 Bo-Ni Song1†
Bo-Ni Song1† Xueyimu Aou1†Rong-Ming Tian1Jing Cai1Wei-Yan Tan1
Xueyimu Aou1†Rong-Ming Tian1Jing Cai1Wei-Yan Tan1 Chang-Kun Liu1,2
Chang-Kun Liu1,2 Xing-Jin He1
Xing-Jin He1 Song-Dong Zhou1*
Song-Dong Zhou1*- 1Key Laboratory of Bio-Resources and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China
- 2College of Resources Environment and Chemistry, Chuxiong Normal University, Chuxiong, China
Background: The genus Cyclorhiza is endemic to China and belongs to the Apiaceae family, which is widely distributed in the Himalaya–Hengduan Mountains (HHM) region. However, its morphology, phylogeny, phylogeography, taxonomy, and evolutionary history were not investigated due to insufficient sampling and lack of population sampling and plastome data. Additionally, we found that Seseli purpureovaginatum was not similar to Seseli members but resembled Cyclorhiza species in morphology, indicating that the taxonomic position of S. purpureovaginatum needs to be re-evaluated.
Methods: First, we observed the morphology of the genus. Second, we newly sequenced four plastomes and conducted comparative analyses. Third, we used the newly sequenced internal transcribed spacer (ITS) and chloroplast DNA (cpDNA) (matK, trnQ-rps16, and trnD-trnT) from 27 populations totaling 244 individuals to explore the genetic diversity and structure. Finally, we performed the phylogenetic analyses based on three datasets (plastome data, ITS sequences, and haplotypes) and estimated the origin and divergence time of the genus.
Results and discussion: The morphology of Cyclorhiza plants and S. purpureovaginatum was highly similar, and their plastomes in structure and features were conserved. The genus possessed high genetic diversity and significant lineage geographic structure, which may be associated with the long-term evolutionary history, complex terrain and habitat, and its sexual reproduction mode. The genus Cyclorhiza originated in the late Eocene (36.03 Ma), which was closely related to the early uplift of the Qinghai–Tibetan Plateau (QTP) and Hengduan Mountains (HDM). The diversification of the genus occurred in the late Oligocene (25.43 Ma), which was largely influenced by the colonization of the newly available climate and terrain. The phylogenetic results showed that Cyclorhiza species clustered into a separate clade and S. purpureovaginatum nested within Cyclorhiza. Cyclorhiza waltonii was sister to Cyclorhiza peucedanifolia, and Cyclorhiza puana clustered with S. purpureovaginatum. Thus, based on the morphology, plastome analyses, and phylogenetic evidence, S. purpureovaginatum should be transferred to Cyclorhiza. All these evidences further supported the monophyly of the genus after including S. purpureovaginatum. Finally, we clarified the generic limits of Cyclorhiza and provided a species classification key index for the genus. In conclusion, the study comprehensively investigated the morphology, phylogeography, phylogeny, taxonomy, and evolution of the genus Cyclorhiza for the first time.
1 Introduction
The genus Cyclorhiza (M. L. Sheh & R. H. Shan) is an endemic genus (Apiaceae) to China with high medicinal value. It was established by M. L. Sheh and R. H. Shan in 1980, with Cyclorhiza waltonii (H. Wolff) M. L. Sheh & R. H. Shan designated as the type species (Sheh and Shan, 1980). Its members are widely distributed in the Himalayan–Hengduan Mountains (HHM) region, which grow in open broad-leaved forests, scrublands, alpine meadows, and bamboo thickets at altitudes ranging from 1,800 to 4,600 m (Pimenov, 2017; Zhou et al., 2021) (Figure 1). The main morphological features of its members are characterized by carrot-like roots with prominent annular scars when old, absence of bracts/bracteoles, and laterally compressed mericarps (Sheh and Watson, 2005) (Figure 2).

Figure 1. Habitats of Cyclorhiza members. (A) Sunny hillside grassland (Daocheng, Sichuan). (B, C) Oak forest (Minlin, Xizang; Jianchuan Yunnan). (D) Pine forest (Linzhi, Xizang). (E, F) Stone walls and crevices (Jiangzi, Xizang; Lasa, Xizang). (G) Scrub (Kangding Sichuan). (H–L) Dry rocky hillside (Derong, Sichuan; Luhuo, Sichuan; Jiangzi, Xizang; Angren, Xizang; and Xinlong, Sichuan).

Figure 2. The distinctive characteristic features of Cyclorhiza members. (A) Cyclorhiza waltonii. (B) Cyclorhiza peucedanifolia. (C) Cyclorhiza puana. (D, E) Leaf. (F, G) Flower. (H, I) Root.
The taxonomy of the genus is quite complex. In 1894, C. B. Clarke discovered a new species and classified it under the genus Seseli L., naming it as Seseli waltonii C. B. Clarke. However, in 1929, H. Wolff found that its morphology was similar to that of Ligusticum L. members and thus transferred it to the genus Ligusticum L., naming it Ligusticum waltonii (C. B. Clarke) Wolff (Wolff, 1925, 1929). In 1980, M. L. Sheh and R. H. Shan observed the topotypes of this species and found that the morphological characteristics of this species differed significantly from those of the genera Seseli L. and Ligusticum L (Sheh and Shan, 1980; Pu and Watson, 2005; Sheh and Watson, 2005; Sheh et al., 2005). For example, the mericarps commissure of this species was contracted, the seed face was deeply concave, and bracts or bracteoles were absent; in contrast, the mericarps commissure of Seseli L. members were not contracted, the seed face was plane, and bracteoles were numerous (Sheh and Shan, 1980; Sheh and Watson, 2005; Sheh et al., 2005). The flowers of this species were yellow with slightly laterally compressed mericarps, and mericarps were pentagonal in cross-section with vitta 1 in each furrow, whereas the flowers of the Ligusticum L. members were white, purple, violet, or pale pinkish, with dorsally compressed mericarps and vittae (1–)2–5 in each furrow and vittae 2–10 on commissure (Sheh and Shan, 1980; Pu and Watson, 2005; Sheh and Watson, 2005; Sheh et al., 2005). Therefore, they established a new genus, Cyclorhiza M. L. Sheh & R. H. Shan, to accommodate this species and designated C. waltonii (H. Wolff) M. L. Sheh & R. H. Shan as the type species of the genus (Sheh and Shan, 1980). Originally, the genus Cyclorhiza only included C. waltonii and a variety, Cyclorhiza waltonii var. major M. L. Sheh & Shan (Sheh and Shan, 1980). In 1997, Constance L. found that Arracacia peucedanifolia Franch was similar to Cyclorhiza members in morphology and transferred it to the genus Cyclorhiza, naming it as Cyclorhiza peucedanifolia (Franch.) Constance (1997). Later, Sheh and Watson (2005) treated C. waltonii var. major as the synonym of C. peucedanifolia. In 2021, Zhou et al. discovered a new species of the genus: Cyclorhiza puana J. Zhou & Z. W. Liu. in Luohuo (Sichuan) (Zhou et al., 2021). Thus, the genus currently comprises three species (C. waltonii, C. peucedanifolia, and C. puana). Interestingly, during our fieldwork, we found that Seseli purpureovaginatum R. H. Shan & M. L. Sheh was not similar to Seseli members but resembled Cyclorhiza members in overall morphology (Supplementary Figure S1). By making detailed field investigations, checking the specimen, and collecting material in type locality, we found that S. purpureovaginatum indeed shared some morphological features with Cyclorhiza members. For example, they are herbaceous and glabrous, with erect stems branching above, no bracts and bracteoles, and yellow petals. Therefore, we hypothesized that S. purpureovaginatum may not belong to the genus Seseli, and its taxonomic position needs to be re-evaluated. From the above, we noticed that the generic limits of Cyclorhiza and the species relationships and species identification based on morphological traits faced challenges. It is necessary to obtain more evidences to re-evaluate the generic limits of Cyclorhiza and further clarify their interspecific boundaries.
Previous studies have confirmed that the fruit and pollen characteristics were the two most important morphological characteristics in the classification system of the Apiaceae (Song et al., 2024a). They have been widely used in taxonomic studies of many notorious genera of Apiaceae, such as Sanicula L. (Song et al., 2024a), Angelica L. (Yang and Liao, 2023), Bupleurum L. (Zhou et al., 2018), Peucedanum L. (Zhang and He, 2009), and Cnidium Cusson (Li et al., 1993). However, the morphological and micromorphological studies of fruits and pollens on the genus Cyclorhiza have not been performed comprehensively, and only three micromorphological studies have been reported so far (Sheh and Shan, 1980; Shu and Sheh, 2001; Zhou et al., 2021). Namely, Sheh & Shan observed and described the fruit morphology of C. waltonii and C. peucedanifolia in 1980 (Sheh and Shan, 1980). In 2001, Shu and Sheh observed the micromorphological features of pollens of C. waltonii and C. peucedanifolia (Shu and Sheh, 2001). Zhou et al. (2021) discovered a new species (C. puana) and described its main morphological characteristics, including the fruits’ features. Although these morphological and micromorphological studies have greatly filled the gaps and provided valuable references for the genus Cyclorhiza, they only described the fruit and pollen features of some species. In addition, previous studies have not performed a comprehensive morphological/micromorphological comparison of fruits and pollens for all Cyclorhiza species, and the value of morphological/micromorphological studies for the taxonomy and evolution of this genus was also unclear. Therefore, it is urgent to further investigate the morphological/micromorphological features of this genus based on expanding sampling.
The HHM region, one of Earth’s 34 biodiversity hotspots, is characterized by its unique geology and dramatic topography (Sun et al., 2017). It is also considered to be the diversification center of many organisms and acted as the primary source area for dispersal to many areas of the world during the last 30 million years (Donoghue and Smith, 2004). The Himalayas define the southern margin of the QTP, whereas the HHM in southwest China forms the southeastern boundary of the plateau (Zhang et al., 2002). Therefore, the HHM region was regarded as the largest evolutionary front of the North Temperate Zone (Wu, 1988; Hughes and Atchison, 2015). Previous studies mainly focused on the species-level diversification caused by the uplift of the QTP, with few studies specifically devoted to an endemic genus of the HHM region (Liu et al., 2006; Wang et al., 2009; Xia et al., 2022). The genus Cyclorhiza is endemic to HHM region (Zhou et al., 2021); thus, it is an ideal object for studying how species genetic diversity responds to historical events of regional environmental change. However, no systematic biogeographic and phylogenetic study on the genus Cyclorhiza has been conducted, nor has the evolutionary history of Cyclorhiza been studied. Therefore, we tried to explore the genetic distribution pattern, genetic diversity and structure, origin, and diversification of the genus from phylogenetic and phylogeographic analyses.
With the advancements in next-generation sequencing (NGS) and bioinformatics technologies, plastome data have been widely and successfully used to generate high supports and resolutions of plant phylogenies at order, family, and genus levels, such as in the order Saxifragales Bercht. & J. Presl (Jia et al., 2024), family Apiaceae (Wen et al., 2021; Liu et al., 2022; Ren et al., 2022; Gui et al., 2023; Qin et al., 2023; Song et al., 2023; Chen et al., 2024; Guo et al., 2024; Song et al., 2024b, c), Loranthaceae Juss (Tang et al., 2024), Liliaceae (Zhou et al., 2024a), subfamily Nolinoideae (Ji et al., 2023), genus Paris L (Zhou et al., 2024b), and Polygonatum Mill (Wang et al., 2023). However, there is no phylogenetic framework related to the genus Cyclorhiza based on plastome data yet. Previous molecular studies related to the Cyclorhiza mainly used single- or multiple-locus DNA sequence data, such as ITS sequence cpDNA (rpl16 and rps16 intron), plastid DNA rpl16 and rps16 intron (Downie et al., 2000; Zhou et al., 2009), yet these studies failed to provide sufficient information to support the improvement of taxonomy for Cyclorhiza due to involved limited Cyclorhiza species. Therefore, in order to comprehensively understand the phylogeny, phylogeography, phylogenomics, and taxonomy of the genus, we used plastome data in this current study. Our major aims were to 1) compare the morphological/micromorphological features of the Cyclorhiza members; 2) characterize the plastomes of Cyclorhiza plants and select highly variable hotspot regions as candidate DNA barcodes for species authentication of the genus; 3) investigate the genetic diversity and genetic structure, origin, and diversification of the genus based on materials from 244 individuals from 27 populations; and 4) reconstruct the phylogenetic framework of the genus based on three datasets (ITS sequence, plastome data, and haplotype) and clarify the generic limits of the genus Cyclorhiza and the interspecific relationships within the genus.
2 Materials and methods
2.1 Plant materials and morphological observations
We collected 244 individuals from 27 populations of four species between 2021 and 2024. These individuals grow in western Sichuan, northwestern Yunnan, and southern Xizang, most of them with altitudes above 3,000 m (Figure 3; Supplementary Table S1). The collection of each species covered the type locality and the entire distribution area, and we randomly collected five to fifteen individuals from each population. We immediately stored the fresh young basal leaves and dried them with silica gel. We deposited the voucher specimens in the herbarium of Sichuan University (Chengdu, China, SZ) (Supplementary Table S1).
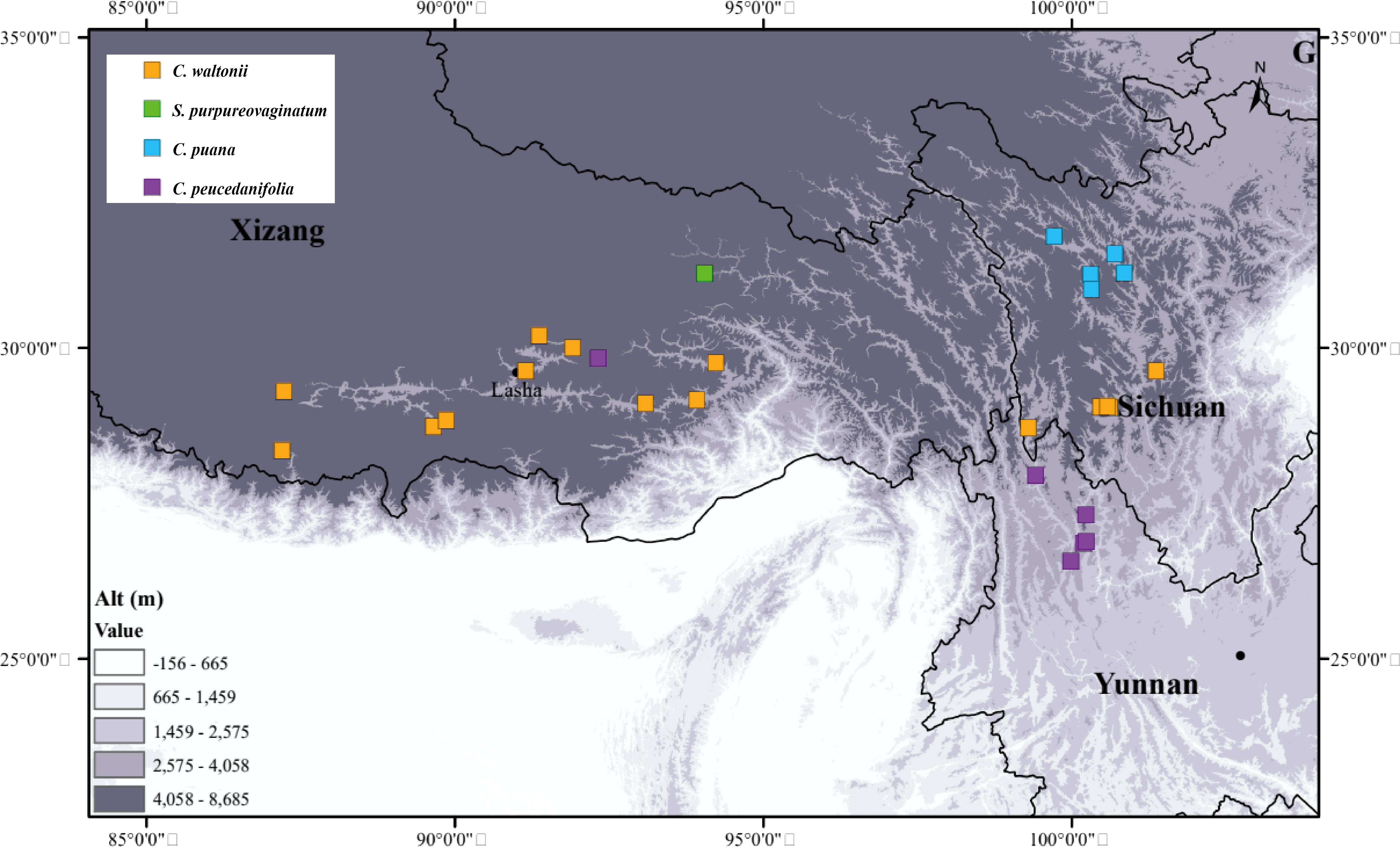
Figure 3. Population sampling distribution points involved in this study.
In addition, we also collected their mature fruits and pollens in the field to observe their micromorphological characteristics. First, we selected 30 representative fruit samples of each species to observe their overall structure and anatomy using a stereo microscope (SMZ25, Nikon Corp., Tokyo, Japan). Second, we selected 10 dry, mature, and full anthers of each species and pasted them on the double-sided conductive adhesive; we gently poked the pollen capsule with a clean dissecting needle to release the pollens, coated them using gold spray, and used a JSM-7500F scanning electron microscope to observe and photograph the overall view of pollen, the equatorial view, the polar view, germ furrow, and the exine ornamentation. We used the software MATO (Liu et al., 2023) to measure the 30 representative fruit samples and 10 pollens for each species, and then we calculated the average value. The description of fruit and pollen characteristics referred to the studies of Kljuykov et al. (2004), Ostroumova (2021), and Shu and Sheh (2001). Furthermore, we also obtained other morphological characteristics of Cyclorhiza members based on extensive documentation, specimen information, and fieldwork.
2.2 DNA extraction, amplification, sequencing, assembly, and annotation
The total genomic DNA of newly collected samples was extracted from silica gel-dried leaves using the modified cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987). The quality and quantity of genomic DNA were tested via 1% agarose gel electrophoresis. For plastome data, the DNA library with an average insert size of 300–400 bp was constructed using the TruSeq DNA Sample Preparation Kits (Illumina) referred to the manufacturer’s protocol (Illumina, San Diego, CA, USA). The DNA library was sequenced using the Illumina HiSeq2500 platform at Novogene (Beijing, China), applying the paired-end 150-bp reads. Then, the software fastP v0.15.0 (-n 10, -q 15) (Chen et al., 2018) was used to filter the raw data and yield clean data (at least 5 GB). For the yielded clean data, the GetOrganelle pipeline (Jin et al., 2020) was used to assemble the complete plastomes, setting the plastome sequence of Changium smyrnioides H. Wolff (MN092718) as a reference. The complete plastomes were initially annotated by the Plastid Genome Annotator (PGA) software (Qu et al., 2019), with C. smyrnioides H. Wolff (MN092718) as a reference, and then the start and stop codons and intron positions were manually checked and corrected in Geneious v9.0.2 (Kearse et al., 2012). Finally, the well-annotated plastomes were displayed using the online program OrganellarGenomeDRAW (OGDRAW) (Stephan et al., 2019).
For the ITS region, the sequence of 244 individuals from 27 populations was amplified using 30-µL polymerase chain reaction (PCR) system, including 2 µL total DNA, 1.5 µL forward primers ITS-4 (5′-TCCTCCGCTTATTGATATGC-3′), 1.5 µL reverse primers ITS-5 (5′-GGAAGTAAAAGTCGTAACAAGG-3′), 15 µL Taq MasterMix (CWBio, Beijing, China), and 10 µL of ddH2O. The PCR amplification program was as follows: 4 minutes of initial denaturation at 94°C; 36 cycles consisting of denaturation at 94°C for 45 seconds, annealing at 52°C for 70 seconds, extension at 72°C for 90 seconds, a final extension at 72°C for 10 minutes; and stored at 4°C freezer (White, 1990). Finally, the PCR products were sent to Sangon (Shanghai, China) for sequencing. The newly generated 244 ITS sequences were assembled and edited using the software Geneious v9.0.2 (Kearse et al., 2012) to gain the consensus sequences.
For cpDNA fragments, we first amplified and sequenced 10 cpDNA regions (rpl16, rpl32–trnL, trnD–trnT, trnH–psbA, trnQ–rps16, trnS–trnG, rbcL, rps16–trnK, matK, and rps16) of 27 individuals from 27 populations to screen out cpDNA fragments that exhibited significant differences at the population level. The primer sequences for these 10 regions are provided in the Supplementary Table S2. Finally, we selected three cpDNA fragments (matK, trnQ–rps16, and trnD–trnT) and amplified these three fragments of all individuals. The PCR amplification system for each cpDNA fragment was 30 µL, including 2 µL total DNA, 1.5 µL forward primers and 1.5 µL reverse primers, 15 µL Taq MasterMix (CWBio, Beijing, China), and 10 µL of ddH2O. The PCR amplification program was as follows: initial denaturation at 94°C for 4 minutes, followed by 35 cycles of denaturation at 94°C for 45 seconds, annealing at 52°C for 45 seconds, extension at 72°C for 1 minute, and a final extension step at 72°C for 7 minutes. Finally, we sent the amplified products to Sangon (Shanghai, China) for bidirectional sequencing. We used the software Geneious v9.0.2 (Kearse et al., 2012) to assemble and edit the newly generated cpDNA fragment sequences and gain the consensus sequences.
All new plastome data, ITS sequences, and cpDNA fragments were submitted to the National Center for Biotechnology Information (NCBI), and the accession numbers were shown in Supplementary Table S3.
2.3 Plastome comparison and hotspots identification
First, four types of repeat sequences (forward, reverse, complement, and palindromic) were detected using the online program REPuter. The parameters were as follows: maximum computed repeats >90%, minimal repeat size ≥30 bp, and a hamming distance = 3 (Kurtz et al., 2001). The simple sequence repeats (SSRs) were also analyzed using the Perl script MISA (https://webblast.ipk-gatersleben.de/misa/), and the minimum number of repeat units parameter was set to ten repetitions for mononucleotides, five repetitions for dinucleotides, four repetitions for trinucleotides, and three repetitions for tetranucleotides, pentanucleotides, and hexanucleotides (Beier et al., 2017). Second, the boundaries between inverted repeat and single-copy (IR/SC) were visualized using the online tool IRscope (https://irscope.shinyapps.io/irapp) (Amiryousef et al., 2018) after manual adjustment. Third, the sequence divergence of whole plastomes was detected and visualized using the online program mVISTA viewer in Shuffle-LAGAN mode (Frazer et al., 2004), with C. waltonii as a reference. Fourth, the DNA rearrangements were detected using Mauve Alignment (Darling et al., 2004) implemented in Geneious v9.0.2 (Kearse et al., 2012), with other parameters set as the default values. Fifth, the CodonW v1.4.2 program (Peden, 1999) was employed to compute the relative synonymous codon usage (RSCU) (Sharp and Li, 1986) of these shared coding sequences (CDSs) (>300 bp). Finally, DNAsp v5.0 (Librado and Rozas, 2009) was used to calculate the nucleotide diversity values (Pi).
2.4 Genetic diversity and structure
First, we used DNAsp v5.0 (Librado and Rozas, 2009) to calculate and count the haplotype types and numbers, and we subsequently estimated the nucleotide diversity (π) (Nei and Li, 1979) and haplotype diversity (Hd) (Nei and Tajima, 1981) of each population and the whole population. Second, we employed PERMUT (Pons and Petit, 1996) to compute the total diversity (HT), within-population diversity (HS), and population differentiation indices (GST and NST) (Grivet and Petit, 2002). We also used U-statistics to compare GST and NST values between populations. Additionally, we performed analyses of molecular variance (AMOVA) with 1,000 permutations using ARLEQUIN v3.5 (Excoffier and Lischer, 2010) to detect the genetic variation among species FSC, among populations within species FST, and within population FCT. Finally, we used the PopART 1.7 software (Polzin and Daneshmand, 2003) to construct the network of ITS haplotypes and cpDNA haplotypes.
2.5 Phylogenetic analyses and divergence time estimation
To better investigate the phylogeny of the genus Cyclorhiza, the phylogenetic trees were reconstructed based on three datasets: dataset 1 was the 39 complete plastomes, dataset 2 was 92 ITS sequences, and dataset 3 was haplotypes (28 ITS haplotypes and 35 cpDNA haplotypes) (Supplementary Table S4). Among them, the Bupleurum L. species were chosen as the outgroup referred to the previous research (Zhou et al., 2021). The ITS sequences and haplotype sequences were straightway aligned with MAFFT v7.221 (Katoh and Standley, 2013) to gain the matrix. For plastome data, 79 commonly shared protein coding sequences (CDSs) of 39 species were manually extracted in Geneious v9.0.2 (Kearse et al., 2012) and aligned with MAFFT v7.221 (Katoh and Standley, 2013). Then, the alignments were trimmed using trimAI (Capella-Gutierrez et al., 2009) and finally concatenated as a super matrix using PhyloSuite v1.2.2 (Zhang et al., 2020). Two methods [maximum likelihood (ML) and Bayesian inference (BI)] were employed to construct the phylogenetic trees. For the ML method, RAxML v8.2.8 (Stamatakis, 2014) with the GTRGAMMA model and 1,000 bootstrap replicates was suggested to estimate the support value [bootstrap value (BS)] for each node referred to in the RAxML manual. For the BI method, MrBayes v3.2.7 (Ronquist et al., 2012) was utilized for Bayesian inference, and the best-fit nucleotide substitution model (GTR + F + I + G4) for the matrix of the CDS dataset, (SYM + G4) for the ITS dataset, (GTR + G) for ITS haplotype, and (GTR + I + G) for cpDNA haplotype were determined using Modeltest v3.7 (Posada and Crandall, 1998). Two independent Markov chain Monte Carlo (MCMC) runs of 10 million generations were performed with sampling every 1,000 generations. The MCMC runs finished when the average standard deviation of the splitting frequency fell below 0.01. The initial 25% of trees were discarded as burn-in, and the remaining trees were used to generate the consensus trees and calculate posterior probabilities (PP). Finally, all results of ML and BI phylogenetic analyses were visualized and edited using FigTree v1.4.2 (Rambaut and Drummond, 2015).
To explore the origin and diversification of the genus, the phylogenetic tree of ITS haplotypes was selected to estimate the divergence time. Bayesian relaxed clock analysis in the program Bayesian Evolutionary Analysis Sampling Trees (BEAST v1.10.4) (Suchard et al., 2018) was performed. Pollen calibration points of Bupleurum, as referred to in previous studies (Calvino et al., 2016; Wen et al., 2021), were selected to constrain the phylogenetic tree, lognormal distribution was applied, and the set offset value was 33.9 and the mean value was 2.389, as referred to in published studies (Banasiak et al., 2013; Wen et al., 2021). BEAUti was employed to set criteria under the uncorrelated relaxed molecular clock model and a Yule tree prior. Modeltest v3.7 (Posada and Crandall, 1998) was used to detect the best-fit nucleotide substitution model (GTR + G). MCMC analysis was run for 10 million generations with parameters sampled every 10,000 generations after discarding the first 20% of generations as burn-in. The convergence of the stationary distribution was accessed by effective sample size (ESS) values (>200) using the software Tracer v.1.7.1 (Rambaut et al., 2018). Maximum clade credibility (MCC) tree was produced using the TreeAnotator v2.1.2 software (Rambaut and Drummond, 2014), and the result was visualized using FigTree v1.4.42 (Rambaut and Drummond, 2015) and Interactive Tree Of Life (iTOL) (Letunic and Bork, 2007).
3 Results
3.1 The morphological and micromorphological analyses
We found that the key external morphological characteristics of Cyclorhiza members were highly similar, such as being herbaceous and glabrous, carrot-like roots with prominent annular scars when old, stem fistulose, erect, branched above, base clothed in purplish-brown remnant sheaths, absence bracts, and bracteoles and with yellow petals (Figure 2; Table 1). In addition, we also observed the fruit micromorphological features (fruit appearance, fruit size, and anatomical characteristics) of the genus and detected that their fruit shapes were ovoid or ellipsoid, smooth and glabrous, slightly laterally compressed, mericarps subpentagonal in cross-section, ribs 5, filiform, prominent, acute-ridged, almost narrowly winged, vittae 1–2 in each furrow and 2–4 on commissure, with the endosperm commissural face deeply sulcate or concave (Figure 4). However, we observed some differences among them, such as the fruit size ranging from (3–5.5) × (1–2.5) mm to (4–7) × (2–3.5) mm (Table 1). Moreover, the pollen of Cyclorhiza species also shared some similarities, such as the equatorial view was rombiformis, the polar view was subtriangular and had an angular germinal aperture, and the exine ornamentation of the equatorial view was short rod-like pseudo-cerebroid (Figure 5). However, the pollen size was different, the polar axis was 24.52–34.63 μm, the equatorial axis was 18.84–25.65 μm, and the ratio of the polar axis to the equatorial axis was 1.12–1.35 μm (Table 1). Furthermore, we found that the external morphological and micromorphological features (fruit and pollen) of S. purpureovaginatum were very similar to those of Cyclorhiza plants (Supplementary Figures S1; Figures 2, 4, 5; Table 1).
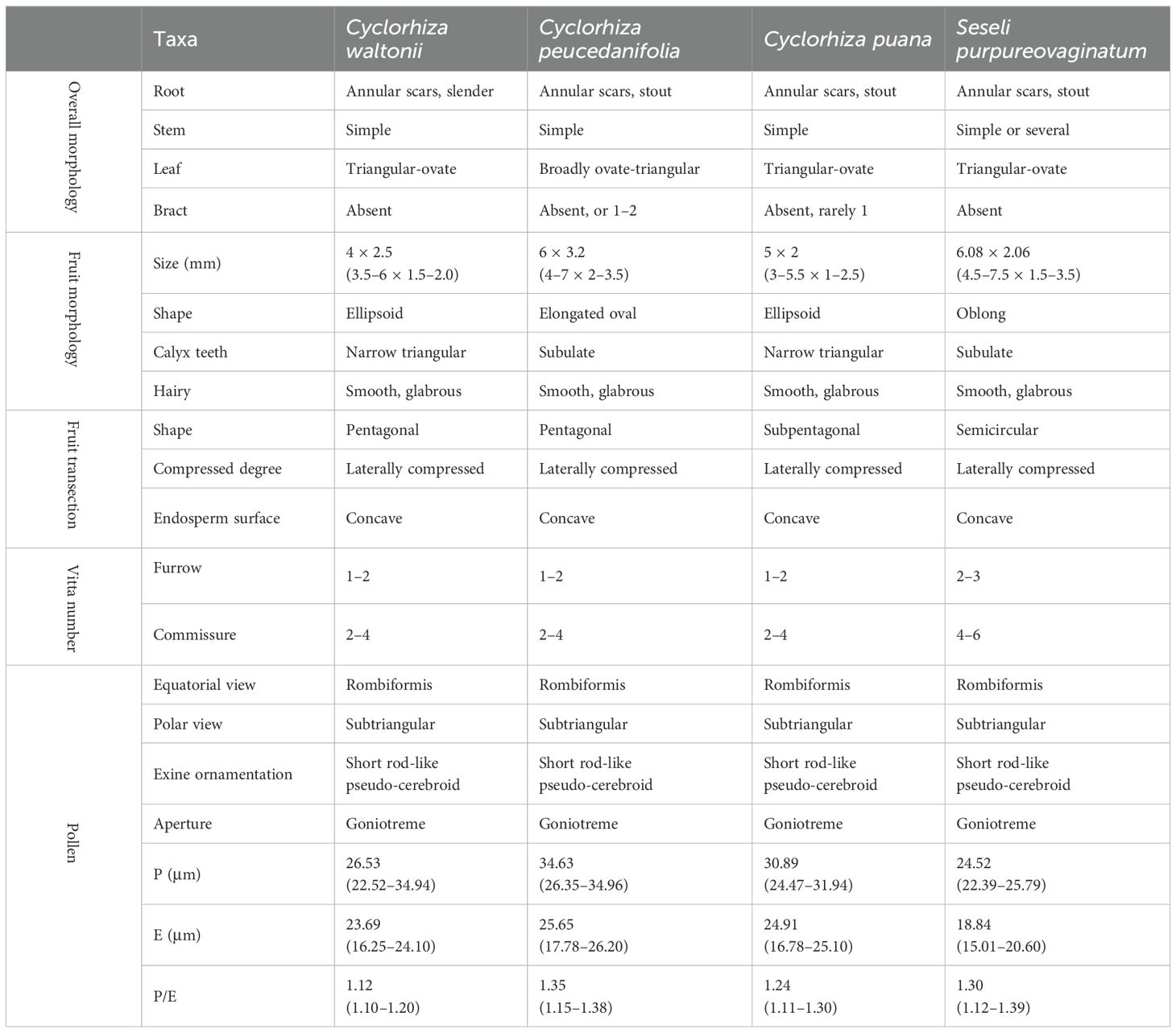
Table 1. The morphological and micromorphological characteristics of four plants.

Figure 4. The fruits’ features of four members. 1: Cyclorhiza waltonii. 2: Cyclorhiza peucedanifolia. 3: Cyclorhiza puana. 4: Seseli purpureovaginatum. (A) Cremocarp. (B) Dorsal view. (C) Commissural side view. (D) Transverse section view. Scale bar=1mm (A–C); Scale bar=0.5mm (D).

Figure 5. The pollen features of four members. 1: Cyclorhiza waltonii. 2: Cyclorhiza peucedanifolia. 3: Cyclorhiza puana. 4: Seseli purpureovaginatum. (A) Pollen grain. (B) Equatorial view. (C) Germ furrow. (D) Polar view. (E) Exine ornamentation.
3.2 Plastome features, plastome comparison, and hotspots identification
The size of four plastomes ranged from 155,169 bp (C. peucedanifolia) to 156,014 bp (C. waltonii). All plastomes exhibited a typical quadripartite structure, including one large single copy (LSC) region (85,079–85,461 bp) and one small single copy (SSC) region (17,794–17,915 bp) separated by two inverted repeats (IRs) regions (26,080–26,369 bp). The total Guanine and Cytosine (GC) content ranged from 37.7% to 37.8%, with the IR regions having the highest GC content (42.8%–43.0%) compared to the GC content in the LSC region (35.8%–35.9%) and SSC region (31.3%–31.5%) (Supplementary Figure S2; Table 2). All plastomes encoded 129 genes, comprising 84 protein-coding genes (PCGs), 37 transfer RNA (tRNA) genes, and eight ribosomal RNA (rRNA) genes (Supplementary Table S5).
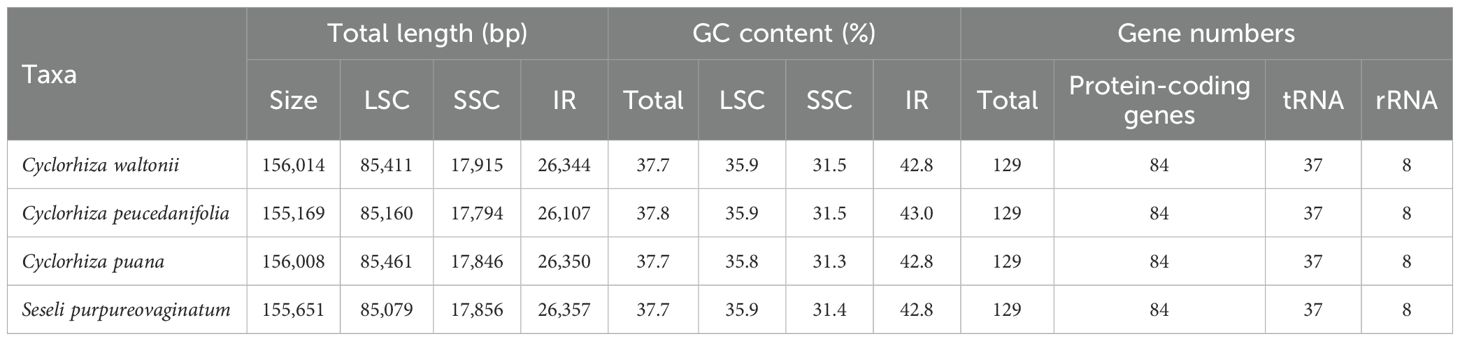
Table 2. Features of the four plastomes.
In this study, the forward, palindromic, reverse, and complementary repeats were detected in these four species, and the total number of repeats was 214. Among these repeats, forward repeats were the most abundant (112), followed by palindromic repeats (98) and reverse repeats (4). Forward repeats and palindromic repeats were detected in all plastomes; reverse repeats were found in C. waltonii, C. peucedanifolia, and S. purpureovaginatum; and complementary repeats were not detected in any of the four plastomes. Moreover, the majority of these repeats were found in intergenic or intron regions (e.g., rrn5–rrn4.5, rrn4.5–rrn5, petN–psbM, ycf2 intron, and ycf3 intron) (Supplementary Figure S3A; Supplementary Table S6). In addition, the total number of SSRs ranged from 48 (C. waltonii) to 59 (S. purpureovaginatum). The number of mono-repeats, di-repeats, tri-repeats, tetra-repeats, and hexa-repeats were 145, 39, 14, 20, and 1, respectively. All plastomes had the mono-repeats, di-repeats, tri-repeats, and tetra-repeats. The penta-repeats were not detected in all plastomes, and hexa-repeats only appeared in S. purpureovaginatum. SSRs were mostly distributed in the LSC region compared to the SSC and IR regions, and the majority of SSRs were distributed in the non-coding regions (Supplementary Figure S3B; Supplementary Table S6).
The IR boundaries of these four plastomes were highly consistent, and the adjacent genes were identical. The rps19 gene was located at the junction of LSC/IRb, with a length of 57 bp in the IRb region. The IRb/SSC junction region was located between the ycf1 gene and the ndhF gene. The length of the ycf1 gene located in the IRb region was 1,827–1,851 bp, and that of the ndhF gene was 17–67 bp away from the IRb/SSC borders. The ycf1 gene crossed the SSC/IRa boundary and had a length of 1,835–1,885 bp in the IRa region. The IRa/LSC boundary was located between the rpl2 gene and trnH gene, and the rpl2 gene and trnH gene were 115 bp and 3 bp away from the IRa/LSC borders, respectively (Supplementary Figure S4). Using the mVISTA program, we discovered that these four plastomes were highly conserved. Specifically, the protein-coding regions were more conserved than the non-coding regions, and the IR regions were more conserved than the LSC and SSC regions (Supplementary Figure S5). Furthermore, the Mauve result also revealed that these four plastomes were highly conserved and that no gene rearrangement or loss was found (Supplementary Figure S6).
To characterize the codon usage patterns across the four plastomes, we extracted and concatenated 53 protein-coding genes from each species. These protein sequences encoded 21,214–21,228 codons, with C. peucedanifolia being the most and C. waltonii being the least. Among them, Leu was the most abundant (2,230–2,240) and encoded by six codons (UUA, UUG, CUU, CUC, CUA, and CUG), whereas Trp was the least abundant (380–382) in all plastomes. The RSCU values of all codons ranged from 0.34 to 1.98 across the four plastomes, with RSCU values of 30 codons in each species being greater than 1 (Supplementary Figure S7; Supplementary Table S7).
Furthermore, we computed the nucleotide diversity (Pi) of protein-coding genes, non-coding regions, and introns within the LSC, SSC, and IR regions (Figure 6; Supplementary Table S8). We identified five protein-coding genes (cemA, matK, ndhF, rpl20, and ycf4) with relatively high nucleotide diversity (Pi > 0.01) (Figure 6A). Ten non-coding regions (ndhE–ndhG, petA–psbJ, trnE–trnT, atpI–rps2, rpoC2–rpoC1, trnT–psbD, psbK–psbI, trnH–psbA, accD–psaI, and rrn5–trnR) also possessed high nucleotide diversity (Pi > 0.02) (Figure 6B).
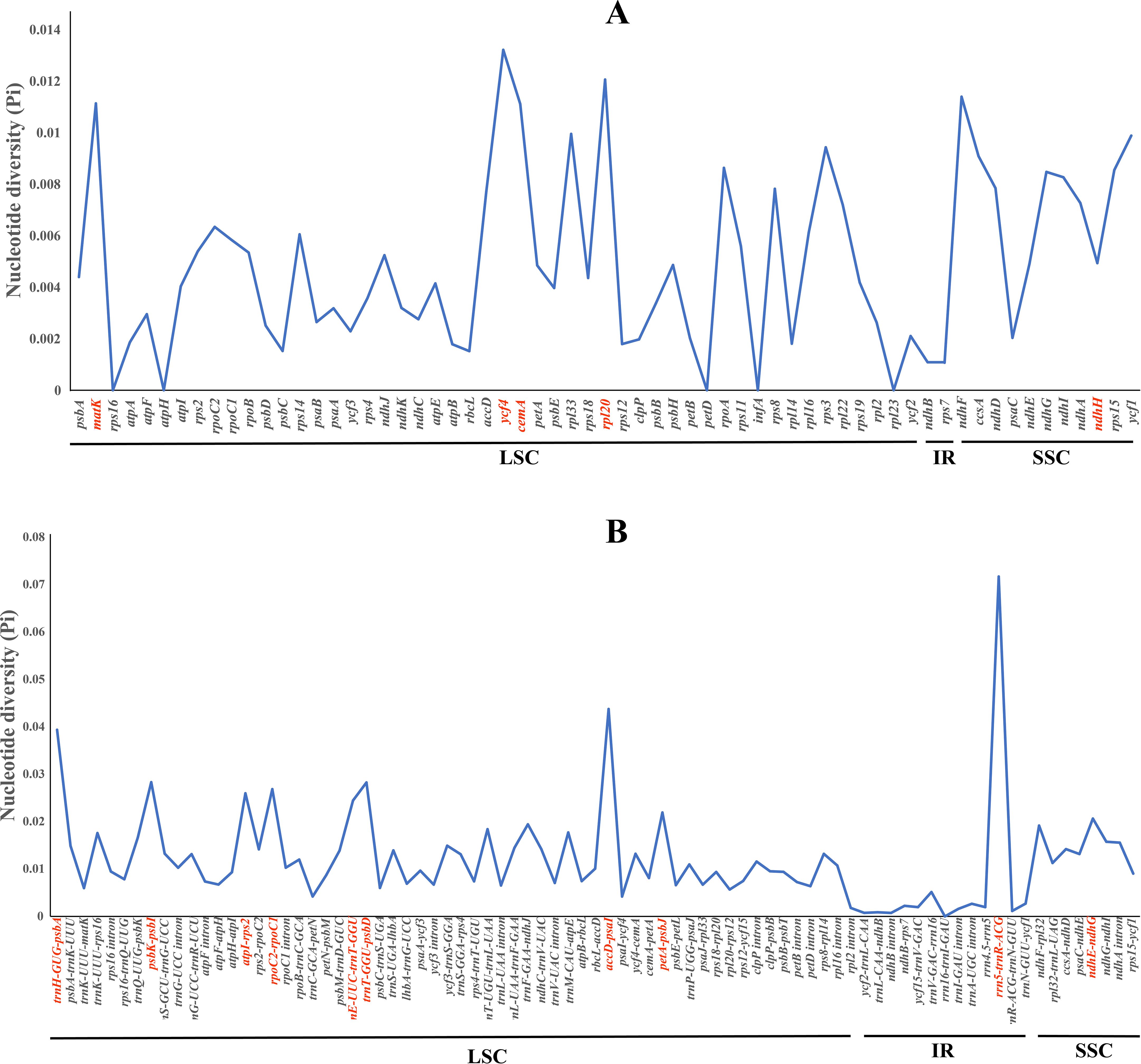
Figure 6. The nucleotide diversity (Pi) values among the four plastomes. (A) Coding regions. (B) Non-coding regions.
3.3 Genetic diversity and structure
In this study, ITS sequence and three cpDNA fragments (matK, trnQ–rps16, and trnD–trnT) were used to analyze 244 individuals from 27 populations. First, the haplotype distribution and genetic diversity were analyzed based on ITS data. The results showed that the aligned sequences of ITS were 600 bp, and GC content ranged from 53.1% to 55.3% (average 54.5%). A total of 66 polymorphic sites and 28 nuclear gene haplotypes (N1–N28) were detected, including six in C. peucedanifolia (N1–N6), fourteen in C. waltonii (N7–N20), six in C. puana (N21–N26), and two in S. purpureovaginatum (N27–N28), and no shared haplotype existed among different species. All species formed a monophyletic group in the ITS haplotype network (Figure 7A; Supplementary Table S9). The haplotype diversity (Hd) was 0.941, and nucleotide diversity (π) was 0.03051 of ITS in the genus. As for these four species, the haplotype diversity (Hd) and nucleotide diversity (π) were respectively 0.867 and 0.02090 in C. waltonii, 0.788 and 0.00354 in C. peucedanifolia, 0.770 and 0.00525 in C. puana, 0.556 and 0.00093 in S. purpureovaginatum. The haplotype diversity (Hd) ranged from 0.000 to 0.002, and nucleotide diversity (π) ranged from 0.000 to 0.0103 of the population (Supplementary Table S9). Additionally, ITS data correlation analysis showed that the total gene diversity (HT) value (0.963) was higher than the average gene diversity within populations (HS) (0.335) at the genus level. The number of substitution types (NST, 0.935) was higher than inter-population differentiation (GST, 0.653). The gene flow (Nm) detected among all 27 populations was 0.06, and C. peucedanifolia possessed the highest Nm value (0.06) compared to C. waltonii (0.05) and C. puana (0.02) (Table 3). ITS AMOVA detected 62.51% of genetic variation occurring between species, 32.53% occurring between populations, and 4.97% occurring within populations. In addition, the Cyclorhiza species exhibited higher genetic variation between populations than within populations (Table 4).
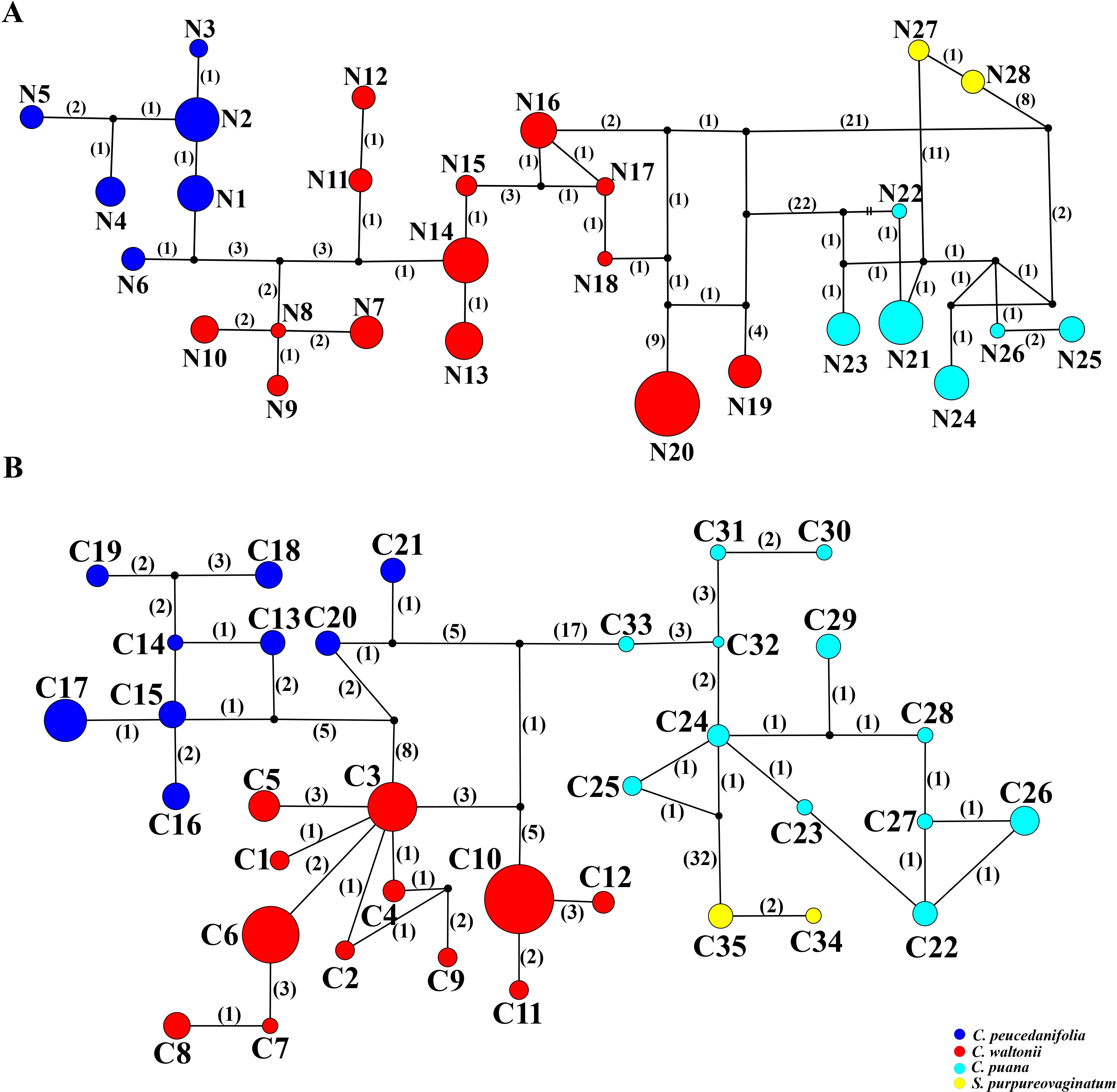
Figure 7. TCS networks for 28 ITS haplotypes and 35 cpDNA haplotypes. Each color represents a species. The size of circles in the network corresponds to the frequency of each haplotype. Numbers on the branches indicate the number of mutations between haplotypes. Small solid black circles denote hypothetic unsampled or extinct ancestral haplotypes. (A) ITS haplotypes. (B) cpDNA haplotypes.
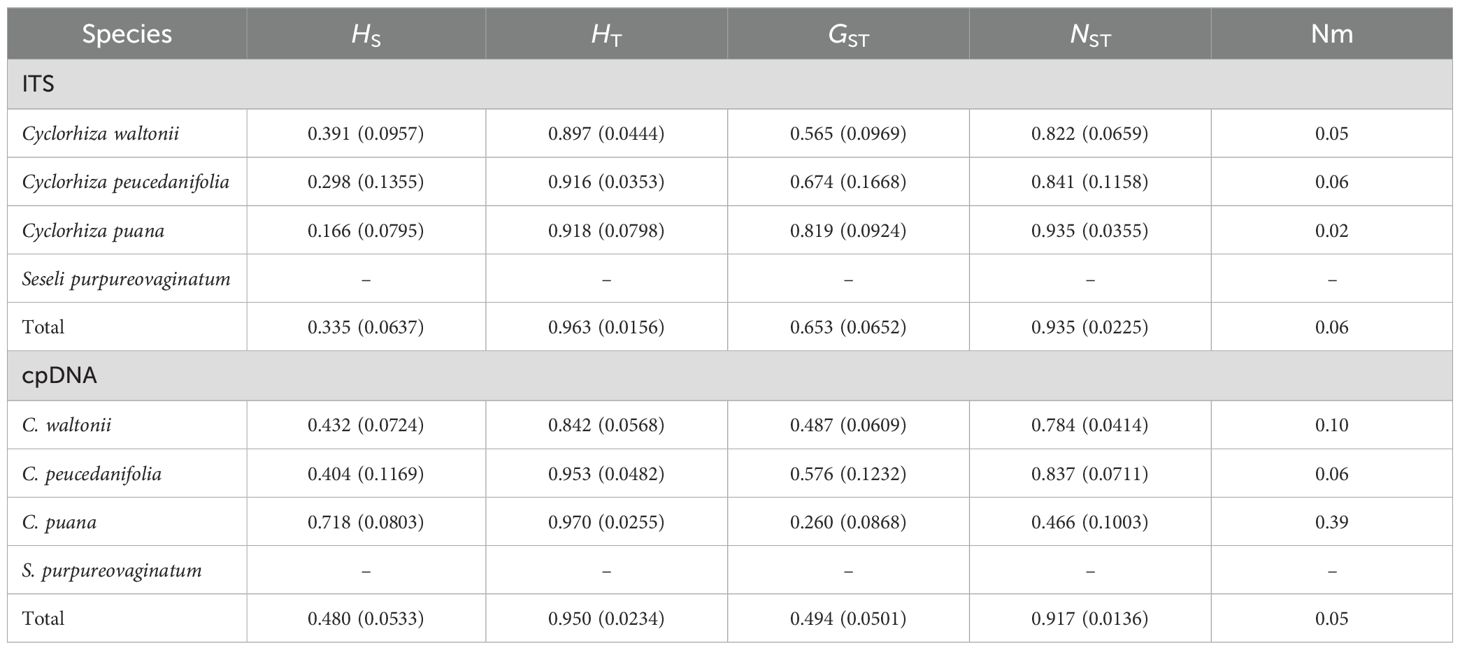
Table 3. Genetic diversity and genetic differentiation based on ITS and cpDNA datasets.
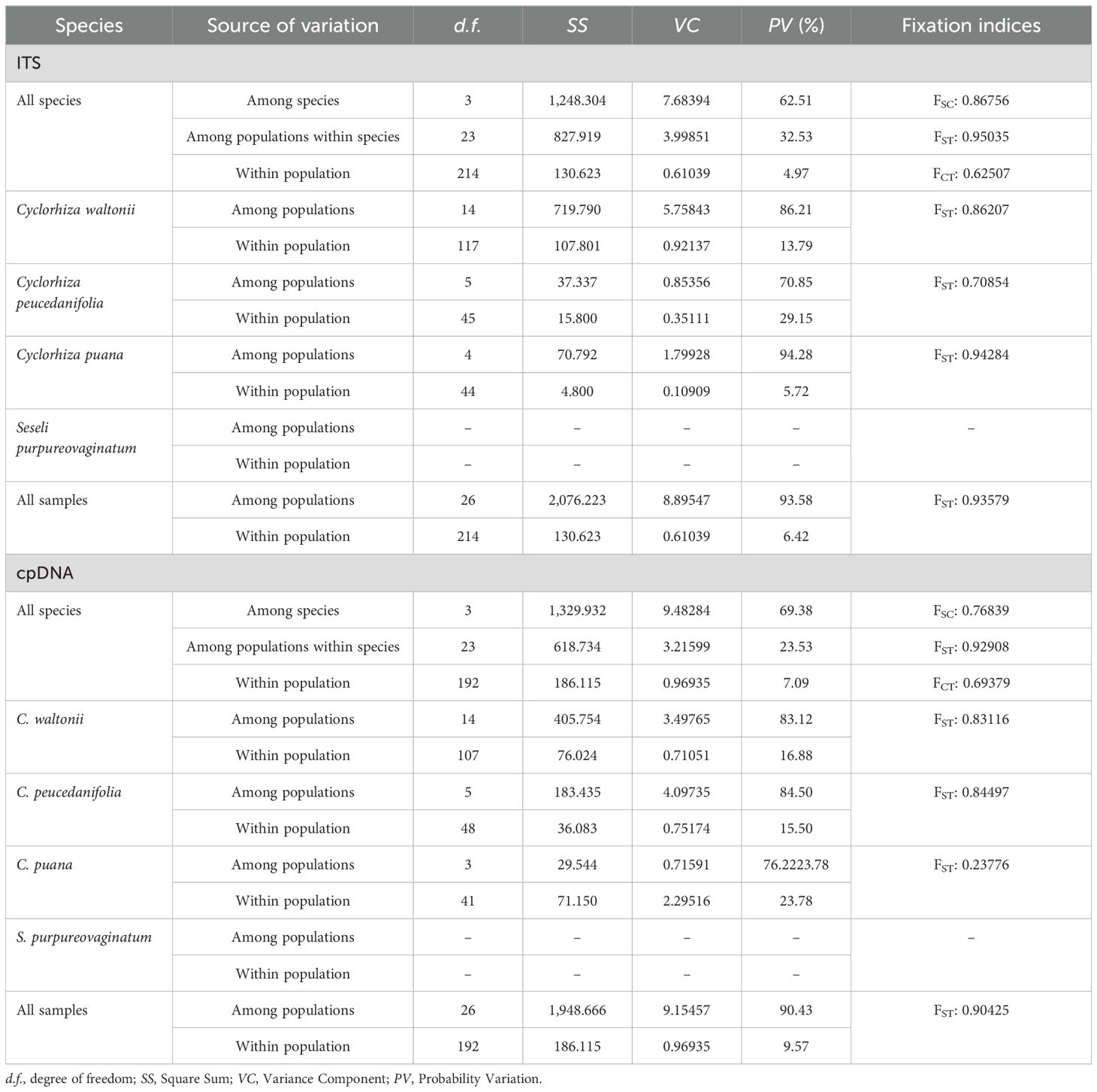
Table 4. Analysis of molecular variance (AMOVA) based on ITS and cpDNA data.
Moreover, the haplotype distribution and genetic diversity were analyzed based on three cpDNA fragment data. The total length of the aligned sequences was 2,116 bp (matK: 745 bp, trnQ-rps16: 662 bp, trnD-trnT: 709 bp). A total of 90 polymorphic sites and 35 chloroplast haplotypes (C1–C35) were detected, including twelve in C. waltonii (C1–C12), nine in C. peucedanifolia (C13–C21), twelve in C. puana (C22–C33), and two in S. purpureovaginatum (C34–C35), and no shared haplotype existed among different species (Figure 7B; Supplementary Table S9). Simultaneously, all species form a monophyletic group in the haplotype network (Figure 7B). At the genus Cyclorhiza level, the haplotype diversity (Hd) was 0.9324, and nucleotide diversity (π) was 0.00927. At the species level, the haplotype diversity (Hd) and nucleotide diversity (π) were respectively 0.815 and 0.00379 in C. waltonii, 0.869 and 0.00391 in C. peucedanifolia, 0.916 and 0.00289 in C. puana, and 0.476 and 0.00045 in S. purpureovaginatum. The haplotype diversity (Hd) ranged from 0.000 to 0.867, and nucleotide diversity (π) ranged from 0.000 to 0.00354 of the population (Supplementary Table S9). At the genus Cyclorhiza level, the total gene diversity (HT) value (0.950) was higher than the average gene diversity within populations (HS) (0.480). In addition, the number of substitution types (NST, 0.917) was higher than inter-population differentiation (GST, 0.494). The gene flow (Nm) detected among all 27 populations was 0.05, and C. puana possessed the highest Nm value (0.39) compared to C. waltonii (0.10) and C. peucedanifolia (0.06) (Table 3). cpDNA AMOVA detected 69.38% of genetic variation occurring between species, 23.53% occurring between populations, and 7.09% occurring within populations. Furthermore, the Cyclorhiz species exhibited higher genetic variation between populations than within populations (Table 4).
3.4 Phylogeny reconstruction and divergence time estimation
We used three molecular datasets: 39 complete plastomes, 92 ITS sequences, and haplotypes (28 ITS haplotypes and 35 cpDNA haplotypes) to reconstruct the phylogenetic trees (Figures 8, 9). All topologies were highly consistent, which strongly supported that the Cyclorhiza species clustered together, belonging to the Komarovia clade. S. purpureovaginatum was nested within the genus Cyclorhiza and also located in the Komarovia clade, while other Seseli members clustered in the Selineae clade and were distant from S. purpureovaginatum. In addition, the phylogenetic results also robustly indicated that C. waltonii was sister to C. peucedanifolia (CDS trees: PP = 1.00, BS = 100; ITS tree: PP = 1.00, BS = 100), and C. puana clustered with S. purpureovaginatum (CDS trees: PP = 1.00, BS = 100; ITS tree: PP = 1.00, BS = 100) (Figure 8). Furthermore, the haplotype phylogenetic trees showed that C. waltonii populations were sisters to the C. peucedanifolia populations (ITS haplotypes: PP = 0.96, BS = 99; cpDNA haplotypes: PP = 0.65, BS = 65), and C. puana populations were sisters to S. purpureovaginatum populations (ITS haplotypes: PP = 1.00, BS = 100; cpDNA haplotypes: PP = 1.00, BS = 100) (Figure 9), and the haplotype phylogenetic trees were consistent with the haplotype network (Figures 7, 9).

Figure 8. Phylogeny from maximum likelihood (ML) and Bayesian inference (BI) analyses. The bootstrap values (BS) of ML and posterior probabilities (PP) of BI are listed at each node (* represents the node = 100/1.00; - represents the node < 50/0.50). (A) CDS tree. (B) ITS tree.
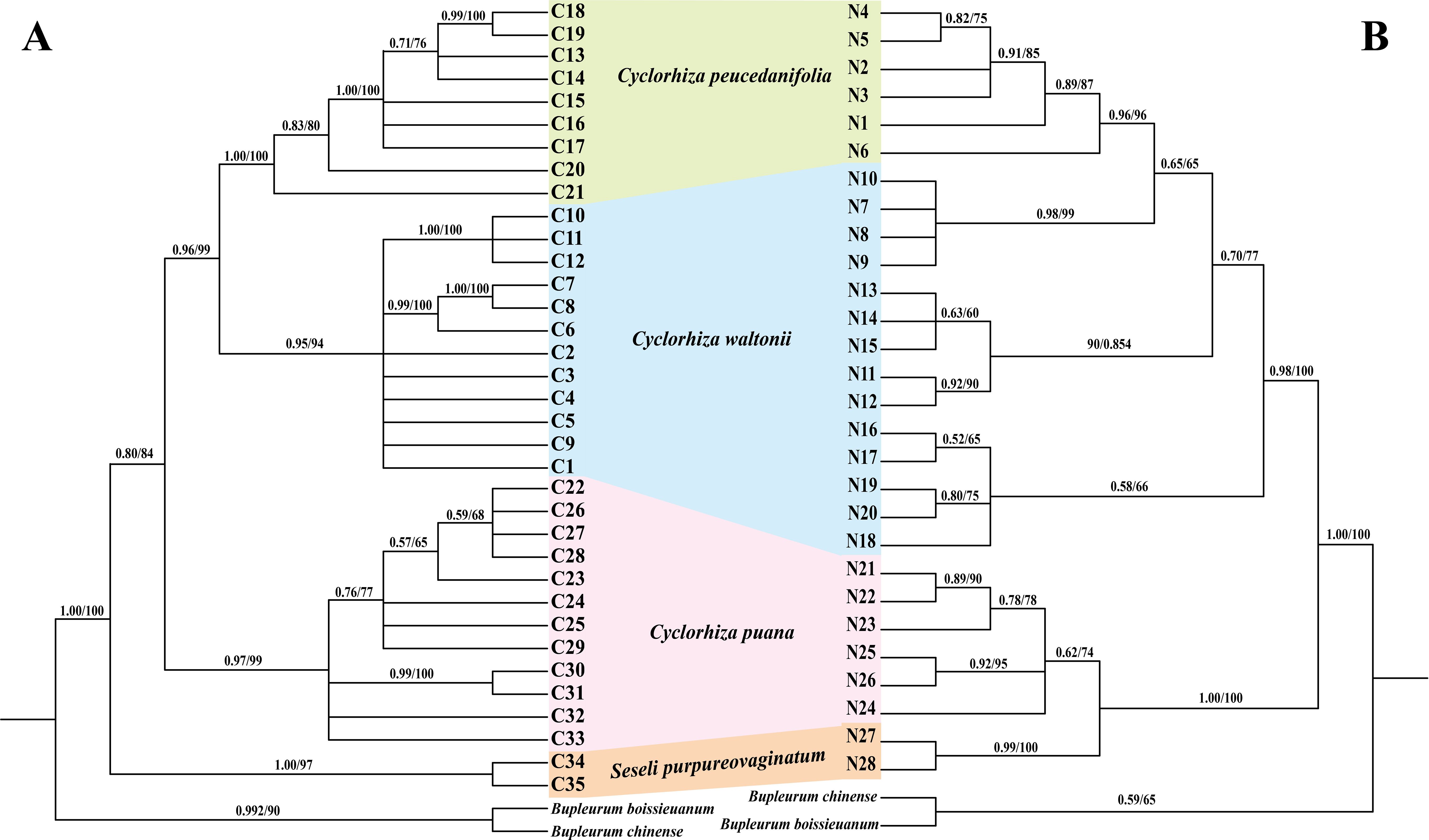
Figure 9. Phylogeny from maximum likelihood (ML) and Bayesian inference (BI) analyses. The bootstrap values (BS) of ML and posterior probabilities (PP) of BI are listed at each node. Haplotypes from different species are marked with different colors. (A) cpDNA haplotypes. (B) ITS haplotypes.
The molecular dating analyses showed that the genus Cyclorhiza originated in the late Eocene [36.03 Ma; 95% highest posterior density (HPD): 34.69–38.08 Ma] and the diversification of the genus occurred at late Oligocene, with the age 25.43 Ma (95% HPD: 13.43–36.33 Ma) (Figure 10). In detail, the divergence time between C. waltonii and C. peucedanifolia was estimated to be 19.21 Ma (95% HPD: 8.21–32.30 Ma) at the early Miocene period, and the divergence time between C. puana and S. purpureovaginatum was estimated to be 15.36 Ma (95% HPD: 4.95–29.03 Ma) at the middle Miocene period. Moreover, the populations of these species continued to differentiate between the Miocene and Pleistocene periods (Figure 10).
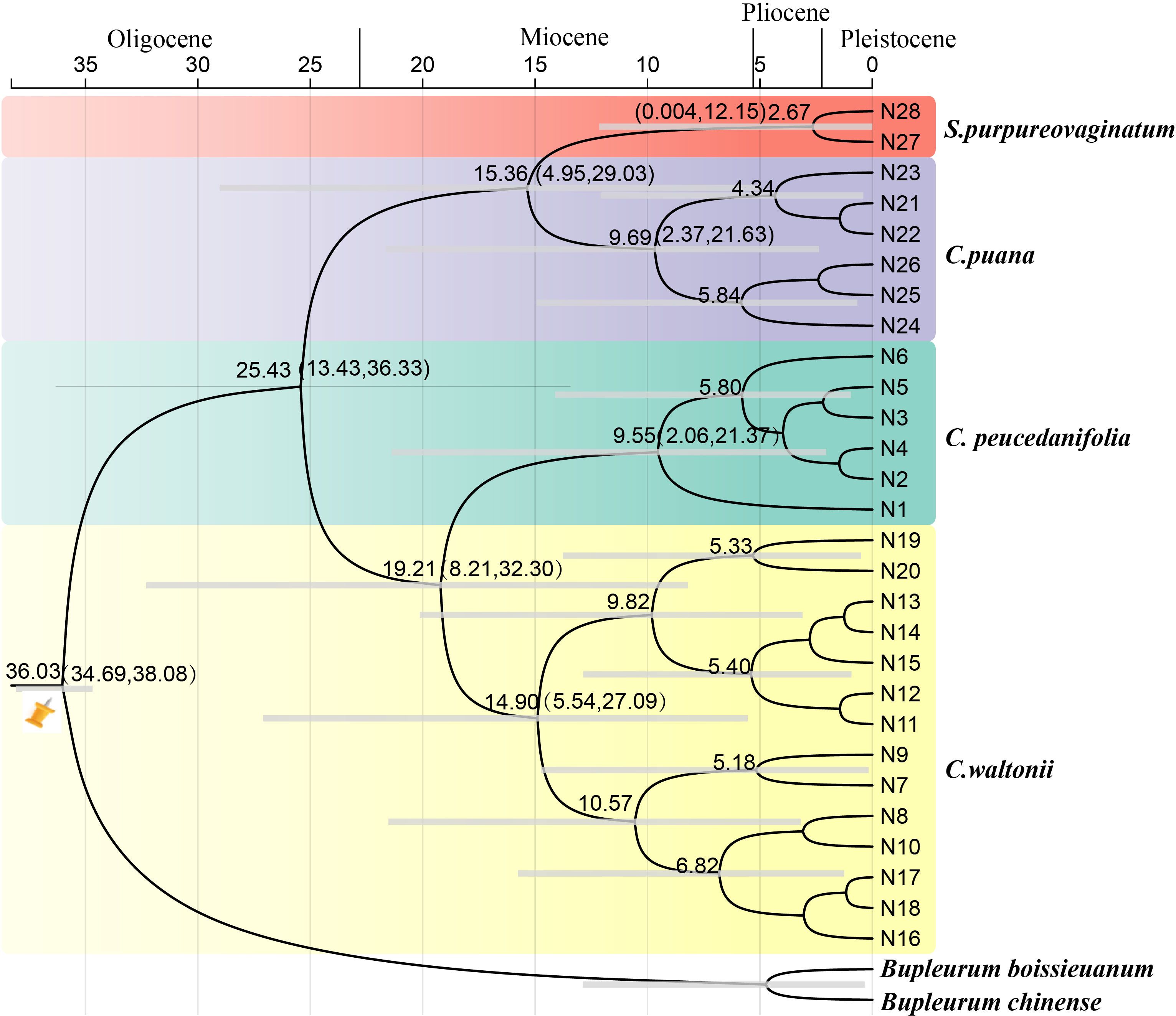
Figure 10. The divergence time estimation based on ITS sequence. The maximum credibility tree from the divergence times estimated with BEAST. The 95% highest posterior density (HPD) estimates for each well-supported clade are represented by bars. Orange nails indicate the calibration points for the molecular dating.
4 Discussion
4.1 The micromorphological taxonomic value of fruits and pollens
Cremocarp is a unique fruit in the Apiaceae family, which is widely used in taxonomic studies of many genera of Apiaceae, such as in Sanicula L. (Song et al., 2024a, c), Ferula L. (Qin et al., 2023), Sinocarum Wolff ex Shan & Pu (Xiao et al., 2021), Seseli L. (Cai et al., 2022), and Acronema Edgew. (Chen et al., 2024). In this study, the fruit micromorphology and anatomical features of the Cyclorhiza species were highly similar. However, some differences (fruit size and calyx teeth) were detected, such as C. peucedanifolia having the largest fruit size, while C. puana had the smallest fruit size, and the calyx teeth of C. peucedanifolia were subulate, whereas those of C. waltonii and C. puana were narrow triangular. These significant differences allow them to be easily distinguishable from each other. Moreover, we also found that the fruit of S. purpureovaginatum shared some similarities with Cyclorhiza species, such as a long-ellipsoid shape, smooth and glabrous, slightly laterally compressed, with the endosperm commissural face concave, ribs prominent, ribs equal, and shortly keeled, which was consistent with the C. puana; its subulate calyx teeth was accordant with C. peucedanifolia, and its vitta numbers overlapped with those of the Cyclorhiza species (Table 1). All these findings implied that S. purpureovaginatum belonged to the genus Cyclorhiza and should be transferred from the genus Seseli to Cyclorhiza. Therefore, the fruit’s micromorphological and anatomical features implied that the genus Cyclorhiza was a monophyletic group after including S. purpureovaginatum.
The morphology and micromorphology of pollen are mainly controlled by genes and are rarely influenced by the environment, thus possessing strong conservation and stability (Peng et al., 2018; Zhou et al., 2022). The pollen morphology (pollen size, outer wall ornamentation, and aperture characteristics) is of great significance in revealing the origin, evolution, and phylogenetic relationships of plants (Palynological Section, 1960; Wang and Wang, 1983; Zhang and Wang, 2007). In taxonomic studies of the Apiaceae family, the rich and diverse pollen characteristics can also provide a valuable reference for exploring the phylogenetic relationships and evolutionary status between species (Qin and Shen, 1990; Meng et al., 2004; Chen et al., 2007; Wang et al., 2012; Zhang et al., 2018; Jardine et al., 2022). In this study, we found that the pollen of Cyclorhiza species exhibited highly similar characteristics, such as rombiformis equatorial view, subtriangle polar view, short rod-like pseudo-cerebroid of exine ornamentation of the equatorial view, and angular germinal aperture, which were accordant with the study of Shu and Sheh (2001). Nevertheless, the pollen size of three Cyclorhiza species displayed differences. For example, C. peucedanifolia had the largest size, while C. waltonii had the smallest size, so they can be clearly distinguished from each other by pollen size. Meanwhile, the rombiformis pollen shape and angular germinal aperture indicated that Cyclorhiza was a relatively primitive group in the Apiaceae family (Zhang et al., 2013). Furthermore, we found that the pollen of S. purpureovaginatum were highly similar to those of Cyclorhiza species, implying that S. purpureovaginatum was indeed a member of the Cyclorhiza and should be transferred to the genus. Thus, pollen morphology once again indicated that the genus Cyclorhiza was a natural group after including S. purpureovaginatum.
4.2 Plastome evolution
All plastomes displayed a typical quadripartite structure that may be related to the stability of plastome function (Liu et al., 2022). No gene rearrangement or loss was detected in these four plastomes, which was consistent with other genera of Apiaceae (Wen et al., 2021; Liu et al., 2022; Ren et al., 2022; Gui et al., 2023; Qin et al., 2023; Song et al., 2023; Chen et al., 2024; Guo et al., 2024; Song et al., 2024b, c). Moreover, the genome size, GC content, IR boundaries, patterns of codon bias, and SSRs were very similar among these plastomes, which showed that these four plastomes were highly conserved. These findings also suggested that S. purpureovaginatum was indeed a member of the genus Cyclorhiza and implied the monophyly of the Cyclorhiza after including S. purpureovaginatum.
4.3 Genetic diversity and structural analyses
The genetic diversity of species plays an important role in their adaptability and survival ability, which is the result of multiple factors working together (Soltis, 1991). The higher the level of genetic diversity of a species, the more advantageous it is for species to adapt to complex and changing environments. Conversely, species with lower levels of genetic diversity have more difficulty adapting to environmental changes, which may even lead to species extinction (Avise and Hamrick, 1996; Petit et al., 2005). In this study, we found that both datasets showed high levels of overall genetic diversity (ITS: HT = 0.963, cpDNA: HT = 0.950) in the genus Cyclorhiza, and the population genetic differentiation coefficient was significantly higher than the geographic population differentiation coefficient, which indicated that the genus Cyclorhiza had a significant lineage geographic structure. Thus, we speculated that the reasons for the high level of genetic diversity in the genus Cyclorhiza may include the following. i) Long-term evolutionary history: as a basal group in the subfamily Apioideae of the family Apiaceae (Zhou et al., 2009; Wen et al., 2021), the genus Cyclorhiza had undergone many geological and climatic events over its long evolutionary history, leading to the accumulation of a large amount of genetic variation. ii) Complex terrain and habitat: Cyclorhiza plants are widely distributed in HHM region (Sheh and Shan, 1980; Zhou et al., 2021). These regions have complex terrains, including alpine meadows, alpine meadows, broad-leaved forests, rocky landforms, river valleys, and streams, as well as unique climate phenomena such as the Foehn effect (Zheng et al., 2021), which often occurs in these areas. The diversity of terrain and climate promotes the differentiation between populations and increases the likelihood of genetic drift, thereby accumulating rich genetic variation. iii) Sexual reproduction mode: the Cyclorhiza plants belong to sexual reproduction, which can maintain the genetic stability of the species, increase the possibility of genetic variation, and further promote the production of genetic diversity.
The diversity of haplotypes (ITS: 0.941, cpDNA: 0.9324) was relatively high at the level of the genus Cyclorhiza and three Cyclorhiza species (C. waltonii, ITS: 0.867, cpDNA: 0.815, C. peucedanifolia, ITS: 0.788, cpDNA: 0.869, and C. puana, ITS: 0.770, cpDNA: 0.916). However, S. purpureovaginatum had a low diversity of haplotypes (ITS: 0.556, cpDNA: 0.476) (Table 4), which may be caused by S. purpureovaginatum possessing only one population, and the data were not comprehensive enough. Therefore, it is necessary to increase the sample size of the S. purpureovaginatum population to avoid this problem in future research. TCS network showed that there were no shared haplotypes among these four species, and most haplotypes were limited to one or adjacent populations. We speculated that the species of this genus remained isolated during both glacial and interglacial periods due to a combination of species specificity and environmental climate factors. On the one hand, Cyclorhiza plants are distributed in the HHM region (Sheh and Shan, 1980; Pimenov, 2017; Zhou et al., 2021). Numerous high-altitude mountain ranges are separated by river canyons, and the rise of monsoons on the mountains has caused significant climate change, including the Foehn effect, which severely hindered gene exchange between species of the genus Cyclorhiza. On the other hand, the limited pollen/seed dispersal ability of the Cyclorhiza species can also affect this process. Similar results were also found in the genus Chamaesium H. Wolff (Zheng et al., 2021).
The AMOVA results based on ITS data and cpDNA fragments indicated that the majority of genetic variation occurred between species (ITS 62.51%, cpDNA: 69.38%), while genetic variation within populations (ITS: 4.97%, cpDNA: 7.09%) was rare. The phenomenon was subjected to geographic barriers and species specificity in the HHM region, which increased the genetic variation between different species (Liu et al., 2006). Meanwhile, this region possessed complex and diverse habitats that increased the selection pressure, and fragmented habitats may limit gene flow between populations (Zheng et al., 2021). In addition, the limited ability of pollen/seed dispersal in the genus Cyclorhiza restricted the gene exchange between populations and increased their genetic differentiation level.
4.4 Phylogenetic analyses
The phylogenetic analyses based on plastome data, ITS sequences, and haplotypes indicated that Cyclorhiza species clustered into a separate clade, belonging to the Komarovia clade, which was consistent with the previous studies (Zhou et al., 2008). Interestingly, S. purpureovaginatum nested within Cyclorhiza, belonging to the Komarovia clade, and other members of Seseli were located in the Selineae clade, which was distant from the Komarovia clade (Figures 8, 9). The previous molecular studies (ITS, rps16 intron, rpl16 intron, and plastome) had proved that Seseli was not a monophyletic group, and its taxonomy has faced extreme challenges (Downie et al., 2000; Spalik et al., 2004; Zhou et al., 2009; Downie et al., 2010; Cai et al., 2022). To further investigate the phylogeny and taxonomy of Seseli, our team established “a narrow sense” of Seseli based on molecular, including the type species of Seseli (S. tortuosum), which was located in the Selineae clade. The morphological data also supported “a narrow sense” of Seseli, such as leaf segments linear to lanceolate, bracts nearly absent, bracteoles linear to lanceolate, rays unequal, calyx teeth very minute, mericarps ovoid or oblong, and ribs prominent (Cai et al., 2022). In this study, our phylogenetic analyses showed S. purpureovaginatum was distant from the generitype species S. tortuosum, and it does not belong to “a narrow sense” of Seseli. It also had different features from S. tortuosum, such as being herbaceous and glabrous, with erect stems branching above, no bracts and bracteoles, and yellow petals. Therefore, the taxonomic positions of S. purpureovaginatum need to be re-evaluated. Therefore, the phylogenetic results strongly suggested that S. purpureovaginatum should be transferred from the genus Seseli to Cyclorhiza. The morphological data and plastome analyses also confirmed the rationality of phylogenetic analyses (Supplementary Figures S2–S6; Tables 1, 2). The molecular evidence, morphological data, and plastome analyses justified the monophyly of the genus Cyclorhiza after including S. purpureovaginatum. Furthermore, we also clarified the relationship among these four species: C. waltonii was sister to C. peucedanifolia, and C. puana clustered with S. purpureovaginatum. The haplotype phylogenetic trees (ITS and cpDNA) justified that the populations of each species gathered together without crossing each other. It also justified that the inter-population differentiation may be due to limited gene flow, which was accordant with the previous studies (Zhang et al., 2013; Meng et al., 2017; Xie et al., 2018).
Finally, we clarified the generic limits of Cyclorhiza based on the robust phylogenetic framework, morphological characteristics, and plastome comparative analyses, that is, herbaceous and glabrous, carrot-like roots with prominent annular scars when old, stem fistulose, erect, branched above, base clothed in purplish-brown remnant sheaths, absence of bracts and bracteoles, yellow petals, fruits ovoid or ellipsoid, smooth, slightly laterally compressed, mericarps subpentagonal in cross-section, and seed face deeply sulcate or concave. The abovementioned morphological characteristics are unique to Cyclorhiza, which can be easily distinguished from other genera of Apiaceae. Hence, we provided a species classification key index for the genus Cyclorhiza.
4.5 Origin and diversification of Cyclorhiza
The HHM region is a natural laboratory for investigating the processes of ecological speciation (Yu et al., 2023). Due to the fact that the genus Cyclorhiza is endemic to the HHM region (Pimenov, 2017; Zhou et al., 2021), it is of great value to study its origin and differentiation. Our analysis based on date estimation revealed that the genus occurred in the late Eocene (36.03 Ma, 95% HPD: 34.69–38.08 Ma). This period was associated with the early uplift of the southern QTP (Barba-Montoya et al., 2018), which led to the development of alpine dwelling habitats and the independent evolution of plant lineages, such as Chamaesieae, Bupleureae, and Pleurospermeae (Wen et al., 2021). A previous study demonstrated that the initial uplift of the QTP occurred in the middle Eocene to late Eocene (45–35 Ma) (Barba-Montoya et al., 2018), and the monolithic uplift of the QTP led to the uplift of HDM. The uplift events have caused drastic habitat fragmentation and heterogeneity, which played important roles in promoting the formation and differentiation of species, such as in Sanicula L. (Song et al., 2024b) and Saxifraga Tourn. ex L. (Ebersbach et al., 2017). Therefore, we hypothesized that the origin of the genus Cyclorhiza was closely related to the early uplift of the QTP and HDM. In addition, the diversification of Cyclorhiza occurred at the late Oligocene, with the age 25.43 Ma (95% HPD: 13.43–36.33 Ma). During the Oligocene (33.9–23.03 Ma), the climate seemed to be temperate, and many regions were nearly tropical. Grasslands expanded and forested regions dwindled (Britannica, 2020). As the uplift continued, the QTP and its adjacent mountain acted as orographic barriers to the Asian atmospheric circulation, which directly led to the formation of the monsoon climate (Zheng et al., 2021). Therefore, we speculated that the differentiation of the genus Cyclorhiza was largely influenced by the colonization of the newly available climate and terrain.
4.6 Taxonomic treatment
Cyclorhiza purpureovaginata (R. H. Shan & M. L. Sheh) J.Cai, X.Y.M.Aou & S.D.Zhou, comb. nov.
≡ Seseli purpureovaginatum R. H. Shan & M. L. Sheh in Acta Phytotax. Sin. 18 (3): 377.
Type: China. Xizang: Biru County, Baiga Valley, in sunny mountain slopes and crevices, ca. 3800 m, 9 September 1976, Qinghai-Xizang Exped. 11346 (holotype PE! barcode PE00935541) (Supplementary Figure S1) (Wolff, 1925; Sheh and Shan, 1980).
Description: Perennial herbs, 25–50 cm tall, monocarpic, glabrous throughout. The stem is solitary, mostly upright, sturdy, and sparsely branched near the top. Basal leaves are numerous and characterized by dark purple scarious margins. The leaf blade is ovate, measuring 6–10 cm in length and 3–5 cm in width, divided into two pinnate segments. The pinnae are shortly petiolulate, and the ultimate segments are linear or linear-elliptic, 4–10 mm in length and 1.5–5 mm in width. The ultimate segments resemble the basal leaves. The synflorescence is dichotomously branched, with a few loose compound umbels that are 2.5–5 cm in diameter. No bracts or bracteoles are present, and three to five rays measure 1.5–3 cm. The calyx teeth are very small or absent. The petals are smooth and white. The stylopodium is cone-shaped, and the styles are short. The fruit is pale yellow and oblong, with a rounded-pentagonal cross-section, measuring 3.5–5 mm in length and 2–3 mm in width. It is glabrous with prominent ribs of equal size and a slight keel. Each furrow contains 2–3 vittae, while there are 4 vittae on the commissure.
Distribution and habitat in China: S. purpureovaginatum is endemic to southwestern China (east Xizang). It grows in alpine meadows on sunny mountain slopes at an elevation of 3,800–4,000 m.
Additional specimens examined: China-Xizang: Biru Xian, alt. 3,800 m, 9 September 1976, Qinghai-Xizang Exped. 11346 (holotype PE); Baiga Village, 31°20′N, 94°04′E, 3,986 m, 31 July 2022, J.Cai & J.Q.Lei CJ202208040101 (SZ).
Key to species of Cyclorhiza
1. 1a. The leaf sheath is a deep, dark purple, with slightly keeled ribs, containing 2–3 vittae in each furrow and 4–6 on the commissure.………………………………C. purpureovaginata
1b. The leaf sheath is a light shade of dark purple, with prominent, almost narrowly winged filiform ribs. There are 1–2 vittae in each furrow and 2–3 on the commissure………2
2a. The final leaf segments are shaped from ovate-oblong to linear-lanceolate, measuring 20 to 60 mm in length and 3 to 10 mm in width.…………………………………C. peucedanifolia
2b. Ultimate leaf segments linear, 2–20 × 0.5–6 mm…………3
3a. Ultimate leaf segments 4–20 × 2–6 mm; rays unequal; stylopodium low-conic; seed face deeply sulcate........C. waltonii
3b. Ultimate leaf segments 2–4 × 0.5–1 mm; rays subequal; stylopodium ob-conic; seed face slightly concave.....…C. puana
5 Conclusion
In this study, we found that Cyclorhiza plants and S. purpureovaginatum shared similar morphological characteristics in root, stem, bracts, bracteoles, petals, fruits, and pollens. These evidence strongly supported the monophyly of the genus Cyclorhiza after including S. purpureovaginatum. In addition, we first sequenced and assembled the Cyclorhiza plastomes and performed comprehensive comparative analyses for the genus. The results revealed that the genome size, GC content, IR boundaries, patterns of codon bias, and SSRs were very similar among these plastomes, which showed that these four plastomes were highly conserved. These findings also suggested that S. purpureovaginatum was indeed a member of the genus Cyclorhiza and implied the monophyly of the Cyclorhiza after including S. purpureovaginatum. Nevertheless, we selected 15 mutation hotspots regions (cemA, matK, ndhF, rpl20, ycf4, ndhE–ndhG, petA–psbJ, trnE–trnT, atpI–rps2, rpoC2–rpoC1, trnT–psbD, psbK–psbI, trnH–psbA, accD–psaI, and rrn5–trnR) as potentially strong DNA barcodes in Cyclorhiza species identification. The phylogenetic analyses of three datasets (plastome data, ITS sequences, and haplotypes) showed that Cyclorhiza species clustered into a separate clade, belonging to the Komarovia clade, and S. purpureovaginatum nested within Cyclorhiza. In detail, C. waltonii formed a clade with C. peucedanifolia, and C. puana clustered with S. purpureovaginatum. Thus, phylogenetic analyses also supported the monophyly of the genus Cyclorhiza after transferring S. purpureovaginatum into the genus. Then, we clarified the generic limits of Cyclorhiza based on morphological evidence, plastome comparative analyses, and phylogenetic analyses and provided a species classification key index for the genus. Furthermore, phylogeography analyses showed that the genus Cyclorhiza possessed high genetic diversity, and the population genetic differentiation coefficient was significantly higher than the geographic population differentiation coefficient, which indicated that the genus Cyclorhiza had a significant lineage geographic structure. Finally, the divergence time estimation showed that the genus Cyclorhiza originated in the late Eocene, which was closely related to the early uplift of the QTP and HDM. The diversification of the genus occurred in the late Oligocene, which was largely influenced by the colonization of the newly available climate and terrain. In conclusion, the study investigated the morphology, phylogeography, phylogeny, taxonomy, and evolution of the genus Cyclorhiza for the first time and provided valuable references for other genera of Apiaceae.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
B-NS: Conceptualization, Formal analysis, Investigation, Methodology, Software, Writing – original draft. XA: Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft. R-MT: Data curation, Investigation, Resources, Software, Writing – original draft. JC: Data curation, Investigation, Methodology, Writing – original draft. W-YT: Investigation, Methodology, Software, Writing – original draft. C-KL: Methodology, Software, Writing – review & editing. X-JH: Supervision, Writing – review & editing. S-DZ: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 32170209, 32070221, and 32470216).
Acknowledgments
We are grateful to Dengfeng Xie, Huanhuan Qin, Zixuan Li, Xianlin Guo, and Yulin Xiao for their help in sample collection. We also thank Herbarium PE, KUN, SZ, NAS, CDBI, and XJU for providing the specimens and the consultation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1504734/full#supplementary-material
Supplementary Figure 1 | The features of Seseli purpureovaginatum. (A) isolectotype (E00935541); (B) plant; (C) leaf; (D) fruit.
Supplementary Figure 2 | Plastome maps of Cyclorhiza members and S. purpureovaginatum. Genes shown outside of the outer layer circle are transcribed clockwise, while those insides are transcribed counterclockwise. The genes belonging to diferent functional groups are color-coded. The dark gray area of the inner circle denotes the GC content of plastome.
Supplementary Figure 3 | Analyses of repeats in the four plastomes. (A) Total number of four repeat types; (B) Total number of SSRs.
Supplementary Figure 4 | Comparison of IR boundaries among four plastomes.
Supplementary Figure 5 | mVISTA alignment for four plastomes with C. waltonii as the reference.
Supplementary Figure 6 | Mauve alignment of four plastomes. Local collinear blocks within each alignment are represented by blocks of the same color connected with lines.
Supplementary Figure 7 | The RSCU values of all concatenated protein-coding genes for four plastomes. Color key: higher RSCU values were denoted in red, while lower RSCU values were indicated in blue. * represented the terminator codons.
References
Amiryousef, A., Hyvonen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Avise, J. C., Hamrick, J. K. (1996). Conservation genetics. Case histories from nature. Science 272, 364. doi: 10.1126/science.272.5260.364.b
Banasiak, L., Piwczynski, M., Ulinski, T., Downie, S. R., Watson, M. F., Shakya, B., et al. (2013). Dispersal patterns in space and time: a case study of Apiaceae subfamily Apioideae. J. Biogeogr. 40, 1324–1335. doi: 10.1111/jbi.12071
Barba-Montoya, J., Reis, M. D., Schneider, H., Donoghue, P. C. J., Yang, Z. (2018). Constraining uncertainty in the timescale of angiosperm evolution and the veracity of a cretaceous Terrestrial Revolution. New Phytol. 218, 819–834. doi: 10.1111/nph.15011
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Britannica. (2020). The Editors of Encyclopaedia. “Oligocene Epoch summary”. In: Encyclopedia Britannica. Available online at: https://www.britannica.com/summary/Oligocene-Epoch (Accessed August 25, 2024).
Cai, J., Qin, H. H., Lei, J. Q., Liu, C. K., He, X. J., Zhou, S. D. (2022). The phylogeny of Seseli (Apiaceae, Apioideae): insights from molecular and morphological data. BMC Plant Biol. 22, 534. doi: 10.1186/s12870-022-03919-9
Calvino, C. I., Teruel, F. E., Downie, S. R. (2016). The role of the Southern Hemisphere in the evolutionary history of Apiaceae, a mostly north temperate plant family. J. Biogeogr. 43, 398–409. doi: 10.1111/jbi.12651
Capella-Gutierrez, S., Silla-Martınez, J. M., Gabaldon, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chen, L., Song, B. N., Yang, L., Wang, Y., Wang, Y. Y., Aou, X., et al. (2024). Phylogeny, adaptive evolution, and taxonomy of Acronema (Apiaceae): evidence from plastid phylogenomics and morphological data. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1425158
Chen, S. F., Zhou, Y. Q., Chen, Y. R., Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chen, W. W., He, X. J., Zhang, X. M., Pu, J. X. (2007). Pollen morphology of the genus Angelica from Southwest China and its systematic evolution analysis. Acta Bot. Boreali-Occident. Sin. 27, 1364–1372. doi: 10.3321/j.issn:1000-4025.2007.07.013
Constance, L. (1997). An instance of East-West confusion in Chinese Umbelliferae, or Arracacia out of Asia! Edin. J. Bot. 54, 99–104. doi: 10.1017/S0960428600003899
Darling, A. C., Mau, B., Blattner, F. R., Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Donoghue, M. J., Smith, S. A. (2004). Patterns in the assembly of the temperate forest around the Northern Hemisphere. Phil. Trans. R. Soc Lond. B 359, 1633–1644. doi: 10.1098/rstb.2004.1538
Downie, S. R., Katz-Downie, D. S., Watson, M. F. (2000). A phylogeny of the flowering plant family Apiaceae based on chloroplast DNA rpl16 and rpoC1 intron sequences: towards a suprageneric classification of subfamily Apioideae. Amer J. Bot. 87, 273–292. doi: 10.2307/2656915
Downie, S. R., Spalik, K., Katz-Downie, D. S., Reduron, J.-P. (2010). Major clades within Apiaceae subfamily Apioideae as inferred by phylogenetic analysis of nrDNA ITS sequences. Plant Divers. Evol. 128, 111. doi: 10.1127/1869-6155/2010/0128-0005
Doyle, J. J., Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15. doi: 10.1016/0031-9422(80)85004-7
Ebersbach, J., Muellner-Riehl, A. N., Michalak, I., Tkach, N., Hoffmann, M. H., Röser, M., et al. (2017). In and out of the Qinghai-Tibet Plateau: divergence time estimation and historical biogeography of the large arctic-alpine genus Saxifraga L. J. Biogeogr. 44, 900–910. doi: 10.1111/jbi.12899
Excoffier, L., Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Grivet, D., Petit, R. J. (2002). Phylogeography of the common ivy (Hedera sp.) in Europe: genetic differentiation through space and time. Mol. Ecol. 11, 1351–1362. doi: 10.1046/j.1365-294X.2002.01522.x
Gui, L. J., Xie, D. F., Peng, C., Ren, T., Yu, L. Y., Zhou, S. D., et al. (2023). Chloroplast genomes and ribosomal DNA provide insights into divergence and morphological evolution of alpine Tongoloa. J. Syst. Evol. 1–13. doi: 10.1111/jse.13028
Guo, X. L., Gou, W., Price, M., Jiang, Q. P., Peng, C., Zhou, S. D., et al. (2024). Reinterpreting the phylogenetic position and taxonomic revision of the genus Pterocyclus (Apiaceae, Apioideae) based on nrITS, complete plastid genome, and morphological evidence. J. Syst. Evol. 62, 384–402. doi: 10.1111/jse.12958
Hughes, C. E., Atchison, G. W. (2015). The ubiquity of alpine plant radiations: from the Andes to the Hengduan Mountains. New Phytol. 207, 275–282. doi: 10.1111/nph.13230
Jardine, P. E., Palazzesi, L., Tellería, M. C., Barreda, V. D. (2022). Why does pollen morphology vary? Evolutionary dynamics and morphospace occupation in the largest angiosperm order (Asterales). New Phytol. 234, 1075–1087. doi: 10.1111/nph.18024
Ji, Y. H., Landis, J. B., Yang, J., Wang, S. Y., Zhou, N., Luo, Y., et al. (2023). Phylogeny and evolution of Asparagaceae subfamily Nolinoideae: new insights from plastid phylogenomics. Ann. Bot. 131, 301–312. doi: 10.1093/aob/mcac144
Jia, L. B., Wang, S. Y., Hu, J. J., Miao, K., Huang, Y. J., Ji, Y. H. (2024). Plastid phylogenomics and fossil evidence provide new insights into the evolutionary complexity of the ‘woody clade’ in Saxifragales. BMC Plant Biol. 24, 277. doi: 10.1186/s12870-024-04917-9
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31. doi: 10.1186/s13059-020-02154-5
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–9. doi: 10.1093/bioinformatics/bts199
Kljuykov, E. V., Liu, M., Ostroumova, T. A., Pimenov, M. G., Tilney, P. M., Wyk, B. V., et al. (2004). Towards a standardised terminology for taxonomically important morphological characters in the Umbelliferae. S. Afr. J. Bot. 70, 488–96. doi: 10.1016/S0254-6299(15)30233-7
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Letunic, I., Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Li, L. B., Yuan, C. Q., Ting, C., Cheo, T. (1993). A taxonomic study on the genus Cnidium in China. Acta Bot. Boreali-Occident. Sin. 13, 63–69 + 89-90.
Librado, P., Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, C. K., Lei, J. Q., Jiang, Q. P., Zhou, S. D., He, X. J. (2022). The complete plastomes of seven Peucedanum plants: comparative and phylogenetic analyses for the Peucedanum genus. BMC Plant Biol. 22, 1–14. doi: 10.1186/s12870-022-03488-x
Liu, J. Q., Wang, Y. J., Wang, A. L., Hideaki, O., Abbott, R. J. (2006). Radiation and diversification within the Ligularia-Cremanthodium-Parasenecio complex (Asteraceae) triggered by uplift of the Qinghai-Tibetan Plateau. Mol. Phylogenet. Evol. 38, 31–49. doi: 10.1016/j.ympev.2005.09.010
Liu, L. J., Wang, Q., Zhang, Z., Yu, Y. (2023). MATO: An updated tool for capturing and analyzing cytotaxonomic and morphological data. Innov. Life. 1, 100010. doi: 10.59717/j.xinn-life.2023.100010
Meng, D. Y., Zhou, S. D., He, X. J., Ma, Y. H., Wang, C. B. (2004). Pollen morphology of the genus Peucedanum from Sichuan and its systematic significance. Acta Bot. Boreali-Occident. Sin. 24, 2341–2345. doi: 10.3321/j.issn:1000-4025.2004.12.026
Meng, H. H., Su, T., Gao, X. Y., Li, J., Jiang, X. L., Sun, H., et al. (2017). Warm-cold colonization: response of oaks to uplift of the Himalaya-Hengduan Mountains. Mol. Ecol. 26, 3276–3294. doi: 10.1111/mec.14092
Nei, M., Li, W. H. (1979). Mathematical-model for studying genetic-variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 76, 5269–5273. doi: 10.1073/pnas.76.10.5269
Nei, M., Tajima, F. (1981). DNA polymorphism detectable by restriction endonucleases. Genetics 97, 145–163. doi: 10.1093/genetics/97.1.145
Ostroumova, T. A. (2021). Fruit micromorphology of Siberian Apiaceae and its value for taxonomy of the family. Turczaninowia 24, 120–143. doi: 10.14258/turczaninowia.24.2.13
Palynological Section. (1960). Institute of Botany, Chinese Academy of Sciences Pollen morphology of Chinese plants (Beijing: Science Press).
Peng, H. W., Zhou, S. D., He, X. J. (2018). Pollen morphology of 26 taxa from 15 genera of malvaceae in China and its systematic significance. Acta Bot. Boreali-Occident. Sin. 38, 1832–1845. doi: 10.7606/j.issn.1000-4025.2018.10.1832
Petit, R. J., Duminil, J., Fineschi, S., Salvini, D., Vendramin, G. (2005). Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 14, 689–701. doi: 10.1111/j.1365-294X.2004.02410.x
Pimenov, M. G. (2017). Updated checklist of Chinese Umbelliferae: Nomenclature, synonymy, typification, distribution. Turczaninowia 20, 106–239. doi: 10.14258/turczaninowia.21.1.10
Polzin, T., Daneshmand, S. V. (2003). On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 31, 12–20. doi: 10.1016/s0167-6377(02)00185-2
Pons, O., Petit, R. J. (1996). Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144, 1237–1245. doi: 10.1093/genetics/144.3.1237
Posada, D., Crandall, K. A. (1998). Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Pu, F. D., Watson, M. F. (2005). Ligusticum L. Flora of China Vol. 14 (Beijing: Science Press), 140–151.
Qin, H. H., Cai, J., Liu, C. K., Zhou, R. X., Price, M., Zhou, S. D., et al. (2023). The plastid genome of twenty-two species from Ferula, Talassia, and Soranthus: comparative analysis, phylogenetic implications, and adaptive evolution. BMC Plant Biol. 23, 9. doi: 10.1186/s12870-022-04027-4
Qin, X. M., Shen, G. M. (1990). Taxonomic study on the genus Ferula and its similar genera in Xinjiang. Arid Zone Res. 4, 23–33. doi: 10.13866/j.azr.1990.04.004
Qu, X. J., Moore, M. J., Li, D. Z., Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 50. doi: 10.1186/s13007-019-0435-7
Rambaut, A., Drummond, A. J. (2014). TreeAnnotator v2.1.2 (Edinburgh: University of Edinburgh, Institute of Evolutionary Biology).
Rambaut, A., Drummond, A. (2015). FigTree, version 1.4.2. Available online at: http://tree.bio.ed.ac.uk/software/fgtree/.
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., Suchard, M. A. (2018). Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Ren, T., Xie, D., Peng, C., Gui, L. J., Price, M., Zhou, S. D., et al. (2022). Molecular evolution and phylogenetic relationships of Ligusticum (Apiaceae) inferred from the whole plastome sequences. BMC Ecol. Evo. 22, 55. doi: 10.1186/s12862-022-02010-z
Ronquist, F., Teslenko, M., Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–42. doi: 10.1093/sysbio/sys029
Sharp, P. M., Li, W. H. (1986). An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 24, 28–38. doi: 10.1007/bf02099948
Sheh, M. L., Pimenov, M. G., Kljuykov, E. V., Watson, M. F. (2005). “Seseli L,” in Flora of China, vol. 14 . Eds. Wu, Z. Y., Raven, P. H., Hong, D. Y. (Science Press, Beijing), 122–130.
Sheh, M. L., Shan, R. H. (1980). Cyclorhiza and Chuanminshen-Two newly proposed genera in Umbelliferae (Apiaceae). J. Syst. Evol. 18, 45–49.
Sheh, M. L., Watson, M. F. (2005). “Cyclorhiza,” in Flora of China, vol. 14 . Eds. Sheh, M. L., Shan, R. H. (Science Press & Missouri Botanical Garden Press, Beijing, China), 53.
Shu, P., Sheh, M. L. (2001). Pollen Atlas of Umbelliferae Plants in China (Shanghai: Shanghai Science and Technology Press).
Soltis, P. S. (1991). Genetic variation in endemic and widespread plant species. Aliso 13, 215–223. doi: 10.5642/aliso.19911301.10
Song, B. N., Liu, C. K., Ren, T., Xiao, Y. L., Chen, L., Xie, D. F., et al. (2024c). Plastid phylogenomics contributes to the taxonomic revision of taxa within the genus Sanicula L. and acceptance of two new members of the genus. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1351023
Song, B. N., Liu, C. K., Xie, D. F., Xiao, Y. L., Tian, R. M., Li, Z. X., et al. (2023). Plastid phylogenomic analyses reveal the taxonomic position of peucedanum franchetii. Plants 12, 97. doi: 10.3390/plants12010097
Song, B. N., Liu, C. K., Zhao, A. Q., Tian, R. M., Xie, D. F., Xiao, Y. L., et al. (2024b). Phylogeny and diversification of genus Sanicula L. (Apiaceae): novel insights from plastid phylogenomic analyses. BMC Plant Biol. 24, 70. doi: 10.1186/s12870-024-04750-0
Song, B. N., Yong, F., Liu, C. K., Wang, Y., Yang, L., Chen, L., et al. (2024a). The micromorphology and its taxonomic value of the genus sanicula L. @ in China (Apiaceae). Plants 13, 1635. doi: 10.3390/plants13121635
Spalik, K., Reduron, J.-P., Downie, S. R. (2004). The phylogenic position of Peucedanum sensu lato and allied genera and their placement in tribe Selineae (Apiaceae, subfamily Apioideae). Pl. Syst. Evol. 243, 189–210. doi: 10.1007/s00606-003-0066-2
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stephan, G., Pascal, L., Ralph, B. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, 59–64. doi: 10.1093/nar/gkz238
Suchard, M. A., Lemey, P., Baele, G., Ayres, D. L., Drummond, A. J., Rambaut, A. (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016. doi: 10.1093/ve/vey016
Sun, H., Zhang, J. W., Deng, T., Bouford, D. E. (2017). Origins and evolution of plant diversity in the Hengduan Mountains, China. Plant Divers. 39, 161–166. doi: 10.1016/j.pld.2017.09.004
Tang, L. L., Wang, T. L., Hou, L. X., Zhang, G. F., Deng, M., Guo, X. R., et al. (2024). Comparative and phylogenetic analyses of Loranthaceae plastomes provide insights into the evolutionary trajectories of plastome degradation in hemiparasitic plants. BMC Plant Biol. 24, 406. doi: 10.1186/s12870-024-05094-5
Wang, L. Y., Ikeda, H., Liu, T. L., Wang, Y. J., Liu, J. Q. (2009). Repeated range expansion and glacial endurance of Potentilla glabra (Rosaceae) in the Qinghai-Tibetan Plateau. J. Integr. Plant Biol. 51, 698–706. doi: 10.1111/j.1744-7909.2009.00818.x
Wang, S. Y., Zhou, N., Shi, N. X., Zhang, G. F., Liu, H. Y., Guo, X. R., et al. (2023). Testing and using complete plastomes for authentication of medicinal Polygonatum species (Asparagaceae). Ind. Crops Prod. 197, 116557. doi: 10.1016/j.indcrop.2023.116557
Wang, Z. X., Tan, J. B., Peng, L., He, X. J. (2012). Pollen morphology of sixteen Pimpinella species (Apiaceae) and the systematic analysis. Acta Bot. Boreali-Occident. Sin. 32, 1592–1598. doi: 10.3969/j.issn.1000-4025.2012.08.014
Wen, J., Xie, D. F., Price, M., Ren, T., Deng, Y. Q., Gui, L. J., et al. (2021). Backbone phylogeny and evolution of Apioideae (Apiaceae): New insights from phylogenomic analyses of plastome data. Mol. Phylogenet. Evol. 161, 107–183. doi: 10.1016/j.ympev.2021.107183
White, T. J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols (California: Academic Press Incorporation) 31, 315–322. doi: 10.1016/B978-0-12-372180-8.50042-1
Wolff, H. (1925). Neue umbelliferen-gattungen aus ostasien. Notizblatt Des. Königl botanischen Gartens und Museums zu Berlin. 9, 275–280. doi: 10.2307/3994550
Wolff, H. J. R. (1929). Umbelliferae Asiatiae novae relictae. II. Repertorium Novarum Specierum Regni Vegetabilis 27, 179–192. doi: 10.1002/fedr.4870270909
Xia, M. Q., Liu, Y., Liu, J. J., Chen, D. H., Shi, Y., Chen, Z. X., et al. (2022). Out of the Himalaya-Hengduan Mountains: Phylogenomics, biogeography and diversification of Polygonatum Mill. (Asparagaceae) in the Northern Hemisphere. Mol. Phylogenet. Evol. 169, 107431. doi: 10.1016/j.ympev.2022.107431
Xiao, Y. P., Guo, X. L., Price, M., Gou, W., Zhou, S. D., He, X. J. (2021). New insights into the phylogeny of Sinocarum (Apiaceae, Apioideae) based on morphological and molecular data. PhytoKeys 175, 13–32. doi: 10.3897/phytokeys.175.60592
Xie, C., Xie, D. F., Zhong, Y., Guo, X. L., Liu, Q., Zhou, S. D., et al. (2018). The effect of Hengduan Mountains Region (HMR) uplift to environmental changes in the HMR and its eastern adjacent area: tracing the evolutionary history of Allium section Sikkimensia (Amaryllidaceae). Mol. Phylogenet. Evol. 130, 380–396. doi: 10.1016/j.ympev.2018.09.011
Yang, M. L., Liao, C. Y. (2023). Palynological research on the Angelica species (Apiaceae) from North America. Acta Bot. Boreali-Occident. Sin. 43, 1236–1250. doi: 10.7606/j.issn.1000-4025.20230716
Yu, J. R., Niu, Y. T., You, Y. C., Cox, C. J., Barrett, R. L., Blasi, A. T., et al. (2023). Integrated phylogenomic analyses unveil reticulate evolution in Parthenocissus (Vitaceae), highlighting speciation dynamics in the Himalayan-Hengduan Mountains. New Phytol. 238, 888–903. doi: 10.1111/nph.18580
Zhang, D., Gao, F., Jakovlic, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, S. Y., Liao, C. Y., Li, M. L., Chen, Y., Zhou, B. (2018). Research on pollen morphologies of Ostericum Hoffm. (Apiaceae) of eight species from seventeen populations. Acta Bot. Boreali-Occident. Sin. 38, 2224–2235. doi: 10.7606/j.issn.1000-4025.2018.12.2224
Zhang, X. M., He, X. J. (2009). Leaf epidermal characters of six species in Peucedanum and its taxonomic significance. Chin. Agric. Sci. Bull. 25, 177–180. doi: 10.11924/j.issn.1000-6850.2009-1130
Zhang, X. P., Wang, Q. (2007). Recent advance in study of pollen morphology and systematics of the family asteraceae. J. Anhui Normal Univ., 326–330. doi: 10.3969/j.issn.1001-2443.2007.03.026
Zhang, Y. L., Li, B. Y., Zheng, D. (2002). A discussion on the boundary and area of the Tibetan Plateau in China. Geogr. Res. 21, 1–18. doi: 10.3321/j.issn:1000-0585.2002.01.001
Zhang, Y., Liu, Q. X., Wang, L. S., Shu, P., Song, C. F. (2013). Pollen morphological characteristics of Pleurospermum Hoffm. (Apiaceae) and its evolution significance. J. Plant Resour. 22, 29–37. doi: 10.3969/j.issn.1674-7895.2013.04.04
Zheng, H. Y., Guo, X. L., Price, M., He, X. J., Zhou, S. D. (2021). Effects of mountain uplift and climatic oscillations on phylogeography and species divergence of chamaesium (Apiaceae). Front. Plant Sci. 12. doi: 10.3389/fpls.2021.673200
Zhou, B. Q., Yang, G. H., Lu, H., Gen, Y. L., Wang, X., Liu, W., et al. (2022). Study on palynology and genetic relationship of mugua (Chaenomelis fructus) based on pollen microscopic characteristics. J. Shandong Univ Tradit Chin. Med. 46, 633–641. doi: 10.16294/j.cnki.1007-659x.2022.05.016
Zhou, J., Gong, X., Downie, S. R., Peng, H. (2009). Towards a more robust molecular phylogeny of Chinese Apiaceae subfamily Apioideae: Additional evidence from nrDNA ITS and cpDNA intron (rpl16 and rps16) sequences. Mol. Phylogenet. Evol. 53, 56–68. doi: 10.1016/j.ympev.2009.05.029
Zhou, J., Niu, J. M., Wang, X. Y., Wang, P., Guo, M. J., Liu, Z. W. (2021). Cyclorhiza puana (Apiaceae), a new species from Sichuan, China. PhytoKeys 182, 57–66. doi: 10.3897/phytokeys.182.67009
Zhou, J., Peng, H., Downie, S. R., Liu, Z. W., Gong, X. (2008). A molecular phylogeny of Chinese Apiaceae subfamily Apioideae inferred from nuclear ribosomal DNA internal transcribed spacer sequences. Taxon 57, 402–416. doi: 10.2307/25066012
Zhou, N., Miao, K., Hou, L. X., Liu, H. Y., Chen, J. H., Ji, Y. H. (2024b). Phylotranscriptomic analyses reveal the evolutionary complexity of Paris L. (Melanthiaceae), a morphologically distinctive genus with significant pharmaceutical importance. Ann. Bot., mcae156. doi: 10.1093/aob/mcae156
Zhou, N., Miao, K., Liu, C. K., Jia, L. B., Hu, J. J., Huang, Y. J., et al. (2024a). Historical biogeography and evolutionary diversification of Lilium (Liliaceae): New insights from plastome phylogenomics. Plant Divers. 46, 219–228. doi: 10.1016/j.pld.2023.07.009
Keywords: Cyclorhiza, morphology, phylogeography, phylogenomics, phylogeny
Citation: Song B-N, Aou X, Tian R-M, Cai J, Tan W-Y, Liu C-K, He X-J and Zhou S-D (2025) Morphology, phylogeography, phylogeny, and taxonomy of Cyclorhiza (Apiaceae). Front. Plant Sci. 15:1504734. doi: 10.3389/fpls.2024.1504734
Received: 01 October 2024; Accepted: 29 November 2024;
Published: 08 January 2025.
Edited by:
Xiaohua Jin, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Ziyoviddin Yusupov, Academy of Sciences Republic of Uzbekistan (UzAS), UzbekistanBo Li, Chinese Academy of Sciences (CAS), China
Copyright © 2025 Song, Aou, Tian, Cai, Tan, Liu, He and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Song-Dong Zhou, enNkQHNjdS5lZHUuY24=
†These authors have contributed equally to this work