 Yubang Gao
Yubang Gao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Plant Sci. , 05 March 2025
Sec. Functional and Applied Plant Genomics
Volume 16 - 2025 | https://doi.org/10.3389/fpls.2025.1528404
Isodon lophanthoides (Figure 1A) is a perennial herb of the Lamiaceae family distributed across China, India, Myanmar, Nepal, and Vietnam (Wen et al., 2011; Zhang et al., 2022). I. lophanthoides contains a variety of bioactive compounds, such as terpenoids, flavonoids, phenolics, and polysaccharides (Lin et al., 2008; Wen et al., 2011; Zhou et al., 2014). I. lophanthoides is traditionally used to alleviate symptoms of acute jaundice hepatitis, arthritis, cholecystitis, enteritis, pharyngitis, ascariasis, and leprosy (Jiang et al., 2000). This herb is utilized in the preparation of therapeutic teas and instant granules. Additionally, it is used as an ingredient in soups and cooking. This plant plays a significant role in traditional Chinese medicine. It is cultivated extensively as a commercial raw material for the medicinal product “Xihuangcao”. The absence of genomic resources for I. lophanthoides has severely limited its genetic improvement and research on its active components. In this study, we assembled the first chromosome-level genome of I. lophanthoides and identified key genes involved in terpene biosynthesis. This work provides a valuable foundation for genetic improvement and exploring its active compounds’ biosynthetic pathways.
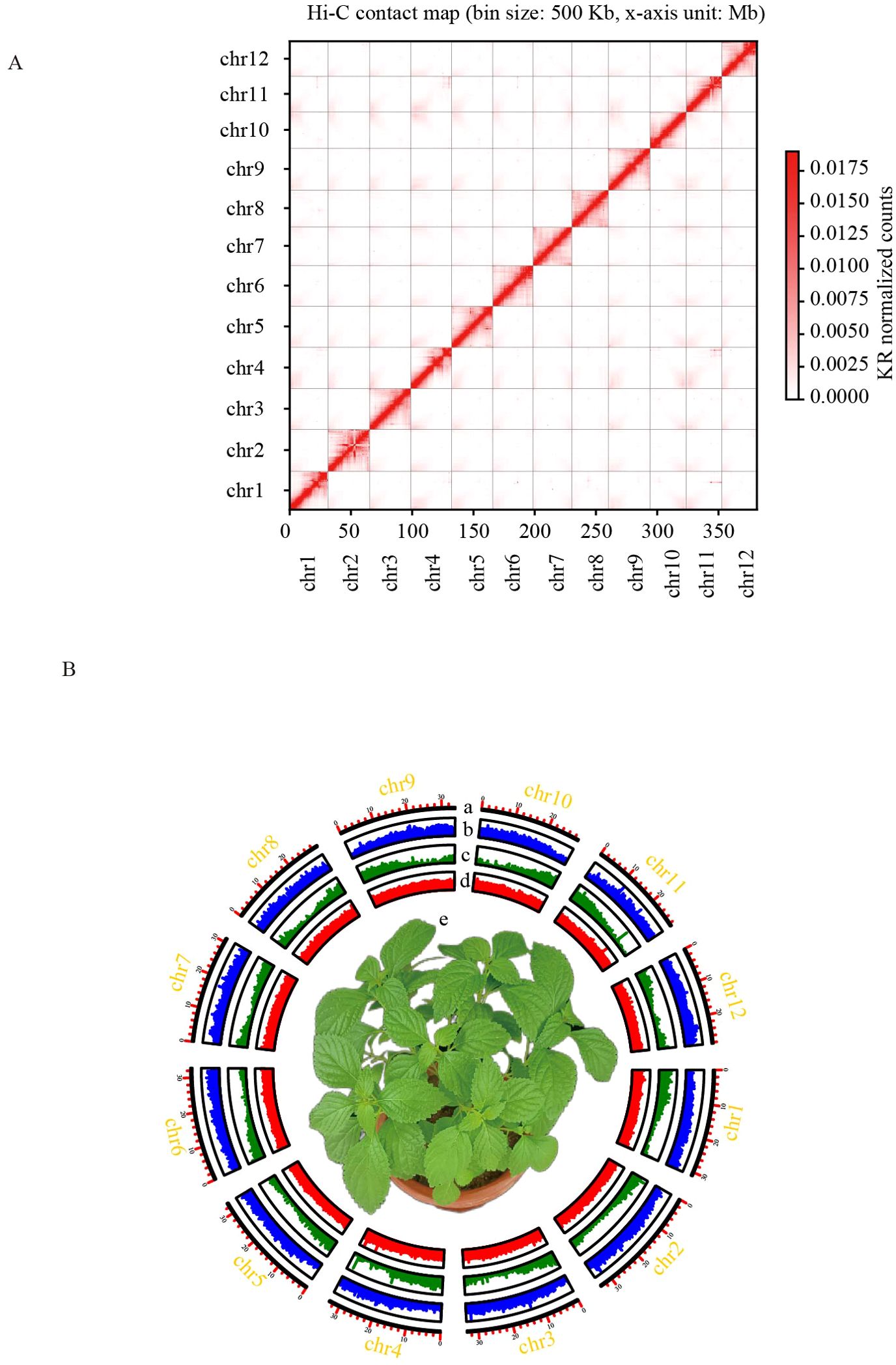
Figure 1. Chromosome-scale assembly of the I. lophanthoides genome. (A) Contact map of I. lophanthoides genome. (B) Circos plot displaying the 12 chromosomes in the I. lophanthoides genome. a. Length of each pseudochromosome (Mb). b. Distribution of repetitive sequences. c. Distribution of gene density. d. Distribution of the GC content. e. The phenotype of I. lophanthoides (The flower pot size was 15 cm).
Young leaves of I. lophanthoides, cultivated at the Artemisia Engineering Technology Center of Nanyang Normal University, were collected to extract high-quality DNA for genome sequencing. After DNA extraction, ultrasonic shearing was applied. The sequencing library was prepared through end-repair, adapter ligation, and amplification, followed by sequencing on the DNB-Seq T7 platform. For long reads, the sequencing library was prepared using the Oxford Nanopore ligation sequencing kit (SQK-LSK109). Sequencing was then performed on an R9 flow cell on the PromethION platform. For Hi-C reads, DNA was fixed in a 4% formaldehyde solution. Digestion was performed with the MboI enzyme, and digested fragments were labeled with biotin-14-dCTP. The crosslinked fragments were then blunt-end repaired and sequenced on the DNB-Seq T7 platform.
The k-mer method was used to estimate genome size and heterozygosity before genome assembly. The k-mer distribution was calculated from short reads using Jellyfish (Marcais and Kingsford, 2012) with k-mer length set to 21. The genome size and heterozygosity rate was estimated using the GenomeScope2 (Ranallo-Benavidez et al., 2020).
Genome assembly was conducted using NextDenovo (Hu et al., 2024) with the overlap-layout-consensus algorithm and default parameters. NextPolish (Hu et al., 2020) was used to polish the genome assembly, applying two rounds of long-read and four rounds of short-read data correction. Hi-C reads were aligned to contigs using Juicer (Durand et al., 2016) and BWA (Jung and Han, 2022), after which the 3D-DNA pipeline (Dudchenko et al., 2017) corrected misassemblies and ordered contigs, integrating them into scaffolds. Manual inspection of scaffolds was then performed using Juicebox Assembly Tools. The final chromosome-length scaffolds were constructed using the 3D-DNA pipeline, with all computational tools run using default parameters. Misassemblies were identified and corrected based on irregular contact patterns in Hi-C data.
Repeat elements in genomes were identified using RepeatModeler (Flynn et al., 2020), and the repeat library was then processed with RepeatMasker (Tarailo-Graovac and Chen, 2009) to annotate repeats across the genome. Transposable elements (TEs) were classified using TEsorter (Zhang et al., 2022). Simple sequence repeat (SSR) markers were predicted using MISA (Beier et al., 2017). Protein-coding genes in the I. lophanthoides genome were identified using an integrative strategy that combined ab initio prediction, protein homology searches, and RNA sequencing data. For ab initio prediction, we used Augustus (Stanke et al., 2006), SNAP (Korf, 2004), GlimmerHMM (Majoros et al., 2004), and GeneMark-ET (Brůna et al., 2020) to identify gene structures in the repeat-masked genome. For protein homology prediction, protein data from sequenced Lamiaceae species were downloaded from the NCBI database and aligned for homology assessment. Additionally, HISAT2 (Kim et al., 2019) was used to map RNA-seq data (PRJNA679679) from various tissues to the genome. PASA was used to predict open reading frames. EVidenceModeler (Haas et al., 2008) integrated results from the three methods, enabling a unified gene prediction. Functional annotation was performed using BLAST (Ye et al., 2006) against NR, SwissProt, eggNOG, InterPro, GO, and KEGG databases. Functional annotations for protein-coding genes were integrated using the above methods.
Protein sequences of A. trichopoda, O. sativa, V. vinifera, T. cacao, A. thaliana, S. lycopersicum, C. canephora, T. grandis, L. japonicus, S. miltiorrhiza, I. rubescens, and A. decumbens were downloaded for subsequent analyses. OrthoVenn3 (Sun et al., 2023) was used for orthology, phylogenetic, and gene family analyses. Pairwise sequence similarity was determined using BLASTP and OrthoMCL (Li et al., 2003) Markov clustering. Phylogenetic trees were constructed using FastTree2 (Price et al., 2010) with the maximum likelihood method and the JTT+CAT model, with node reliability assessed by the SH test. A divergence tree was constructed using single-copy genes and fossil evidence. Divergence times between A. thaliana and T. cacao, S. lycopersicum and C. canephora, A. thaliana and V. vinifera, A. trichopoda and V. vinifera, and L. japonicus and T. grandis were estimated using r8s (Sanderson, 2003). CAFE (Mendes et al., 2020) was used to compare cluster size differences between ancestors and each species to determine gene family expansions and contractions. A random birth-and-death model was applied to assess gene family changes across lineages in the phylogenetic tree. Conditional likelihood was used as the test statistic, with p-values of ≤ 0.01 considered significant.
I. lophanthoides protein sequences were compared to identify homologous blocks. The MCScanX (Wang et al., 2012) pipeline was applied with default settings to map homologous blocks within species. The YN model in KaKs_Calculator 2.0 (Wang et al., 2010) was used to calculate nonsynonymous (Ka) and synonymous (Ks) substitution rates, as well as their ratio (Ka/Ks), for duplicate gene pairs.
DNA was isolated from I. lophanthoides samples cultivated in the laboratory. Genome size and heterozygosity were estimated using DNB short-read sequencing data. The estimated genome size from short reads was 365,686,342 bp, with a heterozygosity rate of 0.64% (k-mer length = 21). DNA from the same plant was used to assemble the I. lophanthoides genome with a combination of Nanopore and Hi-C technologies (Supplementary Table S1). Assembly with Nanopore long reads produced a genome with a total length of 379,974,750 bp, containing 70 contigs (N50 = 17,265,197 bp). After Hi-C scaffolding, 378,710,417 bp (99.67%) of the sequence was placed into 12 linkage groups (Figure 1A). These linkage groups corresponded to the 12 chromosomes of I. lophanthoides (N50 = 32,786,395 bp). BUSCO assessment showed that the assembly covered 98% of the single-copy orthologs in the embryophyta_odb10 database (1,614 genes; Supplementary Table S2). The consensus quality value (QV) was 35.77, indicating that the genome is highly accurate. The genome’s LAI value is 13.78, reaching the level of the reference genome.
50.52% of the genome assembly consisted of repetitive elements, with half of this proportion (30% of the genome) being retrotransposons. This retrotransposon content is similar to that in I. rubescens. In the I. lophanthoides genome, 9.38% of the copies were identified as Copia elements, and 9.93% as Gypsy elements. We further classified transposable elements (TEs) using Tesort (Zhang et al., 2022), identifying 5,880 Helitrons, 4,015 LINEs, 94,428 LTRs, and 13,042 TIRs. Additionally, 153,599 SSR markers were predicted using MISA (Beier et al., 2017).
EVidenceModeler was used to integrate outputs from transcriptome data, ab initio predictions, and homology-based predictions. A total of 30,641 genes were identified, of which 28,541 were protein-coding (Figure 1B). These genes contained an average of 4.8 exons, with an average coding sequence (CDS) length of 1,112 bp (Supplementary Table S3). Functional annotation of 26,492 protein-coding genes (92.8%) was achieved using GO, NR, KEGG, TAIR, and InterProScan databases. A total of 40 genes were associated with terpene metabolism, including 12 genes in the MEA pathway and 28 in the MEP pathway (Supplementary Table S4). Non-coding RNA prediction identified 297 rRNAs, 541 tRNAs, 101 miRNAs, and 341 snRNAs.
To determine the evolutionary relationships between I. lophanthoides, I. rubescens, and other plant species, a phylogenetic tree was constructed using a total of 427,238 proteins from 12 plant species (Supplementary Table S5). These proteins were clustered into 35,165 orthogroups, of which 282 were single-copy genes (Supplementary Table S6). With known divergence times added, the phylogenetic tree indicated that the common ancestor of I. lophanthoides and I. rubescens diverged approximately 12.988 million years ago (MYA) (Figure 2A). In I. lophanthoides, 48 gene families showed significant expansion and 208 showed significant contraction. The number of expanded gene families was smaller than in I. rubescens. Compared with other Lamiaceae species, I. lophanthoides had the fewest unique gene families (Figure 2B). A transposon burst occurred in I. rubescens gene families around 1 MYA (Figure 2C). The Ks method was used to analyze orthologous gene pairs, revealing no lineage-specific whole-genome duplication events other than the shared peak in Lamiaceae (Figure 2D). Further analysis of selection-affected genes identified 323 genes under positive selection and 2,832 under negative selection (Figure 2E). Genes under positive selection were enriched in processes such as “response to salicylic acid” (Supplementary Figure S3).
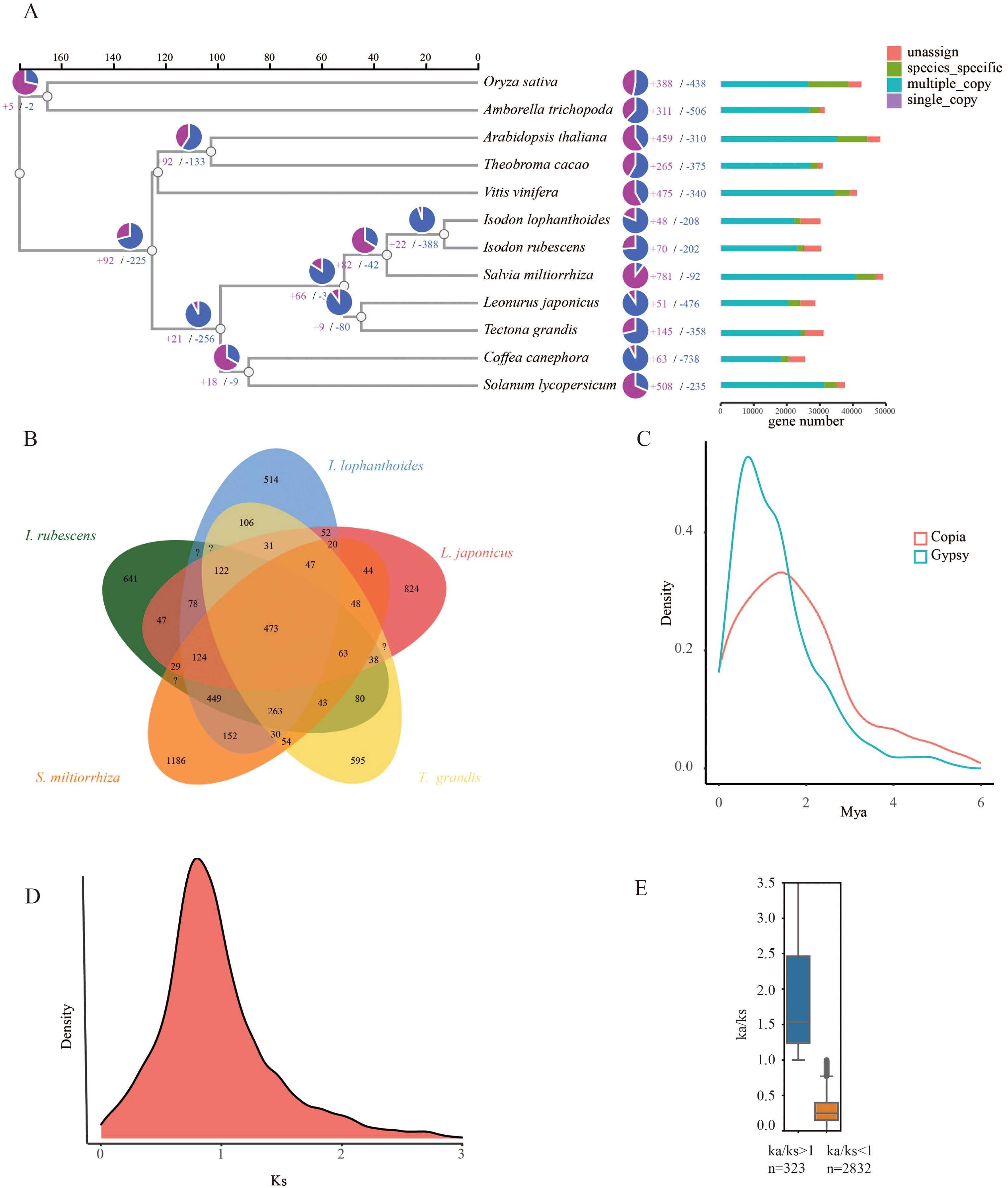
Figure 2. Evolutionary analysis of the I. lophanthoides genome. (A) A phylogenetic tree based on shared single-copy gene families, gene family expansions, and contractions among I. lophanthoides and ten other species. The bar chart on the right displays gene family clustering in I. lophanthoides and ten other plant species. (B) Venn Diagram Representation of Gene Family Overlaps and Specificities Among I. lophanthoides, I. rubescens L. japonicus, T. grandis, and S. miltiorrhiza in Labiatae. (C) Density plot showing the burst of LTR-RTs in I. lophanthoides. (D) Ks value distribution plot for orthologous gene sets of I. lophanthoides. (E) Ka/Ks value distribution plot for orthologous gene sets of I. lophanthoides.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SRR29855129, SRR28822717, SRR29849713 https://figshare.com/, https://figshare.com/s/791b7bef4735829aaf3e.
YG: Data curation, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The Foundation of Nanyang Normal University (2023ZX011; 2024PY019), the Key Scientific Research Project of Higher Education Institutions in Henan Province (23B180002), and the Natural Science Foundation of Henan Province (242300420501) provided funding for this project.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1528404/full#supplementary-material
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. J. B. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics. 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Brůna, T., Lomsadze, A., Borodovsky, M. (2020). GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics Bioinf. 2, lqaa026. doi: 10.1093/nargab/lqaa026
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aEgypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S., Huntley, M. H., Lander, E. S., et al. (2016). Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98. doi: 10.1016/j.cels.2016.07.002
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22. doi: 10.1186/gb-2008-9-1-r7
Hu, J., Fan, J., Sun, Z., Liu, S. (2020). NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255. doi: 10.1093/bioinformatics/btz891
Hu, J., Wang, Z., Sun, Z., Hu, B., Ayoola, A. O., Liang, F., et al. (2024). NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 1–19. doi: 10.1186/s13059-024-03252-4
Jiang, B., Lu, Z.-Q., Zhang, H.-J., Zhao, Q.-S., Sun, H.-D. (2000). Diterpenoids from isodon lophanthoides. Fitoterapia 71, 360–364. doi: 10.1016/S0367-326X(00)00126-X
Jung, Y., Han, D. (2022). BWA-MEME: BWA-MEM emulated with a machine learning approach. Bioinformatics 38, 2404–2413. doi: 10.1093/bioinformatics/btac137
Kim, D., Paggi, J. M., Park, C., Bennett, C., Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Li, L., Stoeckert, C. J., Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lin, C.-Z., Zhang, C.-X., Xiong, T.-Q., Feng, Y.-L., Zhao, Z.-X., Liao, Q.-F., et al. (2008). Gerardianin A, a new abietane diterpenoid from Isodon lophanthoides var. gerardianus. J. Asian Natural Products Res. 10, 841–844. doi: 10.1080/10286020802156564
Majoros, W. H., Pertea, M., Salzberg, S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Mendes, F. K., Vanderpool, D., Fulton, B., Hahn, M. W. (2020). CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518. doi: 10.1093/bioinformatics/btaa1022
Price, M. N., Dehal, P. S., Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One 5, e9490. doi: 10.1371/journal.pone.0009490
Ranallo-Benavidez, T. R., Jaron, K. S., Schatz, M. C. (2020). GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11 (1), 1432. doi: 10.1038/s41467-020-14998-3
Sanderson, M. J. (2003). r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302. doi: 10.1093/bioinformatics/19.2.301
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Sun, J., Lu, F., Luo, Y., Bie, L., Xu, L., Wang, Y. (2023). OrthoVenn3: an integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res. 51, W397–W403. doi: 10.1093/nar/gkad313
Tarailo-Graovac, M., Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 25, 4.10. doi: 10.1002/0471250953.2009.25.issue-1
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinf. 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49. doi: 10.1093/nar/gkr1293
Wen, L., Lin, L., You, L., Yang, B., Jiang, G., Zhao, M. (2011). Ultrasound-assited extraction and structural identification of polysaccharides from Isodon lophanthoides var. gerardianus (Bentham) H. Hara. Carbohydr. Polymers 85, 541–547. doi: 10.1016/j.carbpol.2011.03.003
Ye, J., Mcginnis, S., Madden, T. L. (2006). BLAST: improvements for better sequence analysis. Nucleic Acids Res. 34, W6–W9. doi: 10.1093/nar/gkl164
Zhang, R. G., Li, G. Y., Wang, X. L., Dainat, J., Wang, Z. X., Ou, S., et al. (2022). TEsorter: an accurate and fast method to classify LTR-retrotransposons in plant genomes. Horticulture Res. 9, uhac017. doi: 10.1093/hr/uhac017
Keywords: genome, Chinese herbal medicine, Isodon lophanthoides, nanopore sequence, Hi-C assembly
Citation: Gao Y (2025) Chromosome-level assembly of the Isodon lophanthoides genome. Front. Plant Sci. 16:1528404. doi: 10.3389/fpls.2025.1528404
Received: 14 November 2024; Accepted: 12 February 2025;
Published: 05 March 2025.
Edited by:
Yi-Hong Wang, University of Louisiana at Lafayette, United StatesReviewed by:
Kang Zhang, Chinese Academy of Agricultural Sciences, ChinaCopyright © 2025 Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yubang Gao, Z2FveXViYW5nQHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.