 Long Tong
Long Tong Qingping Zeng
Qingping Zeng Yuan Guo
Yuan Guo Yanjie Li
Yanjie Li Hongyan Li
Hongyan Li Lijie Chen
Lijie Chen Xia Liu
Xia Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 12 February 2025
Sec. Plant Systems and Synthetic Biology
Volume 16 - 2025 | https://doi.org/10.3389/fpls.2025.1514703
Introduction: The basic helix-loop-helix (bHLH) proteins are a large family of transcription factors that are essential to physiology, metabolism, and development. However, the available information is limited about the bHLH gene family in Chimonobambusa utilis, which is widely cultivated in China because of its high-quality and economic value. C. utilis cultivars exhibit five natural color variations in their shoot sheaths, but the molecular mechanism behind this color diversity remains unclear.
Methods: De novo assembly was employed to obtain gene sequences. To identify pathways related to color formation, GO enrichment analysis was performed on the 44,255 functionally annotated unigenes.
Results: The transcriptomic analysis of C. utilis yielded a total of 195,977 transcripts and 75,137 unigenes after removing redundancy. The enrichment results revealed that four pathways were most strongly associated with color formation. Phylogenetic, conserved motif, and protein–protein interaction analyses, along with qRT–PCR validation, confirmed CubHLH17's role in red sheath color.
Discussion: This research not only deepens insights into the functional roles of CubHLH genes but also lays the foundation for genetic improvement of bamboo species. We suggest that these findings will contribute to both scientific research and commercial bamboo cultivation through gene editing technology in the future.
China has rich bamboo germplasm resources and a large bamboo industry with more than 500 species and the bamboo forest area of more than 7 million hectares. China is a major producer and exporter of bamboo shoots (Yu et al., 2023). Chimonobambusa utilis (Keng) P.C. Keng (family Poaceae), a famous bamboo species having high economic value, is mainly distributed in the Jinfo Mountain (Nanchuan district, Chongqing) of China. This species has gradually formed a population with five different shoot sheath colors and relatively stable genetic traits during a long evolutionary process (Yang et al., 2022). C. utilis bamboo shoots, being tasty and crisp, and having rich fiber, high amino acid content, and vibrant colors, are known as the “king of bamboo shoots.” Because of their high quality, these shoots are preferred by consumers in both domestic and foreign lands (Chen et al., 2019; Yang et al., 2022). Bamboo shoot sheaths, a distinguishing characteristic of bamboo species, protect the young bamboo shoots and significantly influence their growth and quality. The color of bamboo shoots is a key agronomic characteristic that directly affects price and market competitiveness (Wang et al., 2020; Chen et al., 2021a; Yu et al., 2023).
The basic helix–loop–helix (bHLH) transcription factors constitute the second largest family in plants, and together with MYB and WD40, they regulate the biosynthesis of anthocyanins (Yu et al., 2024; Zhai et al., 2024) and participate in diverse physiological and biochemical processes in plants, including secondary metabolite biosynthesis, signal transduction, growth and development, and responses to biotic and abiotic stresses (Makkena and Lamb, 2013; Song et al., 2021; Chen et al., 2017). The bHLH domain consists of approximately 50–60 amino acids with two functionally distinct regions: a basic region and a HLH region (Ke et al., 2020). The basic region, located at the N-terminus, comprises approximately 17 amino acids (Murre et al., 1989; Feller et al., 2011). The HLH region includes two amphipathic helices made up of hydrophobic residues connected by a variable loop, which serves as a dimerization domain (Alsamman et al., 2023). Anthocyanins are pigments that determine the color of plant leaves or flowers (Streisfeld and Kohn, 2005; Cooley and Willis, 2009). The bHLH transcription factor was first discovered in maize, where it was found to be involved in anthocyanin biosynthesis (Ludwig et al., 1989). Subsequent studies have reported that bHLH transcription factors are involved in the biosynthesis of anthocyanin in various plants such as cherry (Wang et al., 2019), kiwifruit (Li, 2021), grape (Li et al., 2021a), colored-leaf poplar (Populus deltoides) (Wang et al., 2022), and pear (Han et al., 2023). Four CcBHLH genes in Cinnamomum camphora were identified as potentially involved in anthocyanin biosynthesis (Gong et al., 2023). The CmBHLH2 gene was identified in chrysanthemum and is positively correlated with anthocyanin content, and its upregulation of CmDFR promotes anthocyanin accumulation (Xiang et al., 2015). Although the bHLH gene family has been reported in several species, it has not yet been studied in bamboo (C. utilis).
High-throughput sequencing technologies and a growing number of molecular tools have made it possible to accurately identify genes associated with plant organ coloration and to understand the developmental mechanisms in detail. In this study, we employed de novo assembly to obtain the gene sequences of C. utilis. Gene Ontology (GO) enrichment analysis was performed on all unigenes, and genes enriched in color-related pathways were selected for differential expression analysis. Potential CubHLH candidates associated with color variation were identified based on transcriptome data analysis. Moreover, the physicochemical properties, protein structures, subcellular localization, protein–protein interaction network, and phylogenetic relationships of the bHLH family members were analyzed. Finally, candidate CubHLH17 genes related to bamboo sheath color variation were identified via qRT-PCR. These studies not only increase our understanding of the functions of the CubHLH gene family but also provide important scientific insights for bamboo genetic improvement and industrial applications, such as aesthetic appeal, quality, and market value, thereby contributing to both scientific research and commercial bamboo cultivation.
We selected C. utilis, which has five sheath colors: red, green, brown, yellow, and black. For ease of description, we simplified the cultivar names and used the following abbreviations: black-sheath cultivar, Bsh; brown-sheath cultivar, Brsh; red-sheath cultivar, Rsh; green-sheath cultivar, Gsh; and yellow-sheath cultivar, Ysh. Bamboo sheath samples were collected from Jinfo Mountain, Nanchuan District, Chongqing, China (29.012040° N, 107.195879° E), where the plant grows at an altitude of 2,129 m. On 19 September 2022, bamboo shoots of varying sheath colors were collected approximately 20 cm above the ground. The outermost layer of the sheath was removed, and the sheath material was cut approximately 5 cm from the tip of the bamboo shoot with scissors. The five bamboo sheath colors of C. utilis were collected with three biological replicates (15 samples in total). Fresh sheaths sampled for transcriptome sequencing quickly placed in liquid nitrogen containers and then stored in the laboratory at −80°C until RNA extraction. Total RNA was extracted using TRIzol reagent (Invitrogen, Waltham, USA). The purity of total RNA was assessed using electrophoresis in a 1.2% agarose gel and then verified based on absorbance values (260 nm/280 nm >1.8) using a Nanodrop 2000 Instrument (Thermo Fisher Scientific Inc., USA). An Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) was used to assess RNA integrity. High-quality RNAs were subsequently subjected to high-throughput sequencing on the Illumina HiSeq 2500 platform.
The NGS QC Toolkit (http://www.nipgr.res.in/ngsqctoolkit.html) (Patel and Jain, 2012) was utilized to filter the raw data before assembly, and the adaptor sequences, reads with N ratios greater than 5%, and low-quality reads were removed. Finally, clean, high-quality reads were obtained. Trinity software v2.11.0 (Grabherr et al., 2011) was used for de novo assembly to generate transcripts and unigenes. Functional annotations and protein domain predictions were performed by aligning against the Nr, Swiss-Prot, GO, COG/KOG, KEGG, and Pfam databases using BLASTP and HMMER v3.3.2 (Mistry et al., 2013). Pearson correlation coefficients were calculated to assess the repetitiveness among samples, and a correlation heatmap was generated.
Gene Ontology (GO) enrichment analysis of the unigenes was performed using the GO database (http://www.geneontology.org). GO enrichment analysis was conducted using the clusterProfiler v4.0 R package (Wu et al., 2021). The significance of each GO term was evaluated using Fisher’s test, and the p-value was adjusted for multiple hypothesis testing with the Benjamini−Hochberg method. GO terms with an adjusted p-value ≤ 0.05 were considered significantly enriched. WEGO software (Ye et al., 2006) was used to visualize the GO classifications of all unigenes. The DESeq2 R package v1.16.1 (Love et al., 2014) was used to analyze differential expression between the two groups, with differentially expressed genes identified based on FDR-adjusted p-value < 0.01 and log2-fold change ≥ 2. Conserved motif analysis of the CubHLH gene family was conducted using the online tool MEME (https://meme-suite.org/meme/), and the motifs were visualized in TBtools (Chen et al., 2020).
We downloaded the sequence of bHLH proteins in Arabidopsis from the PlantTFDB v4.0 database (Jin et al., 2017). The profile hidden Markov models (HMMs) of the DNA-binding domains for the bHLH gene family (PF00010) were downloaded from the Pfam database (http://pfam-legacy.xfam.org/) and searched against the C. utilis protein sequence using HMMER (Finn et al., 2011) to identify candidate bHLH genes of C. utilis. The E-value cutoff was set to 10−5. To verify the reliability of the BLAST and HMMER intersection results, the completeness of the candidate gene domains was then checked using the Conserved Domain Database (CDD) from NCBI (https://www.ncbi.nlm.nih.gov/).
The physicochemical properties of the CubHLH gene family, including amino acid number, molecular weight, and isoelectric point were analyzed using TBtools (Chen et al., 2020). The subcellular localization of the CubHLH gene family was predicted using the Plant mPLoc tool (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/). Additionally, the secondary and tertiary structures of the C. utilis bHLH family proteins were predicted using the SOPMA online tool (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html).
Sequence alignment was carried out with MUSCLE. A phylogenetic tree of the CubHLH gene family was constructed using MEGA v7.0 (Hall, 2013) with the maximum likelihood (ML) method, and bootstrap analysis was set to 1,000 replicates. The protein–protein interaction networks were constructed for the identified CubHLH candidate genes using the online tool STRING v11.5 (https://string-db.org/), with A. thaliana selected as the reference species model and a confidence interaction score ≥0.7.
The RNA sequencing reads were aligned to the unigene library using Bowtie2 v2.4.4. Using the R script, the average expression of C. utilis genes in different color bamboo shoot sheaths was calculated based on normalized expression (fragments per kilobase per million mapped reads, FPKM), and RSEM (Li and Dewey, 2011) was used to estimate gene expression levels and then obtain the FPKM values. A heatmap was created using TBtools (Chen et al., 2020).
Complementary DNA (cDNA) was synthesized using the Synthesis SuperMix for qPCR (One-Step gDNA Remover) Kit (Wuhan Servicebio Technology Co., Ltd., Wuhan, China). qRT-PCR was performed using 2×Universal Blue SYBR Green qPCR Master Mix (Wuhan Servicebio Technology Co., Ltd., Wuhan, China) in a 20-μL reaction volume on a CFX Connect Real-Time PCR Detection System (Bio-Rad). The primers were designed using Primer v5.0 software. The TIP41 (Tonoplasm Intrinsic Protein 41) gene was used as an internal reference gene and was obtained from a previous report (Fan et al., 2013). Supplementary Table S1 lists the primers used in qRT-PCR. The 2−ΔΔCT method (Livak and Schmittgen, 2001) was employed to calculate the relative expression level of the candidate gene, and the expression levels of green bamboo shoot sheaths were used as the reference control. All qRT-PCR experiments were conducted with three biological replicates.
Statistical analysis of the bamboo sheath samples was performed in Excel 2021 using Student’s t-tests (p < 0.05). Statistical significance was determined via GraphPad Prism v9.5.1, and the data are presented as the mean ± standard error (SE). GraphPad Prism was used to generate the figures.
Upon visual inspection, the color differences among the five bamboo varieties were obvious (Figure 1). High-throughput sequencing was performed with three biological replicates for each of the 15 samples. A total of 131.38 Gb of clean data were obtained, with at least 7.68 Gb per sample. The GC content ranged between 46% and 49%, with no N bases detected, indicating data integrity (Supplementary Table S2). The Q20 values for all samples exceeded 96%, the Q30 base percentage was 91.27% or higher, and the cycle Q20 reached 100% in each sample, indicating excellent sequencing quality and minimal impact from sequencing cycles. After de novo assembly using high-quality reads, we obtained 195,977 transcripts and 75,137 unigenes after removing redundancy. The N50 value is an important indicator of assembly quality, reflecting the length continuity and integrity of a sequence. The results of this study showed that the N50 value of the transcripts was 1,704, and the N50 value of the unigenes was 1,370, indicating good assembly quality and suitability for subsequent analysis.

Figure 1. The phenotypes of different shoot sheaths color in Chimonobambusa utilis. Bsh represents black sheaths, Brsh represents brown sheaths, Rsh represents red sheaths, Gsh represents green sheaths, and Ysh represents yellow sheaths.
The unigenes were comprehensively annotated using multiple databases, including Nr, Swiss-Prot, GO, COG, KOG, and KEGG. The results indicated that 44,255 unigenes were successfully annotated with functional information. Additionally, protein domains within these unigenes were identified via alignment using the Pfam database and HMMER software, providing an essential basis for further investigations into gene functions and metabolic pathways. Pearson’s correlation coefficients revealed that a strong correlation between biological replicates within each group and showed that the samples were highly reproducible, particularly the correlation coefficient for the black-sheath (Bsh) and the brown-sheath (Brsh) samples, which was close to 0.92 (Supplementary Figure S1), indicating a high degree of consistency among the samples. The correlation coefficients for the yellow-sheath (Ysh) and red-sheath (Rsh) samples were 0.85 and 0.88 (Supplementary Figure S1), respectively, further confirming the reliability of the data. No significant batch effects or experimental biases were observed.
To identify pathways related to color, GO enrichment analysis was performed on the 44,255 functionally annotated unigenes. The enrichment results revealed that four pathways were most strongly associated with color (Figure 2). Pigmentation is a pathway that is directly related to color and involves the production, distribution, and deposition of pigments (Todesco et al., 2022). The transporter activity pathway is the site where pigment molecules are synthesized and accumulate. The genes in this pathway may be involved in regulating the spatial distribution of pigments (Wiriyasermkul et al., 2020). The third pathway, the catalytic activity pathway, may be enriched with genes encoding enzymes that catalyze pigment synthesis and are the core steps involved in regulating pigment synthesis (Zhang et al., 2022). Finally, the metabolic process pathway is enriched with genes related to the synthesis of secondary metabolites (such as flavonoids and anthocyanins), which determine color presentation (Li et al., 2021b). Focusing on the above core pathways, which included a total of 13,854 genes, we effectively located genes related to color.
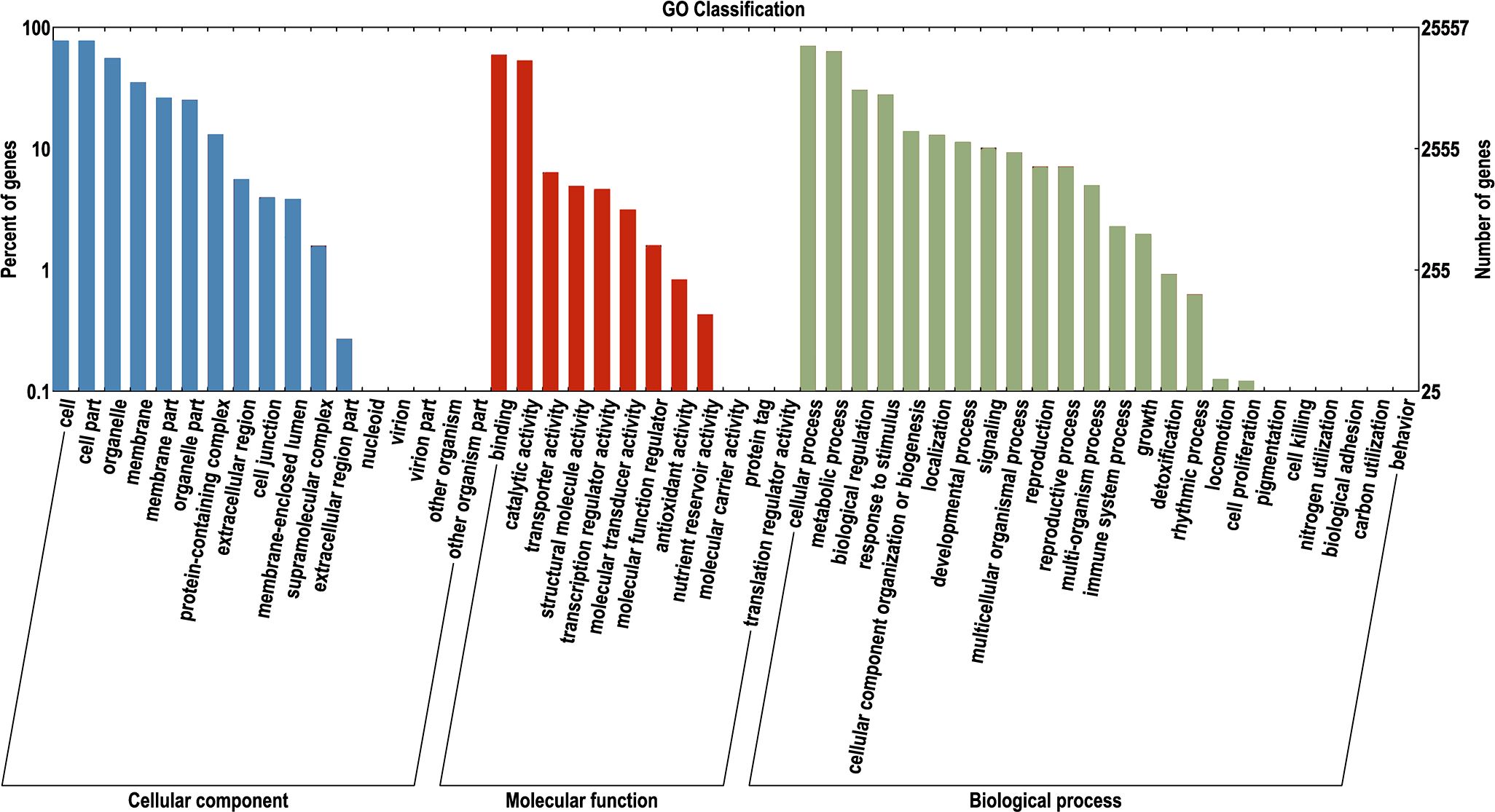
Figure 2. The results of GO enrichment analysis. The x-axis indicates the categories, with blue representing enrichment in cellular component, red representing enrichment in molecular function, and green representing enrichment in biological process. The y-axis represents the percentage of enriched items.
Differential expression analysis showed that a total of 4,185 genes were affected, of which 2,175 genes were upregulated and 2,010 genes were downregulated (Figure 3A). Further filtering of the differential gene expression analysis identified 168 significantly differentially expressed genes (Figure 4). There were obvious differences in the expression of these genes among the different samples. For example, TRINITY_DN9650_c0_g1 showed extremely high expression levels in the Rsh sample, whereas expression was not detected in the Brsh group. It is speculated that this gene may be involved in the biosynthetic pathway or pigment formation mechanism related to the coloration of the Rsh group. The expression of the TRINITY_DN4362_c0_g1 gene in the Ysh sample was significantly upregulated compared with that in the Gsh sample, indicating that it may play an important role in the biological processes or metabolic pathways of the Ysh group. Additionally, the expression of TRINITY_DN23972_c0_g1 showed a similar trend, indicating its potential function in specific physiological activities of the Ysh sample. In contrast, some genes exhibited significant downregulation trends between different color groups. For example, TRINITY_DN19324_c0_g2 showed high expression in the Gsh sample and almost no expression in the Rsh group, indicating that this gene may play a specific role in the biological process of the Gsh sample. The TRINITY_DN22383_c0_g1 gene was significantly downregulated in the Rsh group compared with that of the Brsh group (Figure 4), further highlighting the considerable differences in gene expression between these different sheath color samples.
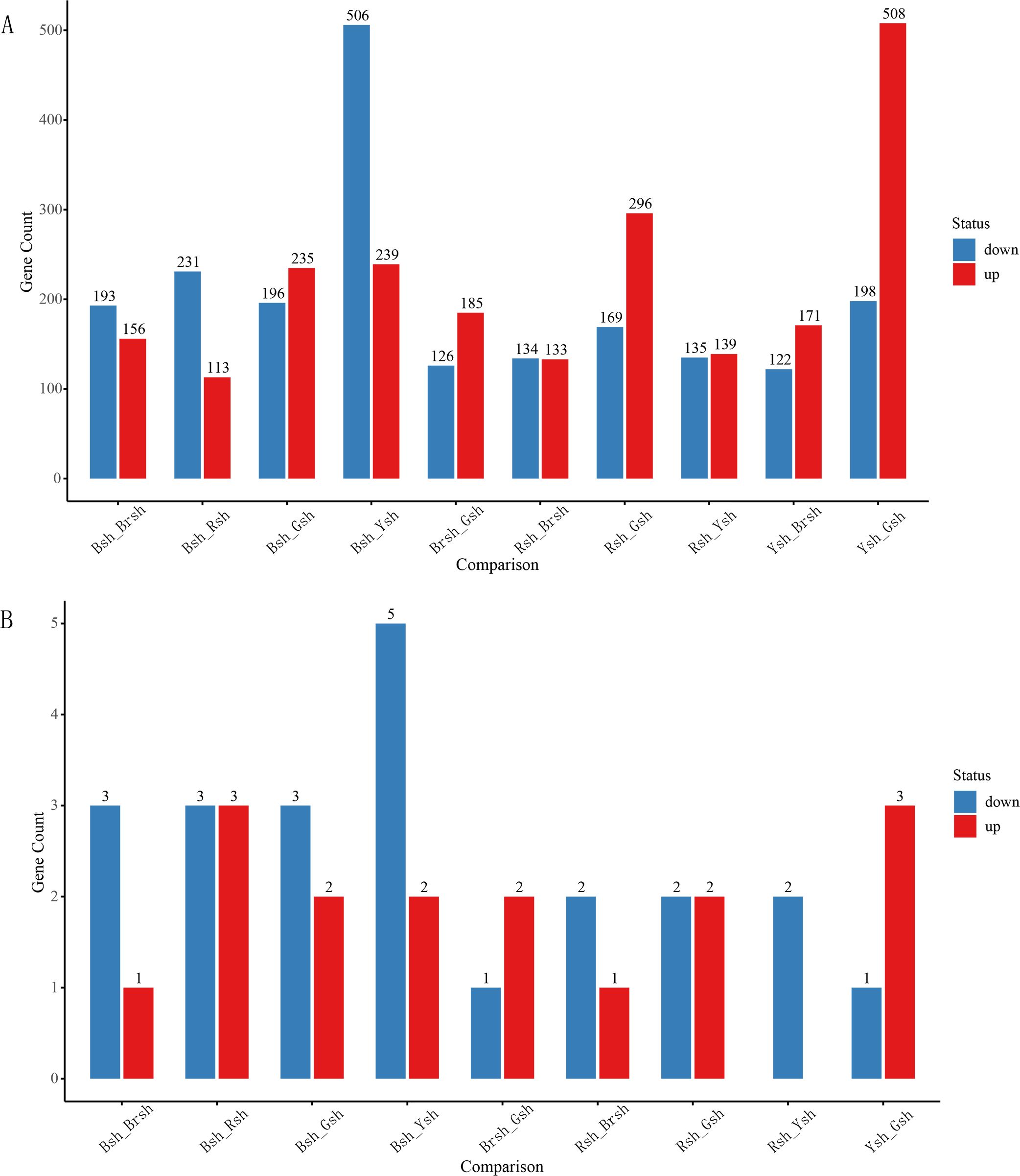
Figure 3. Differential gene analysis. (A) Visualization of the differential analysis based on the GO enrichment results of unigenes, where red indicates upregulation and blue indicates downregulation. The x-axis represents different groups in the differential analysis, and the y-axis represents the specific number of significant differential results. (B) Differential analysis of CubHLH gene family members, where red indicates upregulation and blue indicates downregulation. The x-axis represents different groups in the differential analysis, and the y-axis represents the specific number of significant differential results.
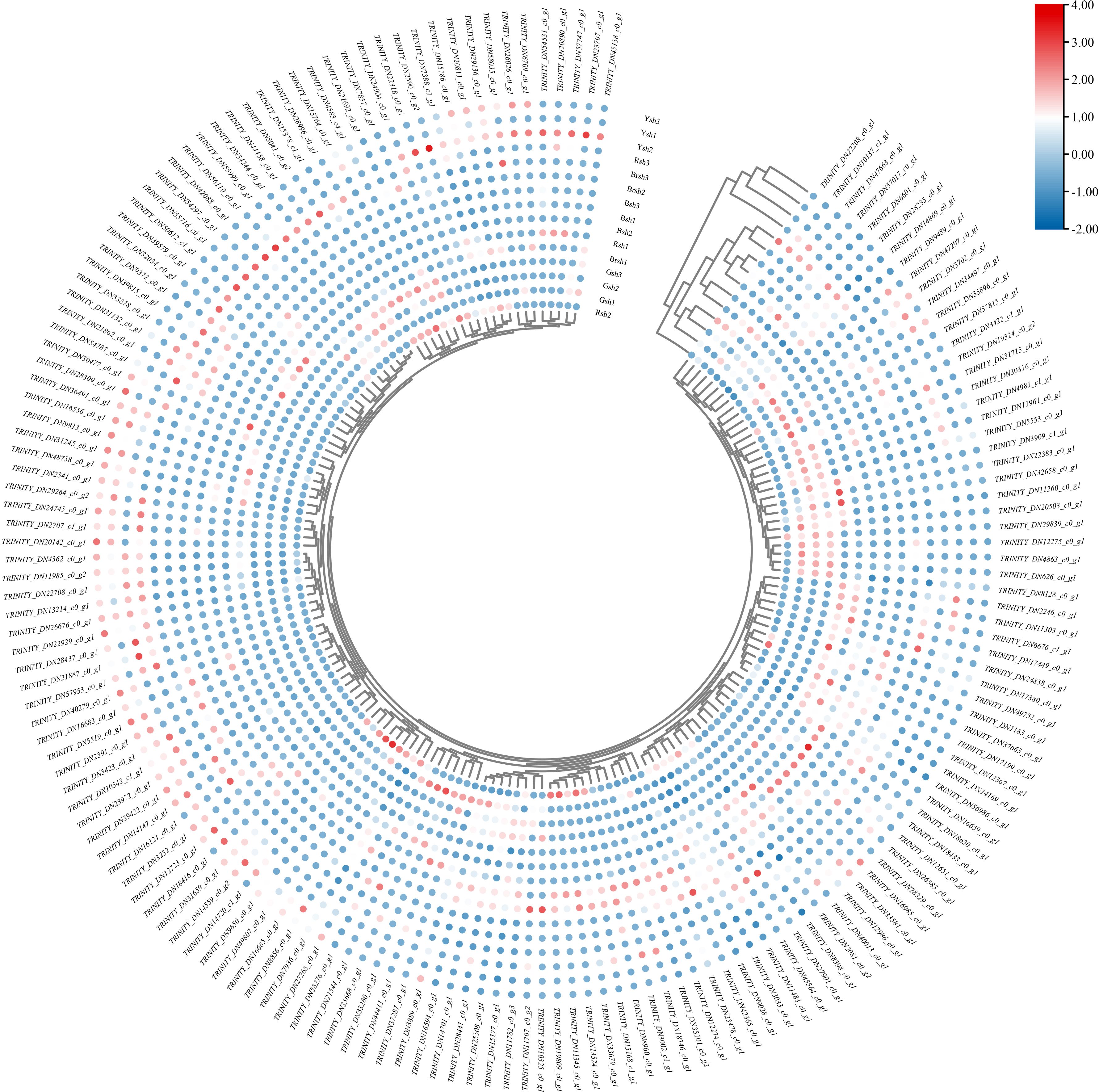
Figure 4. The heatmap of 168 significantly differentially expressed genes. The expression levels decrease gradually from red to blue. Each row represents the expression level of each gene across different samples. The scale bar indicates the log2-transformed FPKM values.
Based on the protein database from the C. utilis transcriptome data, candidate genes were screened using the hmmbuild and hmmsearch tools in HMMER. The sequences were then submitted to NCBI for validation via batch CD-Search. After sequences with incomplete domains or those lacking the bHLH domain were manually removed, a total of 44 CubHLH gene family members were identified and designated CubHLH1 to CubHLH44 (Supplementary Table S3). The specific corresponding original IDs can be found in Supplementary Table S4. Physicochemical property analysis revealed that the amino acid lengths of the CubHLH gene family ranged from 100 to 618 amino acids, with molecular weights ranging from 10,777.15 to 67,522.22 Da (Supplementary Table S5). The isoelectric points (pIs) ranged from 4.61 to 10.46 (Supplementary Table S5). There were 30 proteins with pIs > 7 and 14 with pIs < 7, which indicates that 30 of these proteins are basic amino acids and 14 are acidic amino acids. The instability index ranged from 34.12 to 88.18, and the aliphatic index was between 52.96 and 103.7. The grand average of hydropathy values (GRAVY) of the CubHLH gene family was negative (Supplementary Table S5), indicating that the CubHLH family proteins are hydrophilic. Subcellular localization prediction of CubHLH gene family members showed that, with the exception of CubHLH37, which is located in the chloroplast, the remaining 43 genes were all localized in the nucleus (Supplementary Table S5). Using the SOPMA online tool to predict the secondary structure of the CubHLH gene family proteins (Supplementary Figure S2) revealed that the α-helix content ranged from 16.98% to 42.5%, the extended strand content ranged from 1.8% to 15%, and the β-sheet content ranged from 0.75% to 11%, with CubHLH30 lacking a β-sheet. The random coil content ranged from 30.18% to 70.11% (Supplementary Table S6).
To investigate the potential role of the bHLH gene family in the color formation of C. utilis bamboo shoot sheaths, differential expression analysis was performed on all candidate genes. Differentially expressed genes were analyzed between bamboo shoot sheath samples of different colors (Bsh, Gsh, Brsh, Ysh, and Rsh). A pairwise comparison was conducted, resulting in a total of 10 groups (Supplementary Table S7; Figure 3B). The results showed that no significant differentially expressed genes were identified between the Ysh and Brsh groups. However, CubHLH3, CubHLH4, and CubHLH27 may play a role in the positive regulation of pigment synthesis in different bamboo sheath color samples. Notably, the CubHLH3 and CubHLH4 genes were upregulated in multiple groups, whereas the CubHLH27 gene was specifically upregulated in the Bsh samples (Supplementary Table S7) and is potentially related to the accumulation of black pigments. Conversely, the CubHLH1, CubHLH2, CubHLH7, CubHLH22, and CubHLH41genes, which are downregulated, may negatively regulate color expression by inhibiting certain pigment biosynthesis pathways. The CubHLH7 gene was downregulated in multiple groups (Supplementary Table S7), suggesting that it may suppress the expression of nonblack pigments in black bamboo shoot sheath samples. In addition, CubHLH31 and CubHLH30 showed a dual pattern of upregulation and downregulation in different comparison groups (Supplementary Table S7). We speculate that these genes may balance the synthesis and degradation of pigments through complex tissue-specific or developmental stage-specific regulatory mechanisms, ultimately affecting the color diversity of the bamboo shoot sheath and the development of bamboo sheath color.
The results of the physicochemical property analysis revealed that most of the upregulated genes have isoelectric points primarily in the neutral to slightly acidic range, have moderate molecular weights, and possess negative hydropathy indices, indicating that they are hydrophilic proteins. These proteins likely exert their transcriptional regulatory functions by binding to other proteins or DNA. The CubHLH7 gene, which is downregulated in multiple groups, was found to have a relatively high isoelectric point and is also a hydrophilic protein, which is consistent with its localization in the nucleus (Supplementary Table S5). Therefore, based on the differential expression and physicochemical property analyses, it can be inferred that the upregulated CubHLH gene family members promote pigment synthesis primarily through their transcriptional regulatory functions in the nucleus, whereas downregulated genes such as CubHLH7 may negatively regulate color expression by inhibiting specific pigment biosynthesis pathways. These findings suggest that the CubHLH gene family plays a complex regulatory role in the development of shoot sheath color in bamboo.
Conserved motif prediction and visualization analysis were performed on the 44 CubHLH gene family members (Figure 5A). A total of 10 motifs, designated motif1 to motif10, were identified. Each gene contained 1–5 motifs, among which motif 1 and motif 2 were the most numerous and were located primarily at the N-terminus (5′ end) of the protein. Specifically, motif 1 was present in 40 CubHLH genes, and motif 2 was found in 38 CubHLH genes. CubHLH3, CubHLH9, CubHLH15, and CubHLH26 contained only motif 2, whereas CubHLH6, CubHLH11, CubHLH19, CubHLH37, and CubHLH38 contained only motif 1. Some motifs existed only in specific genes, such as motif 4, which was present only in CubHLH13 and CubHLH43 of the vIIb subfamily. These specific motifs may confer unique functions to genes. Within the same evolutionary subgroup, most genes exhibited similar conserved motif compositions, suggesting that they may share similar biological functions. For example, CubHLH21, CubHLH34, and CubHLH41 from the vIIIc subfamily all contained motifs 1, 2, 3, and 9, whereas CubHLH17 from the IIIf subfamily contained motifs 1, 2, and 8. Given that the CubHLH17 gene of the IIIf subfamily is closely related to anthocyanin biosynthesis, the conserved motif composition of this type of gene may be related to its function in pigment synthesis pathways.
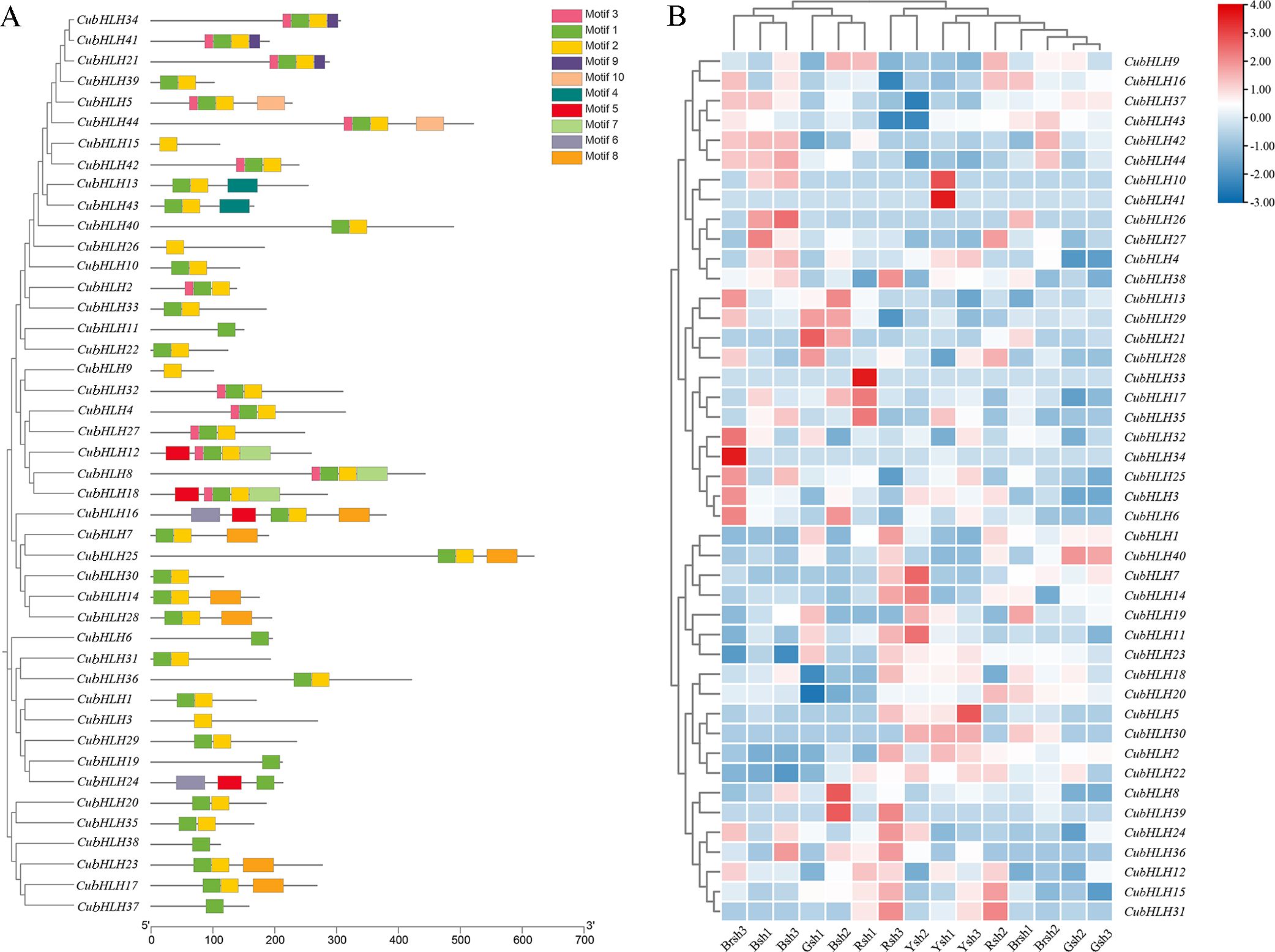
Figure 5. The predicted motif and expression profiles analysis of CubHLHs. (A) The predicted motifs for the candidate genes of the CubHLH gene family. The motifs are denoted by different colored boxes. (B) The heatmap of expression profiles of the CubHLH family candidate genes. The scale indicates the log2-transformed FPKM values, with a color spectrum from red to blue representing gene expression levels that range from high to low, respectively.
The expression levels of the CubHLH genes were analyzed in bamboo shoot sheath samples of different colors, including black (Bsh1, Bsh2, and Bsh3), green (Gsh1, Gsh2, and Gsh3), brown (Brsh1, Brsh2, and Brsh3), yellow (Ysh1, Ysh2, and Ysh3), and red (Rsh1, Rsh2, and Rsh3) (Figure 5B). Clustering analysis of CubHLH gene expression levels among the samples revealed significant variation in expression levels between different samples for each gene.
To explore the evolutionary relationships within the CubHLH gene family, a phylogenetic tree was constructed using the 44 CubHLH protein sequences and the protein sequences of Arabidopsis bHLH (Figure 6). Following the classification method of Arabidopsis (Heim et al., 2003; An et al., 2014), one to three representative genes from different subgroups were selected to construct a phylogenetic tree together with the CubHLH genes, resulting in a tree containing 49 AtbHLH and 44 CubHLH genes. These genes were ultimately classified into 26 subgroups. The results showed that the distribution of CubHLHs in the 26 subgroups was quite different, among which CubHLH16 and CubHLH24 belonged to the independent SI subfamily and did not cluster with any Arabidopsis group, which may reflect unique gene expansion or the generation of novel functions in C. utilis.
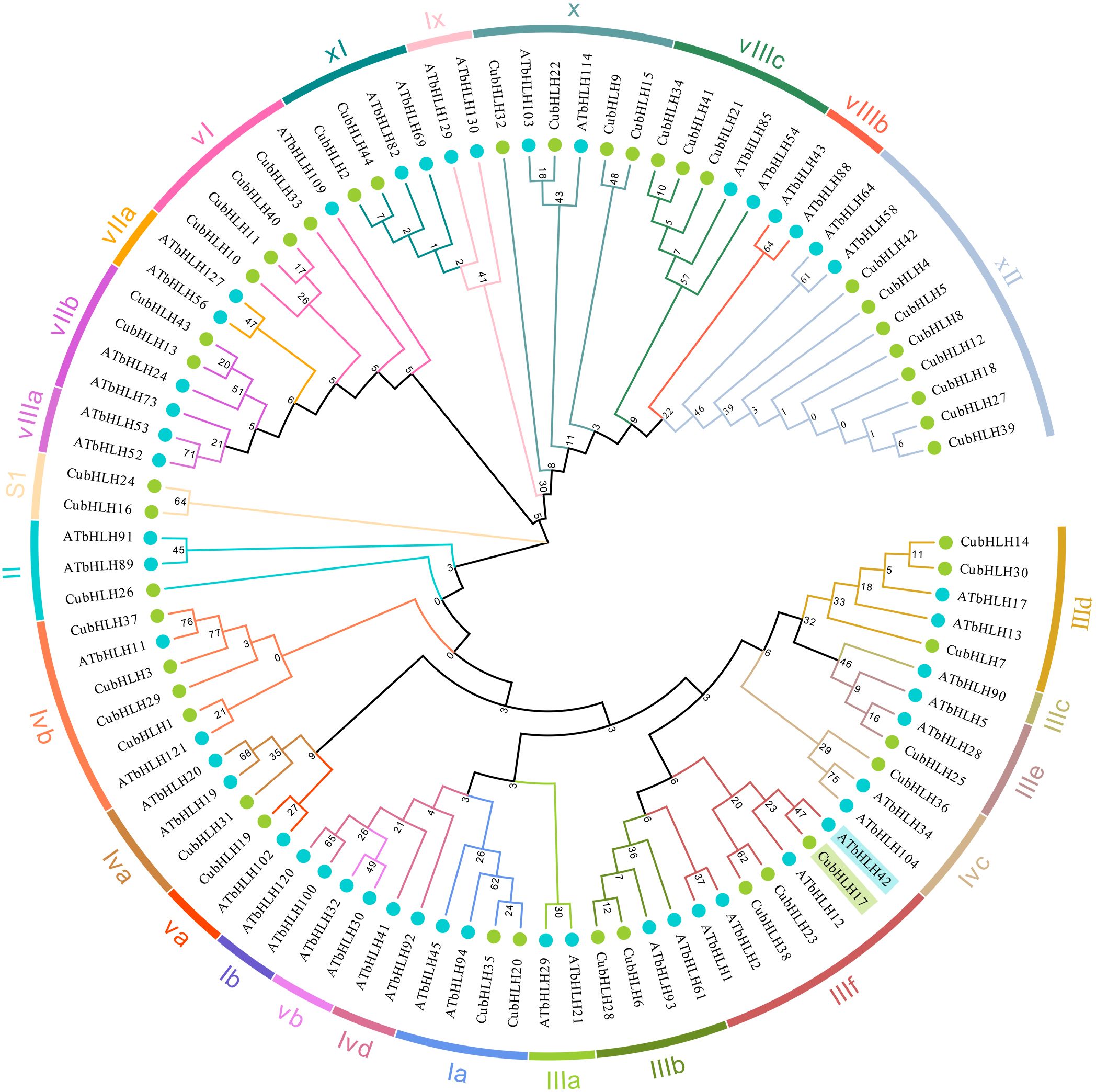
Figure 6. Phylogenetic tree analysis of CubHLHs. The 26 subgroups are marked in different colors on the periphery of the circle. The names of each subgroup, along with their respective gene IDs, are annotated around the outer perimeter of the tree for clarity. The blue circles represent the genes from the Arabidopsis thaliana bHLH family, and green circles represent the genes from the C. utilis bHLH family.
No corresponding CubHLH genes were found in some subgroups, such as Ib, Vb, Ivd, IIIa, IIIc, VIIIb, Ix, VIIa, and VIIIa, which may be the result of gene loss or evolutionary divergence. Notably, CubHLH genes were most abundant in subfamily XII, with a total of eight genes, whereas the remaining genes were more evenly distributed in other subgroups, with each group containing one to four genes. Prior research indicates that AtbHLH12 and AtbHLH42 in the IIIf subfamily are closely related to anthocyanin biosynthesis and that the accumulation of anthocyanins directly affects color expression in plants (Li et al., 2021a; Makkena and Lamb, 2013). Phylogenetic analysis showed that CubHLH17 clustered with the IIIf subfamily, which shares the same branch as AtbHLH42 (Figure 6), suggesting that CubHLH17 may have functions similar to those of the homologous genes in Arabidopsis thaliana and could be involved in anthocyanin biosynthesis in C. utilis bamboo shoot sheaths.
To further analyze the function of the CubHLH gene family in the coloration of C. utilis, this study selected CubHLH17 gene, which is highly related to anthocyanin synthesis, for protein interaction network analysis. Using STRING for prediction and selecting Arabidopsis as the model organism, the results showed that CuBHLH17 strongly interacts with four proteins and is the most closely associated with bHLH30 (Figure 7A). A Venn diagram was drawn based on the 168 highly color-related genes identified via GO enrichment and the CubHLH gene family members (Figure 7B), and the results indicated that CubHLH30 was identified by both approaches, implying that it may have some functions in the bamboo shoot sheath. The clustering of these proteins suggests that they may have similar or related functions. CubHLH17 may participate in specific biological processes through interactions with these proteins, providing important clues for further functional studies.
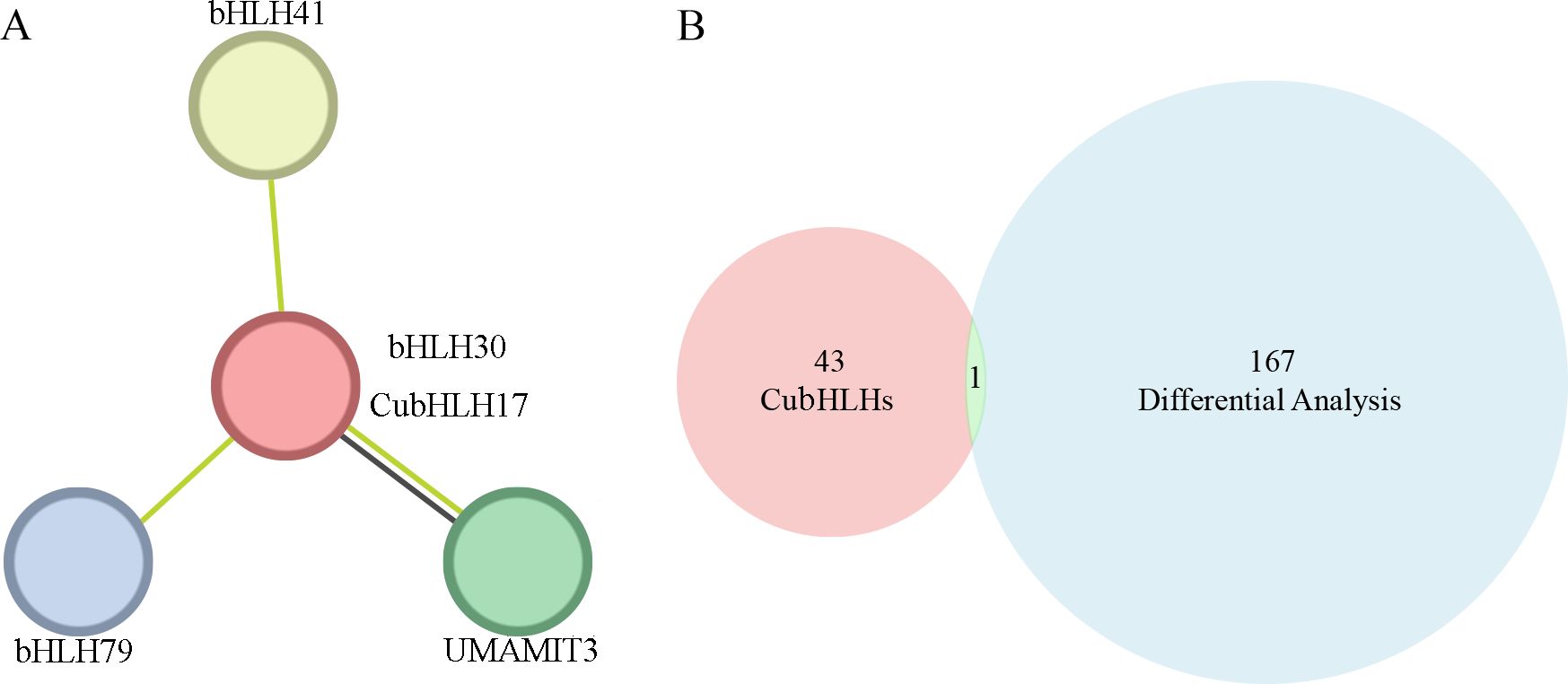
Figure 7. Protein–protein interaction network of CubHLH17 proteins in C.) utilis from the model plant Arabidopsis thaliana homologous genes. (A) Green lines represent associations proteins identified through text mining, and black lines represent co-expression relationships. The red circle in the center indicates that bHLH30 is directly related to CubHLH17 in the protein interaction network. (B) The Venn diagram depicts the overlap between two gene sets. The red circles showed the CubHLH gene family members, while the blue circles represent the genes identified through GO enrichment and differential expression analysis based on unigenes. The intersection in the middle represents the common genes identified by both datasets.
Based on the results of the phylogenetic tree and protein interaction network analysis, we selected the color-related CubHLH17 gene and used real-time quantitative qRT-PCR technology to analyze its expression in bamboo shoots of different colors (Figure 8). The results indicated that CubHLH17 was expressed in all samples (Figure 8A), suggesting that it may play a regulatory role in the process of shoot sheath color formation in bamboo. A t-test revealed a significant difference in CubHLH17 expression between the Rsh group and the Gsh group (Figure 8B). CubHLH17 showed higher expression levels in Rsh, suggesting that CubHLH17 may play a role in the development of specific colors. Protein−protein interaction analysis showed that CubHLH17 is associated with bHLH30 (Figure 7A), which is involved in red coloration. These results provide some evidence that CubHLH17 may be involved in the regulation of red bamboo shoot sheath formation. In future studies, all color-related candidate genes identified in our study, along with CubHLH17, will be integrated with other omics approaches to further explore their key regulatory roles in the coloration of C. utilis.
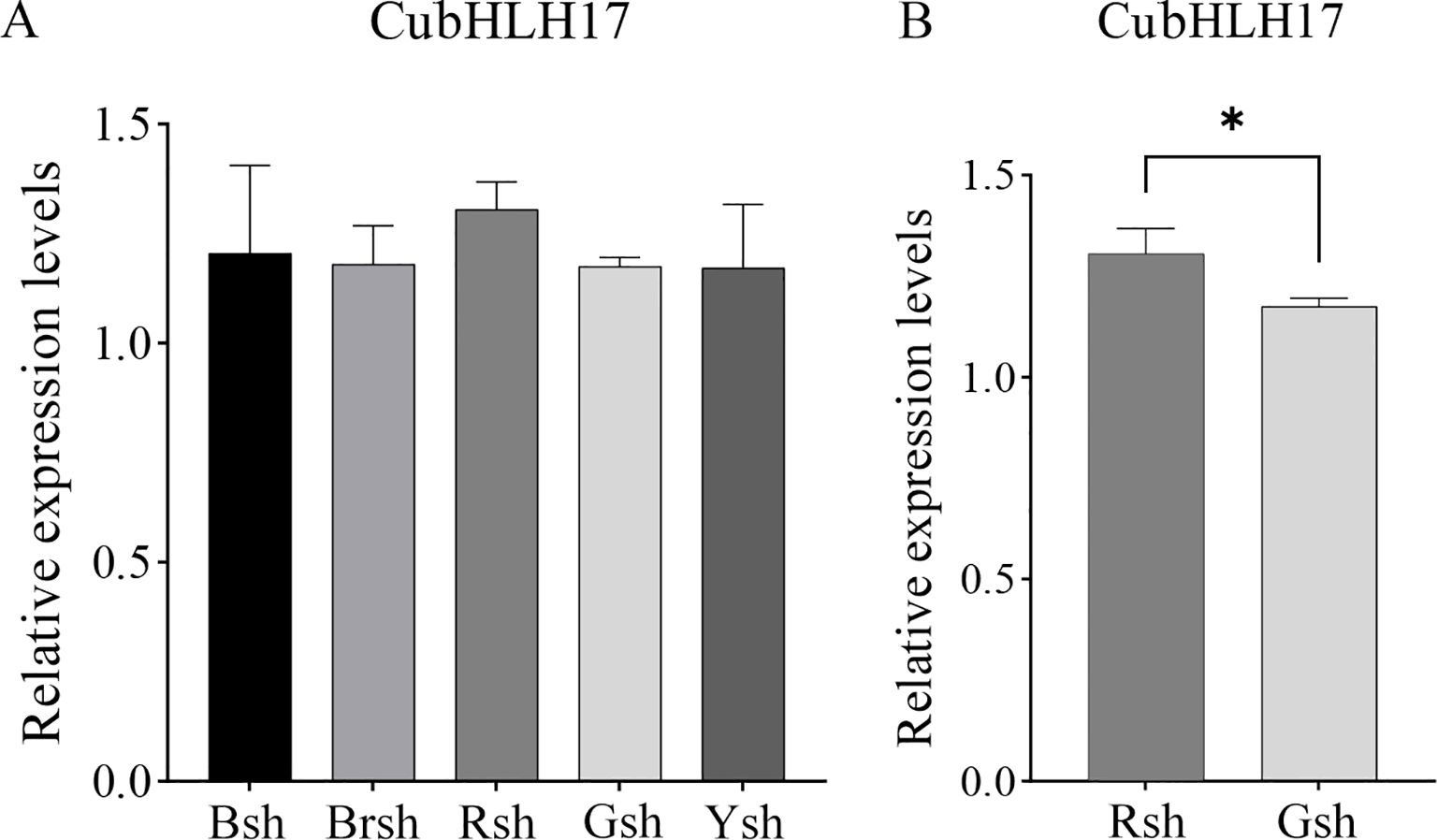
Figure 8. qRT-PCR analysis of the expression of transcription factor. (A) The relative expression levels of transcription factor coding gene of CubHLH17. Black bamboo sheath (Bsh), brown bamboo sheath (Brsh), green bamboo sheath (Gsh), red bamboo sheath (Rsh), and yellow bamboo sheath (Ysh). (B) The relative expression levels of CubHLH17 between the Rsh and Gsh group. The internal reference gene was TIP41, the expression of green bamboo sheath as a reference. Bars are means of three replicates ± SE. Asterisks (*) indicate the statistical significance of the difference between the Rsh and Gsh group. (*p < 0.05).
Elucidating the molecular mechanisms responsible for the coloration of bamboo shoot sheaths is essential for enhancing quality and market competition. Previous studies have shown that bHLH transcription factors play a key regulatory role in anthocyanin biosynthesis in plants (Zhao et al., 2021; Jiang et al., 2022; Liu et al., 2023). We identified 168 significantly differentially expressed genes based on de novo transcriptome assembly data and then selected the bHLH gene family related to coloration for analysis. A total of 44 CubHLH gene family members were identified in the shoot sheaths of C. utilis. Compared with the 602 bHLH genes identified in Brassica napus (Ke et al., 2020), 159 genes found in Solanum lycopersicum (Sun et al., 2015), 85 bHLH genes in Ginkgo biloba (Zhou et al., 2020), 120 bHLHs in Camellia sinensis (Cui et al., 2018), and 128 bHLH genes in Betula platyphylla (Zhao et al., 2023), the number of bHLH genes in C. utilis is relatively small. This difference may reflect the evolutionary characteristics of different species and the different degrees of gene family expansion. Differential expression analysis was performed on all candidate gene families, identifying 12 significantly differentially expressed genes. By comparing all the candidate genes with the 168 genes identified in this study, the CubHLH30 gene was selected. Previous studies have shown that bHLH30 is one of the key transcription factors involved in anthocyanin biosynthesis and is closely related to the synthesis of cyanidin-3-O-galactoside, which is the main pigment responsible for red coloration in fruits. bHLH30 promotes the synthesis of anthocyanins associated with color expression by regulating the expression of structural genes (Ye et al., 2023).
Furthermore, we found that CubHLH17 also clustered within the IIIf subfamily based on the phylogenetic tree constructed using A. thaliana and motif prediction, which has been shown to be closely associated with anthocyanin biosynthesis in other species. For example, three candidate ZjbHLH genes in Ziziphus jujuba belong to the III subfamily and are highly related to anthocyanin biosynthesis (Shi et al., 2019). In Cinnamomum camphora, CcbHH001, CcbHH022, CcbHH118, and CcbHH134 are classified into the IIIf subfamily and are homologous to AtbHLH042, which is associated with anthocyanin biosynthesis (Gong et al., 2023). The SmbHLH1 and SmbHLH117 genes in Solanum melongena have been shown to be highly related to anthocyanin biosynthesis (Tian et al., 2019). CmbHLH2 has been confirmed to be involved in anthocyanin synthesis in Chrysanthemum morifolium (Xiang et al., 2015). Similarly, the VuMYB90-2, VuMYB90-3, VuMYB4-40, VuCPC, VuMYB, bHLH, and WD proteins of Vigna unguiculata, coordinate the regulation of anthocyanin and flavonoid accumulation (Li et al., 2020). In addition, a protein interaction network was constructed with A. thaliana, and CubHLH17 was found to be most closely associated with bHLH30. To verify whether the CubHLH17 gene has a regulatory role similar to that of bHLH30, we used qRT-PCR to verify the expression of CubHLH17. The results showed that the CubHLH17 gene was significantly different between the Rsh and Gsh samples (Figure 8B), indicating that this gene may be related to the development of red bamboo shoot color and may play a key role in anthocyanin regulation, which provides the necessary transcription factor background for color-related biosynthesis pathways.
Pigment synthesis and accumulation are complex biological processes that involve the coordinated regulation of multiple genes, transcription factors, and environmental factors. Although the CubHLH17 gene was stably expressed in all samples and significantly expressed in the red bamboo sheath, its ultimate function may not be produced independently, and it may require interaction with other regulatory factors. bHLH transcription factors typically form regulatory complexes with other transcription factors, such as MYB and WD40 repeat proteins, which affect the synthesis and accumulation of pigments through synergistic effects (Gonzalez et al., 2007; Xu et al., 2015; Chen et al., 2021b). Hence, we speculate that CubHLH17 has an important influence on the development of bamboo shoot color through interactions with other factors. The stable expression of CubHLH17 in samples of different colors indicates that it may be an essential component for maintaining normal operation of the pigment synthesis pathway. In the future, with the improvement in bamboo genome big data, we can further explore the interactions between CubHLH17 and other key transcription factors and provide a new perspective for revealing the molecular regulatory network of bamboo sheath color changes through multilevel functional studies. We can also use plant genome editing tools such as the CRISPR-Cas9 system (Wu et al., 2024), RNA interference (Niu et al., 2008; Spada et al., 2024), and artificial microRNA technologies (Zhang et al., 2023) to target CubHLH17 and transform it into a bamboo variety with potential industrial application value in terms of aesthetics, quality, and market value, thereby contributing to scientific research and commercial bamboo cultivation. In summary, the discovery and identification of CubHLH17 contributes to a deeper understanding of the color variation of bamboo sheaths and provides important genetic insights for the improvement and industrial application of the bamboo sheath. We believe that this research has the potential to improve the color and quality of bamboo through gene editing, thereby driving innovation and progress in the bamboo industry.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
LT: Data curation, Formal analysis, Investigation, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. QZ: Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Writing – review & editing. YG: Data curation, Formal analysis, Software, Validation, Visualization, Writing – original draft. YL: Formal analysis, Investigation, Resources, Software, Writing – review & editing. HL: Investigation, Resources, Software, Validation, Writing – review & editing. LC: Formal analysis, Investigation, Resources, Writing – review & editing. XL: Conceptualization, Investigation, Methodology, Project administration, Resources, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Key research projects of Yibin of Sichuan Province (YBZD2024-1), Chongqing Science and Technology Forestry Major Special Project (ZD2022-4). Chongqing Nanchuan District Forestry Bureau Industrial Development Project (HH-90735).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1514703/full#supplementary-material.
Supplementary Figure 1 | Heatmap of pairwise correlations between different samples or variables. Both the horizontal and vertical axes represent individual samples/variables. Blue indicates low correlation (close to 0.5) and red indicates high correlation (close to 1.0). The value in each cell represents the correlation coefficient between individual samples or variables. The dendrograms on the top and left show hierarchical clustering based on correlation features, with shorter branch lengths indicating higher similarity between samples.
Supplementary Figure 2 | The secondary structure prediction of CubHLH proteins. Listed sequentially from top to bottom as CubHLH1 to CubHLH44.
Alsamman, A., Abdelsattar, M., Allali, A. E., Radwan, K. H., Nassar, A. E., Mousa, K., et al. (2023). Genome-wide identification, characterization, and validation of the bHLH transcription factors in grass pea. Front. Genet. 14. doi: 10.3389/fgene.2023.1128992
An, R., Liu, X.-Y., Wang, R., Wu, H.-C., Liang, S., Shao, J.-X., et al. (2014). The over-expression of two transcription factors, ABS5/bHLH30 and ABS7/MYB101, leads to upwardly curly leaves. PloS One 9, e107637. doi: 10.1371/journal.pone.0107637
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Xia, R. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–2020. doi: 10.1016/j.molp.2020.06.009
Chen, C., Zhou, G., Chen, J., Liu, X.-H., Lu, X.-Y., Chen, H.-M., et al. (2021b). Integrated metabolome and transcriptome analysis unveils novel pathway involved in the formation of yellow peel in Cucumber. Int. J. Mol. Sci. 22, 1494. doi: 10.3390/ijms22031494
Chen, G.-J., Fang, C.-C., Ran, C.-X., Tan, Y., Yu, Q.-Q., Kan, J.-Q. (2019). Comparison of different extraction methods for polysaccharides from bamboo shoots (Chimonobambusa quadrangularis) processing by-products. Int. J. Biol. Macromol. 130, 903–914. doi: 10.1016/j.ijbiomac.2019.03.038
Chen, M., Ju, Y., Ahmad, Z. S., Yin, Z.-F., Ding, Y.-L., Que, F., et al. (2021a). Multi-analysis of sheath senescence provides new insights into bamboo shoot development at the fast growth stage. Tree Physiol. 41, 491–507. doi: 10.1093/treephys/tpaa140
Chen, S.-Y., Yang, W.-T., Jia, Q.-M., Wang, W., Zhang, N., Wang, X.-T., et al. (2017). Pleurotus ostreatus bHLH transcription factors regulate plant growth and development when expressed in Arabidopsis. J. Plant Interact. 12, 542–549. doi: 10.1080/17429145.2017.1400124
Cooley, A. M., Willis, J. H. (2009). Genetic divergence causes parallel evolution of flower color in Chilean Mimulus. New Phytol. 183, 729–739. doi: 10.1111/j.1469-8137.2009.02858.x
Cui, X., Wang, Y.-X., Liu, Z.-W., Wang, W.-L., Li, H., Zhuang, J. (2018). Transcriptome-wide identification and expression profile analysis of the bHLH family genes in Camellia sinensis. Funct. Integr. Genomics 18, 489–503. doi: 10.1007/s10142-018-0608-x
Fan, C.-J., Ma, J.-M., Guo, Q.-R., Li, X.-T., Wang, H., Lu, M.-Z. (2013). Selection of reference genes for quantitative real-time PCR in bamboo (Phyllostachys edulis). PloS One 8, e56573. doi: 10.1371/journal.pone.0056573
Feller, A., Machemer, K., Braun, E. L., Grotewold, E. (2011). Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 66, 94–116. doi: 10.1111/j.1365-313X.2010.04459.x
Finn, R. D., Clements, J., Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Gong, X., Shen, T.-F., Li, X.-Q., Lin, H.-B., Chen, C.-H., Li, H.-H., et al. (2023). Genome-wide characterization and analysis of bHLH transcription factors related to anthocyanin biosynthesis in Cinnamomum camphora (‘Gantong 1’). Int. J. Mol. Sci. 24, 3498. doi: 10.3390/ijms24043498
Gonzalez, A., Zhao, M.-Z., Leavitt, J. M., Lloyd, A. M. (2007). Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 53, 814–827. doi: 10.1111/j.1365-313X.2007.03373.x
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Hall, B. (2013). Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 30, 1229–1235. doi: 10.1093/molbev/mst012
Han, W.-J., Zhang, Q., Suo, Y.-J., Li, H.-W., Diao, S.-F., Sun, P., et al. (2023). Identification and expression analysis of the bHLH gene family members in Diospyros kaki. Horticulturae 9, 380. doi: 10.3390/horticulturae9030380
Heim, M. A., Jakoby, M., Werber, M., Martin, C., Weisshaar, B., Bailey, P. C. (2003). The basic helix-loop-helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 20, 735–747. doi: 10.1093/molbev/msg088
Jiang, H.-M., Liu, L.-L., Shan, X.-Z., Wen, Z.-H., Zhang, X.-L., Yao, X.-W., et al. (2022). Genome-wide identification and expression analysis of the bHLH gene family in cauliflower (Brassica oleracea L.). Physiol. Mol. Biol. Plants. 28, 1737–1751. doi: 10.1007/s12298-022-01238-9
Jin, J.-P., Tian, F., Yang, D.-C., Meng, Y.-Q., Kong, L., Luo, J.-C., et al. (2017). PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045. doi: 10.1093/nar/gkw982
Ke, Y.-Z., Wu, Y.-W., Zhou, H.-J., Chen, P., Wang, M.-M., Liu, M.-M., et al. (2020). Genome-wide survey of the bHLH super gene family in Brassica napus. BMC Plant Biol. 20, 115. doi: 10.1186/s12870-020-2315-8
Li, G.-S. (2021). Comparative analysis of ‘Jinyan’ and ‘Hongyang’ kiwifruit transcriptomes. Acta Hortic. Sin. 48, 1183–1196. doi: 10.16420/j.issn.0513-353x.2020-0510
Li, Y., Chen, Q.-Y., Xie, X.-D., Cai, Y., Li, J.-F., Feng, Y.-L., et al. (2020). Integrated metabolomics and transcriptomics analyses reveal the molecular mechanisms underlying the accumulation of anthocyanins and other flavonoids in cowpea pod (Vigna unguiculata L.). J. Agric. Food Chem. 68, 9260–9275. doi: 10.1021/acs.jafc.0c01851
Li, B., Dewey, C.-N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12, 323. doi: 10.1186/1471-2105-12-323
Li, X., Li, Y., Zhao, M.-H., Hu, Y.-B., Meng, F.-J., Song, X.-S., et al. (2021b). Molecular and metabolic insights into anthocyanin biosynthesis for leaf color change in chokecherry (Padus virginiana). Int. J. Mol. Sci. 22, 10697. doi: 10.3390/ijms221910697
Li, M., Sun, L., Gu, H., Cheng, D.-W., Guo, X.-Z., Chen, R., et al. (2021a). Genome-wide characterization and analysis of bHLH transcription factors related to anthocyanin biosynthesis in spine grapes (Vitis davidii). Sci. Rep. 11, 6863. doi: 10.1038/s41598-021-85754-w
Liu, Z., Fu, X.-A., Xu, H., Zhang, Y.-X., Shi, Z.-D., Zhou, G.-Z., et al. (2023). Comprehensive analysis of bHLH transcription factors in Ipomoea aquatica and its response to anthocyanin biosynthesis. In. J. Mol. Sci. 24, 5652. doi: 10.3390/ijms24065652
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. doi: 10.1186/s13059-020-02196-9
Ludwig, S. R., Habera, L. F., Dellaporta, S. L., Wessler, S. R. (1989). Lc, a member of the maize R gene family responsible for tissue-specific anthocyanin production, encodes a protein similar to transcriptional activators and contains the myc-homology region. Proc. Natl. Acad. Sci. U.S.A. 86, 7092–7096. doi: 10.1073/pnas.86.18.7092
Makkena, S., Lamb, R. S. (2013). The bHLH transcription factor SPATULA is a key regulator of organ size in Arabidopsis thaliana. Plant Signal Behav. 8, e24140. doi: 10.4161/psb.24140
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121. doi: 10.1093/nar/gkt263
Murre, C., McCaw, P. S., Baltimore, D. (1989). A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell 56, 777–783. doi: 10.1016/0092-8674(89)90682-X
Niu, B.-L., Shen, W.-F., Liu, Y., Weng, H.-B., He, L.-H., Mu, J.-J., et al. (2008). Cloning and RNAi-mediated functional characterization of MaLac2 of the pine sawyer, Monochamus alternatus. Insect. Mol. Biol. 17, 303–312. doi: 10.1111/j.1365-2583.2008.00803.x
Patel, R. K., Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS One 7, e30619. doi: 10.1371/journal.pone.0030619
Shi, Q.-Q., Li, X., Du, J.-T., Li, X.-G. (2019). Anthocyanin synthesis and the expression patterns of bHLH transcription factor family during development of the Chinese jujube fruit (Ziziphus jujuba Mill.). Forests 10, 346. doi: 10.3390/f10040346
Song, M.-Y., Wang, H.-M., Wang, Z., Huang, H.-T., Chen, S.-W., Ma, H.-Q. (2021). Genome-wide characterization and analysis of bHLH transcription factors related to anthocyanin biosynthesis in fig (Ficus carica L.). Front. Plant Sci. 12. doi: 10.3389/fpls.2021.730692
Spada, M., Pugliesi, C., Fambrini, M., Pecchia, S. (2024). Challenges and opportunities arising from host–Botrytis cinerea interactions to outline novel and sustainable control strategies: The key role of RNA interference. Int. J. Mol. Sci. 25, 6798. doi: 10.3390/ijms25126798
Streisfeld, M. A., Kohn, J. R. (2005). Contrasting patterns of floral and molecular variation across a cline in Mimulus aurantiacus. Evolution 59, 2548–2559. doi: 10.1111/j.0014-3820.2005.tb00968.x
Sun, H., Fan, H.-J., Ling, H.-Q. (2015). Genome-wide identification and characterization of the bHLH gene family in tomato. BMC Genomics 16, 9. doi: 10.1186/s12864-014-1209-2
Tian, S.-Y., Li, L.-J., Wei, M., Yang, F.-J. (2019). Genome-wide analysis of basic helix-loop–helix superfamily members related to anthocyanin biosynthesis in eggplant (Solanum melongena L.). PeerJ 7, e7768. doi: 10.7717/peerj.7768
Todesco, M., Bercovich, N., Kim, A., Imerovski, I., Owens, G. L., Dorado Ruiz, Ó., et al. (2022). Genetic basis and dual adaptive role of floral pigmentation in sunflowers. Elife 11, e72072. doi: 10.7554/eLife.72072
Wang, X.-J., Peng, X.-Q., Shu, X.-C., Li, Y.-H., Wang, Z., Zhuang, W.-B. (2022). Genome-wide identification and characterization of PdbHLH transcription factors related to anthocyanin biosynthesis in colored-leaf poplar (Populus deltoides). BMC Genomics 23, 244. doi: 10.1186/s12864-022-08460-5
Wang, S.-G., Zhan, H., Li, P.-C., Chu, C.-H., Li, J., Wang, C.-M. (2020). Physiological mechanism of internode bending growth after the excision of shoot sheath in Fargesia yunnanensis and its implications for understanding the rapid growth of bamboos. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.00418
Wang, F., Zhang, X.-P., Yang, Q.-Z., Zhao, Q.-F. (2019). Exogenous melatonin delays postharvest fruit senescence and maintains the quality of sweet cherries. Food Chem. 301, 125311. doi: 10.1016/j.foodchem.2019.125311
Wiriyasermkul, P., Moriyama, S., Nagamori, S. S. (2020). Membrane transport proteins in melanosomes: Regulation of ions for pigmentation. Biochim. Biophys. Acta Biomembr. 1862, 183318. doi: 10.1016/j.bbamem.2020.183318
Wu, T.-Z., Hu, E.-Q., Xu, S.-B., Chen, M.-J., Guo, P.-F., Dai, Z.-H., et al. (2021). clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141. doi: 10.1016/j.xinn.2021.100141
Wu, S.-T., Kyaw, H., Tong, Z.-J., Yang, Y.-R., Wang, Z.-W., Zhang, L.-Y., et al. (2024). A simple and efficient CRISPR/Cas9 system permits ultra-multiplex genome editing in plants. Crop J. 12, 569–582. doi: 10.1016/j.cj.2024.01.010
Xiang, L.-L., Liu, X.-F., Li, X., Yin, X.-R., Grierson, D., Li, F., et al. (2015). A novel bHLH transcription factor involved in regulating anthocyanin biosynthesis in Chrysanthemums (Chrysanthemum morifolium Ramat.). PloS One 10, e0143892. doi: 10.1371/journal.pone.0143892
Xu, W.-J., Dubos, C., Lepiniec, L. (2015). Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 20, 176–185. doi: 10.1016/j.tplants.2014.12.001
Yang, J.-L., Gao, G.-B., Zhang, F.-S., Zheng, J., Wu, L.-R. (2022). Quality analysis and evaluation of five cultivars of Chimonobambusa quadrangularis shoots with color shell from Jinfoshan mountain. Food science. 43, 303–308. doi: 10.7506/spkx1002-6630-20210414-199
Ye, L.-X., Bai, F.-X., Zhang, L., Luo, M.-M., Gao, L., Wang, Z., et al. (2023). Transcriptome and metabolome analyses of anthocyanin biosynthesis in post-harvest fruits of a full red-type kiwifruit (Actinidia arguta) ‘Jinhongguan’. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1280970
Ye, J., Fang, L., Zheng, H.-K., Zhang, Y., Chen, J., Zhang, Z.-J., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297. doi: 10.1093/nar/gkl031
Yu, M.-X., Bai, M.-Z., Chen, M.-M., Zhang, G.-Z., Zhao, Y., Ma, Q.-Q., et al. (2024). Identification of bHLH transcription factors and screening of anthocyanin-related genes in Lagerstroemia indica. Genetica 152, 179–197. doi: 10.1007/s10709-024-00215-2
Yu, L., Yue, J.-J., Dai, Y.-X., Zhang, L., Wang, Q., Yuan, J.-L. (2023). Characterization of color variation in bamboo sheath of Chimonobambusa hejiangensis by UPLC-ESI-MS/MS and RNA sequencing. BMC Plant Biol. 23, 466. doi: 10.1186/s12870-023-04494-3
Zhai, X.-G., Wang, X., Yang, X.-Z., Huang, Q.-X., Wu, D.-D., Wang, Y., et al. (2024). Genome-wide identification of bHLH transcription factors and expression analysis under drought stress in Pseudoroegneria libanotica at germination. Physiol. Mol. Biol. Plants. 30, 467–481. doi: 10.1007/s12298-024-01433-w
Zhang, B.-H., Huang, S.-Q., Meng, Y.-X., Chen, W.-L. (2023). Gold nanoparticles (AuNPs) can rapidly deliver artificial microRNA (AmiRNA)-ATG6 to silence ATG6 expression in Arabidopsis. Plant Cell Rep. 42, 1191–1201. doi: 10.1007/s00299-023-03026-5
Zhang, J., Sun, H.-H., Guo, S.-G., Ren, Y., Li, M.-Y., Wang, J.-F., et al. (2022). ClZISO mutation leads to photosensitive flesh in watermelon. Theor. Appl. Genet. 135, 1565–1578. doi: 10.1007/s00122-022-04054-7
Zhao, L.-F., Bi, W.-Y., Jia, Y.-Q., Shi, J.-J., Chi, Y., Yu, M.-Y., et al. (2023). Genome-wide characterization of bHLH family genes and expression analysis in response to osmotic stress in Betula platyphylla. Plants 12, 3687. doi: 10.3390/plants12213687
Zhao, W., Liu, Y.-H., Li, L., Meng, H.-J., Yang, Y., Dong, Z.-B., et al. (2021). Genome-wide identification and characterization of bHLH transcription factors related to anthocyanin biosynthesis in red walnut (Juglans regia L.). Front. Genet. 12. doi: 10.3389/fgene.2021.632509
Keywords: Chimonobambusa utilis, anthocyanin, bHLH gene family, qRT-PCR, protein interaction network
Citation: Tong L, Zeng Q, Guo Y, Li Y, Li H, Chen L and Liu X (2025) Functional characterization in Chimonobambusa utilis reveals the role of bHLH gene family in bamboo sheath color variation. Front. Plant Sci. 16:1514703. doi: 10.3389/fpls.2025.1514703
Received: 21 October 2024; Accepted: 22 January 2025;
Published: 12 February 2025.
Edited by:
Gursharn Singh Randhawa, Sardar Bhagwan Singh Post Graduate Institute of Biomedical Science and Research, IndiaReviewed by:
Kashmir Singh, Panjab University, IndiaCopyright © 2025 Tong, Zeng, Guo, Li, Li, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Liu, bGl1eGlhdmlwOEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.