

- 1 Department of Cellular and Systems Neurobiology, Max Planck Institute of Neurobiology, Martinsried, Germany
- 2 Biochemistry, Adolf-Butenandt-Institute, Ludwig-Maximilians-University, Munich, Germany
- 3 Department of Neurology, Neurocenter, University of Freiburg, Freiburg, Germany
Alzheimer’s disease (AD) is a protein conformational disorder characterized by two major neuropathological features: extracellular accumulations of amyloid-β peptides in the form of plaques and intracellular tangles, consisting of hyperphosphorylated tau proteins. Several morphological and functional changes are associated with these lesions in the diseased brain, such as dendritic and synaptic alterations, as well as microglial and astroglial recruitment and their activation. The availability of transgenic mouse models that mimic key aspects of the disease in conjunction with recent advances in two-photon imaging facilitate the study of fundamental aspects of AD pathogenesis and allow for longitudinally monitoring the efficacy of therapeutic interventions. Here, we review the ambitious efforts to understand the relationship between the main neuropathological hallmarks of AD and their associated structural and functional abnormalities by means of in vivo two-photon imaging.
Introduction
Longevity is a long-cherished dream of mankind, but the downside of an aging population is currently one of the big challenges society is facing. Alzheimer’s disease (AD), as the most common form of dementia in the elderly, currently affects ~35 million people worldwide (Prince et al., 2011). It is a fatal, progressive, neurodegenerative disease characterized by memory impairment and cognitive decline. When Alois Alzheimer first described the disease in 1906/07 (Alzheimer, 1907), he had already identified the pathological hallmarks that still, to this day, constitute the major diagnostic criteria for AD (Jack et al., 2011). The most prominent of which are senile plaques, representing extracellular proteinaceous aggregates that mainly consist of the amyloid-β peptide (Aβ; Glenner and Wong, 1984; Masters et al., 1985). Aβ is derived from the sequential cleavage of the amyloid-β precursor protein (APP) by the β- and γ-secretases (Haass, 2004; Haass and Selkoe, 2007; Lichtenthaler et al., 2011). Neurofibrillary tangles (NFT) constitute the second major hallmark of AD (Kosik et al., 1986; Nukina and Ihara, 1986). These intracellular aggregates of the hyperphosphorylated microtubule-associated protein tau form intrasomal inclusions as well as neuropil threads, and are thought to be causally related to neuronal apoptosis (Dickson, 2004; Spires-Jones et al., 2009b; Wakasaya et al., 2011).
Interestingly, although plaques and tangles pathologically define the disease, it is the loss of synapses that correlates strongly with the dementia typical of AD (Masliah et al., 1991; Terry et al., 1991; Scheff et al., 2006). Research over the past decade has thus focused on the mechanisms causing synaptic deterioration, including the abnormal accumulation of Aβ and tau. The resulting decrease of synapse density is believed to eventually lead to neuronal network disruption and cognitive decline.
Traditional histological analyses of affected brains only allow for a static assessment of pathology. The advent of two-photon microscopy (Denk et al., 1990) has enabled in vivo long-term imaging studies (Holtmaat et al., 2009), from which the field of AD research has benefited tremendously. In vivo imaging of transgenic mice through a cranial window not only allows to monitor general neuropathological features of AD-like lesions (Table 1; Figure 1), but also to address questions regarding the kinetics and temporal sequence of events (Figure 2; Bacskai et al., 2001; Spires et al., 2005; Spires-Jones et al., 2011). Two-photon microscopy furthermore facilitates direct observation of the effects of new treatment approaches. Although none of the mouse models available to date perfectly recapitulates the human AD neuropathology in its entirety, they still represent a valuable tool to precisely study key aspects of the disease, such as the occurrence and growth of amyloid plaques, the onset and progression of dendritic spine pathology, or the induction of neuronal apoptosis (Radde et al., 2008).
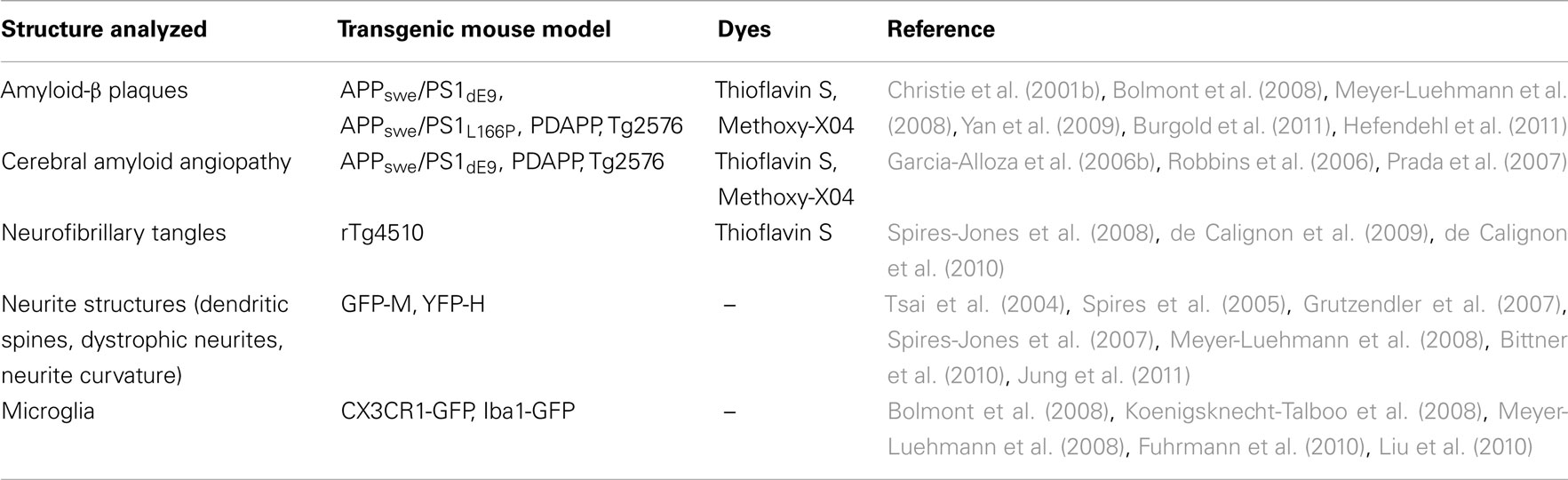
Table 1. Structures analyzed in mouse models of Alzheimer’s disease by in vivo two-photon microscopy.
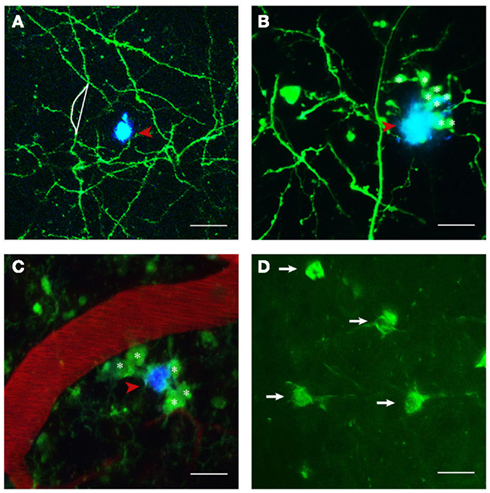
Figure 1. Structures imaged in APP transgenic mice in vivo. (A) Maximum projection of a z-stack with GFP expressing neuronal structures pseudocolored in green and Methoxy-XO4 labeled amyloid plaques shown in blue (indicated by red arrowhead). Neurite curvature (depicted by white lines) corresponds to the ratio of the length of the curved neuritic stretch to the straight end-to-end length of the same stretch (here R = 1.08). (B) Dystrophic neurites (marked with asterisks) in the close vicinity of a plaque (red arrowhead). (C) Activated microglia (CX3CR1-GFP+/−; denoted by asterisks) encompassing a plaque (red arrowheads) next to a blood vessel stained with Texas red. (D) Neurofibrillary tangles labeled with Thioflavin S (white arrows). Scale bars represent 30 μm (A), 20 μm (B,C), and 15 μm (D), respectively.
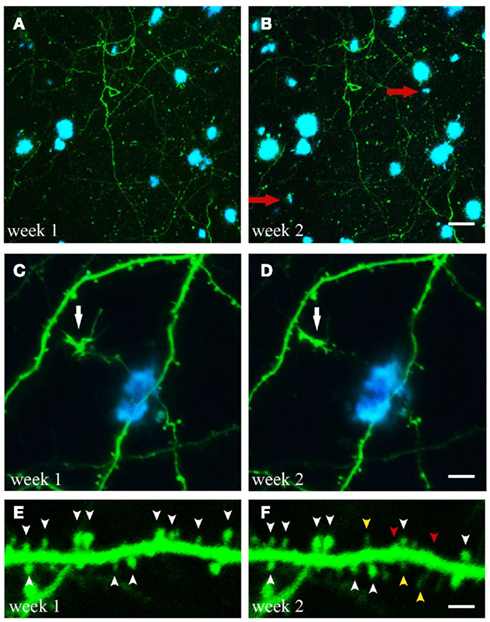
Figure 2. Time-lapse imaging of structural alterations. (A,B) Maximum projections of z-stacks with GFP expressing neuronal structures and Methoxy-XO4 labeled amyloid plaques. Over the course of 1 week two new plaques emerged within the region analyzed (marked with red arrows). (C,D) (White arrow). (E,F) Spine dynamics of an apical dendritic tuft (layer V pyramidal neuron of GFP-M mouse). Spines present at both imaging time points are indicated by white arrowheads, spines lost over the course of 1 week are labeled with red arrowheads, spines newly formed during the same period are marked with yellow arrowheads. Scale bars represent 25 μm (B), 10 μm (D), and 2 μm (F), respectively.
We here review some of the key findings and concepts that have emerged from the rapidly growing body of literature, on structural and functional alterations in transgenic mouse models of AD as revealed by in vivo two-photon microscopy.
Structural in vivo Imaging
Imaging Amyloid Deposits in APP Transgenic Mice
Amyloid-β plaques have been studied extensively as one of the major hallmarks of AD, and as a consequence many treatment studies are aimed at reducing the formation of plaques (Shah et al., 2008; Chopra et al., 2011; Kurz and Perneczky, 2011). Extracellular amyloid plaques mainly consist of aggregated Aβ peptides that are derived from a larger APP. To release Aβ, APP is first cleaved at its ectodomain by β-secretase, and subsequently is cleaved intramembranously by γ-secretase (Haass, 2004; Steiner, 2004; Lichtenthaler et al., 2011). Depending on the exact cleavage site, the released Aβ peptide can vary in length, with longer Aβ peptides being more prone to aggregation (Yan and Wang, 2006). According to in vitro models, Aβ aggregation follows a nucleation polymerization model, where Aβ monomers form higher molecular-weight oligomers and protofibrils, before they finally aggregate into insoluble amyloid plaques (Harper and Lansbury, 1997).
In order to monitor amyloid plaques in vivo, dyes binding to beta-pleated sheet protein structures, such as Thioflavin S or Methoxy-XO4, are applied either topically or peripherally, with the latter agent being able to cross the blood-brain-barrier (Christie et al., 2001b; Klunk et al., 2002; Bacskai et al., 2003). As opposed to dyes binding to beta-pleated sheet protein structures, which only allow for the visualization of the compact forms of plaques, fluorescently labeled antibodies against Aβ additionally mark diffuse plaques.
Crucial aspects addressed by longitudinal two-photon imaging are the process of de novo plaque formation as well as the kinetics of cerebral amyloidosis. The appearance of new plaques is a rare, but rapid event that is age-dependent (Meyer-Luehmann et al., 2008; Burgold et al., 2011; Hefendehl et al., 2011; Figures 2A,B). Previous studies assessing the kinetics of plaque growth have yielded somewhat inconsistent findings. Whereas some studies reported no significant change in plaque size when imaged over the course of 14 days (Meyer-Luehmann et al., 2008) and up to 150 days (Christie et al., 2001b), others found an increase in plaque size only for small plaques (<10 μm radius) over the course of 4 weeks (Bolmont et al., 2008). More recent studies observed growth for all plaques analyzed, and reported on either growth rates independent of plaque size (Hefendehl et al., 2011) or a higher growth rate for newly deposited (i.e., small) compared to pre-existing plaques (Yan et al., 2009; Burgold et al., 2011). Factors potentially contributing to these inconsistencies could be of biological or methodological nature. Among others, the type of the transgenic mouse model as well as sex and age of the animals differed between the studies (Table 2). Furthermore, the dye applied in order to label amyloid plaques, Methoxy-XO4, is in itself known to interfere with the process of plaque deposition (Cohen et al., 2009). Therefore the imaging frequency, i.e., the frequency of dye re-application, is likely to contribute to the variability in observed plaque growth (Burgold et al., 2011).
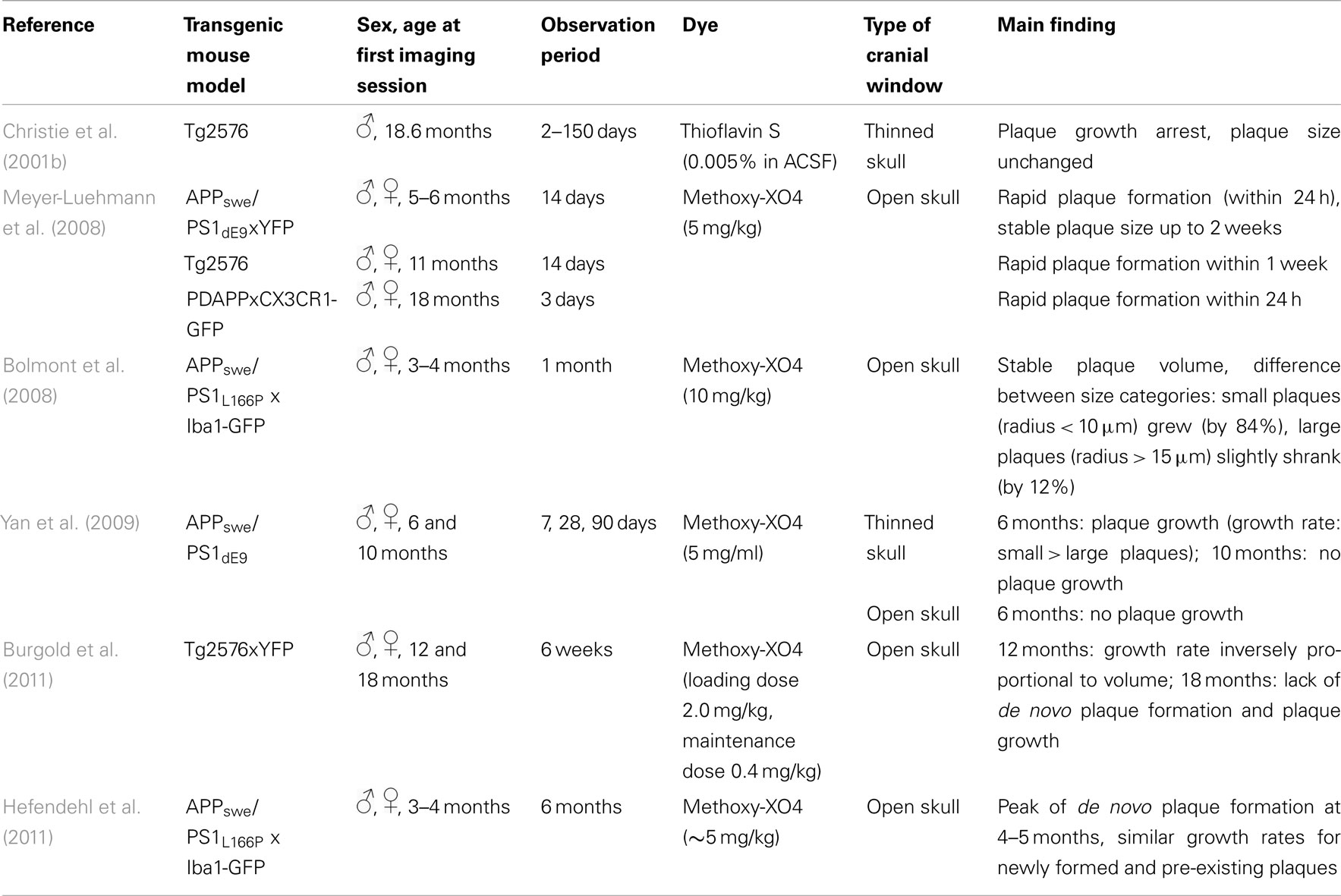
Table 2. Studies analyzing amyloid plaque formation and growth in viv o.
It has also been suggested that the type of cranial window, used to perform in vivo imaging, might be an additional confounding variable. Yan et al. (2009) compared thinned – vs. open skull preparations and only detected significant plaque growth with the thinned skull preparation. Two recent studies, however, observed clear plaque growth with the open skull preparation, suggesting the equivalence of both approaches (Burgold et al., 2011; Hefendehl et al., 2011). Furthermore it should be mentioned that image processing and the applied detection stringency, i.e., the set detection threshold, can also affect the results. Notably, plaque pathology reaches a ceiling phase, lacking apparent growth, and de novo plaque formation (Burgold et al., 2011). In summary, longitudinal in vivo two-photon imaging revealed the rapid appearance and slow, but steady growth of plaques with varying growth rates influenced by such factors, as the transgenic mouse models, age of the animals, initial size of plaques (pre-existing vs. newborn plaques), or type of cranial window. These studies shed light on the kinetics of plaque pathology progression, thereby providing basic knowledge for further therapeutic intervention studies.
In addition to amyloid plaques, extracellular Aβ deposits often occur within the walls of cerebral and leptomeningeal blood vessels, called cerebral amyloid angiopathy (CAA). CAA results in a loss of vascular smooth muscle cells and induces microhemorrhages (Van Dorpe et al., 2000; Christie et al., 2001a; Winkler et al., 2001; Fryer et al., 2003). Many of the APP transgenic mouse models develop not only Aβ deposits in the parenchyma but also CAA (Calhoun et al., 1999; Van Dorpe et al., 2000; Christie et al., 2001a; Robbins et al., 2006). However, some APP transgenic mice, e.g., APPswe/PS1dE9, are not an ideal model to study CAA, as these mice develop CAA at a much slower rate when compared to other APPswe transgenic mice (Garcia-Alloza et al., 2006b).
The temporal progression of CAA was first approximated by imaging postmortem intact whole brains of transgenic mice of different ages, revealing a consistent, and stereotyped age-dependent spatial distribution of CAA (Domnitz et al., 2005). A far more precise approach to analyze the kinetics of CAA progression is longitudinal two-photon imaging of the same individual vessel segment. Thus in vivo imaging of APP transgenic mice at weekly intervals, starting at the age of 8–11 months, showed that the emergence of CAA is a multifocal event in form of band-like Aβ deposits (Robbins et al., 2006). Furthermore, in contrast to the accumulation of amyloid plaques in the parenchyma, CAA progression tends to propagate from already existing deposits (Robbins et al., 2006). Therapeutic interventions, such as passive immunization, was able to halt CAA progression and even to reduce vascular amyloid in APP transgenic mice, as demonstrated by immunohistochemical analysis (Schroeter et al., 2008; Cattepoel et al., 2011). While chronic antibody treatment over 14 days monitored by two-photon microscopy resulted in a robust CAA regression, a single antibody exposure only led to moderate CAA regression, thus corroborating histological studies (Prada et al., 2007).
Imaging Neurites in the Vicinity of Amyloid Plaques
Albeit pathologically defining the disease and being a diagnostic criterion, the occurrence of amyloid plaques only poorly correlates with the cognitive decline characteristic of AD (Terry et al., 1991; Guillozet et al., 2003). A much better correlate is the loss of synapses (Terry et al., 1991; Masliah et al., 1994). Amyloid plaques are associated with numerous structural and functional pathological features, such as neurite curvature (D’Amore et al., 2003; Garcia-Alloza et al., 2006a; Meyer-Luehmann et al., 2008; Figure 1A) and breakage (Tsai et al., 2004; Figures 2C,D), neuritic dystrophies (D’Amore et al., 2003; Tsai et al., 2004; Figure 1B), synapse loss (Tsai et al., 2004; Spires et al., 2005), accumulation of glial cells (Bolmont et al., 2008; Figure 1C), and altered neuronal response properties (Busche et al., 2008). Conventional immunohistochemical techniques have been extensively used to elucidate the spatial relationship between amyloid plaques and synaptic markers and found a decrease in synaptic density, both in AD patients and in transgenic animal models, with synapse loss being most pronounced in the immediate vicinity of amyloid plaques (DeKosky et al., 1996; Grutzendler et al., 2007; Knobloch and Mansuy, 2008; Penzes et al., 2011). These studies however do not allow for a detailed analysis of the dynamics of synaptic changes (reviewed by Giannakopoulos et al., 2009).
In vivo two-photon microscopy has therefore been employed to unravel the temporal sequence of pathological events as well as their dynamics. To this end, mouse models of AD were crossbred with mice expressing variants of GFP in neuronal subsets, such as the GFP-M or YFP-H line (Feng et al., 2000; Tsai et al., 2004; Meyer-Luehmann et al., 2008). Fluorophore expression in these mice is regulated by the Thy-1 promoter, which in the cerebral cortex results in a sparse labeling of mainly layer V neurons (Feng et al., 2000). Alternatively, neuronal structures can be labeled by the injection of viral constructs, inducing fluorophore expression (Spires et al., 2005).
Since Aβ is known to bind to molecular components of the postsynapse (NMDA-, EphB receptors; Lacor et al., 2007; Cisse et al., 2011), imaging studies primarily focused on the analysis of dendritic spines, which are the main site of excitatory synaptic input to cortical pyramidal cells (Tsai et al., 2004; Spires et al., 2005; Spires-Jones et al., 2007). Typically, the distal tufts of apical dendrites of layer V pyramidal neurons are repeatedly imaged. Despite being relatively stable in adulthood when compared to development, dendritic spines are generally flexible and motile structures, thereby allowing for continuous remodeling of neuronal circuits (Majewska et al., 2006; Bhatt et al., 2009). Even under baseline conditions (i.e., without any particular sensory deprivation paradigm, often employed to study synaptic plasticity) a certain fraction of spines is constantly added and removed, while overall spine density remains mostly unaffected (Trachtenberg et al., 2002; Holtmaat et al., 2005, 2009; see however Hofer et al., 2009).
In APPswe/PS1M146L mice, Tsai et al. (2004) found an ongoing enhanced spine formation and elimination within the peri-plaque region (i.e., within 15 μm of the plaque border) when imaging over the course of 2 days for up to 5 weeks. In an APP transgenic mouse model (Tg2576) only older animals (18–24 months) showed increased spine elimination and decreased formation compared to age-matched wildtype control animals (Spires-Jones et al., 2007). Spine elimination rate exceeded the formation rate, measured over the relatively short time of 1 h, thereby presumably resulting in the described spine loss (Spires-Jones et al., 2007). A recent longitudinal imaging study (Bittner et al., 2010) for the first time analyzed spine changes in a triple transgenic mouse model of AD (3xTg-AD), bearing in addition to the APPswe and PS1M146V, also the tauP301L mutation (Oddo et al., 2003). Amyloid plaque-dependent as well as – independent events were found to underlie dendritic spine loss (Bittner et al., 2010). Bittner et al. found that spine loss in hippocampus and frontal cortex only became apparent after plaques were deposited, and was most pronounced in the vicinity of plaques as observed by immunohistochemistry. In contrast, somatosensory cortex in the 3xTg-AD mouse model is devoid of amyloid plaques even in old mice. Longitudinal in vivo two-photon imaging of apical dendrites in somatosensory cortex also revealed the loss of spines, but exclusively on dystrophic dendrites, which were positive for both hyperphosphorylated tau as well as intraneuronal Aβ (Bittner et al., 2010).
Transgenic mouse models, bearing PS1 mutations only, do not exhibit plaque pathology, hence allow for the separate analysis of the impact of PS1 mutations on dendritic spines independent of amyloid pathology. The impact of additional overexpression of either wildtype PS1 or the familial AD-PS1A246E (FAD–PS1) mutation on dendritic spines was analyzed by another study (Jung et al., 2011). Whereas apical tuft dendrites showed an increased spine density for both types of PS1 overexpression, basal dendrites only exhibited a higher spine density in the FAD–PS1 mice (Jung et al., 2011). Interestingly, neither spine dynamics nor shape was affected by the different PS1 overexpression, thus the authors suggest a gain-of-function role for PS1 on dendritic spine plasticity (Jung et al., 2011).
Besides the manifold observations of dendritic spine alterations in AD mouse models, additional structural changes were characterized by in vivo two-photon imaging, such as neuritic dystrophies, neurite curvature, and – breakage or dendritic shaft atrophy (Tsai et al., 2004; Garcia-Alloza et al., 2006a). Axonal dystrophies, seen in the immediate vicinity of plaques, also undergo constant formation and elimination over the course of days to weeks (Tsai et al., 2004). Interestingly, plaque pathology (i.e., Aβ deposition) precedes neuritic pathology, as curved as well as dystrophic neurites only occurred days to weeks after a plaque was newly formed (Meyer-Luehmann et al., 2008). These results further support the notion of amyloid plaques acting as a mediator of neurite and spine pathology, which is most likely caused by the combination of a high oligomeric Aβ concentration associated with plaques as well as their space occupying nature (Koffie et al., 2009) and is at least partially due to calcineurin activation (Wu et al., 2010).
Knowing the kinetics of plaque pathology and the above-mentioned associated structural changes allows for monitoring the effects of treatment approaches aiming at the reduction of amyloid burden. Immunotherapy has been proven to rapidly clear plaques (Bacskai et al., 2001), reduce the number and size of plaque-associated neuritic dystrophies (Brendza et al., 2005), and to acutely increase spine formation in APP transgenic mice (Spires-Jones et al., 2009a). Curved neurites around a plaque seem to be positively affected by antioxidants (Garcia-Alloza et al., 2006a, 2010), but not by γ-secretase inhibition (Garcia-Alloza et al., 2009). Unfortunately, previous treatment studies have limited their analysis to a small subset of neuropathological features seen in APP transgenic mice, thereby hindering the evaluation of the efficacy of the treatment approach per se. Future imaging studies should therefore aim at monitoring multiple neuropathological events simultaneously.
Imaging Microglia in APP Transgenic Mice
Apart from the above-mentioned neuritic alterations associated with amyloid plaque deposition, the immediate plaque vicinity is characterized by an accumulation of glial cells. Resting microglia are uniformly distributed throughout the brain. They are considered the surveillants of the brain, as their processes are highly motile, constantly sampling the microenvironment, whereas the soma itself remains stationary (Nimmerjahn et al., 2005). Once microglia detect cues of a brain insult, they rapidly become activated in a multistaged process, allowing them to migrate to the lesion site, release cytokines, and phagocytose material (Kettenmann et al., 2011). In AD, as well as in mouse models of AD, microglial cells are associated with amyloid plaques (Itagaki et al., 1989; Stalder et al., 1999). A recent in vivo imaging study demonstrated that microglia migrate rapidly to newly formed plaques (Meyer-Luehmann et al., 2008), a process which is enhanced by immunotherapy targeting aggregated Aβ (Koenigsknecht-Talboo et al., 2008). Their precise contribution to AD pathophysiology, however, remains controversial, as they are ascribed both a protective and a detrimental role (Morgan et al., 2005; Johnston et al., 2011).
Multiple studies have addressed the question, whether microglia cells are of crucial relevance in the formation and growth of amyloid plaques. There is some evidence from immunohistochemical – and in vivo imaging experiments, suggesting that microglia are able to phagocytose Aβ, thus restricting plaque growth (Simard et al., 2006; Bolmont et al., 2008). However, this is in contrast to an earlier electron microscopy study, in which the authors failed to detect any phagocytosed Aβ within microglia (Stalder et al., 2001) and a previous in vivo imaging study, where microglia lacked intracellular dense core plaque material (labeled with Methoxy-XO4), but were positive for protofibrillar Aβ (Liu et al., 2010). Surprisingly, microglial depletion was found to neither affect plaque count nor plaque size, thereby questioning a causal involvement of microglia at least in the pathogenesis of amyloid-β deposition (Grathwohl et al., 2009).
A number of in vivo studies have tried to unravel the influence of microglial activation, mediated by the chemokine receptor CX3CR1, on amyloid pathology, and associated neurotoxicity (Cardona et al., 2006; Fuhrmann et al., 2010; Liu et al., 2010). Notably, CX3CR1-deficient APP transgenic mice showed gene dose dependent reduction of amyloid deposition (Lee et al., 2010). Disruption of neuron–microglia interaction via fractalkine signaling seems to alter microglia activation and phagocytic capacity, thereby resulting in an enhanced phagocytosis of protofibrillar Aβ (Lee et al., 2010; Liu et al., 2010).
In a separate in vivo imaging study, triple transgenic mice (3xTg-AD; Oddo et al., 2003), crossbred to mice lacking the CX3CR1 receptor, showed no neuron loss, indicating a crucial role of the microglial chemokine receptor in mediating neuronal apoptosis (Fuhrmann et al., 2010). Interestingly, at the age of 4–6 months, 3xTg-AD animals do neither exhibit neurofibrillary tangle nor amyloid plaque pathology. This gives rise to the question of what the causing agent inducing the expression of fractalkine in subsequent apoptotic neurons is. One likely candidate is intracellular Aβ, as mice at the age analyzed do not yet possess hyperphosphorylated tau proteins. But why are other mouse models, which also exhibit intracellular Aβ, devoid of neuronal apoptosis? And furthermore, it remains unsolved whether microglia are causally involved in the apoptotic process or just recruited to the site of insult. In other words can interference with the CX3CR1 pathway prevent neuronal loss? Taken together, the role of microglia cells in the course of AD pathology is far from clear. Hopefully chronic two-photon imaging studies will help to resolve some of the open questions regarding the precise nature of microglial involvement in AD pathology.
Imaging tau Pathology
Whereas in vivo imaging of amyloid-β pathology has been feasible for quite some time, monitoring the second pathological hallmark of AD, namely neurofibrillary tangles in vivo, was only accomplished recently (Spires-Jones et al., 2008). NFT are thought to occur and spread downstream of abnormalities of Aβ during the course of AD (Gotz et al., 2004; Oddo et al., 2004; Santacruz et al., 2005; Tanzi, 2005; Sperling et al., 2011), but their correlation to the cognitive decline is much better than that observed for amyloid-β pathology, arguing for the importance of NFT in the degenerative process (Grober et al., 1999; Giannakopoulos et al., 2003). Aggregation of hyperphosphorylated tau protein into NFT has been suggested to contribute to neuronal loss in tau transgenic mice (Andorfer et al., 2005; Ramsden et al., 2005; Santacruz et al., 2005). The timing of hyperphosphorylated tau protein aggregation into tangles and the question whether NFT directly lead to neuronal cell death are fundamental issues that are ideally suited for in vivo two-photon imaging techniques.
The rTg4510 transgenic mouse model, which reversibly expresses human tauP301L, is characterized by the development of NFT, neuronal loss, and memory impairment (Ramsden et al., 2005; Santacruz et al., 2005; Spires et al., 2006). These mice start to build up NFT at 2.5 months of age. Spires-Jones and co-workers repeatedly imaged rTg4510 mice to study the kinetics of NFT formation as well as the causal relationship between NFT and neuronal cell death. Surprisingly, they found that neurofibrillary tangle formation, caspase activation, and even the disruption of membrane integrity do not immediately induce neuronal cell death (Spires-Jones et al., 2008; de Calignon et al., 2009). Instead, neurofibrillary tangle formation occurred within 1 day after caspase activation, and the respective neurons remained healthy for several days (de Calignon et al., 2010). The authors thus propose that tangles may not be harmful or toxic per se (as opposed to soluble hyperphosphorylated tau), but rather serve a protective role against caspase activation and subsequent apoptosis. However, given the short follow-up period of 5 days, this hypothesis needs further evaluation in order to rule out a slower process of apoptosis. Moreover, it is unclear whether tangle bearing neurons maintain their physiological functional properties, although some evidence from in vitro experiments of rTg4510 cortical slice cultures points to no apparent functional differences between tangle – and non-tangle bearing neurons (Rocher et al., 2010).
These studies challenge our current view on the causal involvement of one of the main neuropathological hallmarks of AD and ultimately raise the question as to which neurons actually undergo apoptosis in mouse models of tauopathies including the rTg4510 transgenic or the 3xTg-AD mice. Do these neurons represent rare cases in which caspase activation does not result in the induction of tangle formation? And if so, how can the occurrence of NFT correlate with the degree of cognitive decline in AD? Can tangles be regarded as proxy for the concentration of soluble tau? Besides all these open questions the recent findings unraveled the temporal sequence of events, clearly establishing caspase activation as the initiating event in the process of tangle formation.
Functional in vivo Imaging
Imaging Neuronal Activity
Given the structural changes accompanying the deposition of amyloid plaques, it does not come as a surprise that neuronal function is also altered in neuronal structures neighboring plaques. Conventional electrophysiological methods to study the functional properties of neurons either yield a low spatial resolution (recordings of groups of neurons, e.g., field recordings or EEG) or only allow for single cell analysis (e.g., patch clamp). One elegant way of analyzing the spatio-temporal dynamics of cell populations, with single cell resolution, is in vivo two-photon imaging using calcium indicators. Calcium indicators take advantage of the fact that electrical activity of an excitable cell is tightly coupled to changes in intracellular calcium concentration (Tsien, 1981; Stosiek et al., 2003). Two main classes of indicators are available: synthetic dyes (e.g., OGB-1, Fluo-4, Rhod-2), which are applied by the bolus loading technique (Stosiek et al., 2003; Garaschuk et al., 2006), and genetically encoded calcium indicators (e.g., GCaMP, yellow chameleon, troponin C-based indicators), which are delivered to the brain by viral vectors or electroporation (Grewe and Helmchen, 2009). Genetically encoded calcium indicators allow for chronic functional imaging. In addition to being a proxy of neuronal activity, calcium plays an important role as intracellular signaling molecule in a number of processes, such as synaptic plasticity (transmitter release, excitability) and development (dendritic development, synaptogenesis, growth cone behavior; Ryglewski et al., 2007).
Calcium homeostasis and signaling are known to be disrupted in AD (Berridge, 2011). Hence, in vivo calcium imaging can be employed to address both the detection of altered calcium concentrations under baseline (i.e., resting) conditions, as well as changes in neuronal/glial activity. Two papers recently addressed the relationship between amyloid plaques and either intracellular calcium concentration or neuronal activity (Busche et al., 2008; Kuchibhotla et al., 2008). Garaschuk and co-workers for the first time described aberrant neuronal activity in the immediate vicinity of amyloid plaques in APP23xPS45 mice (Busche et al., 2008). Using Oregon Green BAPTA-1 AM (OGB-1 AM) as a calcium indicator, they found significantly more cells (50% in APP23xPS45 vs. 12% in WT) with extremely low or very high activity levels in frontal cortex (layer II/III) of APP23xPS45 compared to non-transgenic wildtype mice. When analyzing the spatial distribution of these neurons, they observed hyperactive cells exclusively within a 60 μm radius from the next amyloid plaque. Silent cells and such with regular firing rates were distributed throughout the cortex, with a relative increase of silent cells further away from plaques. Pre-depositing mice on the other hand did not show any abnormal neuronal activity patterns. In order to disentangle the mechanism underlying the hyperactivity, the authors used a pharmacological approach and increased or decreased GABAergic inhibition. Hyperactive cells responded to both treatments, but exhibited a lower relative frequency increase after application of a GABAA receptor antagonist compared to silent or regularly firing cells. Hence, the authors suggest that impaired synaptic inhibition underlies the observed hyperactivity (Busche et al., 2008).
A nicely complementing study more specifically investigated calcium concentrations within dendrites and spines using the ratiometric genetically encoded calcium indicator yellow chameleon (YC3.6; Kuchibhotla et al., 2008). A large proportion of dendrites in the immediate vicinity of amyloid plaques exhibited an overload of resting calcium concentration (Kuchibhotla et al., 2008). As a consequence, a disruption of spino-dendritic compartmentalization, a feature thought to be necessary for proper synaptic network integration (Augustine et al., 2003; Bloodgood and Sabatini, 2007), was observed. Furthermore, the study provided a link between structural and functional alterations, since more than half of the neurites with elevated calcium concentration were beaded, i.e., dystrophic (Kuchibhotla et al., 2008).
Taken together, these studies pinpoint amyloid plaques as mediators of functional alterations observed in AD mouse models, both on the cellular as well as on the subcellular level. Furthermore, they link structural changes to altered function, which presumably underlies the neuronal network dysfunction prominent in AD. To date, no study has addressed chronic changes in neuronal activity. Recently developed genetically encoded calcium indicators allow for repeated functional imaging, thereby providing a valuable tool to monitor the induction and progression of the above-mentioned functional neuronal alterations.
Imaging Astrocytic Activity
Another cell type known to exhibit large calcium signals are astrocytes. Astrocytes are involved in numerous physiological processes, such as neurovascular coupling, provision of nutrients to neurons, balance of extracellular ion concentration, and the uptake and release of neurotransmitters (“tripartite synapse”; Araque et al., 1999; Verkhratsky, 2010; Verkhratsky and Parpura, 2010). They also play a crucial role under pathological conditions, such as stroke, epilepsy, traumatic brain injuries, multiple sclerosis, and neurodegeneration (Verkhratsky et al., 2010; Sidoryk-Wegrzynowicz et al., 2011). Astrocytes are specifically implicated in AD pathology as they are, along with microglia, recruited to amyloid plaques (Itagaki et al., 1989; Nagele et al., 2004; Tuppo and Arias, 2005). Reactive astrogliosis may be triggered by factors released by affected cells as well as amyloid-β deposits, and results in an enhanced gap-junction coupling between cells (Pihlaja et al., 2008; Peters et al., 2009). The particular role of astrocytes is not yet fully understood. Whereas active glial cells might convey detrimental effects on neurons by releasing cytokines, interleukins, reactive oxygen species, or TNFα (Johnstone et al., 1999; Akiyama et al., 2000; Koistinaho et al., 2002), astrocytes are also linked to the clearance of plaques, as they can phagocytose deposited Aβ (Wyss-Coray et al., 2003; Koistinaho et al., 2004; Pihlaja et al., 2008).
To investigate the functional response of astrocytes to amyloid plaques in APPswe/PS1dE9 mice in vivo, Kuchibhotla et al. (2009) co-injected the calcium indicator OGB-1 AM and sulforhodamine 101(SR 101) in order to counterstain astrocytes (Nimmerjahn et al., 2004). They observed a globally elevated resting calcium concentration within the astrocytic network, independent of amyloid plaque proximity. Synchronous calcium transients were much more frequent in APP/PS1 than in non-transgenic control mice, and appeared in a coordinated fashion across long distances, but were uncoupled from neuronal activity (Kuchibhotla et al., 2009). Furthermore, the authors succeeded in detecting calcium waves between astrocytes in vivo, exclusively in mice already bearing amyloid plaques. These waves seem to originate in the plaque vicinity and travel radially over ~200 μm, thereby indicating a focal neuropathological insult (Scemes and Giaume, 2006). In conjunction with in vitro studies, these findings emphasize the active contribution of astrocytes to AD neuropathology (Rodriguez et al., 2009).
Conclusion
While great progress towards understanding the molecular mechanisms and genetic pre-dispositions of AD has been achieved within the past two decades, therapeutic options that halt or reverse the disease are still not available to date. Further research, unraveling underlying cellular and molecular mechanisms, and validating newly developed treatment approaches is thus needed. Performing transcranial in vivo two-photon microscopy on transgenic mouse models of AD has shed much light upon the kinetics and temporal sequence of neuropathological AD hallmarks and their associated structural and functional alterations in the living intact brain. In vivo imaging data stress the relevance of amyloid plaques as a focal source of neurotoxicity, as numerous pathological features, such as the occurrence of dystrophic neurites, neurite breakage, spine loss, clusters of hyperactive neurons as well as the initiation of intracellular calcium waves among astrocytes, are tightly spatially and temporally linked to plaque deposition. Furthermore, recent studies challenge our current understanding of the toxic nature of NFT by proposing a protective role for these intracellular aggregates. Future research will hopefully provide more insight into the causality of the observed pathological features as well as the efficacy of newly developed treatment approaches by, e.g., combining and correlating longitudinal imaging approaches and behavioral studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We like to thank Lary Walker, Georg Keller, and Mark Hübener for excellent comments on the manuscript. Figure 1D was kindly provided by Alix de Calignon. This work was supported by the Emmy Noether Program of the German Research Foundation (DFG; Melanie Meyer-Luehmann), the Hans and Ilse Breuer Foundation (Melanie Meyer-Luehmann), the Graduate School of Systemic Neurosciences (Sabine Liebscher), and the International Max Planck Research School for Molecular and Cellular Life Sciences (Sabine Liebscher).
References
Akiyama, H., Arai, T., Kondo, H., Tanno, E., Haga, C., and Ikeda, K. (2000). Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis. Assoc. Disord. 14(Suppl. 1), S47–S53.
Alzheimer, A. (1907). Über eine eigenartige Erkrankung der Hirnrinde. Allgeime Zeitschrift für Psychiatrie und Psychisch-Gerichtliche Medizin 64, 146–148.
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454.
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215.
Augustine, G. J., Santamaria, F., and Tanaka, K. (2003). Local calcium signaling in neurons. Neuron 40, 331–346.
Bacskai, B. J., Hickey, G. A., Skoch, J., Kajdasz, S. T., Wang, Y., Huang, G. F., Mathis, C. A., Klunk, W. E., and Hyman, B. T. (2003). Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 12462–12467.
Bacskai, B. J., Kajdasz, S. T., Christie, R. H., Carter, C., Games, D., Seubert, P., Schenk, D., and Hyman, B. T. (2001). Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat. Med. 7, 369–372.
Bhatt, D. H., Zhang, S., and Gan, W. B. (2009). Dendritic spine dynamics. Annu. Rev. Physiol. 71, 261–282.
Bittner, T., Fuhrmann, M., Burgold, S., Ochs, S. M., Hoffmann, N., Mitteregger, G., Kretzschmar, H., Laferla, F. M., and Herms, J. (2010). Multiple events lead to dendritic spine loss in triple transgenic Alzheimer’s disease mice. PLoS ONE 5, e15477. doi:10.1371/journal.pone.0015477
Bloodgood, B. L., and Sabatini, B. L. (2007). Ca(2+) signaling in dendritic spines. Curr. Opin. Neurobiol. 17, 345–351.
Bolmont, T., Haiss, F., Eicke, D., Radde, R., Mathis, C. A., Klunk, W. E., Kohsaka, S., Jucker, M., and Calhoun, M. E. (2008). Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 28, 4283–4292.
Brendza, R. P., Bacskai, B. J., Cirrito, J. R., Simmons, K. A., Skoch, J. M., Klunk, W. E., Mathis, C. A., Bales, K. R., Paul, S. M., Hyman, B. T., and Holtzman, D. M. (2005). Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J. Clin. Invest. 115, 428–433.
Burgold, S., Bittner, T., Dorostkar, M. M., Kieser, D., Fuhrmann, M., Mitteregger, G., Kretzschmar, H., Schmidt, B., and Herms, J. (2011). In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 121, 327–335.
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K. H., Haass, C., Staufenbiel, M., Konnerth, A., and Garaschuk, O. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689.
Calhoun, M. E., Burgermeister, P., Phinney, A. L., Stalder, M., Tolnay, M., Wiederhold, K. H., Abramowski, D., Sturchler-Pierrat, C., Sommer, B., Staufenbiel, M., and Jucker, M. (1999). Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. U.S.A. 96, 14088–14093.
Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M., Huang, D., Kidd, G., Dombrowski, S., Dutta, R., Lee, J. C., Cook, D. N., Jung, S., Lira, S. A., Littman, D. R., and Ransohoff, R. M. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 9, 917–924.
Cattepoel, S., Hanenberg, M., Kulic, L., and Nitsch, R. M. (2011). Chronic intranasal treatment with an anti-Abeta(30-42) scFv antibody ameliorates amyloid pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 6, e18296. doi:10.1371/journal.pone.0018296
Chopra, K., Misra, S., and Kuhad, A. (2011). Current perspectives on pharmacotherapy of Alzheimer’s disease. Expert Opin. Pharmacother. 12, 335–350.
Christie, R., Yamada, M., Moskowitz, M., and Hyman, B. (2001a). Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am. J. Pathol. 158, 1065–1071.
Christie, R. H., Bacskai, B. J., Zipfel, W. R., Williams, R. M., Kajdasz, S. T., Webb, W. W., and Hyman, B. T. (2001b). Growth arrest of individual senile plaques in a model of Alzheimer’s disease observed by in vivo multiphoton microscopy. J. Neurosci. 21, 858–864.
Cisse, M., Halabisky, B., Harris, J., Devidze, N., Dubal, D. B., Sun, B., Orr, A., Lotz, G., Kim, D. H., Hamto, P., Ho, K., Yu, G. Q., and Mucke, L. (2011). Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52.
Cohen, A. D., Ikonomovic, M. D., Abrahamson, E. E., Paljug, W. R., Dekosky, S. T., Lefterov, I. M., Koldamova, R. P., Shao, L., Debnath, M. L., Mason, N. S., Mathis, C. A., and Klunk, W. E. (2009). Anti-amyloid effects of small molecule Abeta-binding agents in PS1/APP mice. Lett. Drug Des. Discov. 6, 437.
D’Amore, J. D., Kajdasz, S. T., McLellan, M. E., Bacskai, B. J., Stern, E. A., and Hyman, B. T. (2003). In vivo multiphoton imaging of a transgenic mouse model of Alzheimer disease reveals marked thioflavine-S-associated alterations in neurite trajectories. J. Neuropathol. Exp. Neurol. 62, 137–145.
de Calignon, A., Fox, L. M., Pitstick, R., Carlson, G. A., Bacskai, B. J., Spires-Jones, T. L., and Hyman, B. T. (2010). Caspase activation precedes and leads to tangles. Nature 464, 1201–1204.
de Calignon, A., Spires-Jones, T. L., Pitstick, R., Carlson, G. A., and Hyman, B. T. (2009). Tangle-bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J. Neuropathol. Exp. Neurol. 68, 757–761.
DeKosky, S. T., Scheff, S. W., and Styren, S. D. (1996). Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 5, 417–421.
Denk, W., Strickler, J. H., and Webb, W. W. (1990). Two-photon laser scanning fluorescence microscopy. Science 248, 73–76.
Dickson, D. W. (2004). Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: cause or effect? J. Clin. Invest. 114, 23–27.
Domnitz, S. B., Robbins, E. M., Hoang, A. W., Garcia-Alloza, M., Hyman, B. T., Rebeck, G. W., Greenberg, S. M., Bacskai, B. J., and Frosch, M. P. (2005). Progression of cerebral amyloid angiopathy in transgenic mouse models of Alzheimer disease. J. Neuropathol. Exp. Neurol. 64, 588–594.
Feng, G., Mellor, R. H., Bernstein, M., Keller-Peck, C., Nguyen, Q. T., Wallace, M., Nerbonne, J. M., Lichtman, J. W., and Sanes, J. R. (2000). Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51.
Fryer, J. D., Taylor, J. W., Demattos, R. B., Bales, K. R., Paul, S. M., Parsadanian, M., and Holtzman, D. M. (2003). Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J. Neurosci. 23, 7889–7896.
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., Haass, C., Laferla, F. M., Kretzschmar, H., and Herms, J. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413.
Garaschuk, O., Milos, R. I., and Konnerth, A. (2006). Targeted bulk-loading of fluorescent indicators for two-photon brain imaging in vivo. Nat. Protoc. 1, 380–386.
Garcia-Alloza, M., Borrelli, L. A., Hyman, B. T., and Bacskai, B. J. (2010). Antioxidants have a rapid and long-lasting effect on neuritic abnormalities in APP:PS1 mice. Neurobiol. Aging 31, 2058–2068.
Garcia-Alloza, M., Dodwell, S. A., Meyer-Luehmann, M., Hyman, B. T., and Bacskai, B. J. (2006a). Plaque-derived oxidative stress mediates distorted neurite trajectories in the Alzheimer mouse model. J. Neuropathol. Exp. Neurol. 65, 1082–1089.
Garcia-Alloza, M., Robbins, E. M., Zhang-Nunes, S. X., Purcell, S. M., Betensky, R. A., Raju, S., Prada, C., Greenberg, S. M., Bacskai, B. J., and Frosch, M. P. (2006b). Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 24, 516–524.
Garcia-Alloza, M., Subramanian, M., Thyssen, D., Borrelli, L. A., Fauq, A., Das, P., Golde, T. E., Hyman, B. T., and Bacskai, B. J. (2009). Existing plaques and neuritic abnormalities in APP:PS1 mice are not affected by administration of the gamma-secretase inhibitor LY-411575. Mol. Neurodegener. 4, 19.
Giannakopoulos, P., Herrmann, F. R., Bussiere, T., Bouras, C., Kovari, E., Perl, D. P., Morrison, J. H., Gold, G., and Hof, P. R. (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500.
Giannakopoulos, P., Kovari, E., Gold, G., Von Gunten, A., Hof, P. R., and Bouras, C. (2009). Pathological substrates of cognitive decline in Alzheimer’s disease. Front. Neurol. Neurosci. 24, 20–29.
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890.
Gotz, J., Schild, A., Hoerndli, F., and Pennanen, L. (2004). Amyloid-induced neurofibrillary tangle formation in Alzheimer’s disease: insight from transgenic mouse and tissue-culture models. Int. J. Dev. Neurosci. 22, 453–465.
Grathwohl, S. A., Kalin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., Odenthal, J., Radde, R., Eldh, T., Gandy, S., Aguzzi, A., Staufenbiel, M., Mathews, P. M., Wolburg, H., Heppner, F. L., and Jucker, M. (2009). Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363.
Grewe, B. F., and Helmchen, F. (2009). Optical probing of neuronal ensemble activity. Curr. Opin. Neurobiol. 19, 520–529.
Grober, E., Dickson, D., Sliwinski, M. J., Buschke, H., Katz, M., Crystal, H., and Lipton, R. B. (1999). Memory and mental status correlates of modified Braak staging. Neurobiol. Aging 20, 573–579.
Grutzendler, J., Helmin, K., Tsai, J., and Gan, W. B. (2007). Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1097, 30–39.
Guillozet, A. L., Weintraub, S., Mash, D. C., and Mesulam, M. M. (2003). Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch. Neurol. 60, 729–736.
Haass, C. (2004). Take five – BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 23, 483–488.
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112.
Harper, J. D., and Lansbury, P. T. Jr. (1997). Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66, 385–407.
Hefendehl, J. K., Wegenast-Braun, B. M., Liebig, C., Eicke, D., Milford, D., Calhoun, M. E., Kohsaka, S., Eichner, M., and Jucker, M. (2011). Long-term in vivo imaging of beta-amyloid plaque appearance and growth in a mouse model of cerebral beta-amyloidosis. J. Neurosci. 31, 624–629.
Hofer, S. B., Mrsic-Flogel, T. D., Bonhoeffer, T., and Hubener, M. (2009). Experience leaves a lasting structural trace in cortical circuits. Nature 457, 313–317.
Holtmaat, A., Bonhoeffer, T., Chow, D. K., Chuckowree, J., De Paola, V., Hofer, S. B., Hubener, M., Keck, T., Knott, G., Lee, W. C., Mostany, R., Mrsic-Flogel, T. D., Nedivi, E., Portera-Cailliau, C., Svoboda, K., Trachtenberg, J. T., and Wilbrecht, L. (2009). Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat. Protoc. 4, 1128–1144.
Holtmaat, A. J., Trachtenberg, J. T., Wilbrecht, L., Shepherd, G. M., Zhang, X., Knott, G. W., and Svoboda, K. (2005). Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291.
Itagaki, S., Mcgeer, P. L., Akiyama, H., Zhu, S., and Selkoe, D. (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 24, 173–182.
Jack, C. R. Jr., Albert, M. S., Knopman, D. S., Mckhann, G. M., Sperling, R. A., Carrillo, M. C., Thies, B., and Phelps, C. H. (2011). Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 257–262.
Johnston, H., Boutin, H., and Allan, S. M. (2011). Assessing the contribution of inflammation in models of Alzheimer’s disease. Biochem. Soc. Trans. 39, 886–890.
Johnstone, M., Gearing, A. J., and Miller, K. M. (1999). A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J. Neuroimmunol. 93, 182–193.
Jung, C. K., Fuhrmann, M., Honarnejad, K., Van Leuven, F., and Herms, J. (2011). Role of presenilin1 in structural plasticity of cortical dendritic spines in vivo. J. Neurochem. 119, 1064–1073.
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553.
Klunk, W. E., Bacskai, B. J., Mathis, C. A., Kajdasz, S. T., Mclellan, M. E., Frosch, M. P., Debnath, M. L., Holt, D. P., Wang, Y., and Hyman, B. T. (2002). Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J. Neuropathol. Exp. Neurol. 61, 797–805.
Knobloch, M., and Mansuy, I. M. (2008). Dendritic spine loss and synaptic alterations in Alzheimer’s disease. Mol. Neurobiol. 37, 73–82.
Koenigsknecht-Talboo, J., Meyer-Luehmann, M., Parsadanian, M., Garcia-Alloza, M., Finn, M. B., Hyman, B. T., Bacskai, B. J., and Holtzman, D. M. (2008). Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J. Neurosci. 28, 14156–14164.
Koffie, R. M., Meyer-Luehmann, M., Hashimoto, T., Adams, K. W., Mielke, M. L., Garcia-Alloza, M., Micheva, K. D., Smith, S. J., Kim, M. L., Lee, V. M., Hyman, B. T., and Spires-Jones, T. L. (2009). Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U.S.A. 106, 4012–4017.
Koistinaho, M., Kettunen, M. I., Goldsteins, G., Keinanen, R., Salminen, A., Ort, M., Bures, J., Liu, D., Kauppinen, R. A., Higgins, L. S., and Koistinaho, J. (2002). Beta-amyloid precursor protein transgenic mice that harbor diffuse A beta deposits but do not form plaques show increased ischemic vulnerability: role of inflammation. Proc. Natl. Acad. Sci. U.S.A. 99, 1610–1615.
Koistinaho, M., Lin, S., Wu, X., Esterman, M., Koger, D., Hanson, J., Higgs, R., Liu, F., Malkani, S., Bales, K. R., and Paul, S. M. (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 10, 719–726.
Kosik, K. S., Joachim, C. L., and Selkoe, D. J. (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 83, 4044–4048.
Kuchibhotla, K. V., Goldman, S. T., Lattarulo, C. R., Wu, H. Y., Hyman, B. T., and Bacskai, B. J. (2008). Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59, 214–225.
Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T., and Bacskai, B. J. (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215.
Kurz, A., and Perneczky, R. (2011). Amyloid clearance as a treatment target against Alzheimer’s disease. J. Alzheimers Dis. 24(Suppl. 2), 61–73.
Lacor, P. N., Buniel, M. C., Furlow, P. W., Clemente, A. S., Velasco, P. T., Wood, M., Viola, K. L., and Klein, W. L. (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807.
Lee, S., Varvel, N. H., Konerth, M. E., Xu, G., Cardona, A. E., Ransohoff, R. M., and Lamb, B. T. (2010). CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 177, 2549–2562.
Lichtenthaler, S. F., Haass, C., and Steiner, H. (2011). Regulated intramembrane proteolysis – lessons from amyloid precursor protein processing. J. Neurochem. 117, 779–796.
Liu, Z., Condello, C., Schain, A., Harb, R., and Grutzendler, J. (2010). CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J. Neurosci. 30, 17091–17101.
Majewska, A. K., Newton, J. R., and Sur, M. (2006). Remodeling of synaptic structure in sensory cortical areas in vivo. J. Neurosci. 26, 3021–3029.
Masliah, E., Mallory, M., Hansen, L., Deteresa, R., Alford, M., and Terry, R. (1994). Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci. Lett. 174, 67–72.
Masliah, E., Terry, R. D., Alford, M., Deteresa, R., and Hansen, L. A. (1991). Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer’s disease. Am. J. Pathol. 138, 235–246.
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., Mcdonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249.
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., De Calignon, A., Rozkalne, A., Koenigsknecht-Talboo, J., Holtzman, D. M., Bacskai, B. J., and Hyman, B. T. (2008). Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724.
Morgan, D., Gordon, M. N., Tan, J., Wilcock, D., and Rojiani, A. M. (2005). Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J. Neuropathol. Exp. Neurol. 64, 743–753.
Nagele, R. G., Wegiel, J., Venkataraman, V., Imaki, H., and Wang, K. C. (2004). Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 25, 663–674.
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318.
Nimmerjahn, A., Kirchhoff, F., Kerr, J. N., and Helmchen, F. (2004). Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat. Methods 1, 31–37.
Nukina, N., and Ihara, Y. (1986). One of the antigenic determinants of paired helical filaments is related to tau protein. J. Biochem. 99, 1541–1544.
Oddo, S., Billings, L., Kesslak, J. P., Cribbs, D. H., and Laferla, F. M. (2004). Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43, 321–332.
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., Metherate, R., Mattson, M. P., Akbari, Y., and Laferla, F. M. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421.
Penzes, P., Cahill, M. E., Jones, K. A., Vanleeuwen, J. E., and Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293.
Peters, O., Schipke, C. G., Philipps, A., Haas, B., Pannasch, U., Wang, L. P., Benedetti, B., Kingston, A. E., and Kettenmann, H. (2009). Astrocyte function is modified by Alzheimer’s disease-like pathology in aged mice. J. Alzheimers Dis. 18, 177–189.
Pihlaja, R., Koistinaho, J., Malm, T., Sikkila, H., Vainio, S., and Koistinaho, M. (2008). Transplanted astrocytes internalize deposited beta-amyloid peptides in a transgenic mouse model of Alzheimer’s disease. Glia 56, 154–163.
Prada, C. M., Garcia-Alloza, M., Betensky, R. A., Zhang-Nunes, S. X., Greenberg, S. M., Bacskai, B. J., and Frosch, M. P. (2007). Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J. Neurosci. 27, 1973–1980.
Prince, M. B., Bryce, R., and Ferri, C. (2011). World Alzheimer Report 2011: The Benefits of Early Diagnosis and Intervention. London: Institute of Psychiatry, King’s College London.
Radde, R., Duma, C., Goedert, M., and Jucker, M. (2008). The value of incomplete mouse models of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 35(Suppl. 1), S70–S74.
Ramsden, M., Kotilinek, L., Forster, C., Paulson, J., Mcgowan, E., Santacruz, K., Guimaraes, A., Yue, M., Lewis, J., Carlson, G., Hutton, M., and Ashe, K. H. (2005). Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 25, 10637–10647.
Robbins, E. M., Betensky, R. A., Domnitz, S. B., Purcell, S. M., Garcia-Alloza, M., Greenberg, C., Rebeck, G. W., Hyman, B. T., Greenberg, S. M., Frosch, M. P., and Bacskai, B. J. (2006). Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J. Neurosci. 26, 365–371.
Rocher, A. B., Crimins, J. L., Amatrudo, J. M., Kinson, M. S., Todd-Brown, M. A., Lewis, J., and Luebke, J. I. (2010). Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp. Neurol. 223, 385–393.
Rodriguez, J. J., Olabarria, M., Chvatal, A., and Verkhratsky, A. (2009). Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 16, 378–385.
Ryglewski, S., Pflueger, H. J., and Duch, C. (2007). Expanding the neuron’s calcium signaling repertoire: intracellular calcium release via voltage-induced PLC and IP3R activation. PLoS Biol. 5, e66. doi:10.1371/journal.pbio.0050066
Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., Guimaraes, A., Deture, M., Ramsden, M., Mcgowan, E., Forster, C., Yue, M., Orne, J., Janus, C., Mariash, A., Kuskowski, M., Hyman, B., Hutton, M., and Ashe, K. H. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481.
Scemes, E., and Giaume, C. (2006). Astrocyte calcium waves: what they are and what they do. Glia 54, 716–725.
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384.
Schroeter, S., Khan, K., Barbour, R., Doan, M., Chen, M., Guido, T., Gill, D., Basi, G., Schenk, D., Seubert, P., and Games, D. (2008). Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J. Neurosci. 28, 6787–6793.
Shah, R. S., Lee, H. G., Xiongwei, Z., Perry, G., Smith, M. A., and Castellani, R. J. (2008). Current approaches in the treatment of Alzheimer’s disease. Biomed. Pharmacother. 62, 199–207.
Sidoryk-Wegrzynowicz, M., Wegrzynowicz, M., Lee, E., Bowman, A. B., and Aschner, M. (2011). Role of astrocytes in brain function and disease. Toxicol. Pathol. 39, 115–123.
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P., and Rivest, S. (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502.
Sperling, R. A., Jack, C. R. Jr., and Aisen, P. S. (2011). Testing the right target and right drug at the right stage. Sci. Transl. Med. 3, 111cm133.
Spires, T. L., Meyer-Luehmann, M., Stern, E. A., Mclean, P. J., Skoch, J., Nguyen, P. T., Bacskai, B. J., and Hyman, B. T. (2005). Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 25, 7278–7287.
Spires, T. L., Orne, J. D., Santacruz, K., Pitstick, R., Carlson, G. A., Ashe, K. H., and Hyman, B. T. (2006). Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 168, 1598–1607.
Spires-Jones, T. L., De Calignon, A., Matsui, T., Zehr, C., Pitstick, R., Wu, H. Y., Osetek, J. D., Jones, P. B., Bacskai, B. J., Feany, M. B., Carlson, G. A., Ashe, K. H., Lewis, J., and Hyman, B. T. (2008). In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J. Neurosci. 28, 862–867.
Spires-Jones, T. L., De Calignon, A., Meyer-Luehmann, M., Bacskai, B. J., and Hyman, B. T. (2011). Monitoring protein aggregation and toxicity in Alzheimer’s disease mouse models using in vivo imaging. Methods 53, 201–207.
Spires-Jones, T. L., Meyer-Luehmann, M., Osetek, J. D., Jones, P. B., Stern, E. A., Bacskai, B. J., and Hyman, B. T. (2007). Impaired spine stability underlies plaque-related spine loss in an Alzheimer’s disease mouse model. Am. J. Pathol. 171, 1304–1311.
Spires-Jones, T. L., Mielke, M. L., Rozkalne, A., Meyer-Luehmann, M., De Calignon, A., Bacskai, B. J., Schenk, D., and Hyman, B. T. (2009a). Passive immunotherapy rapidly increases structural plasticity in a mouse model of Alzheimer disease. Neurobiol. Dis. 33, 213–220.
Spires-Jones, T. L., Stoothoff, W. H., De Calignon, A., Jones, P. B., and Hyman, B. T. (2009b). Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 32, 150–159.
Stalder, M., Deller, T., Staufenbiel, M., and Jucker, M. (2001). 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol. Aging 22, 427–434.
Stalder, M., Phinney, A., Probst, A., Sommer, B., Staufenbiel, M., and Jucker, M. (1999). Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am. J. Pathol. 154, 1673–1684.
Stosiek, C., Garaschuk, O., Holthoff, K., and Konnerth, A. (2003). In vivo two-photon calcium imaging of neuronal networks. Proc. Natl. Acad. Sci. U.S.A. 100, 7319–7324.
Tanzi, R. E. (2005). Tangles and neurodegenerative disease – a surprising twist. N. Engl. J. Med. 353, 1853–1855.
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., Deteresa, R., Hill, R., Hansen, L. A., and Katzman, R. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580.
Trachtenberg, J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E., and Svoboda, K. (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794.
Tsai, J., Grutzendler, J., Duff, K., and Gan, W. B. (2004). Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 7, 1181–1183.
Tsien, R. Y. (1981). A non-disruptive technique for loading calcium buffers and indicators into cells. Nature 290, 527–528.
Tuppo, E. E., and Arias, H. R. (2005). The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 37, 289–305.
Van Dorpe, J., Smeijers, L., Dewachter, I., Nuyens, D., Spittaels, K., Van Den Haute, C., Mercken, M., Moechars, D., Laenen, I., Kuiperi, C., Bruynseels, K., Tesseur, I., Loos, R., Vanderstichele, H., Checler, F., Sciot, R., and Van Leuven, F. (2000). Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am. J. Pathol. 157, 1283–1298.
Verkhratsky, A., Olabarria, M., Noristani, H. N., Yeh, C. Y., and Rodriguez, J. J. (2010). Astrocytes in Alzheimer’s disease. Neurotherapeutics 7, 399–412.
Verkhratsky, A., and Parpura, V. (2010). Recent advances in (patho)physiology of astroglia. Acta Pharmacol. Sin. 31, 1044–1054.
Wakasaya, Y., Kawarabayashi, T., Watanabe, M., Yamamoto-Watanabe, Y., Takamura, A., Kurata, T., Murakami, T., Abe, K., Yamada, K., Wakabayashi, K., Sasaki, A., Westaway, D., Hyslop, P. S., Matsubara, E., and Shoji, M. (2011). Factors responsible for neurofibrillary tangles and neuronal cell losses in tauopathy. J. Neurosci. Res. 89, 576–584.
Winkler, D. T., Bondolfi, L., Herzig, M. C., Jann, L., Calhoun, M. E., Wiederhold, K. H., Tolnay, M., Staufenbiel, M., and Jucker, M. (2001). Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci. 21, 1619–1627.
Wu, H. Y., Hudry, E., Hashimoto, T., Kuchibhotla, K., Rozkalne, A., Fan, Z., Spires-Jones, T., Xie, H., Arbel-Ornath, M., Grosskreutz, C. L., Bacskai, B. J., and Hyman, B. T. (2010). Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 30, 2636–2649.
Wyss-Coray, T., Loike, J. D., Brionne, T. C., Lu, E., Anankov, R., Yan, F., Silverstein, S. C., and Husemann, J. (2003). Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 9, 453–457.
Yan, P., Bero, A. W., Cirrito, J. R., Xiao, Q., Hu, X., Wang, Y., Gonzales, E., Holtzman, D. M., and Lee, J. M. (2009). Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J. Neurosci. 29, 10706–10714.
Keywords: amyloid plaques, neurofibrillary tangles, dendritic spines, glial cells, multiphoton imaging, microglia, calcium imaging, CAA
Citation: Liebscher S and Meyer-Luehmann M (2012) A peephole into the brain: neuropathological features of Alzheimer’s disease revealed by in vivo two-photon imaging. Front. Psychiatry 3:26. doi: 10.3389/fpsyt.2012.00026
Received: 03 January 2012; Paper pending published: 23 January 2012;
Accepted: 07 March 2012; Published online: 02 April 2012.
Edited by:
Silke Vogelgesang, University of Greifswald, GermanyReviewed by:
Romy Baguhl, University of Greifswald, GermanyOlga Garaschuk, University of Tübingen, Germany
Copyright: © 2012 Liebscher and Meyer-Luehmann. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Melanie Meyer-Luehmann, Department of Neurology, Neurocenter, University of Freiburg, Breisacherstr. 64, 79106 Freiburg, Germany. e-mail:bWVsYW5pZS5tZXllci1sdWVobWFubkB1bmlrbGluaWstZnJlaWJ1cmcuZGU=