (20) and the ESDP cohorts (24) have also suggested a biological gradient between exposure load and psychotic outcome. More recent evidence also supports this dose–response effect in a sample of individuals with sub-clinical psychotic symptoms; this sample showed that among heaviest users (>5 per day) the relative risk, after adjusting for confounders (“sex, age, social exclusion, alcohol, cannabis use before age 17, and heavy non-cannabis drug use”), of experiencing auditory hallucinations was 5.4 and relative risk for first-rank symptoms was 11.6 (316).
The specificity of the association between cannabis and psychotic disorders is low. In a prospective study of 3-year follow up, of all patients who developed psychosis (assessed by BPRS), only 21% had any use of cannabis at baseline. Furthermore, of those who used cannabis at baseline, only 8 in 312 subjects (2.6%) developed psychosis (20). Similar data was reported from the Swedish military conscript cohort (197). While the association between cannabis and schizophrenia is not specific, it is stronger and more consistent than the association between cannabis and anxiety or depressive disorders. Odds ratios for the development of anxiety or depressive disorders with exposure to cannabis typically range from 0.7 to 1.5 with many studies yielding statistically insignificant results; in contrast, a meta-analysis of multiple longitudinal prospective studies found a statistically significant, adjusted odds ratio of 2.09 (95% CI 1.54–2.84) for psychosis outcome among heaviest cannabis users with all but one of the six high-quality, longitudinal studies showing a statistically significant outcome. These longitudinal studies controlled for about 60 different potential confounders, including personality traits, socio-economic markers, other substance use, and other mental health problems (25). In a longitudinal study of over 18,000 patients hospitalized for substance-induced psychosis, the 8-year cumulative risk of conversion to schizophrenia was 46% when the offending substance was cannabis. In contrast, the conversion rate to schizophrenia over the same period of time for alcohol-induced psychosis was 5%. Notably, the risk for the development of schizophrenia when the diagnosis was amphetamine-induced psychosis was 30% (188).
While not all epidemiological studies have detected an association between cannabis use and psychosis, most longitudinal studies (described in detail previously) show a statistically significant increased risk of psychosis outcome in those who use cannabis heavily. Among each study’s heaviest users, the following longitudinal studies have demonstrated a significantly increased risk of any psychosis outcome: the Swedish military conscript cohort (heaviest users being those who had used marijuana >50 times) (197–199), the NEMESIS cohort (weekly users) (20), EDSP cohort (daily users) (24), Epidemiological Catchment Area study (daily users) (317), Dunedin cohort (19, 200), and the CDHS cohort (daily users) (23). The National Psychiatric Morbidity Survey found an increased odds ratio (adjusted for alcohol consumption, gender, IQ score, marital status, and others) that was not statistically significant (even in their heaviest using subjects, those with cannabis dependence) (318). Among these same studies, an analysis of those who had ever used marijuana (even if just once), the Epidemiological Catchment Area study, EDSP study, and the NEMESIS cohort showed increased risk of any psychosis outcome but this risk was not statistically significant.
The precise pathophysiology of psychosis or psychotic disorders remains unclear; therefore, a biologically plausible mechanism whereby exposure to cannabis can increase the risk for psychosis or a psychotic disorder is yet to be established. THC, the principal active component of cannabis, is a partial agonist at CB1Rs where it has modest affinity (Ki = 35–80 nmol) and low intrinsic activity (319). CB1Rs are G-protein-mediated receptors that are distributed with high density in the cerebral cortex (particularly frontal regions), basal ganglia, hippocampus, anterior cingulate cortex, and cerebellum; these brain regions have been implicated in the putative neural circuitry of psychosis. The primary effect of cannabinoids is the modulation of neurotransmitter release via activation of presynaptic CB1Rs. Thus cannabinoids, by activating CB1Rs, can modulate the release of a number of neurotransmitters already implicated in psychosis, including dopamine, glutamate or GABA.
The dopamine hypothesis, which postulates that positive symptoms of psychosis may be attributed to disturbed and hyperactive dopaminergic activity, remains one of the more enduring and dominant hypotheses of schizophrenia (320). CB1R-mediated increases in mesolimbic dopaminergic activity may explain the positive psychotic symptoms induced by THC. Converging preclinical evidence suggests interactions between cannabinoid (CB1) and dopamine (DA) systems [reviewed in Ref. (321, 322)]. CB1 and D2 receptors are co-expressed in several brain regions (323) and there is signal transduction convergence in these regions (324). Cannabinoids have been shown to induce firing of dopaminergic mesolimbic neurons and induce DA release in the striatum in animals (100, 321, 325–329). Cannabinoids regulate DA firing via a CB1-GABAergic-mediated disinhibition of DA neuronal activation. However, the results of in vivo imaging studies of THC-induced striatal dopamine release in humans have been mixed (96, 330–332). The effect of cannabinoids on striatal dopamine release may be differentially affected by biological vulnerability for psychosis. While chronic cannabis use was found to be associated with decreased striatal dopamine synthesis in healthy individuals (332), THC was found to increase striatal dopamine release in first-degree relatives of individuals with psychotic disorder (333).
The effects of cannabinoids on dopaminergic systems in the PFC might account for some of their acute cognitive deficits. It is well-know that either too much or too little dopaminergic activity in the PFC is associated with impairments in PFC-related cognitive functions leading to an inverted “U” (bell shaped) relationship between dopamine levels and working memory efficiency (334, 335). Systemic administration of cannabinoids has been reported to increase prefrontal cortical DA release or turnover in several studies (100, 336–339). This may explain how cannabinoids produce acute impairments in PFC-related cognitive functions including working memory and attention.
Cannabinoids might induce psychosis and cognitive impairments via actions on GABAergic systems. Higher order cognitive processes, including working memory, are associated with θ (4–7 Hz) and γ (30–80 Hz) oscillations in the PFC. Deficits in working memory are a hallmark of schizophrenia and are associated with reduced cortical θ and γ band power. Cortical θ and γ oscillations are dependent on inhibition of pyramidal neurons. This inhibition is driven by specific cholecystokinin (CCKb cells) and parvalbumin (PVb cells) containing GABAergic interneurons. In several brain regions, CB1Rs are present on the terminals of axons in cholecystokinin (CCK)-containing GABA interneurons that target the perisomatic regions of pyramidal cells (340). Activation of CB1R reduces GABA release, which in turn releases the inhibition effects on pyramidal cells. While admittedly speculative, the disinhibition of pyramidal cells may lead to cortical oscillation deficits and working memory impairments.
While the acute effects of cannabinoids on DA, GABA, and glutamate neurotransmission may explain some of the acute positive, negative, and cognitive symptoms of cannabinoids, the mechanism by which exposure to cannabinoids might cause schizophrenia has not yet been established. If schizophrenia is a neurodevelopmental illness (341, 342), then the observation that early cannabis exposure is associated with a greater risk for the development of schizophrenia may offer some clues to the underlying biological mechanisms. Consistent with the human epidemiological data, animal studies suggest that early (adolescent) but not later (adult) exposure to cannabinoids is associated with persistent impaired social behaviors, including psychotic-like behaviors, cognitive, and sensorimotor gating deficits in adults (253–257).
Adolescence and young adulthood are critical phases for cerebral development. Brain development continues into young adulthood (up to 25 years) (343) and therefore, any factors that interfere with brain development during this time may have far reaching consequences. During this period of neuronal plasticity, there is sprouting and pruning of synapses, myelinization, changes in neurotransmitter concentrations and their receptor levels in brain areas necessary for behavioral and cognitive functions (344). The endocannabinoid system plays an important role in several processes important in neurodevelopment including neurogenesis, neural specification, neural maturation, neuronal migration, axonal elongation, glia formation, and positioning of inhibitory GABAergic interneurons and excitatory glutamatergic neurons (259–262, 345–349). Perturbation of the endocannabinoid system in the rapidly changing brain, as is the case in adolescence, by excessive or non-physiological stimulation, as may be the case with exposure to exogenous cannabinoids, may have far reaching consequences. This would be especially so in the presence of already altered neurodevelopmental processes. Therefore, by disrupting the endocannabinoid system and interfering with neurodevelopmental processes, exogenous cannabinoids may provide a biologically plausible mechanism by which exposure to cannabinoids during adolescence may increase the risk for the development of schizophrenia.
In the general population, the strength of association between any cannabis exposure and the development of psychosis is modest. A systematic review of 35 longitudinal studies found the relative risk of developing schizophrenia after any cannabis exposure to be 1.4 after adjusting for about 60 potentially confounding variables, including personality traits, socio-economic markers, other substance use, and other mental health problems (25). However, as discussed above, in heavy users (as well as those who begin use at earlier ages), the risk can be much greater. A follow up of the original Swedish military conscript after 35 years yielded an adjusted relative risk of 3.7 for the development of a psychotic disorder (199).
Indirect but compelling evidence is seen in conversion of cannabis-induced psychosis to schizophrenia. Longitudinal studies have found that the risk of developing schizophrenia is nearly 50% in patients admitted for cannabis-induced psychosis (181, 188). Such findings suggest that genetic (or other predisposing) susceptibility to cannabis-induced psychosis may explain why the cannabis–schizophrenia association does not fulfill all causality criteria. That is, in a sub-population of individuals with a history of childhood abuse and genetic vulnerability, the association between cannabis and schizophrenia may be significantly stronger and more specific than in the general population. Individuals with neurobiological vulnerabilities who develop acute psychosis, which persists for a limited period after cannabis intoxication may be those who, with prolonged exposure, are more likely to develop permanent psychotic disorders.
As noted above, direct experimental evidence for acute and transient psychosis caused by cannabis intoxication is compelling (89, 95). In some individuals, this effect persists after the acute intoxication period has ended. In randomized, placebo-controlled, experimental settings, acute psychosis in response to THC intoxication is quite common and reproducible (89, 95, 350). Positive (paranoia, grandiose delusions, fragmented thinking, and perceptual alterations), negative (blunted affect, emotional withdrawal, and psychomotor slowing), and cognitive symptoms (impairments of abstraction, attention, executive function, and memory) have been well-documented. Thus, the main symptom clusters of schizophrenia are seen acutely with THC intoxication. Occasionally, immediate psychosis precipitated by cannabis persists beyond the period of intoxication and may require intervention, though most of these data come from case reports and small series rather than experimental evidence.
Unlike studying acute effects, an experimental approach to characterize the effects of chronic, heavy, and early cannabis exposure is neither ethical nor feasible. An alternative approach is to compare a group with chronic, heavy early cannabis use to controls. Such samples do exist and have been discussed in detail previously.
There is substantial coherence between the laboratory study findings and epidemiological findings regarding the acute effects of cannabinoids. Cannabinoids induce a range of psychosis-like effects in laboratory studies and epidemiological studies are replete with reports of psychosis following the consumption of cannabinoids. Similarly, cannabinoids have been shown to exacerbate symptoms in individuals with a psychotic disorder and epidemiological studies have shown that cannabis use by schizophrenia patients is associated with a negative impact on the expression and course of the illness. However, as an experimental approach to characterize the effects of chronic, heavy, and early cannabis exposure is neither ethical nor feasible, it is impossible to determine coherence between laboratory and epidemiological studies with regard to the consequences of chronic, early, and heavy cannabis use and psychosis.
Several parallels can be drawn between the cannabis–psychosis association and other associations in medicine that have been accepted to be causal in nature. For instance, excess salt consumption has been shown to be a well-established cause of hypertension (351), yet not all people who consume more than 2 g of salt daily have hypertension. Similarly, most people who smoke cigarettes do not develop lung cancer; further, there are types of lung cancer (i.e., adenocarcinoma), which develop in the absence of smoking. Yet smoking is understood to be the single most important modifiable causal component in the development of lung cancer.
It is unlikely that schizophrenia is a homogenous disorder with a single pathophysiology; instead, it is more likely a syndrome with distinct neurobiological etiologies. Similarly, the term “lung cancer” comprises several different histological types, including adenocarcinoma, squamous cell carcinoma, and small-cell carcinoma. The risk that smoking confers in the development of cancer varies considerably as the sub-type of cancer becomes more specific. For instance, the risk of developing various types of cancer (including liver, kidney, cervical, myeloid leukemia, gastric, nasopharyngeal, nasal, or esophageal adenocarcinoma) among current smokers may be relatively low, with estimates of the relative risk being ~1.5–2.0 (352). In a large meta-analysis, the relative risk of developing any lung cancer among current smokers is much higher at 8.43 (95% CI 7.63–9.31); the relative risk of developing squamous cell carcinoma of the lung is even higher, recently cited at 16.43 (95% CI 12.66–21.32) (353). Viewed from another perspective of the analogy, it is estimated that tobacco smoke is responsible for ~21% of all types of cancer-related deaths worldwide (354) and 87% of all deaths related to lung cancer (2013). By comparison, it is estimated that 8–14% cases of schizophrenia may be due to cannabis use (25, 207). Therefore, the magnitude of the risk for schizophrenia conferred by cannabis exposure is significantly lower than the risk of lung cancer conferred by smoking. It is unlikely that there is any single cause of an illness as heterogeneous as schizophrenia. As research progresses and our understanding of the biological causes of mental illness advances, cannabis-induced psychotic disorder may emerge as a distinct sub-type among the different disorders that constitute what we now classify broadly as schizophrenia.
In summary, the relationship between cannabinoids and psychosis fulfills many but not all of the traditional criteria for causality. Given the evidence presented above, it is likely that cannabis is an important component cause in the development of psychotic disorders (16, 205). This causal role is likely magnified when cannabis exposure occurs at an earlier age, in greater quantities, and over a longer time-course. Further, as discussed elsewhere in this review, specific populations (i.e., those with a genetic vulnerability or a history of childhood abuse) may be particularly susceptible to the causal effects of cannabis. Notably, although meta-analytical studies suggest that cannabis might account for between 8 and 14% of schizophrenia cases (25, 207), the fourfold increase in the rates of cannabis use over the last four decades (198, 355) has not resulted in a commensurate 40–70% increase in prevalence of schizophrenia. Some studies suggest that the rates of schizophrenia may be decreasing (356), while others suggest the contrary (357, 358). The discrepancy between the recent changes in the rates of cannabis consumption and relative stability of schizophrenia rates are difficult to explain in the context of the findings reviewed above; one possible explanation is that schizophrenia rates are lagging behind increased rates of cannabis consumption. Again, it is important to note that schizophrenia is likely a very heterogeneous illness, comprised of multiple sub-types. It is unlikely that there is a single causative factor. As proposed by Rounsaville, it is possible that a cannabis-induced psychotic disorder comprises one of the distinct sub-types of the schizophrenia-spectrum (191).
In summary, acute exposure to both natural and synthetic cannabinoids can produce a full range of transient symptoms, cognitive deficits, and psychophysiological abnormalities that bear a striking resemblance to some of the features of schizophrenia. Also clear is that, in individuals with an established psychotic disorder, cannabinoids can exacerbate symptoms, trigger relapse, and have negative consequences on the course of the illness. Finally, exposure to cannabinoids in adolescence confers a higher risk for psychosis outcomes in later life and the risk is dose-related. However, it should be remembered that the majority of individuals who consume cannabis do not experience any kind of psychosis.
The findings from research reviewed above have profound implications for public health. Aside from alcohol, cannabis is currently the most prevalent drug used worldwide. In the United States, the legal status of cannabis for medical and recreational purposes is changing rapidly. Pertinent findings that are likely to impact public health include high conversion rates from cannabis-induced psychosis to schizophrenia; global and specific domains of cognitive impairment resulting from cannabis use, which may be irreversible; the effects of acute intoxication; the precipitation of psychotic disorders in genetically vulnerable populations, including individuals with a history of childhood abuse or family history of psychotic disorders; and the increased risk of negative effects of cannabis use in prolonged and early exposure. Additional high-quality epidemiological studies are needed to further characterize the extent to which cannabis causes these negative effects or unmasks them in a vulnerable subset of the population.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ONDCP. Marijuana: the greatest cause of illegal drug abuse. In: The Marijuana Factbook. Washington, DC: Executive Office of the President (2008). p. 1–20.
2. SAMHSA. Results from the 2010 National Survey on Drug Use and Health: Summary of National Findings. Rockville, MD: Substance Abuse and Mental Health Services Administration (2011).
3. Johnston LD, O’Malley PM, Bachman HG, Schulenberg JE. Monitoring the Future National Results on Adolescent Drug Use: Overview of Key Findings, 2011. Ann Arbor, MI: Institute for Social Research, The University of Michigan (2012).
4. Mehmedic Z, Chandra S, Slade D, Denham H, Foster S, Patel AS, et al. Potency trends of Delta9-THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. J Forensic Sci (2010) 55:1209–17. doi: 10.1111/j.1556-4029.2010.01441.x
5. Procon.Org. Medical Marijuana (2011). Available from: http://medicalmarijuana.procon.org/view.resource.php?resourceID=000881
6. Kleber HD, DuPont RL. Physicians and medical marijuana. Am J Psychiatry (2012) 169:564–8. doi:10.1176/appi.ajp.2012.12030373
8. Spaderna M, Addy PH, D’Souza D. Spicing things up: the effects of synthetic cannabinoids. Psychopharmacology (Berl) (2013) 228(4):525–40. doi:10.1007/s00213-013-3188-4
9. NIDA. Info Facts: Spice (2011). Available from: http://www.drugabuse.gov/publications/infofacts/Spice
10. Vardakou I, Pistos C, Spiliopoulou C. Spice drugs as a new trend: mode of action, identification and legislation. Toxicol Lett (2010) 197:157–62. doi:10.1016/j.toxlet.2010.06.002
11. Johnson LA, Johnson RL, Alfonzo C. Spice: a legal marijuana equivalent. Mil Med (2011) 176:718–20. doi:10.7205/MILMED-D-10-00356
12. Every-Palmer S. Synthetic cannabinoid JWH-018 and psychosis: an explorative study. Drug Alcohol Depend (2011) 117:152–7. doi:10.1016/j.drugalcdep.2011.01.012
13. Hu X, Primack BA, Barnett TE, Cook RL. College students and use of K2: an emerging drug of abuse in young persons. Subst Abuse Treat Prev Policy (2011) 6:16. doi:10.1186/1747-597X-6-16
14. Vandrey R, Dunn KE, Fry JA, Girling ER. A survey study to characterize use of Spice products (synthetic cannabinoids). Drug Alcohol Depend (2012) 120:238–41. doi:10.1016/j.drugalcdep.2011.07.011
15. SAMHSA. Drug Abuse Warning Network, 2010: National Estimates of Drug-Related Emergency Department Visits. Rockville, MD: Center for Behavioral Health Statistics and Quality (formerly the Office of Applied Studies), Substance Abuse and Mental Health Services Administration (SAMHSA) (2012).
16. D’Souza DC, Sewell RA, Ranganathan M. Cannabis and psychosis/schizophrenia: human studies. Eur Arch Psychiatry Clin Neurosci (2009) 259:413–31. doi:10.1007/s00406-009-0024-2
17. Sewell RA, Ranganathan M, D’Souza DC. Cannabinoids and psychosis. Int Rev Psychiatry (2009) 21:152–62. doi:10.1080/09540260902782802
18. Radhakrishnan R, Addy PH, Sewell RA, Skosnik PD, Ranganathan M, D’Souza DC. Cannabis, cannabinoids and the link with psychosis. In: Madras B, Kuhar M, editors. The Effects of Drug Abuse on the Human Nervous System. San Diego, CA: Academic Press (Elsevier) (2014). p. 423–74.
19. Arseneault L, Cannon M, Poulton R, Murray R, Caspi A, Moffitt TE. Cannabis use in adolescence and risk for adult psychosis: longitudinal prospective study. BMJ (2002) 325:1212–3. doi:10.1136/bmj.325.7374.1212
20. van Os J, Bak M, Hanssen M, Bijl RV, De Graaf R, Verdoux H. Cannabis use and psychosis: a longitudinal population-based study. Am J Epidemiol (2002) 156:319–27. doi:10.1093/aje/kwf043
21. Weiser M, Knobler HY, Noy S, Kaplan Z. Clinical characteristics of adolescents later hospitalized for schizophrenia. Am J Med Genet (2002) 114:949–55. doi:10.1002/ajmg.10647
22. Ferdinand RF, Sondeijker F, Van Der Ende J, Selten JP, Huizink A, Verhulst FC. Cannabis use predicts future psychotic symptoms, and vice versa. Addiction (2005) 100:612–8. doi:10.1111/j.1360-0443.2005.01070.x
23. Fergusson DM, Horwood LJ, Ridder EM. Tests of causal linkages between cannabis use and psychotic symptoms. Addiction (2005) 100:354–66. doi:10.1111/j.1360-0443.2005.01001.x
24. Henquet C, Krabbendam L, Spauwen J, Kaplan C, Lieb R, Wittchen HU, et al. Prospective cohort study of cannabis use, predisposition for psychosis, and psychotic symptoms in young people. BMJ (2005) 330:11. doi:10.1136/bmj.38267.664086.63
25. Moore TH, Zammit S, Lingford-Hughes A, Barnes TR, Jones PB, Burke M, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet (2007) 370:319–28. doi:10.1016/s0140-6736(07)61162-3
26. McGrath J, Welham J, Scott J, Varghese D, Degenhardt L, Hayatbakhsh MR, et al. Association between cannabis use and psychosis-related outcomes using sibling pair analysis in a cohort of young adults. Arch Gen Psychiatry (2010) 67:440–7. doi:10.1001/archgenpsychiatry.2010.6
27. Genetic Risk and Outcome in Psychosis (GROUP) Investigators. Evidence that familial liability for psychosis is expressed as differential sensitivity to cannabis: an analysis of patient-sibling and sibling-control pairs. Arch Gen Psychiatry (2011) 68:138–47. doi:10.1001/archgenpsychiatry.2010.132
28. Kuepper R, Van Os J, Lieb R, Wittchen HU, Hofler M, Henquet C. Continued cannabis use and risk of incidence and persistence of psychotic symptoms: 10 year follow-up cohort study. BMJ (2011) 342:d738. doi:10.1136/bmj.d738
29. Chopra GS. Studies on psycho-clinical aspects of long-term marihuana use in 124 cases. Int J Addict (1973) 8:1015–26.
30. Chopra GS, Smith JW. Psychotic reactions following cannabis use in East Indians. Arch Gen Psychiatry (1974) 30:24–7. doi:10.1001/archpsyc.1974.01760070014002
31. Mechoulam R. Plant cannabinoids: a neglected pharmacological treasure trove. Br J Pharmacol (2005) 146:913–5. doi:10.1038/sj.bjp.0706415
32. Zuardi AW, Crippa JA, Hallak JE, Bhattacharyya S, Atakan Z, Martin-Santos R, et al. A critical review of the antipsychotic effects of cannabidiol: 30 years of a translational investigation. Curr Pharm Des (2012) 18:5131–40. doi:10.2174/138161212802884681
34. Marshall C. The active principle of Indian help: a preliminary communication. Lancet (1897) 1:235–8. doi:10.1016/S0140-6736(01)95658-2
35. Smith DE. Acute and chronic toxicity of marijuana. J Psychedelic Drugs (1968) 2:37–47. doi:10.1080/02791072.1969.10524413
36. Grossman W. Adverse reactions associated with cannabis products in India. Ann Intern Med (1969) 70:529–33. doi:10.7326/0003-4819-70-3-529
37. Talbott JA, Teague JW. Marihuana psychosis: acute toxic psychosis associated with the use of cannabis derivatives. JAMA (1969) 210:299–302. doi:10.1001/jama.210.2.299
42. D’Souza DC. Cannabinoids and psychosis. Int Rev Neurobiol (2007) 78:289–326. doi:10.1016/S0074-7742(06)78010-2
43. Reilly D, Didcott P, Swift W, Hall W. Long-term cannabis use: characteristics of users in an Australian rural area. Addiction (1998) 93:837–46. doi:10.1046/j.1360-0443.1998.9368375.x
44. Green B, Kavanagh D, Young R. Being stoned: a review of self-reported cannabis effects. Drug Alcohol Rev (2003) 22:453–60. doi:10.1080/09595230310001613976
45. Peters BD, De Koning P, Dingemans P, Becker H, Linszen DH, De Haan L. Subjective effects of cannabis before the first psychotic episode. Aust N Z J Psychiatry (2009) 43:1155–62. doi:10.3109/00048670903179095
46. Compton MT, Broussard B, Ramsay CE, Stewart T. Pre-illness cannabis use and the early course of nonaffective psychotic disorders: associations with premorbid functioning, the prodrome, and mode of onset of psychosis. Schizophr Res (2011) 126:71–6. doi:10.1016/j.schres.2010.10.005
47. Mechoulam R, Gaoni Y. A total synthesis of Dl-Delta-1-tetrahydrocannabinol, the active constituent of hashish. J Am Chem Soc (1965) 87:3273–5. doi:10.1021/ja00952a620
48. Cronin CM, Sallan SE, Gelber R, Lucas VS, Laszlo J. Antiemetic effect of intramuscular levonantradol in patients receiving anticancer chemotherapy. J Clin Pharmacol (1981) 21:43S–50S. doi:10.1002/j.1552-4604.1981.tb02572.x
49. Diasio RB, Ettinger DS, Satterwhite BE. Oral levonantradol in the treatment of chemotherapy-induced emesis: preliminary observations. J Clin Pharmacol (1981) 21:81S–5S. doi:10.1002/j.1552-4604.1981.tb02579.x
50. Heim ME, Romer W, Queisser W. Clinical experience with levonantradol hydrochloride in the prevention of cancer chemotherapy-induced nausea and vomiting. J Clin Pharmacol (1981) 21:86S–9S. doi:10.1002/j.1552-4604.1981.tb02580.x
51. Jain AK, Ryan JR, McMahon FG, Smith G. Evaluation of intramuscular levonantradol and placebo in acute postoperative pain. J Clin Pharmacol (1981) 21:320S–6S. doi:10.1002/j.1552-4604.1981.tb02610.x
52. Laszlo J, Lucas VS Jr, Hanson DC, Cronin CM, Sallan SE. Levonantradol for chemotherapy-induced emesis: phase I-II oral administration. J Clin Pharmacol (1981) 21:51S–6S. doi:10.1002/j.1552-4604.1981.tb02573.x
53. Kenny JB, Wilkinson PM. Levonantradol effectiveness in cancer patients resistant to conventional anti-emetics. Clin Oncol (1982) 8:335–9.
54. Stuart-Harris RC, Mooney CA, Smith IE. Levonantradol: a synthetic cannabinoid in the treatment of severe chemotherapy-induced nausea and vomiting resistant to conventional anti-emetic therapy. Clin Oncol (1983) 9:143–6.
55. Heim ME, Queisser W, Altenburg HP. Randomized crossover study of the antiemetic activity of levonantradol and metoclopramide in cancer patients receiving chemotherapy. Cancer Chemother Pharmacol (1984) 13:123–5. doi:10.1007/BF00257128
56. Sheidler VR, Ettinger DS, Diasio RB, Enterline JP, Brown MD. Double-blind multiple-dose crossover study of the antiemetic effect of intramuscular levonantradol compared to prochlorperazine. J Clin Pharmacol (1984) 24:155–9. doi:10.1002/j.1552-4604.1984.tb01824.x
57. Stambaugh JE Jr, McAdams J, Vreeland F. Dose ranging evaluation of the antiemetic efficacy and toxicity of intramuscular levonantradol in cancer subjects with chemotherapy-induced emesis. J Clin Pharmacol (1984) 24:480–5. doi:10.1002/j.1552-4604.1984.tb02756.x
58. Citron ML, Herman TS, Vreeland F, Krasnow SH, Fossieck BE Jr, Harwood S, et al. Antiemetic efficacy of levonantradol compared to delta-9-tetrahydrocannabinol for chemotherapy-induced nausea and vomiting. Cancer Treat Rep (1985) 69:109–12.
59. Volkow ND, Fowler JS, Wolf AP, Gillespi H. Metabolic studies of drugs of abuse. NIDA Res Monogr (1990) 105:47–53.
60. Marinol product monograph (Roxane—US), Rev 6/96. In: PDR Physicians’ Desk Reference. 52nd ed. Montvale, NJ: Medical Economics Company (1998). p. 2544–6.
61. Leweke FM, Schneider U, Thies M, Munte TF, Emrich HM. Effects of synthetic delta9-tetrahydrocannabinol on binocular depth inversion of natural and artificial objects in man. Psychopharmacology (Berl) (1999) 142:230–5. doi:10.1007/s002130050884
62. Wesnes K, Annas P, Edgar C, Deeprose C, Karlsten R, Philipp A, et al. Nabilone produces marked impairments to cognitive function and changes in subjective state in healthy volunteers. J Psychopharmacol (2009) 24(11):1659–69. doi:10.1177/0269881109105900
63. Machado Rocha FC, Stefano SC, De Cassia Haiek R, Rosa Oliveira LM, Da Silveira DX. Therapeutic use of Cannabis sativa on chemotherapy-induced nausea and vomiting among cancer patients: systematic review and meta-analysis. Eur J Cancer Care (Engl) (2008) 17:431–43. doi:10.1111/j.1365-2354.2008.00917.x
64. Tramer MR, Carroll D, Campbell FA, Reynolds DJ, Moore RA, McQuay HJ. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ (2001) 323:16–21. doi:10.1136/bmj.323.7303.16
65. Auwarter V, Dresen S, Weinmann W, Müller M, Putz M, Ferreiros N. “Spice” and other herbal blends: harmless incense or cannabinoid designer drugs? J Mass Spectrom (2009) 44:832–7. doi:10.1002/jms.1558
66. Uchiyama N, Kikura-Hanajiri R, Kawahara N, Haishima Y, Goda Y. Identification of a cannabinoid analog as a new type of designer drug in a herbal product. Chem Pharm Bull (Tokyo) (2009) 57:439–41. doi:10.1248/cpb.57.439
67. Dresen S, Ferreiros N, Putz M, Westphal F, Zimmermann R, Auwarter V. Monitoring of herbal mixtures potentially containing synthetic cannabinoids as psychoactive compounds. J Mass Spectrom (2010) 45:1186–94. doi:10.1002/jms.1811
68. Uchiyama N, Kikura-Hanajiri R, Ogata J, Goda Y. Chemical analysis of synthetic cannabinoids as designer drugs in herbal products. Forensic Sci Int (2010) 198:31–8. doi:10.1016/j.forsciint.2010.01.004
69. Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) (2011) 49:760–4. doi:10.3109/15563650.2011.609822
70. Schneir AB, Cullen J, Ly BT. “Spice” girls: synthetic cannabinoid intoxication. J Emerg Med (2011) 40:296–9. doi:10.1016/j.jemermed.2010.10.014
71. Uchiyama N, Kikura-Hanajiri R, Goda Y. Identification of a novel cannabimimetic phenylacetylindole, cannabipiperidiethanone, as a designer drug in a herbal product and its affinity for cannabinoid CB and CB receptors. Chem Pharm Bull (Tokyo) (2011) 59:1203–5. doi:10.1248/cpb.59.1203
72. RTL. Synthetic Cannabinoid Urine Test (2012). Available from: http://www.redwoodtoxicology.com/documents/services/3369_jwh_sellsheet.pdf
73. Seely KA, Lapoint J, Moran JH, Fattore L. Spice drugs are more than harmless herbal blends: a review of the pharmacology and toxicology of synthetic cannabinoids. Prog Neuropsychopharmacol Biol Psychiatry (2012) 39:234–43. doi:10.1016/j.pnpbp.2012.04.017
74. Müller H, Huttner HB, Kohrmann M, Wielopolski JE, Kornhuber J, Sperling W. Panic attack after spice abuse in a patient with ADHD. Pharmacopsychiatry (2010) 43:152–3. doi:10.1055/s-0029-1243252
75. American Association of Poison Control Centers. (2011). Synthetic Marijuana. Available from: http://www.aapcc.org/alerts/synthetic-marijuana/
82. Vearrier D, Osterhoudt KC. A teenager with agitation: higher than she should have climbed. Pediatr Emerg Care (2010) 26:462–5. doi:10.1097/PEC.0b013e3181e4f416
83. Benford DM, Caplan JP. Psychiatric sequelae of Spice, K2, and synthetic cannabinoid receptor agonists. Psychosomatics (2011) 52:295. doi:10.1016/j.psym.2011.01.004
84. Mir A, Obafemi A, Young A, Kane C. Myocardial infarction associated with use of the synthetic cannabinoid K2. Pediatrics (2011) 128:e1622–7. doi:10.1542/peds.2010-3823
85. Every-Palmer S. Warning: legal synthetic cannabinoid-receptor agonists such as JWH-018 may precipitate psychosis in vulnerable individuals. Addiction (2010) 105:1859–60. doi:10.1111/j.1360-0443.2010.03119.x
86. Müller H, Sperling W, Kohrmann M, Huttner HB, Kornhuber J, Maler JM. The synthetic cannabinoid Spice as a trigger for an acute exacerbation of cannabis induced recurrent psychotic episodes. Schizophr Res (2010) 118:309–10. doi:10.1016/j.schres.2009.12.001
87. Hurst D, Loeffler G, McLay R. Psychosis associated with synthetic cannabinoid agonists: a case series. Am J Psychiatry (2011) 168:1119. doi:10.1176/appi.ajp.2011.11010176
88. Cohen J, Morrison S, Greenberg J, Saidinejad M. Clinical presentation of intoxication due to synthetic cannabinoids. Pediatrics (2012) 129(4):1064–7. doi:10.1542/peds.2011-1797
89. D’Souza DC, Perry E, Macdougall L, Ammerman Y, Cooper T, Wu YT, et al. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology (2004) 29:1558–72. doi:10.1038/sj.npp.1300496
90. Fusar-Poli P, Crippa JA, Bhattacharyya S, Borgwardt SJ, Allen P, Martin-Santos R, et al. Distinct effects of {delta}9-tetrahydrocannabinol and cannabidiol on neural activation during emotional processing. Arch Gen Psychiatry (2009) 66:95–105. doi:10.1001/archgenpsychiatry.2008.519
91. Bhattacharyya S, Morrison PD, Fusar-Poli P, Martin-Santos R, Borgwardt S, Winton-Brown T, et al. Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology (2010) 35:764–74. doi:10.1038/npp.2009.184
92. Englund A, Morrison PD, Nottage J, Hague D, Kane F, Bonaccorso S, et al. Cannabidiol inhibits THC-elicited paranoid symptoms and hippocampal-dependent memory impairment. J Psychopharmacol (2013) 27:19–27. doi:10.1177/0269881112460109
93. Juckel G, Roser P, Nadulski T, Stadelmann AM, Gallinat J. Acute effects of Delta9-tetrahydrocannabinol and standardized cannabis extract on the auditory evoked mismatch negativity. Schizophr Res (2007) 97:109–17. doi:10.1016/j.schres.2007.08.015
94. Kaufmann RM, Kraft B, Frey R, Winkler D, Weiszenbichler S, Backer C, et al. Acute psychotropic effects of oral cannabis extract with a defined content of Delta9-tetrahydrocannabinol (THC) in healthy volunteers. Pharmacopsychiatry (2010) 43:24–32. doi:10.1055/s-0029-1237397
95. Morrison PD, Zois V, McKeown DA, Lee TD, Holt DW, Powell JF, et al. The acute effects of synthetic intravenous Delta9-tetrahydrocannabinol on psychosis, mood and cognitive functioning. Psychol Med (2009) 39:1607–16. doi:10.1017/S0033291709005522
96. Barkus E, Morrison PD, Vuletic D, Dickson JC, Ell PJ, Pilowsky LS, et al. Does intravenous Delta9-tetrahydrocannabinol increase dopamine release? A SPET study. J Psychopharmacol (2011) 25:1462–8. doi:10.1177/0269881110382465
97. Morrison PD, Stone JM. Synthetic delta-9-tetrahydrocannabinol elicits schizophrenia-like negative symptoms which are distinct from sedation. Hum Psychopharmacol (2011) 26:77–80. doi:10.1002/hup.1166
98. Bhattacharyya S, Crippa JA, Allen P, Martin-Santos R, Borgwardt S, Fusar-Poli P, et al. Induction of psychosis by Delta9-tetrahydrocannabinol reflects modulation of prefrontal and striatal function during attentional salience processing. Arch Gen Psychiatry (2012) 69:27–36. doi:10.1001/archgenpsychiatry.2011.161
99. Martin-Santos R, Crippa JA, Batalla A, Bhattacharyya S, Atakan Z, Borgwardt S, et al. Acute effects of a single, oral dose of d9-tetrahydrocannabinol (THC) and cannabidiol (CBD) administration in healthy volunteers. Curr Pharm Des (2012) 18:4966–79. doi:10.2174/138161212802884780
100. Chen J, Paredes W, Lowinson JH, Gardner EL. Delta 9-tetrahydrocannabinol enhances presynaptic dopamine efflux in medial prefrontal cortex. Eur J Pharmacol (1990) 190:259–62. doi:10.1016/0014-2999(90)94136-L
101. Kleinloog D, Liem-Moolenaar M, Jacobs G, Klaassen E, De Kam M, Hijman R, et al. Does olanzapine inhibit the psychomimetic effects of Delta(9)-tetrahydrocannabinol? J Psychopharmacol (2012) 26:1307–16. doi:10.1177/0269881112446534
102. Liem-Moolenaar M, Te Beek ET, De Kam ML, Franson KL, Kahn RS, Hijman R, et al. Central nervous system effects of haloperidol on THC in healthy male volunteers. J Psychopharmacol (2010) 24:1697–708. doi:10.1177/0269881109358200
103. D’Souza DC, Braley G, Blaise R, Vendetti M, Oliver S, Pittman B, et al. Effects of haloperidol on the behavioral, subjective, cognitive, motor, and neuroendocrine effects of Delta-9-tetrahydrocannabinol in humans. Psychopharmacology (Berl) (2008) 198:587–603. doi:10.1007/s00213-007-1042-2
104. D’Souza DC, Abi-Saab WM, Madonick S, Forselius-Bielen K, Doersch A, Braley G, et al. Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biol Psychiatry (2005) 57:594–608. doi:10.1016/j.biopsych.2004.12.006
105. Tysk L. Time estimation by healthy subjects and schizophrenic patients: a methodological study. Percept Mot Skills (1983) 56:983–8. doi:10.2466/pms.1983.56.3.983
106. Andreasen NC, Nopoulos P, O’Leary DS, Miller DD, Wassink T, Flaum M. Defining the phenotype of schizophrenia: cognitive dysmetria and its neural mechanisms. Biol Psychiatry (1999) 46:908–20. doi:10.1016/S0006-3223(99)00152-3
107. Davalos DB, Kisley MA, Ross RG. Effects of interval duration on temporal processing in schizophrenia. Brain Cogn (2003) 52:295–301. doi:10.1016/S0278-2626(03)00157-X
108. Carroll CA, O’Donnell BF, Shekhar A, Hetrick WP. Timing dysfunctions in schizophrenia span from millisecond to several-second durations. Brain Cogn (2009) 70:181–90. doi:10.1016/j.bandc.2009.02.001
109. Conrad DG, Elsmore TF, Sodetz FJ. 9-Tetrahydrocannabinol: dose-related effects on timing behavior in chimpanzee. Science (1972) 175:547–50. doi:10.1126/science.175.4021.547
110. Schulze GE, McMillan DE, Bailey JR, Scallet A, Ali SF, Slikker W Jr, et al. Acute effects of delta-9-tetrahydrocannabinol in rhesus monkeys as measured by performance in a battery of complex operant tests. J Pharmacol Exp Ther (1988) 245:178–86.
111. McClure GY, McMillan DE. Effects of drugs on response duration differentiation. VI: differential effects under differential reinforcement of low rates of responding schedules. J Pharmacol Exp Ther (1997) 281:1368–80.
112. Han CJ, Robinson JK. Cannabinoid modulation of time estimation in the rat. Behav Neurosci (2001) 115:243–6. doi:10.1037/0735-7044.115.1.243
113. Tinklenberg JR, Kopell BS, Melges FT, Hollister LE. Marihuana and alcohol, time production and memory functions. Arch Gen Psychiatry (1972) 27:812–5. doi:10.1001/archpsyc.1972.01750300074013
114. Hicks RE, Gualtieri CT, Mayo JP Jr, Perez-Reyes M. Cannabis, atropine, and temporal information processing. Neuropsychobiology (1984) 12:229–37. doi:10.1159/000118144
115. Mathew RJ, Wilson WH, Turkington TG, Coleman RE. Cerebellar activity and disturbed time sense after THC. Brain Res (1998) 797:183–9. doi:10.1016/S0006-8993(98)00375-8
116. McDonald J, Schleifer L, Richards JB, De Wit H. Effects of THC on behavioral measures of impulsivity in humans. Neuropsychopharmacology (2003) 28:1356–65. doi:10.1038/sj.npp.1300176
117. Stone JM, Morrison PD, Nottage J, Bhattacharyya S, Feilding A, McGuire PK. Delta-9-tetrahydrocannabinol disruption of time perception and of self-timed actions. Pharmacopsychiatry (2010) 43:236–7. doi:10.1055/s-0030-1255030
118. Sewell RA, Schnakenberg A, Elander J, Radhakrishnan R, Williams A, Skosnik PD, et al. Acute effects of THC on time perception in frequent and infrequent cannabis users. Psychopharmacology (Berl) (2012) 226(2):401–13. doi:10.1007/s00213-012-2915-6
119. Koethe D, Gerth CW, Neatby MA, Haensel A, Thies M, Schneider U, et al. Disturbances of visual information processing in early states of psychosis and experimental delta-9-tetrahydrocannabinol altered states of consciousness. Schizophr Res (2006) 88:142–50. doi:10.1016/j.schres.2006.07.023
120. Emrich HM, Weber MM, Wendl A, Zihl J, Von Meyer L, Hanisch W. Reduced binocular depth inversion as an indicator of cannabis-induced censorship impairment. Pharmacol Biochem Behav (1991) 40:689–90. doi:10.1016/0091-3057(91)90383-D
121. Leweke FM, Schneider U, Radwan M, Schmidt E, Emrich HM. Different effects of nabilone and cannabidiol on binocular depth inversion in man. Pharmacol Biochem Behav (2000) 66:175–81. doi:10.1016/S0091-3057(00)00201-X
122. Semple DM, Ramsden F, McIntosh AM. Reduced binocular depth inversion in regular cannabis users. Pharmacol Biochem Behav (2003) 75:789–93. doi:10.1016/S0091-3057(03)00140-0
123. Miller LL, McFarland D, Cornett TL, Brightwell D. Marijuana and memory impairment: effect on free recall and recognition memory. Pharmacol Biochem Behav (1977) 7:99–103. doi:10.1016/0091-3057(77)90191-5
124. Hooker WD, Jones RT. Increased susceptibility to memory intrusions and the Stroop interference effect during acute marijuana intoxication. Psychopharmacology (Berl) (1987) 91:20–4. doi:10.1007/BF00690920
125. Marks DF, MacAvoy MG. Divided attention performance in cannabis users and non-users following alcohol and cannabis separately and in combination. Psychopharmacology (Berl) (1989) 99:397–401. doi:10.1007/BF00445566
126. Heishman SJ, Huestis MA, Henningfield JE, Cone EJ. Acute and residual effects of marijuana: profiles of plasma THC levels, physiological, subjective, and performance measures. Pharmacol Biochem Behav (1990) 37:561–5. doi:10.1016/0091-3057(90)90028-G
127. Leweke M, Kampmann C, Radwan M, Dietrich DE, Johannes S, Emrich HM, et al. The effects of tetrahydrocannabinol on the recognition of emotionally charged words: an analysis using event-related brain potentials. Neuropsychobiology (1998) 37:104–11. doi:10.1159/000026487
128. Hart CL, Van Gorp W, Haney M, Foltin RW, Fischman MW. Effects of acute smoked marijuana on complex cognitive performance. Neuropsychopharmacology (2001) 25:757–65. doi:10.1016/S0893-133X(01)00273-1
129. Ranganathan M, D’Souza DC. The acute effects of cannabinoids on memory in humans: a review. Psychopharmacology (Berl) (2006) 188:425–44. doi:10.1007/s00213-006-0508-y
130. Lichtman AH, Varvel SA, Martin BR. Endocannabinoids in cognition and dependence. Prostaglandins Leukot Essent Fatty Acids (2002) 66:269–85. doi:10.1054/plef.2001.0351
131. Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science (2002) 296:678–82. doi:10.1126/science.1063545
132. Heinrichs RW, Zakzanis KK. Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology (1998) 12:426–45. doi:10.1037/0894-4105.12.3.426
133. Henquet C, Rosa A, Krabbendam L, Papiol S, Fananas L, Drukker M, et al. An experimental study of catechol-o-methyltransferase Val158Met moderation of delta-9-tetrahydrocannabinol-induced effects on psychosis and cognition. Neuropsychopharmacology (2006) 31:2748–57. doi:10.1038/sj.npp.1301197
134. Luck SJ, Mathalon DH, O’Donnell BF, Hamalainen MS, Spencer KM, Javitt DC, et al. A roadmap for the development and validation of event-related potential biomarkers in schizophrenia research. Biol Psychiatry (2010) 70(1):28–34. doi:10.1016/j.biopsych.2010.09.021
135. Polich J, Criado JR. Neuropsychology and neuropharmacology of P3a and P3b. Int J Psychophysiol (2006) 60:172–85. doi:10.1016/j.ijpsycho.2005.12.012
136. Bramon E, Rabe-Hesketh S, Sham P, Murray RM, Frangou S. Meta-analysis of the P300 and P50 waveforms in schizophrenia. Schizophr Res (2004) 70:315–29. doi:10.1016/j.schres.2004.01.004
137. de Wilde OM, Bour LJ, Dingemans PM, Koelman JH, Linszen DH. A meta-analysis of P50 studies in patients with schizophrenia and relatives: differences in methodology between research groups. Schizophr Res (2007) 97:137–51. doi:10.1016/j.schres.2007.04.028
138. Rentzsch J, Penzhorn A, Kernbichler K, Plockl D, Gomez-Carrillo De Castro A, et al. Differential impact of heavy cannabis use on sensory gating in schizophrenic patients and otherwise healthy controls. Exp Neurol (2007) 205(1):241–9. doi:10.1016/j.expneurol.2007.02.004
139. Solowij N, Michie PT. Cannabis and cognitive dysfunction: parallels with endophenotypes of schizophrenia? J Psychiatry Neurosci (2007) 32:30–52.
140. Patterson JV, Hetrick WP, Boutros NN, Jin Y, Sandman C, Stern H, et al. P50 sensory gating ratios in schizophrenics and controls: a review and data analysis. Psychiatry Res (2008) 158:226–47. doi:10.1016/j.psychres.2007.02.009
141. Dissanayake DW, Zachariou M, Marsden CA, Mason R. Auditory gating in rat hippocampus and medial prefrontal cortex: effect of the cannabinoid agonist WIN55,212-2. Neuropharmacology (2008) 55:1397–404. doi:10.1016/j.neuropharm.2008.08.039
142. Zachariou M, Dissanayake DW, Coombes S, Owen MR, Mason R. Sensory gating and its modulation by cannabinoids: electrophysiological, computational and mathematical analysis. Cogn Neurodyn (2008) 2:159–70. doi:10.1007/s11571-008-9050-4
143. Patrick G, Straumanis JJ, Struve FA, Fitz-Gerald MJ, Leavitt J, Manno JE. Reduced P50 auditory gating response in psychiatrically normal chronic marihuana users: a pilot study. Biol Psychiatry (1999) 45:1307–12. doi:10.1016/S0006-3223(98)00155-3
144. Patrick G, Struve FA. Reduction of auditory P50 gating response in marihuana users: further supporting data. Clin Electroencephalogr (2000) 31:88–93.
145. Edwards CR, Skosnik PD, Steinmetz AB, O’Donnell BF, Hetrick WP. Sensory gating impairments in heavy cannabis users are associated with altered neural oscillations. Behav Neurosci (2009) 123:894–904. doi:10.1037/a0016328
146. Roth WT, Cannon EH. Some features of the auditory evoked response in schizophrenics. Arch Gen Psychiatry (1972) 27:466–71. doi:10.1001/archpsyc.1972.01750280034007
147. Duncan CC. Event-related brain potentials: a window on information processing in schizophrenia. Schizophr Bull (1988) 14:199–203. doi:10.1093/schbul/14.2.199
148. Braff DL. Information processing and attention dysfunctions in schizophrenia. Schizophr Bull (1993) 19:233–59. doi:10.1093/schbul/19.2.233
149. Turetsky BI, Colbath EA, Gur RE. P300 subcomponent abnormalities in schizophrenia: I. Physiological evidence for gender and subtype specific differences in regional pathology. Biol Psychiatry (1998) 43:84–96. doi:10.1016/S0006-3223(97)00258-8
150. Jeon YW, Polich J. Meta-analysis of P300 and schizophrenia: patients, paradigms, and practical implications. Psychophysiology (2003) 40:684–701. doi:10.1111/1469-8986.00070
151. Bramon E, McDonald C, Croft RJ, Landau S, Filbey F, Gruzelier JH, et al. Is the P300 wave an endophenotype for schizophrenia? A meta-analysis and a family study. Neuroimage (2005) 27:960–8. doi:10.1016/j.neuroimage.2005.05.022
152. Turetsky BI, Calkins ME, Light GA, Olincy A, Radant AD, Swerdlow NR. Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr Bull (2007) 33:69–94. doi:10.1093/schbul/sbl060
153. Ilan AB, Gevins A, Coleman M, Elsohly MA, De Wit H. Neurophysiological and subjective profile of marijuana with varying concentrations of cannabinoids. Behav Pharmacol (2005) 16:487–96. doi:10.1097/00008877-200509000-00023
154. Roser P, Juckel G, Rentzsch J, Nadulski T, Gallinat J, Stadelmann AM. Effects of acute oral Delta9-tetrahydrocannabinol and standardized cannabis extract on the auditory P300 event-related potential in healthy volunteers. Eur Neuropsychopharmacol (2008) 18:569–77. doi:10.1016/j.euroneuro.2008.04.008
155. D’Souza DC, Fridberg DJ, Skosnik PD, Williams A, Roach B, Singh N, et al. Dose-related modulation of event-related potentials to novel and target stimuli by intravenous Delta(9)-THC in humans. Neuropsychopharmacology (2012) 37:1632–46. doi:10.1038/npp.2012.8
156. Solowij N, Michie PT, Fox AM. Effects of long-term cannabis use on selective attention: an event-related potential study. Pharmacol Biochem Behav (1991) 40:683–8. doi:10.1016/0091-3057(91)90382-C
157. Solowij N, Michie PT, Fox AM. Differential impairments of selective attention due to frequency and duration of cannabis use. Biol Psychiatry (1995) 37:731–9. doi:10.1016/0006-3223(94)00178-6
158. Skosnik PD, Park S, Dobbs L, Gardner WL. Affect processing and positive syndrome schizotypy in cannabis users. Psychiatry Res (2008) 157:279–82. doi:10.1016/j.psychres.2007.02.010
159. de Sola S, Tarancon T, Pena-Casanova J, Espadaler JM, Langohr K, Poudevida S, et al. Auditory event-related potentials (P3) and cognitive performance in recreational ecstasy polydrug users: evidence from a 12-month longitudinal study. Psychopharmacology (Berl) (2008) 200:425–37. doi:10.1007/s00213-008-1217-5
160. Kempel P, Lampe K, Parnefjord R, Hennig J, Kunert HJ. Auditory-evoked potentials and selective attention: different ways of information processing in cannabis users and controls. Neuropsychobiology (2003) 48:95–101. doi:10.1159/000072884
161. Naatanen R, Alho K. Generators of electrical and magnetic mismatch responses in humans. Brain Topogr (1995) 7:315–20. doi:10.1007/BF01195257
162. Rinne T, Alho K, Ilmoniemi RJ, Virtanen J, Naatanen R. Separate time behaviors of the temporal and frontal mismatch negativity sources. Neuroimage (2000) 12:14–9. doi:10.1006/nimg.2000.0591
163. Pesa N, Hermens DF, Battisti RA, Kaur M, Hickie IB, Solowij N. Delayed preattentional functioning in early psychosis patients with cannabis use. Psychopharmacology (Berl) (2012) 222:507–18. doi:10.1007/s00213-012-2676-2
164. Perez VB, Woods SW, Roach BJ, Ford JM, McGlashan TH, Srihari VH, et al. Automatic auditory processing deficits in schizophrenia and clinical high-risk patients: forecasting psychosis risk with mismatch negativity. Biol Psychiatry (2013) 75(6):459–69. doi:10.1016/j.biopsych.2013.07.038
165. Roser P, Della B, Norra C, Uhl I, Brüne M, Juckel G. Auditory mismatch negativity deficits in long-term heavy cannabis users. Eur Arch Psychiatry Clin Neurosci (2010) 260(6):491–8. doi:10.1007/s00406-010-0097-y
166. Rentzsch J, Buntebart E, Stadelmeier A, Gallinat J, Jockers-Scherubl MC. Differential effects of chronic cannabis use on preattentional cognitive functioning in abstinent schizophrenic patients and healthy subjects. Schizophr Res (2011) 130:222–7. doi:10.1016/j.schres.2011.05.011
167. Greenwood LM, Broyd SJ, Croft R, Todd J, Michie PT, Johnstone S, et al. Chronic effects of cannabis use on the auditory mismatch negativity. Biol Psychiatry (2013) 75(6):449–58. doi:10.1016/j.biopsych.2013.05.035
168. Hajos M, Hoffmann WE, Kocsis B. Activation of cannabinoid-1 receptors disrupts sensory gating and neuronal oscillation: relevance to schizophrenia. Biol Psychiatry (2008) 63:1075–83. doi:10.1016/j.biopsych.2007.12.005
169. Bocker KB, Hunault CC, Gerritsen J, Kruidenier M, Mensinga TT, Kenemans JL. Cannabinoid modulations of resting state EEG theta power and working memory are correlated in humans. J Cogn Neurosci (2010) 22:1906–16. doi:10.1162/jocn.2009.21355
170. Skosnik PD, Krishnan GP, Vohs JL, O’Donnell BF. The effect of cannabis use and gender on the visual steady state evoked potential. Clin Neurophysiol (2006) 117:144–56. doi:10.1016/j.clinph.2005.09.024
171. Skosnik PD, D’Souza DC, Steinmetz AB, Edwards CR, Vollmer JM, Hetrick WP, et al. The effect of chronic cannabinoids on broadband EEG neural oscillations in humans. Neuropsychopharmacology (2012) 37:2184–93. doi:10.1038/npp.2012.65
172. Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci (2010) 11:100–13. doi:10.1038/nrn2774
173. Uhlhaas PJ, Roux F, Rodriguez E, Rotarska-Jagiela A, Singer W. Neural synchrony and the development of cortical networks. Trends Cogn Sci (2010) 14:72–80. doi:10.1016/j.tics.2009.12.002
174. Bernhardson G, Gunne LM. Forty-six cases of psychosis in cannabis abusers. Int J Addict (1972) 7:9–16.
175. Tennant FS Jr, Groesbeck CJ. Psychiatric effects of hashish. Arch Gen Psychiatry (1972) 27:133–6. doi:10.1001/archpsyc.1972.01750250117016
176. Harding T, Knight F. Marihuana-modified mania. Arch Gen Psychiatry (1973) 29:635–7. doi:10.1001/archpsyc.1973.04200050048008
177. Rottanburg D, Robins AH, Ben-Arie O, Teggin A, Elk R. Cannabis-associated psychosis with hypomanic features. Lancet (1982) 2:1364–6. doi:10.1016/S0140-6736(82)91270-3
178. Carney MW, Bacelle L, Robinson B. Psychosis after cannabis abuse. Br Med J (Clin Res Ed). (1984) 288:1047. doi:10.1136/bmj.288.6427.1381-a
179. Mathers DC, Ghodse AH, Caan AW, Scott SA. Cannabis use in a large sample of acute psychiatric admissions. Br J Addict (1991) 86:779–84. doi:10.1111/j.1360-0443.1991.tb03104.x
180. Wylie AS, Scott RT, Burnett SJ. Psychosis due to “skunk”. BMJ (1995) 311:125. doi:10.1136/bmj.311.6997.125
181. Arendt M, Rosenberg R, Foldager L, Perto G, Munk-Jorgensen P. Cannabis-induced psychosis and subsequent schizophrenia-spectrum disorders: follow-up study of 535 incident cases. Br J Psychiatry (2005) 187:510–5. doi:10.1192/bjp.187.6.510
182. Kolansky H, Moore WT. Effects of marihuana on adolescents and young adults. JAMA (1971) 216:486–92. doi:10.1001/jama.1971.03180290062009
183. Thacore VR, Shukla SR. Cannabis psychosis and paranoid schizophrenia. Arch Gen Psychiatry (1976) 33:383–6. doi:10.1001/archpsyc.1976.01770030081012
184. Chaudry HR, Moss HB, Bashir A, Suliman T. Cannabis psychosis following bhang ingestion. Br J Addict (1991) 86:1075–81. doi:10.1111/j.1360-0443.1991.tb01874.x
185. Cohen SI. Cannabis consumption and schizophrenia. Br J Psychiatry (1994) 165:410–1. doi:10.1192/bjp.165.3.410
186. Basu D, Malhotra A, Bhagat A, Varma VK. Cannabis psychosis and acute schizophrenia. a case-control study from India. Eur Addict Res (1999) 5:71–3. doi:10.1159/000018968
187. Hall W, Solowij N. Adverse effects of cannabis. Lancet (1998) 352:1611–6. doi:10.1016/S0140-6736(98)05021-1
188. Niemi-Pynttari JA, Sund R, Putkonen H, Vorma H, Wahlbeck K, Pirkola SP. Substance-induced psychoses converting into schizophrenia: a register-based study of 18,478 Finnish inpatient cases. J Clin Psychiatry (2013) 74:e94–9. doi:10.4088/JCP.12m07822
189. Pierre JM. Psychosis associated with medical marijuana: risk vs. benefits of medicinal cannabis use. Am J Psychiatry (2010) 167:598–9. doi:10.1176/appi.ajp.2010.09121762
190. Aggarwal M, Banerjee A, Singh SM, Mattoo SK, Basu D. Substance-induced psychotic disorders: 13-year data from a de-addiction centre and their clinical implications. Asian J Psychiatr (2012) 5:220–4. doi:10.1016/j.ajp.2011.11.008
191. Rounsaville BJ. DSM-V research agenda: substance abuse/psychosis comorbidity. Schizophr Bull (2007) 33:947–52. doi:10.1093/schbul/sbm054
192. Rolfe M, Tang CM, Sabally S, Todd JE, Sam EB, Hatib N’Jie AB. Psychosis and cannabis abuse in The Gambia. A case-control study. Br J Psychiatry (1993) 163:798–801. doi:10.1192/bjp.163.6.798
193. Degenhardt L, Hall W. The association between psychosis and problematical drug use among Australian adults: findings from the National Survey of Mental Health and Well-Being. Psychol Med (2001) 31:659–68. doi:10.1017/S0033291701003865
194. Degenhardt L, Hall W, Lynskey M. Alcohol, cannabis and tobacco use among Australians: a comparison of their associations with other drug use and use disorders, affective and anxiety disorders, and psychosis. Addiction (2001) 96:1603–14. doi:10.1046/j.1360-0443.2001.961116037.x
195. Agosti V, Nunes E, Levin F. Rates of psychiatric comorbidity among U.S. residents with lifetime cannabis dependence. Am J Drug Alcohol Abuse (2002) 28:643–52. doi:10.1081/ADA-120015873
196. Stefanis NC, Delespaul P, Henquet C, Bakoula C, Stefanis CN, Van Os J. Early adolescent cannabis exposure and positive and negative dimensions of psychosis. Addiction (2004) 99:1333–41. doi:10.1111/j.1360-0443.2004.00806.x
197. Andreasson S, Allebeck P, Engstrom A, Rydberg U. Cannabis and schizophrenia. A longitudinal study of Swedish conscripts. Lancet (1987) 2:1483–6. doi:10.1016/S0140-6736(87)92620-1
198. Zammit S, Allebeck P, Andreasson S, Lundberg I, Lewis G. Self reported cannabis use as a risk factor for schizophrenia in Swedish conscripts of 1969: historical cohort study. BMJ (2002) 325:1199. doi:10.1136/bmj.325.7374.1199
199. Manrique-Garcia E, Zammit S, Dalman C, Hemmingsson T, Andreasson S, Allebeck P. Cannabis, schizophrenia and other non-affective psychoses: 35 years of follow-up of a population-based cohort. Psychol Med (2012) 42:1321–8. doi:10.1017/s0033291711002078
200. Caspi A, Moffitt TE, Cannon M, McClay J, Murray R, Harrington H, et al. Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol Psychiatry (2005) 57:1117–27. doi:10.1016/j.biopsych.2005.01.026
201. Johnson BA, Smith BL, Taylor P. Cannabis and schizophrenia. Lancet (1988) 1:592–3. doi:10.1016/S0140-6736(88)91389-X
202. Negrete JC. Cannabis and schizophrenia. Br J Addict (1989) 84:349–51. doi:10.1111/j.1360-0443.1989.tb00576.x
203. Johnson BA. Psychopharmacological effects of cannabis. Br J Hosp Med (1990) 43:114–6, 118–20, 122.
204. Mirken B, Earleywine M. The cannabis and psychosis connection questioned: a comment on Fergusson et al. 2005. Addiction (2005) 100:714–5; author reply 715–6. doi:10.1111/j.1360-0443.2005.01117.x
205. Arseneault L, Cannon M, Witton J, Murray RM. Causal association between cannabis and psychosis: examination of the evidence. Br J Psychiatry (2004) 184:110–7. doi:10.1192/bjp.184.2.110
206. Macleod J, Oakes R, Copello A, Crome I, Egger M, Hickman M, et al. Psychological and social sequelae of cannabis and other illicit drug use by young people: a systematic review of longitudinal, general population studies. Lancet (2004) 363:1579–88. doi:10.1016/S0140-6736(04)16200-4
207. Henquet C, Murray R, Linszen D, Van Os J. The environment and schizophrenia: the role of cannabis use. Schizophr Bull (2005) 31:608–12. doi:10.1093/schbul/sbi027
208. Semple DM, McIntosh AM, Lawrie SM. Cannabis as a risk factor for psychosis: systematic review. J Psychopharmacol (2005) 19:187–94. doi:10.1177/0269881105049040
209. Minozzi S, Davoli M, Bargagli AM, Amato L, Vecchi S, Perucci CA. An overview of systematic reviews on cannabis and psychosis: discussing apparently conflicting results. Drug Alcohol Rev (2010) 29:304–17. doi:10.1111/j.1465-3362.2009.00132.x
210. Gage SH, Zammit S, Hickman M. Stronger evidence is needed before accepting that cannabis plays an important role in the aetiology of schizophrenia in the population. F1000 Med Rep (2013) 5:2. doi:10.3410/M5-2
211. Kolansky H, Moore WT. Effects of marihuana on adolescents and young adults. J Psychiatr Nurs Ment Health Serv (1971) 9:9–16.
212. Halikas J, Weller R, Morse C. Effects of regular marijuana use on sexual performance. J Psychoactive Drugs (1982) 14:59–70. doi:10.1080/02791072.1982.10471911
213. Millman RB, Sbriglio R. Patterns of use and psychopathology in chronic marijuana users. Psychiatr Clin North Am (1986) 9:533–45.
214. Rubin V, Comitas L. Ganja in Jamaica: A Medical Anthropological Study of Chronic Marihuana Use. The Hague: Mouton Publishers (1975).
216. Nunn JA, Rizza F, Peters ER. The incidence of schizotypy among cannabis and alcohol users. J Nerv Ment Dis (2001) 189:741–8. doi:10.1097/00005053-200111000-00002
217. Bersani G, Orlandi V, Kotzalidis G, Pancheri P. Cannabis and schizophrenia: impact on onset, course, psychopathology and outcomes. Eur Arch Psychiatry Clin Neurosci (2002) 252:86–92. doi:10.1007/s00406-002-0366-5
218. Compton MT, Furman AC, Kaslow NJ. Lower negative symptom scores among cannabis-dependent patients with schizophrenia-spectrum disorders: preliminary evidence from an African American first-episode sample. Schizophr Res (2004) 71:61–4. doi:10.1016/j.schres.2004.01.005
219. Pope HG Jr, Gruber AJ, Yurgelun-Todd D. The residual neuropsychological effects of cannabis: the current status of research. Drug Alcohol Depend (1995) 38:25–34. doi:10.1016/0376-8716(95)01145-O
220. Pope HG Jr, Yurgelun-Todd D. The residual cognitive effects of heavy marijuana use in college students. JAMA (1996) 275:521–7. doi:10.1001/jama.275.7.521
221. Pope HG Jr, Gruber AJ, Hudson JI, Huestis MA, Yurgelun-Todd D. Neuropsychological performance in long-term cannabis users. Arch Gen Psychiatry (2001) 58:909–15. doi:10.1001/archpsyc.58.10.909
222. Bolla KI, Brown K, Eldreth D, Tate K, Cadet JL. Dose-related neurocognitive effects of marijuana use. Neurology (2002) 59:1337–43. doi:10.1212/01.WNL.0000031422.66442.49
223. Lundqvist T. Cognitive consequences of cannabis use: comparison with abuse of stimulants and heroin with regard to attention, memory and executive functions. Pharmacol Biochem Behav (2005) 81:319–30. doi:10.1016/j.pbb.2005.02.017
224. Solowij N, Battisti R. The chronic effects of cannabis on memory in humans: a review. Curr Drug Abuse Rev (2008) 1:81–98. doi:10.2174/1874473710801010081
225. Crean RD, Crane NA, Mason BJ. An evidence based review of acute and long-term effects of cannabis use on executive cognitive functions. J Addict Med (2011) 5:1–8. doi:10.1097/ADM.0b013e31820c23fa
226. Fontes MA, Bolla KI, Cunha PJ, Almeida PP, Jungerman F, Laranjeira RR, et al. Cannabis use before age 15 and subsequent executive functioning. Br J Psychiatry (2011) 198:442–7. doi:10.1192/bjp.bp.110.077479
227. Addy PH, Radhakrishnan R, Cortes JA, D’Souza DC. Comorbid alcohol, cannabis, and cocaine use disorders in schizophrenia: epidemiology, consequences, mechanisms, and treatment. Focus (2012) 2:140–53. doi:10.1176/appi.focus.10.2.140
228. Pope HG Jr, Gruber AJ, Yurgelun-Todd D. Residual neuropsychologic effects of cannabis. Curr Psychiatry Rep (2001) 3:507–12. doi:10.1007/s11920-001-0045-7
229. Jager G, Kahn RS, Van Den Brink W, Van Ree JM, Ramsey NF. Long-term effects of frequent cannabis use on working memory and attention: an fMRI study. Psychopharmacology (Berl) (2006) 185:358–68. doi:10.1007/s00213-005-0298-7
230. Fried PA, Watkinson B, Gray R. Neurocognitive consequences of marihuana – a comparison with pre-drug performance. Neurotoxicol Teratol (2005) 27:231–9. doi:10.1016/j.ntt.2004.11.003
231. Solowij N. Do cognitive impairments recover following cessation of cannabis use? Life Sci (1995) 56:2119–26. doi:10.1016/0024-3205(95)00197-E
232. Pope HG Jr. Cannabis, cognition, and residual confounding. JAMA (2002) 287:1172–4. doi:10.1001/jama.287.9.1172
233. Pope HG Jr, Gruber AJ, Hudson JI, Huestis MA, Yurgelun-Todd D. Cognitive measures in long-term cannabis users. J Clin Pharmacol (2002) 42:41S–7S. doi:10.1002/j.1552-4604.2002.tb06002.x
234. Pope HG Jr, Gruber AJ, Hudson JI, Cohane G, Huestis MA, Yurgelun-Todd D. Early-onset cannabis use and cognitive deficits: what is the nature of the association? Drug Alcohol Depend (2003) 69:303–10. doi:10.1016/S0376-8716(02)00334-4
235. Lyons MJ, Bar JL, Panizzon MS, Toomey R, Eisen S, Xian H, et al. Neuropsychological consequences of regular marijuana use: a twin study. Psychol Med (2004) 34:1239–50. doi:10.1017/S0033291704002260
236. Verdejo-Garcia AJ, Lopez-Torrecillas F, Aguilar de Arcos F, Perez-Garcia M. Differential effects of MDMA, cocaine, and cannabis use severity on distinctive components of the executive functions in polysubstance users: a multiple regression analysis. Addict Behav (2005) 30:89–101. doi:10.1016/j.addbeh.2004.04.015
237. Verdejo-Garcia A, Bechara A, Recknor EC, Perez-Garcia M. Executive dysfunction in substance dependent individuals during drug use and abstinence: an examination of the behavioral, cognitive and emotional correlates of addiction. J Int Neuropsychol Soc (2006) 12:405–15. doi:10.1017/S1355617706060486
238. Stefanis C, Liakos A, Boulougouris J, Fink M, Freedman AM. Chronic hashish use and mental disorder. Am J Psychiatry (1976) 133:225–7.
239. Wig NN, Varma VK. Patterns of long-term heavy cannabis use in north India and its effects on cognitive functions: a preliminary report. Drug Alcohol Depend (1977) 2:211–9. doi:10.1016/0376-8716(77)90028-X
240. Culver CM, King FW. Neuropsychological assessment of undergraduate marihuana and LSD users. Arch Gen Psychiatry (1974) 31:707–11. doi:10.1001/archpsyc.1974.01760170093015
241. Carlin AS, Trupin EW. The effect of long-term chronic marijuana use on neuropsychological functioning. Int J Addict (1977) 12:617–24.
242. Meier MH, Caspi A, Ambler A, Harrington H, Houts R, Keefe RS, et al. Persistent cannabis users show neuropsychological decline from childhood to midlife. Proc Natl Acad Sci U S A (2012) 109:E2657–64. doi:10.1073/pnas.1206820109
243. Potvin S, Joyal CC, Pelletier J, Stip E. Contradictory cognitive capacities among substance-abusing patients with schizophrenia: a meta-analysis. Schizophr Res (2008) 100:242–51. doi:10.1016/j.schres.2007.04.022
244. Rabin RA, Zakzanis KK, George TP. The effects of cannabis use on neurocognition in schizophrenia: a meta-analysis. Schizophr Res (2011) 128:111–6. doi:10.1016/j.schres.2011.02.017
245. Ralevski E, Gianoli M, Russo M, Dawn R, Radhakrishnan R. Cognitive impairment in schizophrenia and alcoholism: a review of the evidence and findings on the effects of pharmacological treatments on cognitive functioning in patients with dual diagnosis. J Dual Diagn (2012) 8:205–17. doi:10.1080/15504263.2012.696534
246. Yucel M, Bora E, Lubman DI, Solowij N, Brewer WJ, Cotton SM, et al. The impact of cannabis use on cognitive functioning in patients with schizophrenia: a meta-analysis of existing findings and new data in a first-episode sample. Schizophr Bull (2012) 38:316–30. doi:10.1093/schbul/sbq079
247. Ferraro L, Russo M, O’Connor J, Wiffen BD, Falcone MA, Sideli L, et al. Cannabis users have higher premorbid IQ than other patients with first onset psychosis. Schizophr Res (2013) 150:129–35. doi:10.1016/j.schres.2013.07.046
248. Rabin RA, Zakzanis KK, Daskalakis ZJ, George TP. Effects of cannabis use status on cognitive function, in males with schizophrenia. Psychiatry Res (2013) 206:158–65. doi:10.1016/j.psychres.2012.11.019
249. Sanchez-Torres AM, Basterra V, Rosa A, Fananas L, Zarzuela A, Ibanez B, et al. Lifetime cannabis use and cognition in patients with schizophrenia spectrum disorders and their unaffected siblings. Eur Arch Psychiatry Clin Neurosci (2013) 263(8):643–53. doi:10.1007/s00406-013-0404-5
250. Dragt S, Nieman DH, Schultze-Lutter F, Van Der Meer F, Becker H, De Haan L, et al. Cannabis use and age at onset of symptoms in subjects at clinical high risk for psychosis. Acta Psychiatr Scand (2012) 125:45–53. doi:10.1111/j.1600-0447.2011.01763.x
251. Schimmelmann BG, Conus P, Cotton SM, Kupferschmid S, Karow A, Schultze-Lutter F, et al. Cannabis use disorder and age at onset of psychosis – a study in first-episode patients. Schizophr Res (2011) 129:52–6. doi:10.1016/j.schres.2011.03.023
252. Large M, Sharma S, Compton MT, Slade T, Nielssen O. Cannabis use and earlier onset of psychosis: a systematic meta-analysis. Arch Gen Psychiatry (2011) 68:555–61. doi:10.1001/archgenpsychiatry.2011.5
253. O’Shea M, Singh ME, McGregor IS, Mallet PE. Chronic cannabinoid exposure produces lasting memory impairment and increased anxiety in adolescent but not adult rats. J Psychopharmacol (2004) 18:502–8. doi:10.1177/0269881104047277
254. Cha YM, White AM, Kuhn CM, Wilson WA, Swartzwelder HS. Differential effects of delta9-THC on learning in adolescent and adult rats. Pharmacol Biochem Behav (2006) 83:448–55. doi:10.1016/j.pbb.2006.03.006
255. Quinn HR, Matsumoto I, Callaghan PD, Long LE, Arnold JC, Gunasekaran N, et al. Adolescent rats find repeated Delta(9)-THC less aversive than adult rats but display greater residual cognitive deficits and changes in hippocampal protein expression following exposure. Neuropsychopharmacology (2008) 33:1113–26. doi:10.1038/sj.npp.1301475
256. Schneider M. Puberty as a highly vulnerable developmental period for the consequences of cannabis exposure. Addict Biol (2008) 13:253–63. doi:10.1111/j.1369-1600.2008.00110.x
257. Schneider M, Schomig E, Leweke FM. Acute and chronic cannabinoid treatment differentially affects recognition memory and social behavior in pubertal and adult rats. Addict Biol (2008) 13:345–57. doi:10.1111/j.1369-1600.2008.00117.x
258. Gonzalez-Pinto A, Alberich S, Barbeito S, Gutierrez M, Vega P, Ibanez B, et al. Cannabis and first-episode psychosis: different long-term outcomes depending on continued or discontinued use. Schizophr Bull (2011) 37:631–9. doi:10.1093/schbul/sbp126
259. Berghuis P, Dobszay MB, Wang X, Spano S, Ledda F, Sousa KM, et al. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc Natl Acad Sci U S A (2005) 102:19115–20. doi:10.1073/pnas.0509494102
260. Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urban GM, et al. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science (2007) 316:1212–6. doi:10.1126/science.1137406
261. Harkany T, Guzman M, Galve-Roperh I, Berghuis P, Devi LA, Mackie K. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol Sci (2007) 28:83–92. doi:10.1016/j.tips.2006.12.004
262. Mulder J, Aguado T, Keimpema E, Barabas K, Ballester Rosado CJ, Nguyen L, et al. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc Natl Acad Sci U S A (2008) 105:8760–5. doi:10.1073/pnas.0803545105
263. Gaffuri AL, Ladarre D, Lenkei Z. Type-1 cannabinoid receptor signaling in neuronal development. Pharmacology (2012) 90:19–39. doi:10.1159/000339075
264. Wenner P. The effects of endocannabinoid signaling on network activity in developing and motor circuits. Ann N Y Acad Sci (2013) 1279:135–42. doi:10.1111/nyas.12068
265. Stefanis NC, Dragovic M, Power BD, Jablensky A, Castle D, Morgan VA. Age at initiation of cannabis use predicts age at onset of psychosis: the 7- to 8-year trend. Schizophr Bull (2013) 39:251–4. doi:10.1093/schbul/sbs188
266. Degenhardt L, Hall W, Lynskey M. Testing hypotheses about the relationship between cannabis use and psychosis. Drug Alcohol Depend (2003) 71:37–48. doi:10.1016/S0376-8716(03)00064-4
267. Cunha PJ, Rosa PG, Ayres Ade M, Duran FL, Santos LC, Scazufca M, et al. Cannabis use, cognition and brain structure in first-episode psychosis. Schizophr Res (2013) 147:209–15. doi:10.1016/j.schres.2013.04.009
268. Keshavan MS, Kulkarni S, Bhojraj T, Francis A, Diwadkar V, Montrose DM, et al. Premorbid cognitive deficits in young relatives of schizophrenia patients. Front Hum Neurosci (2010) 3:62. doi:10.3389/neuro.09.062.2009
269. Brewer WJ, Francey SM, Wood SJ, Jackson HJ, Pantelis C, Phillips LJ, et al. Memory impairments identified in people at ultra-high risk for psychosis who later develop first-episode psychosis. Am J Psychiatry (2005) 162:71–8. doi:10.1176/appi.ajp.162.1.71
270. Rogosch FA, Oshri A, Cicchetti D. From child maltreatment to adolescent cannabis abuse and dependence: a developmental cascade model. Dev Psychopathol (2010) 22:883–97. doi:10.1017/s0954579410000520
271. Griffith-Lendering MF, Huijbregts SC, Mooijaart A, Vollebergh WA, Swaab H. Cannabis use and development of externalizing and internalizing behaviour problems in early adolescence: a TRAILS study. Drug Alcohol Depend (2011) 116:11–7. doi:10.1016/j.drugalcdep.2010.11.024
272. McGuire P, Jones P, Harvey I, Williams M, McGuffin P, Murray R. Morbid risk of schizophrenia for relatives of patients with cannabis-associated psychosis. Schizophr Res (1995) 15:277–81. doi:10.1016/0920-9964(94)00053-B
273. Arendt M, Mortensen PB, Rosenberg R, Pedersen CB, Waltoft BL. Familial predisposition for psychiatric disorder: comparison of subjects treated for cannabis-induced psychosis and schizophrenia. Arch Gen Psychiatry (2008) 65:1269–74. doi:10.1001/archpsyc.65.11.1269
274. Boydell J, Dean K, Dutta R, Giouroukou E, Fearon P, Murray R. A comparison of symptoms and family history in schizophrenia with and without prior cannabis use: implications for the concept of cannabis psychosis. Schizophr Res (2007) 93:203–10. doi:10.1016/j.schres.2007.03.014
275. Houston JE, Murphy J, Adamson G, Stringer M, Shevlin M. Childhood sexual abuse, early cannabis use, and psychosis: testing an interaction model based on the National Comorbidity Survey. Schizophr Bull (2008) 34:580–5. doi:10.1093/schbul/sbm127
276. Harley M, Kelleher I, Clarke M, Lynch F, Arseneault L, Connor D, et al. Cannabis use and childhood trauma interact additively to increase the risk of psychotic symptoms in adolescence. Psychol Med (2010) 40:1627–34. doi:10.1017/S0033291709991966
277. Konings M, Stefanis N, Kuepper R, De Graaf R, Ten Have M, Van Os J, et al. Replication in two independent population-based samples that childhood maltreatment and cannabis use synergistically impact on psychosis risk. Psychol Med (2012) 42:149–59. doi:10.1017/S0033291711000973
278. Kuepper R, Henquet C, Lieb R, Wittchen HU, Van Os J. Non-replication of interaction between cannabis use and trauma in predicting psychosis. Schizophr Res (2011) 131:262–3. doi:10.1016/j.schres.2011.06.012
279. Duncan AE, Sartor CE, Scherrer JF, Grant JD, Heath AC, Nelson EC, et al. The association between cannabis abuse and dependence and childhood physical and sexual abuse: evidence from an offspring of twins design. Addiction (2008) 103:990–7. doi:10.1111/j.1360-0443.2008.02210.x
280. Read J, Van Os J, Morrison AP, Ross CA. Childhood trauma, psychosis and schizophrenia: a literature review with theoretical and clinical implications. Acta Psychiatr Scand (2005) 112:330–50. doi:10.1111/j.1600-0447.2005.00634.x
281. Zammit S, Spurlock G, Williams H, Norton N, Williams N, O’Donovan MC, et al. Genotype effects of CHRNA7, CNR1 and COMT in schizophrenia: interactions with tobacco and cannabis use. Br J Psychiatry (2007) 191:402–7. doi:10.1192/bjp.bp.107.036129
282. Zammit S, Owen MJ, Evans J, Heron J, Lewis G. Cannabis, COMT and psychotic experiences. Br J Psychiatry (2011) 199:380–5. doi:10.1192/bjp.bp.111.091421
283. Costas J, Sanjuan J, Ramos-Rios R, Paz E, Agra S, Tolosa A, et al. Interaction between COMT haplotypes and cannabis in schizophrenia: a case-only study in two samples from Spain. Schizophr Res (2011) 127:22–7. doi:10.1016/j.schres.2011.01.014
284. van Winkel R. Family-based analysis of genetic variation underlying psychosis-inducing effects of cannabis: sibling analysis and proband follow- up. Arch Gen Psychiatry (2011) 68:148–57. doi:10.1001/archgenpsychiatry.2010.152
285. Di Forti M, Iyegbe C, Sallis H, Kolliakou A, Falcone MA, Paparelli A, et al. Confirmation that the AKT1 (rs2494732) genotype influences the risk of psychosis in cannabis users. Biol Psychiatry (2012) 72:811–6. doi:10.1016/j.biopsych.2012.06.020
286. Papaleo F, Crawley JN, Song J, Lipska BK, Pickel J, Weinberger DR, et al. Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J Neurosci (2008) 28:8709–23. doi:10.1523/JNEUROSCI.2077-08.2008
287. Tunbridge EM, Harrison PJ, Weinberger DR. Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry (2006) 60:141–51. doi:10.1016/j.biopsych.2005.10.024
288. Henquet C, Rosa A, Delespaul P, Papiol S, Fananas L, Van Os J, et al. COMT ValMet moderation of cannabis-induced psychosis: a momentary assessment study of “switching on” hallucinations in the flow of daily life. Acta Psychiatr Scand (2009) 119:156–60. doi:10.1111/j.1600-0447.2008.01265.x
289. Kantrowitz JT, Nolan KA, Sen S, Simen AA, Lachman HM, Bowers MB Jr. Adolescent cannabis use, psychosis and catechol-O-methyltransferase genotype in African Americans and Caucasians. Psychiatr Q (2009) 80:213–8. doi:10.1007/s11126-009-9108-4
290. Estrada G, Fatjo-Vilas M, Munoz MJ, Pulido G, Minano MJ, Toledo E, et al. Cannabis use and age at onset of psychosis: further evidence of interaction with COMT Val158Met polymorphism. Acta Psychiatr Scand (2011) 123:485–92. doi:10.1111/j.1600-0447.2010.01665.x
291. Pelayo-Teran JM, Suarez-Pinilla P, Chadi N, Crespo-Facorro B. Gene-environment interactions underlying the effect of cannabis in first episode psychosis. Curr Pharm Des (2012) 18:5024–35. doi:10.2174/138161212802884609
292. Vinkers CH, Van Gastel WA, Schubart CD, Van Eijk KR, Luykx JJ, van Winkel R, et al. The effect of childhood maltreatment and cannabis use on adult psychotic symptoms is modified by the COMT ValMet polymorphism. Schizophr Res (2013) 150(1):303–11. doi:10.1016/j.schres.2013.07.020
293. Alemany S, Arias B, Fatjo-Vilas M, Villa H, Moya J, Ibanez MI, et al. Psychosis-inducing effects of cannabis are related to both childhood abuse and COMT genotypes. Acta Psychiatr Scand (2013) 129(1):54–62. doi:10.1111/acps.12108
294. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature (1995) 378:785–9. doi:10.1038/378785a0
295. Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol (2001) 2:760–8. doi:10.1038/35096067
296. Sánchez MG, Ruiz-Llorente L, Sánchez AM, Díaz-Laviada I. Activation of phosphoinositide 3-kinase/PKB pathway by CB(1) and CB(2) cannabinoid receptors expressed in prostate PC-3 cells. Involvement in Raf-1 stimulation and NGF induction. Cell Signal (2003) 15:851–9. doi:10.1016/S0898-6568(03)00036-6
297. Ozaita A, Puighermanal E, Maldonado R. Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J Neurochem (2007) 102:1105–14. doi:10.1111/j.1471-4159.2007.04642.x
298. Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat Genet (2004) 36:131–7. doi:10.1038/ng1296
299. Thiselton DL, Vladimirov VI, Kuo P-H, McClay J, Wormley B, Fanous A, et al. AKT1 is associated with schizophrenia across multiple symptom dimensions in the Irish study of high density schizophrenia families. Biol Psychiatry (2008) 63:449–57. doi:10.1016/j.biopsych.2007.06.005
300. van Winkel R, Van Beveren NJ, Simons C. AKT1 moderation of cannabis-induced cognitive alterations in psychotic disorder. Neuropsychopharmacology (2011) 36:2529–37. doi:10.1038/npp.2011.141
301. Bhattacharyya S, Atakan Z, Martin-Santos R, Crippa JA, Kambeitz J, Prata D, et al. Preliminary report of biological basis of sensitivity to the effects of cannabis on psychosis: AKT1 and DAT1 genotype modulates the effects of delta-9-tetrahydrocannabinol on midbrain and striatal function. Mol Psychiatry (2012) 17:1152–5. doi:10.1038/mp.2011.187
302. Talkowski ME, Kirov G, Bamne M, Georgieva L, Torres G, Mansour H, et al. A network of dopaminergic gene variations implicated as risk factors for schizophrenia. Hum Mol Genet (2008) 17:747–58. doi:10.1093/hmg/ddm347
303. Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet (2002) 71:877–92. doi:10.1086/342734
304. Munafo MR, Attwood AS, Flint J. Neuregulin 1 genotype and schizophrenia. Schizophr Bull (2008) 34:9–12. doi:10.1093/schbul/sbm129
305. Boucher AA, Arnold JC, Duffy L, Schofield PR, Micheau J, Karl T. Heterozygous neuregulin 1 mice are more sensitive to the behavioural effects of Delta9-tetrahydrocannabinol. Psychopharmacology (Berl) (2007) 192:325–36. doi:10.1007/s00213-007-0721-3
306. Decoster J, Van Os J, Kenis G, Henquet C, Peuskens J, De Hert M, et al. Age at onset of psychotic disorder: cannabis, BDNF Val66Met, and sex-specific models of gene-environment interaction. Am J Med Genet B Neuropsychiatr Genet (2011) 156B:363–9. doi:10.1002/ajmg.b.31174
307. Ho B-C, Wassink TH, Ziebell S, Andreasen NC. Cannabinoid receptor 1 gene polymorphisms and marijuana misuse interactions on white matter and cognitive deficits in schizophrenia. Schizophr Res (2011) 128:66–75. doi:10.1016/j.schres.2011.02.021
308. Hill AB. The environment and disease: association or causation? Proc R Soc Med (1965) 58:295–300.
309. Allebeck P, Adamsson C, Engstrom A, Rydberg U. Cannabis and schizophrenia: a longitudinal study of cases treated in Stockholm County. Acta Psychiatr Scand (1993) 88:21–4. doi:10.1111/j.1600-0447.1993.tb03408.x
310. Linszen DH, Dingemans PM, Lenior ME. Cannabis abuse and the course of recent-onset schizophrenic disorders. Arch Gen Psychiatry (1994) 51:273–9. doi:10.1001/archpsyc.1994.03950040017002
311. Hambrecht M, Hafner H. Substance abuse and the onset of schizophrenia. Biol Psychiatry (1996) 40:1155–63. doi:10.1016/s0006-3223(95)00609-5
312. Green AI, Tohen MF, Hamer RM, Strakowski SM, Lieberman JA, Glick I, et al. First episode schizophrenia-related psychosis and substance use disorders: acute response to olanzapine and haloperidol. Schizophr Res (2004) 66:125–35. doi:10.1016/j.schres.2003.08.001
313. Morgan VA, Waterreus A, Jablensky A, Mackinnon A, McGrath JJ, Carr V, et al. People living with psychotic illness in 2010: the second Australian national survey of psychosis. Aust N Z J Psychiatry (2012) 46:735–52. doi:10.1177/0004867412449877
314. Moore E, Mancuso SG, Slade T, Galletly C, Castle DJ. The impact of alcohol and illicit drugs on people with psychosis: the second Australian National Survey of Psychosis. Aust N Z J Psychiatry (2012) 46:864–78. doi:10.1177/0004867412443900
315. Henquet C, Van Os J, Kuepper R, Delespaul P, Smits M, Campo JA, et al. Psychosis reactivity to cannabis use in daily life: an experience sampling study. Br J Psychiatry (2010) 196:447–53. doi:10.1192/bjp.bp.109.072249
316. Ruiz-Veguilla M, Barrigon ML, Hernandez L, Rubio JL, Gurpegui M, Sarramea F, et al. Dose-response effect between cannabis use and psychosis liability in a non-clinical population: evidence from a snowball sample. J Psychiatr Res (2013) 47:1036–43. doi:10.1016/j.jpsychires.2013.03.003
317. Tien AY, Anthony JC. Epidemiological analysis of alcohol and drug use as risk factors for psychotic experiences. J Nerv Ment Dis (1990) 178:473–80. doi:10.1097/00005053-199017880-00001
318. Wiles NJ, Zammit S, Bebbington P, Singleton N, Meltzer H, Lewis G. Self-reported psychotic symptoms in the general population: results from the longitudinal study of the British National Psychiatric Morbidity Survey. Br J Psychiatry (2006) 188:519–26. doi:10.1192/bjp.bp.105.012179
319. Compton DR, Johnson MR, Melvin LS, Martin BR. Pharmacological profile of a series of bicyclic cannabinoid analogs: classification as cannabimimetic agents. J Pharmacol Exp Ther (1992) 260:201–9.
320. Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry (1991) 148:1474–86.
321. Gardner EL. Endocannabinoid signaling system and brain reward: emphasis on dopamine. Pharmacol Biochem Behav (2005) 81:263–84. doi:10.1016/j.pbb.2005.01.032
322. Laviolette SR, Grace AA. cannabinoids potentiate emotional learning plasticity in neurons of the medial prefrontal cortex through basolateral amygdala inputs. J Neurosci (2006) 26:6458–68. doi:10.1523/JNEUROSCI.0707-06.2006
323. Hermann H, Marsicano G, Lutz B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience (2002) 109:451–60. doi:10.1016/S0306-4522(01)00509-7
324. Meschler JP, Howlett AC. Signal transduction interactions between CB1 cannabinoid and dopamine receptors in the rat and monkey striatum. Neuropharmacology (2001) 40:918–26. doi:10.1016/S0028-3908(01)00012-0
325. French ED. delta9-Tetrahydrocannabinol excites rat VTA dopamine neurons through activation of cannabinoid CB1 but not opioid receptors. Neurosci Lett (1997) 226:159–62. doi:10.1016/S0304-3940(97)00278-4
326. French ED, Dillon K, Wu X. Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. Neuroreport (1997) 8:649–52. doi:10.1097/00001756-199702100-00014
327. Tanda G, Pontieri FE, Di Chiara G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science (1997) 276:2048–50. doi:10.1126/science.276.5321.2048
328. Gessa GL, Melis M, Muntoni AL, Diana M. Cannabinoids activate mesolimbic dopamine neurons by an action on cannabinoid CB1 receptors. Eur J Pharmacol (1998) 341:39–44. doi:10.1016/S0014-2999(97)01442-8
329. Fadda P, Scherma M, Spano MS, Salis P, Melis V, Fattore L, et al. Cannabinoid self-administration increases dopamine release in the nucleus accumbens. Neuroreport (2006) 17:1629–32. doi:10.1097/01.wnr.0000236853.40221.8e
330. Bossong MG, Van Berckel BN, Boellaard R, Zuurman L, Schuit RC, Windhorst AD, et al. Delta 9-tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology (2009) 34:759–66. doi:10.1038/npp.2008.138
331. Stokes PR, Mehta MA, Curran HV, Breen G, Grasby PM. Can recreational doses of THC produce significant dopamine release in the human striatum? Neuroimage (2009) 48:186–90. doi:10.1016/j.neuroimage.2009.06.029
332. Bloomfield MA, Morgan CJ, Egerton A, Kapur S, Curran HV, Howes OD. Dopaminergic function in cannabis users and its relationship to cannabis-induced psychotic symptoms. Biol Psychiatry (2014) 75:470–8. doi:10.1016/j.biopsych.2013.05.027
333. Kuepper R, Ceccarini J, Lataster J, Van Os J, Van Kroonenburgh M, Van Gerven JM, et al. Delta-9-tetrahydrocannabinol-induced dopamine release as a function of psychosis risk: 18F-fallypride positron emission tomography study. PLoS One (2013) 8:e70378. doi:10.1371/journal.pone.0070378
334. Goldman-Rakic PS. Regional and cellular fractionation of working memory. Proc Natl Acad Sci U S A (1996) 93:13473–80. doi:10.1073/pnas.93.24.13473
335. Robbins TW. Chemical neuromodulation of frontal-executive functions in humans and other animals. Exp Brain Res (2000) 133:130–8. doi:10.1007/s002210000407
336. Jentsch JD, Andrusiak E, Tran A, Bowers MB Jr, Roth RH. Delta 9-tetrahydrocannabinol increases prefrontal cortical catecholaminergic utilization and impairs spatial working memory in the rat: blockade of dopaminergic effects with HA966. Neuropsychopharmacology (1997) 16:426–32. doi:10.1016/S0893-133X(97)00018-3
337. Diana M, Melis M, Gessa GL. Increase in meso-prefrontal dopaminergic activity after stimulation of CB1 receptors by cannabinoids. Eur J Neurosci (1998) 10:2825–30. doi:10.1111/j.1460-9568.1998.00292.x
338. Pistis M, Porcu G, Melis M, Diana M, Gessa GL. Effects of cannabinoids on prefrontal neuronal responses to ventral tegmental area stimulation. Eur J Neurosci (2001) 14:96–102. doi:10.1046/j.0953-816x.2001.01612.x
339. Pistis M, Ferraro L, Pira L, Flore G, Tanganelli S, Gessa GL, et al. Delta(9)-tetrahydrocannabinol decreases extracellular GABA and increases extracellular glutamate and dopamine levels in the rat prefrontal cortex: an in vivo microdialysis study. Brain Res (2002) 948:155–8. doi:10.1016/S0006-8993(02)03055-X
340. Eggan SM, Lewis DA. Immunocytochemical distribution of the cannabinoid CB1 receptor in the primate neocortex: a regional and laminar analysis. Cereb Cortex (2007) 17:175–91. doi:10.1093/cercor/bhj136
341. Weinberger DR. On the plausibility of “the neurodevelopmental hypothesis” of schizophrenia. Neuropsychopharmacology (1996) 14:1S–11S. doi:10.1016/0893-133X(95)00199-N
342. Rapoport JL, Addington AM, Frangou S, Psych MR. The neurodevelopmental model of schizophrenia: update 2005. Mol Psychiatry (2005) 10:434–49. doi:10.1038/sj.mp.4001642
343. Crews F, He J, Hodge C. Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol Biochem Behav (2007) 86:189–99. doi:10.1016/j.pbb.2006.12.001
344. Rice D, Barone S Jr. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect (2000) 108(Suppl 3):511–33. doi:10.1289/ehp.00108s3511
345. Fernandez-Ruiz J, Berrendero F, Hernandez ML, Ramos JA. The endogenous cannabinoid system and brain development. Trends Neurosci (2000) 23:14–20. doi:10.1016/S0166-2236(99)01491-5
346. Jin K, Xie L, Kim SH, Parmentier-Batteur S, Sun Y, Mao XO, et al. Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol Pharmacol (2004) 66:204–8. doi:10.1124/mol.66.2.204
347. Aguado T, Palazuelos J, Monory K, Stella N, Cravatt B, Lutz B, et al. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J Neurosci (2006) 26:1551–61. doi:10.1523/JNEUROSCI.3101-05.2006
348. Galve-Roperh I, Aguado T, Palazuelos J, Guzman M. The endocannabinoid system and neurogenesis in health and disease. Neuroscientist (2007) 13:109–14. doi:10.1177/1073858406296407
349. Watson S, Chambers D, Hobbs C, Doherty P, Graham A. The endocannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol Cell Neurosci (2008) 38:89–97. doi:10.1016/j.mcn.2008.02.001
350. D’Souza DC, Ranganathan M, Braley G, Gueorguieva R, Zimolo Z, Cooper T, et al. Blunted psychotomimetic and amnestic effects of delta-9-tetrahydrocannabinol in frequent users of cannabis. Neuropsychopharmacology (2008) 33:2505–16. doi:10.1038/sj.npp.1301643
351. Frisoli TM, Schmieder RE, Grodzicki T, Messerli FH. Salt and hypertension: is salt dietary reduction worth the effort? Am J Med (2012) 125:433–9. doi:10.1016/j.amjmed.2011.10.023
352. Sasco AJ, Secretan MB, Straif K. Tobacco smoking and cancer: a brief review of recent epidemiological evidence. Lung Cancer (2004) 45(Suppl 2):S3–9. doi:10.1016/j.lungcan.2004.07.998
353. Lee PN, Forey BA, Coombs KJ. Systematic review with meta-analysis of the epidemiological evidence in the 1900s relating smoking to lung cancer. BMC Cancer (2012) 12:385. doi:10.1186/1471-2407-12-385
354. Brawley OW. Avoidable cancer deaths globally. CA Cancer J Clin (2011) 61:67–8. doi:10.3322/caac.20108
355. Aust R, Sharp C, Goulden C. Prevalence of Drug Use: Key Findings from the 2001/2002 British Crime Survey, Findings 182. London: Home Office Research, Development and Statistics Directorate (2002).
356. Der G, Gupta S, Murray RM. Is schizophrenia disappearing? Lancet (1990) 335:513–6. doi:10.1016/0140-6736(90)90745-Q
357. Ajdacic-Gross V, Lauber C, Warnke I, Haker H, Murray RM, Rossler W. Changing incidence of psychotic disorders among the young in Zurich. Schizophr Res (2007) 95:9–18. doi:10.1016/j.schres.2007.06.001
Keywords: cannabis, psychosis, spice, synthetic cannabinoids, schizophrenia, psychophysiology, schizotypy
Citation: Radhakrishnan R, Wilkinson ST and D’Souza DC (2014) Gone to pot – a review of the association between cannabis and psychosis. Front. Psychiatry 5:54. doi: 10.3389/fpsyt.2014.00054
Received: 23 October 2013; Accepted: 02 May 2014;
Published online: 22 May 2014.
Edited by:
Elizabeth Clare Temple, Federation University Australia, AustraliaReviewed by:
Thomas Hillemacher, Hannover Medical School, GermanyCopyright: © 2014 Radhakrishnan, Wilkinson and D’Souza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deepak Cyril D’Souza, Psychiatry Service, 116A, VA Connecticut Healthcare System, 950 Campbell Avenue, West Haven, CT 06516, USA e-mail:ZGVlcGFrLmRzb3V6YUB5YWxlLmVkdQ==
†Rajiv Radhakrishnan and Samuel T. Wilkinson share first-authorship on this paper.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.