


- Department of Brain and Cognitive Sciences, Picower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, MA, USA
Development of the nervous system proceeds through a set of complex checkpoints which arise from a combination of sequential gene expression and early neural activity sculpted by the environment. Genetic and environmental insults lead to neurodevelopmental disorders which encompass a large group of diseases that result from anatomical and physiological abnormalities during maturation and development of brain circuits. Rett syndrome (RTT) is a neurological disorder of genetic origin, caused by mutations in the X-linked gene methyl-CpG binding protein 2 (MeCP2). It features a range of neuropsychiatric abnormalities including motor dysfunctions and mild to severe cognitive impairment. Here, we discuss key questions and recent studies describing animal models, cell-type specific functions of methyl-CpG binding protein 2 (MeCP2), defects in neural circuit plasticity, and attempts to evaluate possible therapeutic strategies for RTT. We also discuss how genes, proteins, and overlapping signaling pathways affect the molecular etiology of apparently unrelated neuropsychiatric disorders, an understanding of which can offer novel therapeutic strategies for a range of autism spectrum disorders (ASDs).
Introduction
Rett syndrome (RTT) [Online Mendelian Inheritance in Man (OMIM) #312750], first reported by the Austrian physician Andreas Rett, is a monogenic, postnatal developmental disorder that affects normal brain development during early childhood in females, with an incidence of 1 in 10,000–15,000 live births (Rett, 1966; Chahrour and Zoghbi, 2007). Patients with RTT appear to reach developmental milestones seemingly normally until about 6–18 months after birth when signs and symptoms of the disease begin to appear. This typically includes severe mental retardation, stereotypic hand movements, motor coordination deficits, epileptic seizures, language and learning disabilities, and mild to severe cognitive impairments (Hagberg et al., 1983; Nomura, 2005; Chahrour and Zoghbi, 2007). Other clinical hallmarks include ataxia, spasticity, respiratory abnormalities, and autonomic dysfunction (Williamson and Christodoulou, 2006). Loss-of-function mutations in the MECP2, which encodes a transcriptional regulator and epigenetic modifier MeCP2, have been found to be the primary genetic component in 90% of patients suffering from RTT (Lewis et al., 1992; Amir et al., 1999; Guy et al., 2011). Interestingly, only one copy of MECP2 is active in females due to random X-chromosome inactivation (Adler et al., 1995). RTT is not a heritable disorder and most mutations in MECP2 arise de novo in germ cells, usually on the paternal side (Trappe et al., 2001). Growing evidence implicates alteration of MECP2 expression in the etiology of several other neuropsychiatric disorders including Angelman syndrome, childhood schizophrenia, and congenital encephalopathy in boys (Schule et al., 2008). Originally, RTT was considered to be a disorder of early postnatal life; however, one recent study has shown that inducible deletion of MECP2 in adults recapitulates the germ-line knock out phenotype in mice (McGraw et al., 2011).
The link between MECP2 and RTT is fascinating because RTT is one of a small group of ASDs that gives us the opportunity to study mutations in a single gene and how they affect sequential phenotypic checkpoints of brain development leading to neuropathological endpoints in psychiatric disorders (Ben-Ari and Spitzer, 2010). In this review, we will first briefly discuss RTT and its genetic basis and the role of MeCP2 in brain development and plasticity. Second, we will provide a detailed description of mouse models of RTT and discuss their use to study molecular, synaptic, and circuit pathophysiology and to test novel therapeutic approaches to reverse neurological deficits. Finally, we will examine how overlapping signaling pathways affect the molecular etiology of apparently unrelated neuropsychiatric disorders like Fragile-X syndrome (FXS) and tuberous sclerosis (TSC) and how such an understanding can be utilized to design novel therapeutic strategies.
MeCP2: Multi-Functional Global Regulator with a Local Function?
The human MECP2 gene consists of four exons resulting in expression of two protein isoforms due to alternative splicing of exon 2. These splice variants differ only in their N-termini, and include the more abundant MeCP2-e1 isoform (encoded by MECP2B or Mecp2α in mice) as well as the MeCP2-e2 isoform (encoded by MECP2A or Mecp2β in mice; Mnatzakanian et al., 2004; Kriaucionis et al., 2006). Gene expression studies show that different brain regions are enriched with different splice variants; MeCP2-e2 is prevalent in dorsal thalamus and layer 5 of the cortex while MeCP2-e1 is detected in the hypothalamus (Dragich et al., 2007). Recent results suggest that MeCP2-e2 isoform is upregulated in Aβ-treated cortical neurons and promotes neuronal death in post-mitotic neurons, a pathway normally inhibited by forkhead protein FOXG1 (Dastidar et al., 2012). Furthermore, MECP2 has a long conserved 3′UTR which contains multiple poly-adenylation sites, which can additionally generate four different transcripts.
MeCP2 is a highly conserved basic nuclear protein, initially found to be associated with methylated DNA (5′ CpG islands) regulating gene silencing and chromatin remodeling (Nan et al., 1993; Hendrich and Bird, 1998). MeCP2 is also an “intrinsically disordered” protein with long stretches of unorganized segments, without standard three-dimensional secondary structure, proposed to participate in the formation of a flexible scaffold required for multiple biological interactions (Adkins and Georgel, 2011). The genome-wide distribution of MeCP2 has consequences for global epigenetic state. While MeCP2 is expressed in various tissues throughout the body, including lung and spleen, it is abundant in mature, post-mitotic neurons in the adult brain. Expression of neuronal MeCP2 in mice is developmentally regulated. Initially, MeCP2 expression is high during embryonic development, with a low level at birth during neuronal maturation and synaptogenesis. This is followed by a consistent increase over the first three postnatal weeks reaching a plateau with a subsequent increase later in adult life (Shahbazian et al., 2002b; Kishi and Macklis, 2004). The role of MeCP2 in the development and maturation of the nervous system compared to the maintenance of adult neurons is not yet fully understood.
Structurally, MeCP2 is known to have two important functional domains: methyl-CpG binding domain (MBD) and the transcriptional repressor domain (TRD; Nan et al., 1993, 1997). There is also an additional C-terminal nuclear localization signal (NLS) which helps traffic the protein into the nucleus. One way to analyze MeCP2 function is to identify partner proteins which interact with these defined protein structural domains. The MBD specifically binds to methylated CpG di-nucleotides in DNA and also to unmethylated four-way DNA junctions suggesting a role for MeCP2 in regulating higher-order chromatin structure (Galvao and Thomas, 2005). The TRD is involved in transcriptional repression via the recruitment of transcriptional co-repressors and chromatin remodeling proteins. MeCP2’s role as transcriptional repressor has been suggested based on the observation that MeCP2 specifically inhibits transcription from methylated promoters (Nan et al., 1997) while TRD recruits co-repressors like SIN3A and histone deacetylases (HDACs) 1 and 2 causing global compaction of chromatin by promoting nucleosome clustering. There is also evidence that MeCP2 through its TRD binds to Y-box binding protein-1 (YB1), a protein implicated in transcriptional activation regulation of splicing in reporter constructs (Young et al., 2005). Although precise protein–protein interaction mechanisms and consequences are yet unknown, it is clear that MeCP2 plays an important role in globally regulating chromatin structure and transcription of its immediate downstream targets. The abundance of MeCP2 and its binding sites in the brain makes it an unlikely candidate to be a gene-specific classical transcriptional regulator. It is possible that specificity of MeCP2 function can be significantly controlled post-translationally in a cell-type specific manner (Cohen et al., 2011). Our knowledge of the set of neuronal MeCP2 target genes is incomplete, however, such knowledge would be of great benefit in determining the molecular basis of pathology and for potential therapeutic measures.
Mouse Models for RTT
To understand the detailed mechanism of RTT, several animal models have been generated (Calfa et al., 2011b). MeCP2 null and conditional mutant mouse models with cell-type or area-specific loss of MeCP2 in the brain show phenotypic features that resemble some features of RTT patient symptoms. All these models have been generated by mutating the mouse endogenous Mecp2 gene or by the introduction of the human MECP2 gene with a representative RTT mutation (see Table 1). These diverse approaches have produced mouse lines with gene products that range from a complete absence of the protein (Guy et al., 2001) to expression of a truncated but non-functional MeCP2 protein (Chen et al., 2001; Shahbazian et al., 2002a; Goffin et al., 2012). In addition, mouse models have been created with germ-line or conditional deletions upon recombination with different tissue, cell-type, or developmental stage specific Cre-lines (Chen et al., 2001). Behavioral face validity of these mouse models, or how well they recapitulate the usual RTT symptoms, is varied. Several of the mouse models show symptoms common to those present in RTT patients such as stereotypies, ataxia, akinesia, and breathing irregularities. Nevertheless, behavioral phenotypes, specifically anxiety and socialization are more heterogeneous across lines (Table 1). This behavioral heterogeneity can be attributed to variability in mouse line genetic backgrounds and the levels of expression of the MeCP2 protein due to the type of mutation. Social choice paradigms such as the social approach and social recognition tests show different results when comparing common inbred strains. Some strains (C57BL/6J, FVB/NJ, AKR/J) show a high preference for socializing with new stranger animals, whereas other strains show low preference or even direct avoidance (A/J, BALB, 129S1). Similarly the time they spend in the closed arms of the plus-maze test, a measurement that positively correlates with the anxiety level of the animal, is also variable. There seems to be an association between higher degree of anxiety and reduced social interest so mouse background has to be taken into account when comparing behavior phenotypes (Moy et al., 2007, 2008, 2009). Interestingly, the recent literature has started to address whether different translation types and levels of MeCP2 lead to variation in the anxiety and social phenotype: the complete lack of protein might produce a stronger phenotype with less anxiety-like behavior whereas a truncated protein could generate mouse lines with increased stress (Chao and Zoghbi, 2012). Understanding the correlation between functional interaction of the different Mecp2 transcripts produced by these models with other anxiety-related genes like corticotrophin releasing hormone (Crh) will be capital, not only to explain the mice phenotypic variance but for clinical applicability (De Filippis et al., 2010; Kerr et al., 2010; Goffin et al., 2012).

Table 1. Molecular, physiological, defects and behavioral abnormalities found in various mouse models of RTT.
RTT as a Disorder of Synaptic and Neural Circuit Maturation
In many RTT patients and in mouse models of RTT, MECP2 is absent in nearly half (female heterozygous) or every cell (male hemizygous) throughout the body. However, experiments with targeted brain-specific deletion of Mecp2 (using Nestin-cKO mice) showing similar phenotype as that of Mecp2 null in all tissues support a crucial role for brain-specific Mecp2 defects in the pathogenesis and profound neurological phenotype of RTT (Chen et al., 2001; Guy et al., 2001). Autopsy studies on post-mortem human brain shows an overall decrease in the size and reduction in weight and volume of the brain (Armstrong, 2005). However, the RTT brains does not show any obvious sign of degeneration, atrophy, or cell death.
Structural Changes
Although there is no gross structural change in RTT brain, mouse models of RTT show delayed neuronal maturation and synaptogenesis (Fukuda et al., 2005). Several recent studies have shown functional, morphological, and molecular alterations in different mouse models of RTT. At the cellular level, neuronal soma size is reduced in the absence of MeCP2 with increased packing density (Armstrong, 2005). Synaptic structural and morphological defects include reduced dendritic branching, spine density, and reduced spine morphology (Kishi and Macklis, 2004; Belichenko et al., 2009). Presynaptically, loss of MeCP2 affects the number of axonal boutons and axonal arborization in general and their targeting (Belichenko et al., 2009), which suggests a decrease in the number of synapses in RTT brains. However, in line with the structural changes, many subtle alterations in synaptic transmission lead to the neuronal maturation defects that impact circuit-level plasticity in these mouse models of RTT.
Excitatory: Inhibitory Synaptic Balance
Analysis of spontaneous miniature excitatory and inhibitory postsynaptic currents (mEPSCs and mIPSCs respectively) in MeCP2 KO mice indicates a change in the excitatory/inhibitory (E/I) balance as revealed by increased excitatory and reduced inhibitory synaptic transmission in the hippocampus and cortex (Dani et al., 2005; Nelson et al., 2006; Chao et al., 2007), whereas E/I balance from Mecp2Tg1 (with human MeCP2 over-expressing mice) shows the opposite effect (Collins et al., 2004). Whole-cell patch-clamp recordings from thick-tufted layer 5 pyramidal neurons in primary somatosensory cortex (S1) show that spontaneous EPSC and spontaneous action potential firing are reduced in MeCP2 KO male mice (Tropea et al., 2009). Additionally, quadruple whole-cell patch-clamp recordings from layer 5 pyramidal neurons of four-week-old MeCP2 KO mouse slices show that excitatory synaptic connectivity is also reduced with weakening of individual connections (Dani and Nelson, 2009). The evidence for functional deficits supported by immunofluorescence staining of PSD95 in MeCP2 KO mice showing a significant reduction in layer 5 neurons in motor cortex (M1; Tropea et al., 2009). On the other hand, extracellular field EPSPs recorded from layer 2/3 with evoked stimulation in layer 4 in primary sensory and motor areas are unaffected by the expression of a truncated MeCP2 protein (Moretti et al., 2006). Lastly, inhibitory synaptic transmission might also be altered in mouse models of RTT, as evidence indicates that there are both pre- and postsynaptic defects of GABAergic neurotransmission in the brainstem (Medrihan et al., 2008). Developmentally, MeCP2 deficiency might have a distinct developmental effect on circuit-level function. Initial phases of synaptic development and pruning are normal at retino-geniculate synapses [from postnatal day (P)9–21 in MeCP2 KO mice compared to wild-type (WT) control], whereas the circuit becomes abnormal and immature in a subsequent experience-dependent phase (P27–34; Noutel et al., 2011).
Plasticity Defects
Several studies have shown that long-term potentiation and depression (LTP and LTD respectively), cellular mechanisms of long-term synaptic plasticity and learning and memory, are impaired in MeCP2 KO animals. LTP evoked by theta-burst stimulation in L2/3 of S1 is reduced in magnitude in eight-week-old MeCP2 KO mice compared to wild-type controls, an impairment that can be reversed by environmental enrichment (Lonetti et al., 2010). Also, LTP is impaired in hippocampal Schaffer’s collateral (SC)–CA1 synapses despite basal synaptic transmission being intact (Asaka et al., 2006). Hippocampal slices from adult MeCP2 KO mice show a clear decrease in the magnitude of EPSP with high frequency stimulation (HFS) but not with more physiological theta-burst stimulation (TBS). Interestingly, these experiments reveal a decrease only in the maintenance phase and not in the induction phases of LTP. Considering that distinct protein machinery and signaling pathways are involved in the induction and maintenance phase of LTP, it is possible that MeCP2 KO animals have selective impairment in plasticity induction. In contrast to HFS and TBS, a more physiologically plausible plasticity paradigm studied between paired L5 cortical pyramidal neurons using spike-timing-dependent protocols revealed no change in LTP in MeCP2 KO mice (Dani and Nelson, 2009). However, in this case, fewer synaptically-coupled connections were found in MeCP2 KO mice and individual connections were weaker. This suggests that loss of Mecp2 function reduces connectivity of excitatory synapses which precedes deficits in plasticity (Dani and Nelson, 2009). In support of this stance, a recent study using in utero injection of short hairpin RNA to knock down Mecp2 and glutamate uncaging by laser-scanning photostimulation to map neocortical circuit found that Mecp2 deficiency leads to significant reduction of mainly local excitatory input strength in superficial cortical layers (Wood et al., 2009). Along with LTP deficits, LTD, on the other hand, is also abolished in SC–CA1 synapses in adult MeCP2 KO animals (>6 weeks) with no obvious changes in 3–5 week old KO animals (Asaka et al., 2006). Furthermore, mouse model (MeCP2308, expressing a truncated form of MeCP2 protein showed similar impairments in synaptic plasticity with reduced LTP and LTD at SC–CA1 synapses in mice at 5–6 months of age, and exhibits increased basal synaptic transmission and decreased paired-pulse facilitation (PPF; Shahbazian et al., 2002a; Moretti et al., 2006). Conversely, a mouse model with mild Mecp2 over-expression (MeCP2Tg1) shows an enhanced LTP with normal basal synaptic transmission (Collins et al., 2004).
The molecular composition of synaptic NMDA receptor subunits showed an interesting trend in these MeCP2 KO animals: GluN2B-to-GluN2A subunit switching, which regulates the channel kinetics and biophysical properties of excitatory synapses in the developing brain, shows a delayed postnatal maturation and may be responsible for the molecular pathology of synaptic defects in RTT (Asaka et al., 2006).
Recent evidence points to the hypothesis that the neurological deficits found in RTT arise from a recoverable failure of synaptic and circuit development in the brain (Tropea et al., 2009). A plausible hypothesis, complementary to that of reduced excitatory synapse maturation, is that the level or nature of inhibition is altered in mouse models of RTT, so that cortical circuits persist in an immature state. Consistent with this hypothesis, it has been shown that the potential to trigger visual cortical plasticity by closing one eye persists into adulthood in MeCP2 KO mice, well past the critical period for such plasticity in wild-type mice (Tropea et al., 2009).
Role of MeCP2 in Homeostatic Synaptic Scaling
Synaptic scaling is a form of homeostatic plasticity in which average neuronal activity levels are modulated to allow for dynamic adjust of synaptic strength to promote stability of neuronal circuits (Turrigiano and Nelson, 2004). Recent evidence shows how MeCP2 mediates activity-dependent synaptic scaling in rat hippocampal cultures (Qiu et al., 2012). Increase in neuronal activity upon bicuculline treatment leads to an increased level of MeCP2 expression, which in turn binds to the GluR2 promoter and recruits a repressor complex to inhibit its expression and availability of these molecules at the synapse. Regulating AMPA receptor GluR2 subunit expression is one direct way to mediate an adaptive response that regulates synaptic strength and prevents recurrent circuit excitation.
Inhibition and RTT
Epilepsy is often seen in RTT patients and often difficult to treat (Steffenburg et al., 2001). Since most cases of RTT are caused by mutations in the MECP2 gene, it is assumed that convulsions are based on genetic mechanisms, however, balance of excitation and inhibition is also believed to play a critical role in the progression of the disease during early development. A new study has looked at dysfunctions of neuronal and network excitability using a combination of voltage-sensitive dye imaging and electrophysiology in hippocampal slices from symptomatic Mecp2 mutant animals (Calfa et al., 2011a). They found that CA1 and CA3 regions of the hippocampus are highly hyper-excitable and network excitability in CA3 may contribute to the hippocampal dysfunction and limbic seizures observed in Mecp2 mutant mice and RTT patients.
Whereas the loss of MeCP2 is known to alter excitatory synaptic transmission and plasticity (Nelson et al., 2006), little is known about how MeCP2 regulates the development and plasticity of inhibitory GABAergic circuits and how they are altered in RTT. In a recent study, using Viaat-Cre/MeCP2−/y (vesicular inhibitory amino acid transporter) mouse model, it has been shown that loss of MeCP2 from a subset of forebrain GABAergic neurons also recapitulates many features of RTT (Chao et al., 2010). MeCP2-deficient GABAergic neurons show reduced inhibitory quantal size, consistent with a presynaptic reduction in glutamic acid decarboxylase 1 (Gad1) and glutamic acid decarboxylase 2 (Gad2) levels, and GABA immunoreactivity. However, in this study, the effect of Mecp2 deletion from particular interneuron subclasses on inhibitory circuit plasticity in vivo has not been assessed.
Glia and RTT
Since its discovery, RTT has been regarded primarily as a neuro-pathophysiological disorder. This conclusion was mainly based on the finding that selective re-expression of Mecp2 in post-mitotic neurons (using neuron-specific Tau locus) is sufficient to rescue RTT (Luikenhuis et al., 2004). However, recent studies have shown that MeCP2 in glial cells plays a very important role in neuropathology of RTT. Unlike previous reports, it is now clear that MeCP2 is also expressed in astrocytes and in vitro co-culture studies have shown that MeCP2 null astrocytes can have non-cell-autonomous effects on both wild-type and MeCP2 null neurons (Ballas et al., 2009). Interestingly, a recent study has shown that re-expression of Mecp2, preferentially in astrocytes, significantly improves several of the hallmark mouse behavioral phenotypes of RTT mice (locomotion, anxiety levels, respiratory abnormalities, and lifespan) compared to globally null mice (Lioy et al., 2011).
Microglia may also influence the onset and progression of RTT. Aberrant microglial activity has been found in mouse models of RTT. Elevated levels of glutamate, released from microglia, may cause abnormal stunted dendritic morphology, microtubule disruption, and damage of postsynaptic glutamatergic components making microglial glutamate synthesis or release a potential therapeutic target for RTT (Maezawa and Jin, 2010; Derecki et al., 2012).
Molecular Pathophysiology of RTT and Therapeutic Approaches
Using mouse genetic models of RTT, key molecular signaling pathways that contribute to the deficits in synaptic function and maturation have been studied. Once identified, these mouse models have also been used to experimentally validate possible therapeutic avenues using genetic, pharmacological, and behavioral approaches. So far, three major approaches have been investigated: pharmacological treatment aimed to restore signaling pathway activity, supplementary diets and reinforcement therapies, and genetic manipulation that re-establish Mecp2 gene expression.
Molecular Pathophysiology and Effect on Signaling Pathways
The specific role of MeCP2 in transcription and translational control might vary depending on the different molecules recruited and protein–protein interactions. This complexity, for example, is shown in the regulation of one of the most important targets of MeCP2 in the central nervous system: brain-derived neurotrophic factor (BDNF). MeCP2 regulates BDNF expression by binding to promoter IV and repressing its transcription until MeCP2 is phosphorylated and released via a neuronal activity-dependent mechanism. This mechanism might explain the activity-dependent increase of BDNF, however it is unclear how it would cause the lower levels of this growth factor observed in RTT patients and mouse models when compared with healthy individuals or WT animals (Chang et al., 2006). A recent study (Abuhatzira et al., 2007) has suggested that BDNF protein down-regulation may be mediated by dis-inhibition of complex repressor REST/Co-REST. The translation of this complex is suppressed in the presence of MeCP2; in MeCP2 null mice the complex is overexpressed. The REST/Co-REST complex binds to a position between BDNF promoters I and II, located upstream of promoter IV, thereby overriding the direct effects of MeCP2 on the downstream promoter. BDNF is critical for neuronal development, synaptic maturation, and plasticity through the activation of specific neurotrophic tyrosine kinase receptor type 2 (TrkB), which, in turn, activates signal transduction pathways such as PLCγ, PI3K/Akt, and MAPK/ERK that regulate protein synthesis and neural function by activating PSD95 (Yoshii and Constantine-Paton, 2007). This pathway is of central importance to the expression and amelioration of the RTT phenotype. Similarly the PI3K-Akt-PSD95 pathway is known to drive the up-regulation of PSD95 by insulin (Lee et al., 2005), and this pathway is also stimulated by IGF-1 (Zheng and Quirion, 2004). Although there is not yet a clear picture of how cellular and molecular changes in RTT neurons in turn regulate their physiological properties, there is increasing evidence showing that the molecular signaling pathways and aberrant neuronal protein synthesis at the synapses are a possible explanation for several features in the pathology of RTT. The regulation of protein synthesis via the PI3K pathway has been proven to be crucial in synaptic function, dendrite structure, and plasticity (Jaworski et al., 2005; Kumar et al., 2005; Cuesto et al., 2011). All of these functions have been shown to be compromised in RTT (Belichenko et al., 2009; Noutel et al., 2011). Recently, direct evidence has demonstrated the dysregulation of the entire Akt/mTOR axis in MeCP2 null mice, giving a molecular theoretical framework for the mechanism of action of genes regulated by MeCP2, like BDNF (Ricciardi et al., 2011).
The importance of the PI3K pathway is reflected in the number of therapies designed for RTT that aim to restore its activity through the direct application or augmented endogenous synthesis of growth factors such as BDNF or IGF-1 (Tropea et al., 2009; Kline et al., 2010; Lonetti et al., 2010; Castro et al., 2011). These therapies target the tyrosine kinase receptors and hyper-activation of their subsequent downstream cascade that will cause increased protein synthesis that in the end impact synaptic maturation and function (Yoshii and Constantine-Paton, 2007).
Genetic Restoration of MeCP2
All evidence indicates that RTT has an extremely subtle pathophysiology compared to other severe neurodegenerative disorders. Genetic intervention and pharmacological treatments have been shown to rescue certain phenotypes of RTT, indicating that some deficits are reversible. Several genetic manipulation approaches have been tested for the reversal of behavioral impairments in RTT mouse models (see Table 2 for a summary of the major findings). One recent unbiased, high-content, small molecule screen in primary cortical neurons derived from the Angelman syndrome mouse model revealed a possible new therapeutic avenue that may be applicable to RTT (Huang et al., 2012). This study showed inhibitors of topoisomerase I and II, enzymes that regulate DNA supercoiling, could be used to unsilence the dormant paternal copy of the Ube3a gene (the primary genetic cause of Angelman Syndrome) in several regions of the nervous system. The resulting unsilencing lead to the expression of a functionally normal Ube3a protein, the level of which remained elevated in a subset of spinal cord neurons weeks after drug treatment. These finding highlight a remarkable potential for a short-term treatment that could lead to long-term effects on gene expression and possible reestablishment of proper neuronal UBE3A function. Whether a similar high-throughput screen can identify compounds that can be used to unsilence and reactivate X-inactivated copy of Mecp2 is an open question and an area of active research by several laboratories.
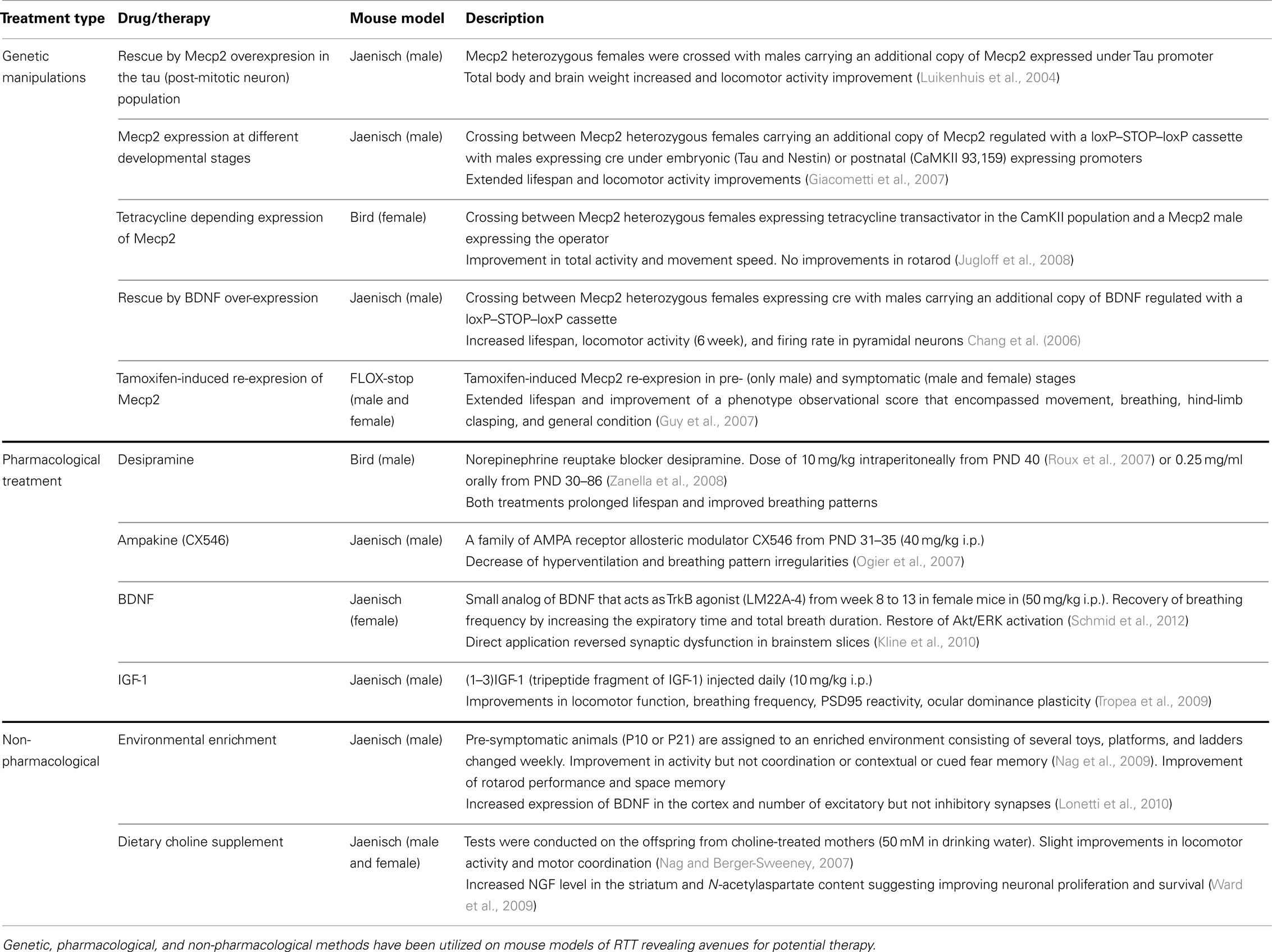
Table 2. Current therapeutic approaches employing RTT mouse models.
Convergence of Evidence for a Subset of Neurodevelopmental Disorders
Rett syndrome, Fragile-X syndrome (FXS), and TSC are examples of monogenic disorders that show compromised synaptic plasticity (Tavazoie et al., 2005; Moretti et al., 2006; Talos et al., 2008). Although these diseases have diverse genetic origin and phenotypes, they share common intermediates in the signaling pathways that will influence the availability of new proteins for functional and structural changes at the synapse. It is widely accepted that protein synthesis, including local protein synthesis at synapses, is required for several forms of synaptic plasticity (Sutton and Schuman, 2006). This local protein synthesis enables synapses to control synaptic strength independently of the cell body via rapid translation from pre-existing mRNAs and therefore, the mechanisms and the signaling pathways regulating translation are likely to be intimately involved in modulating synaptic strength (Figure 1). The PI3K/Akt/mTOR axis, ERK, and PKC γ,ζ signaling pathways have been shown to be heavily involved in controlling all the steps of the protein synthesis process (Lin et al., 1994; Beretta et al., 1996; Dufner et al., 1999; Hou and Klann, 2004) and are dysregulated at different levels in RTT, FXS, and TSC (Inoki et al., 2002; Manning et al., 2002; Gross et al., 2010; Ricciardi et al., 2011).
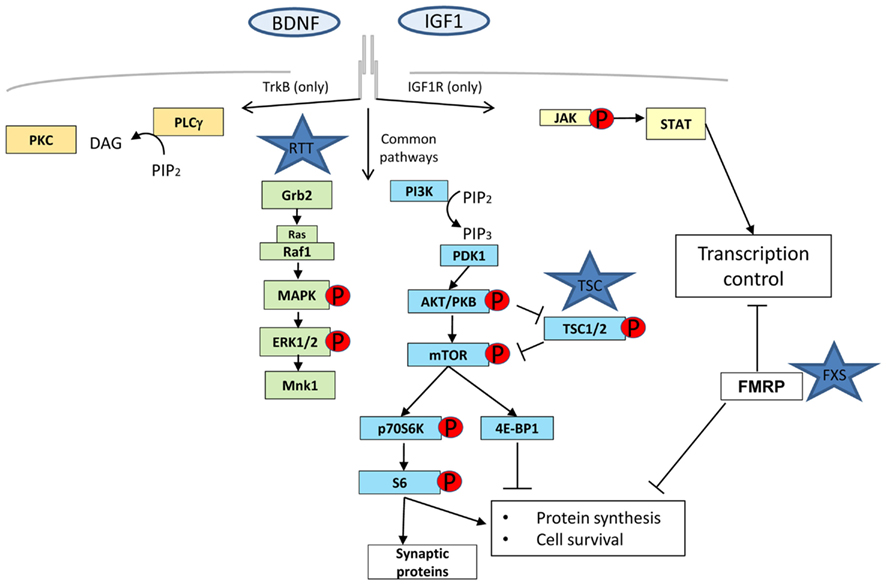
Figure 1. The four major molecular pathways activated by tyrosine kinase receptor B (TrkB) and IGF-1 receptor (IGF-1R). Phospholipase gamma (PLCγ) in orange, mitogen activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) in green, Phosphatidylinositol-3 kinase (PI3K)/Akt in blue, and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) in yellow. MAPK and PI3K are common transduction pathways for these two receptors whereas PLCγ is only specific for TrkB and JAK/STAT for IGF-1R. FMRP, although not directly involved in any signaling pathway is represented to show its influence in the different stages of protein synthesis: transcription, through mRNA stability control and translation.
FXS gene product FMRP may play several roles not associated directly with protein translation like trafficking and half-life stability of mRNA, but its major function is to act as a brake of protein translation by attaching to poly-ribosomes through ERK-mediated phosphorylation signaling (Mazroui et al., 2002; Gallagher et al., 2004; Qin et al., 2005). The FXS mouse model, a knock out of Fmr1 gene, is characterized by de-repression of protein translation but interestingly also has elevated PI3K/Akt/mTOR and ERK (Hou et al., 2006) activity levels through a negative feedback loop with PIKE (Gross et al., 2010; Sharma et al., 2010). TSC1 and 2 gene products (Hamartin and Tuberin respectively) form a complex that are more directly implicated in the protein synthesis pathway as a downstream target of PI3K that in turn becomes activated upon the binding of growth factors (e.g., IGF or BDNF). Activated PI3K leads to the recruitment of PDK1 and the serine/threonine protein kinase Akt, and subsequent phosphorylation/activation of Akt by PDK1. Activated Akt negatively regulates TSC complex by directly phosphorylating TSC2.
It is thus remarkable that although they work in opposite directions (FXS and TSC by protein synthesis up-regulation and RTT through down-regulation), all of these syndromes share at least certain common signaling pathways and a pathophysiological point of convergence in the synapse (Figure 1), where tight regulation is necessary for proper function. This convergence opens up the possibility that these three disorders and maybe other ASDs (such as Angelman disorder with a disrupted protein synthesis control caused by a defective UBE3A) have related biological foundations which can be exploited in the design of new therapeutic strategies (Auerbach et al., 2011).
New Avenues in RTT Research
Almost two decades of research on RTT has led to the development of an intriguing story of how a single transcription factor can play a crucial role in neuronal development, synaptic maturation, and plasticity. Although the primary function of MeCP2 in normal brain development remains unclear, it is becoming increasingly evident that there is a complex interplay of genes and environment which results in the synaptic and circuit-level defects in brain function. Restoration of MeCP2 expression in three-week-old brain resulted in improvements in LTP and neuro-anatomical parameters (Guy et al., 2007), illustrating that it is possible to improve the symptoms of this neuropsychiatric disorder. Disease models using induced pluripotent stem cells (iPS) from RTT patients’ fibroblasts have opened up a new avenue of drug discovery for therapeutic treatment of RTT (Kim et al., 2011; Marchetto et al., 2011). Recent data also suggests that immune system, whether it is adaptive (T cells) or innate (microglia), profoundly impact normal brain function and plasticity (Derecki et al., 2010; Graeber and Streit, 2010; Tremblay et al., 2011). Therefore, bone marrow transplant from healthy animals into mutant Mecp2 male animals is being investigated as an approach for amelioration of RTT symptoms (Derecki et al., 2012). Interestingly, several new functions of MeCP2 are only beginning to be understood including their role in dynamic genome regulation in neurons (Muotri et al., 2010). With the exciting new discoveries of genome-editing techniques using zinc-finger nucleases, an interesting new possibility would be to generate and use rat models to study RTT. Future studies with cell-type specific manipulation of MeCP2 to identify and examine the circuit-level contributions to function promise to elucidate further mechanisms of disease progression and provide new potential therapeutic targets for RTT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Travis Emery and Jeremy C. Petravicz for critically reading the manuscript. This work is supported by a Simons Foundation postdoctoral fellowship (Abhishek Banerjee) and grants from the NIH and Simons Foundation (Mriganka Sur).
References
Abuhatzira, L., Makedonski, K., Kaufman, Y., Razin, A., and Shemer, R. (2007). MeCP2 deficiency in the brain decreases BDNF levels by REST/CoREST-mediated repression and increases TRKB production. Epigenetics 2, 214–222.
Adkins, N. L., and Georgel, P. T. (2011). MeCP2: structure and function. Biochem. Cell Biol. 89, 1–11.
Adler, D. A., Quaderi, N. A., Brown, S. D., Chapman, V. M., Moore, J., Tate, P., and Disteche, C. M. (1995). The X-linked methylated DNA binding protein, Mecp2, is subject to X inactivation in the mouse. Mamm. Genome 6, 491–492.
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188.
Asaka, Y., Jugloff, D. G., Zhang, L., Eubanks, J. H., and Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 21, 217–227.
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68.
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317.
Belichenko, P. V., Wright, E. E., Belichenko, N. P., Masliah, E., Li, H. H., Mobley, W. C., and Francke, U. (2009). Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J. Comp. Neurol. 514, 240–258.
Ben-Ari, Y., and Spitzer, N. C. (2010). Phenotypic checkpoints regulate neuronal development. Trends Neurosci. 33, 485–492.
Beretta, L., Gingras, A. C., Svitkin, Y. V., Hall, M. N., and Sonenberg, N. (1996). Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 15, 658–664.
Blue, M. E., Kaufmann, W. E., Bressler, J., Eyring, C., O’Driscoll, C., Naidu, S., and Johnston, M. V. (2011). Temporal and regional alterations in NMDA receptor expression in Mecp2-null mice. Anat. Rec. (Hoboken) 294, 1624–1634.
Calfa, G., Hablitz, J. J., and Pozzo-Miller, L. (2011a). Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J. Neurophysiol. 105, 1768–1784.
Calfa, G., Percy, A. K., and Pozzo-Miller, L. (2011b). Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp. Biol. Med. (Maywood) 236, 3–19.
Castro, J., Kwok, S., Garcia, R., and Sur, M. (2011). Effects of recombinant human IGF1 treatment in a mouse model of Rett syndrome. Paper Presentation in Society for Neuroscience. Poster number cited 59.17/DD26.
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437.
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348.
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., Neul, J. L., Gong, S., Lu, H. C., Heintz, N., Ekker, M., Rubenstein, J. L., Noebels, J. L., Rosenmund, C., and Zoghbi, H. Y. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269.
Chao, H. T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65.
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331.
Cohen, S., Gabel, H. W., Hemberg, M., Hutchinson, A. N., Sadacca, L. A., Ebert, D. H., Harmin, D. A., Greenberg, R. S., Verdine, V. K., Zhou, Z., Wetsel, W. C., West, A. E., and Greenberg, M. E. (2011). Genome-Wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 72, 72–85.
Collins, A. L., Levenson, J. M., Vilaythong, A. P., Richman, R., Armstrong, D. L., Noebels, J. L., David Sweatt, J., and Zoghbi, H. Y. (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13, 2679–2689.
Cuesto, G., Enriquez-Barreto, L., Carames, C., Cantarero, M., Gasull, X., Sandi, C., Ferrus, A., Acebes, A., and Morales, M. (2011). Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J. Neurosci. 31, 2721–2733.
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565.
Dani, V. S., and Nelson, S. B. (2009). Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J. Neurosci. 29, 11263–11270.
Dastidar, S. G., Bardai, F. H., Ma, C., Price, V., Rawat, V., Verma, P., Narayanan, V., and D’Mello, S. R. (2012). Isoform-specific toxicity of Mecp2 in postmitotic neurons: suppression of neurotoxicity by FoxG1. J. Neurosci. 32, 2846–2855.
De Filippis, B., Ricceri, L., and Laviola, G. (2010). Early postnatal behavioral changes in the Mecp2-308 truncation mouse model of Rett syndrome. Genes Brain Behav. 9, 213–223.
Derecki, N. C., Cronk, J. C., Lu, Z., Xu, E., Abbott, S. B., Guyenet, P. G., and Kipnis, J. (2012). Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109.
Derecki, N. C., Privman, E., and Kipnis, J. (2010). Rett syndrome and other autism spectrum disorders – brain diseases of immune malfunction? Mol. Psychiatry 15, 355–363.
Dragich, J. M., Kim, Y. H., Arnold, A. P., and Schanen, N. C. (2007). Differential distribution of the MeCP2 splice variants in the postnatal mouse brain. J. Comp. Neurol. 501, 526–542.
Dufner, A., Andjelkovic, M., Burgering, B. M., Hemmings, B. A., and Thomas, G. (1999). Protein kinase B localization and activation differentially affect S6 kinase 1 activity and eukaryotic translation initiation factor 4E-binding protein 1 phosphorylation. Mol. Cell. Biol. 19, 4525–4534. doi:10.1371/journal.pone.0011534
Fukuda, T., Itoh, M., Ichikawa, T., Washiyama, K., and Goto, Y. (2005). Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J. Neuropathol. Exp. Neurol. 64, 537–544.
Gallagher, S. M., Daly, C. A., Bear, M. F., and Huber, K. M. (2004). Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J. Neurosci. 24, 4859–4864.
Galvao, T. C., and Thomas, J. O. (2005). Structure-specific binding of MeCP2 to four-way junction DNA through its methyl CpG-binding domain. Nucleic Acids Res. 33, 6603–6609.
Gemelli, T., Berton, O., Nelson, E. D., Perrotti, L. I., Jaenisch, R., and Monteggia, L. M. (2006). Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol. Psychiatry 59, 468–476.
Giacometti, E., Luikenhuis, S., Beard, C., and Jaenisch, R. (2007). Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc. Natl. Acad. Sci. U.S.A. 104, 1931–1936.
Goffin, D., Allen, M., Zhang, L., Amorim, M., Wang, I. T. J., Reyes, A.-R. S., Mercado-Berton, A., Ong, C., Cohen, S., Hu, L., Blendy, J. A., Carlson, G. C., Siegel, S. J., Greenberg, M. E., and Zhou, Z. (2012). Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat. Neurosci. 15, 274–283.
Graeber, M. B., and Streit, W. J. (2010). Microglia: biology and pathology. Acta Neuropathol. 119, 89–105.
Gross, C., Nakamoto, M., Yao, X., Chan, C. B., Yim, S. Y., Ye, K., Warren, S. T., and Bassell, G. J. (2010). Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 30, 10624–10638.
Guy, J., Cheval, H., Selfridge, J., and Bird, A. (2011). The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652.
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147.
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326.
Hagberg, B., Aicardi, J., Dias, K., and Ramos, O. (1983). A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann. Neurol. 14, 471–479.
Hendrich, B., and Bird, A. (1998). Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 18, 6538–6547.
Hou, L., Antion, M. D., Hu, D., Spencer, C. M., Paylor, R., and Klann, E. (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454.
Hou, L., and Klann, E. (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 24, 6352–6361.
Huang, H. S., Allen, J. A., Mabb, A. M., King, I. F., Miriyala, J., Taylor-Blake, B., Sciaky, N., Dutton, J. W. Jr., Lee, H. M., Chen, X., Jin, J., Bridges, A. S., Zylka, M. J., Roth, B. L., and Philpot, B. D. (2012). Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 481, 185–189.
Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan, K. L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657.
Jaworski, J., Spangler, S., Seeburg, D. P., Hoogenraad, C. C., and Sheng, M. (2005). Control of dendritic arborization by the phosphoinositide-3’-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 25, 11300–11312.
Jugloff, D. G., Vandamme, K., Logan, R., Visanji, N. P., Brotchie, J. M., and Eubanks, J. H. (2008). Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Hum. Mol. Genet. 17, 1386–1396.
Kerr, B., Alvarez-Saavedra, M., Saez, M. A., Saona, A., and Young, J. I. (2008). Defective body-weight regulation, motor control and abnormal social interactions in Mecp2 hypomorphic mice. Hum. Mol. Genet. 17, 1707–1717.
Kerr, B., Silva, P. A., Walz, K., and Young, J. I. (2010). Unconventional transcriptional response to environmental enrichment in a mouse model of Rett syndrome. PLoS ONE 5, e11534.
Kim, K. Y., Hysolli, E., and Park, I. H. (2011). Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 14169–14174.
Kishi, N., and Macklis, J. D. (2004). MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 27, 306–321.
Kline, D. D., Ogier, M., Kunze, D. L., and Katz, D. M. (2010). Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J. Neurosci. 30, 5303–5310.
Kriaucionis, S., Paterson, A., Curtis, J., Guy, J., Macleod, N., and Bird, A. (2006). Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol. Cell. Biol. 26, 5033–5042.
Kumar, V., Zhang, M. X., Swank, M. W., Kunz, J., and Wu, G. Y. (2005). Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 25, 11288–11299.
Lee, C. C., Huang, C. C., Wu, M. Y., and Hsu, K. S. (2005). Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J. Biol. Chem. 280, 18543–18550.
Lewis, J. D., Meehan, R. R., Henzel, W. J., Maurer-Fogy, I., Jeppesen, P., Klein, F., and Bird, A. (1992). Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69, 905–914.
Lin, T. A., Kong, X., Haystead, T. A., Pause, A., Belsham, G., Sonenberg, N., and Lawrence, J. C. Jr. (1994). PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science 266, 653–656.
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., Hirrlinger, P. G., Kirchhoff, F., Bissonnette, J. M., Ballas, N., and Mandel, G. (2011). A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500.
Lonetti, G., Angelucci, A., Morando, L., Boggio, E. M., Giustetto, M., and Pizzorusso, T. (2010). Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 null mice. Biol. Psychiatry 67, 657–665.
Luikenhuis, S., Giacometti, E., Beard, C. F., and Jaenisch, R. (2004). Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc. Natl. Acad. Sci. U.S.A. 101, 6033–6038.
Maezawa, I., and Jin, L. W. (2010). Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 30, 5346–5356.
Manning, B. D., Tee, A. R., Logsdon, M. N., Blenis, J., and Cantley, L. C. (2002). Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162.
Marchetto, M. C., Brennand, K. J., Boyer, L. F., and Gage, F. H. (2011). Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum. Mol. Genet. 20, R109–R115.
Mazroui, R., Huot, M. E., Tremblay, S., Filion, C., Labelle, Y., and Khandjian, E. W. (2002). Trapping of messenger RNA by fragile X mental retardation protein into cytoplasmic granules induces translation repression. Hum. Mol. Genet. 11, 3007–3017.
McGill, B. E., Bundle, S. F., Yaylaoglu, M. B., Carson, J. P., Thaller, C., and Zoghbi, H. Y. (2006). Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 103, 18267–18272.
McGraw, C. M., Samaco, R. C., and Zoghbi, H. Y. (2011). Adult neural function requires MeCP2. Science 333, 186.
Medrihan, L., Tantalaki, E., Aramuni, G., Sargsyan, V., Dudanova, I., Missler, M., and Zhang, W. (2008). Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 99, 112–121.
Mnatzakanian, G. N., Lohi, H., Munteanu, I., Alfred, S. E., Yamada, T., Macleod, P. J., Jones, J. R., Scherer, S. W., Schanen, N. C., Friez, M. J., Vincent, J. B., and Minassian, B. A. (2004). A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 36, 339–341.
Moretti, P., Bouwknecht, J. A., Teague, R., Paylor, R., and Zoghbi, H. Y. (2005). Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 14, 205–220.
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., Armstrong, D., Arancio, O., Sweatt, J. D., and Zoghbi, H. Y. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327.
Moy, S. S., Nadler, J. J., Young, N. B., Nonneman, R. J., Grossman, A. W., Murphy, D. L., D’Ercole, A. J., Crawley, J. N., Magnuson, T. R., and Lauder, J. M. (2009). Social approach in genetically engineered mouse lines relevant to autism. Genes Brain Behav. 8, 129–142.
Moy, S. S., Nadler, J. J., Young, N. B., Nonneman, R. J., Segall, S. K., Andrade, G. M., Crawley, J. N., and Magnuson, T. R. (2008). Social approach and repetitive behavior in eleven inbred mouse strains. Behav. Brain Res. 191, 118–129.
Moy, S. S., Nadler, J. J., Young, N. B., Perez, A., Holloway, L. P., Barbaro, R. P., Barbaro, J. R., Wilson, L. M., Threadgill, D. W., Lauder, J. M., Magnuson, T. R., and Crawley, J. N. (2007). Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav. Brain Res. 176, 4–20.
Muotri, A. R., Marchetto, M. C., Coufal, N. G., Oefner, R., Yeo, G., Nakashima, K., and Gage, F. H. (2010). L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446.
Nag, N., and Berger-Sweeney, J. E. (2007). Postnatal dietary choline supplementation alters behavior in a mouse model of Rett syndrome. Neurobiol. Dis. 26, 473–480.
Nag, N., Moriuchi, J. M., Peitzman, C. G., Ward, B. C., Kolodny, N. H., and Berger-Sweeney, J. E. (2009). Environmental enrichment alters locomotor behaviour and ventricular volume in Mecp2 1lox mice. Behav. Brain Res. 196, 44–48.
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481.
Nan, X., Meehan, R. R., and Bird, A. (1993). Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 21, 4886–4892.
Nelson, E. D., Kavalali, E. T., and Monteggia, L. M. (2006). MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr. Biol. 16, 710–716.
Nomura, Y. (2005). Early behavior characteristics and sleep disturbance in Rett syndrome. Brain Dev. 27(Suppl. 1), S35–S42.
Noutel, J., Hong, Y. K., Leu, B., Kang, E., and Chen, C. (2011). Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron 70, 35–42.
Ogier, M., Wang, H., Hong, E., Wang, Q., Greenberg, M. E., and Katz, D. M. (2007). Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J. Neurosci. 27, 10912–10917.
Panayotis, N., Pratte, M., Borges-Correia, A., Ghata, A., Villard, L., and Roux, J. C. (2011). Morphological and functional alterations in the substantia nigra pars compacta of the Mecp2-null mouse. Neurobiol. Dis. 41, 385–397.
Pratte, M., Panayotis, N., Ghata, A., Villard, L., and Roux, J. C. (2011). Progressive motor and respiratory metabolism deficits in post-weaning Mecp2-null male mice. Behav. Brain Res. 216, 313–320.
Qin, M., Kang, J., Burlin, T. V., Jiang, C., and Smith, C. B. (2005). Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J. Neurosci. 25, 5087–5095.
Qiu, Z., Sylwestrak, E. L., Lieberman, D. N., Zhang, Y., Liu, X. Y., and Ghosh, A. (2012). The Rett syndrome protein MeCP2 regulates synaptic scaling. J. Neurosci. 32, 989–994.
Rett, A. (1966). On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 116, 723–726.
Ricceri, L., De Filippis, B., Fuso, A., and Laviola, G. (2011). Cholinergic hypofunction in MeCP2-308 mice: beneficial neurobehavioural effects of neonatal choline supplementation. Behav. Brain Res. 221, 623–629.
Ricciardi, S., Boggio, E. M., Grosso, S., Lonetti, G., Forlani, G., Stefanelli, G., Calcagno, E., Morello, N., Landsberger, N., Biffo, S., Pizzorusso, T., Giustetto, M., and Broccoli, V. (2011). Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 20, 1182–1196.
Roux, J. C., Dura, E., Moncla, A., Mancini, J., and Villard, L. (2007). Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur. J. Neurosci. 25, 1915–1922.
Santos, M., Silva-Fernandes, A., Oliveira, P., Sousa, N., and Maciel, P. (2007). Evidence for abnormal early development in a mouse model of Rett syndrome. Genes Brain Behav. 6, 277–286.
Schaevitz, L. R., Moriuchi, J. M., Nag, N., Mellot, T. J., and Berger-Sweeney, J. (2010). Cognitive and social functions and growth factors in a mouse model of Rett syndrome. Physiol. Behav. 100, 255–263.
Schmid, D. A., Yang, T., Ogier, M., Adams, I., Mirakhur, Y., Wang, Q., Massa, S. M., Longo, F. M., and Katz, D. M. (2012). A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J. Neurosci. 32, 1803–1810.
Schule, B., Armstrong, D. D., Vogel, H., Oviedo, A., and Francke, U. (2008). Severe congenital encephalopathy caused by MECP2 null mutations in males: central hypoxia and reduced neuronal dendritic structure. Clin. Genet. 74, 116–126.
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., Armstrong, D., Paylor, R., and Zoghbi, H. (2002a). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254.
Shahbazian, M. D., Antalffy, B., Armstrong, D. L., and Zoghbi, H. Y. (2002b). Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 11, 115–124.
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T., Mcbride, S. M., Klann, E., and Zukin, R. S. (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702.
Stearns, N. A., Schaevitz, L. R., Bowling, H., Nag, N., Berger, U. V., and Berger-Sweeney, J. (2007). Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience 146, 907–921.
Steffenburg, U., Hagberg, G., and Hagberg, B. (2001). Epilepsy in a representative series of Rett syndrome. Acta Paediatr. 90, 34–39.
Sutton, M. A., and Schuman, E. M. (2006). Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127, 49–58.
Talos, D. M., Kwiatkowski, D. J., Cordero, K., Black, P. M., and Jensen, F. E. (2008). Cell-specific alterations of glutamate receptor expression in tuberous sclerosis complex cortical tubers. Ann. Neurol. 63, 454–465.
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J., and Sabatini, B. L. (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734.
Trappe, R., Laccone, F., Cobilanschi, J., Meins, M., Huppke, P., Hanefeld, F., and Engel, W. (2001). MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am. J. Hum. Genet. 68, 1093–1101.
Tremblay, M. E., Stevens, B., Sierra, A., Wake, H., Bessis, A., and Nimmerjahn, A. (2011). The role of microglia in the healthy brain. J. Neurosci. 31, 16064–16069.
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., Mccurry, C., Fu, D. D., Flannery, R., Jaenisch, R., and Sur, M. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A 106, 2029–2034.
Turrigiano, G. G., and Nelson, S. B. (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 5, 97–107.
Ward, B. C., Kolodny, N. H., Nag, N., and Berger-Sweeney, J. E. (2009). Neurochemical changes in a mouse model of Rett syndrome: changes over time and in response to perinatal choline nutritional supplementation. J. Neurochem. 108, 361–371.
Williamson, S. L., and Christodoulou, J. (2006). Rett syndrome: new clinical and molecular insights. Eur. J. Hum. Genet. 14, 896–903.
Wood, L., Gray, N. W., Zhou, Z., Greenberg, M. E., and Shepherd, G. M. (2009). Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J. Neurosci. 29, 12440–12448.
Yoshii, A., and Constantine-Paton, M. (2007). BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat. Neurosci. 10, 702–711.
Young, J. I., Hong, E. P., Castle, J. C., Crespo-Barreto, J., Bowman, A. B., Rose, M. F., Kang, D., Richman, R., Johnson, J. M., Berget, S., and Zoghbi, H. Y. (2005). Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. U.S.A. 102, 17551–17558.
Zanella, S., Mebarek, S., Lajard, A. M., Picard, N., Dutschmann, M., and Hilaire, G. (2008). Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir. Physiol. Neurobiol. 160, 116–121.
Keywords: RTT, visual cortex, synapse, plasticity, development
Citation: Banerjee A, Castro J and Sur M (2012) Rett syndrome: genes, synapses, circuits, and therapeutics. Front. Psychiatry 3:34. doi: 10.3389/fpsyt.2012.00034
Received: 16 December 2011; Accepted: 28 March 2012;
Published online: 08 May 2012.
Edited by:
Daniela Tropea, Trinity College Dublin, IrelandReviewed by:
Anne E. West, Duke University Medical Center, USAOmar Khwaja, Harvard Medical School, USA
Copyright: © 2012 Banerjee, Castro and Sur. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mriganka Sur, Department of Brain and Cognitive Sciences, Picower Institute for Learning and Memory, Massachusetts Institute of Technology, 43 Vassar Street, Cambridge, MA 02139, USA. e-mail:bXN1ckBtaXQuZWR1