 Mengru Dou1,2,3
Mengru Dou1,2,3 Yuhang Li1,2,3Yu Hao1,2,3Kangzhuang Zhang1,2,3Xiao Yin1,2,3
Yuhang Li1,2,3Yu Hao1,2,3Kangzhuang Zhang1,2,3Xiao Yin1,2,3 Zinuo Feng1,2,3Xi Xu1,2,3Qi Zhang1,2,3Wenwu Bao1,2Xi Chen1,2,3
Zinuo Feng1,2,3Xi Xu1,2,3Qi Zhang1,2,3Wenwu Bao1,2Xi Chen1,2,3 Guotian Liu1,2,3
Guotian Liu1,2,3 Yuejin Wang1,2,3
Yuejin Wang1,2,3 Ling Tian4*
Ling Tian4* Yan Xu1,2,3*
Yan Xu1,2,3*- 1State Key Laboratory of Crop Stress Biology in Arid Areas, Northwest Agriculture & Forestry University, Yangling, Shaanxi, China
- 2College of Horticulture, Northwest A&F University, Yangling, Shaanxi, China
- 3Key Laboratory of Horticultural Plant Biology and Germplasm Innovation in Northwest China, Ministry of Agriculture, Yangling, Shaanxi, China
- 4School of Management, Shenzhen Polytechnic University, Shenzhen, Guangdong, China
Introduction: Grape is of high economic value. Colletotrichum viniferum, a pathogen causing grape ripe rot and leaf spot, threatens grape production and quality.
Methods: This study investigates the interplay between C. viniferum by Cytological study and transcriptome sequencing.
Results: Different grapevine germplasms, V. vinifera cv. Thompson Seedless (TS), V. labrusca accession Beaumont (B) and V. piasezkii Liuba-8 (LB-8) were classified as highly sensitive, moderate resistant and resistant to C. viniferum, respectively. Cytological study analysis reveals distinct differences between susceptible and resistant grapes post-inoculation, including faster pathogen development, longer germination tubes, normal appressoria of C. viniferum and absence of white secretions in the susceptible host grapevine. To understand the pathogenic mechanisms of C. viniferum, transcriptome sequencing was performed on the susceptible grapevine “TS” identifying 236 differentially expressed C. viniferum genes. These included 56 effectors, 36 carbohydrate genes, 5 P450 genes, and 10 genes involved in secondary metabolism. Fungal effectors are known as pivotal pathogenic factors that modulate plant immunity and affect disease development. Agrobacterium-mediated transient transformation in Nicotiana benthamiana screened 10 effectors (CvA13877, CvA01508, CvA05621, CvA00229, CvA07043, CvA05569, CvA12648, CvA02698, CvA14071 and CvA10999) that inhibited INF1 (infestans 1, P. infestans PAMP elicitor) induced cell death and 2 effectors (CvA02641 and CvA11478) that induced cell death. Additionally, transcriptome analysis of “TS” in response to C. viniferum identified differentially expressed grape genes related to plant hormone signaling (TGA, PR1, ETR, and ERF1/2), resveratrol biosynthesis genes (STS), phenylpropanoid biosynthesis genes (PAL and COMT), photosynthetic antenna proteins (Lhca and Lhcb), transcription factors (WRKY, NAC, MYB, ERF, GATA, bHLH and SBP), ROS (reactive oxygen species) clearance genes (CAT, GSH, POD and SOD), and disease-related genes (LRR, RPS2 and GST).
Discussion: This study highlights the potential functional diversity of C. viniferum effectors. Our findings lay a foundation for further research of infection mechanisms in Colletotrichum and identification of disease response targets in grape.
1 Introduction
Grape (Vitis vinifera L.), an ancient and economically significant, is consumed worldwide (Dong et al., 2023). However, grapes are susceptible to various pathogens, including Colletotrichum viniferum, particularly under high temperatures and humidity. This pathogen affects grape cultivars such as Pione, Kyoho, Ives, and Merlot (Echeverrigaray et al., 2020; Yokosawa et al., 2020), resulting in significant economic losses of approximately 67% in the Mid-Atlantic region of the United States and around 37% in Northeast China (Cosseboom and Hu, 2022; Ji et al., 2021). C. viniferum infects grapevine leaves, shoots, stems, and berries, causing grape ripe rot and leaf spot, with particularly severe damage to berries (Pan et al., 2016).
Colletotrichum species are identified and categorized in China and globally according to morphological and molecular features. Pathogenic species identified include C. acutatum, C. aenigma, C. capsici, C. fruticola, C. gloeosporioides, and C. viniferum, among others (Lei et al., 2016; Oo and Oh, 2017; Echeverrigaray et al., 2020). With the continuous development of sequencing techniques, high-throughput sequencing technology has been widely adopted. Several Colletotrichum species from various host plants have had their complete genomes sequenced, including C. lindemuthianum (Gutiérrez et al., 2016), C. acutatum (Han et al., 2016), C. graminicola (Buiate et al., 2017), C. sublineola (Buiate et al., 2017), C. asianum (Meng et al., 2019), C. higginsianum (Ayako et al., 2019), and C. fioriniae (Xu et al., 2024). Fungal genome sequencing has greatly enhanced our understanding of the interactions between hosts and pathogens. However, the genomic sequencing of C. viniferum CvYL2a on grape has only recently been published (Dou et al., 2022).
Different grape varieties exhibit varying resistance levels to Colletotrichum. Researchers have classified the disease resistance abilities of various grape cultivars and their hybrids (Shiraishi et al., 2007). A comprehensive study of grape ripe rot resistance using natural field assessment, field inoculation, and indoor fruit in vitro analysis highlighted the exceptional resistance of Chinese wild grapes as valuable disease-resistant germplasm resources (He and Ren, 1990). Subsequent research revealed that cultivars such as Emerald Seedless, Tano Red, and Rem46-77 (Aestivalis GVIT 0970) are susceptible to grape ripe rot, while Agawan, Huangguan, and Xiangfei are resistant (Kim and Oh, 2019). The diversity in resistance levels provides valuable germplasm resources for investigating grape ripe rot resistance mechanisms, crucial for breeding and evaluating new resistant grapevine cultivars, and developing innovative disease control methods. Several investigations have analyzed the infection processes of various Colletotrichum spp. isolates in host fruits, such as apple infected by C. fructicola, Guava infected by C. gloeosporioides, blueberry, strawberry, and almond affected by C. acutatum (Arroyo et al., 2005; Diéguez-Uribeondo et al., 2005; Wharton and Schilder, 2008; Moraes et al., 2013; Shang et al., 2020). However, limited research has focused on grape pathogenesis and the histological differences between susceptible and resistant varieties. Currently, control of Colletotrichum spp. mainly relies on fungicide use, which negatively impacts soil, food safety, and the environment. Therefore, it’s critical to develop more effective and environmentally friendly disease management strategies by studying potential plant-pathogen interactions with a more profound comprehensive approach.
Transcriptome sequencing is the most straightforward way to investigating gene expression levels, with numerous studies exploring transcriptome sequencing in host-pathogen interactions involving Colletotrichum species. Transcriptome analysis of the host included strawberry infected by C. siamense, sugarcane infected by C. falcatum, and Asian ginseng (Panax ginseng) infected by C. panacicola (Nandakumar et al., 2021; Zheng et al., 2022; Xia et al., 2023). Similarly, transcriptome analyses of Colletotrichum spp. have explored pathogenicity mechanisms in different host-pathogen interactions, such as C. camelliae on tea, C. falcatum on sugarcane, and C. fructicola on apple (Liang et al., 2018; Prasanth et al., 2022; He et al., 2023). However, real-time integration of transcriptomic data from both hosts and Colletotrichum spp. during infection remains limited. While several studies have investigated grapevine transcriptomes in response to Colletotrichum spp. infection, the transcriptomic dynamics of Colletotrichum itself are missing (Lei et al., 2022; Shen et al., 2022; Yu et al., 2023).
Colletotrichum spp. seriously threatens the yield and quality of grapes, especially affecting high-quality grape varieties with poor resistance. Fortunately, most wild grape germplasm resources in China are resistant to this fungus. Through histochemical staining and ultrastructural observation, V. vinifera cv. Thompson Seedless (TS), V. labrusca accession Beaumont (B) and V. piasezkii Liuba-8 (LB-8) were identified as highly sensitive, moderately resistant and resistant to C. viniferum, respectively. The transient transformation assays in Nicotiana benthamiana showed that Colletotrichum spp. effectors CoNIS1, Cte1, Cg2LysM and CgCFEM1 inhibited plant immune response, CtNUDIX and ChCEC3 induced cell death similar to HR response (Bhadauria et al., 2013; Tsushima et al., 2021; Zhao et al., 2023; Feng et al., 2024; Yuan et al., 2024). Effectors play different roles during infection. Currently, there are no studies about C. viniferum effector on grape, and the infection mechanism remains unclear. To uncover the infection processes of C. viniferum, particularly the roles of effectors during infection and the responses of grapevine to C. viniferum, we conducted a study focusing on pathogenesis, selecting the susceptible grapes “TS” for transcriptome analysis. Our aim was to screen C. viniferum pathogenic genes by transcriptome sequencing, leading to the identification of 85 effectors. These effectors were subsequently screened on Nicotiana benthamiana to examine their roles in the infection process. Simultaneously, we analyzed the grapevines’ responses to C. viniferum, providing insights into the interaction between C. viniferum and grapes and shedding light on its pathogenic mechanism.
2 Materials and methods
2.1 Fungal strain, plant materials, and inoculation
The C. viniferum strain CvYL2a, previously obtained and purified in our research facility, was cultured on potato dextrose agar (PDA) under dark conditions at 25°C. C. viniferum conidia were induced by transferring mycelia from PDA to a Carboxymethylcellulose (CMC) sodium fluid medium and incubating them at 150 rpm and 25°C for 3-5 days. Conidia were separated from mycelia by filtering with two-layer sterilization gauze and collected after centrifugation at 5000 rpm for 10 min. After two washes with sterile distilled water (SDW), the conidia were standardized to a 5 × 106 conidia/mL concentration, as determined using a hemocytometer and a light microscope.
For conidia inoculation, undamaged fully expanded leaves were collected from the apex of “TS”, “B” and “LB-8” grown in the grapevine germplasm resource vineyard at Northwest A&F University in Yangling, Shaanxi, China. When inoculation was carried out in June 2020, average outdoor day/night temperature ranged between 32 and 15°C. The vineyard management is under regular management with fungicides and insecticides, and organic and compound fertilizers. After sterilizing the leaf surfaces spraying with 75% ethanol for 30 seconds and rinsing them three times with SDW, circular leaf discs with a diameter of 15 mm were prepared, and placed in sterile Petri dishes 90 mm in diameter with moist double-layer filter papers. These leaf discs were inoculated with 20 µL of the above concentration of conidia suspension, and incubated at 25°C with a relative humidity of 90% and a photoperiod of 16 h light/8 h dark. Leaves placed in trays were sprayed with the same concentration of conidial suspension under the same conditions. The experiment was conducted with nine leaf discs, the diameter of each leaf disc necrosis was measured three times. The experiment was repeated three times.
2.2 Staining and light microscope imaging
The leaf disks were immersed in a solution containing ethanol and trichloromethane (3:1, vol/vol) along with trichloroacetic acid (0.45% wt/vol) for 3 days, followed by clearing in chloral hydrate (250 g/100 mL water) for another 3 days.
For trypan blue staining, leaf discs underwent a 30-minute staining process using a solution comprising trypan blue (20 mg), lactic acid (20 mL), glycerol (20 mL), phenol (20 mL), and ddH2O (20 mL). For DAB (Diaminobenzidine) staining, leaf discs were immersed in 5 mL centrifuge tubes filled with DAB staining Zsolution (1 mg DAB/mL ddH2O, pH 3.8) for 20 min under vacuum in the dark, followed by incubation at 25°C for 8 h in tin foil bags, and examined with 200 × or 400 × magnification using a light microscope (BX-53, Olympus). The resolution of the photographs was 150 dpi.
2.3 Sample preparation and scanning electron microscopy
Grape leaves inoculated with C. viniferum CvYL2a were cut into 5 × 5 mm pieces and initially fixed in 4% (vol/vol) glutaraldehyde in 0.1 M phosphate buffer (pH 6.8) at 4°C overnight. After 4 × 10 min rises in the same buffer, the samples were subjected to dehydration in a series of ethanol solutions ranging from 30% to 90% (vol/vol), each step lasting for 15 min, followed by three cycles of absolute ethyl alcohol dehydration. Finally, these samples were dried using a CO2 critical point dryer before being coated with gold and observed with SEM (Nano SEM-450, America).
2.4 Sample preparation and transmission electron microscopy
Grape leaves inoculated with C. viniferum CvYL2a were cut into 1 × 1 mm pieces and fixed in 4% (vol/vol) glutaraldehyde in 0.1 M phosphate buffer (pH 6.8) at 4°C overnight. After 4 × 10 min washes in the same buffer, the samples underwent post-fixation using a solution containing 1% osmium tetroxide (OsO4) at 4°C for 2 h and were subjected to dehydration as described in Section 2.3. Next, these samples were infiltrated with London Resin Company Ltd (LR) White Resin (Basingstoke, UK) in a series of proportions, 3:1 (vol/vol) for 2 h, 1:1 (vol/vol) for 8 h, 1:3 (vol/vol) for 12 h, and pure resin for 48 h, with a change every 24 h. Finally, they were immersed in pure resin and polymerized at 55°C for 48 h.
For semi-thin sections of 1µm thickness, samples, cut using a glass knife, were treated with 0.3% aqueous toluidine blue in 1.89% sodium tetraborate and observed with bright-field microscope (Olympus BX-51, Japan). Ultra-thin sections (90 nm) were prepared using a diamond knife and treated with uranyl acetate and lead citrate before being examined via a transmission electron microscope (HT7700, Japan).
2.5 Measurement of reactive oxygen species production rate
Plants rapidly produce reactive oxygen species (ROS) that directly inhibit the growth of pathogens during pathogens infection (Li et al., 2021). The DAB staining method is commonly used to detect H2O2 production (Huckelhoven et al., 1999; Ding et al., 2019). H2O2 production in both resistant and sensitive leaves was observed after DAB staining using a light microscopy. The rate was determined as the percentage of appressoria inducing H2O2 production in leaf cells to the overall count of appressoria. A total of 100 appressoria were randomly selected for assessment from each leaf, and this analysis was conducted on three blades at each time point. The experiment was performed with three independent biological replicates.
2.6 Gene family and phylogenetic evolution analyses
The gene sets of 14 fungal species were filtered, and similarity relationships between protein sequences of all species were analyzed using DIAMOND (Buchfink et al., 2015), which were further clustered using OrthoFinder2 (Emms and Kelly, 2019). To identify common single copy orthologous genes, multi-sequence alignment of genes within each single-copy homologous gene family was conducted using MUSCLE (Edgar, 2004). The evolutionary tree was constructed using RAxML with the maximum likelihood model (Stamatakis, 2014). Divergence times were estimated by integrating the constructed evolutionary tree with data from the TimeTree website and analyzed using the MCMCTree program in the software r8s and PAML software packages (Sanderson, 2003; Hedges et al., 2006). Data from 14 genomes used in comparative genomics can be found in the National Center for Biotechnology Information (NCBI) data library. These links and fungal strain names were shown in Supplementary Table 8.
2.7 RNA-seq and data analysis
Frozen tissue samples were obtained from C. viniferum CvYL2a-infested “TS” leaves at five different time points (0, 6, 12, 24, and 48 hours post inoculation (hpi)). Total RNA from these samples was extracted by Novogene Corporation Inc using the TRIzol Reagent (Invitrogen, Cat no. 15596026) and confirmed for integrity by 1% agarose gel electrophoresis. RNA purity and concentration were assessed using Nanodrop (Thermo Scientific, USA) and Agilent 5400 (Agilent Technologies, USA), respectively. Sequencing libraries of 150 bp paired-end reads were prepared and sequenced on an Illumina Novaseq platform. Clean reads were filtered by trimming the adapter sequences and low-quality reads of raw reads. High-quality reads were aligned to the C. viniferum CvYL2a (Dou et al., 2022) and the grapevine (12 × PN40024) reference genomes using HISAT2 (Mortazavi et al., 2008). Gene expression levels were quantified by calculating the fragments per kilobase of transcript per million mapped readings (FPKM). Differential expression genes (DEGs) were identified by comparing the inoculated samples at each infection time point with the 0 h samples using DESeq2 software (Love et al., 2014), with criteria set at Log2|(fold change)| > 4 and P-value < 0.05 via Benjamini and Hochberg’s approach. The gene ontology (GO) terms were categorized using GOSeq (Joung et al., 2010), and KEGG (http://www.genome.jp/kegg/) was used to explore the metabolic pathways of DEGs. Visualization of the results was done using venn diagrams and heatmaps created using TBtools v0.664 software (Chen et al., 2018).
2.8 qRT-PCR analysis
45 total RNA from three different grape germplasms, five inoculation periods (0, 6, 12, 24, and 48 hpi) with C. viniferum infection, and three replicates per period were extracted following the manufacturer’s instruction (R6827-01, Omega Bio-tek, USA). TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix (AT311-02, Trans gen, China) was used for the synthesis of first-strand cDNA. Primers are listed in Supplementary Table 2. Real-time PCR amplification was conducted using TransScript® II Green One-Step qRT-PCR SuperMix (AQ311-01, Trans gen, China).
2.9 Determination of endogenous hormone concentrations
According to the description of the method of extracting grape leaves hormones (Li et al., 2021b), 100 mg of TS leaves infected C. viniferum per period were promptly crushed in liquid nitrogen, followed by extraction with 1 mL of solvent (glacial acetic acid: methanol: isopropanol = 1: 20: 79 (v/v/v)). The concentration of hormones including salicylic acid (SA), ethylene (ET) synthesis precursor ACC and jasmonates (JA) in the extract was quantified by liquid chromatography-quadrupole ion trap-mass spectrometry (LC-QTRAP-MS, USA). There were three biological replicates for each hormone.
2.10 Plasmid construction and Agrobacterium transformation
SignalP and TMHMM software were used to select genes with signal peptide domain and TMHM without transmembrane domain as candidate effectors. 85 putative C. viniferum effectors were amplified, using cDNA as templates, with their predicted N-terminal signal sequences removed. The source of the cDNA was RNA extracted from grape leaves inoculated with C. viniferum. Specific primers designed (Supplementary Table 2) for each effector can be found in Appendix T. Subsequently, these sequences were cloned into vector pCAMBIA2300-GFP (Green fluorescent protein). Recombinant plasmids were transformed into Agrobacterium tumefaciens strain GV3101 using the liquid nitrogen freezing-thawing method. Agrobacteria carrying effector-GFP constructs were cultured in LB liquid medium, collected after centrifugation at 5000 rpm for 3 min, and resuspended in infiltration buffer (10 mM MgCl2, 10 mM MES (pH 5.7), 200µM acetosyringone) to achieve an OD600 of 0.6. The suspension was incubated for 3 h at 28°C in dark before infiltrating the leaves. Healthy leaves of 4 to 5 week-old N. benthamiana were infiltrated using a 1-mL needless syringe. INF1 (infestans 1, P. infestans PAMP elicitor) and GFP served as positive and negative controls, respectively. For experiments designed to suppress cell death triggered by INF1-GFP, leaves were infiltrated with effectors 12 h before INF1-GFP infiltration. Finally, leaf symptoms of cell death were evaluated through photography.
3 Results
3.1 Evolution of gene family in C.viniferum CvYL2a reveals unique features
To analyze the evolutionary relationships among C. viniferum CvYL2a and 13 other fungi, we clustered their proteins using OrthoFinder2. This process identified 16678 orthogroups containing 177914 proteins (Supplementary 1). From them, we selected 1155 single-copy orthologous families for phylogenetic analysis (Supplementary 2). Our findings indicate a close evolutionary relationship between C. viniferum CvYL2a and C. viniferum CGW01. CvYL2a appears to have diverged 3.2 million years ago within the genus Colletotrichum, which initially diverged approximately 45.2 million years ago (Figure 1). Additionally, we identified 33 specific genes in C. viniferum CvYL2a grouped into 13 categories. Notably, 20 of these genes (A08618, A10003, A09924, A08597, A01565, A09902, A09903, A09928, A11559, A09926, A09909, A01566, A10007, A09984, A09985, A10008, A14055, A10009, A11149 and A11147) lack significant homologous sequences in the NCBI database (Supplementary Table 1).
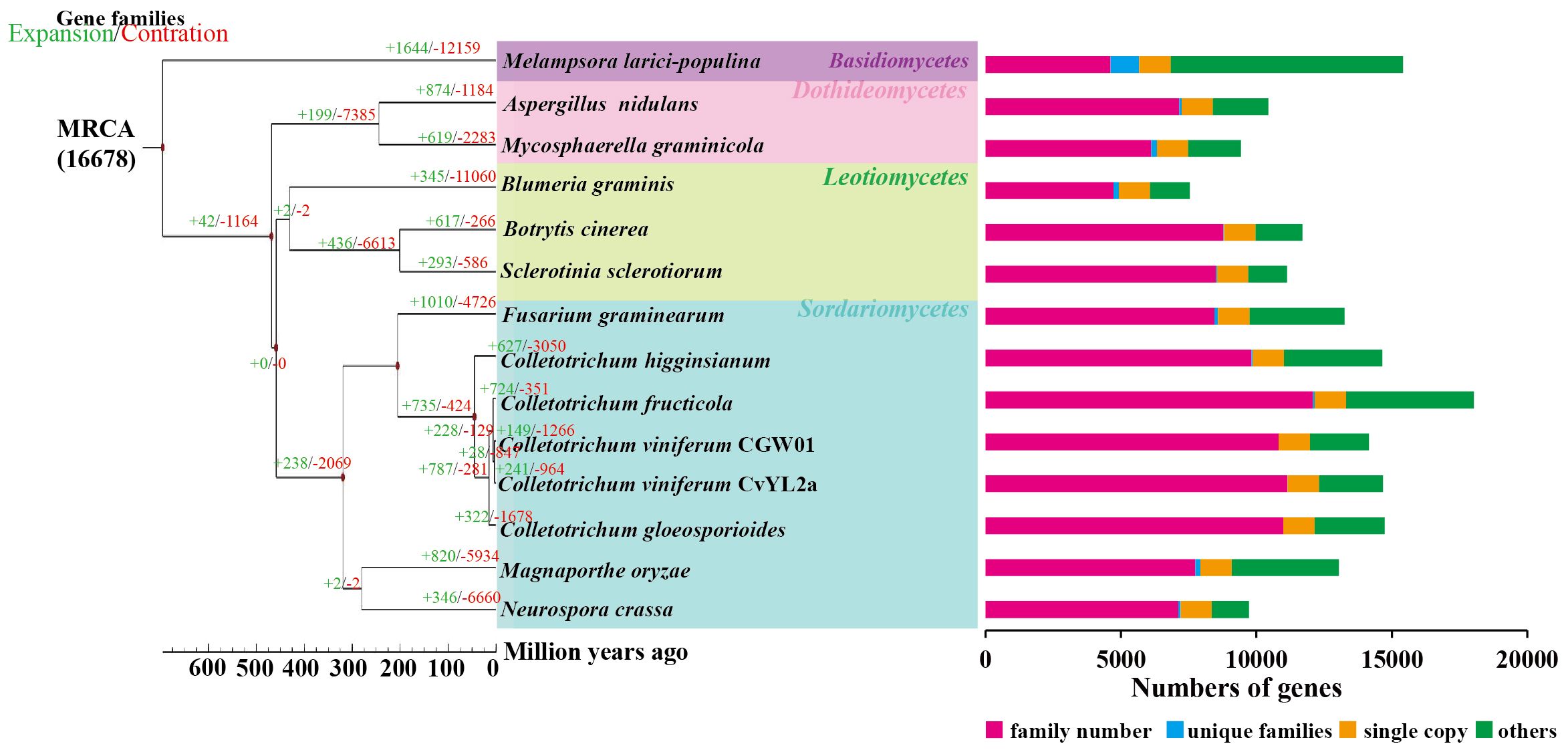
Figure 1. Phylogenetic relationship. The maximum likelihood phylogenetic tree was constructed using PhyML, based on the concatenated alignment of 1155 conserved single-copy orthologs from all species. Divergence time estimation for each species was performed utilizing the phylogenetic tree, software r8s, and the mcmctree program in the PAML software package. Time-correction points were obtained by integrating data from the TimeTree website.
3.2 Symptoms of C. viniferum infection in three grape germplasms
C. viniferum infection on grape leaves results in necrotic lesions, a typical symptom of grape ripe rot disease. To study the differences in resistance among the three grape germplasm resources to C. viniferum, we used leaf disc assays to observe necrotic lesion development. Small necrotic lesions appeared on “TS” leaves within 24 hpi, progressively expanding to a diameter of 14.81 cm by 120 hpi (Figures 2A, C). In contrast, “B” showed smaller and milder lesions from 72 to 120 hpi. Remarkably, “LB-8” exhibited minimal or no lesions at 120 hpi. When leaves were sprayed with the spore suspension, similar symptoms were observed. Brown grape leaf spots initially appeared at 12 hpi on “TS”, with spots enlarging and coalescing by 24 hpi, and large brown speckles became visible at 48 hpi. On the contrary, “B” and “LB-8” did not exhibit any disease necrotic spots before 24 hpi and small brown spots were observed only by 48 hpi (Figure 2B). Based on the grape ripe rot resistance grading standard (Gao et al., 2016), “TS”, “B” and “LB-8” were classified as high-sensitive (HS), moderate resistance (MR) and resistant (R) to C. viniferum, respectively.
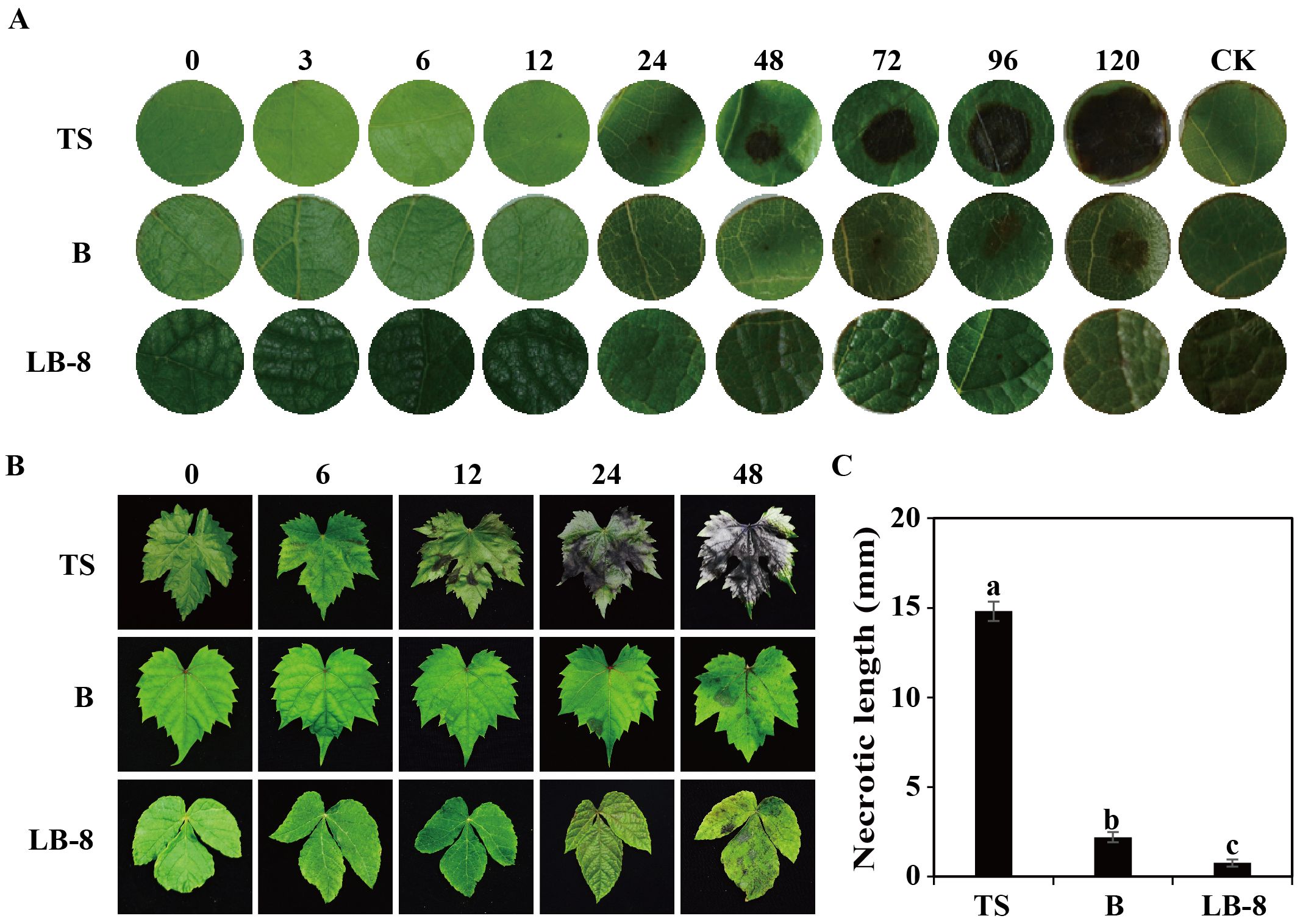
Figure 2. Ripe rot symptoms on three grapevine germplasms infected with C. viniferum. (A) Images captured at various time points [0, 3, 6, 12, 24, 48, 72, 96, and 120 hours post inoculation (hpi)] after applying 20 µl of fungi suspension (5 × 106 conidia/ml) and incubating at 25°C. (B) Images were taken at 0, 6, 12, 24, and 48 hpi. Minimal Lesions were observed on “B” and “LB-8”, whereas necrosis rapidly developed on “TS” from 12 hpi. One representative leaf of three replicates is shown for each time point. (C) Symptoms evaluation through measurement of necrotic length at 120 hpi. Data represent the means ± SE of three independent experiments with three replicates taken from different plants, each containing ten replicates for each genotype. Distinct letters indicate significant differences (ANOVA test with Tukey’s comparisons, P < 0.05). TS, “V. vinifera cv. Thompson Seedless”; B, “V. labrusca accession Beaumont”; LB-8, “V. piasezkii Liuba-8”.
To study the differences in the infection process in three grape germplasms, we observed cytological changes under a light microscope after toluidine blue staining. From 6 to 24 hpi, conidia were distributed across the epidermal cells of “TS” leaves. By 48 hpi, epidermal cells began to collapse, reaching complete destruction at 72 hpi. At 96 hpi, the palisade mesophyll cells were also destroyed and leaf tissues had completely collapsed by 120 hpi. Compared to “TS”, “B” exhibited delayed symptom onset, with epidermal cells becoming concave at 72 hpi, and noticeable damage observed by 120 hpi. Remarkably, the epidermal cells of “LB-8” remained intact at 120 hpi (Figure 3A).
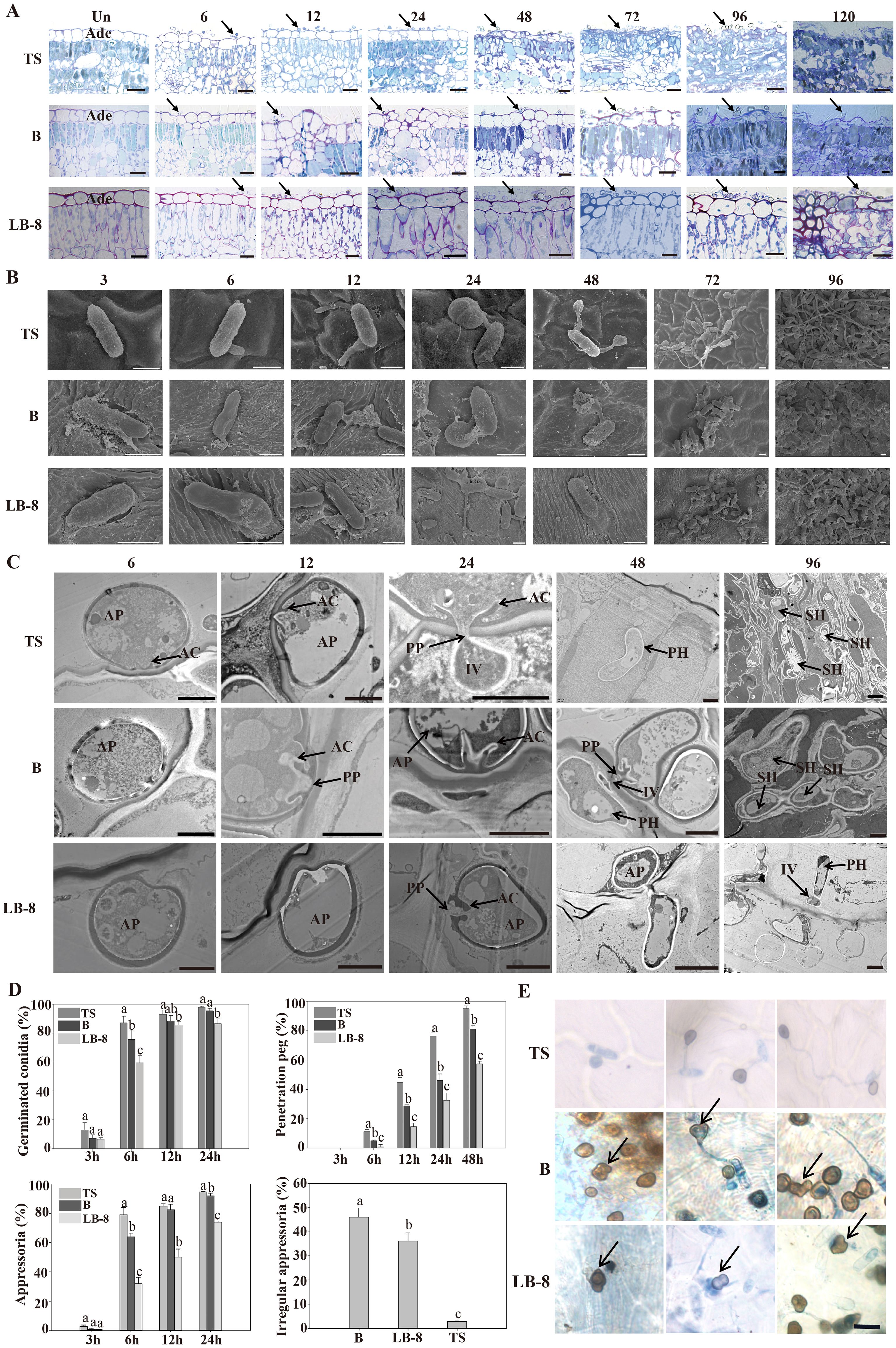
Figure 3. Histological and ultrastructural study of resistant and susceptible grapevine leaves infected by CvYL2a. (A) Cytological study of C. viniferum CvYL2a infected grape leaves. Un: Un-inoculated with C. viniferum; Ade, adaxial epidermis; scale bar, 20 μm. (B) SEM imaging of conidia development on leaves infected by C. viniferum. Scale bar = 5 μm (C) Ultrastructure of host-parasite interactions on “TS”, “B”, and “LB-8” leaves under TEM. AP, appressorium; AC, appressorial cone; Cu, cuticle; FCW, fungal cell wall; HCW, host cell wall; HPM, host plasma membrane; IV, infection vesicle; PP, penetration peg. Scale bar = 2 μm. (D) The percentages of C. viniferum germinated conidia, appressoria, and penetration peg on three grape germplasms at different time points post-inoculation, along with the presence of irregular appressoria on the three grape genotypes at 48 hpi, results are presented as mean ± SE, n=9. Different letters indicate significant differences (ANOVA test with Tukey’s comparisons, P < 0.05). (E) Occurrence of more mature appressorium deformities on “TS”, “B” and “LB-8” inoculated with C. viniferum CvYL2a. Deformed appressorium are marked by the arrow. Scal bar = 10 μm.
3.3 Developmental differences of C. viniferum on the leaves of three grape germplasm resources
To study developmental differences of C. viniferum on the leaves of three grape genotypes, we performed scanning and transmission electron microscopy observations. Initially, conidia formed a central septum at 3 hpi, dividing the conidia into two equal-sized parts on the leaves of all three grapevines. Additionally, the conidia on “B” and “LB-8” leaves were surrounded by abundant white secretions compared to “TS” (Figure 3B). Previous studies of downy mildew infection have shown that resistant grape varieties also produced white secretions (Yin et al., 2017). Subsequently, these conidia germinated and formed germination tubes from one or both ends of the conidia (Figure 3B). Although conidial germination in all three grapevines started at 3 hpi and significantly increased by 6 hpi, “TS” exhibited the highest germination rate at 24 hpi (Figure 3D). Interestingly, at 72 hpi, “B” and “LB-8” elongated germination tubes were shorter compared to “TS”, a difference that persisted even at 120 hpi (Figure 3B).
Next, appressoria developed from conidia, either directly or from the elongated germination tube, resulting in the production of several appressoria from a single conidium (Figure 3B). Appressorium formation became evident by 6 hpi on the leaves of all grape germplasms, with the highest appressorium formation rate occurring on “TS” by 24 hpi (Figure 3D). The shape of the appressoria is generally round or irregular in some cases (Velho et al., 2016). Irregular appressoria, such as downward concave hearts, gourds, and outward protruding convex morphology, were easily found with a proportion over 30% on “B” and “LB-8” (Figures 3D, E). This was possibly due to appressorium collapse caused by secretions from “B” and “LB-8” (Figure 3B) (Yin et al., 2017) or dense hair on grape leaves (Figure 3C; Supplementary Figure 1) (Kortekamp and Zyprian, 1999; Han et al., 2021). The paraxial surface of the B and LB-8 blades is covered with hairs, while the TS surface is hair-free.
The appressoria on “LB-8” appeared deflated and irregularly shaped compared to those on “TS”.
Transmission electron microscopy revealed that on “TS”, the appressoria formed penetration peg, infected vesicles and primary hyphae, and the primary hyphae further formed secondary hyphae (Figure 3C). The formation of penetration pegs started at 6 hpi on all grapevines. However, the formation ratio was significantly lower on “LB-8” compared to the other two germplasms, with nearly half of the appressoria on “LB-8” failing to produce penetration pegs. Despite a similar developmental trend across different germplasms, penetration pegs formed more rapidly on “TS” than on “B” and “LB-8” at each time point post-inoculation (Figure 3D). While “B” closely resembled “TS” in the penetration process, “LB-8” exhibited distinctive characteristics. “B” demonstrated a prolonged biotrophic stage compared to “TS”, with no observable secondary hyphae on “LB-8” (Figure 3C).
3.4 Expression of pathogenicity genes
To analyze the expression dynamics of C. viniferum pathogenic genes, we selected the susceptible grape “TS” characterized by rapid C. viniferum development for transcriptome analysis. A total of 95.29 Gb of sequence data was produced, resulting in approximately 42 million clean readings from each sample (Figures 4E, F). The clean reads were then mapped to the C. viniferum CvYL2a genome, with percentages ranging from 0.43% to 16.19% (Supplementary Table 2). The shared DEGs among the 6, 12, 24, and 48 hpi samples increased over infection period. Specifically, compared to 0 hpi samples, there were 852, 1029, 1115, and 1141 up-regulated DEGs (|log2 (fold change)| > 4, P < 0.05), and 83, 78, 269, and 309 down-regulated DEGs detected at 6, 12, 24, and 48 hpi, respectively (Figure 4A; Supplementary Table 19–22). In addition, there exhibited 236 CvYL2a DEGs consistent differential expression across all infection time points (Figure 4C).
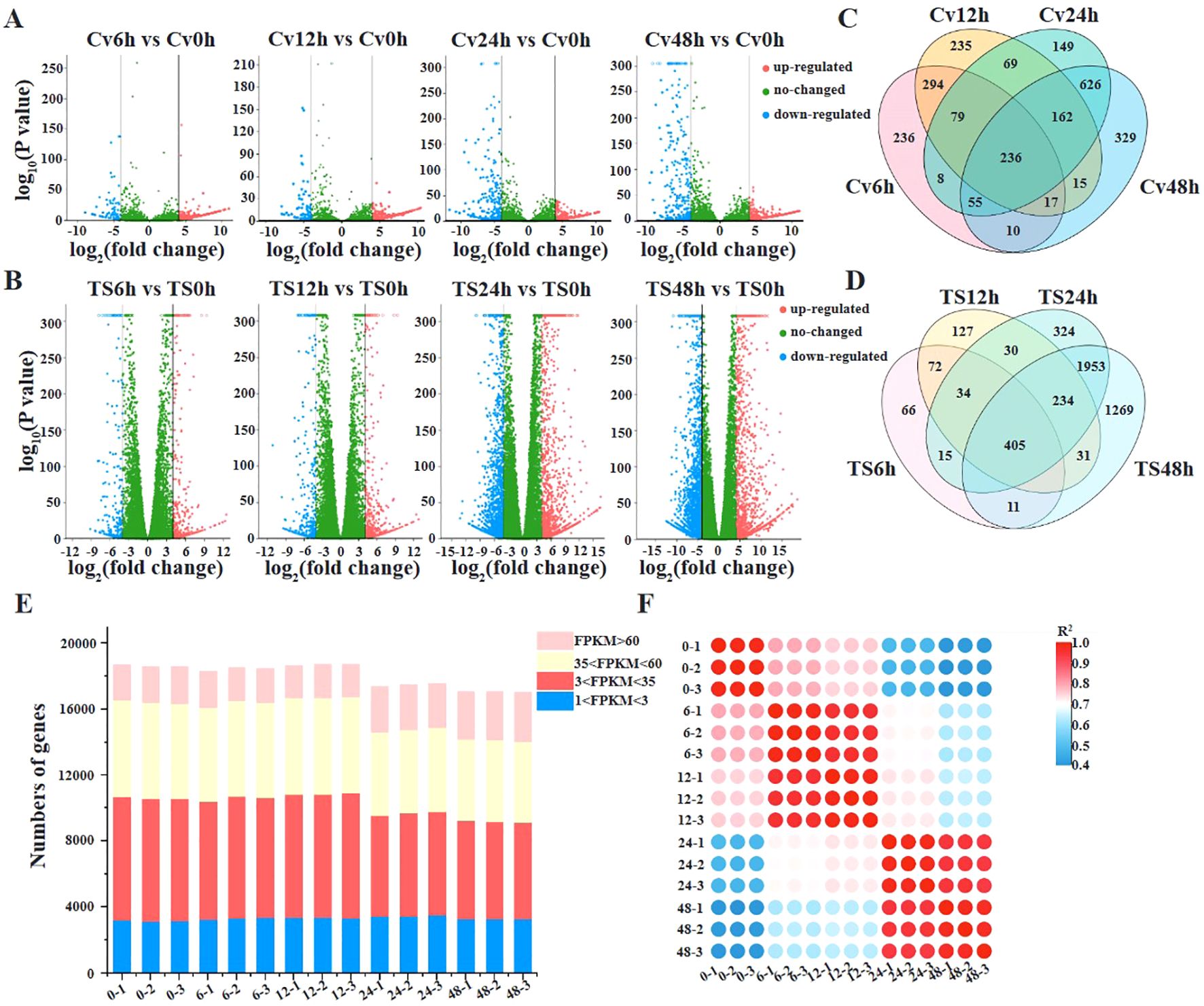
Figure 4. Transcriptome analyses of grapevines infected by C. viniferum. (A, B)Volcano plots displaying differentially expressed genes in C. viniferum and grapevine “TS”, respectively. Cv0h, Cv6h, Cv12h, Cv24h and Cv48h represent C. viniferum transcriptome, while TS0h, TS6h, TS12h, TS24h and TS48h represent “TS” transcriptome prepared from grape leaves post-inoculation with C. viniferum at 0, 6, 12, 24 and 48 hpi, respectively. (C), Overlap of upregulated and downregulated DEGs in C. viniferum. (D), Overlap of upregulated and downregulated DEGs in “TS”. (E), Number of transcripts detected in 15 samples. Transcripts with an FPKM (fragments per kilobase of gene per million mapped reads) >1 are considered to be detected. Detected transcripts are further categorized as low (1<FPKM<3), moderated (3<FPKM<60), or high (FPKM>60) detection levels. (F), Correlation of gene expression levels among grape leaves infected with C. viniferum at 0, 6, 12, 24, and 48 hpi.
Transcriptome data was searched using SignalP, CAZy (Carbohydrate-Active Enzyme) database, AntiSMASH program, and the P450 database. All C. viniferum secondary metabolism gene clusters were shown in Supplementary Table 10. The transcriptome revealed significant differential expression of 56 effector genes, 36 Cazy genes, 5 P450 genes and 10 genes in secondary metabolism gene clusters at one or more infection stages (Supplementary Table 5). For further investigation into the functions of differentially expressed effectors, 20 cysteine-rich small effectors, 9 virulence-related effectors, and 28 effectors randomly chosen from the genome, along with 28 up-regulated effectors from both the genome and the transcriptome were selected (Supplementary Table 6) and assessed their abilities to induce or inhibit plant cell death. Agrobacterium tumefaciens carrying a GFP expression vector inserted with one of these 87 effectors was injected into N. benthamiana leaves. 12 h later, the hypersensitive response-inducing INF1 was injected at the same location. Among the effectors, ten inhibited INF1-induced cell death, while two induced cell death in N. benthamiana leaves. Subcellular localizations of 12 effectors in N. benthamiana were shown in Supplementary Figure 2. These findings suggest that these 12 effectors play active roles in pathogen-host interactions (Figure 5G).
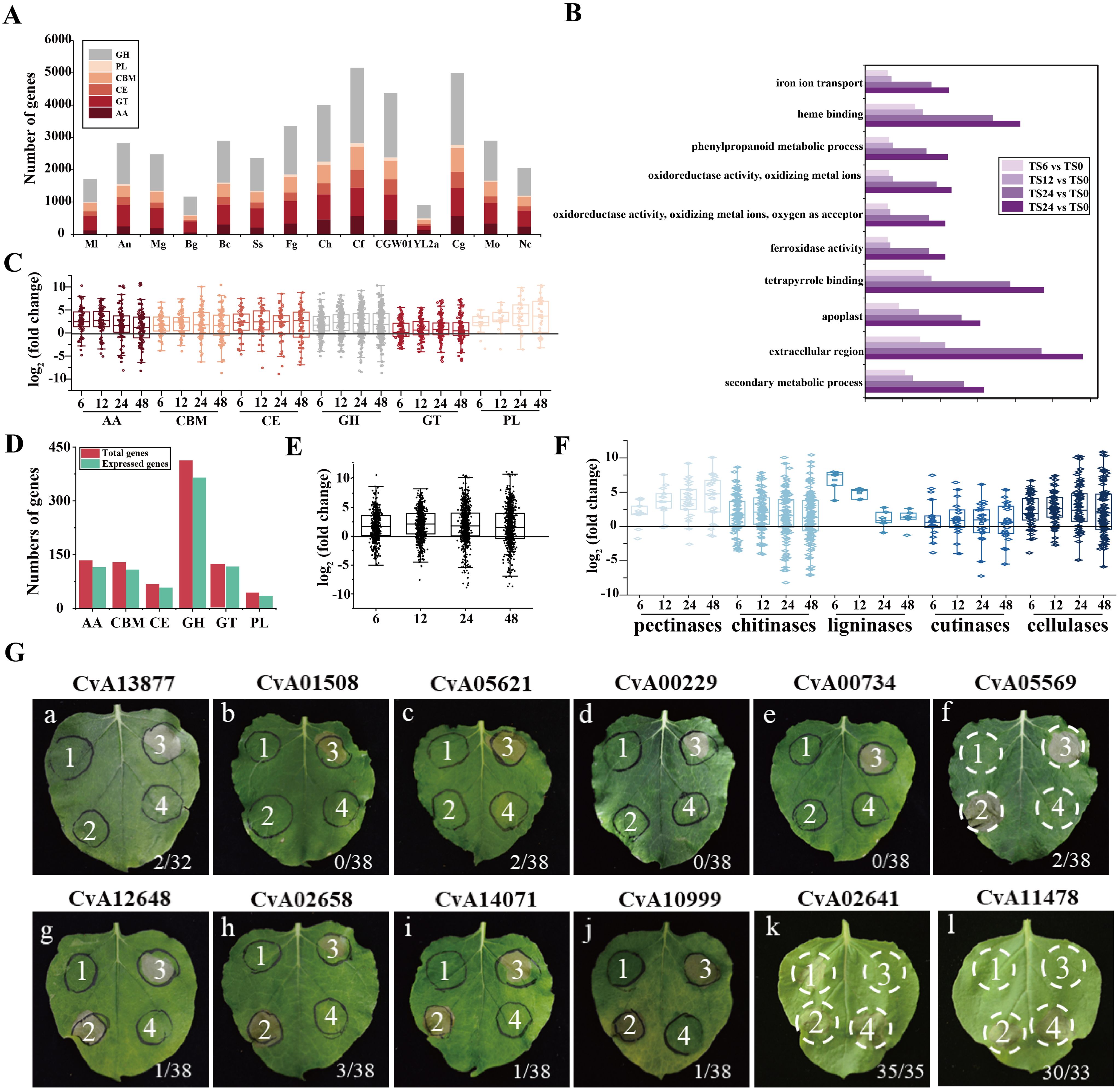
Figure 5. Gene Ontology and C. viniferum pathogenicity genes. (A) Repertoires of the carbohydrate-active enzymes (CAZymes) in C. viniferum and other 13 fungal genomes. GH, glycoside hydrolase; CE, carbohydrate esterase; AA, auxiliary activities; CBM, carbohydrate-binding module; PL, polysaccharide lyase. (B) Functional categorization of regulated differentially expressed genes (DEGs) in the C. viniferum-infected leaves of TS based on Gene Ontology (GO). P-value < 0.05. (C) Box plot of CAZymes gene expression of each family at 6, 12, 24, and 48 hpi. (D) CAZymes genes expressed post inoculation with C. vinifera. (E) Box plot displays expressed CAZymes (n = 530) in C. viniferum at 6, 12, 24, and 48 hpi. (F) Box plot of expressed CWDEs of C. viniferum in grape leaves at 6, 12, 24, and 48 hpi. (G) Cell death-suppression by INF1 and cell death-inducing activity of C. viniferum effectors on plant cell death induced. The label on the panel represents the name of the effector. a-j panels: the ability of 10 putative C. viniferum effectors to suppress cell death triggered by INF1. A. tumefaciens infiltration sites in N. benthamiana leaves expressing each of the 10 effectors were challenged with A. tumefaciens expressing the INF1 elicitin. A. tumefaciens strain carrying GFP was used as a control. Representative cell death symptoms were photographed at 7 days after INF1-GFP infiltration. k and l panels: 2 putative C. viniferum effectors CvA02641 and CvA11478 induced cell death. a-e panels received the following infiltration: 1, GFP; 2, effector-GFP; 3, GFP and INF1-GFP; 4, effector-GFP and INF1-GFP. f-j panels showed leaves infiltrated with 1, GFP; 2, INF1-GFP; 3, GFP and INF1-GFP; 4, effector-GFP and INF1-GFP. k and l panels were leaves infiltrated with 1, GFP; 2, effector-GFP; 3, GFP; 4, INF1-GFP. Ratios represent the proportion of infiltration sites displaying the cell death phenotype.
We compared the CAZyme library across 14 fungal species and identified 909 CAZymes in the predicted proteome of C. viniferum CvYL2a. These CAZymes were distributed across six major CAZymes superfamilies, including 413 glycoside hydrolases (GHs), 121 glycosyl transferases (GTs), 44 polysaccharide lyases (PLs), 68 carbohydrate esterases (CEs), 134 auxiliary activities (AAs), and 129 carbohydrate binding modules (CBMs). Compared to other fungi, CvYL2a had fewer genes distributed in each superfamily. The expressed carbohydrate genes in CvYL2a during infection account for 87.79% of the total carbohydrate genes (Figures 5C–E). Within the CAZyme library, we identified 316 cell wall degrading enzymes (CWDEs), including 29 pectinases, 148 chitinases, 6 ligninases 26 cutinases, and 107 cellulases. Of these CWDEs, 273 (86.39%) genes were expressed during infection, with 103 being up-regulated and 21 down-regulated (Figure 5F; Supplementary Table 9).
3.5 Grapevine responses to C.viniferum infection
To study the host response to C. viniferum, clean reads also were mapped to the V. vinifera PN40024 genome, with mapping percentages of 93.90 ± 0.06%, 93.82 ± 0.06%, 93.67 ± 0.12%, 91.59 ± 0.20%, and 77.90 ± 0.53%, respectively (Supplementary Table 3). In the V. vinifera transcriptome, we detected 422, 570, 1359, and 1503 up-regulated DEGs along with 261, 377, 1702, and 2480 down-regulated DEGs. Moreover, 405 common DEGs were identified throughout the infection process (Figures 4B, D). DEGs functional annotations with different time point were shown in Supplementary Tables 11–14. GO terms at different time point were analyzed (Supplementary Tables 15–18). Gene ontology analysis revealed their involvement in biological processes like iron ion transport, phenylpropane metabolism and secondary metabolism, cell components like apoplast and extracellular domain; and molecular functions including heme binding, tetrapyrrole binding and oxidoreductase activity (Figure 5B). Additionally, KEGG pathway enrichment analysis showed significant enrichment in phenylpropanoid-guided flavonoid and resveratrol biosynthesis (34 DEGs), phenylpropanoid biosynthesis (18 DEGs), photosynthesis antenna proteins (16 DEGs) (Figures 6–8).
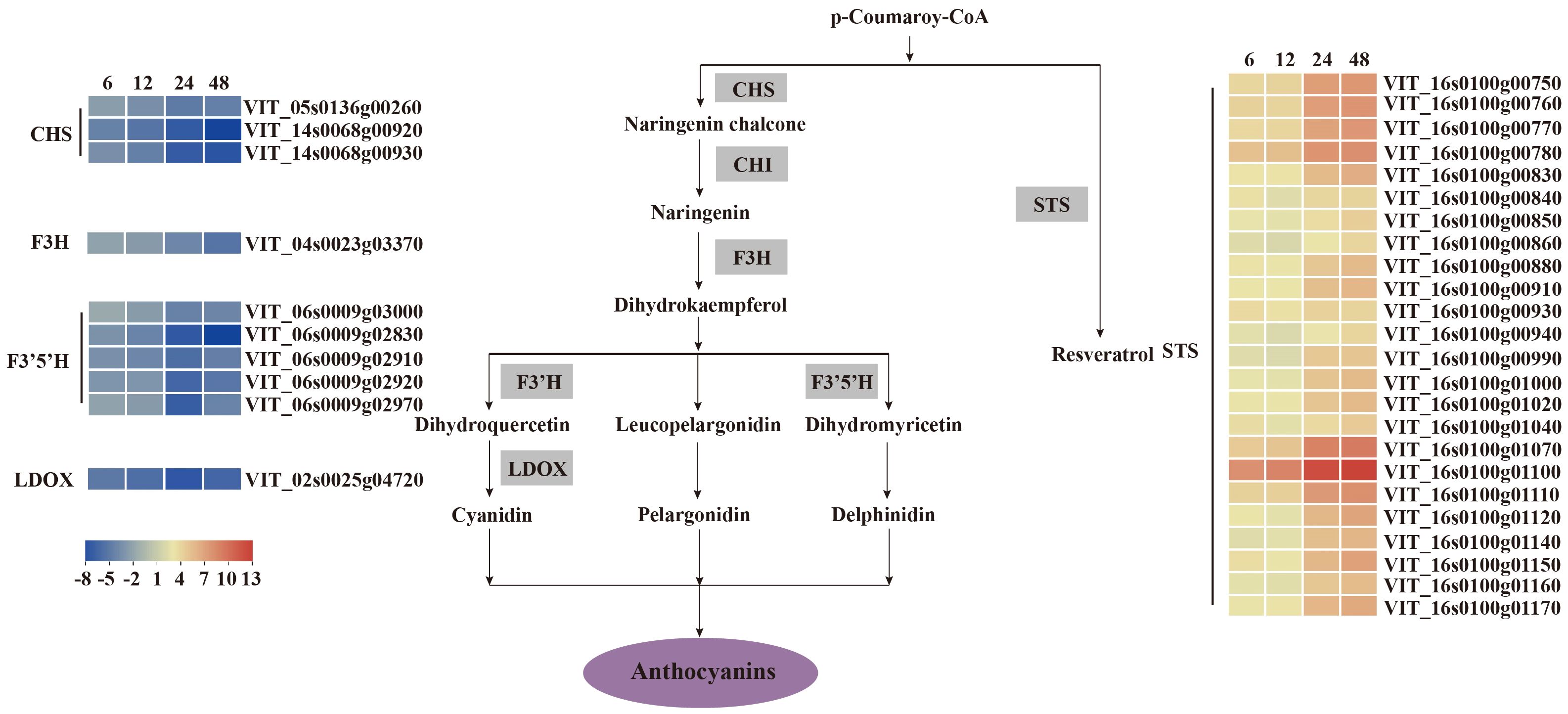
Figure 6. V. vinifera pathways and related gene expression for flavonoid biosynthesis in C. viniferum-infected leaves of Vitis vinifera cv. Thompson Seedless. Red indicates upregulation and blue indicates downregulation (P < 0.05) relative to the expression levels at 0 hpi. STS, stilbene synthase; CHS, chalcone synthase; CHI, chalcone isomerase; F3’H, flavonoid 3’ hydroxylase; F3’5’H, flavonoid 3’,10’-hydroxylase; F3H, flavanone 3-hydroxylase; LDOX, leucoanthocyanidin dioxygenase.
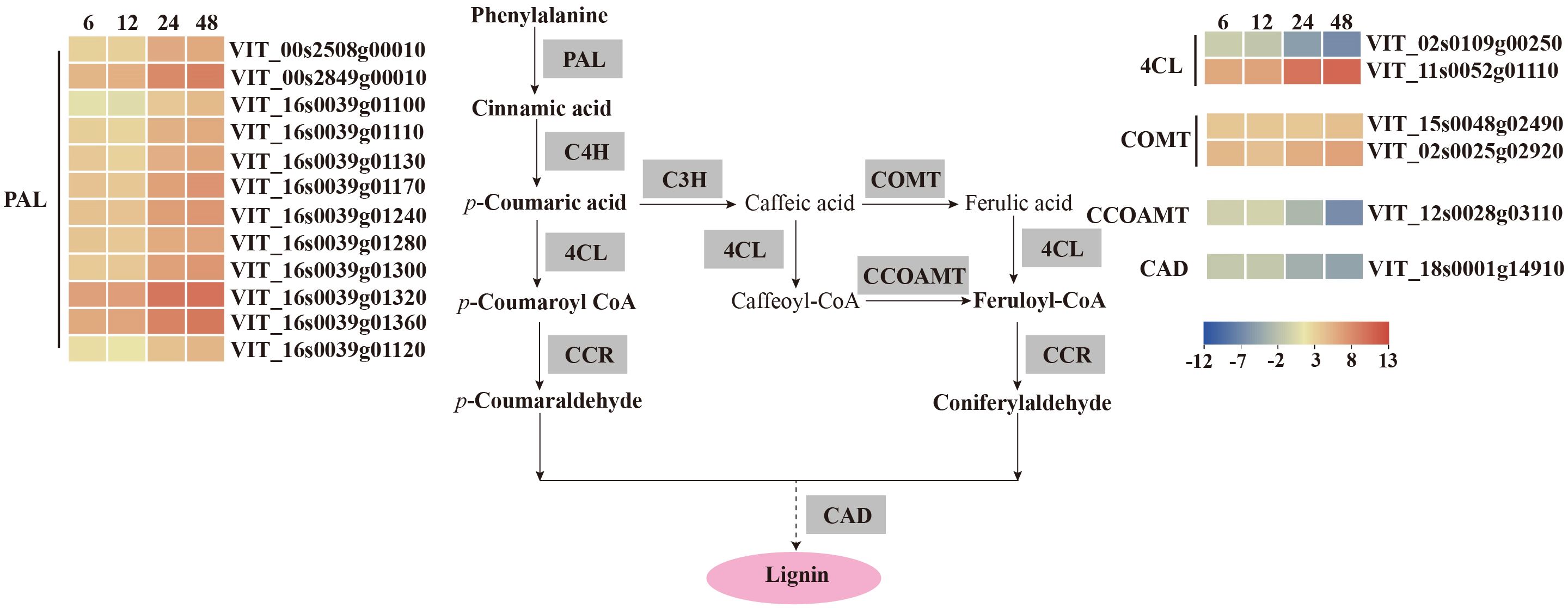
Figure 7. V. vinifera pathways and related gene expression for phenylpropanoid biosynthesis in C. viniferum-infected leaves of Vitis vinifera cv. Thompson Seedless. Red indicates upregulation and blue indicates downregulation, (P < 0.05) relative to the expression levels at 0 hpi. PAL, phenylalanine ammonia-lyase; C4H, cinnamate 4-hydroxylase; C3H, p-coumarate 3-hydroxylase; COMT, caffeic acid 3-O-methyltransferase; 4CL, 4 - coumarate -CoA ligase; CCOAMT, caffeoyl-CoA O-methyltransferase; CAD, cinnamyl alcohol dehydrogenase.

Figure 8. Gene expression of photosynthesis-antenna proteins in C. viniferum-infected leaves of Vitis vinifera cv. Thompson Seedless. Blue indicate downregulation, respectively; and unshaded represents no significant change (P < 0.05) relative to the expression levels at 0 hpi. Lhca, chlorophyll a binding protein of Light-harvesting chlorophyII; Lhcb, chlorophyll b binding protein of Light-harvesting chlorophyII.
Our analysis demonstrated distinct patterns of gene expression in several pathways, including phenylpropanoid-guided flavonoid and resveratrol biosynthesis, phenylpropanoid biosynthesis, and photosynthesis-antenna proteins. Notably, RNA-seq analysis showed downregulation of genes associated with anthocyanins synthesis, including flavonoid 3’,10’-hydroxylase (F3’5’H), flavanone 3-hydroxylase (F3H), chalcone synthase (CHS), and leucoanthocyanidin dioxygenase (LDOX). Conversely, the stilbene synthase (STS) gene associated with the synthesis of resveratrol was significantly upregulated (Figure 6). In the phenylpropanoid biosynthesis pathway, while caffeoyl-CoA O-methyltransferase (CCOAMT) and phenylalanine ammonia-lyase (PAL) were upregulated, cinnamyl alcohol dehydrogenase (CAD), a gene related to lignin synthesis, was downregulated (Figure 7). Grape genes involved in the photosynthesis-associated pathway encoded light-harvesting chlorophyll a/b-binding proteins (Lhc). The Lhc superfamily contains eight subfamilies: early light-induced protein (ELIP), ferrochelatase II (FCII), Lhca, Lhcb, one-helix protein (OHP), photosystem II subunit S (PsbS), photosystem II protein 33 (Psb33), and stress-enhanced protein (SEP) (Klimmek et al., 2006). All differentially expressed photosynthesis antenna genes including 5 Lhca and 11 Lhcb were downregulated in susceptible V. vinifera “TS” (Figure 8).
Plant disease resistance is significantly influenced by the involvement of transcription factors. Recent research has highlighted the involvement of various transcription factor families in regulating signal transduction in plant SA, ET and JA pathways responding to different pathogen infections, such as WRKY (WRKY domain-containing transcription factors) family, MYB (v-myb avian myeloblastosis viral oncogene homolog) family, and ERF (thylene responsive factors) family (Liu et al., 2021; Gao et al., 2022; Zhou et al., 2022). Our study found that numerous transcription factors, including 10 WRKYs, 18 NACs (NAM, ATAF1/2, and CUC2), 38 MYBs, 53 ERFs, 6 GATAs (GATA-binding transcription factors), 2 bHLHs (basic helix-loop-helix proteins), one SBP (quamosa promoter promoter binding proteins) were differentially expressed during C.viniferum infection at four stages (Figure 9). Gene expression associated with JA, SA, and ET signaling pathways significantly increased upon C. viniferum infection (Figure 10). Our results showed that the JA synthesis-related genes including LOX (VIT_00s0265g00170), AOS (VIT_03s0063g01820, VIT03s0063g01860 and VIT03s0063g01830), AOC (VIT_01s0011g03090, VIT_18s0041g02040, VIT_18s0041g02010, VIT_18s0041g02260, VIT_18s0041g02020, VIT_18s0041g02060, VIT_18s0041g02050), OPCL (VIT_00s0662g00010), and KAT (VIT_05s0051g00720), as well as the SA-synthesis-related genes PAL (VIT_16s0039g01170, VIT_00s2508g00010, VIT_16s0039g01280, VIT_00s2849g00010, VIT_16s0039g01100, VIT_16s0039g01300, VIT_16s0039g01110, VIT_16s0039g01130, VIT_16s0039g01240, VIT_16s0039g01320, VIT_16s0039g01120 and VIT_16s0039g01360) and ACS (VIT_15s0046g02220), a new transcript AOC1 (novel.1296) for ethylene synthesis were upregulated during C. viniferum infection of grape leaves (Figure 10). We measured the hormones levels of C. viniferum infection and found that SA, JA, and ET synthesis precursor ACC significantly increased (Figure 10). The findings suggest the involvement of JA, SA, ET and ABA in regulating the response to C. viniferum during the late stage of infection.
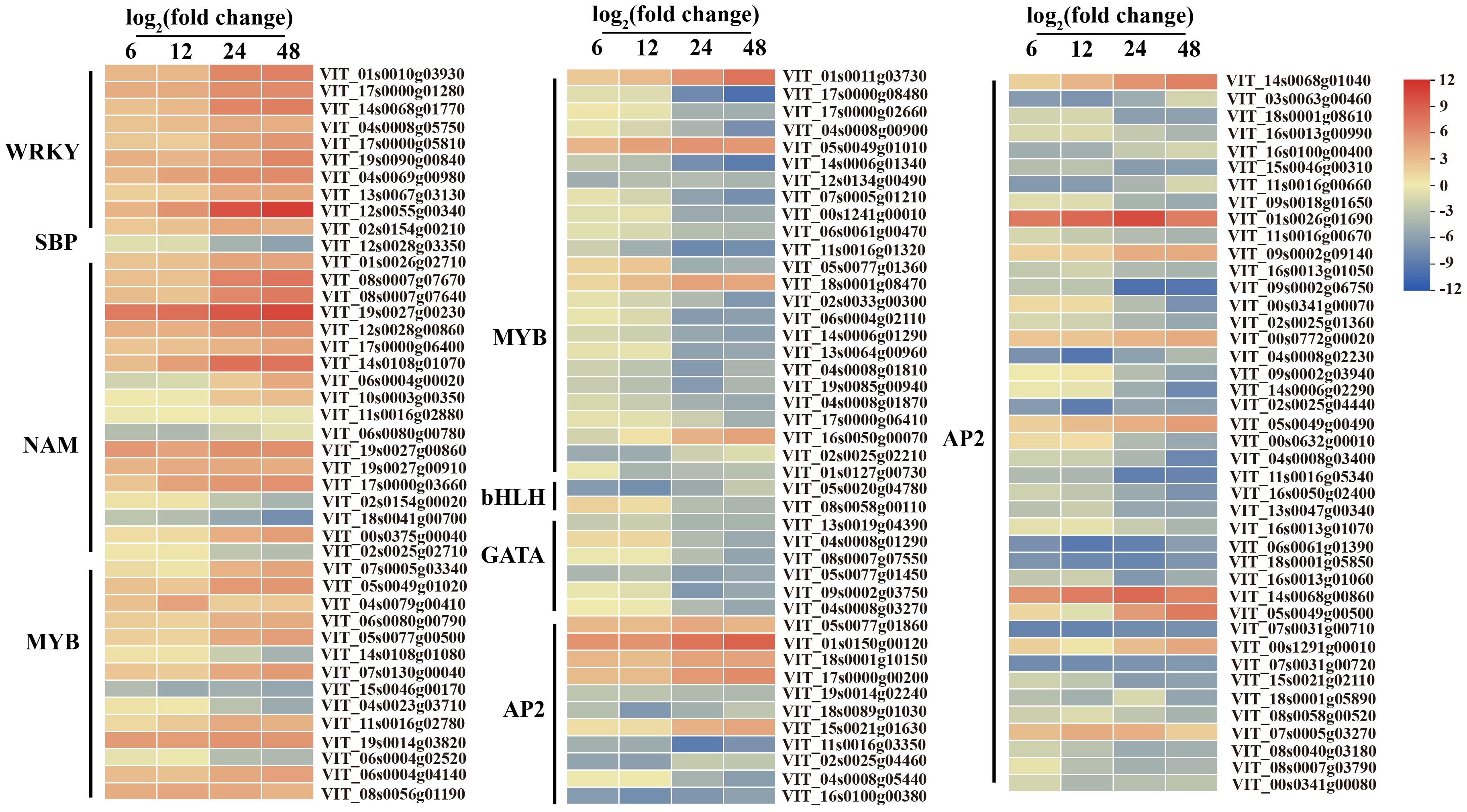
Figure 9. Expression patterns of differentially expressed transcription factors at 6, 12, 24, and 48 hpi in C. viniferum-infected V. vinifera leaves. WRKY, WRKY domain-containing transcription factors, SBP, quamosa promoter promoter binding proteins, NAM, No apical meristem, MYB, v-myb avian myeloblastosis viral oncogene homolog, bHLH, basic helix-loop-helix proteins, GATA, GATA-binding transcription factors, AP2, APETALA2 responsive factor. Red and blue indicate upregulation and downregulation, respectively, and yellow represents no significant change (P < 0.05) relative to the expression levels at 0 hpi.
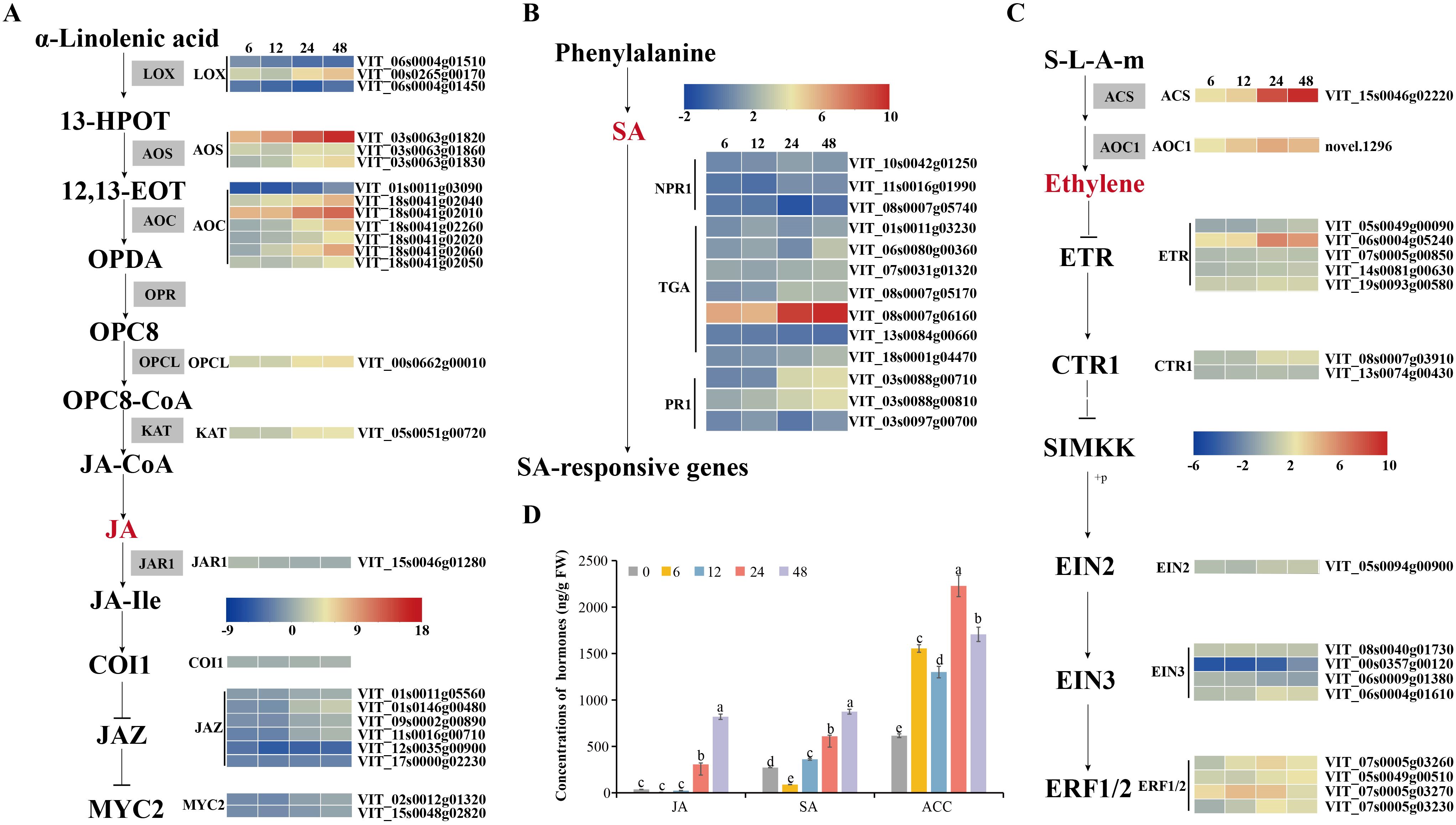
Figure 10. Hormonal content of grape leaves during infection and expression patterns of differentially expressed genes assigned to hormone signaling. (A–C) V. vinifera jasmonates (JA) biosynthesis, salicylic acid (SA) and ethylene (ET) pathways and related gene expression in C. viniferum-infected leaves at 6, 12, 24, and 48 hpi. Red and blue indicate upregulation and downregulation (P < 0.05), respectively, relative to the expression levels at 0 hpi. 12,13-EOT: 12,13(S)-epoxylinolenic acid; 13-HPOT: 13(S)-hydroperoxylinolenic acid; AOS: allene oxide synthase; LOX: lipoxygenase; AOC: allene oxide cyclase; OPR: 12-oxophytodienoate reductase; OPC:8, 3-oxo-2-(cis-29-pentenyl)-cyclopentane-1-octanoic acid; OPCL: 3-oxo-2-(cis-29-pentenyl)-cyclopentane-1-octanoic acid CoA Ligase; OPDA: 12-oxocis-10,15-phytodienoic acid; COI1, coronatine-insensitive protein 1; JAZ, jasmonate ZIM domain-containing protein; MYC2, transcription factor MYC2; ACS: 1-aminocyclopropane-1-carboxylate synthase 3; ACO1: 1-aminocyclopropane-1-carboxylate oxidase; ETR: ethylene receptor; CTR1: serine/threonine-protein kinase CTR1; EIN2: ethylene-insensitive protein 2; EIN3: ethylene-insensitive protein 3; EBF1/2: EIN3-binding F-box protein; ERF: ethylene-responsive transcription factor; NPR1: nonexpressor of pathogenesis-related genes 1; TGA: transcription factor TGA; PR1: pathogenesis-related protein 1. (D) Hormonal content of grape leaves in C. viniferum-infected leaves at 0, 6, 12, 24, and 48 hpi. Distinct letters indicate significant differences (ANOVA test with Tukey’s comparisons, P < 0.05). ACC: 1-aminocyclopropane-1-carboxylic acid, precursor of ethylene.
To measure the production and accumulation of hydrogen peroxide (H2O2), we used diaminobenzidine (DAB) staining, which relies on the peroxidase-catalyzed oxidation of oxygen released by H2O2, resulting in tan deposits on the plant surface. Fewer cells produced H2O2 in “TS” leaves after C. viniferum inoculation. However, “B” and “LB-8” exhibited obvious browning due to DAB oxidation. “B” showed lighter staining and fewer stained cells after C. viniferum inoculation, while “LB-8” displayed the opposite pattern. At 6 hpi, none of the three grape leaves significantly produced H2O2. However, by 12 hpi, cells beneath the appressorium in “LB-8” exhibited tan staining, with a significant increase observed from 24 hpi to 48 hpi. In “B” leaves, the proportion increased from 6.00% to 12.12%, while “LB-8” reached 25.53%. “TS” leaves showed slight staining only at 48 hpi. These results suggest that C. viniferum invasion is more extensive during the initial 2 days after inoculation, with “TS” leaves exhibiting the lowest H2O2 production rate (Figures 11A–D). We identified 76 DEGs involved in antioxidation during C. viniferum infection in grapes. Among these, one-third (22) of the peroxidase (POD), and four-fifths of glutathione peroxidase (GSH) related DEGs were up-regulated. Furthermore, three catalase (CAT) DEGs showed increased expression, and one was down-regulated. These findings underscore the significant role of ROS scavenging by POD, CAT and GSH in the grapevine responses to C. viniferum (Figure 11F).
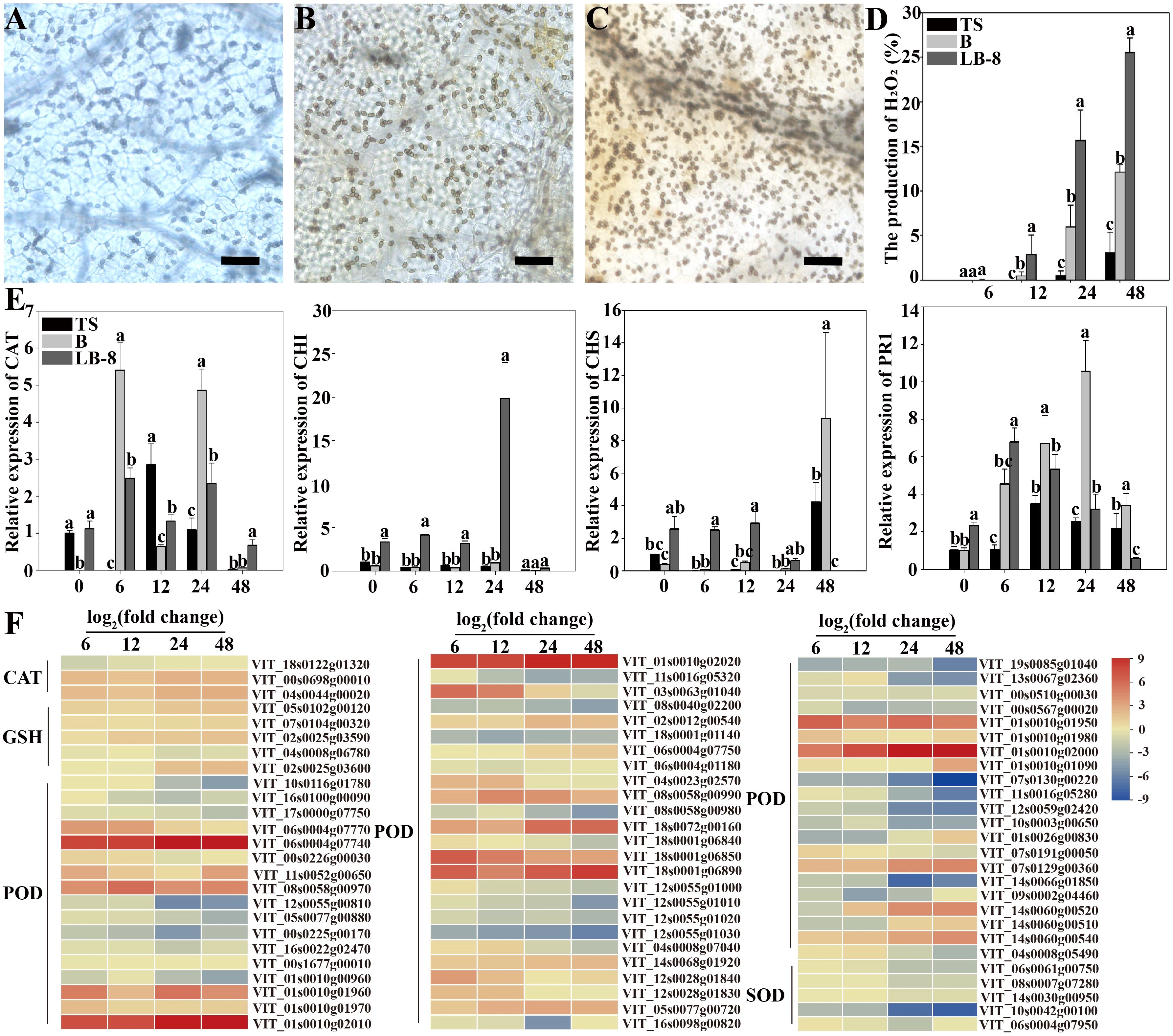
Figure 11. Responses of grapevines to C viniferum CvYL2a. (A-C) 2-day DAB staining of leaves from three grape varieties: (A) TS, (B) B, and (C) LB-8 inoculated with C. viniferum CvYL2a. Scal bar = 25 μm. (D) The production rate of H2O2 after inoculation of leaves from three grape varieties with C. viniferum CvYL2a, results are presented as mean ± SE, n = 9. Different letters indicate significant differences (ANOVA test with Tukey’s comparisons, P < 0.05). (E) Quantitative analysis of defense-related genes in “TS”, “B”, and “LB-8” plants after inoculation with C. viniferum. The gene expression levels were analyzed using quantitative real-time polymerase chain reaction (RT-qPCR). The natural logarithm transformed normalized-expression values were plotted for each gene at five time points (0, 6, 12, 24 and 48 hpi). Results are presented as mean ± SE, n = 3. Different letters indicate significant differences (ANOVA test with Tukey’s comparisons, P < 0.05). CAT, Catalase. CHI, Chalcone isomerase; CHS, Chalcone synthase; PR1, pathogenesis-related 1. (F) DEGs associated with reactive oxygen species (ROS) scavenging in C viniferum-infected “TS”. CAT, Catalase; GSH, Glutathione peroxidase; POD, Peroxidase; SOD, Superoxide dismutase.
One notable transcriptomic characteristic observed in grape leaves following C. viniferum infection is the up-regulation of defense-related genes. To further substantiate our transcriptome sequencing results, we performed qRT-PCR analysis of four defense-related genes at different time points post inoculation with C. viniferum. Two genes, CAT (Catalase) and PR1 (pathogenesis-related 1), showed higher expression levels at 6 hpi in “B” grape leaves. In “TS”, the expression levels of defense-related genes increased slowly. In contrast, two other genes CHI (Chalcone isomerase) and CHS (Chalcone synthase) displayed rapid upregulation during infection in “LB-8” compared to “B” and “TS” (Figure 11E). Additionally, among 72 identified LRR (Leucine-rich repeat receptor) proteins in infected grape leaves, 23 were up-regulated. Two disease-resistant proteins (DRP) were detected, RPS2 (VIT_11s0016g01860) was upregulated and DRP (VIT_01s0026g01420) was down-regulated. Among two glutathione S-transferase (GST) genes, VIT_12s0028g00920 was up-regulated, whereas VIT_04s0079g00690 was down-regulated (Supplementary Table 8). These genes collectively play an important role in disease resistance responses.
4 Discussion
Colletotrichum spp. are highly destructive fungal pathogens to various plants, such as grapes, peppers, and lentils (Lei et al., 2016; Bhadauria et al., 2019; Huo et al., 2021). They can invade various parts of plants, including leaves, stems, shoots, tendrils, and berries Wang et al., 2021). Among these pathogens, C. viniferum poses a particularly serious risk to grape production. Previous research has demonstrated varying degrees of grapevine resistance to C. viniferum, indicating that different plant materials exhibit distinct reactions towards the same pathogen (Baroncelli et al., 2014; Echeverrigaray et al., 2020).
Histological observation is an effective method for studying the differences in cytology and ultrastructure of grapes infected with different germplasm resources by C. viniferum. Transcriptome analysis has become a common approach to studying pathogenic infection hosts. In this study, these two methods were used to reveal the interaction between grape and C. viniferum, providing a basis for the selection of candidate fungal pathogenic genes including CAZymes, effectors, P450 and grapevine response genes including plant hormone signaling, metabolite synthesis, expression of transcription factors, ROS clear genes and disease-associated genes.
4.1 C. viniferum developed rapidly on susceptible grapes
Current studies on Colletotrichum spp. in grapes have mainly focused on evolutionary and taxonomic aspects, with limited histological examination of the infection process of Colletotrichum spp., especially C. viniferum (Whitelaw-Weckert et al., 2010; Yan et al., 2015). Observations of Colletotrichum spp. on grape morphology, including conidia, appressorium, and hyphae, remain relatively scarce (Melksham et al., 2002; Peng et al., 2013; Oo and Oh, 2017). To evaluate the variance in C. viniferum infection on the differences of grapevines, the histopathology of the leaves of high-susceptible grape germplasm “TS”, medium-resistant grape germplasm “B”, and resistant grape germplasm “LB-8” were observed by transmission electron microscopy and scanning electron microscopy. By comparing the resistance levels of these three germplasm grapes to C. viniferum, the differences between susceptible grapes and resistant grapevine germplasm were studied. The results of this study can provide a theoretical basis for the later study of the pathogenesis of C. viniferum.
Our study revealed significant differences in the response of susceptible and resistant grapevines to C. viniferum infection. One notable distinction was the fast disintegration of upper epidermal cells in highly susceptible grapes compared to medium-resistant and resistant grapes. Additionally, visible damage on the leaf surface formed earlier in the susceptible grapevine (Figures 2, 3A). After C. viniferum infection, white secretions appeared on the leaves of “B” and “LB-8”, while “TS” did not (Figure 3B), presumably a response, similar to previous studies on downy mildew infection of resistant grapes, which showed that “Langao-5” and “Liuba-8” resistant grape germplasms rapidly produced white secretion to limit downy mildew in the early stage of inoculation (Yin et al., 2017). We observed variations in the formation rate of appressoria and penetration pegs among different grapes. “TS” exhibited faster formation of appressoria and penetration pegs compared to “B” and “LB-8”. This pattern is consistent with findings in other pathosystems, such as C. acutatum-almond pathosystem (Diéguez-Uribeondo et al., 2005). Interestingly, “B” and “LB-8” displayed more than 30% malformed appressoria, which we attribute to two factors. Firstly, the secretion from resistant grapes “B” and “LB-8” may interfere with appressorium formation (Figure 3B), a response observed in grapevines against downy mildew (Yu et al., 2012; Yin et al., 2017). Secondly, the dense hair on grapevine leaves may have compressed and deformed appressoria (Figure 3C), acting as a primary barrier against fungal infection (Kortekamp and Zyprian, 1999).
Colletotrichum spp. infections typically undergo a biotrophic phase in the early stage, followed by a necrotrophic stage after the emergence of secondary hyphae (Shang et al., 2020; Li et al., 2021a). The period of the brief biotrophic stage of Colletotrichum spp. infection varied by host and other factors. For instance, when C. fructicola infects apple leaves, its short biotrophic stage continues for at least 36 hpi, and then it transforms into the necrotrophic stage at 48 hpi (Shang et al., 2020). Similarly, our observations indicate variations in the duration of the biotrophic phase among grape germplasm resources. For “TS”, the brief biotrophic phase lasted 24 h before transitioning to the necrotrophic phase by the emergence of secondary hyphae and the disintegration of epidermal cells. However, “B” exhibited a longer biotrophic stage than “TS” before progressing to the necrotrophic stage. Notably, secondary hyphae were not seen on “LB-8”, indicating that C. viniferum infection on the resistant grape “LB-8” maintains in the biotrophic stage (Figure 3C).
4.2 C. viniferum pathogenicity genes may play an important role in infection
Transcriptome sequencing offers a direct way to explore gene expression levels, and numerous studies have investigated transcriptome sequencing of Colletotrichum spp.-infected hosts, encompassing both fungal and host transcriptomes. Transcriptome analyses of Colletotrichum spp. includes C. fructicola on apples (Liang et al., 2018), C. falcatum on sugarcane (Prasanth et al., 2022), and C. camelliae on tea (He et al., 2023). However, the transcriptome of C. viniferum on grapes has not been studied. To study the pathogenic mechanism of C. viniferum, we conducted transcriptome analysis using the susceptible grape “TS”. Through transcriptome analysis, we identified a range of differentially expressed pathogenic factors, including 35 CAZymes, 56 effectors, and 5 P450 genes. Secondary metabolites are related to pathogenicity of fungi during infection. We also identified 67 secondary metabolic gene clusters, and only 10 genes (A01092, A01212, A01323, A07559, A08496, A11116, A13125, A13129, A13635, A13736) distributed in 9 secondary metabolite clusters (Cluster2, 21, 25, 26, 32, 33, 35, 57 and 65) had significant differences in expression levels during infection, these 10 genes may play core roles, and the remaining other genes on these 9 clusters may play accessory roles in fungal pathogenicity. (Supplementary Tables 4, 10).
The numbers of major CAZyme superfamilies and CAZymes in CvYL2a were lower than those in other fungi (Figure 5A). Some of these enzymes were responsible for plant cell wall degradation, such as pectinase and cellulase. During C. viniferum infection of “TS”, the expression levels of these genes significantly increased, indicating their important roles in grape infection (Figure 5F). Furthermore, fungal and oomycete effectors are known as pivotal pathogenic factors that modulate plant immunity and affect disease development (Nie et al., 2019; Chen et al., 2021; Yin et al., 2022). In this study, two effectors induced HR responses in plants, potentially linked to plant resistance R genes in plants. Moreover, ten effectors were found to inhibit INF1-induced cell death, indicating their ability to evade immune recognition by grapevines and promote pathogen infection (Figure 5G).
4.3 Grape responses to C. viniferum include plant hormone signaling, metabolite synthesis, expression of transcription factors, ROS clear genes, and disease-associated genes
To explore grapevine responses to C. viniferum, we analyzed differentially expressed grapes genes involved in plant hormone signaling and metabolite synthesis, transcription factors, ROS clearance genes, and disease-associated genes. Plant hormones are important in plant responses to pathogens (Zhang et al., 2017; Li et al., 2019a). In this study, the level of SA increased at 6-12 hpi, JA increased at 24-48 hpi, and ACC, synthetic precursor of ET, increased at 12-24 hpi (Figure 10D). Our experiment indicated that the early stage of infection corresponds to the biotrophic stage, while the later stage corresponds to the necrotrophic stage. This is consistent with previous findings that SA-dependent responses are associated with biotrophic organisms, while ET and JA-dependent responses are associated with necrotic organisms (Glazebrook, 2005). The expression levels of hormone synthesis-related genes, downstream transcription factors (MYC, ERF, and TGA), and disease-related genes (PR1) were upregulated in the transcriptome. This finding suggests that these hormones may activate downstream transcription factors and disease-resistant genes. Hormonal networks that regulate downstream gene expression during plant-pathogen interactions have long been discovered. For example, SA activates TGA and PR1 (Shimizu et al., 2022), JA activates MYC (Schmiesing et al., 2016), and ethylene activates ERF (Broekaert et al., 2006). Additionally, other differentially expressed transcription factors (WRKY, SBP, NAM, MYB, and GATA) may also play critical roles in plant disease resistance and warrant further experimental studies (Figure 9). We hypothesize that the induction of JA, ET, and SA signaling pathways, along with the activation of transcription factors, are essential components of the grape response to C. viniferum.
Phenylpropanoid-guided flavonoid and resveratrol biosynthesis pathways, phenylpropanoid biosynthesis pathways, and photosynthetic antenna proteins have been implicated in plant resistance to pathogens (Jantasuriyarat et al., 2005; Manickavelu et al., 2010; Nabavi et al., 2020; Huang et al., 2021). Transcriptome analysis revealed upregulation of resveratrol synthesis genes during C. viniferum infection (Figure 6). Resveratrol is important in the fight against pathogen infections (Vestergaard and Ingmer, 2019; Zhou et al., 2024). This suggests a coordinated response wherein plants synthesize more resveratrol to resist C. viniferum infection. In phenylpropanoid biosynthesis, the downregulation of CAD genes, a key enzyme in lignin synthesis, was observed. CAD7, a negative regulator of plant immunity targeted by Avr3a effectors, promotes pathogen infection by inhibiting plant PAMP-triggered immunity (Li et al., 2019b). This raises the possibility that CAD could serve as a target protein for fungal effectors, leading to the loss of lignin synthesis activity and further promoting pathogen infection (Figure 7). Conversely, all differentially expressed photosynthetic antenna proteins were downregulated. Lhca/b-binding protein in the photosynthetic pathway is one of the most abundant proteins in plant chloroplasts. Genes encoding the Lhca/b protein have been identified in interaction between susceptible plants and pathogens (Jantasuriyarat et al., 2005; Manickavelu et al., 2010). The process of inducing plant response systems requires a lot of energy, which increases the need for photosynthesis in plants (Swarbrick et al., 2006). However, this study showed down regulation of Lhca and Lhcb during infection (Figure 8). Similarly, previous experiments have shown that tomato seedlings inoculated with DC3000 were also downregulated the expression of photosynthesis-related genes (Ishiga et al., 2009). This suggests that plants may limit the carbon sources available to pathogens or protect plant cells from oxidative damage by downregulating photosynthesis.
Earlier research has highlighted the diverse roles of plant response genes, such as chalcone synthase, pathogenesis-related proteins, catalase, and chitinase, in conferring resistance or tolerance to fungal pathogens (Nandeeshkumar et al., 2008; Jaulneau et al., 2010; Feng et al., 2011). In this study, we observed an earlier response in plants “LB-8” and “B” compared to “TS”, characterized by higher expression levels of response genes (Figure 11E). Another notable difference is the accumulation of reactive oxygen species. Hydrogen peroxide increases significantly during host-pathogen interactions (Wang et al., 2010). Our results suggest that plant ROS clearance genes are upregulated during “TS” infection (Figure 11F), consistent with previous findings on pathogenic infections (Segal and Wilson, 2018). Peroxidase activity in grapes during C. viniferum infection may contribute to lignin breakdown and ROS scavenging (Figure 7). The most studied function of LRR protein in plants is to play an important role in disease resistance. LRR-encoding genes act as pattern recognition receptors (PRRs) to sense pathogen-associated molecular patterns (PAMPs) and as R genes involved in the immune responses of plants (Kunze et al., 2004; Meyers et al., 2005). Additionally, genes associated with grape response, including LRR, disease resistance genes, and glutathione S-transferase, were upregulated (Supplementary Table 6). Despite the elevated expression levels of these genes, susceptible grapevines remain infected with C. viniferum and develop lesions. This phenomenon parallels observations in susceptible grapevines V. vinifera cv. Red globe infected with Elsinoë ampelina (Li et al., 2021c).
Currently, only three grape transcriptome datasets related to Colletotrichum spp. infection are available (Lei et al., 2022; Shen et al., 2022; Yu et al., 2023). However, these three studies exclusively analyzed grape transcripts, leaving the changes in both Colletotrichum and grape transcriptomes during the infection process remain unexplored. Therefore, our research employed simultaneous transcriptome analyses to examine fungal genes associated with pathogenesis and grape genes associated with defense responses. We proposed a model depicting the interaction between grape and C. viniferum (Figure 12), providing a basis for further research on the pathogenesis of C. viniferum and the interaction between grapevines and C. viniferum.
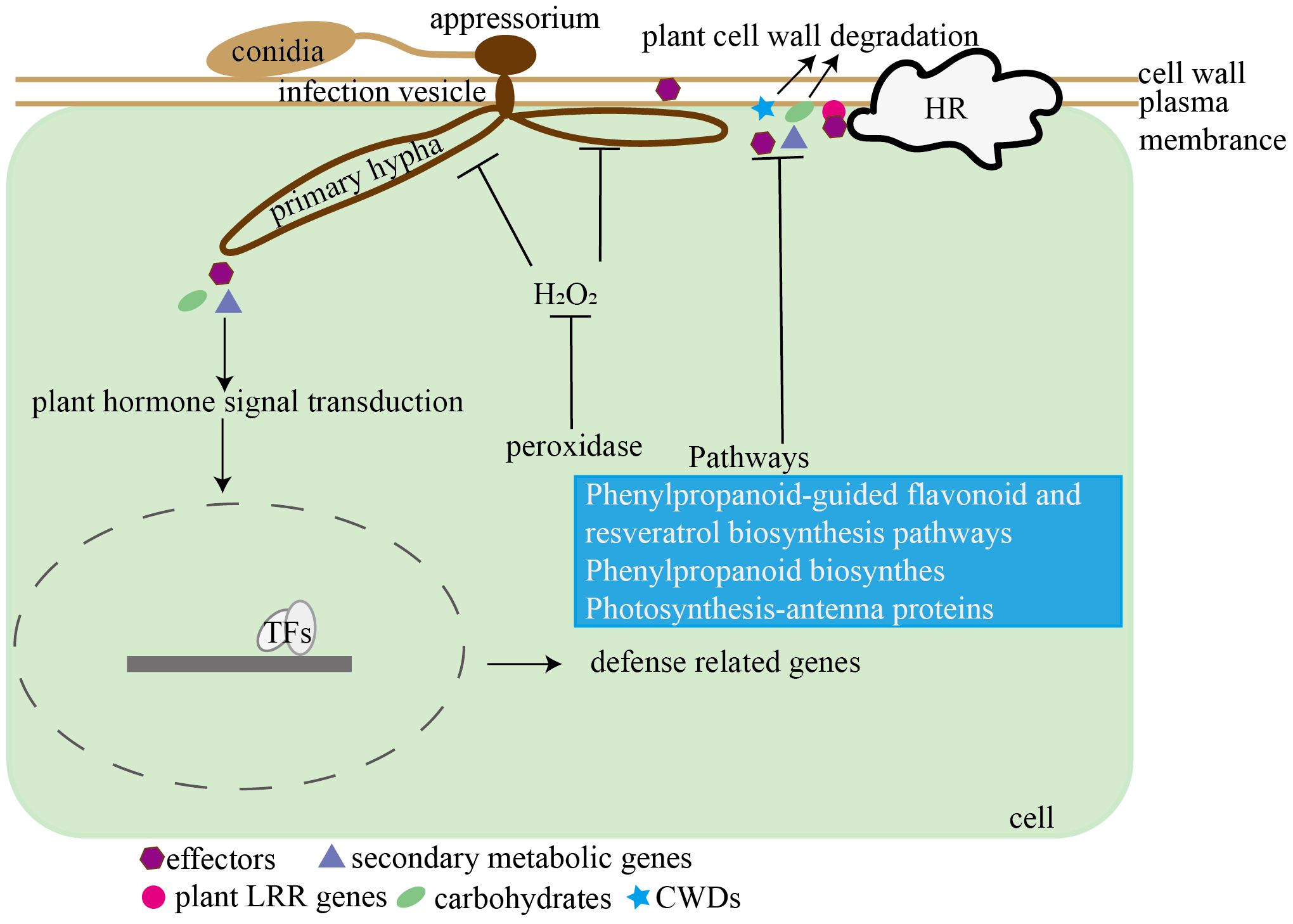
Figure 12. Hypothetical model of the interaction between C. viniferum and V. vinifera. C. viniferum infects grapevines through pathogenic genes including effectors, carbohydrates, secondary metabolites, and plant cell wall degradation enzymes, while grapes inhibit C. viniferum infection by LRRs recognizing pathogenic fungi and activating ROS accumulation.
5 Conclusion
In summary, our comparative analysis of susceptible and resistant grapevines through light and electron microscopy revealed distinct differences in infection dynamics. Notably, the diameter of C. viniferum lesions was the largest on “TS”, followed by “B”, and smallest on “LB-8”. Susceptible grapes exhibited no white secretion, normal appressoria shape, and the formation of secondary hyphae, facilitating C. viniferum infection on grapes. We propose a model illustrating the interaction between grapes and C. viniferum (Figure 12). C. viniferum employs a variety of virulence strategies, including effectors, CWDEs, and secondary metabolites to target various cellular processes for nutrient acquisition. Conversely, grapevine responses to C. viniferum involves hormone signaling, disease-related genes expression, metabolic pathways activation, ROS accumulation, and transcription factors regulation. Our findings hold significance for the selection of candidate fungal pathogenic genes and plant disease response genes. Furthermore, they laid the groundwork for further investigation into the pathogenic mechanisms of C. viniferum and the intricacies of the grape and C. viniferum interaction.
Data availability statement
All raw RNA-Seq read data in this study can be found in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) with the accession numbers PRJNA1022593 and PRJNA1022588. The genomic datasets in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
MD: Data curation, Validation, Visualization, Writing – original draft, Writing – review & editing, Conceptualization, Formal analysis, Methodology, Investigation. YL: Validation, Writing – review & editing, Investigation. YH: Validation, Writing – review & editing. KZ: Validation, Writing – review & editing. XY: Writing – review & editing, Methodology. ZF: Validation, Writing – review & editing. XX: Validation, Writing – review & editing. QZ: Visualization, Writing – review & editing. WB: Visualization, Writing – review & editing. XC: Writing – review & editing, Visualization. GL: Writing – review & editing. YW: Writing – review & editing. LT: Writing – review & editing, Funding acquisition. YX: Funding acquisition, Writing – review & editing, Conceptualization, Project administration, Resources, Supervision.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Project of Agricultural Breeding in Ningxia Hui Autonomous Region, China (NXNYYZ20210104), and Shenzhen Polytechnic University Research Project (6024330001K).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1446288/full#supplementary-material
References
Arroyo, F. T., Moreno, J., García-Herdugo, G., Santos, B. D. L., Barrau, C., Porras, M., et al. (2005). Ultrastructure of the early stages of Colletotrichum acutatum infection of strawberry tissues. Can. J. Bot. 83, 491–500. doi: 10.1139/b05-022
Ayako, T., Pamela, G., Naoyoshi, K., Mari, N., Yoshitaka, T., Yoshihiro, N., et al. (2019). Genomic plasticity mediated by transposable elements in the plant pathogenic fungus Colletotrichum higginsianum. Genome Biol. Evol. 11, 1487–1500. doi: 10.1093/gbe/evz087
Baroncelli, R., Sanz-Martín, J. M., Rech, G. E., Sukno, S. A., Thon, M. R. (2014). Draft genome sequence of Colletotrichum sublineola, a destructive pathogen of Cultivated Sorghum. Genome Announc. 2, e00540–e00514. doi: 10.1128/genomeA.00540-14
Bhadauria, V., Banniza, S., Vandenberg, A., Selvaraj, G., Wei, Y. (2013). Overexpression of a novel biotrophy-specific Colletotrichum truncatum effector, CtNUDIX, in hemibiotrophic fungal phytopathogens causes incompatibility with their host plants. Eukaryot. Cell. 12, 2–11. doi: 10.1128/EC.00192-12
Bhadauria, V., MacLachlan, R., Pozniak, C., Cohen-Skalie, A., Li, L., Halliday, J., et al. (2019). Genetic map-guided genome assembly reveals a virulence-governing minichromosome in the lentil anthracnose pathogen Colletotrichum lentis. New Phytol. 221, 431–445. doi: 10.1111/nph.15369
Broekaert, W. F., Delauré, S. L., De Bolle, M. F., Cammue, B. P. (2006). The role of ethylene in host-pathogen interactions. Annu. Rev. Phytopathol. 44, 393–416. doi: 10.1146/annurev.phyto.44.070505.143440
Buchfink, B., Xie, C., Huson, D. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Buiate, E. A. S., Xavier, K. V., Moore, N., Torres, M. F., Farman, M. L., Schardl, C. L., et al. (2017). A comparative genomic analysis of putative pathogenicity genes in the host-specific sibling species Colletotrichum graminicola and Colletotrichum sublineola. BMC Genomics 18, 67. doi: 10.1186/s12864-016-3457-9
Chen, C. J., Xia, R., Chen, H., He, Y. H. (2018). TBtools, a toolkit for biologists integrating various biological data handling tools with a user-friendly interface. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, T. T., Peng, J., Yin, X., Li, M. J., Xiang, G. Q., Wang, Y. J., et al. (2021). Importin-αs are required for the nuclear localization and function of the Plasmopara viticola effector PvAVH53. Hortic. Res-England. 8, 46. doi: 10.1038/s41438-021-00482-6
Cosseboom, S. D., Hu, M. (2022). Ontogenic susceptibility of grapevine clusters to ripe rot, caused by the Colletotrichum acutatum and C. gloeosporioides species complexes. Phytopathol. 112, 1956–1964. doi: 10.1094/PHYTO-01-22-0004-R
Diéguez-Uribeondo, J., Forster, H., Soto-Estrada, A., Adaskaveg, J. E. (2005). Subcuticular-intracellular hemibiotrophic and intercellular necrotrophic development of Colletotrichum acutatum on almond. Phytopathology 95, 751–758. doi: 10.1094/PHYTO-95-0751
Ding, L. N., Li, M., Guo, X. J., Tang, M. Q., Cao, J., Wang, Z., et al. (2019). Arabidopsis GDSL1 overexpression enhances rapeseed Sclerotinia sclerotiorum resistance and the functional identification of its homolog in Brassica napus. Plant Biotechnol. J. 18, 1255–1270. doi: 10.1111/pbi.13289
Dong, Y., Duan, S., Xia, Q., Liang, Z., Dong, X., Margaryan, K., et al. (2023). Dual domestications and origin of traits in grapevine evolution. Science 379, 892–901. doi: 10.1126/science.add8655
Dou, M. R., Hao, Y., Yang, J., Yuan, X. J., Yin, X., Jiao, Y. T., et al. (2022). Genome sequence resource for Colletotrichum viniferum, the cause of grapevine ripe rot in China. Mol. Plant Microbe In. 35, 90–93. doi: 10.1094/MPMI-04-21-0077-A
Echeverrigaray, S., Scariot, F. J., Fontanella, G., Favaron, F., Sella, L., Santos, M. C., et al. (2020). Colletotrichum species causing grape ripe rot disease in Vitis labrusca and V. vinifera varieties in the highlands of southern Brazil. Plant Pathol. 69, 1504–1512. doi: 10.1111/ppa.13240
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Feng, L., Dong, M., Huang, Z., Wang, Q., An, B., He, C., et al. (2024). CgCFEM1 is required for the full virulence of Colletotrichum gloeosporioides. Int. J. Mol. Sci. 25, 2937. doi: 10.3390/ijms25052937
Feng, J. X., Cao, L., Li, J., Duan, C. J., Luo, X. M., Le, N., et al. (2011). Involvement of OsNPR1/NH1 in rice basal resistance to blast fungus Magnaporthe oryzae. Eur. J. Plant Pathol. 131, 221–235. doi: 10.1007/s10658-011-9801-7
Gao, X. M., Fan, X. C., Zhang, G. H., Zhang, Y., Jiang, J. F., Sun, H. S., et al. (2016). Identification and screening of hybrid progenies with resistance to grape anthracnose. Acta Agriculturae Boreali-Occidentalis Sin. 25, 882–888. doi: 10.7606/j.issn.1004-1389.2016.06.014
Gao, H., Jiang, L. Y., Du, B. H., Ning, B., Ding, X. D., Zhang, C. Z., et al. (2022). GmMKK4-activated GmMPK6 stimulates GmERF113 to trigger resistance to Phytophthora sojae in soybean. Plant J. 111, 473–495. doi: 10.1111/tpj.15809
Glazebrook, J. (2005). Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 43, 205–227. doi: 10.1146/annurev.phyto.43.040204.135923
Gutiérrez, P., Alzate, J., Yepes, M. S., Marin, M. (2016). Complete mitochondrial genome sequence of the common bean anthracnose pathogen Colletotrichum lindemuthianum. Mitochondrial DNA A 27, 136–137. doi: 10.3109/19401736.2013.878912
Han, J. H., Chon, J. K., Ahn, J. H., Choi, I. Y., Lee, Y. H., Kim, K. S. (2016). Whole genome sequence and genome annotation of Colletotrichum acutatum, causal agent of anthracnose in pepper plants in South Korea. Genomics Data 8, 45–46. doi: 10.1016/j.gdata.2016.03.007
Han, R., Yin, W., Ahmad, B., Gao, P., Li, Z., Wang, X. (2021). Pathogenesis and immune response in resistant and susceptible cultivars of grapevine (Vitis spp.) against Elsinoë ampelina infection. Phytopathology. 111, 799–807. doi: 10.1094/PHYTO-03-20-0079-R
He, S., Qiao, X., Zhang, S., Xia, J., Wang, L., Liu, S. (2023). Urate oxidase from tea microbe Colletotrichum camelliae is involved in the caffeine metabolism pathway and plays a role in fungal virulence. Front. Nutr. 9. doi: 10.3389/fnut.2022.1038806
He, P. C., Ren, Z. B. (1990). Study on the resistances of wild Vitis species native to China to grape ripe rot. J. Fruit Sci. 7, 7–12. doi: 10.13925/j.cnki.gsxb.1990.01.002
Hedges, S. B., Dudley, J., Kumar, S. (2006). TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972. doi: 10.1093/bioinformatics/btl505
Huang, X. X., Wang, Y., Lin, J. S., Chen, L., Li, Y. J., Liu, Q., et al. (2021). The novel pathogen-responsive glycosyltransferase UGT73C7 mediates the redirection of phenylpropanoid metabolism and promotes SNC1-dependent Arabidopsis immunity. Plant J. 107, 149–165. doi: 10.1111/tpj.15280
Huckelhoven, R., Fodor, J., Kogel, K. H. (1999). Hypersensitive cell death and papilla formation in barley attacked by the powdery mildew fungus are associated with hydrogen peroxide but not with salicylic acid accumulation. Plant Physiol. 119, 1251–1260. doi: 10.1104/pp.119.4.1251
Huo, J., Wang, Y., Hao, Y., Yao, Y., Wang, Y., Zhang, K., et al. (2021). Genome sequence resource for Colletotrichum scovillei, the cause of anthracnose disease of chili. Mol. Plant Microbe In. 34, 122–126. doi: 10.1094/MPMI-03-20-0055-A
Ishiga, Y., Uppalapati, S. R., Ishiga, T., Elavarthi, S., Martin, B., Bender, C. L. (2009). The phytotoxin coronatine induces light-dependent reactive oxygen species in tomato seedlings. New Phytol. 181, 147–160. doi: 10.1111/j.1469-8137.2008.02639.x
Jantasuriyarat, C., Gowda, M., Haller, K., Hatfield, J., Lu, G. D., Stahlberg, E., et al. (2005). Large-scale identification of expressed sequence tags involved in rice and rice blast fungus interaction. Plant Physiol. 138, 105–115. doi: 10.1104/pp.104.055624
Jaulneau, V., Cazaux, M., Hoi, J. W. S., Fournier., S., Esquerré-Tugayé, M., Jacquet, C., et al. (2010). Host and nonhost resistance in Medicago-Colletotrichum interactions. Mol. Plant Microbe Interact. 23, 1107–1117. doi: 10.1094/MPMI-23-9-1107
Ji, T., Salotti, I., Dong, C., Li, M., Rossi, V. (2021). Modeling the effects of the environment and the host plant on the ripe rot of grapes, caused by the Colletotrichum species. Plants 10, 2288. doi: 10.3390/plants10112288
Joung, J. G., Corbett, A. M., Fellman, S. M., Tieman, D. M., Klee, H. J., Giovannoni, J. J., et al. (2010). Plant MetGenMAP: an integrative analysis system for plant systems biology. Plant Physiol. 151, 1758–1768. doi: 10.1104/pp.109.145169
Kim, M. R., Oh, S. K. (2019). Analysis of varietal difference and genetic diversity of grapevine cultivars through the leaf inoculation of Colletotrichum spp. J. Agric. Life Sci. 52, 49–60. doi: 10.14397/jals.2018.52.6.49
Klimmek, F., Sjödin, A., Noutsos, C., Leister, D., Jansson, S. (2006). Abundantly and rarely expressed Lhc protein genes exhibit distinct regulation patterns in plants. Plant Physiol. 140, 793–804. doi: 10.1104/pp.105.073304
Kortekamp, A., Zyprian, E. (1999). Leaf hairs as a basic protective barrier against downy mildew of grape. J. Phytopathol. 147, 453–459. doi: 10.1111/j.1439-0434.1999.tb03850.x
Kunze, G., Zipfel, C., Robatzek, S., Niehaus, K., Boller, T., Felix, G. (2004). The N terminus of bacterial elongation factor Tu elicits innate immunity in Arabidopsis plants. Plant Cell 16, 3496–3507. doi: 10.1105/tpc.104.026765
Lei, Y., Tang, X. B., Jayawardena, R. S., Yan, J. Y., Wang, X. D., Liu, M., et al. (2016). Identification and characterization of Colletotrichum species causing grape ripe rot in southern China. Mycosphere 7, 1177–1191. doi: 10.5943/mycosphere/si/2c/8
Lei, Y., Yuan, X. J., Chen, T., Yuan, Y., Liu, X. M., Tang, X. B., et al. (2022). Transcriptome analysis of berries of spine grape (Vitis davidii Föex) infected by Colletotrichum viniferum during symptom development. Horticulturae 8, 843. doi: 10.3390/horticulturae8090843
Li, N., Han, X., Feng, D., Yuan, D. Y., Huang, L. J. (2019a). Signaling crosstalk between salicylic acid and ethylene/jasmonate in plant defense: do we understand what they are whispering. Int. J. Mol. Sci. 20, 671. doi: 10.3390/ijms20030671
Li, Z. Q., Jiao, Y. T., Zhang, C., Dou, M. R., Weng, K., Wang, Y. J., et al. (2021b). VvHDZ28 positively regulates salicylic acid biosynthesis during seed abortion in Thompson Seedless. Plant Biotechnol. J. 19, 1824–1838. doi: 10.1111/pbi.13596
Li, M., Liu, J., Zhou, G. Y. (2021a). Histopathological and ultrastructural observations of Camellia oleifera infected with Colletotrichum fructicola. Australas. Plant Pathol. 50, 523–531. doi: 10.1007/s13313-021-00811-2
Li, Z., Wang, Y., Fan, Y., Ahmad, B., Wang, X., Zhang, S., et al. (2021c). Transcriptome analysis of the grape-Elsinoë ampelina pathosystem reveals novel effectors and a robust defense response. Mol. Plant Microbe In. 34, 110–121. doi: 10.1094/MPMI-08-20-0227-R
Li, T. T., Wang, Q. H., Feng, R. R., Li, L. C., Ding, L. W., Fan, G. J., et al. (2019b). Negative regulators of plant immunity derived from cinnamyl alcohol dehydrogenases are targeted by multiple Phytophthora Avr3a-like effectors. New Phytol. doi: 10.1111/nph.16139
Li, P., Zhao, L., Qi, F., Htwe, N. M. P. S., Li, Q., Zhang, D., et al. (2021). The receptor-like cytoplasmic kinase RIPK regulates broad-spectrum ROS signaling in multiple layers of plant immune system. Mol. Plant 14, 1652–1667. doi: 10.1016/j.molp.2021.06.010
Liang, X., Shang, S., Dong, Q., Wang, B., Zhang, R., Gleason, M. L., et al. (2018). Transcriptomic analysis reveals candidate genes regulating development and host interactions of Colletotrichum fructicola. BMC Genomics 19, 557. doi: 10.1186/s12864-018-4934-0
Liu, T. L., Chen, T. Z., Kan, J. L., Yao, Y., Guo, D. S., Yang, Y. W., et al. (2021). The GhMYB36 transcription factor confers resistance to biotic and abiotic stress by enhancing PR1 gene expression in plants. Plant Biotechnol. J. 20, 722–735. doi: 10.1111/pbi.13751
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Manickavelu, A., Kawaura, K., Oishi, K., Shin-I, T., Kohara, Y., Yahiaoui, N., et al. (2010). Comparative gene expression analysis of susceptible and resistant near-isogenic lines in common wheat infected by Puccinia triticina. DNA Res. 17, 211–222. doi: 10.1093/dnares/dsq009
Melksham, K. J., Weckert, M. A., Steel, C. C. (2002). An unusual bunch rot of grapes in sub-tropical regions of Anstralia caused by Colletotrichum acutatum. Australas. Plant Pathol. 31, 193–194. doi: 10.1071/AP02013
Meng, Y. A., Gleason, M. L., Zhang, R., Sun, G. Y. (2019). Genome sequence resource of the wide-host-range anthracnose pathogen Colletotrichum siamense. Mol. Plant Microbe In. 32, 931–934. doi: 10.1094/MPMI-01-19-0010-A
Meyers, B. C., Kaushik, S., Nandety, R. S. (2005). Evolving disease resistance genes. Curr. Opin. Plant Biol. 8, 129–134. doi: 10.1016/j.pbi.2005.01.002
Moraes, S. R. G., Tanaka, F. A. O., Massola, N. S. (2013). Histopathology of Colletotrichum gloeosporioides on guava fruits (Psidium guajava L.). Rev. Bras. Frutic. 35, 657–664. doi: 10.1590/S0100-29452013000200039
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nabavi, S., Šamec, D., Tomczyk, M., Milella, L., Russo, D., Habtemariam, S., et al. (2020). Flavonoid biosynthetic pathways in plants: Versatile targets for metabolic engineering. Biotechnol. Adv. 38, 107316. doi: 10.1016/j.biotechadv.2018.11.005
Nandakumar, M., Malathi, P., Sundar, A. R., Viswanathan, R. (2021). Expression analyses of resistance-associated candidate genes during sugarcane-Colletotrichum falcatum went interaction. Sugar Tech 23, 1056–1063. doi: 10.1007/s12355-021-00976-6
Nandeeshkumar, P., Ramachandrakini, K., Prakash, H. S., Niranjana, S. R., Shekar Shetty, H. (2008). Induction of resistance against downy mildew on sunflower by rhizobacteria. J. Plant Interact. 3, 255–262. doi: 10.1080/17429140802245697
Nie, J. J., Yin, Z. Y., Li, Z. P., Wu, Y. X., Huang, L. L. (2019). A small cysteine-rich protein from two kingdoms of microbes is recognized as a novel pathogen-associated molecular pattern. New Phytol. 222, 995–1011. doi: 10.1111/nph.15631
Oo, M. M., Oh, S. K. (2017). Identification and characterization of new record of grape ripe rot disease caused by Colletotrichum viniferum in Korea. Mycobiology 45, 421–425. doi: 10.5941/MYCO.2017.45.4.421
Pan, F. Y., Huang, Y., Lin, L., Zhou, Y. M., Wei, R. F., Guo, W. F., et al. (2016). First report of Colletotrichum capsici causing grape ripe rot in Guangxi, China. Plant Dis. 100, 2531–2531. doi: 10.1094/PDIS-05-16-0691-PDN
Peng, L. J., Sun, T., Yang, Y. L., Cai, L., Hyde, K. D., Bahkali, A. H., et al. (2013). Colletotrichum species on citrus leaves in Guizhou and Yunnan provinces, China. Mycoscience 54, 29–41. doi: 10.1094/PDIS-06-23-1057-PDN
Prasanth, C. N., Viswanathan, R., Malathi, P., Sundar, A. R. (2022). Carbohydrate active enzymes (CAZy) regulate cellulolytic and pectinolytic enzymes in Colletotrichum falcatum causing red rot in sugarcane. 3 Biotech. 12, 48. doi: 10.1007/s13205-022-03113-6
Sanderson, M. J. (2003). r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301. doi: 10.1093/bioinformatics/19.2.301
Schmiesing, A., Emonet, A., Gouhier-Darimont, C., Reymond, P. (2016). Arabidopsis MYC transcription factors are the target of hormonal salicylic acid/jasmonic acid cross talk in response to Pieris brassicae egg extract. Plant Physiol. 170, 2432–2443. doi: 10.1104/pp.16.00031
Segal, L. M., Wilson, R. A. (2018). Reactive oxygen species metabolism and plant-fungal interactions. Fungal Genet. Biol. 110, 1–9. doi: 10.1016/j.fgb.2017.12.003
Shang, S. P., Liang, X. F., Liu, G. L., Zhang, S., Lu, Z. X., Zhang, R., et al. (2020). Histological and ultrastructural characterization of the leaf infection events of Colletotrichum fructicola on Malus domestica ‘Gala’. Plant Pathol. 69, 538–548. doi: 10.1111/ppa.13141
Shen, C. Q., Sun, L., Zhang, Y., Jiang, J. F., Liu, C. H., Fan, X. C. (2022). Transcriptomic analysis of different resistant leaves infected by Colletotrichum gloeosporioides in grape. J. Fruit Sci. 39, 730–742. doi: 10.13925/j.cnki.gsxb.20210573
Shimizu, K., Suzuki, H., Uemura, T., Nozawa, A., Desaki, Y., Hoshino, R., et al. (2022). Immune gene activation by NPR and TGA transcriptional regulators in the model monocot Brachypodium distachyon. Plant J. 110, 470–481. doi: 10.1111/tpj.15681
Shiraishi, M., Koide, M., Itamura, H., Yamada, M., Mitani, N., Ueno, T. (2007). Screening for resistance to ripe rot caused by Colletotrichum acutatum in grape germplasm. Vitis 46, 196–200. doi: 10.2503/jjshs.75.264
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Swarbrick, P. J., Schulze-Lefert, P., Scholes, J. D. (2006). Metabolic consequences of susceptibility and resistance (race-specific and broad-spectrum) in barley leaves challenged with powdery mildew. Plant Cell Environ. 29, 1061–1076. doi: 10.1111/j.1365-3040.2005.01472.x
Tsushima, A., Narusaka, M., Gan, P., Kumakura, N., Hiroyama, R., Kato, N., et al. (2021). The conserved Colletotrichum spp. effector candidate CEC3 induces nuclear expansion and cell death in plants. Front. Microbiol. 3, 682155. doi: 10.3389/fmicb.2021.682155
Velho, A. C., Rockenbach, M. F., Mondino, P., Stadnika, M. J. (2016). Modulation of oxidative responses by a virulent isolate of Colletotrichum fructicola in apple leaves. Fungal Biol-UK. 120, 1184–1193. doi: 10.1016/j.funbio.2016.07.001
Vestergaard, M., Ingmer, H. (2019). Antibacterial and antifungal properties of resveratrol. Int. J. Antimicrob. Ag. 53, 716–723. doi: 10.1016/j.ijantimicag.2019.02.015
Wang, C. F., Huang, L. L., Zhang, H. C., Han, Q. M., Buchenauer, H., Kang, Z. S. (2010). Cytochemical localization of reactive oxygen species (O2 and H2O2) and peroxidase in the incompatible and compatible interaction of wheat-Puccinia striiformis f. sp. tritici. Physiol. Mol. Plant P. 74, 221–229. doi: 10.1016/j.pmpp.2010.02.002
Wang, M., Luo, X., Wang, H. (2021). First draft genome sequence resource of Colletotrichum liriopes causing leaf anthracnose on Ophiopogon japonicus. Plant Dis. 105, 1179–1182. doi: 10.1094/PDIS-11-20-2326-A
Wharton, P. S., Schilder, A. C. (2008). Novel infection strategies of Colletotrichum acutatum on ripe blueberry fruit. Plant Pathol. 57, 122–134. doi: 10.1111/j.1365-3059.2007.01698.x
Whitelaw-Weckert, M. A., Curtin, S. J., Huang, R., Steel, C. C., Blanchard, C. L., Roffey, P. E. (2010). Phylogenetic relationships and pathogenicity of Colletotrichum acutatum isolates from grape in subtropical Australia. Plant Pathol. 56, 448–463. doi: 10.1111/j.1365-3059.2007.01569.x
Xia, J., Liu, N., Han, J., Sun, J., Xu, T., Liu, S. (2023). Transcriptome and metabolite analyses indicated the underlying molecular responses of Asian ginseng (Panax ginseng) toward Colletotrichum panacicola infection. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1182685
Xu, X. W., Li, J. C., Hai, D., Wang, Y. X., Li, J. Y., Zha, Y. P. (2024). Complete genome sequence of a novel alternavirus isolated from the phytopathogenic fungus Colletotrichum fioriniae. Arch. Virol. 169, 79. doi: 10.1007/s00705-024-06010-w
Yan, J. Y., Jayawardena, M. M. R. S., Goonasekara, I. D., Wang, Y., Zhang, W., Liu, M., et al. (2015). Diverse species of Colletotrichum associated with grapevine anthracnose in China. Fungal Divers. 71, 233–246. doi: 10.1007/s13225-014-0310-9
Yin, X., Fu, Q. Q., Shang, B. X., Wang, Y. J., Liu, R. Q., Chen, T. T., et al. (2022). An RxLR effector from Plasmopara viticola suppresses plant immunity in grapevine by targeting and stabilizing VpBPA1. Plant J. 112, 104–114. doi: 10.1111/tpj.15933
Yin, X., Liu, R. Q., Su, H., Su, L., Guo, Y. R., Wang, Z. J., et al. (2017). Pathogen development and host responses to Plasmopara viticola in resistant and susceptible grapevines: an ultrastructural study. Hortic. Res-England. 4, 17033. doi: 10.1038/hortres.2017.33
Yokosawa, S., Eguchi, N., Sato, T. (2020). Characterization of the Colletotrichum gloeosporioides species complex causing grape ripe rot in Nagano Prefecture, Japan. J. Gen. Plant Pathol. 86, 163–172. doi: 10.1007/s10327-020-00907-5
Yu, D., Wei, W., Fan, Z. Q., Chen, J. Y., You, Y. L., Huang, W. D., et al. (2023). VabHLH137 promotes proanthocyanidin and anthocyanin biosynthesis and enhances resistance to Colletotrichum gloeosporioides in grapevine. Hortic. Res-England. 10, uhac261. doi: 10.1093/hr/uhac261
Yu, Y., Zhang, Y., Yin, L., Lu, J. (2012). The mode of host resistance to Plasmopara viticola infection of grapevines. Phytopathology 102, 1094–1101. doi: 10.1094/PHYTO-02-12-0028-R
Yuan, X., Pan, S. H., Zhang, Q., Dai, F., Zhang, J. X. (2024). A Colletotrichum tabacum effector Cte1 targets and stabilizes NbCPR1 to suppress plant immunity. Mol. Plant Microbe Interact. 37, 477–484. doi: 10.1094/MPMI-11-23-0197-R
Zhang, L., Zhang, F., Melotto, M., Yao, J., He, S. Y. (2017). Jasmonate signaling and manipulation by pathogens and insects. J. Exp. Bot. 68, 1371–1385. doi: 10.1093/jxb/erw478
Zhao, L., Liao, Z., Feng, L., An, B., He, C., Wang, Q., et al. (2023). Colletotrichum gloeosporioides Cg2LysM contributed to virulence toward rubber tree through affecting invasive structure and inhibiting chitin-triggered plant immunity. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1129101
Zheng, R., Shu, B., Luo, C. (2022). Colletotrichum siamense infection caused transcripts involved plant hormone signal transduction and phenylpropanoid biosynthesis varied in strawberry. Not. Bot. Horti Agrobo. 50, 12791. doi: 10.15835/nbha50312791
Zhou, R., Dong, Y. H., Liu, X., Feng, S., Wang, C. X., Ma, X. M., et al. (2022). JrWRKY21 interacts with JrPTI5L to activate the expression of JrPR5L for resistance to Colletotrichum gloeosporioides in walnut. Plant J. 111, 1152–1166. doi: 10.1111/tpj.15883
Keywords: grape, Colletotrichum viniferum, genome annotation, fungal infection strategy, grapevine response
Citation: Dou M, Li Y, Hao Y, Zhang K, Yin X, Feng Z, Xu X, Zhang Q, Bao W, Chen X, Liu G, Wang Y, Tian L and Xu Y (2024) Histological and transcriptomic insights into the interaction between grapevine and Colletotrichum viniferum. Front. Plant Sci. 15:1446288. doi: 10.3389/fpls.2024.1446288
Received: 09 June 2024; Accepted: 24 July 2024;
Published: 16 August 2024.
Edited by:
Brigitte Mauch-Mani, Université de Neuchâtel, SwitzerlandReviewed by:
Hye-Seon Kim, United States Department of Agriculture, United StatesSophie Trouvelot, Université de Bourgogne, France
Copyright © 2024 Dou, Li, Hao, Zhang, Yin, Feng, Xu, Zhang, Bao, Chen, Liu, Wang, Tian and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Tian, dGlhbmxpbmdAc3pwdS5lZHUuY24=; Yan Xu, eWFuLnh1QG53c3VhZi5lZHUuY24=