
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 19 July 2024
Sec. Plant Systematics and Evolution
Volume 15 - 2024 | https://doi.org/10.3389/fpls.2024.1421170
This article is part of the Research Topic Evolution of Crop Genomes and Epigenomes Volume II View all 5 articles
 Sicheng Li1,2†Cuifang Yang1†Zhen Wang2,3Chaohua Xu4Gemin Zhang1Yuxin Huang1Baoqing Zhang1Shan Zhou1Yijing Gao1Wenyi Zong1Weixing Duan1*Xiping Yang2,3*
Sicheng Li1,2†Cuifang Yang1†Zhen Wang2,3Chaohua Xu4Gemin Zhang1Yuxin Huang1Baoqing Zhang1Shan Zhou1Yijing Gao1Wenyi Zong1Weixing Duan1*Xiping Yang2,3*Saccharum complex includes genera Saccharum, Miscanthus, Erianthus, Narenga, and Tripidium. Since the Saccharum complex/Saccharinae constitutes the gene pool used by sugarcane breeders to introduce useful traits into sugarcane, studying the genomic characterization of the Saccharum complex has become particularly important. Here, we assembled graph-based mitochondrial genomes (mitogenomes) of four Saccharinae species (T. arundinaceum, E. rockii, M. sinensis, and N. porphyrocoma) using Illumina and PacBio sequencing data. The total lengths of the mitogenomes of T. arundinaceum, M. sinensis, E. rockii and N. porphyrocoma were 549,593 bp, 514,248 bp, 481,576 bp and 513,095 bp, respectively. Then, we performed a comparative mitogenomes analysis of Saccharinae species, including characterization, organelles transfer sequence, collinear sequence, phylogenetics analysis, and gene duplicated/loss. Our results provided the mitogenomes of four species closely related to sugarcane breeding, enriching the mitochondrial genomic resources of the Saccharinae. Additionally, our study offered new insights into the evolution of mitogenomes at the family and genus levels and enhanced our understanding of organelle evolution in the highly polyploid Saccharum genus.
In the Andropogoneae tribe, there are seed-based commercial crops such as maize, sorghum, and Coix lacryma-jobi (Wall and Ross, 1970; Ranum et al., 2014; Xi et al., 2016) and leaf-based commercial crops like Chrysopogon zizanioides (Chahal et al., 2015). Additionally, crops like sugarcane, which primarily yield from their stalks, are of significant commercial importance (Bell and Garside, 2005). Sugarcane belongs to the genus Saccharum and is the principal constituent of the Saccharum complex, which also includes other genera such as Miscanthus, Erianthus, Narenga, Sclerostachya, and Tripidium (Vasquez et al., 2022). Some of these genera are clearly not closely related to Saccharum (Li et al., 2022). Indeed, recent phylogenetic studies indicate that Tripidium is over 11 million years divergent from Saccharum, and Eriochrysis is even more divergent (Evans and Joshi, 2020). Miscanthus species are traditionally believed to possess a basic chromosome number of n = x = 19, and genome sequencing studies have shown that Miscanthus sinensis is a paleotetraploid comprising the A and B subgenomes (Mitros et al., 2020). Initially considered the closest diploid relative of sugarcane, Narenga porphyrocoma (2n = 30) has been estimated to have diverged from sugarcane approximately 2.5 million years ago (Evans and Joshi, 2020). Erianthus rockii (2n = 4x = 30) is a drought- and cold-tolerant wild relative of sugarcane from China (Qi et al., 2022).
To broaden the genetic base of modern sugarcane cultivars, sugarcane breeders aim to enrich the sugarcane gene pool through intergenic crosses with sugarcane relatives, enhancing yield, stress resistance, and disease resistance. Sugarcane breeders have used species from closely related genera to improve sugarcane varieties. Tripidium, for example, has demonstrated considerable cold hardiness and biomass yields (Mahadevaiah et al., 2023); N. porphyrocoma has excellent characteristics such as high tillering ability, drought tolerance, and mosaic disease resistance (Liu et al., 2018). Wild-type clones of Tripidium arundinaceum and Saccharum spontaneum show the potential to provide resistance to smut and high biomass, fiber, and bioenergy (Anna Durai and Karuppaiyan, 2023; Meena et al., 2024). Given that the Saccharum complex/Saccharinae serves as the gene pool for sugarcane breeders attempting to introgress useful traits into sugarcane, determining the phylogenetic relationships among these genera and species through molecular strategies is of considerable significance and relevance.
Mitochondria are essential for cellular energy production and numerous biological processes, including growth, development, and adaptation to environmental stress, potentially affecting agronomic traits (Mahadevaiah et al., 2023). However, the specific impact may vary depending on plant species and environmental conditions. Plant mitochondria are more complex and have a higher proportion of non-coding regions compared to animal mitochondria (Møller et al., 2021), likely because plants must manage various biotic and abiotic stresses to maintain proper physiological functions. Given their role as the cell’s energy supply system and their predominantly matrilineal inheritance in most plants (Li et al., 2021), mitochondria are an excellent research focus for studying parental relationships in polyploidization and the evolution of the energy supply system (Vallejo-Marín et al., 2016).
Recent studies have identified different conformations of mitochondrial genomes (mitogenomes), including linear structure of Lactuca sativa (Guo et al., 2019), branched structure of some cytoplasmic male sterility (CMS) lines in Zea mays (Zhang et al., 2020b), and numerous circular arrangements, such as Silene noctiflora (Wu et al., 2015) and Ombrophytum subterraneum (Roulet et al., 2020). Mitogenomes in the Poaceae family usually consist of individual ring DNA molecules, as in Oryza minuta (Asaf et al., 2016), Sporobolus alterniflorus (Wang et al., 2021), Avena longiglumis (Liu et al., 2023), Elymus magellanicus (Chen et al., 2023b), and Eleusine indica (Hall et al., 2020). However, there are two distinct circular DNA molecules in the mitogenome of Saccharum species (Evans et al., 2019). With advances in sequencing technology and assembly strategies, the master circular model no longer fully characterizes the actual mitochondrial genome.
Currently, only the mitogenomes of Saccharum spp. have been published (Li et al., 2024), leaving numerous properties of Saccharinae mitogenomes yet to be discovered. In this research, we de novo assembled the complete mitogenome of four Saccharinae species (T. arundinaceum, M. sinensis, E. rockii, and N. porphyrocoma) using a combination of second-generation Illumina sequencing and third-generation PacBio sequencing technologies. Then, mitogenome organization, characteristic, phylogenetic relationship, and comparative genome analyses were performed. Our study revealed reticulate mitochondrial conformations featuring multiple junctions. By comparing the organellar genomes of four Saccharinae species, we aimed to identify structural, sequence, and evolutionary differences within Saccharinae that have significant contributions to the cultivar enhancement of Saccharum cultivars.
Four accessions from the Saccharum complex, including BM87-36 (T. arundinaceum), M022 (M. sinensis), DZM1 (E. rockii), and HBW017 (N. porphyrocoma), were used for mitogenome investigation (Table 1). Leaf samples of the four accessions were collected (September 1, 2023) in Guangxi Key Laboratory of Sugarcane Genetic Improvement (22°50′N, 108°15′E). The extracted total genomic DNA was used for library construction with 150-bp and 15-kb insert sizes and then sequenced on the Illumina NovoSeq 6000 sequencing platform (Illumina, San Diego, CA, USA) and PacBio Revio platform for short and long reads, respectively. Finally, the Illumina and Nanopore high-quality reads were obtained and processed (Supplementary Table S1).

Table 1 Sample information of Tripidium arundinaceum, Miscanthus sinensis, Erianthus rockii, and Narenga porphyrocoma.
A hybrid assembly strategy was used for mitogenome assembly. First, the GetOrganelle (v1.7.6.1) was used to assemble short reads into a corresponding unitig graph with the parameters ‘-R 30 -k 85,105,115,127 -F embplant_mt’, and the contigs that contained the mitochondrial core genes in Andropogoneae were selected. The mitochondrial contigs obtained by GetOrganelle assembly were then used as bait to extract mitochondrial PacBio HiFi reads by Seqkit (v2.2.0); then, they were assembled by flye (2.9.1-b1780) with the parameters ‘–pacbio-hifi –meta -g 500K -t 20’; and the final mitogenome was visualized and adjusted manually by the Bandage (v0.8.1) software (Wick et al., 2015).
The mitogenome of four Saccharinae species was annotated by Geseq (https://chlorobox.mpimp-golm.mpg.de/geseq.html) and IPMGA (http://www.1kmpg.cn/mgavas/) and then manually annotated and corrected based on the reference mitochondrial genome of Sorghum bicolor and Saccharum officinarum (Khon Kaen 3) [National Center for Biotechnology Information (NCBI) accession numbers: NC008360.1 and NC031164.1]. The mitogenome structure map was drawn using OGDRAW (http://ogdraw.mpimp-golm.mpg.de/cgi-bin/ogdraw.pl).
Linkages between graphical contigs were designed within a range of 100–500 bp on either side of each junction site using the Primer 3 program (http://bioinfo.ut.ee/primer3-0.4.0/) (Supplementary Table S2). The DNA isolated from young leaf tissue of each species was used to conduct PCR verification. PCR was carried out in a 20-µL reaction mixture containing 10 µL of 10× reaction buffer, 5 pmol of each primer, 1.25 units of Taq DNA polymerase, and 20 ng of DNA template. The PCR was performed in thermocyclers using the following cycling parameters: 94°C (5 min); 30 cycles of 94°C (30 s), 55°C–57°C (30 s); 72°C (30 s), then 72°C (7 min). PCR products were visualized on agarose gels (2.0%–3.0%) containing Safe gel stain.
Transfer fragments between mitogenome and chloroplast genome (MTPTs) and between mitogenome itself (MTMTs) were identified by Blastn (2.5.0+) (parameters: e-value 1e−10 -outfmt 6). The Circos plot was visualized using the Advanced Circos module in TBtools (Chen et al., 2023a).
To better and comprehensively explore the evolutionary relationship of the Saccharum complex, the 12 mitogenomes of Poaceae (Supplementary Table S3) were downloaded from the NCBI database. A total of 13 shared protein-coding genes (PCGs) among the analyzed species were identified and extracted using PhyloSuite (v1.2.2) (Zhang et al., 2020a). All the PCGs were aligned in batches with MAFFT (v7.313) (Katoh and Standley, 2013) and integrated into PhyloSuite using normal-alignment mode. Maximum likelihood phylogenies were inferred using IQ-TREE under the Edge-unlinked partition model for 50,000 ultrafast bootstraps, and the tree was visualized using iTOL.
Six species related and within Saccharum complex were selected for analysis, including S. bicolor (NC013816.1), T. arundinaceum (in this study), M. sinensis (in this study), E. rockii (in this study), N. porphyrocoma (in this study), and S. spontaneum (Li et al., 2024), to conduct comparative mitogenome analysis and collinearity analysis. To investigate the similarity of mitogenome sequences within the Saccharum complex and closely related species, homologous sequences between the four relatives were detected using Blastn (2.5.0+) (parameters: e-value 1e−10). Homologous sequences less than 0.5 kb were not retained. A multiple synteny plot of those four mitogenomes was generated using TBtools.
A dot plot of pairwise comparison on conserved collinear blocks was generated and plotted using MUMmer (Kurtz et al., 2004). Based on sequence similarity, a Multiple Synteny Plot of the five mitogenomes from this and previous studies with closely related species was plotted using MCScanX in TBtools.
Accurate mitogenomes were obtained by combining Illumina and PacBio HiFi reads. Consistent depths of mapping reads revealed the high-quality gap-free assembly (Supplementary Table S4). First, the mitogenome of four Saccharum complex species was assembled into initial graph-based structures. From PacBio data, 13 contigs of T. arundinaceum, six contigs of M. sinensis, three contigs of N. porphyrocoma, and six contigs of E. rockii were obtained (Figure 1). Primer design and PCR validation were performed based on the junctions of the graphical assembly results (Supplementary Figures S1A–D), which showed that the junctions and product lengths were as expected, as evidenced by the variable structure of the assembly results and other conformations of the mitochondria. Subsequently, by examining the connections between contigs and mapping withthird-generation long sequences, a relatively simplified primary conformation can be obtained (Figure 2; Table 2), with a total length of 549,593 bp, 514,248 bp, 481,576 bp, and 513,095 bp in T. arundinaceum, M. sinensis, E. rockii, and N. porphyrocoma, respectively.
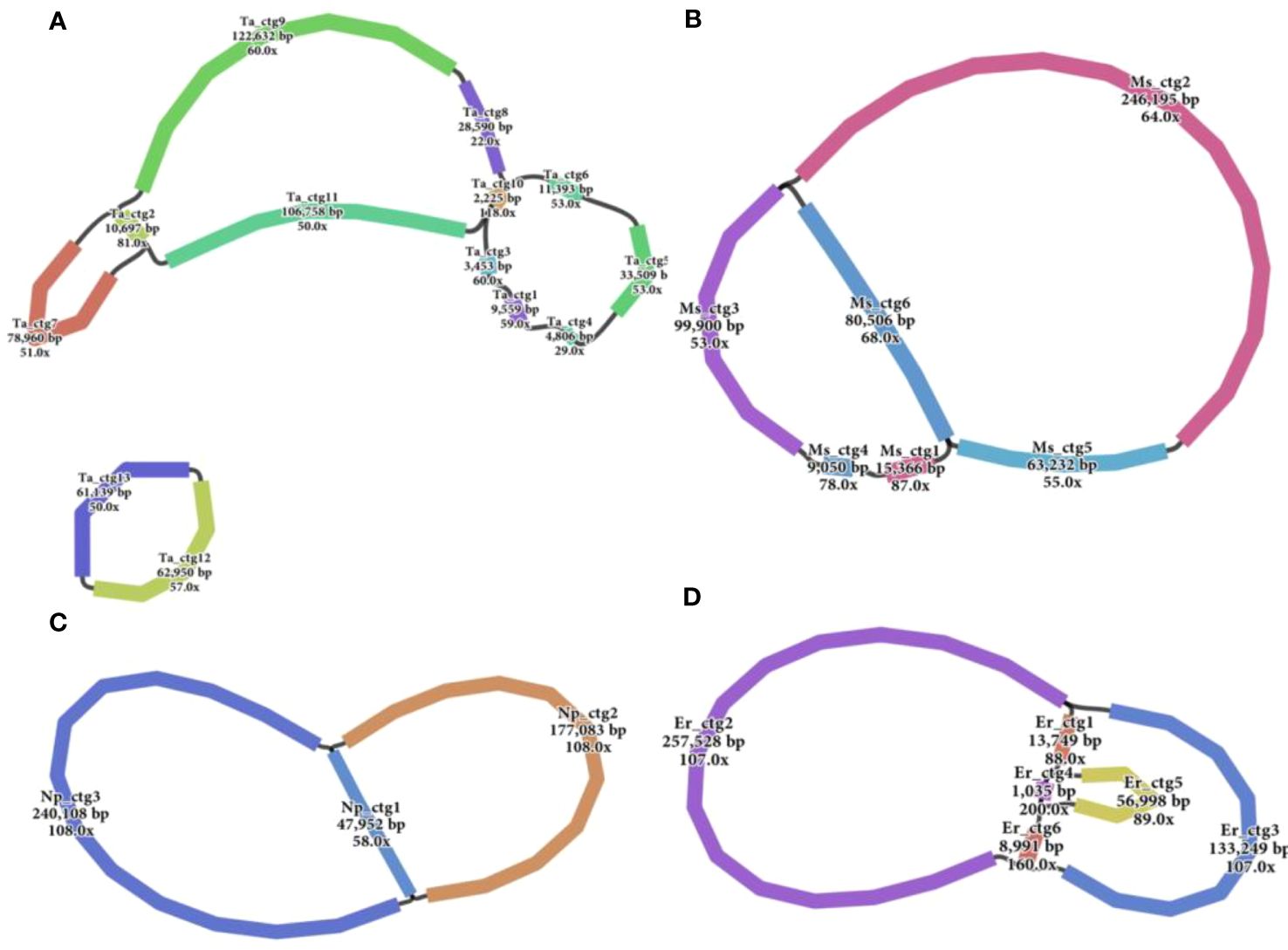
Figure 1 Branched conformation of four Saccharinae species mitogenomes. (A) Tripidium arundinaceum, (B) Miscanthus sinensis, (C) Narenga porphyrocoma, and (D) Erianthus rockii.
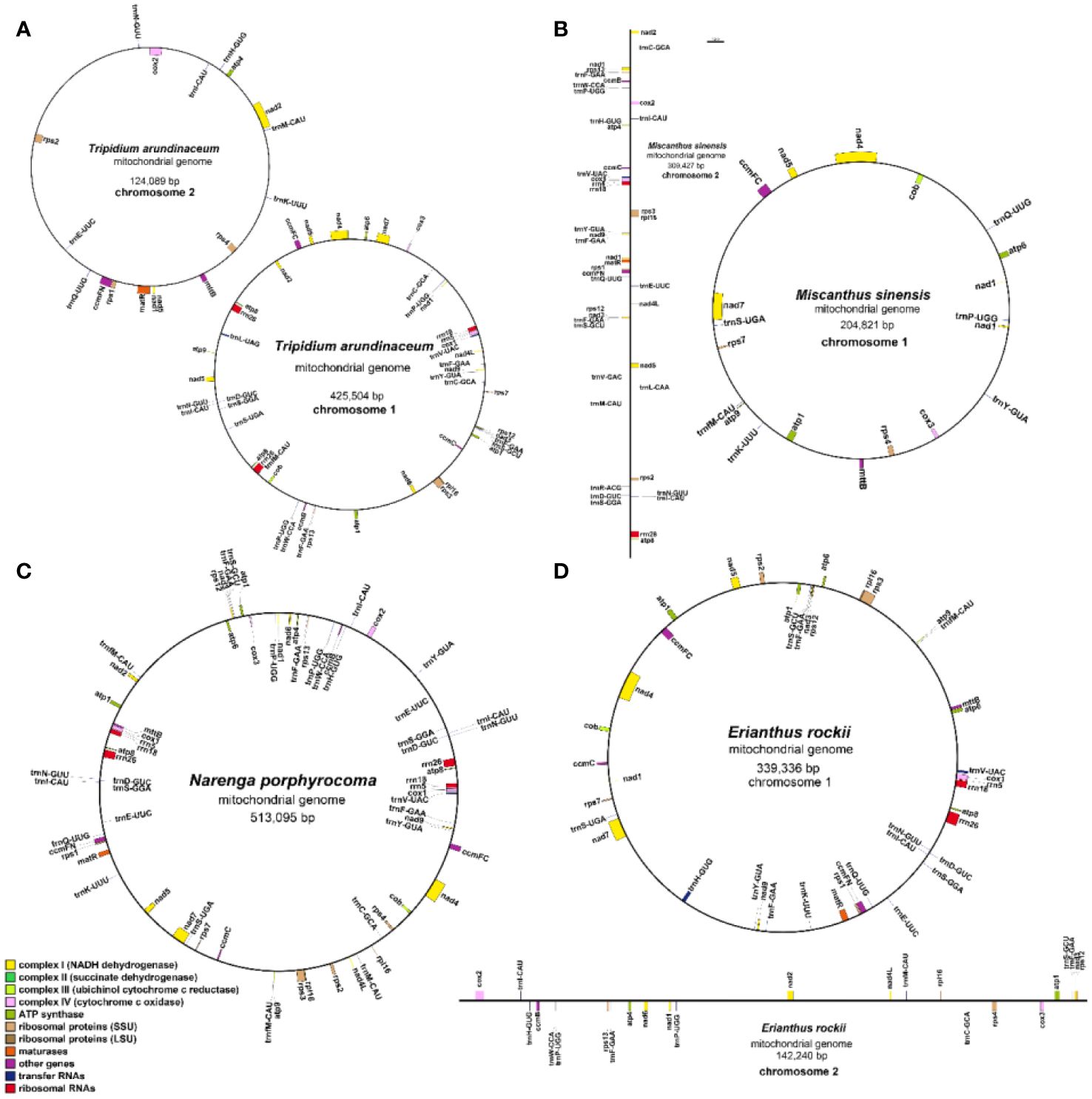
Figure 2 Gene map of the four Saccharinae species mitogenomes. (A) Tripidium arundinaceum, (B) Miscanthus sinensis, (C) Narenga porphyrocoma, and (D) Erianthus rockii.
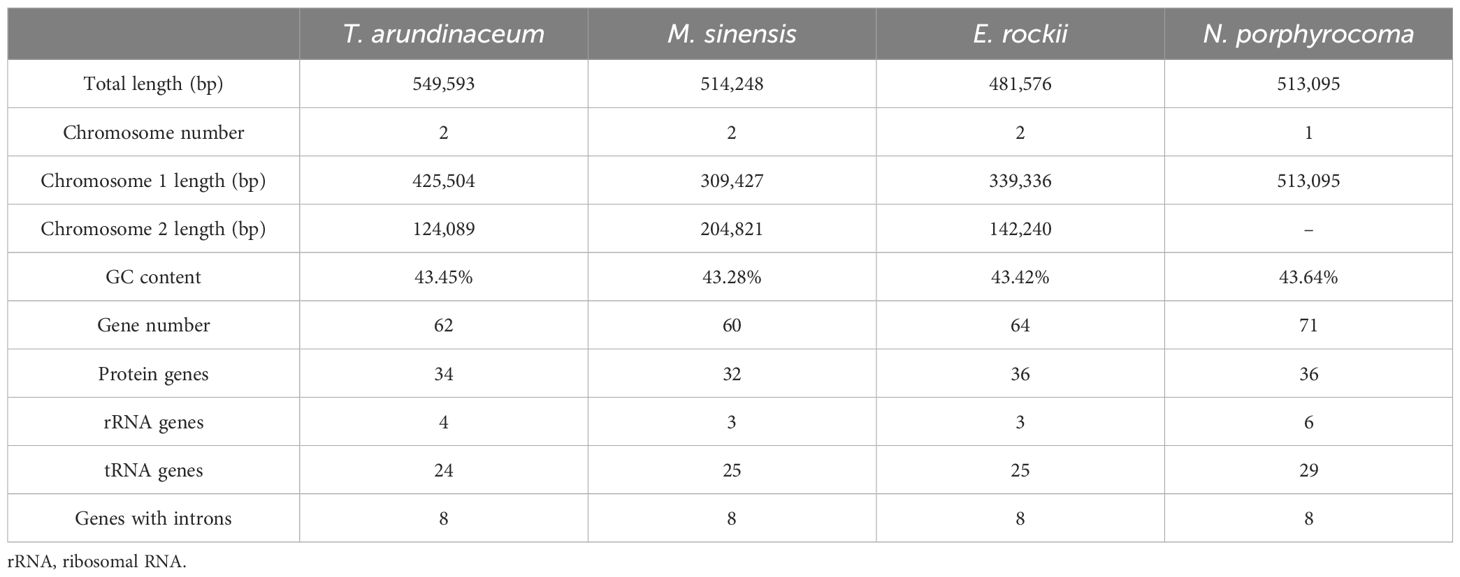
Table 2 Mitogenome assembly of Tripidium arundinaceum, Miscanthus sinensis, Erianthus rockii, and Narenga porphyrocoma.
A total of 51 unique genes were identified from the assembled mitogenome, comprising 32 PCGs, 16 transfer RNA (tRNA) genes, and three ribosomal RNA (rRNA) genes (Table 3). Notably, the gene copy numbers varied among four species: the atp1 gene was duplicated in T. arundinaceum, E. rockii, and N. porphyrocoma, while the atp8 gene exhibited duplication in T. arundinaceum and N. porphyrocoma. Additionally, cox1 was found to be duplicated in T. arundinaceum, and nad3 showed duplication in E. rockii. Eight genes were identified to contain one to four introns, including ccmFc (1), cox2 (1), nad1 (4), nad2 (4), nad4 (3), nad5 (4), nad7 (4), and rps3 (1). It was noteworthy that in all four species, the exons of nad1 and nad5 were identified to be located on different chromosomes, requiring trans-splicing to generate complete transcripts.
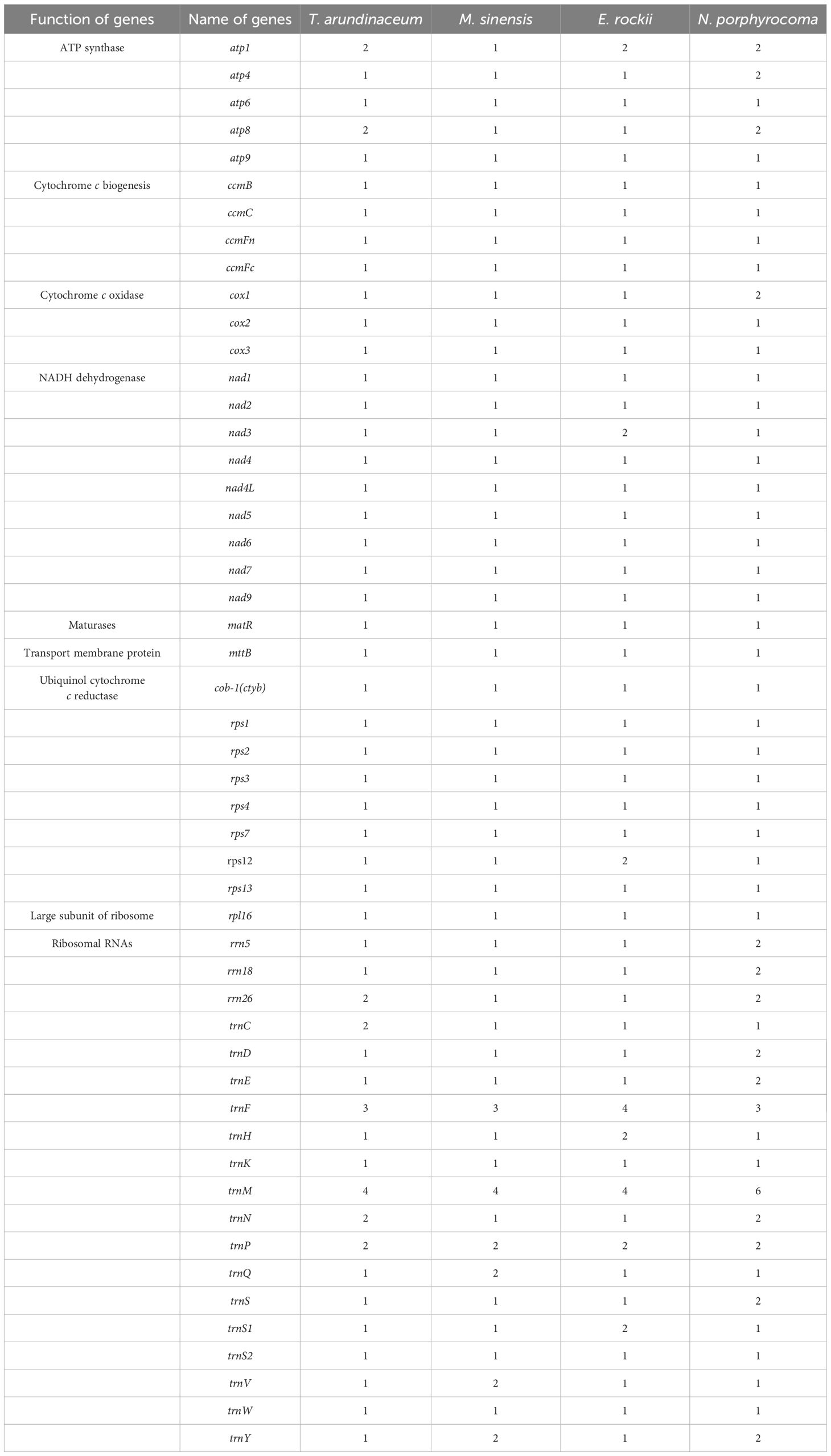
Table 3 Gene content of mitogenome in Tripidium arundinaceum, Miscanthus sinensis, Erianthus rockii, and Narenga porphyrocoma.
During the evolution of mitochondria, chloroplast fragments were transferred to the mitogenome (Wang et al., 2007). Approximately 5%–10% of the sequences in mitogenome that can be identified as homologs are derived from the chloroplast genome (Rodríguez-Moreno et al., 2011). In this study, the chloroplast genomes of four species were reassembled and annotated based on PacBio HiFi reads, and then the transfer sequences between mitochondrial and chloroplast genomes were analyzed.
The four completed chloroplast genomes ranged in length from 141,128 bp to 141,258 bp with two inverted repeat regions (IRA and IRB), separating the large single-copy (LSC) and small single-copy (SSC) regions (Figure 3). Gene annotation showed that there were 130 genes in all assembled chloroplast (cp) genomes, including 83 protein-coding genes (74 single-copy PCGs; two copies of ndhB, rps12, rps19, rpl2, rpl23, rps7, and rps15 in inverted repeat regions), 38 tRNA genes (two copies of trnA-UGC, trnH-GUG, trnI-CAU, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG, trnN-GUU, and trnL-CAA in inverted repeat regions), and eight rRNA genes (two copies of rrn23, rrn4.5, rrn5, and rrn16 in inverted repeat regions) (Supplementary Table S5). The results of reassembly by long read and annotation were similar to previously published second-generation sequencing assembled chloroplast genomes of the Saccharinae (Li et al., 2022).
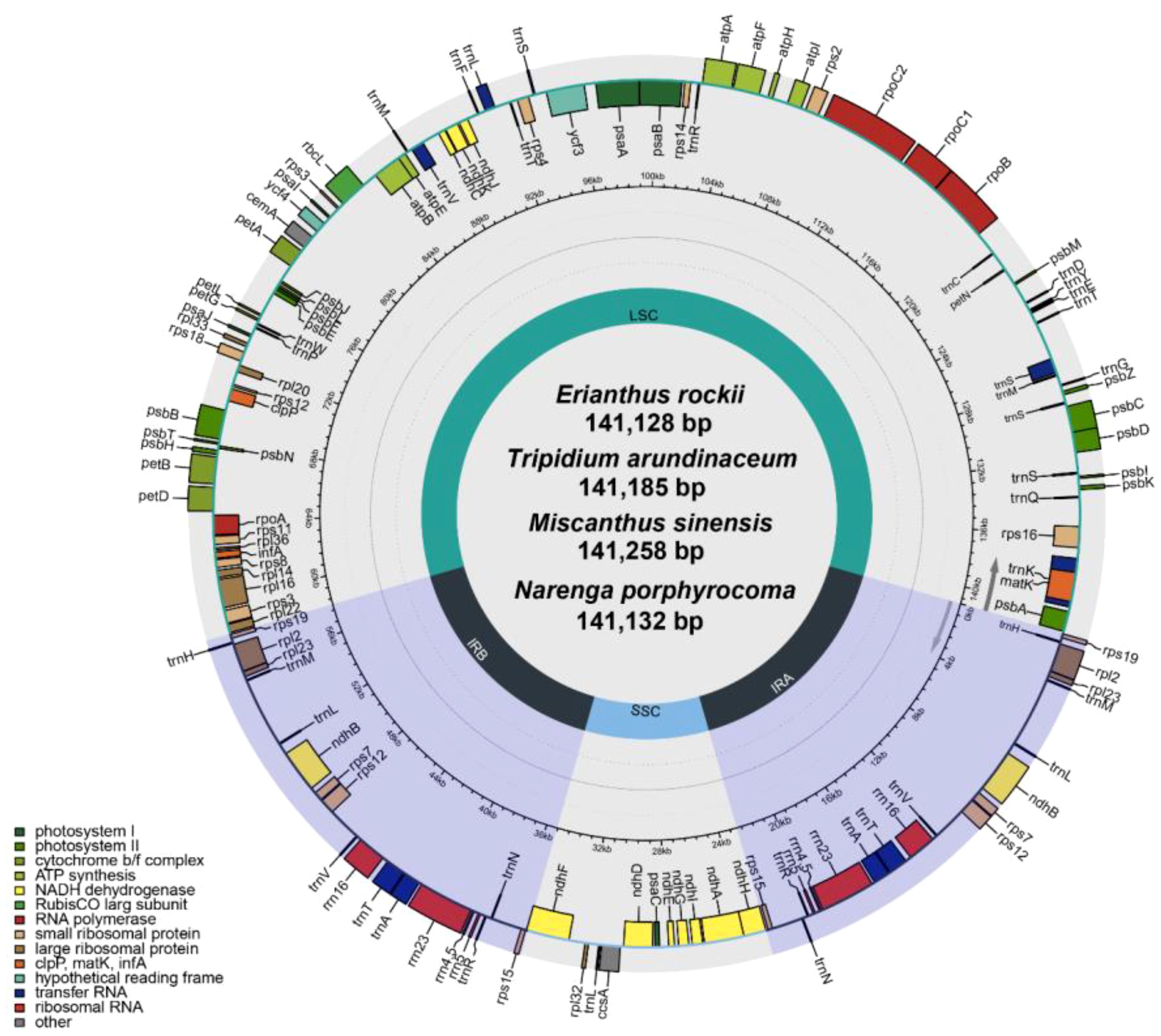
Figure 3 Gene map of the chloroplast genome of four Saccharinae species.
The total fragment lengths of the MTPTs of T. arundinaceum, M. sinensis, N. porphyrocoma, and E. rockii were 37,559 bp, 55,349 bp, 40,117 bp, and 37,541 bp, respectively (Supplementary Tables S6–9). There were 43, 52, 44, and 43 plastid-derived sequences identified, accounting for 6.83%, 10.07%, 7.30%, and 6.83% of the respective mitochondrial DNA (mtDNA), and 26.60%, 39.18%, 7.30%, and 6.83% of the respective chloroplast DNA (cpDNA), respectively (Table 4). In M. sinensis, the transfer length, quantity, and proportion of MTPT fragments were the highest among all organelle genomes. The total length of MTPT fragments in Saccharum was approximately 3 kb, which was approximately 8% and 25% of the length of the mitochondrial and chloroplast genomes, respectively (Li et al., 2024).
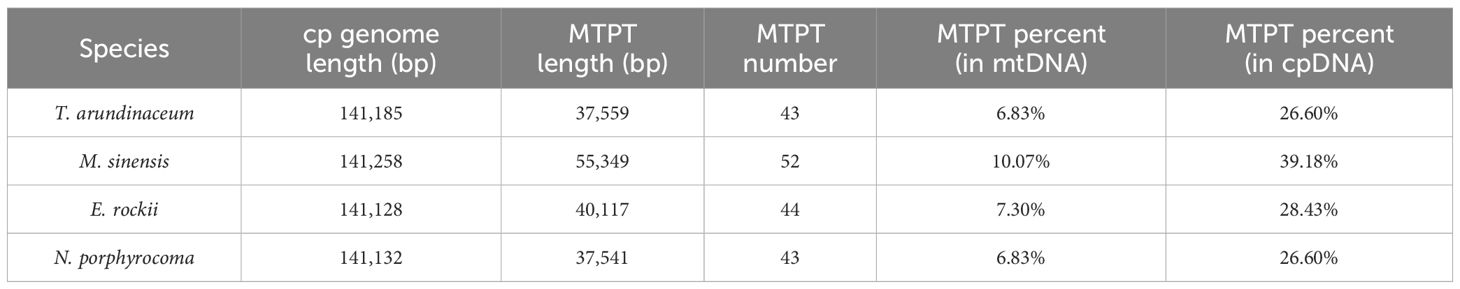
Table 4 Chloroplast genome and MTPT information of Tripidium arundinaceum, Miscanthus sinensis, Erianthus rockii, and Narenga porphyrocoma.
Mitochondrial genes are a valuable source of information for phylogenetic analyses at large-scale taxonomic levels due to their low substitution rates. To determine the phylogenetic position of Saccharinae, we used 13 conserved mitochondrial PCGs (ccmC, cox1, cox2, cox3, cob, matR, nad3, nad4L, nad9, rp3, rps4, rps7, and rps12) shared in 20 Poaceae mitogenomes (Figure 4). Based on a relatively high support rate and the classification outlined by the Angiosperm Phylogeny Group (APG IV), Saccharinae belonged to the PACMAD clade in Poaceae. The four genera, Tripidium, Miscanthus, Erianthus, and Narenga from the core “Saccharum complex”, was monophyletic as sister to Saccharum. Saccharum spp. were clustered into the same clades, and N. porphyrocoma was the most closely related to the Saccharum genus, followed by E. rockii. T. arundinaceum and M. sinensis were not closely related to Saccharum. The phylogenetic relationships of species in the core “Saccharum complex” were consistent with previous chloroplast-based phylogenies (Li et al., 2022).
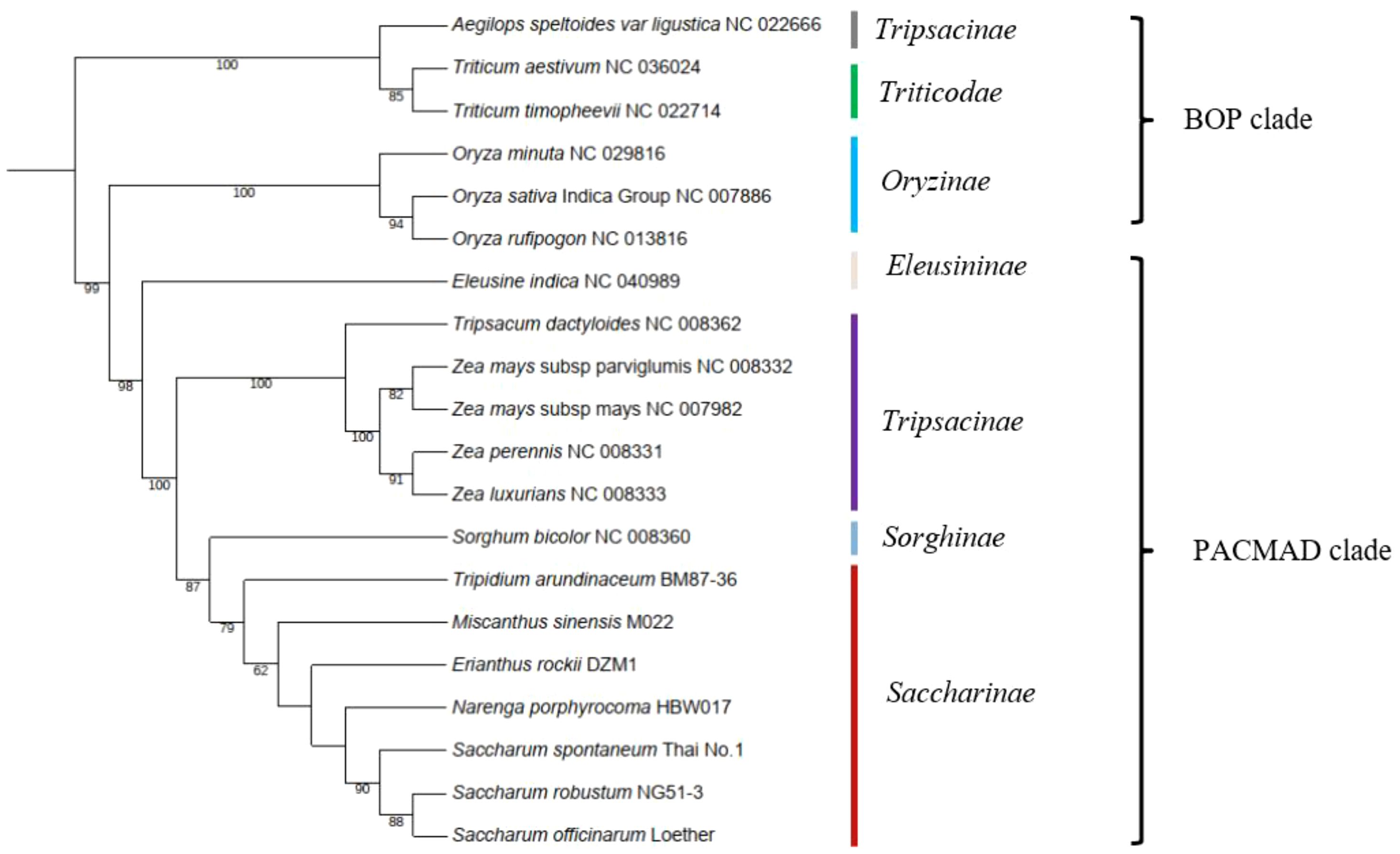
Figure 4 Phylogenetic tree of 20 species in BOP clade and PACMAD clade. The number at each node was the bootstrap probability.
Sequence transfer between species was also explored to understand which sequences were retained in the mitochondrial genome during evolution and how these sequences were recombined between species. There were collinear segments with a total length of 335,303 bp, 392,247 bp, 360,963 bp, 420,218 bp, and 433,309 bp for S. bicolor–T. arundinaceum (350 fragments), T. arundinaceum–M. sinensis (369 fragments), M. sinensis–E. rockii (334 fragments), E. rockii–N. porphyrocoma (260 fragments), and N. porphyrocoma–S. spontaneum (277 fragments), respectively (Figure 5). Collinear sequences over 10 kb in length were retained in each species, and short collinear sequences were often lost after species divergence. In the graph assembly results, short collinear sequences were often nodes connected between long and repetitive contigs, which may have played a role in the conformation of the species but were not retained during evolution.

Figure 5 Mitogenome synteny. Bars indicate the mitogenomes, and the lines show the homologous sequences between the adjacent species. The pink line represents the sequence of inheritance between the six species. Sb, Sorghum bicolor; Ta, Tripidium arundinaceum; Ms, Miscanthus sinensis; Er, Erianthus rockii; Np, Narenga porphyrocoma; Ss, Saccharum spontaneum.
Based on the phylogenetic relationships, we performed a dot-plot analysis of two species with close phylogenetic relationships. The results showed numerous collinear blocks in T. arundinaceum–M. sinensis and M. sinensis–E. rockii, but all were short and fragmented. However, long collinear blocks were found in both E. rockii–N. porphyrocoma and N. porphyrocoma–S. spontaneum. The relatively short divergence time between these two species may not have led to a large-scale reorganization of the mitogenome, and large collinear blocks are still retained between the two species (Figure 6). The mitogenomes in the subtribe taxonomic level had undergone extensive genomic rearrangements with closely related species, and the mitogenome was extremely not conserved in structure.
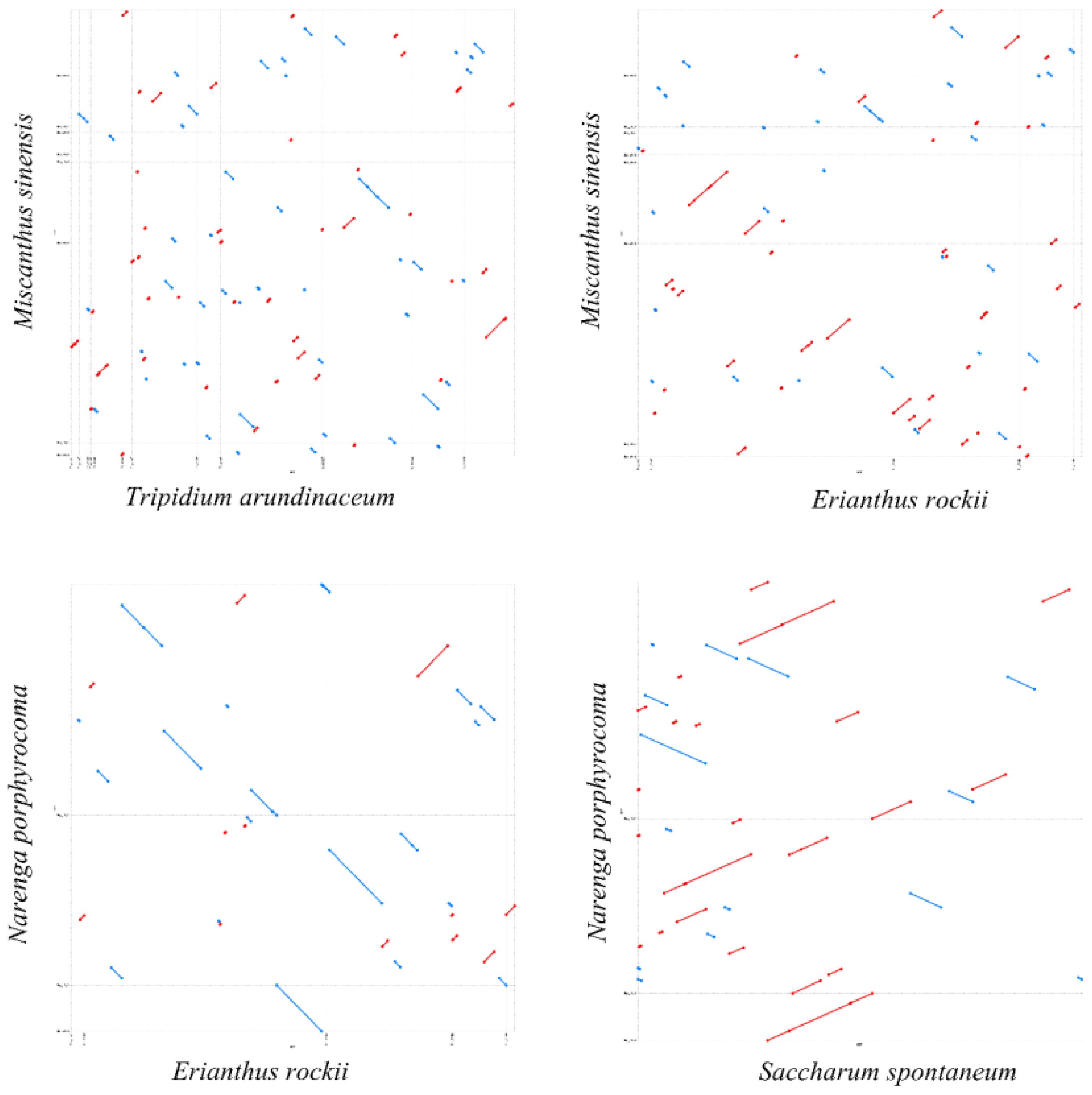
Figure 6 Dot-plot analysis. The blue and red lines represent reverse and forward sequences, respectively. Dot plot of Tripidium arundinaceum with Miscanthus sinensis is shown in the upper left corner, and the rest are dot plots of M. sinensis with Erianthus rockii, E. rockii with Narenga porphyrocoma, and N. porphyrocoma with Saccharum spontaneum.
Gene duplication and loss often occur during the evolution of plants, and the genes that are retained by duplication are crucial for normal life activities. Therefore, gene duplication and loss in the Andropogoneae species were compared here (Figure 7). For the mitochondrial PCGs, it was found that the ATP synthase genes and the cytochrome c synthesis gene were not lost with strong conservation. However, the succinate dehydrogenase proteins (sdh3 and sdh4) were found lost in Poaceae with high volatility and weak conservation during evolution. It was observed that genes within the ATP and COX family may exist in multiple copies in the Andropogoneae mitogenomes. For instance, Z. mays (OP832500) and S. bicolor (NC008360) had two copies of atp1; Coix lacryma (MT471100) harbored two copies of atp4 and atp9; C. zizanioides (MN635785) exhibited a duplication of atp6. Furthermore, cox1 was found in triplicate in C. zizanioides (MN635785) and duplicated in C. lacryma (MT471100). The mttB gene was not found in some accessions of Zea, which may be the impact of the incompetent annotation.
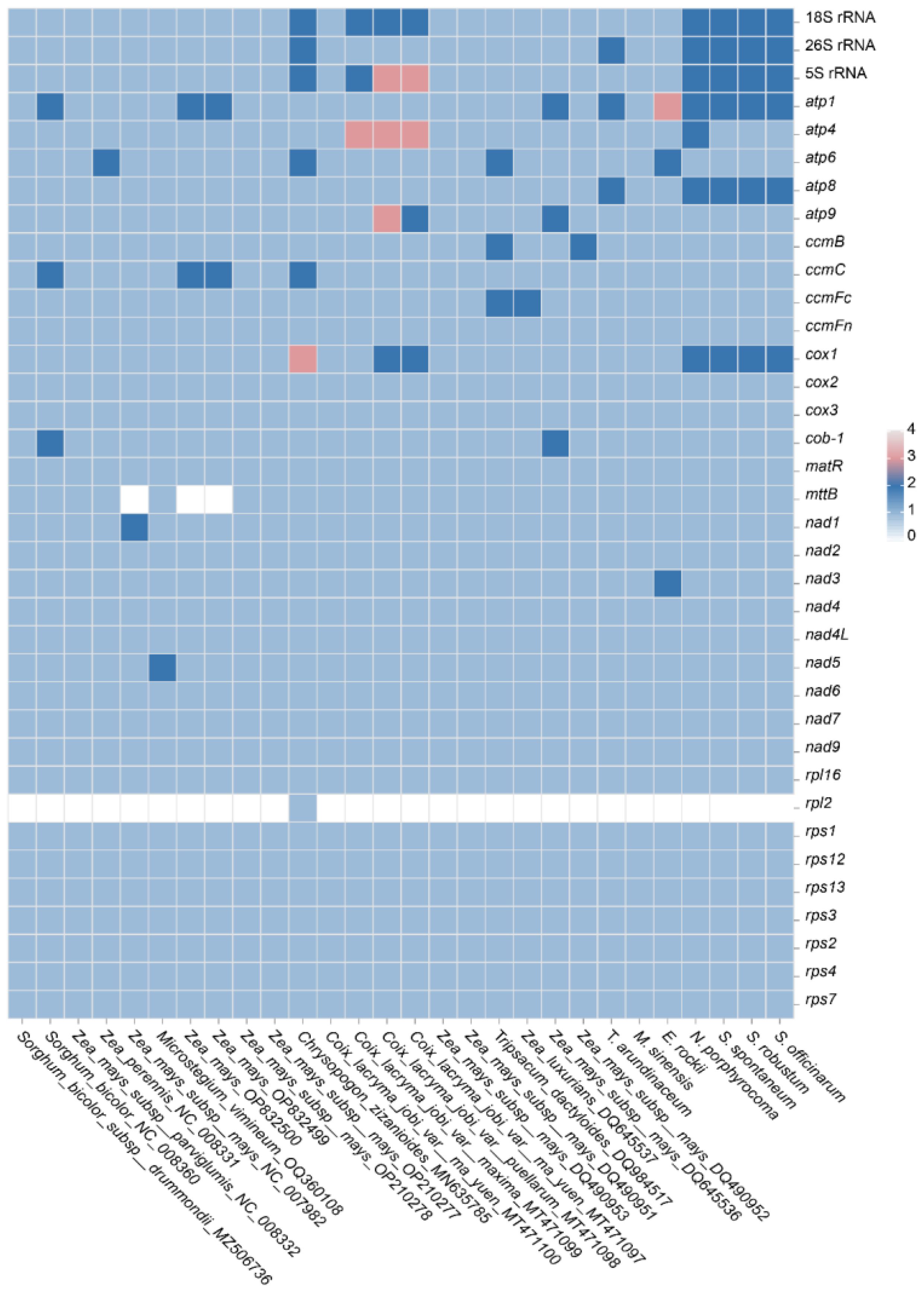
Figure 7 Gene duplication and loss in Andropogoneae species.
In Andropogoneae, most mitogenomes were published in the genus Zea, followed by genera Coix, Saccharum, and Sorghum. The length of mitogenomes ranged from 449,028 bp (S. bicolor subsp. drummondii, MZ506736.1) to 739,719 bp (Z. mays subsp. mays genotype CMS-C, DQ645536.1), with an average length of approximately 500 kb (Table 5). The total lengths in T. arundinaceum, M. sinensis, E. rockii, and N. porphyrocoma were 549,593 bp, 514,248 bp, 481,576 bp, and 513,095 bp, respectively, which were close to the published mitochondrial genome lengths of Andropogoneae. The mitochondrial genome lengths of species that have been domesticated in Andropogoneae, such as domesticated maize under the genus Zea, differ significantly from those of wild species (Zea perennis and Zea luxurians) of maize. In different accessions of C. lacryma-jobi, the length of the genome varied considerably, from 598,321 bp to 673,349 bp.
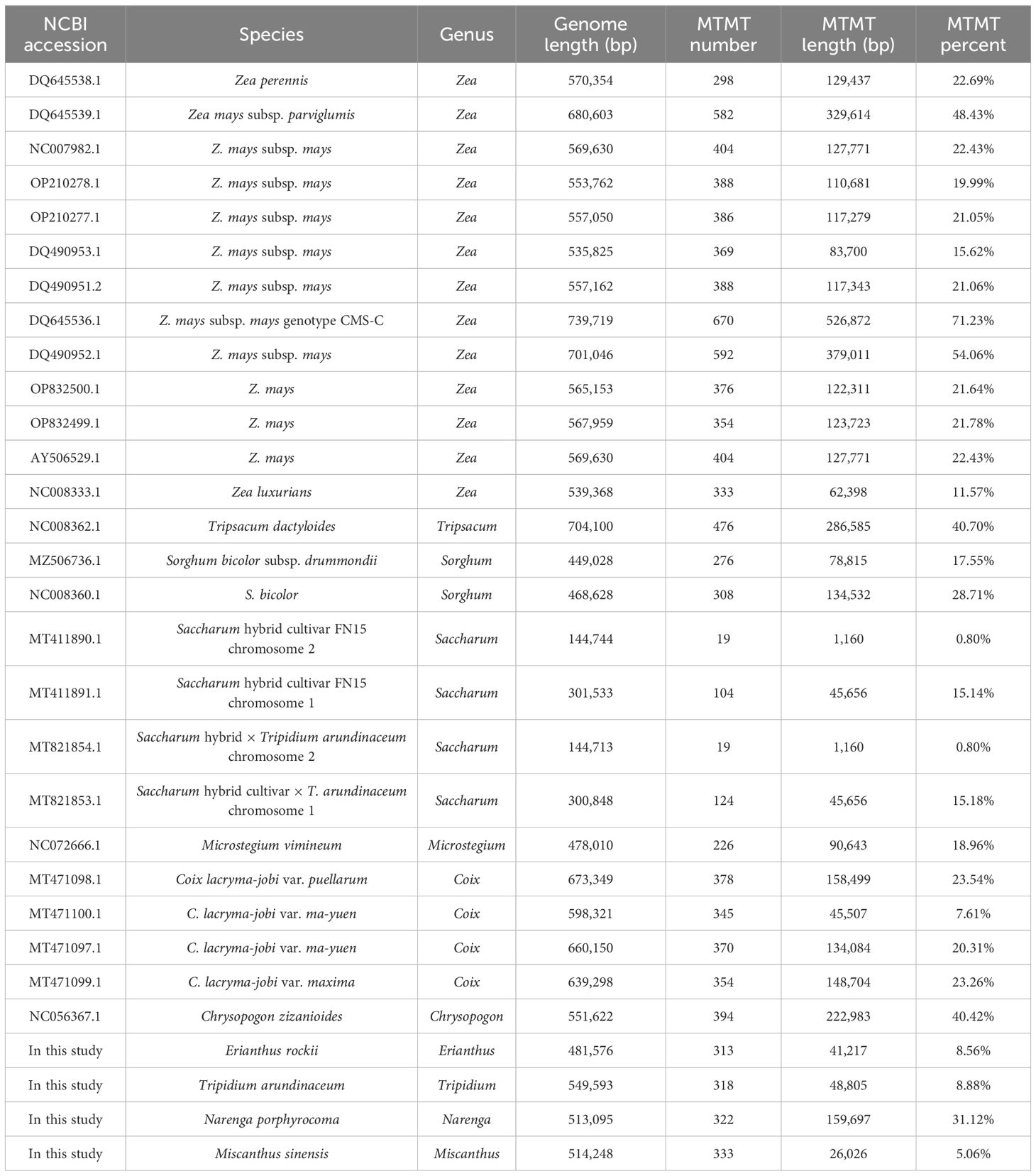
Table 5 Genome length and mitogenome transfer sequence of Andropogoneae species.
Variations in mitochondrial genome sizes mainly result from the transfer of sequences from the nucleus and chloroplasts, as well as the expansion of repetitive sequences within the mitogenomes. We examined the Mitochondrion to Mitochondrion sequences (MTMTs) events and the proportion of the genome they occupied for species in Andropogoneae. Z. mays subsp. mays genotype CMS-C had not only the largest genome length but also the highest percentage of MTMTs (71.23%); meanwhile, the lowest percentage of MTMT sequences (0.80%) was found in chromosome 2 of Saccharum species.
Advances in sequencing and assembly strategies have made it possible for researchers to recover polymorphic conformations of plant mitochondria (Wang et al., 2024). In this study, we assembled the mitogenome of four species in Saccharinae first to provide a more comprehensive description of the mitogenome in Saccharinae, including basic mitochondrial features, structure, and homologous sequences.
In Saccharinae, the mitogenome length averaged approximately 500 kb. Our study revealed that T. arundinaceum had the longest mitogenome (549,593 bp). While the unique PCGs in Saccharinae mitogenomes were similar across the four species and Saccharum, variations in gene copy numbers were observed among them. Duplication of cox1, atp1, and atp8 genes was found in T. arundinaceum; atp1 and nad3 genes in E. rockii; and atp1 and atp8 in N. porphyrocoma, but these genes were single-copy in M. sinensis. Within Andropogoneae, certain PCGs, such as those associated with ATP synthesis (atp1, atp4, and atp8) and the COX subunit complex (cox1), exhibited varying copy numbers from one to three. C4 plants in Andropogoneae, such as sorghum, maize, and sugarcane, are leading players in global agriculture and can survive in hot and dry conditions. Mitochondria play a crucial role in the internal regulation of organisms under harmful environmental conditions such as hypoxia and high temperature (Xiong et al., 2022). Through our analysis of gene duplications and losses in published Andropogoneae mitogenomes, we observed that most plants harbored multiple copies of ATP synthase and COX subunits, crucial for plant respiration. These findings offer insights into how C4 plants maintain cellular physiological functions in response to high temperatures.
The polymorphic structure of plant mitochondrial genomes often results in an uneven distribution of functional genes on each molecule, with many individual molecules lacking functional genes, and others having multiple copies of functional genes due to their presence in repetitive regions. For example, in the species within the N. porphyrocoma and Saccharum species, cox1, atp8, rrn5, rrn18, and rrn26 all have two copies; nad1 and nad5 are usually present on different single molecules in the genomes of the Saccharinae, which are assembled into functional transcripts by trans-spicing. Single molecules lacking functional genes are often shown in graphical results as nodal molecules connecting long contigs, and they may have an important role in shaping the conformation of mitogenomes. We compared mitogenome synteny among six species, sorghum, T. arundinaceum, M. sinensis, E. rockii, N. porphyrocoma, and S. spontaneum, to identify transfer sequences among them. We found that most transfer sequences were long contigs and that short contigs (i.e., single molecules lacking functional genes) were rarely transferred between species, suggesting that they play a role in these species and that these sequences were not homologous by disrupted recombination after species formation.
Most land plant mitogenomes analyzed to date can be represented as a single circular chromosome (called the major cyclic chromosome or master circle) and a set of secondary chromosomes (called the minor cyclic chromosome), which arise by active recombination of large direct repeats (Wang et al., 2024). Few comparative analyses of plant mitochondrial genes have so far revealed the relationship between species evolution and mitochondrial conformation. The simplest structures of the mitochondrial genomes of Saccharum robustum and S. officinarum in the genus Saccharum can be described as two-ring structures (300 kb and 144 kb), and the structure of the S. spontaneum as a 380-kb linear structure and a 100-kb-ring structure (Li et al., 2024). The diploid N. porphyrocoma, closely related to Saccharum, exhibited a single cyclic structure. Its secondary conformation can generate substructures of 225 kb and 288 kb, whose secondary conformation allowed the generation of 225 kb and 288 kb substructures, respectively. In T. arundinaceum, there was no connection between chromosome 1 (425 kb) and chromosome 2 (124 kb), similar to that in S. robustum and S. officinarum. However, the linear structures of M. sinensis and E. rockii can form a closed structure in the graph-based assembly. During speciation, the conformation of plant mitochondria may have undergone fusion and division to accommodate species formation.
Many studies have shown that mtDNA evolves faster in structure but slower in sequence change compared to cpDNA or nuclear DNA, making mtDNA a good tracer for genome evolution (Hu et al., 2023). Changes in gene copy number are common in plant mitochondria, and whole-genome duplication (WGD) events occurring in plant nuclear genomes can affect the relative copy numbers of nuclear, mitochondrial, and chloroplast genomes (Zwonitzer et al., 2024). Increases in organelle genome copy number represent a common response to polyploidization, suggesting that maintenance of nuclear stoichiometry is an important aspect of establishing polyploid lineages.
In the evolution of species, understanding how nucleoplasmic equilibrium evolves after polyploidization requires studying the exchange between mitochondrial and nuclear genomes of their diploid ancestors (Korpelainen, 2004). The ploidy levels of species within the Saccharinae vary significantly, with most of the close relatives of sugarcane being diploid (N. porphyrocoma, M. sinensis, and E. rockii in this study are diploid, while T. arundinaceum is tetraploid), some wild sugarcane species ranging from tetraploid to octoploid (Zhang et al., 2022), and cultivated sugarcane species being over octoploid (Zhang et al., 2019). Chromosome-level genome assemblies have been recently published for the genera Erianthus (Erianthus rufipilus) (Wang et al., 2023) and Saccharum (Healey et al., 2024). The structure and copy number of the mitogenome are closely associated with the balance of genetic material within the cell during polyploidization (Zwonitzer et al., 2024), but the evolutionary mechanisms of the mitochondrial genome following nuclear genome polyploidization remain to be studied. The mitogenomes of the four Saccharinae species assembled and analyzed in this study lay the groundwork for exploring nucleoplasmic interactions between the mitogenome and nuclear genome during WGD events in polyploidization within the Saccharinae.
In this study, we assembled graph-based mitochondrial genomes of four Saccharinae species (T. arundinaceum, E. rockii, M. sinensis, and N. porphyrocoma) closely related to sugarcane and provided a more complete characterization of the mitochondrial genomes instead of the “master circular” structure. Second, we performed a comparative mitogenome analysis of Saccharinae species, including characterization, transfer sequence (MTPTs and MTMTs), collinear sequence, phylogenetic analysis, and gene duplication/loss. Our results provided the mitochondrial genomes of four Saccharum complex species, enriching the mitochondrial genomic resources of the Saccharinae. Second, it provided new insights into the evolution of mitochondrial genomes at low taxonomic levels. Finally, it facilitated our understanding of organelle evolution in the highly polyploid Saccharum genus.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. The data presented in the study are deposited in online repository, accession number Narenga porphyrocoma (PP551632.1), Tripidium arundinaceum (PP664562.1, PP664563.1), Miscanthus sinensis (PP665700.1, PP665701.1), and Erianthus rockii (PP622780.1, PP622781.1). The raw data used in this work were deposited at National Center for Biotechnology Information (NCBI), BioProject: PRJNA1097978.
SL: Data curation, Investigation, Writing – original draft, Writing – review & editing. CY: Writing – review & editing, Conceptualization, Data curation. ZW: Data curation, Formal analysis, Writing – review & editing. CX: Writing – review & editing. GZ: Writing – review & editing. YH: Writing – review & editing. BZ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. SZ: Writing – review & editing. YG: Writing – review & editing. WZ: Writing – review & editing. WD: Funding acquisition, Project administration, Resources, Writing – review & editing. XY: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by the National Key Research and Development Program (2021YFD1200204), the Guangxi Natural Science Foundation (GK AA22117002, 2020GXNSFAA297039 and GK AD21075011), Natural Science Foundation of China (31901510), Fundamental Research Fund of Guangxi Academy of Agriculture Sciences (2021YT004), Chongzuo science and technology project (CK 20220619) and the ‘One Hundred Person’ Project of Guangxi Province.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1421170/full#supplementary-material
Anna Durai, A., Karuppaiyan, R. (2023). “Potential parents for developing climate-resilient sugarcane varieties in India: a Breeding Perspective,” in Agro-industrial perspectives on sugarcane production under environmental stress (Singapore: Springer Nature Singapore), 57–83.
Asaf, S., Khan, A. L., Khan, A. R., Waqas, M., Kang, S.-M., Khan, M. A., et al. (2016). Mitochondrial genome analysis of wild rice (Oryza minuta) and its comparison with other related species. PloS One 11, e0152937. doi: 10.1371/journal.pone.0152937
Bell, M. J., Garside, A. (2005). Shoot and stalk dynamics and the yield of sugarcane crops in tropical and subtropical Queensland, Australia. Field Crops Res. 92, 231–248. doi: 10.1016/j.fcr.2005.01.032
Chahal, K. K., Kaushal, S., Sandhu, A. K. (2015). Chemical composition and biological properties of Chrysopogon zizanioides (L.) Roberty syn. Vetiveria zizanioides (L.) Nash-A Review. Indian J. Nat. Prod. Resour. 6, 251–260.
Chen, C., Wu, Y., Li, J., Wang, X., Zeng, Z., Xu, J., et al. (2023a). TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742. doi: 10.1016/j.molp.2023.09.010
Chen, X., Wu, X., Zhang, J., Zhang, M., You, J., Ru, Z. (2023b). Characterization of the complete mitochondrial genome of Elymus magellanicus (É. Desv.) Á. Löve (Poaceae, Pooideae). Mitochondrial DNA Part B. Resour. 8, 795–798. doi: 10.1080/23802359.2023.2238931
Evans, D. L., Hlongwane, T. T., Joshi, S. V., Pachón, D. M. R. (2019). The sugarcane mitochondrial genome: assembly, phylogenetics and transcriptomics. PeerJ 7, e7558. doi: 10.7717/peerj.7558
Evans, D. L., Joshi, S. V. (2020). On the validity of the saccharum complex and the saccharinae subtribe: a re-assesment. BioRxiv. doi: 10.1101/2020.07.29.226753
Guo, Z., Liu, H., Chen, X., Huang, L., Fan, J., Zhou, J., et al. (2019). Modified-atmosphere packaging maintains the quality of postharvest whole lettuce (Lactuca sativa L. Grand Rapid) by mediating the dynamic equilibrium of the electron transport chain and protecting mitochondrial structure and function. Postharvest Biol. Tec. 147, 206–213. doi: 10.1016/j.postharvbio.2018.09.001
Hall, N. D., Zhang, H., Mower, J. P., Mcelroy, J. S., Goertzen, L. R. (2020). The mitochondrial genome of Eleusine indica and characterization of gene content within Poaceae. Genome Biol. Evol. 12, 3684–3697. doi: 10.1093/gbe/evz229
Healey, A., Garsmeur, O., Lovell, J., Shengquiang, S., Sreedasyam, A., Jenkins, J., et al. (2024). The complex polyploid genome architecture of sugarcane. Nature 628, 804–810. doi: 10.1038/s41586-024-07231-4
Hu, H.-L., Zhang, F., Wang, P., Lu, F.-H. (2023). Evolutionary genetics of wheat mitochondrial genomes. Crop J. 11, 1774–1781. doi: 10.1016/j.cj.2023.09.011
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Bio. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Korpelainen, H. (2004). The evolutionary processes of mitochondrial and chloroplast genomes differ from those of nuclear genomes. Naturwissenschaften 91, 505–518. doi: 10.1007/s00114-004-0571-3
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5, 1–9. doi: 10.1186/gb-2004-5-2-r12
Li, J., Xu, Y., Shan, Y., Pei, X., Yong, S., Liu, C., et al. (2021). Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 254, 1–16. doi: 10.1007/s00425-021-03684-3
Li, S., Duan, W., Zhao, J., Jing, Y., Feng, M., Kuang, B., et al. (2022). Comparative analysis of chloroplast genome in Saccharum spp. and related members of ‘Saccharum Complex’. Int. J. Mol. Sci. 23, 7661. doi: 10.3390/ijms23147661
Li, S., Wang, Z., Jing, Y., Duan, W., Yang, X. (2024). Graph-based mitochondrial genomes of three foundation species in the Saccharum genus. Plant Cell Rep. 43, 191. doi: 10.21203/rs.3.rs-3784122/v1
Liu, Q., Yuan, H., Xu, J., Cui, D., Xiong, G., Schwarzacher, T., et al. (2023). The mitochondrial genome of the diploid oat Avena longiglumis. BMC Plant Biol. 23, 218. doi: 10.1186/s12870-023-04217-8
Liu, X., Zhang, R., Ou, H., Gui, Y., Wei, J., Zhou, H., et al. (2018). Comprehensive transcriptome analysis reveals genes in response to water deficit in the leaves of Saccharum narenga (Nees ex Steud.) hack. BMC Plant Biol. 18, 1–16. doi: 10.1186/s12870-018-1428-9
Mahadevaiah, C., Vignesh, P., Appunu, C., Valarmathi, R., Dhansu, P., Kumar, A., et al. (2023). Physiological Characterization of Tripidium arundinaceum and Sugarcane (Saccharum spp.) Germplasm for Salinity Stress Tolerance at the Formative Stage. Sustainability 15, 6962. doi: 10.3390/su15086962
Meena, M. R., Govindaraj, P., Kumar, R. A., Elayaraja, K., Appunu, C., Kumar, R., et al. (2024). Biomass and energy potential of Erianthus arundinaceus and Saccharum spontaneum-derived novel sugarcane hybrids in rainfed environments. BMC Plant Biol. 24, 1–14. doi: 10.1186/s12870-024-04885-0
Mitros, T., Session, A. M., James, B. T., Wu, G. A., Belaffif, M. B., Clark, L. V., et al. (2020). Genome biology of the paleotetraploid perennial biomass crop Miscanthus. Nat. Commun. 11, 5442. doi: 10.1038/s41467-020-18923-6
Møller, I. M., Rasmusson, A. G., Van Aken, O. (2021). Plant mitochondria–past, present and future. Plant J. 108, 912–959. doi: 10.1111/tpj.15495
Qi, Y., Gao, X., Zeng, Q., Zheng, Z., Wu, C., Yang, R., et al. (2022). Sugarcane breeding, germplasm development and related molecular research in China. Sugar Tech 24, 73–85. doi: 10.1007/s12355-021-01055-6
Ranum, P., Peña-Rosas, J. P., Garcia-Casal, M. N. (2014). Global maize production, utilization, and consumption. Ann. N. Y. Acad. Sci. 1312, 105–112. doi: 10.1111/nyas.12396
Rodríguez-Moreno, L., González, V. M., Benjak, A., Martí, M. C., Puigdomènech, P., Aranda, M. A., et al. (2011). Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin. BMC Genom. 12, 1–14. doi: 10.1186/1471-2164-12-424
Roulet, M. E., Garcia, L. E., Gandini, C. L., Sato, H., Ponce, G., Sanchez-Puerta, M. V. (2020). Multichromosomal structure and foreign tracts in the Ombrophytum subterraneum (Balanophoraceae) mitochondrial genome. Plant Mol. Biol. 103, 623–638. doi: 10.1007/s11103-020-01014-x
Vallejo-Marín, M., Cooley, A. M., Lee, M. Y., Folmer, M., Mckain, M. R., Puzey, J. R. (2016). Strongly asymmetric hybridization barriers shape the origin of a new polyploid species and its hybrid ancestor. Am. J. Bot. 103, 1272–1288. doi: 10.3732/ajb.1500471
Vasquez, S. I., Welker, C. A., Solís Neffa, V. G., Peichoto, M. C. (2022). Morphometric analysis of the Saccharum complex (Poaceae, Andropogoneae). Plant Syst. Evol. 308, 10. doi: 10.1007/s00606-021-01801-z
Wall, J. S., Ross, W. M. (1970). Sorghum production and utilization. Sorghum production utilization.
Wang, D., Wu, Y.-W., Shih, A. C.-C., Wu, C.-S., Wang, Y.-N., Chaw, S.-M. (2007). Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol. Bio. Evol. 24, 2040–2048. doi: 10.1093/molbev/msm133
Wang, J., Kan, S., Liao, X., Zhou, J., Tembrock, L. R., Daniell, H., et al. (2024). Plant organellar genomes: Much done, much more to do. Trends Plant Sci. doi: 10.1016/j.tplants.2023.12.014
Wang, T., Wang, B., Hua, X., Tang, H., Zhang, Z., Gao, R., et al. (2023). A complete gap-free diploid genome in Saccharum complex and the genomic footprints of evolution in the highly polyploid Saccharum genus. Nat. plants. 9, 554–571. doi: 10.1038/s41477-023-01378-0
Wang, Y., Xie, W., Cao, J., He, Y., Zhao, Y., Qu, C., et al. (2021). The complete mitochondrial genome of Sporobolus alterniflorus (loisel.) PM Peterson & Saarela (Poaceae) and phylogenetic analysis. Mitochondrial DNA Part B. Resour. 6, 1303–1305. doi: 10.1080/23802359.2021.1907248
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinform. 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z., Cuthbert, J. M., Taylor, D. R., Sloan, D. B. (2015). The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc. Nat. Acad. Sci. 112, 10185–10191. doi: 10.1073/pnas.1421397112
Xi, X.-J., Zhu, Y.-G., Tong, Y.-P., Yang, X.-L., Tang, N.-N., Ma, S.-M., et al. (2016). Assessment of the genetic diversity of different Job's tears (Coix lacryma-jobi L.) accessions and the active composition and anticancer effect of its seed oil. PloS One 11, e0153269. doi: 10.1371/journal.pone.0153269
Xiong, Y., Yu, Q., Xiong, Y., Zhao, J., Lei, X., Peng, Y., et al. (2022). The complete mitogenome of Elymus sibiricus and insights into its evolutionary pattern based on simple repeat sequences of seed plant mitogenomes. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.802321
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020a). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, H., Wang, B., Li, B., Lin, Y., Yang, H., Ding, D., et al. (2020b). Comparative proteomic analysis of mitochondrial proteins from maize CMS-C sterile, maintainer and restorer anthers. Plant Genom. 13, e20022. doi: 10.1002/tpg2.20022
Zhang, J., Zhang, Q., Li, L., Tang, H., Zhang, Q., Chen, Y., et al. (2019). Recent polyploidization events in three Saccharum founding species. Plant Biotechnol. J. 17, 264–274. doi: 10.1111/pbi.12962
Zhang, Q., Qi, Y., Pan, H., Tang, H., Wang, G., Hua, X., et al. (2022). Genomic insights into the recent chromosome reduction of autopolyploid sugarcane Saccharum spontaneum. Nat. Genet. 54, 885–896. doi: 10.1038/s41588-022-01084-1
Keywords: mitochondrial genome, phylogenetic, Saccharinae, Tripidium arundinaceum, Erianthus rockii, Miscanthus sinensis, Narenga porphyrocoma
Citation: Li S, Yang C, Wang Z, Xu C, Zhang G, Huang Y, Zhang B, Zhou S, Gao Y, Zong W, Duan W and Yang X (2024) Assembly and comparative genome analysis of four mitochondrial genomes from Saccharum complex species. Front. Plant Sci. 15:1421170. doi: 10.3389/fpls.2024.1421170
Received: 22 April 2024; Accepted: 01 July 2024;
Published: 19 July 2024.
Edited by:
Hai Du, Southwest University, ChinaReviewed by:
Youxiong Que, Fujian Agriculture and Forestry University, ChinaCopyright © 2024 Li, Yang, Wang, Xu, Zhang, Huang, Zhang, Zhou, Gao, Zong, Duan and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weixing Duan, duanweixing84@126.com; Xiping Yang, xipingyang@gxu.edu.cn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.