 Thida Soe
Thida Soe Jiali Kong1,2†
Jiali Kong1,2† Liyun Nie
Liyun Nie Jie Wang
Jie Wang Luke R. Tembrock
Luke R. Tembrock Zhiqiang Wu
Zhiqiang Wu- 1Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Key Laboratory of Synthetic Biology, Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, China
- 2State Key Laboratory of Crop Stress Adaptation and Improvement, School of Life Sciences, Henan University, Kaifeng, China
- 3School of Medical, Molecular and Forensic Sciences, Murdoch University, Perth, WA, Australia
- 4College of Agriculture, Center for Genomics and Biotechnology, Fujian Agriculture and Forestry University, Fuzhou, China
- 5Department of Agricultural Biology, Colorado State University, Fort Collins, CO, United States
Typha is a cosmopolitan aquatic plant genus that includes species with widespread global distributions. In previous studies, a revised molecular phylogeny was inferred using seven plastid loci from nine Typha species across different geographic regions. By utilizing complete organellar genomes, we aim to provide a more comprehensive dataset that offers a robust phylogenetic signal for resolving Typha species evolutionary relationships. Here, we assembled T. latifolia and T. domingensis mitochondrial genomes (mitogenomes) using a combination of short-read and long-read data (PacBio, ONT). The mitogenomes of both species are assembled into single circular molecules of 395,136 bp and 395,140 bp in length, respectively, with a similar GC content of 46.7%. A total of 39 protein-coding genes, 17 tRNA genes, and 3 rRNA genes were annotated in both mitogenomes. The plastid genomes (plastomes) of both species possess typical quadripartite structures observed across most plants, with sizes of 161,545 bp and 161,230 bp. The overall average GC content of the plastomes of both species was 36.6%. The comparative analysis of the plastome and mitogenome revealed that 12 mitogenome DNA fragments share similar sequences with in the repeat regions of the corresponding plastomes, suggesting a past transfer of repeat regions into the mitogenome. Additionally, the mitogenomes of the two Typha species exhibited high sequence conservation with several syntenic blocks. Phylogenetic analysis of the organellar genomes of the two Typha species and 10 related species produced congruent phylogenetic trees. The availability of these organellar genomes from two Typha species provide valuable genetic resources for studying the evolution of Typhaceae and will improve taxonomic classifications within the family.
Introduction
Typha (Typhaceae) is a perennial, monocot plant distributed worldwide in wetlands, lakes, and rivers (McNaughton, 1966). The APG II places Typhaceae in the order Poales within the Commelinid clade of the monocots (Ii, 2003). The family was previously consisted of a single genus (Typha). However, APG III included another genus, Sparganium, to this family (Angiosperm Phylogeny, 2009). The two genera have a total of 51 recognized species (Christenhusz and Byng, 2016). Typha latifolia (broadleaf cattail) and Typha domingensis (southern cattail) are prominent species within the genus Typha. T. latifolia has wide leaves and dense flower spikes, thriving in freshwater habitats, where it plays a keystone role by supporting wildlife and aiding nutrient cycling (Pandey and Verma, 2018). It is widely introduced for habitat restoration and soil stabilization due to its rhizome network (Grace and Harrison, 1986; Tornberg et al., 1994). In contrast, T. domingensis, with narrower leaves and fragmented flower spikes, inhabits tropical and subtropical regions and tolerates salinity, making it vital in coastal wetlands (Vivian et al., 2014; Bansal et al., 2019). Typha comprises 38 species, most of which have a widespread global distribution. (https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:30003823-2). However, taxonomic studies of Typha have often been confined to a specific regions, such as India (Saha, 1968), Europe (Volkova and Bobrov, 2022), Iran, Pakistan, Australia (Bokhari, 1983), North America (Kuehn and White, 1999), and China. The taxonomy of Typha has long been debated due to significant morphological variation and frequent interspecific hybridization (Smith, 1987). Since 1987, fifteen new species have been described, through all were based on local populations and lacked molecular data. A number of recent studies have provided complete plastome assemblies, and molecular phylogenies for the genus Typha (Liu et al., 2020). Up to now, only T. latifolia and T. angustifolia nuclear genomes were published in the genus Typha (Widanagama et al., 2022; Liao et al., 2023), and no more complete Typha genus mito genomes are available to date. Exploring the organelle genomes of T. latifolia and T. domingensis will help in the classification of Typha species and provide a genetic resource for further study of nucleocytoplasmic interaction.
Organellar genomes play a crucial role in an organism’s growth and development (Mahapatra et al., 2021). Plant organellar genomes employ high fidelity molecular mechanism to repair DNA damage and maintain of nucleotide sequences integrity, enabling them to withstand genotoxic stresses more effectively than nuclear genomes resulting in lower mutation rates (Maréchal and Brisson, 2010). Extensive analysis of organellar genomes has provided insights into the classification and evolution of various plant taxa (Wolfsberg et al., 2001). By 2023, the NCBI GenBank database contained approximately 13,000 plastomes but only 673 complete plant mitogenomes, with only 285 species having both organellar genomes sequenced (Wang et al., 2024b). The lower number of sequenced mitogenomes compared to plastomes is due to high repeat sequences, recombination, rearrangement rate, and the insertion of plastome sequences into the mitogenome (Johnston, 2019). Given these characteristics, the mitogenome is an effective model for analyzing genomic structure and evolutionary processes (Drouin et al., 2008). A previous molecular phylogenetic study outlined the relationships within Typha using sequences from seven plastid loci (Zhou et al., 2018). However, including additional taxa and genetic markers could enhance the resolution and support of subsequent phylogenetic analyses. Moreover, no studies have described a mitogenome from Typha or explored the exchange of DNA between the plastome and the mitogenome.
In this study, we assembled two Typha species (T. latifolia and T. domingensis) mitogenomes using previously published short-read and long-read (PacBio, ONT) data. We analyzed genomic characteristics, including gene content, repeat detection, codon usage, intracellular sequence transfer, mitogenomic synteny, and organellar phylogeny, to explore intrageneric variation in the maternally inherited organelles. The assembly of complete organellar genomes for these globally important species provides a foundational resource for further detailed studies of taxonomy, metabolic function, evolution, and molecular ecology.
Materials and methods
Data source
All raw sequence resources of both short and long-read from chloroplast and mitochondria were obtained from NCBI (https://www.ncbi.nlm.nih.gov/sra/). The raw sequencing data generated and analyzed during the current study of two Typha species were from BioProject PRJNA751759 and PRJNA742003. The T. latifolia plant was collected in Ontario, Canada, and T. domingenis plant was collected in the United States. Mitogenome and plastome sequences obtained from NCBI website were used for phylogenetic inference of the genus Typha, including the following species: Spirodela polyrhiza (NC_015891.1, NC_017840.1), Butomus umbellatus (NC_051949.1, NC_021399.1), Zea mays NB (NC_001666.2, AY506529.1), Allium cepa (OR783242.1, NC_030100.1), Crocus sativus (NC_041460.1, OL804177.1), Zea perennis (NC_030300.1, NC_008331.1), Phoenix dactylifera (NC_013991.2, NC_016740.1), Oryza sativa Japonica (LC739565.1, BA000029.3), Oryza minuta (MT726938.1, NC_029816.1), and Arabidopsis thaliana (AP000423, JF729202.1).
Organelle genome assembly and annotation
The clean Illumina PE reads from whole-genome sequencing were first randomly selected using SeqKit 0.13.1 (Shen et al., 2016) to create datasets for mitogenome and plastome assembly. A combined strategy was performed to obtain accurate mitogenomes. The obtained Illumina reads were de novo assembled with using five distinct procedures in SPAdes 3.15.2 (Bankevich et al., 2012), wherein K-mer was then adjusted at five different values (51, 71, 91, 101, and 121) and obtained the assembled scaffolds. The coding sequence (CDS) of the Oryza sativa mitogenome (BA000029.3) was utilized as the query sequences to search against the draft scaffolds that were, visualized in Bandage 0.8.1 (Wick et al., 2015) to exclude non-mitogenomic scaffolds. Mitogenomic sequences from Illumina reads were eventually acquired after removing the fragments with abnormal depths as compared to general mitochondrial sequences (10 and more times lower, from the nuclear genome and 100 times and higher from the plastome). The mitochondrial sequences were used to select PacBio/ONT reads with the use of BLAST 2.11.0+ (Ye et al., 2006) with 80% identity, before which PacBio/ONT reads were self-corrected with the use of Nextdenovo 2.3.1 (https://github.com/Nextomics/NextDenovo). The corrected PacBio/ONT reads were de novo assembled using Flye 2.8.3 (Kolmogorov et al., 2019). The graph-based mitogenome was visualized by Bandage v0.8.1. For plastome assembly, SPAdes 3.15.2 was utilized for de novo assembly with five k-mer levels, as above. Bandage software (v.0.8.1) was employed to visualize the connections among contigs. Accurate annotations for the four organellar genomes were obtained using Geseq (https://chlorobox.mpimp-golm.mpg.de/geseq.html) with Oryza sativa (BA000029.3) and Typha latifolia (NC_013823.1) as references for the mitogenome and plastome, respectively. All annotations underwent manual verification and correction. The genome map was generated using OGDRAW1.3.1, (https://chlorobox.mpimpgolm.mpg.de/OGDraw.html).
Repeats detection and codon usage analysis
Dispersed repeats were identified using the REPuter online program (https://bibiserv.cebitec.uni-bielefeld.de/reputer) with the following parameters: hamming distance = three, minimal repeat size = 30 (90% sequence identity or greater), maximum computed repeats = 5,000, and an e-value cutoff = 1e-5 (Kurtz et al., 2001). Simple sequence repeats (SSRs) were detected using MISA (Beier et al., 2017), incorporating motif sizes ranging from one to six nucleotide units, with repeat lower thresholds set to eight, five, four, three, three, and three repeat units for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs, respectively. Additionally, we used the default parameter for Max. Length of sequence between two SSRs to register as compound SSR, set to 100 base pairs, allowing accurate identification of compound SSRs within our data. REPuter was utilized to identify long repeats across the chloroplast genome with default settings. The relative synonymous codon usage (RSCU) of the mitogenome was computed using DAMBE 5.2.73 (Xia and Xie, 2001).
Synteny and transfer fragment analysis
In order to identify transferred fragments between the plastome and mitogenome, BLAST 2.11.0+ was employed to search for homologous fragments with an e-value of 1e-5 and 80% identity cutoff, as described previously (Hong et al., 2021). Interspecies homologous regions were examined to demonstrate mitogenomic synteny between the two Typha species, with fragments shorter than 100 bp excluded from the analysis. The results were visualized using the Circos package within TBtools (Zhang et al., 2013).
Organellar phylogenetical inference
Both the plastome and the mitogenome of two Typha species and 10 related species were subjected to phylogenetic analysis. Considering all the available mitogenomic data in NCBI, the mitogenome and plastome of 10 species that represent various clades or branches along the tree of life, including Spirodela polyrhiza, Butomus umbellatus, Zea mays NB, Allium cepa, Crocus sativus, Zea perennis, Phoenix dactylifera, Oryza sativa Japonica, Oryza minuta (MT726938.1, NC_029816.1), and Arabidopsis thaliana as the outgroup. All shared coding sequences (CDS) were aligned in MEGA 7 (Kumar et al., 2016). The aligned CDSs, consisting of 24 for the mitogenome and 77 for the plastome, were concatenated using Geneious Prime (https://www.geneious.com). Gblocks v.0.91b was utilized to select the optimal multiple sequence alignment regions with default parameters (Castresana, 2000). A Maximum Likelihood (ML) phylogenetic inference was conducted in IQ-Tree 2.1 (Nguyen et al., 2015) using the best evolutionary models, ‘TVM+F+I+G4’ for plastomes and ‘GTR+F+G4’ for mitogenomes, selected based on Bayesian Information Criterion scores. Clade support was assessed with 1,000 bootstrap (BS) replicates. The newick format tree was visualized using iTOL6 (https://itol.embl.de/).
Results
General features of the organellar genomes from two Typha species
To assemble the organellar genomes of T. latifolia and T. domingensis, we employed a combined approach using 1 Gb of Illumina reads, 86 Gb of PacBio reads and 20 Gb of ONT reads per sample. The results showed that the plastomes of T. latifolia and T. domingensis have the typical quadripartite structures like most flowering plants, with genome sizes of 161,545 bp and 161,230 bp respectively (Figures 1A, B). Both the plastomes were comprised of four distinctive parts in which the large single copy (LSC-, 89,131 bp, 88,964 bp) and the small single copy (SSC-, 18,554 bp, 18,503 bp) regions were separated by two inverted repeats (IRs, 26,930 bp, 26,881 bp). The overall average (GC) content of the plastomes was (36.6%) in both species. We discovered a total of 150 distinct genes in T. latifolia, consisting of 96 protein-coding genes, 10 rRNA genes, and 44 tRNA genes. In the assembled plastome of T. domingensis, we detected 141 distinct genes, with 89 protein-coding genes, 8 rRNA genes, and 44 tRNA genes (Supplementary Tables S1, S2). Seventeen genes contained one intron (rpl2, ndhA, ndhB, rps12, rps16, rpl16, rrn23, rpoC1, petB, petD, atpF, trnG-GCC, trnL-UAA, trnV-UAC, trnA-UGC, trnI-GAU, trnK-UUU), and two genes (ycf3, clpP) had two introns. The gene copy number was largely conserved between the two species, although T. latifolia exhibited higher copy numbers for several genes such as trnI-GAU, rrn23S, whereas T. domingensis showed increased copy numbers for trnI-GAU and rps12. We compared the plastomes using a dot plot. The results showed that the two plastomes were highly collinear indicating a conserved genomic architecture (Supplementary Figure S1). In both T. latifolia and T. domingensis mitogenomes, a single circular molecule was resolved, with sizes of 395,136 bp and 395,140 bp, respectively, and a GC content of 46.7% for both (Figures 1C, D). The nucleotide composition of the whole mitogenomes showed only slight variations, A, 27.0%; T, 26.04%; C, 23.03%; and G, 23.04% in T. latifolia and A, 26.04%; T, 27.0%; C, 23.04%; and G, 23.03% in T. domingensis. A total of 59 unique mitochondrial genes were located in T. latifolia and T. domingensis, and the two species shared 59 genes including 39 protein-coding genes (PCGs), three rRNA genes, and 17 tRNA genes. There were eleven genes that contained introns, with the details shown in Table 1. Among the eleven genes containing introns, the distribution is as follows: five genes possess one intron each (ccmFC, rpl2, rps10, rps3, trnL-CAA), one gene contains two introns (cox2), and one gene contains three introns (nad4). Additionally, four genes exhibit four introns each (nad1, nad2, nad5, nad7) in both T. latifolia and T. domingensis mitogenome.
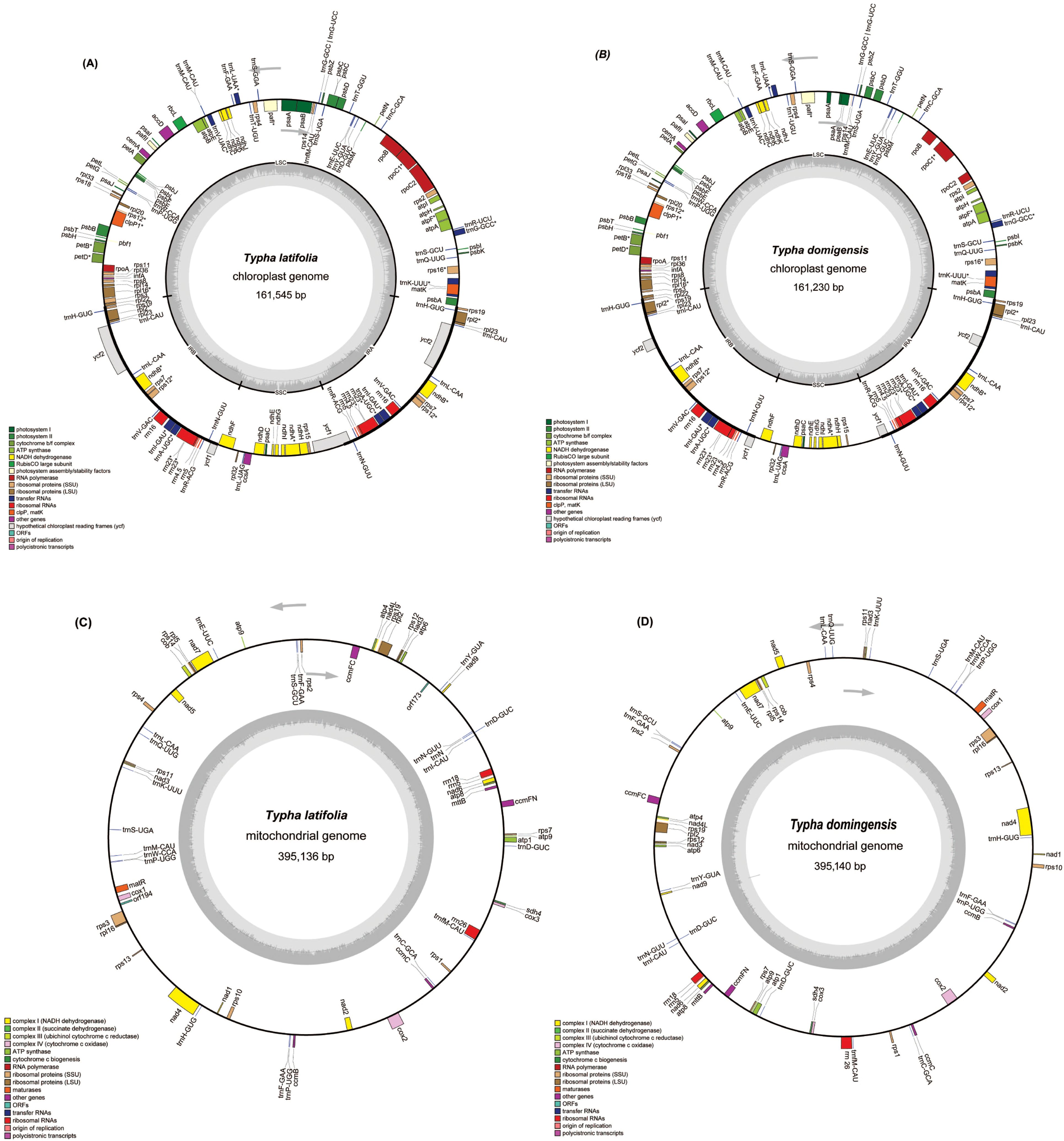
Figure 1. The circular maps of the organelle genomes of two Typha species. (A) The circular map of the plastome in T. latifolia. (B) The circular map of the plastome in T. domingensis. (C) The circular map of the mitogenome in T. latifolia. (D) The circular map of the mitogenome in T. domingensis. Genomic features transcribed clockwise and counter-clockwise are drawn on the inside and outside of the circle, respectively. Genes are color-coded based on their functional groups. GC content is represented on the inner circle by the dark gray plot.
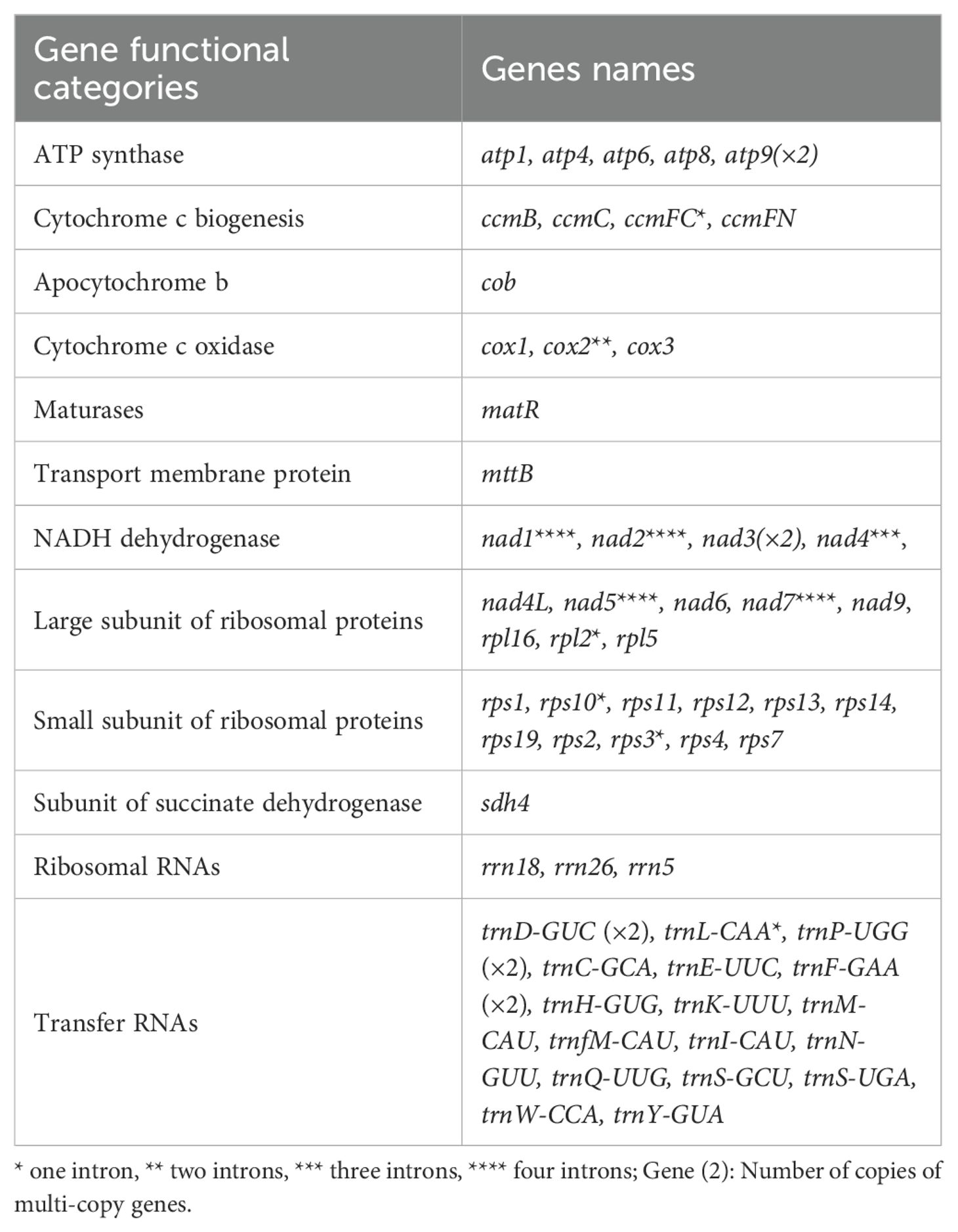
Table 1. Known functional genes in the two Typha mitogenomes.
Repeat analysis
To clarify repeat sequence diversity, different types of repetitive sequences in two Typha species were investigated. Within the plastomes, the predominant SSRs feature a single-nucleotide repeat unit, with A/T repeats being most abundant. The proportion of A/T repeat units comprises 68.68% in T. latifolia and 44.8% in T. domingensis of all identified SSR repeats. However, the distribution of SSRs of different types in the mitogenomes is uniform. In both species, there are a total of 180 mononucleotide SSRs, 28 dinucleotide SSRs, 20 trinucleotide SSRs, 51 tetranucleotide SSRs, 3 pentanucleotide SSRs, and 4 hexanucleotide SSRs. Most of SSRs in the mitogenomes feature a tetranucleotide repeat unit, accounting for 55.0% and 59.0% of all repeat numbers. Additionally, homopolymer SSR A/T were also identified as prevalent SSR types (Figure 2). A total of 58 tandem repeats were identified in T. latifolia and 48 in T. domingensis within the plastomes (Supplementary Tables S3, S5) while 7 tandem repeats were found in T. latifolia and 6 in T. domingensis within the mitogenomes (Supplementary Tables S4, S6). Dispersed repeats across the plastomes and mitogenomes were identified as two types, forward and palindromic matches. While forward and palindromic repeats were predominant, one reverse repeat was also identified (Supplementary Table S9). In the plastomes, the most prevalent and longest repeats are palindromic, with the longest fragment measuring 26,930 bp in T. latifolia and 26,882 bp in T. domingensis. Additionally, 75 and 63 dispersed repeats were identified in the plastomes of T. latifolia and T. domingensis, respectively (Supplementary Tables S8, S10).The most abundant and longest repeats in the mitogenomes are forward repeats, with the longest fragment measuring 686 bp in both genomes. These forward repeats constitute 57.9% and 56.7% of the total repeats in the mitogenome. T. latifolia and T. domingensis mitogenomes possessed 190 and 198 dispersed repeats respectively (Supplementary Tables S7, S9). While repeats exceeding 1000 bp were observed in the plastomes, no such fragments were found in the mitogenomes (Figure 3). Additionally, a total of 32 shared codons encoding 18 amino acids (one was a stop codon, UGA) showed that RSCU exceeded 1 across all two Typha spp. (Table 2).
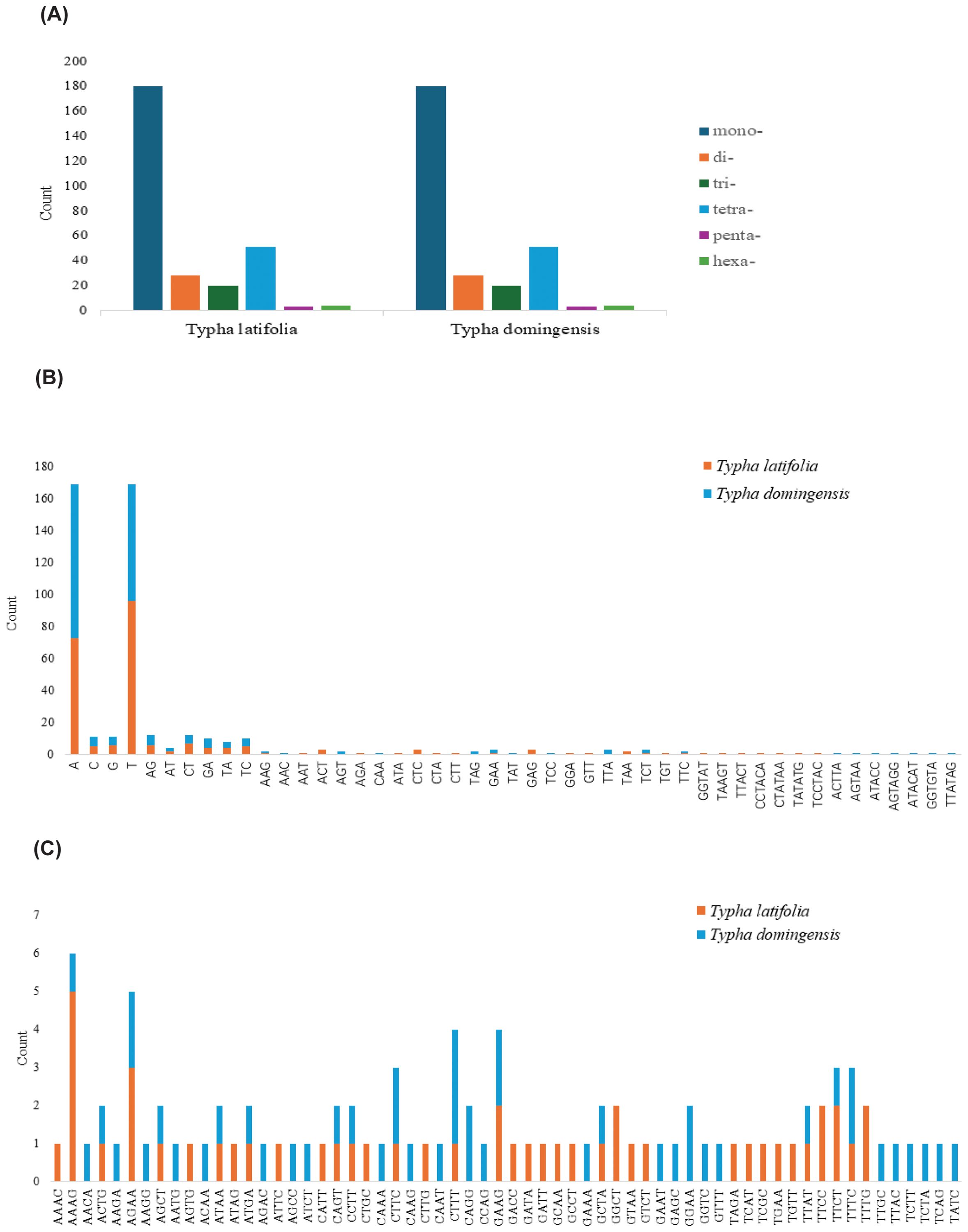
Figure 2. SSRs in two Typha species mitogenomes. (A) Count of six unit-sized SSRs. (B) Count of different unit components from mono- to hexa- except for tetra-nucleotide SSRs. (C) Count of different unit components of tetra-nucleotide SSRs.
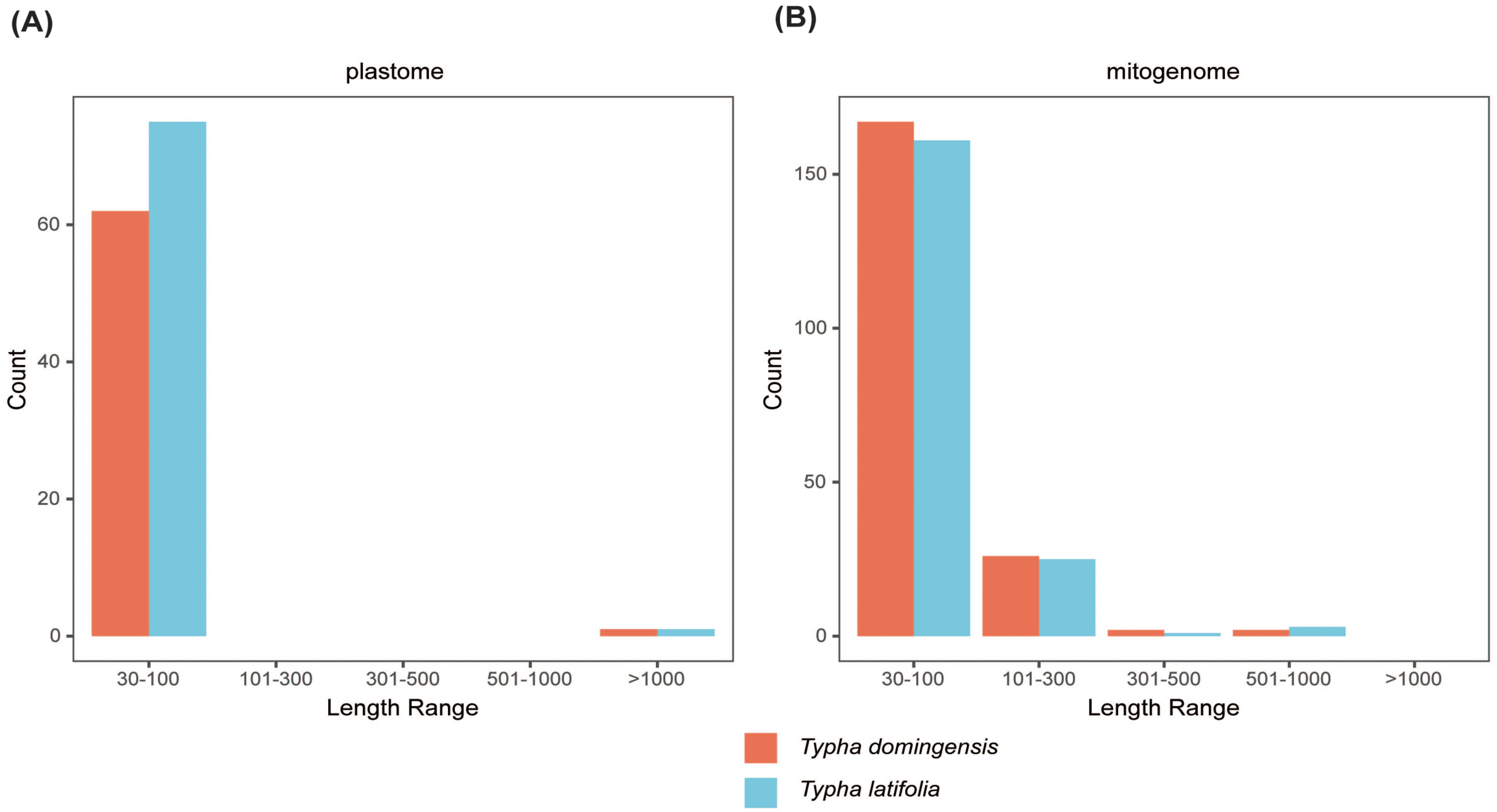
Figure 3. Length of dispersed repeats in two Typha species (A) plastomes and (B) mitochondrial genomes.
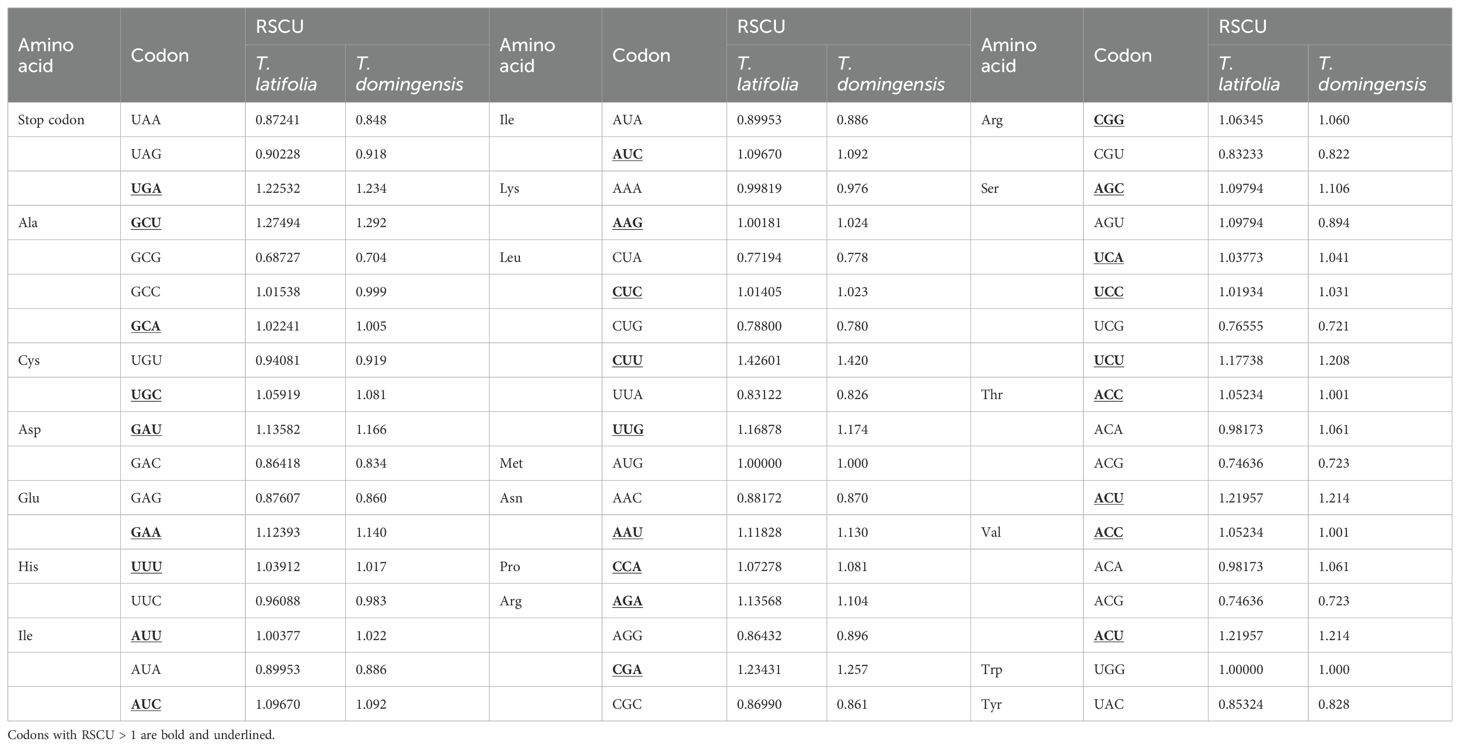
Table 2. RSCU of two Typha mitogenomes.
Synteny and homologous sequences between mitogenome and plastome
Analysis of sequence similarity between the mitogenomes and plastomes of T. latifolia and T. domingensis revealed that 86,496 bp and 73,949 bp in the respective mitogenomes likely originated from past transfer from the plastomes, accounting for 21.9% and 18.7% of the respective mitogenome length (Figure 4). 16 homologous fragments between mitogenome and plastome were detected in both Typha species, with the longest spanning 959 bp in T. latifolia and 976 bp in T. domingensis. Eight transferred DNA fragments into mitogenome were identified in T. latifolia and T. domingensis that were located in plastome IR region half the total transfer fragments. In T. latifolia the transferred fragments encompass thirteen plastome genes (rpl16, ycf2, rpl14, trnW-CCA, trnP-UGG, psbD, rps8, clpP1, trnN-GUU, rrn23, rpl16, ndhE, and trnH-GUG) and six mitochondrial genes (trnW-CCA, trnP-UGG, trnN-GUU, trnH-GUG, rrn26, and trnH-GUG). In T. domingensis, the homologous fragments include ten plastome genes (rps12, cemA, clpP1, rpl2, ndhI, ycfI, trnL-UAA, rpl20, psbC, psbA) and four mitochondrial genes (trnW-CCA, trnP-UGG, trnN-GUU, and trnH-GUG). The synteny analysis of the entire mitogenome in two Typha species revealed extensive interspecific large homologous regions, with two crosses observed. In addition, small homologous regions could be found across the entire mitogenome, which revealed high compositional and low structural conservation (Figure 5).
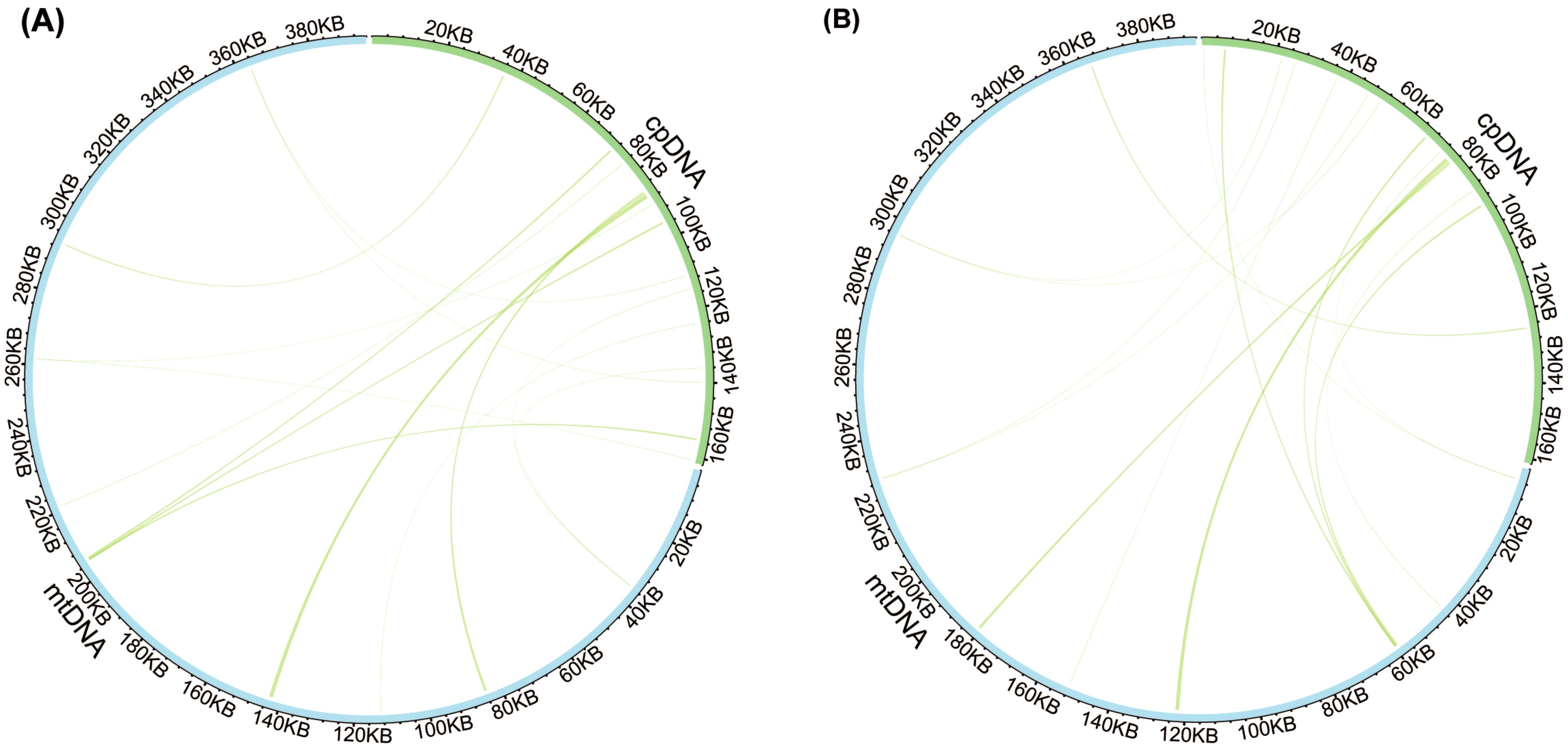
Figure 4. Comparison of the plastome and mitogenome of two Typha species. (A) Comparison of the plastome and mitogenome of T. latifolia. (B) Comparison of the plastome and mitogenome of T. domingensis. The blue and green outer arcs represent the mitogenome (mtDNA) and plastome (cpDNA), respectively, and the inner arcs show the homologous DNA fragments. The scale is shown on the outer arcs, with intervals of 20 kb.
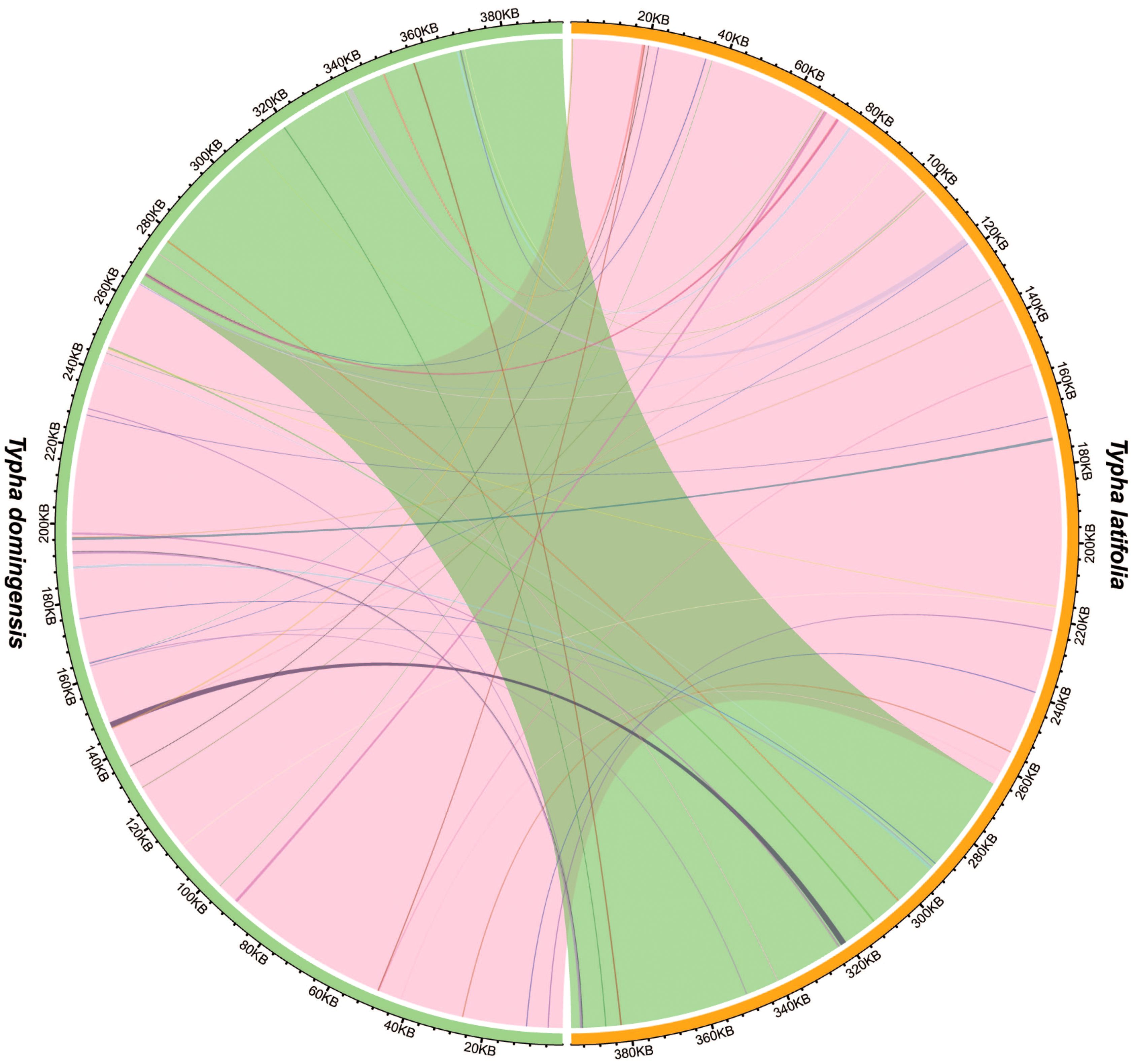
Figure 5. Interspecific mitogenomic synteny indicated by homologous regions. The orange and green outer arcs represent T. latifolia and T. domingensis, respectively, and the inner arcs show the homologous DNA fragments. The scale is shown on the outer arcs, with intervals of 20 kb.
Phylogenetic analysis
To investigate the evolutionary patterns of organellar genomes in two Typha species, we performed a phylogenetic analysis using the organelle genomes of these species along with 10 related species (Arabidopsis thaliana was chosen as outgroup for comparison). Our analysis involved the nucleotide sequences of 77 common genes (accD, atpA, atpB, atpE, atpF, atpH, atpI, ccsA, cemA, clpP1, infA, matK, ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhK, pafI, petA, petB, petD, petG, petL, petN, psaA, psaB, psaC, psaI, psaJ, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbT, psbZ, rbcl, rpl2, rpl14, rpl16, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36, rpoA, rpoB, rpoC1, rpoC2, rps2, rp23, rps4, rps7, rps8, rps11, rps12, rps14, rps15, rps16, rps18, rps19, ycfI, and ycfII) for the plastome-based phylogenetic analysis and 24 common genes (atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, cob, cox1, cox2, cox3, nad1, nad2, nad4, nad4L, nad5, nad6, nad7, nad9, rpl6, rps1, rps3, rps4, rps12) for the mitogenome-based analysis. Notably, the plastome tree exhibited full support for all nodes, except for three nodes with a bootstrap value of 93, 98, and 98. The mitogenome tree showed one node with weak support with a bootstrap value of 65, all others had strong bootstrap support. Despite differences in clade support the topology was identical between the datasets and the two Typha species formed a monophyletic clade (Figure 6). Based on the data of mitogenome and plastome, the genus Typha was closer to the Poaceae clade, which includes maize and rice, consistent with previously published reports (Guisinger et al., 2010).
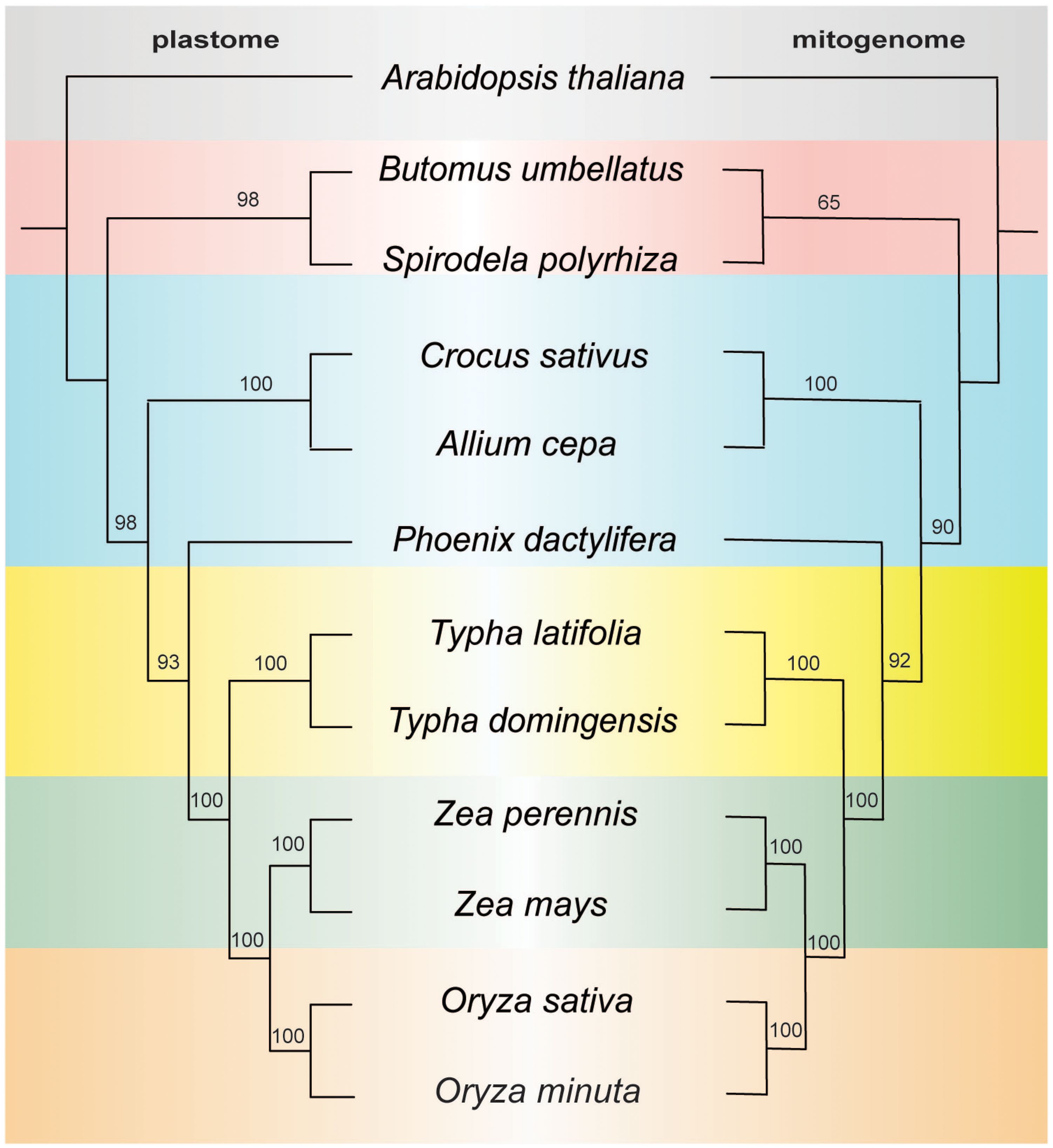
Figure 6. ML tree based on 77 shared plastid genes (left) and 24 shared mitochondrial genes (right). The numbers near nodes indicated the bootstrap value.
Discussion
Typha is an ecologically crucial lineage of plants in marshes and wetlands globally, contributing significantly to bioremediation, ecological functioning, and serving as a source of biofuel materials (Bonanno and Cirelli, 2017). The assembly and annotation of the Typha organellar genomes, is expected to serve as a significant tool in wetland management (Bansal et al., 2019). Here, we present the first published organellar genome assemblies from the Typhaceae family. In the present study, the mitogenomes of T. latifolia (395,136bp) and T. domingensis (395,140bp) are more than twice the size of the respective plastomes (161,545bp and 161,230bp). In plants, the mitogenome is nearly always considerably larger than the plastome (Ye Ning et al., 2017; Cheng et al., 2021). It is also common for some part of the mitogenome to be homologous to the plastome (Hazkani-Covo et al., 2010; Smith, 2011), which are known as so-called intracellular gene transfer (IGT). Persistent IGTs continuously reshape and increase the complexity of plant mitogenomes. For example, these homologous regions within mitogenomes can be hotspots for recombination, possibly generating chimeric open reading frame (ROFs) with new functions (Wang et al., 2024b). Mitochondrial plastid DNAs (MTPTs) occurred at least 300 MYA and been described for numerous species. However, the model of MTPTs was not clear. Early previously reported that cpDNA transfers were randomly and likely occurred at any positions of the cpDNAs (Wang et al., 2007), while transfer hot spots and cold spots chloroplast genome were exist based on the frequency of genes (Wang et al., 2018). We found 16 fragments in the mitogenome of two Typha species, representing approximately 6.6% of the mitogenome, and there are eight transferred DNA fragments were identified that were located in T. latifolia and T. domingensis plastome IR region half the total transfer fragments. This finding was consistent with previously reported that IR region was highest coverage of MTPTs (Nhat Nam et al., 2024). In comparing the genome size between organelles, the difference between plastomes was greater than between mitogenomes. Specifically, the T. latifolia plastome was 315 bp larger than that of T. domingensis primarily due to the expansion and contraction of the single-copy (SC) and inverted repeat (IR) boundaries. In contrast, the mitogenome of T. latifolia was 4 bp smaller than that of T. domingensis. Similar variations in plastome lengths due to the expansion and contraction of SC/IR boundaries have been observed in other plant species, as reported by (Jansen and Ruhlman, 2012; Ruhlman and Jansen, 2014; Mower and Vickrey, 2018).
Plant mitogenomes are commonly described as circular structures, similar to the circular chromosomes present in bacteria and animal mitochondria. However, numerous studies have demonstrated that the basic circular model of genome structure, which is applicable to the majority of animal species, is inadequate for comprehending plant mitochondria, which can possess multiple circular replicons, branched, linear, or mixed forms of genomic structure. For example, the Silene noctiflora mitogenome is arranged into multiple circular chromosomes; thus, one ring cannot rule them all based on mapping to a circular reference (Sloan, 2013; Wu et al., 2015). Both T. latifolia and T. domingensis mitogenomes were assembled into a single circular structure which may explain the similarity in size between the two species examined here. However, future studies should examine more species and investigate the possibility of tissue-specific mitogenome variation, as has been observed in some plants. Here, T. domingensis demonstrates higher copy numbers for trnI-GAU and rps12 in comparison to T. latifolia. The two Typha mitogenomes shared 59 unique genes, including 39 PCGs, three rRNA genes, and 17 tRNA genes, which are highly similar to those of other species such as Oryza sativa (Notsu et al., 2002) and Arabidopsis thaliana (Unseld et al., 1997). Since the mitogenome is in control of oxidative phosphorylation, the fundamental energy production required for living activities, the conservation of gene content throughout several plant lineages suggests that the mitogenome has a stable function (Mackenzie and McIntosh, 1999). A closer relationship between T. latifolia and T. domingensis within Typha was supported by the phylogenetic analysis that followed, which was based on organellar genes. This was consistent with the synteny pattern, which showed that there were more bulks of synteny between these two species, indicating a more similar sequence component and arrangement.
Phylogenomic analysis using our assembled mitogenomes and plastomes showed congruent results. This congruence between plastome and mitogenome topologies has also been found in other plant taxa, such as Saposhnikovia and Moraceae, where both organellar genomes exhibit similar evolutionary histories (Ni et al., 2022) (Lai et al., 2022). However, it is noteworthy that the plastome tree showed higher support for certain clades compared to the mitogenome tree, suggesting that while the overall topologies are congruent, there may be differences in the rates of evolution between organelles. Plants mitogenomes exhibit extensive variation in size and structure, ranging from approximately 66 kb in Viscum scurruloideum to about 12 Mb in Larix sibirica (Wang et al., 2024b). Complex mitogenomes contain a large number of repetitive sequences, as seen in the mitochondrial genome of Panax notoginseng, where 64 pairs of recombination-mediated repeats form a multi-molecular structure comprising a “master circle” that interconverts with numerous “subgenomic circles” (Yang et al., 2023). The types of repetitive sequences found in plant mitochondrial genomes include inverted repeats, tandem repeats, non-tandem repeats, and large repeats (Wynn and Christensen, 2019). These repetitive sequences are involved in genome replication, recombination, insertion, and deletion, playing a crucial role in the evolution of mitochondrial genome size and structure in plants (Wang et al., 2024a) Here, T. latifolia and T. domingensis mitogenomes were assembled into circular structure, with size of 395,136 bp and 395,140 bp. No long repetitive sequences greater than 1,000 bp were detected in the two mitogenomes, which may explain why these genomes were assembled into simple circular structures with a small size.
Conclusions
The mitogenomes of two Typha species were described for the first time in this work, and their plastomes were constructed using the same sequencing data set. Here, T. latifolia and T. domingensis mitogenomes were assembled into circular structure, with size of 395,136 bp and 395,140 bp. 39 protein-coding genes (PCGs) were annotated in the each Typha species mitogenome. Congruent trees with 10 related species were displayed by phylogenomic analysis using the assembled plastome and mitogenome. Based on the data of mitogenome and plastome, the genus Typha was closer to the Poaceae clade, which includes maize and rice. Through sequence comparisons between the mitogenome and plastome, we were able to identify twelve mitochondrial DNA fragments that were similar to those found in the plastome. This study provides valuable information to understand the coordinated evolution of the plastome and the mitogenomes of plant species belonging to the family Typhaceae and monocots in general.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
TS: Conceptualization, Data curation, Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing, Visualization, Methodology, Resources. JK: Conceptualization, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. LN: Writing – review & editing, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Validation, Visualization. JW: Writing – review & editing, Conceptualization, Formal analysis, Methodology, Resources, Software, Validation, Visualization. DP: Conceptualization, Investigation, Methodology, Project administration, Software, Validation, Writing – review & editing. LT: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing, Investigation, Resources. ZW: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing, Validation.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. the Guangdong Pearl River Talent Program (grants 2021QN02N792), and the Chinese Academy of Agricultural Sciences Elite Youth Program (grant 110243160001007).
Acknowledgments
The authors gratefully acknowledge helpful comments from reviewers on earlier versions of this manuscript. We sincerely thank members of Wu Lab for their help in preparing this paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1484531/full#supplementary-material
References
Angiosperm Phylogeny, G. (2009). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Botanical J. Linn. Soc. 161, 105–121. doi: 10.1111/j.1095-8339.2009.00996.x
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bansal, S., Lishawa, S. C., Newman, S., Tangen, B. A., Wilcox, D., Albert, D., et al. (2019). Typha (cattail) invasion in North American wetlands: biology, regional problems, impacts, ecosystem services, and management. Wetlands 39, 645–684. doi: 10.1007/s13157-019-01174-7
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bonanno, G., Cirelli, G. L. (2017). Comparative analysis of element concentrations and translocation in three wetland congener plants: Typha domingensis, Typha latifolia and Typha angustifolia. Ecotoxicology Environ. Saf. 143, 92–101. doi: 10.1016/j.ecoenv.2017.05.021
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Cheng, Y., He, X., Priyadarshani, S., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genomics 22, 1–15. doi: 10.1186/s12864-021-07490-9
Christenhusz, M. J. M., Byng, J. W. (2016). The number of known plants species in the world and its annual increase. Phytotaxa 261, 201–217. doi: 10.11646/phytotaxa.261.3.1
Drouin, G., Daoud, H., Xia, J. (2008). Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 49, 827–831. doi: 10.1016/j.ympev.2008.09.009
Grace, J. B., Harrison, J. S. (1986). The biology of Canadian weeds.: 73. Typha latifolia L., Typha angustifolia L. and Typha xglauca Godr. Can. J. Plant Sci. 66, 361–379. doi: 10.4141/cjps86-051
Guisinger, M. M., Chumley, T. W., Kuehl, J. V., Boore, J. L., Jansen, R. K. (2010). Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in Poaceae. J. Mol. Evol. 70, 149–166. doi: 10.1007/s00239-009-9317-3
Hazkani-Covo, E., Zeller, R. M., Martin, W. (2010). Molecular poltergeists: Mitochondrial DNA copies (numts) in sequenced nuclear genomes. PloS Genet. 6, e1000834. doi: 10.1371/journal.pgen.1000834
Hong, Z., Liao, X., Ye, Y., Zhang, N., Yang, Z., Zhu, W., et al. (2021). A complete mitochondrial genome for fragrant Chinese rosewood (Dalbergia odorifera, Fabaceae) with comparative analyses of genome structure and intergenomic sequence transfers. BMC Genomics 22, 1–13. doi: 10.1186/s12864-021-07967-7
Ii, A. P. G. (2003). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Botanical J. Linn. Soc. 141, 399–436. doi: 10.1046/j.1095-8339.2003.t01-1-00158.x
Jansen, R. K., Ruhlman, T. A. (2012). Plastid genomes of seed plants. Genomics chloroplasts mitochondria 35, 103–126. doi: 10.1007/978-94-007-2920-9_5
Johnston, I. G. (2019). Tension and resolution: dynamic, evolving populations of organelle genomes within plant cells. Mol. Plant 12, 764–783. doi: 10.1016/j.molp.2018.11.002
Kolmogorov, M., Yuan, J., Lin, Y., Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kuehn, M. M., White, B. N. (1999). Morphological analysis of genetically identified cattails Typha latifolia, Typha angustifolia, and Typha ×glauca. Can. J. Bot. 77, 906–912. doi: 10.1139/b99-037
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lai, C., Wang, J., Kan, S., Zhang, S., Li, P., Reeve, W. G., et al. (2022). Comparative analysis of mitochondrial genomes of Broussonetia spp.(Moraceae) reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. Front. Plant Sci. 13, 1052151. doi: 10.3389/fpls.2022.1052151
Liao, Y., Zhao, S., Zhang, W., Zhao, P., Lu, B., Moody, M. L., et al. (2023). Chromosome-level genome and high nitrogen stress response of the widespread and ecologically important wetland plant Typha angustifolia. Front. Plant Sci. 14, 1138498. doi: 10.3389/fpls.2023.1138498
Liu, Z. D., Zhou, X. L., Ma, H. Y., Tian, Y. Q., Shen, S. K. (2020). Characterization of the complete chloroplast genome sequence of wetland macrophyte Typha orientalis (Typhaceae). Mitochondrial DNA Part B 5, 136–137. doi: 10.1080/23802359.2019.1698348
Mackenzie, S., McIntosh, L. (1999). Higher plant mitochondria. The Plant Cell. 11, 4, 571–585. doi: 10.1105/tpc.11.4.571
Mahapatra, K., Banerjee, S., De, S., Mitra, M., Roy, P., Roy, S. (2021). An insight into the mechanism of plant organelle genome maintenance and implications of organelle genome in crop improvement: An update. Front. Cell Dev. Biol. 9, 671698. doi: 10.3389/fcell.2021.671698
Maréchal, A., Brisson, N. (2010). Recombination and the maintenance of plant organelle genome stability. New Phytol. 186, 299–317. doi: 10.1111/j.1469-8137.2010.03195.x
McNaughton, S. J. (1966). Ecotype function in the Typha community-type. Ecol. Monogr. 36, 298–325. doi: 10.2307/1942372
Mower, J. P., Vickrey, T. L. (2018). Structural diversity among plastid genomes of land plants. Adv. botanical Res. 85, 263–292. doi: 10.1016/bs.abr.2017.11.013
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nhat Nam, N., Pham Anh Thi, N., Do, H. D. K. (2024). New insights into the diversity of mitochondrial plastid DNA. Genome Biol. Evol. 16, evae184. doi: 10.1093/gbe/evae184
Ni, Y., Li, J., Chen, H., Yue, J., Chen, P., Liu, C. (2022). Comparative analysis of the chloroplast and mitochondrial genomes of Saposhnikovia divaricata revealed the possible transfer of plastome repeat regions into the mitogenome. BMC Genomics 23, 570. doi: 10.1186/s12864-022-08821-0
Notsu, Y., Masood, S., Nishikawa, T., Kubo, N., Akiduki, G., Nakazono, M., et al. (2002). The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genomics 268, 434–445. doi: 10.1007/s00438-002-0767-1
Pandey, A., Verma, R. K. (2018). Taxonomical and pharmacological status of Typha: A Review. Ann. Plant Sci. 7, 2101–2106. doi: 10.21746/aps.2018.7.3.2
Ruhlman, T. A., Jansen, R. K. (2014). The plastid genomes of flowering plants. Chloroplast biotechnology: Methods Protoc. 1132, 3–38. doi: 10.1007/978-1-62703-995-6_1
Saha, S. (1968). The genus Typha in India—its distribution and uses. Bull. Bot. Soc Bengal 22, 11–18.
Shen, W., Le, S., Li, Y., Hu, F. (2016). SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PloS One 11, e0163962. doi: 10.1371/journal.pone.0163962
Sloan, D. B. (2013). One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’model of plant mitochondrial DNA structure. New Phytol. 200, 978–985. doi: 10.1111/nph.2013.200.issue-4
Smith, S. G. (1987). Typha: Its taxonomy and the ecological significance of hybrids. Archiv für Hydrobiologie 27, 129–138.
Smith, D. R. (2011). Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol. Evol. 3, 743–748. doi: 10.1093/gbe/evr068
Tornberg, T., Bendix, M., Brix, H. (1994). Internal gas transport in Typha latifolia L. and Typha angustifolia L. 2. Convective throughflow pathways and ecological significance. Aquat. Bot. 49, 91–105. doi: 10.1016/0304-3770(94)90031-0
Unseld, M., Marienfeld, J. R., Brandt, P., Brennicke, A. (1997). The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 15, 57–61. doi: 10.1038/ng0197-57
Vivian, L. M., Godfree, R. C., Colloff, M. J., Mayence, C. E., Marshall, D. J. (2014). Wetland plant growth under contrasting water regimes associated with river regulation and drought: implications for environmental water management. Plant Ecol. 215, 997–1011. doi: 10.1007/s11258-014-0357-4
Volkova, P. A., Bobrov, A. A. (2022). Easier than it looks: Notes on the taxonomy of Typha L.(Typhaceae) in East Europe. Aquat. Bot. 176, 103453. doi: 10.1016/j.aquabot.2021.103453
Wang, X. C., Chen, H., Yang, D., Liu, C. (2018). Diversity of mitochondrial plastid DNAs (MTPTs) in seed plants. Mitochondrial DNA Part A 29, 635–642. doi: 10.1080/24701394.2017.1334772
Wang, J., Kan, S., Liao, X., Zhou, J., Tembrock, L. R., Daniell, H., et al. (2024b). Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 29, 754-769. doi: 10.1016/j.tplants.2023.12.014
Wang, H., Wu, Z., Li, T., Zhao, J. (2024a). Highly active repeat-mediated recombination in the mitogenome of the aquatic grass Hygroryza aristata. BMC Plant Biol. 24, 644. doi: 10.1186/s12870-024-05331-x
Wang, D., Wu, Y. W., Shih, A. C. C., Wu, C. S., Wang, Y. N., Chaw, S. M. (2007). Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol. Biol. Evol. 24, 2040–2048. doi: 10.1093/molbev/msm133
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Widanagama, S. D., Freeland, J. R., Xu, X., Shafer, A. B. (2022). Genome assembly, annotation, and comparative analysis of the cattail Typha latifolia. G3 12, jkab401. doi: 10.1093/g3journal/jkab401
Wolfsberg, T. G., Schafer, S., Tatusov, R. L., Tatusova, T. A. (2001). Organelle genome resources at NCBI. Trends Biochem. Sci. 26, 199–203. doi: 10.1016/S0968-0004(00)01773-4
Wu, Z., Cuthbert, J. M., Taylor, D. R., Sloan, D. B. (2015). The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc. Natl. Acad. Sci. 112, 10185–10191. doi: 10.1073/pnas.1421397112
Wynn, E. L., Christensen, A. C. (2019). Repeats of unusual size in plant mitochondrial genomes: Identification, incidence and evolution. G3-Genes Genomes Genet. 9, 549–559. doi: 10.1534/g3.118.200948
Xia, X., Xie, Z. (2001). DAMBE: Software package for data analysis in molecular biology and evolution. J. heredity 92, 371–373. doi: 10.1093/jhered/92.4.371
Yang, H., Ni, Y., Zhang, X., Li, J., Chen, H., Liu, C. (2023). The mitochondrial genomes of Panax notoginseng reveal recombination mediated by repeats associated with DNA replication. Int. J. Biol. Macromolecules 252, 126359. doi: 10.1016/j.ijbiomac.2023.126359
Ye, J., McGinnis, S., Madden, T. L. (2006). BLAST: Improvements for better sequence analysis. Nucleic Acids Res. 34, W6–W9. doi: 10.1093/nar/gkl164
Ye Ning, Y. N., Wang XueLin, W. X., Li Juan, L. J., Bi ChangWei, B. C., Xu YiQing, X. Y., Wu DongYang, W. D., et al. (2017). Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. 5, e3148. doi: 10.7717/peerj.3148
Zhang, H., Meltzer, P., Davis, S. (2013). RCircos: An R package for Circos 2D track plots. BMC Bioinf. 14, 1–5. doi: 10.1186/1471-2105-14-244
Keywords: organellar genomes, phylogeny, comparative analysis, Typha, synteny
Citation: Soe T, Kong J, Nie L, Wang J, Peng D, Tembrock LR and Wu Z (2024) Organelle genome assembly, annotation, and comparative analyses of Typha latifolia and T. domingensis: two keystone species for wetlands worldwide. Front. Plant Sci. 15:1484531. doi: 10.3389/fpls.2024.1484531
Received: 22 August 2024; Accepted: 18 November 2024;
Published: 05 December 2024.
Edited by:
Linchun Shi, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Xinwei Xu, Wuhan University, ChinaDaniele De Luca, Suor Orsola Benincasa University, Italy
Copyright © 2024 Soe, Kong, Nie, Wang, Peng, Tembrock and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiqiang Wu, d3V6aGlxaWFuZ0BjYWFzLmNu; Luke R. Tembrock, bHVrZS50ZW1icm9ja0Bjb2xvc3RhdGUuZWR1
†These authors have contributed equally to this work