 Danni Meng1,2†
Danni Meng1,2† Tianxin Lu1,2,3†Meng He1,2Yuze Ren1,2Mumei Fu1,2Yuxiao Zhang1,2Peifeng Yang1,2Xinyu Lin1,2Yong Yang1,2
Tianxin Lu1,2,3†Meng He1,2Yuze Ren1,2Mumei Fu1,2Yuxiao Zhang1,2Peifeng Yang1,2Xinyu Lin1,2Yong Yang1,2 Ying Zhang4
Ying Zhang4 Yuchen Yang5
Yuchen Yang5 Xiang Jin1,2*
Xiang Jin1,2*- 1Ministry of Education Key Laboratory for Ecology of Tropical Islands, Key Laboratory of Tropical Animal and Plant Ecology of Hainan Province, College of Life Sciences, Hainan Normal University, Haikou, China
- 2Hainan Observation and Research Station of Dongzhaigang Mangrove Wetland Ecosystem, Hainan Normal University, Haikou, China
- 3Key Laboratory of Xinjiang Phytomedicine Resource and Utilization of Ministry of Education, Key Laboratory of Oasis Town and Mountain-Basin System Ecology of Xinjiang Production and Construction Corps, College of Life Sciences, Shihezi University, Shihezi, China
- 4Engineering and Technological Research in Protection and Utilization of Mangrove Rare and Endangered Species, Lingnan Normal University, Zhanjiang, China
- 5State Key Laboratory of Biocontrol, School of Ecology, Sun Yat-sen University, Shenzhen, China
Chloroplast and mitochondrial genomes harbor crucial information that can be utilized for elucidating plant evolution and environmental adaptation. The organellar genomic characteristics of Goodeniaceae, a sister family to Asteraceae, remain unexplored. Here, using a combination of short-read and long-read sequencing technologies, we successfully assembled the complete organellar genomes of two Goodeniaceae species native to China, Scaevola taccada and S. hainanensis. Chloroplast genome collinearity analysis revealed that Scaevola expanded its genome length through inverted repeat expansion and large single copy fragment duplication, resulting in 181,022 bp (S. taccada) and 182,726 bp (S. hainanensis), ~30 kb increase compared to its related species. Mitochondrial genomes of two Scaevola species exhibit multi-ring topology, forming dual mitochondrial chromosomes of 314,251 bp (S. taccada) and 276,175 bp (S. hainanensis). Sequence variation analysis demonstrated substantial chloroplast sequence divergence (Pi = 0.45) and an increase in gene copy number within the genus. Relative synonymous codon usage (RSCU) analysis revealed that Scaevola chloroplast has a higher bias for A/U-ending codons than mitochondria, with chloroplasts RSCU values ranging from 0.32 to 1.94, whereas mitochondrial RSCU values ranging from 0.38 to 1.62. Phylogenetic analyses support the monophyly of the Asteraceae-Goodeniaceae sister group, whereas the extended evolutionary branches of Scaevola, coupled with mitochondrial collinearity analysis, indicate rapid organellar genome evolution of Scaevola. Organellar-nuclear horizontal gene transfer analysis identified specific increased in the copy numbers of photosynthesis-related genes and chloroplast-nuclear transfer events in S. taccada. Our study not only provides insights for understanding environmental adaptation mechanisms of coastal plants, but also contributes to elucidating organellar genome evolution in Scaevola and Goodeniaceae.
1 Introduction
Goodeniaceae is a distinctive taxon within the Asterales order of angiosperms, comprising approximately 11 genera and over 400 species, primarily originating from the Australian continent, where about 95% of species remain endemic (Ghisalberti, 2004). Molecular phylogenetic studies suggest that Goodeniaceae shares a common ancestor with Asteraceae, with their divergence occurring approximately 80 million years ago, highly consistent with the geological timing of Australian continent’s separation from the Gondwana supercontinent (Shen et al., 2023). Scaevola is the only genus within Goodeniaceae that has successfully dispersed beyond Australia, expanding into tropical and subtropical coastal zones through efficient long-distance dispersal mechanisms, demonstrating its remarkable propagation capability and environmental adaptability (Jabaily et al., 2014). S. taccada is widely distributed across the coastlines of Pacific islands and Indian Ocean, and has spread to the Caribbean region as an invasive species, potentially leading to ecological threats to native species in Puerto Rico, such as S. plumieri (Swensen et al., 2024). As a typical coastal pioneer plant, S. taccada exhibits high tolerance to salt spray, drought resistance, and wind-breaking, as well as the capability for sand-stabilizing (Lee et al., 2020; Starman and Lombardini, 2006; Toscano et al., 2020). These advantages make it an ideal plant resources for coastal ecosystem restoration (Walker et al., 1997). Extracts from S. taccada exhibit antiviral and anticancer activities, suggesting potential medicinal value (Locher et al., 1996). Population genetic studies have uncovered complex gene flow patterns and substantial population differentiation in S. taccada, shedding light on the adaptive evolution and dispersal mechanisms of coastal species. In the Hawaiian Islands, Scaevola exhibits a typical case of hybrid speciation (Howarth and Baum, 2005). However, the limited molecular characterization of Scaevola species contrasts with their significant ecological and potential medical values.
Plant organelle genomes are indispensable for elucidating plant origin, evolution, and adaptation, due to their unique genetic traits, including maternal inheritance, high conservation, and abundant copy numbers, as well as their critical physiological functions in photosynthesis and energy metabolism (Wang et al., 2024a). The chloroplast genome is structurally conserved, typically containing 110-130 genes, with only 0.5-1.0 base substitutions per million years, making it an ideal molecular marker for reconstructing higher-order phylogenies (Curtis and Clegg, 1984). For instance, comparative analysis of Asterales chloroplast genomes has revealed early divergence events between Goodeniaceae and Calyceraceae (Panero and Crozier, 2016). Additionally, simple sequence repeat (SSR) markers developed from chloroplast intergenic regions, such as trnL–trnF and psbA–trnH, have been successfully applied in genetic diversity studies of crop wild relatives (Lima et al., 2021; He et al., 2024). Emerging research has demonstrated that chloroplast genomes in halophytes often display unique selection patterns and retrograde signalings. For instance, adaptive evolution of the ndhF gene may play an important role in resistance against high-salt environments (Zheng et al., 2025), while the expression of some salt-tolerance genes is also influenced by organelle genomes (Robles and Quesada, 2019). In contrast, mitochondrial genomes have greater dynamic complexity, including horizontal gene transfer, frequent recombination, and substantial size variation (0.1–10 Mb), which are closely linked to plant regulatory mechanisms for environmental adaptation (Alverson et al., 2010). Mitochondrial genes, such as cox1, nad5, and atp6, exhibit significant positive selection signals under environmental stress (Qiu et al., 2021). However, despite the NCBI database has cataloged approximately 13,000 plant chloroplast genomes, these records predominantly focus on economic crops and model plants, with research on wild plants remaining insufficient. Compared to chloroplast, the number of published plant complete mitochondrial genomes (~673) is one to two orders of magnitude lower (Wang et al., 2024b). The application of long-read sequencing technologies has significantly advanced organelle genome research, especially in resolving complex structures and repetitive sequences in mitogenomes (Liu et al., 2024).
In China, only two Scaevola species are distributed: the widespread S. taccada and the China-Vietnam endemic species S. hainanensis (Flora of China, 2020). Chromosome-level genome assembly reveals that, compared to S. hainanensis, S. taccada has undergone specific gene family expansions and adaptive modifications to withstand coastal environmental stresses, such as intense light, high salinity air, and poor soil conditions in coastal areas (Li et al., 2023). Within the Goodeniaceae family, only one chloroplast genome sequences of Scaevola has been reported, as outgroup to a large-scale study of the Caryophyllales (Yao et al., 2019). Population genetic study based on chloroplast SSR markers of S. taccada has revealed its dispersal pathways of Western Pacific island populations (Banerjee et al., 2022). However, mitochondrial genome studies in Scaevola remains largely unexplored, limiting our comprehensive understanding of its evolutionary diversity.
In this study, we employed short-read and long-read sequencing technologies to assemble the organellar genomes of two Scaevola species native to China. We conducted comparative analyses of the organellar genomes between these two species, including gene contents, repeat distributions, codon usage, genomic structural variations, RNA editing sites and phylogenetic relationships. An in-depth investigation of Scaevola organellar genomes not only contributes to elucidating nuclear-organellar genome co-evolution, but also provides critical insights for understanding the evolutionary divergence between Scaevola and its related species.
2 Materials and methods
2.1 Plant material and DNA sequencing
Young leaves of S. taccada and S. hainanensis were harvest from individuals planted in the plant garden of the college of life sciences in Hainan Normal University. Leaves were immediately frozen in liquid nitrogen and ground into fine powder. Genomic DNA extraction was performed using the CTAB method. DNA quality and quantity were evaluated using NanoDrop (Thermo Fisher Scientific, USA). Sequencing was carried out on the Illumina NovaSeq 6000 (Illumina Inc., USA) to generate paired-end short reads (2 × 150 bp). The Oxford Nanopore PromethION platform (Oxford Nanopore Technologies, UK) was employed to generate long-reads. High-quality short reads were generated by adapter trimming and quality filtering using fastp software, while long reads were used to facilitate the assembly of the complex structural architecture of organellar genomes.
2.2 Organellar genomes assembly
The chloroplast genome was assembled using ptGAUL with the parameters -t 20 -f 3000 (Zhou et al., 2022). The long-read Nanopore data were used for initial contig construction. Illumina short reads were mapped to the draft assembly using BWA v0.7.17 (Li and Durbin, 2009) for base error correction. Assembly errors were corrected through multiple rounds of iterative polishing using Pilon. v1.24 (Walker et al., 2014).
The mitochondrial genomes were assembled using GSAT v1.1.2 (He et al., 2023a), a hybrid assembler that integrates Illumina and Nanopore data. Flye v2.9 (Kolmogorov et al., 2019) was employed as an auxiliary assembly tool to ensure the comprehensiveness of the long-read assembly. Assembly graphs were visualized and validated using Bandage v0.8.1 (Wick et al., 2015). Conflicting regions between assemblies were resolved by cross-verification, and complex structural variations (e.g., repeats or rearrangements) were validated by BLASTN alignment (-task megablast) of Nanopore reads (Camacho et al., 2009). Only structures supported by long-read coverage over 10× were retained.
2.3 Genome annotation and visualization
Chloroplast and mitochondrial genomes were annotated using GeSeq (Tillich et al., 2017) with default parameters. Transfer RNA and ribosomal RNA genes were manually curated by comparison with closely related species. Open reading frames (ORFs) were predicted using ORFfinder (NCBI). The annotated genomes were visualized with OGDRAW (Greiner et al., 2019).
2.4 Sequence features analyses
Simple sequence repeats (SSR) were identified using MISA (http://pgrc.ipk-gatersleben.de/misa/) with the minimum repeat parameters set to 10, 6, 4, 3, 3, and 3 repeats for mono-, di-, tri-, tetra-, penta-, and hexa- nucleotide SSRs, respectively. The online software REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer) was used to detect repeat types and numbers for long sequence repeats, with a maximum computed repeats of 50 and minimal repeat size of 30 bp. Potential RNA editing sites in protein-coding genes were predicted using PREP Suite online tools (http://prep.unl.edu/) with default parameters. MEGA10 was used to calculate relative synonymous codon usage (RSCU) values to quantify the codon usage patterns. And DNaSP5 component of MEGA10 was used to calculate synonymous (Ks) and nonsynonymous (Ka) substitution rates for evaluating selective pressure.
2.5 Synteny and nuclear-organellar DNA transfer analyses
Syntenic relationships between Scaevola and its close relatives were analyzed using SyRI v1.6.3 (Goel et al., 2019), with visualization performed via plotsR. Potential nuclear-cytoplasmic gene transfers were identified by BLASTN alignment of organellar genomes against the nuclear genome, using the following parameters: -word_size 9 -evalue 1e-5 -reward 2 -gapopen 5 -gapextend 2 -penalty -3. Regions with >75% identity and length >100 bp were retained (Richardson and Palmer, 2007). Results were visualized using NGenomeSyn (He et al., 2023b).
2.6 Phylogenetic analysis
Maximum likelihood phylogenetic trees were reconstructed using IQ-TREE V.1.6.8 in PhyloSuite v1.2.3 (Zhang et al., 2020) under the Partition Mode, with 1000 bootstrap replicates. Alignments were performed using the chloroplast and mitochondrial protein-coding genes from Scaevola and related taxa, respectively. Phylogenetic trees were visualized and annotated using iTOL (Letunic and Bork, 2021).
3 Results
3.1 Assembly and sequence features of the organellar genomes of two Scaevola species
Sequencing of DNA extracted from leaves of two Scaevola species yielded 16 Gb, 15 Gb Illumina short-reads and 26.6 Gb and 10.3 Gb Nanopore long-reads for S. taccada and S. hainanensis, respectively. Multi-circular topological structures were generated for the mitochondrial genome assemblies (Figure 1). Numerous studies have reported the existence of multi-circular molecules in plant mitochondria (Butenko et al., 2024; Wang et al., 2025; Wu et al., 2022). Consequently, manual corrections were performed by aligning long-reads data to shared fragments that could potentially exhibit multiple connection patterns. For S. taccada, one shared fragment was identified as a forward repeat, with all possible scaffold arrangements supported by the long-reads. This led to the assembly of three potential mitochondrial genome topological structures (TYPE I, II and III) sharing identical scaffold sequences but different arrangements (Figure 1B). These alternative topological structures affected only the scaffold connection orientations without altering gene content, therefore, they were considered as a single circular chromosome in subsequent analyses. However, for S. hainanensis, two chromosomes with different sequences (resulting from distinct topological structures) were finally identified (Figure 1D) and analyzed as separate chromosomes (chromosome1 and chromosome2) in subsequent analyses.
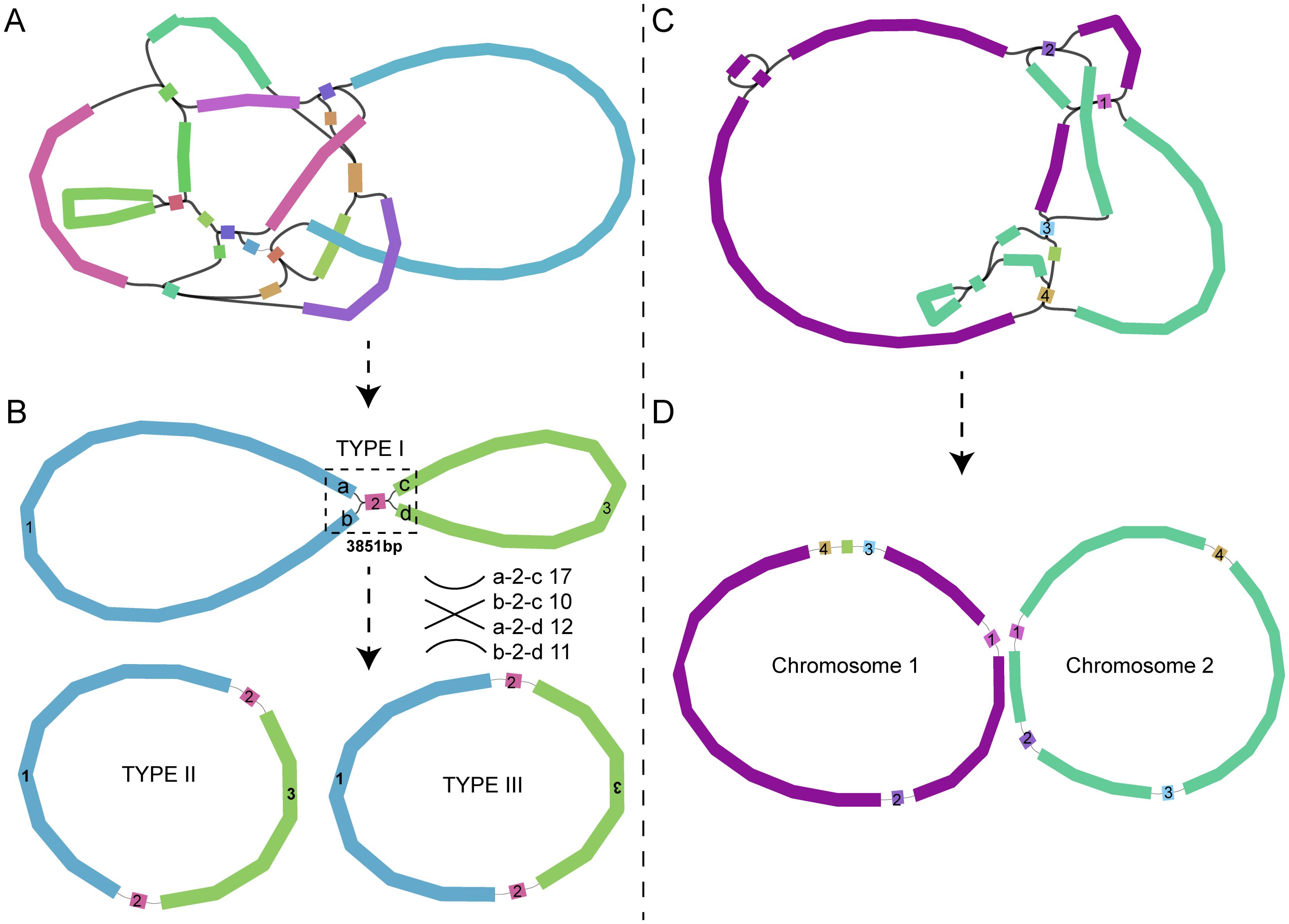
Figure 1. Mitochondrial genome assemblies of S. taccada (A, B) and S. hainanensis (C, D). The draft mitochondrial genomes (A, C) were assembled using Illumina short-reads data, and the final chromosomes (B, D) were further determined using long-reads data. In each of the panel (A, C), the scaffolds connected together (on the same long read) are presented by the same colors.
The statistical information of chloroplast and mitochondrial genomes of two Scaevola species is provide in Tables 1, 2. Generally, the chloroplast genomes of S. taccada and S. hainanensis are 181,022 bp and 182,726 bp in length, which are longer than the typical chloroplast genome of angiosperm (115–165 kb). Accordingly, the protein coding gene numbers are 88 and 86 for S. taccada and S. hainanensis (Table 1). The mitochondrial genomes of S. taccada and S. hainanensis are 314,251 bp and 276,175 bp in length, both encoding 24 proteins (Table 2). Detailed gene names and gene classifications of S. taccada and S. hainanensis are shown in Supplementary Tables S1, S2.
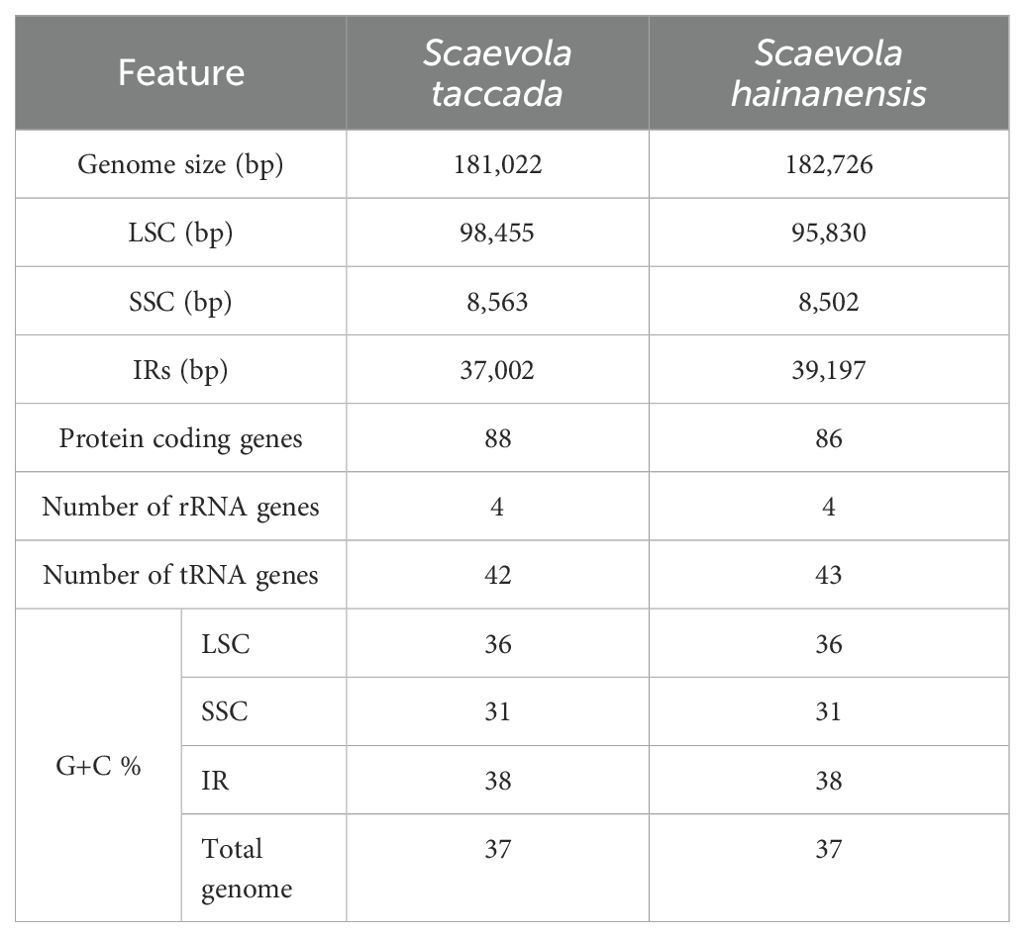
Table 1. Comparison of chloroplast genome features of two Scaevola species.
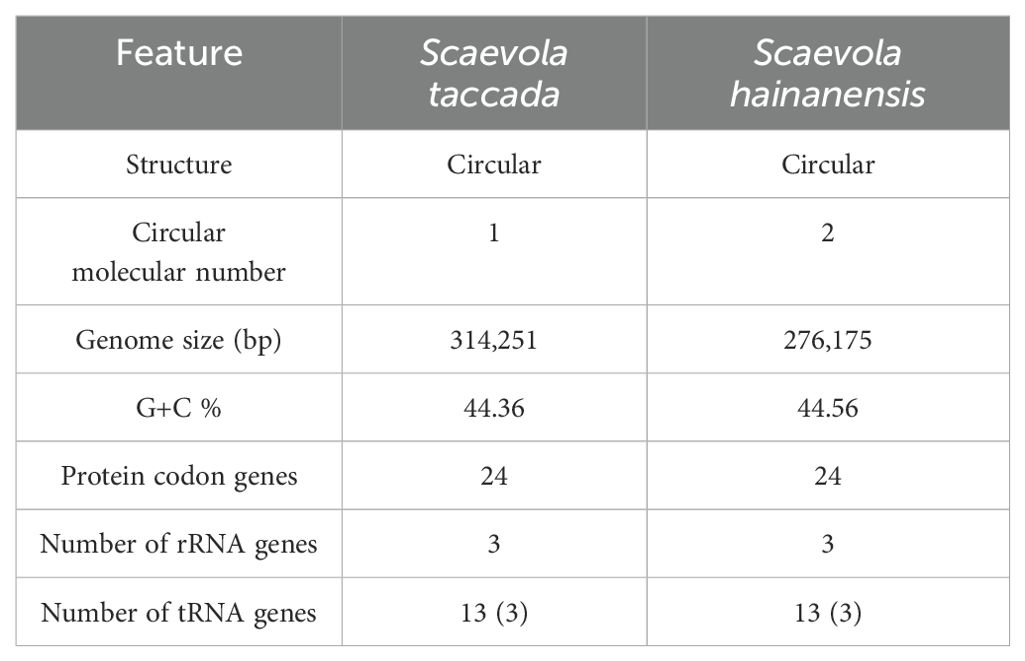
Table 2. The mitochondrial genome data of two Scaevola species.
The circular maps of organellar genomes of two Scaevola species were conducted to illustrate their structure features. The plastomes are consist of the typically conserved four distinctive parts (Figure 2A). The lengths of chloroplast genome of Scaevola are around 30 kb longer than the typical chloroplast genome of the family Asteraceae, which shares a common ancestor with the family Goodeniaceae. Notably, both Scaevola species have a short single copy (SSC) of 8.5 kb, much shorter than that of Asteraceae species, which are already reported as “small SSC” (Cho et al., 2024; Chen et al., 2022). To investigate the reason for this phenomena, we performed collinear analysis between Scaevola plastomes and two Asterales plastomes (Nymphoides peltata from Menyanthaceae and Lactuca sativa from Asteraceae). Results showed that a fragment duplication and rearrangement events in LSC, as well as a duplication event in SSC, together leading to IR expansion and SSC contraction in Scaevola species (Figure 3). The LSC duplication and rearrangement events included some protein coding regions that are important for photosynthesis, resulting in gene duplication in chloroplast genomes of Scaevola, such as rbcL. The copy numbers of several important chloroplast coding genes are also duplicated in Scaevola compared to 13 related species, including accD, matK, ndhF, ndhH, psbA, rbcL, rpl22 and rps15 (Supplementary Figure S1).
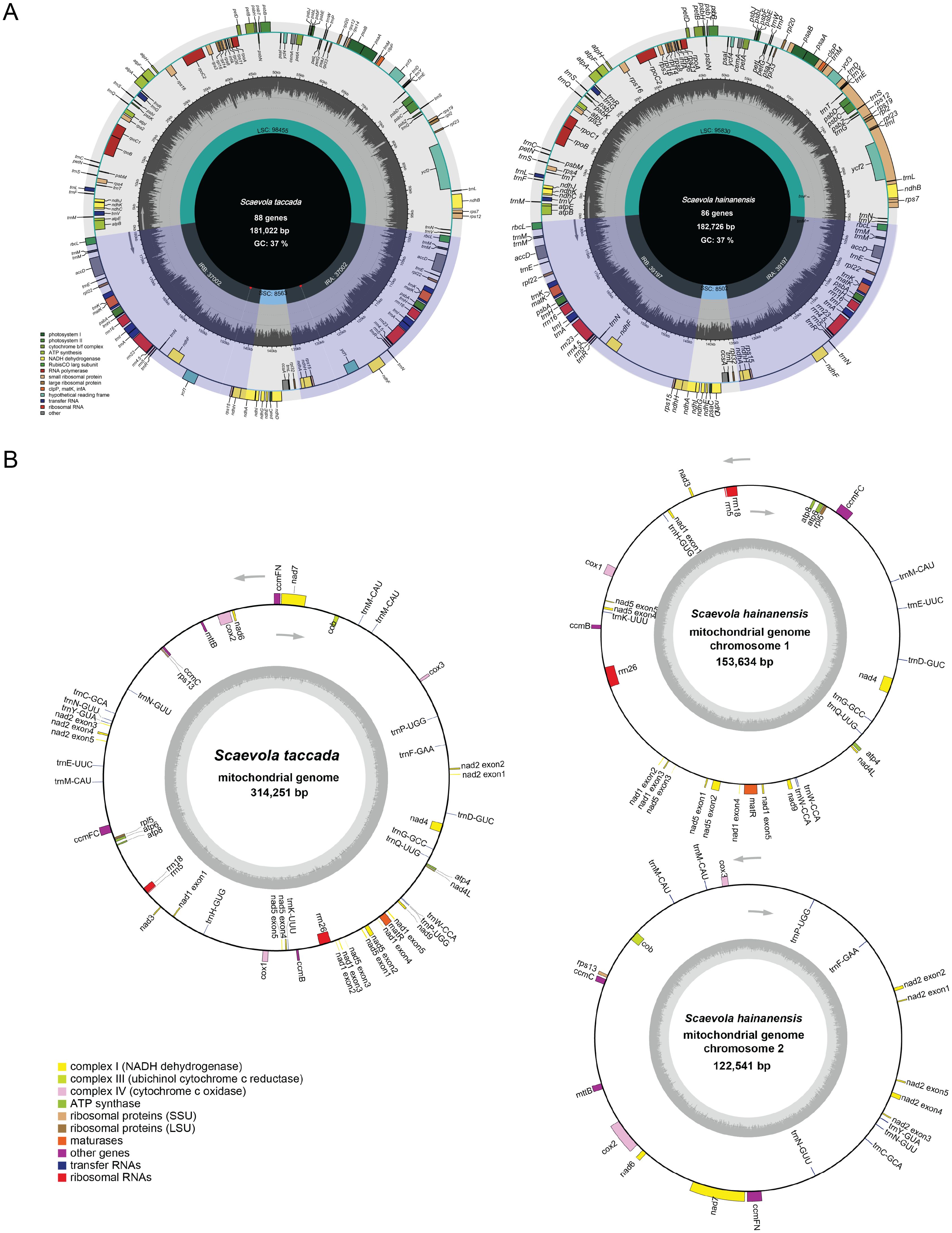
Figure 2. Circular maps of the chloroplast (A) and mitochondrial (B) genomes of S. taccada and S. hainanensis. For each panel, genes outside the circles are transcribed clockwise and genes inside the circle are transcribed counter-clockwise. The dark-gray inner circle represents GC contents and light-gray one represents the AT contents. The coding genes belonging to different classes are presented by different colors.
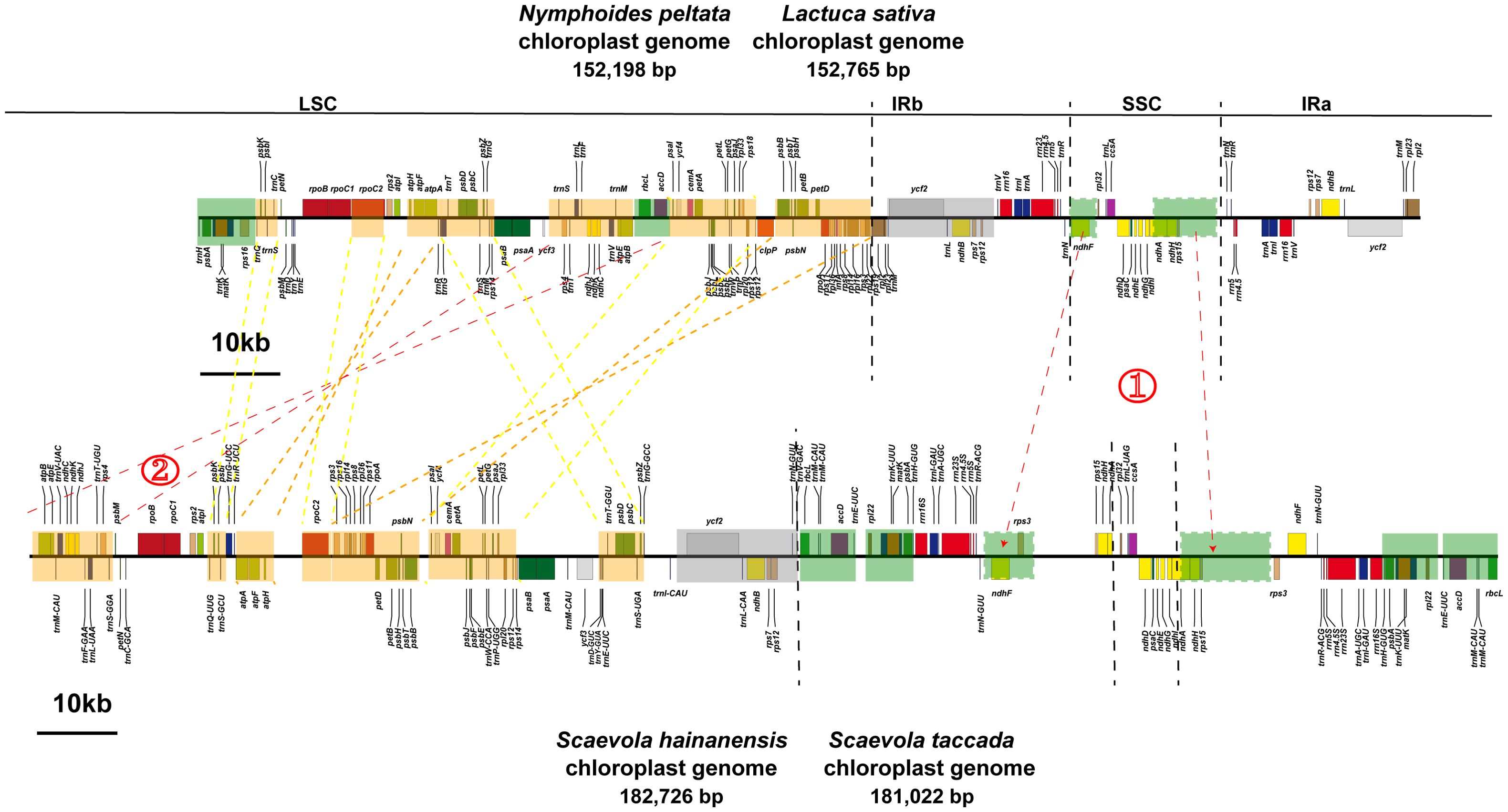
Figure 3. Fragment rearrangement in the LSC region and IR expansion in the chloroplast genomes of two Scaevola species. The orange blocks represent fragment rearrangement in the LSC region, while the green blocks represent IR expansion and SSC contraction.
3.2 Sequence variation and codon usage of organellar genomes of two Scaevola species
The chloroplast genomes of S. taccada and S. hainanensis are conserved in general (identity of 98.17%), with several regions showing substantially high variation. As is shown, the nucleotide diversity (Pi) of the two chloroplast genomes is higher than 0.1 in the genome regions of rpoC2-rps3, rps3-rpl16, trnM-ycf3, rpl2-rpl23 (with the highest Pi value of 0.45), ycf2, accD-trnE, and ycf1 (Supplementary Figure S2).
RSCU of organellar genomes of both Scaevola species were analyzed. A total of 24,050 and 25,742 codons were identified in the protein coding genes (PCGs) of chloroplast genomes of S. taccada and S. hainanensis (Supplementary Table S3), as well as 7,685 and 7,988 codons in their mitochondrial genomes (Supplementary Table S4). Leucine is the most frequently used amino acid (11.26%), followed by isoleucine (8.45%) and serine (7.81%), while cysteine has the lowest abundance, with a proportion of 1.17%. Consistent with previous studies, organellar PCGs of Scaevola showed strong bias on A/U-ending codons, with RSCU values > 1 (Figure 4). The RSCU values of all chloroplast codons ranged from 0.32 (CUC for leucine in S. hainanensis) to 1.94 (UUA for leucine in S. hainanensis). For mitochondrial genome, the RSCU values ranged from 0.38 (UAG for stop codon) to 1.62 (GCU for alanine in S. hainanensis).
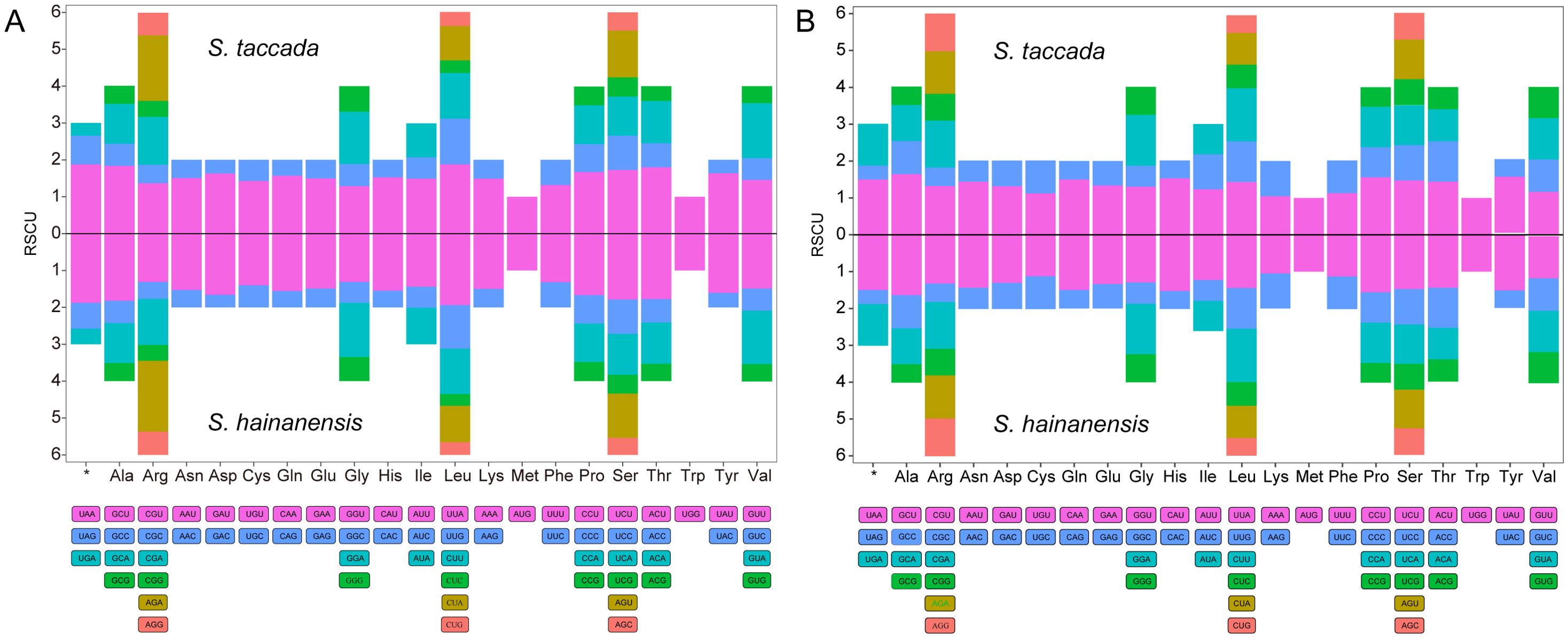
Figure 4. Relative synonymous codon usage (RSCU) histogram of the chloroplast (A) and mitochondrial (B) genomes of two Scaevola species. For each amino acid, different codons are shown in different colors.
Furthermore, repeat sequences in the organellar genomes of two Scaevola species were analyzed. In the mitochondrial genomes, the most abundant SSR is tetrameric repeats (32 and 25 in S. taccada and S. hainanensis). The palindromic dispersed repeats are more in S. hainanensis (80) than in S. taccada (42), while the longest palindromic repeats in S. taccada (3,871 bp) is much longer than in S. hainanensis (169 bp) (Supplementary Table S5; Figure 5).
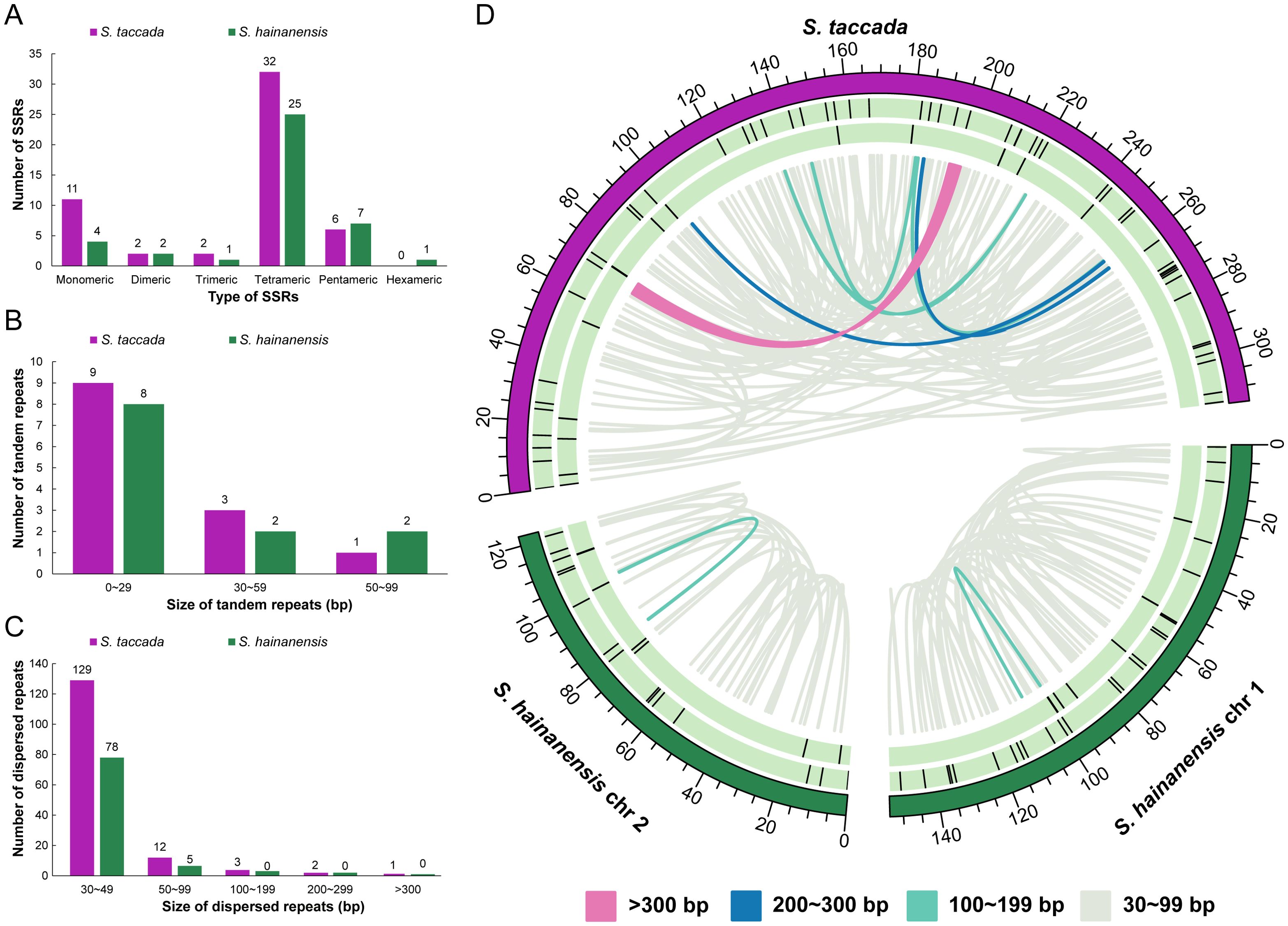
Figure 5. The distribution of repeat elements in two mitochondrial genomes. (A) The frequency of identified SSRs. (B) The frequency of identified tandem repeats. (C) The frequency of identified dispersed repeats. (D) Distribution of repeat elements in mitochondrial genomes. The outer circle represents the SSRs, followed by tandem repeats and the inner lines represent the dispersed repeats.
A total of 289 and 287 potential RNA editing sites were identified in chloroplast genomes of S. taccada and S. hainanensis, with the ndhB gene possessing the most RNA editing sites (22 and 19 in S. taccada and S. hainanensis). Additionally, 347 and 350 RNA editing sites were identified in the mitochondrial genomes of two Scaevola species, with the ccmB gene possessing the most RNA editing sites (32 and 33 in S. taccada and S. hainanensis) (Supplementary Figure S3).
3.3 Phylogenetic and syntenic analyses of organellar genomes of two Scaevola species
Phylogenetic trees were constructed using the chloroplast and mitochondrial genomes of S. taccada and S. hainanensis, and other related species, respectively. Complete chloroplast and mitochondrial genomes of Goodeniaceae are limited to only two species reported in this study. The chloroplast genomes of 5 representative Asteraceae species and 6 Asterales species other than Asteraceae were obtained from NCBI Genbank. In the tree constructed using 62 common chloroplast genes, the two Scaevola species formed a clade and exhibited the closest relationship with Asteraceae species (Figure 6A). For mitochondrial genomes, much fewer sequences are available, leading to a smaller scale of genome dataset for phylogenetic analysis. Seventeen common mitochondrial genes were used to construct a phylogenetic tree of 11 species, including 7 representative Asterales species, 2 Scaevola species, and 2 outgroups (Ilex pubescens and Psychotria viridis). The branches of mitochondrial based phylogenetic trees are longer than those of chloroplast genome based trees, indicating a faster nucleotide substitution rate in mitochondrial genome (Figure 6B). A bigger tree performed with 50 available mitochondrial genomes of Asterales species showed similar branch length (Supplementary Figure S4). The selective pressure analysis further revealed higher Ka/Ks values in mitochondrial-coded genes compared to chloroplast-coded genes (Supplementary Figure S5), indicating that the organelle evolutionary strategies of Scaevola species may be influenced by their environmental pressures, especially concerning the mitochondrial genome.
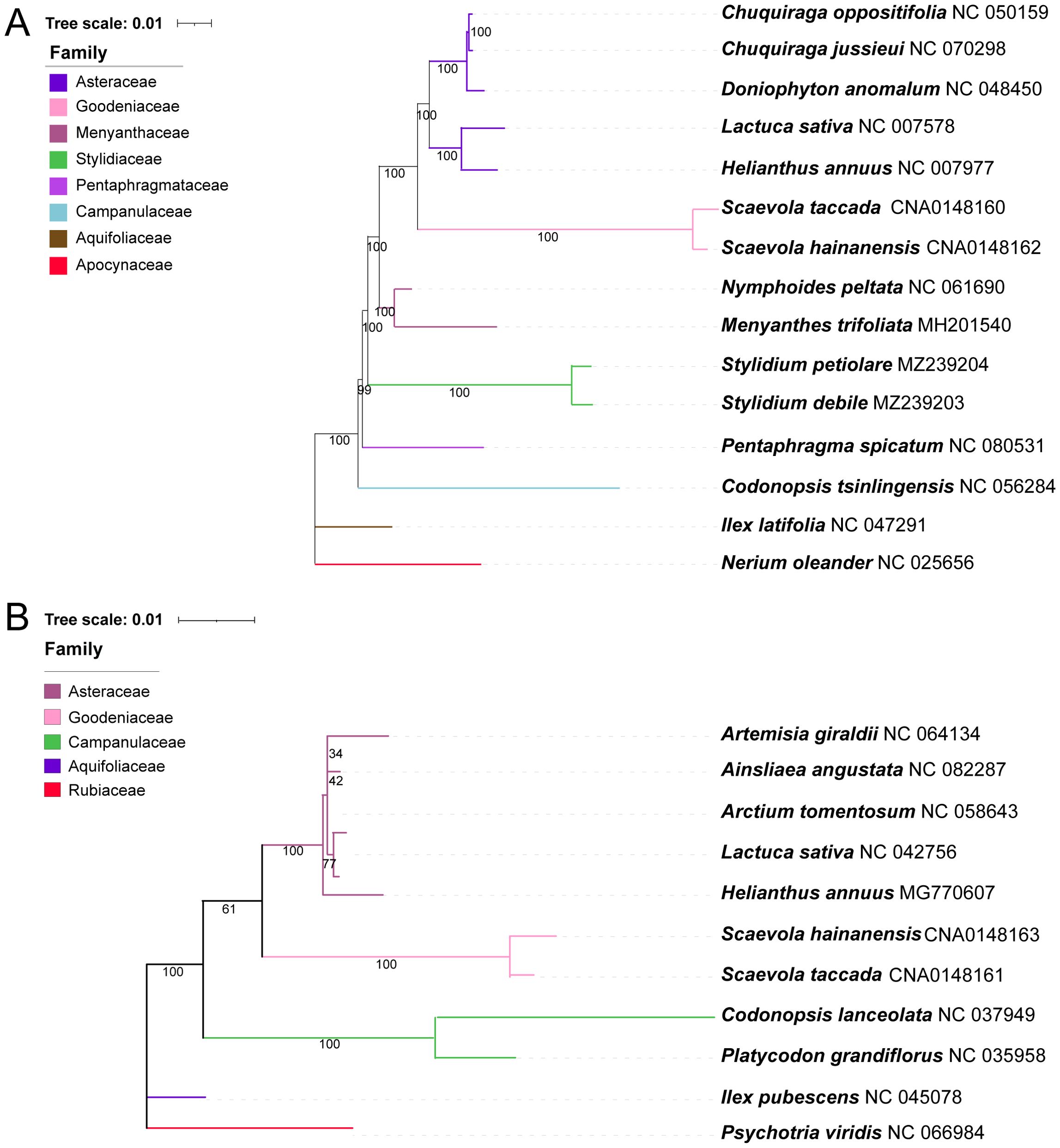
Figure 6. Phylogenetic tree based on 62 common chloroplast genes (A) and 17 conserved protein coding genes of mitochondria (B) using the IQtree. Bootstrap values obtained from 1000 replicates are shown below the clades. Different families are shown in different colors. Nerium oleander and Ilex latifolia were used as outgroups for phylogenetic tree of the chloroplast genes and Ilex pubescens and Psychotria viridis for mitochondria genes.
Syntenic analysis was performed to investigated the mitochondrial genome sequences and structure variations (Figure 7). The mitochondrial genomes of two Scaevola species exhibited high collinearity. However, compared to representative species of related family Asteraceae, only a few collinear segments could be identified, with a large portion of rearrangement, indicating fast evolutionary divergence of mitogenome between Scaevola and its related species. Further, to investigate the horizontal gene transfer between organellar genomes and the nuclear genome, syntenic fragments were also identified by comparing organellar genomes to each other and to nuclear genome (Supplementary Figure S6). Notably, S. taccada showed substantially more syntenic segments over 10 kb between the mitochondrial genome and the nuclear genome. Further examining these fragments in detail helped identify horizontal gene transfer in S. taccada. PCGs within the syntenic segments were labeled outside the circle in Figure 8. Some proteins important for photosynthesis were transferred to the nuclear genome, including rbcL, psaC, psbA, and psbH, while some important mitochondrial coding genes were also transferred to the nuclear genome, including ndh6 and subunits of nad2 complex.

Figure 7. Whole mitogenome collinearity analysis among S. taccada, S. hainanensis, Helianthus annuus and Lactuca sativa. Long strips of different colors represent different plant mitochondrial genomes. Liner blocks represent collinear segments.
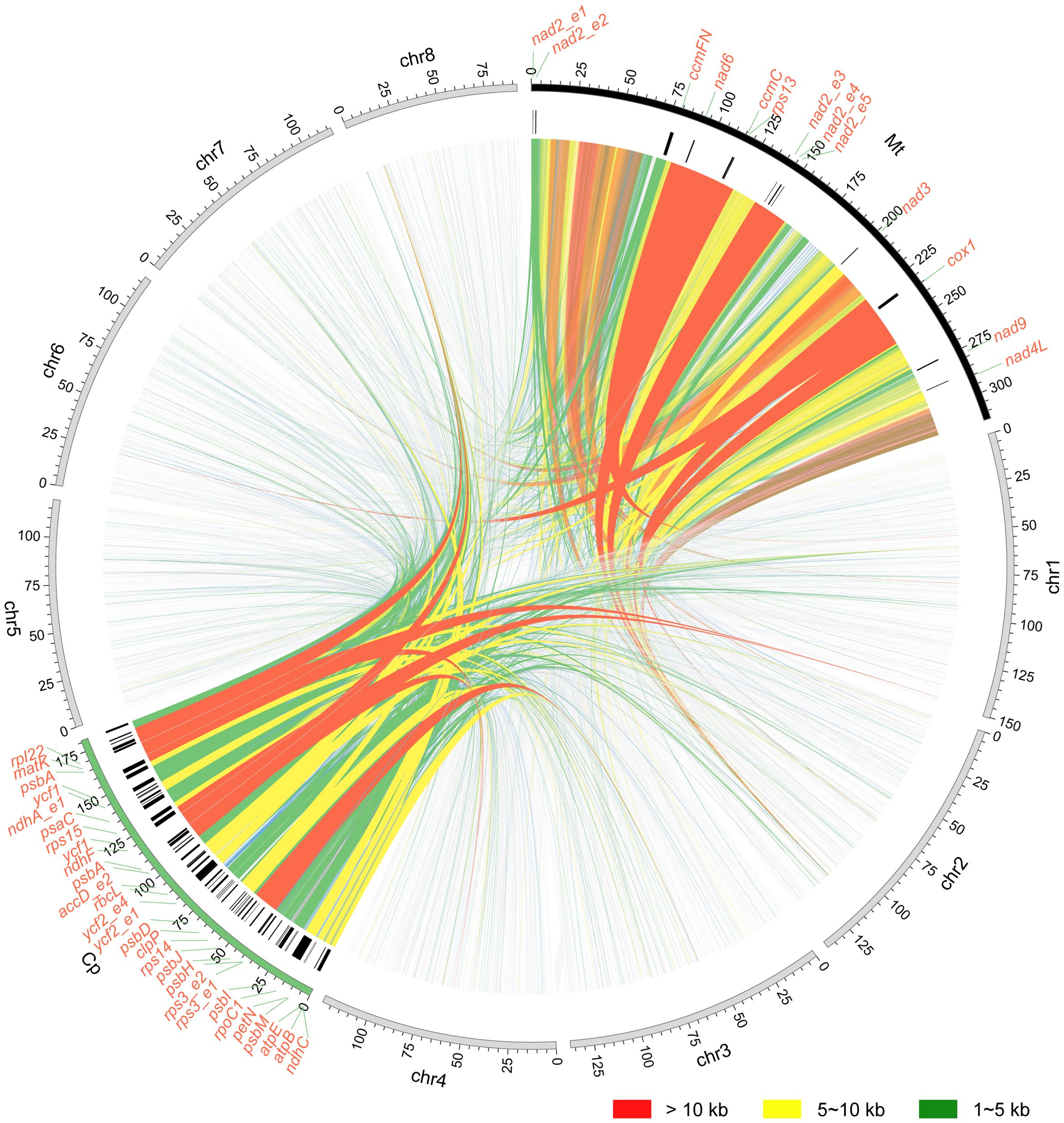
Figure 8. Schematic representations of mitochondrial plastid DNA (MTPT) and nuclear–mitochondrial DNA segments (NUMT) of S. taccada. Collinear segments of different length are presented by different colors: red, >10 kb; yellow, 5–10 kb; green, 1–5 kb. Names of the genes within the collinear segments are shown at the corresponding positions.
4 Discussion
Plants of the Scaevola have important ecological value as pioneer species on tropical coral islands. They also possess high ornamental value, characterized by their symmetrical fan-shaped colorful flowers, so called “fan flowers”. Some species have been introduced to countries outside Australia as horticultural plants. The lack of genomic data has been a major limiting factor in molecular evolution research on Scaevola, despite previous studies developing several markers to investigate the phylogeny of this genus (Ando et al., 2014; Emura et al., 2022). Goodeniaceae shares the most recent common ancestor with Asteraceae, considered as the “most advanced” family of angiosperms. After diverging from Asteraceae approximately 80 million years ago, Goodeniaceae underwent a radiation of approximately 400 species across the Australian continent (Ghisalberti, 2004). However, the complete chloroplast genome of only one species of Goodeniaceae (S. taccada) has been reported in a large-scale study of the Caryophyllales (Yao et al., 2019). This study presents the first complete organellar genomes (chloroplast and mitochondrial) for two Goodeniaceae species. The chloroplast genome of Scaevola is larger than that of representative Asteraceae plants but possesses a very small SSC (Table 1; Figure 2). Our study reveals that this phenomenon is resulted from duplication and rearrangement of LSC fragments, as well as duplication of SSC fragments, causing an IR expansion of approximately 10 kb each (Figure 3). This LSC duplication and IR expansion led to increased copy numbers of some chloroplast encoded genes, including several photosystem II complex members and rbcL gene (Supplementary Table S1; Supplementary Figure S1), which may correlate with the adaptation of Scaevola plants to tropical island habitats characterized by high light intensity and drought conditions. Expanding sampling across Goodeniaceae will be essential to elucidate the evolutionary significance and ecological implications of this organellar genomic architecture.
Compared to chloroplast genomes, plant mitochondrial genomes exhibit greater variation and a broader range of lengths, from 66 kb in Viscum scurruloideum to 12 Mb in Larix sibirica (Wu et al., 2022; Putintseva et al., 2020). The widespread replication events and repetitive fragments in mitogenomes significantly increase the difficulty of their assembly, often resulting in complicated topological multiple circular molecules (Wang et al., 2024a). Long-reads sequencing technologies greatly improve assembly challenges. Benefit from the application of Nanopore long-read sequencing, we simplified the complex topological structure of S. taccada mitochondrial genome into two large circles and one shared fragment (3,871 bp), ultimately obtaining circular molecules with identical sequences but two different connection patterns. While the topological structure of S. hainanensis mitochondrial genome was more complex, finally resolved into two distinct circular molecules with different sequences (sharing four short repetitive sequences), defined as chromosome 1 and chromosome 2 (Figure 1). The current disparity between sequenced mitochondrial (~600) and chloroplast (~13,000 NCBI entries till September 2023) genomes will be diminished with the aid of long-read sequencing technologies.
In this study, the GC contents of Scaevola mitochondrial and chloroplast genomes were 44% and 37%, similar to those reported Asteraceae plants (Tables 1, 2). The mitochondrial genome sizes of the two Scaevola species were 314,251 bp (S. taccada) and 276,175 bp (S. hainanensis), smaller than most reported Asteraceae plants and containing fewer protein-coding genes, while the chloroplast genomes were larger and contained more protein-coding genes (Wang et al., 2024b). These different organelle genome evolutionary patterns between Scaevola and their Asteraceae related species may be related to their survival strategies and adaptations to different habitats. RSCU analysis revealed that Scaevola chloroplast has a higher bias for A/U-ending codons than mitochondria, with many A/U-ending codons in chloroplasts having RSCU values exceeding 1.5 (maximum 1.94), while fewer mitochondrial codons had RSCU values exceeding 1.5 (maximum 1.62) (Figure 4; Supplementary Tables S3, S4). This lower preference for A/U-ending codon may contribute to the higher GC content observed in mitochondrial genomes. Compared to chloroplasts, mitochondria endure greater oxidative stress and therefore face stronger pressure to maintain genomic stability, typically resulting in higher RNA editing sites and efficiency (Hu et al., 2024). In both Scaevola species reported in this study, over 70% of mitochondrial genes possess more than 10 RNA editing sites (maximum 33), while most chloroplast genes have fewer than 5 predicted RNA editing sites (maximum 22), which is consistent with current understanding (Supplementary Figure S3).
Within the order Asterales, the phylogenetic position of Goodeniaceae is relatively well established, with current evidence strongly supporting its sister-group relationship to Asteraceae, as demonstrated by the phylogenetic analyses in this study based on both chloroplast and mitochondrial genomes (Figure 6; Supplementary Figure S4). However, due to the lack of organellar genome information, this study was unable to resolve the currently controversial phylogenetic relationships among different genera within the family Goodeniaceae. In the current era of big data, sampling and sequencing more organelle genomes of representative Goodeniaceae species could help address this challenge. Nevertheless, we noticed that compared to other well-studied genera, such as genus Saussurea in Asteraceae (Mahai et al., 2024), the chloroplast genomes within genus Scaevola exhibited a higher level of sequence divergence, with the highest Pi value reaching 0.45 (Supplementary Figure S2), whereas many chloroplast genome studies show Pi values less than 0.1 (Fang et al., 2024; Jin et al., 2023). Both chloroplast and mitochondrial genome-based phylogenies exhibited longer evolutionary branch lengths for Scaevola (Figure 6; Supplementary Figure S4), indicating high levels of sequence divergence between Goodeniaceae and Asteraceae, within Goodeniaceae, and even within the genus Scaevola. This reflects a faster rate of nucleotide substitution, suggesting that Goodeniaceae may be undergoing rapid radiation, potentially linked to the relatively recent emergence of Asteraceae and Goodeniaceae in the evolutionary history of angiosperms (Zhang et al., 2024). Mitochondrial genome collinearity analysis also revealed strong collinearity between Scaevola plants, with several large fragment insertions and inversions, whereas between Scaevola and related Asteraceae plants, almost no large collinearity blocks were found in their mitochondrial genomes (Figure 7). This indicates that Goodeniaceae and Asteraceae plants have experienced substantial structural variation and rapid mitochondrial genome evolution since their divergence 80 million years ago. Notably, in S. taccada, one of the few Pacific-Indian Ocean widespread species in Goodeniaceae, we observed not only chloroplast genome fragment duplication events leading to increased copy numbers of important photosynthetic genes (such as rbcL, falling into the IR region), but also horizontal transfer of these genes between chloroplast and nuclear genomes (such as rbcL, Figure 8). These phenomena were not observed in the regionally restricted species S. hainanensis, which may be related to S. taccada’s extensive adaptation to the high light intensity, low water retention, high air salinity, and nutrient-poor soil environments of tropical coral islands across the Pacific-Indian Ocean (Li et al., 2023).
5 Conclusions
This study presents the first group of organellar genomes from Scaevola plants, revealing their unique evolutionary patterns. The chloroplast genomes of Scaevola plants have increased in length through IR expansion and LSC duplication, making them approximately 30 kb longer than those of their sister group Asteraceae. Meanwhile, the mitochondrial genomes exhibit multi-circular topological structures (dual-chromosome model). Significantly accelerated variation (maximum Pi = 0.45) and gene copy number expansion were observed in both Scaevola species, whereas their mitochondrial genomes showed size reduction and increased RNA editing sites, suggesting their divergent evolutionary strategies. Phylogenetic analyses confirm the monophyly of the Asteraceae-Goodeniaceae group, yet the remarkable divergences in organellar genome structures between these families suggest independent evolutionary paths. The adaptive expansion of chloroplast genomes (such as increased rbcL copy numbers) and horizontal gene transfer events (chloroplast-to-nuclear genome) may be closely associated with the adaptive radiation of the widespread of S. taccada to tropical coral island habitats, characterized by intense light, high air salinity and drought conditions. These findings provide organelle-level insights into the evolutionary divergence between Scaevola plants and their related species.
Data availability statement
The complete organellar genome sequences generated in this study have been deposited in the China National GeneBank DataBase (CNGBdb; https://db.cngb.org/) under accession numbers CNA0148160, CNA0148161, CNA0148162, and CNA0148163. These annotations are publicly accessible through the CNGBdb repository search interface. Further inquiries can be directed to the corresponding author.
Author contributions
DM: Writing – original draft, Writing – review & editing, Formal Analysis, Visualization. TL: Formal Analysis, Visualization, Writing – original draft, Writing – review & editing. MH: Visualization, Data curation, Writing – review & editing. YR: Visualization, Writing – review & editing, Data curation. MF: Writing – review & editing, Data curation. YZ: Writing – review & editing, Data curation. PY: Data curation, Writing – review & editing. XL: Data curation, Writing – review & editing. YY: Data curation, Writing – review & editing. YZ: Data curation, Writing – review & editing. YCY: Data curation, Writing – review & editing. XJ: Writing – review & editing, Conceptualization, Funding acquisition, Supervision, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Specific Research Fund of the Innovation Platform for Academicians of Hainan Province (Grant Nos. YSPTZX2022011, YSPTZX2025011), and the Undergraduates Training Program for Innovation and Entrepreneurship of Hainan Province.
Acknowledgments
The authors gratefully acknowledge the support from the Innovation Platform for Academicians of Hainan Province. We also sincerely thank to members of Jin Lab for their help in proof reading this paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1587750/full#supplementary-material
References
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Barry, K., Palmer, J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27, 1436–1448. doi: 10.1093/molbev/msq029
Ando, H., Emura, N., Denda, T., Nakahama, N., Inoue-Murayama, M., Isagi, Y. (2014). Development of microsatellite markers for the coastal shrub Scaevola taccada (Goodeniaceae). Appl. Plant Sci. 2, 1300094. doi: 10.3732/apps.1300094
Banerjee, A. K., Wu, H., Guo, W., Ng, W., Li, W., Ma, Y., et al. (2022). Deciphering the global phylogeography of a coastal shrub (Scaevola taccada) reveals the influence of multiple forces on contemporary population structure. J. Syst. Evol. 60, 809–823. doi: 10.1111/jse.12746
Butenko, A., Lukeš, J., Speijer, D., Wideman, J. G. (2024). Mitochondrial genomes revisited: why do different lineages retain different genes? BMC Biol. 22, 15. doi: 10.1186/s12915-024-01824-1
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Chen, C., Miao, Y., Luo, D., Li, J., Wang, Z., Luo, M., et al. (2022). Sequence characteristics and phylogenetic analysis of the artemisia argyi chloroplast genome. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.906725
Cho, M. S., Yang, J., Kim, S. H., Crawford, D. J., Stuessy, T. F., López-Sepúlveda, P., et al. (2024). Plastid phylogenomics of Robinsonia (Senecioneae; Asteraceae), endemic to the Juan Fernández Islands: insights into structural organization and molecular evolution. BMC Plant Biol. 24, 1016. doi: 10.1186/s12870-024-05711-3
Curtis, E., Clegg, T. (1984). Molecular evolution of chloroplast DNA sequences. Mol. Biol. 1, 291–301. doi: 10.1093/oxfordjournals.molbev.a040319
Emura, N., Muranaka, T., Iwasaki, T., Honjo, M. N., Nagano, A. J., Isagi, Y., et al. (2022). Effects of fruit dimorphism on genetic structure and gene flow in the coastal shrub Scaevola taccada. Ann. Bot. 130, 1029–1040. doi: 10.1093/aob/mcac138
Fang, Z., Li, D., Murong, H., He, M., Liu, Y., Liu, J., et al. (2024). Comparative plastome analysis between endangered mangrove species Acanthus ebracteatus and Acanthus relatives provides insights into its origin and adaptive evolution. Ecol. Evo. 14, e70566. doi: 10.1002/ece3.70566
Ghisalberti, E. L. (2004). The goodeniaceae. Fitoterapia 75, 429–446. doi: 10.1016/j.fitote.2004.01.018
Goel, M., Sun, H., Jiao, W. B., Schneeberger, K. (2019). SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 277. doi: 10.1186/s13059-019-1911-0
Greiner, S., Lehwark, P., Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1101/54550o
He, M., Han, X., Qin, X., Bao, J., Li, H., Xie, Q., et al. (2024). Comparative chloroplast genome analyses provide new insights into phylogeny of Taraxacum and molecular markers for distinguishing rubber producing dandelions from their weedy relatives in China. Ind. Crops Prod. 207, 117712. doi: 10.1016/j.indcrop.2023.117712
He, W., Xiang, K., Chen, C., Wang, J., Wu, Z. (2023a). Master graph: an essential integrated assembly model for the plant mitogenome based on a graph-based framework. Brief. Bioinf. 24, bbac522. doi: 10.1093/bib/bbac522
He, W., Yang, J., Jing, Y., Xu, L., Yu, K., Fang, X. (2023b). NGenomeSyn: an easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics 39, btad121. doi: 10.1093/bioinformatics/btad121
Howarth, D. G., Baum, D. A. (2005). Genealogical evidence of homoploid hybrid speciation in an adaptive radiation of Scaevola (Goodeniaceae) in the Hawaiian Islands. Evolution 59, 948–961. doi: 10.1111/j.0014-3820.2005.tb01034.x
Hu, Y. X., Huang, A., Li, Y., Molloy, D. P., Huang, C. (2024). Emerging roles of the C-to-U RNA editing in plant stress responses. Plant Sci. 349, 112263. doi: 10.1016/j.plantsci.2024.112263
Jabaily, R. S., Shepherd, K. A., Gardner, A. G., Gustafsson, M. H. G., Howarth, D. G., Motley, T. J. (2014). Historical biogeography of the predominantly Australian plant family Goodeniaceae. J. Biogeogr. 41, 2057–2067. doi: 10.1111/jbi.12363
Jin, G., Li, W., Song, F., Yang, L., Wen, Z., Feng, Y. (2023). Comparative analysis of complete Artemisia subgenus Seriphidium (Asteraceae: Anthemideae) chloroplast genomes: insights into structural divergence and phylogenetic relationships. BMC Plant Biol. 23, 136. doi: 10.1186/s12870-023-04113-1
Kolmogorov, M., Yuan, J., Lin, Y., Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Lee, J. T., Yen, L. Z., Chu, M. Y., Lin, Y. S., Chang, C. C., Lin, R. S., et al. (2020). Growth characteristics and anti-wind erosion ability of three tropical foredune pioneer species for sand dune stabilization. Sustainability 12, 3353. doi: 10.3390/su12083353
Letunic, I., Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, S., Mao, X., He, Z., Xu, S., Guo, Z., Shi, S. (2023). Chromosomal-scale genome assemblies of two coastal plant species, Scaevola taccada and S. hainanensis—insight into adaptation outside of the common range. Int. J. Mol. Sci. 24, 7355. doi: 10.3390/ijms24087355
Lima, D. F., Goldenberg, R., Forest, F., Cowan, R. S., Lucas, E. J. (2021). Phylogeny and biogeography of Myrcia sect. Aguava (Myrtaceae, Myrteae) based on phylogenomic and Sanger data provide evidence for a Cerrado origin and geographically structured clades. Mol. Phylogenet. Evol. 157, 107043. doi: 10.1016/j.ympev.2020.107043
Liu, L., Long, Q., L., W., Qian, J., Egan, A. N., Shi, Y., et al. (2024). Long repeat sequences mediated multiple mitogenome conformations of mulberries (Morus spp.), an important economic plant in China. Genomics Commun. 1, e005. doi: 10.48130/gcomm-0024-0005
Locher, C. P., Witvrouw, M., De Béthune, M. P., Burch, M. T., Mower, H. F., Davis, H., et al. (1996). Antiviral activity of Hawaiian medicinal plants against human immunodeficiency Virus Type-1 (HIV-1). Phytomedicine 2, 259–264. doi: 10.1016/S0944-7113(96)80052-3
Mahai, R., Sheng, S., Wang, X., Yuan, J., Mu, Z. (2024). Comparative analysis of complete chloroplast genomes of 14 Asteraceae species. Mol. Biol. Rep. 51, 1094. doi: 10.1007/s11033-024-10030-9
Panero, J. L., Crozier, B. S. (2016). Macroevolutionary dynamics in the early diversification of Asteraceae. Mol. Phylogenet. Evol. 99, 116–132. doi: 10.1016/j.ympev.2016.03.007
Putintseva, Y. A., Bondar, E. I., Simonov, E. P., Sharov, V. V., Oreshkova, N. V., Kuzmin, D. A., et al. (2020). Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genomics 21, 654. doi: 10.1186/s12864-020-07061-4
Qiu, T., Zhao, X., Feng, H., Qi, L., Yang, J., Peng, Y., et al. (2021). OsNBL3, a mitochondrion-localized pentatricopeptide repeat protein, is involved in splicing nad5 intron 4 and its disruption causes lesion mimic phenotype with enhanced resistance to biotic and abiotic stresses. Plant Biotechnol. J. 19, 2277–2290. doi: 10.1111/pbi.13659
Richardson, A. O., Palmer, J. D. (2007). Horizontal gene transfer in plants. J.Exp. Bot. 58, 1–9. doi: 10.1093/jxb/erl148
Robles, P., Quesada, V. (2019). Transcriptional and post-transcriptional regulation of organellar gene expression (OGE) and its roles in plant salt tolerance. Int. J. Mol. Sci. 20, 1056. doi: 10.3390/ijms20051056
Shen, F., Qin, Y., Wang, R., Huang, X., Wang, Y., Gao, T., et al. (2023). Comparative genomics reveals a unique nitrogen-carbon balance system in Asteraceae. Nat. Commun. 14, 4334. doi: 10.1038/s41467-023-40002-9
Starman, T., Lombardini, L. (2006). Growth, gas exchange, and chlorophyll fluorescence of four ornamental herbaceous perennials during water deficit conditions. J. Amer. Soc Hortic. Sci. 131, 469–475. doi: 10.21273/JASHS.131.4.469
Swensen, S. M., Gomez, A. M., Piasecki-Masters, C., Chime, N., Wine, A. R., Rodriguez, N. C., et al. (2024). Minimal impacts of invasive Scaevola taccada on Scaevola plumieri via pollinator competition in Puerto Rico. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1281797
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Toscano, S., Branca, F., Romano, D., Ferrante, A. (2020). An evaluation of different parameters to screen ornamental shrubs for salt spray tolerance. Biology 9, 250. doi: 10.3390/biology9090250
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9, e112963. doi: 10.1371/journal.pone.0112963
Walker, R. B., Gessel, S. P., Held, E. E. (1997). The ecosystem study on rongelap atoll. Health Phys. 73, 223–233. doi: 10.1097/00004032-199707000-00019
Wang, J., Kan, S., Liao, X., Zhou, J., Tembrock, L. R., Daniell, H., et al. (2024a). Plant organellar genomes: much done, much more to do. Trends Plant Sci. 29, 754–769. doi: 10.1016/j.tplants.2023.12.014
Wang, S., Qiu, J., Sun, N., Han, F., Wang, Z., Yang, Y., et al. (2025). Characterization and comparative analysis of the first mitochondrial genome of Michelia (Magnoliaceae). Genomics Commun. 2, e001. doi: 10.48130/gcomm-0025-0001
Wang, L., Yang, H., Xu, G., Liu, Z., Meng, F., Shi, L., et al. (2024b). Asteraceae genome database: a comprehensive platform for Asteraceae genomics. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1445365
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z., Liao, X., Zhang, X., Tembrock, L. R., Broz, A. (2022). Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 60, 160–168. doi: 10.1111/jse.12655
Yao, G., Jin, J. J., Li, H. T., Yang, J. B., Mandala, V. S., Croley, M., et al. (2019). Plastid phylogenomic insights into the evolution of Caryophyllales. Mol. Phylogen. Evol. 134, 74–86. doi: 10.1016/j.ympev.2018.12.023
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, G., Yang, J., Zhang, C., Jiao, B., Panero, J. L., Cai, J., et al. (2024). Nuclear phylogenomics of Asteraceae with increased sampling provides new insights into convergent morphological and molecular evolution. Plant Commun. 5, 100851. doi: 10.1016/j.xplc.2024.100851
Zheng, M., Jiang, Y., Ran, Z., Liang, S., Xiao, T., Li, X., et al. (2025). A cyanobacteria-derived intermolecular salt bridge stabilizes photosynthetic NDH-1 and prevents oxidative stress. Commun. Biol. 8, 172. doi: 10.1038/s42003-025-07556-4
Keywords: organelle genomes, Scaevola, Goodeniaceae, Asteraceae, evolutionary divergence
Citation: Meng D, Lu T, He M, Ren Y, Fu M, Zhang Y, Yang P, Lin X, Yang Y, Zhang Y, Yang Y and Jin X (2025) Organelle genomes of two Scaevola species, S. taccada and S. hainanensis, provide new insights into evolutionary divergence between Scaevola and its related species. Front. Plant Sci. 16:1587750. doi: 10.3389/fpls.2025.1587750
Received: 04 March 2025; Accepted: 02 April 2025;
Published: 24 April 2025.
Edited by:
Zhiqiang Wu, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Jie Wang, Murdoch University, AustraliaLiu Rongpeng, Beijing University of Chinese Medicine, China
Yanping Xie, Huaibei Normal University, China
Copyright © 2025 Meng, Lu, He, Ren, Fu, Zhang, Yang, Lin, Yang, Zhang, Yang and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Jin, amlueEBoYWlubnUuZWR1LmNu
†These authors have contributed equally to this work