 Hongxia Qiao1,2,3†Yajuan Chen1,2†Ruiping Wang4†Wei Zhang1Zhang Zhang1,2Fengqiang Yu4Haifeng Yang3*Guiming Liu1,2*
Hongxia Qiao1,2,3†Yajuan Chen1,2†Ruiping Wang4†Wei Zhang1Zhang Zhang1,2Fengqiang Yu4Haifeng Yang3*Guiming Liu1,2* Jiewei Zhang1,2*
Jiewei Zhang1,2*- 1Beijing Academy of Agriculture and Forestry Sciences, Beijing, China
- 2Beijing Key Laboratory of Agricultural Genetic Resources and Biotechnology, Institute of Biotechnology, Beijing, China
- 3College of Forestry, Inner Mongolia Agricultural University, Hohhot, China
- 4Ordos Forestry and Grassland Development Center, Ordos, China
Salix psammophila, commonly known as the sandlive willow, is a vital shrub species within the Salicaceae family, particularly significant for its ecological role in regions susceptible to desertification and sandy soils. In this study, we assembled the complete S. psammophila mitochondrial genome using Pacbio HiFi third-generation sequencing data. The genome was found to be a typical single circular structure, with a total length of 715,555 bp and a GC content of 44.89%. We annotated 33 unique protein-coding genes (PCGs), which included 24 core mitochondrial genes and 9 variable genes, as well as 18 tRNA genes (5 of which were multicopy genes) and 3 rRNA genes. Comparative analysis of the PCGs from the mitochondrial genomes of S. psammophila, Populus deltoides, Populus simonii, Salix wilsonii, and Salix suchowensis revealed that these genes are relatively conserved within the Salicaceae family, with variability primarily occurring in the ribosomal protein genes. The absence of the rps14, which encodes a ribosomal protein, may have played a role in the evolution of stress tolerance in Salicaceae plants. Additionally, we identified 232 SSRs, 19 tandem repeat sequences, and 236 dispersed repeat sequences in the S. psammophila mitochondrial genome, with palindromic and forward repeats being the most abundant. The longest palindromic repeat measured 260 bp, while the longest forward repeat was 86,068 bp. Furthermore, 324 potential RNA editing sites were discovered, all involving C-to-U edits, with the nad4 having the highest number of edits. These findings provide valuable insights into the phylogenetic and genetic research of Salicaceae plants.
Introduction
Salix psammophila is a perennial fast-growing and multi-resistance shrub in the genus Salix of the Salicaceae family, naturally distributed in arid and semi-arid desert areas, produced in the Maowusu Sandland and the Kubuqi Desert, and is the first choice of species for the prevention and control of desertification areas (Bao and Zhang, 2012; Jia et al., 2019; Lu et al., 2019; Yang et al., 2022). In the S. psammophila asexual line, the 17–38 asexual line is an excellent asexual variety, and in the morphology of the performance of the upright, short branchlets, the landscape effect is good. Currently, research on S. psammophila mainly focuses on strain type and sand barriers, while the genetic resources and phylogenetic status of S. psammophila mitochondria are still unclear. In order to further develop and utilize S. psammophila, the 17–38 asexual line of S. psammophila was used as a germplasm resource to assemble and annotate the S. psammophila mitochondrial genome and to analyze the structural characteristics and phylogenetic status of the S. psammophila mitochondrial genome.
The mitochondria are organelles with semi-autonomous genetic characteristics, relatively independent of the nucleus, with a relatively independent genetic system (Gray, 2012; Chen et al., 2017). It is an important organelle in eukaryotic cells that produce ATP by oxidative phosphorylation and is a source of energy for a variety of biochemical processes, playing a crucial role in plant development and reproduction (Wu et al., 2019). Plant mitochondrial genomes have a variety of structures due to the possession of a large number of repetitive sequences, and assembled mitochondrial genomes may be unicyclic, polycyclic, linear, or multibranching in structure (Ye et al., 2017; Cheng et al., 2021). For example, the mitochondrial genome of Populus deltoides and P. simonii has been assembled into three circular chromosomes. In contrast, the mitochondrial genome of P. tremula and P. tremula × P. alba is each organized into a single circular chromosome (Kersten et al., 2016; Bi et al., 2022; Qu et al., 2022). In addition, the size of plant mitochondrial genomes varies greatly, for instance, the small 66-kb Viscum scurruloideum (mistletoe) and the large 11.3-Mb Silene conica (Sloan et al., 2012; Skippington et al., 2015). It has been shown that the main cause of mitochondrial genome amplification in plants is the insertion of foreign sequences and the presence of a large number of repetitive sequences (Richardson and Palmer, 2006; Li, 2009; Smith, 2011). The number of genes in plant mitogenomes also varied, from 19 in the Viscum album to 221 in the pepper (Capsicum annuum) (Jo et al., 2011; Petersen et al., 2015).
Although the size, structure, and number of plant mitogenomes vary greatly, the encoded genes are relatively conserved, such as ATP synthase genes, NADH dehydrogenase genes, cytochrome C reductase genes, and cytochrome C oxidase genes (Bi et al., 2020). In addition, there are protein-coding genes (PCGs) that are lost during the evolution of the plant mitochondrial genome, such as the succinate dehydrogenase genes and the ribosomal protein (RP) genes (Bi et al., 2016). The genes rps12, sdh3, and sdh4 appear to be absent in the monocotyledonous plants, whereas the rps2 gene is notably missing in dicotyledonous plants (Zhang et al., 2012). RPs have important roles in metabolism, cell division, and growth processes (Wang et al., 2013a). RP genes are among the most highly expressed genes in most cells, and their expressions are differentially regulated by environmental and signaling molecules (Vemanna et al., 2019; Petibon et al., 2020; Fakih et al., 2023). Wang et al. explored the changes of ribosomes in phosphate- and iron-deficient Arabidopsis roots and found that most RP genes were related to translation and ribosome assembly, and some were related to low temperature, UV-B, and salt stress (Wang et al., 2013a).
It is well known that plant mitochondrial genomes vary greatly in size, structure, incorporation of exogenous DNA, and mutation rates, and there are a large number of repetitive sequences in the genomes, making it difficult to sequence plant mitochondrial genomes (Petersen et al., 2020). However, with the rapid development of third-generation sequencing technology, especially the emergence of PacBio and Oxford Nanopore, more and more plant mitochondrial genomes have been assembled and submitted to the NCBI database. In this study, we obtained, assembled, and annotated the complete S. psammophila mitochondrial genome for the first time based on Pacbio HiFi third-generation sequencing data. Characteristics of the S. psammophila mitochondrial genome were analyzed, including codon preference, RNA editing events, repetitive sequence analysis, phylogenetic analysis, and sequence migration analysis. These results will further lay the foundation for understanding and studying the evolution and inheritance of mitochondria in Salicaceae plants.
Materials and methods
Plant materials, DNA extraction, and sequencing
The experimental material used in this study was mature leaves of the 3-year-old 17–38 asexual clone of S. psammophila from the Germplasm Resources gene bank of S. psammophila in Ordos Dalad, the Inner Mongolia Autonomous Region of China (E 110°38′59.1″, N 40°14′15.5″). The leaves were collected, snap frozen in liquid nitrogen, and stored in a −80°C refrigerator. The total genomic DNA was extracted using a plant DNA extraction kit and sequenced with Pacbio HiFi sequencing technology following the manufacturer’s kits and protocols (Zhang et al., 2012, 2024).
Assembly and annotation of the mitochondrial genome
We used Pacbio HiFi third-generation sequencing data to assemble the S. psammophila mitochondrial genome. The long-reads data were assembled using the default parameters of the Flye software (v2.9.4) (Kolmogorov et al., 2019) to obtain a graphical mitochondrial genome in GFA format. Use makeblastdb to build a library of all fasta-formatted contigs assembled. Then, the contig fragments containing the mitochondrial genome were identified using the BLASTn program (Chen et al., 2015) and using the plant mitochondrial genome from Arabidopsis thaliana as a reference genome with the parameters “-evalue 1e-5 -outfmt 6 -max_hsps 10 -word_size 7 -task blastn-short”. In addition, we also included two closely related species, S. wilsonii and S. suchowensis, as references for re-assembly. This mitochondrial genome was visualized by Bandage software (v0.8.1) (Wick et al., 2015) and screened for mitochondrial contigs based on BLASTn results to obtain a sketch of the mitochondrial genome of S. psammophila.
A. thaliana (NC_037304), Liriodendron tulipifera (NC_021152.1), P. simonii (MZ905370), and S. wilsonii (NC_064688.1) were used as reference genomes, and the mitochondrial genome of S. psammophila was annotated using Geseq software (v2.03) (Tillich et al., 2017). The tRNA and rRNA of the genome were annotated using Tr4Nascan-SE software (v.2.0.11) (Lowe and Eddy, 1997) and BLASTn software (v2.13.0) (Chen et al., 2015). Finally, each mitochondrial genome annotation was manually corrected using Apollo software (v1.11.8) (Lewis et al., 2002).
Structural analysis and codon preference analysis of the genome
To resolve the obtained repetitive regions in the graphical mitochondrial genome, the long reads were compared to the repetitive sequences to determine whether any long reads crossed the repetitive region, especially the ctg3 repetitive region. Selection of suitable pathways is based on long reads to hypothesize the most likely mitochondrial genome structure formed in S. psammophila.
Genomic PCGs were extracted by the Phylosuite software (v1.1.16) (Zhang et al., 2020) and then analyzed for codon preference by the Mega software (v7.0) (Kumar et al., 2016), and RSCU values were calculated.
Sequence repeat analysis
Microsatellite repeats, tandem repeats, and dispersed repeats were identified using MISA (v2.1) (https://webblast.ipk-gatersleben.de/misa/) (Beier et al., 2017), TRF (v4.09) (https://tandem.bu.edu/trf/trf.unix.help.html) (Benson, 1999), and the REPuter web server (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) (Kurtz, 2001), and the results were analyzed using Excel software and the Circos package (v0.69.9) (Zhang et al., 2013) for visualization.
Sequence transfer analysis and synteny analysis
The chloroplast genome was assembled and annotated using the GetOrganelle software (Jin et al., 2020) and CPGAVAS2 software (Shi et al., 2019). The annotation results were subsequently corrected using the CPGView software (Liu et al., 2023). Homologous fragments of the mitochondrial and chloroplast genomes were analyzed using the BLASTn software (v2.13.0) (Chen et al., 2015), and the results were visualized using the Circos package (v0.69.9) (Zhang et al., 2013).
Six tree species, including S. psammophila, were selected for analysis, and based on the BLASTn results of two–two comparisons between individual mitochondrial genomes, homologous sequences of more than 500 bp were retained as conserved covariate blocks, and Multiple Synteny Plot was plotted.
Prediction of RNA editing sites
We used the sequences of all PCGs encoded by the S. psammophila mitochondrial genome as the input file and used Deepred-mt (Edera et al., 2021) to predict RNA editing sites from C to U of the mitochondrial PCGs, and retained all results with probability values greater than 0.9.
Phylogenetic analysis
Thirty-four mitochondrial genomes from three families, namely, Salicaceae, Euphorbiaceae, and Rhizophoraceae, were selected for phylogenetic analysis based on their affinities, with two Rhizophoraceae mitochondrial genomes set as the outgroup. Common genes were extracted using the PhyloSuite software (v1.1.16) (Zhang et al., 2020). Multiple sequence alignment was performed using MAFFT software (v7.505) (Katoh and Standley, 2013). Then, phylogenetic trees were constructed using the IQ-TREE software (v1.6.12) (Nguyen et al., 2015) based on the maximum likelihood (ML) method with the parameters “–alrt 1000 -B 1000” and the analysis results were visualized using ITOL software (v6) (Letunic and Bork, 2019).
Results
Assembly and annotation of the mitochondrial genome
A total of 265,714 reads were utilized in assembling the S. psammophila mitochondrial genome, with the longest read length reaching 48,394 bp, an average read length of 18,699 bp, an N50 read length of 18,657 bp, and total read bases amounting to 4,968,733,600 bp. After exclusion of duplicated regions, one molecular circular was obtained with a total length of 715,555 bp and 44.89% GC content (Figure 1). We annotated the mitochondrial genome of the S. psammophila and the annotation results are shown in Table 1. A total of 33 unique PCGs were annotated, including 24 unique core genes and 9 variable genes, 18 tRNA genes (5 of which were multicopy genes), and 3 rRNA genes. The core genes include five ATP synthase genes (atp1, atp4, atp6, atp8, and atp9), nine NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9), four cytochrome C biogenesis genes (ccmB, ccmC, ccmFC, and ccmFN), three cytochrome C oxidase genes (cox1, cox2, and cox3), one protein transport subunit gene (mttB), one maturases gene (matR), and one cytochrome b gene (cob). Variable genes include three RP large subunit genes (rpl2, rpl10, and rpl16), five RP small subunit genes (rps1, rps3, rps4, rps7, and rps12), and one succinate dehydrogenase (sdh4).
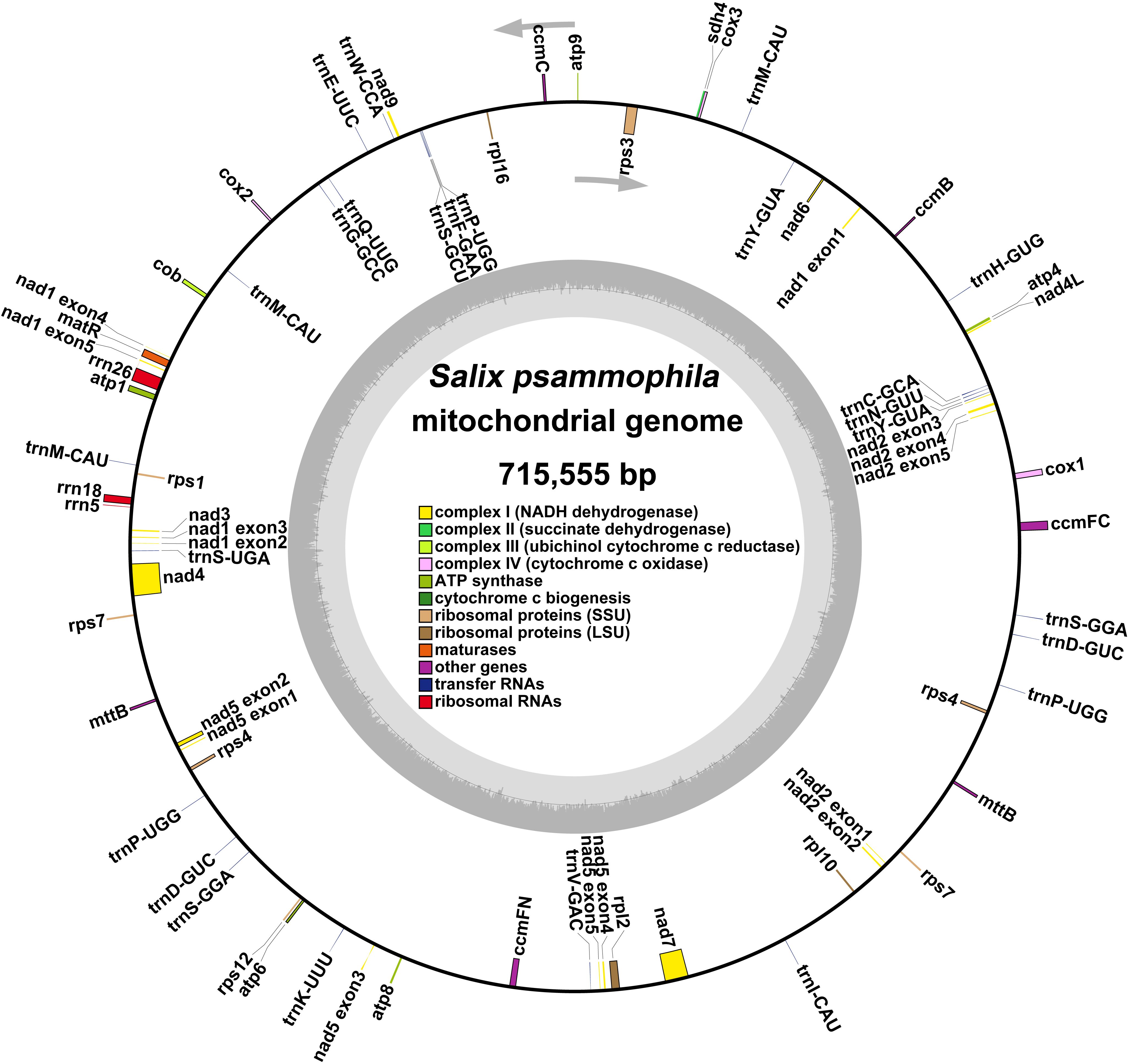
Figure 1. Genome circular map representing the mitochondrial genome of S. psammophila. The colored squares inside and outside the circular map represent various mitochondrial genes. Genes belonging to the same function are represented by the same color.
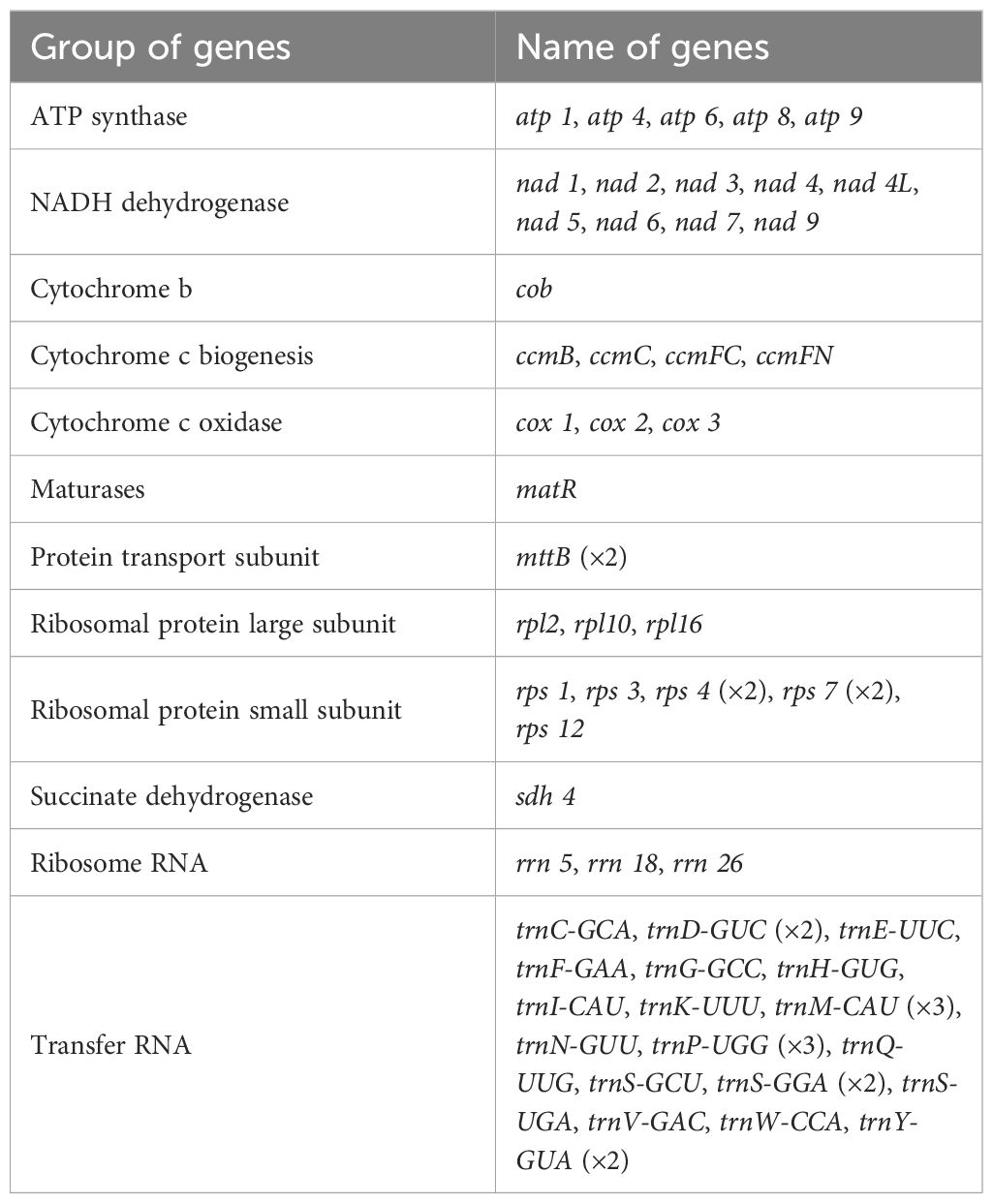
Table 1. Gene composition in the mitogenome of Salix psammophila.
The structure of the mitochondrial genome
A sketch of the assembled S. psammophila mitochondrial genome was visualized using Bandage software (v0.8.1), and the results are shown in Figure 2A. The sketch contains three nodes (see Supplementary Table S2A for details). Each node represents a sequence (contig) obtained by assembly, and two nodes are connected by a black line, which represents the overlapping region between the two sequences. Together, all sequences form a complex multibranched closed genome structure that represents the complete mitochondrial genome sequence of S. psammophila. We use long reads to address several key nodes where branching exists, derive the relevant sequences that are at branching nodes, and map them to long reads. Two sequences connected by a black line appear on the same long reads and are connected at the beginning and end, which means that the long reads support the interconnection of these two sequences. A number of different connections exist on different branch nodes, but we prioritize connections that are supported by more long reads when assembling. The resolution of branching nodes due to repeated sequences (ctg3) based on long reads yields a circular contig sequence (Figure 2B), the resolution of which is summarized in Supplementary Table S2B. In addition, ctg3 can be formed into a circular structure with ctg1 and ctg2, respectively, to form a potential secondary conformation of the two circular (Figure 2C).
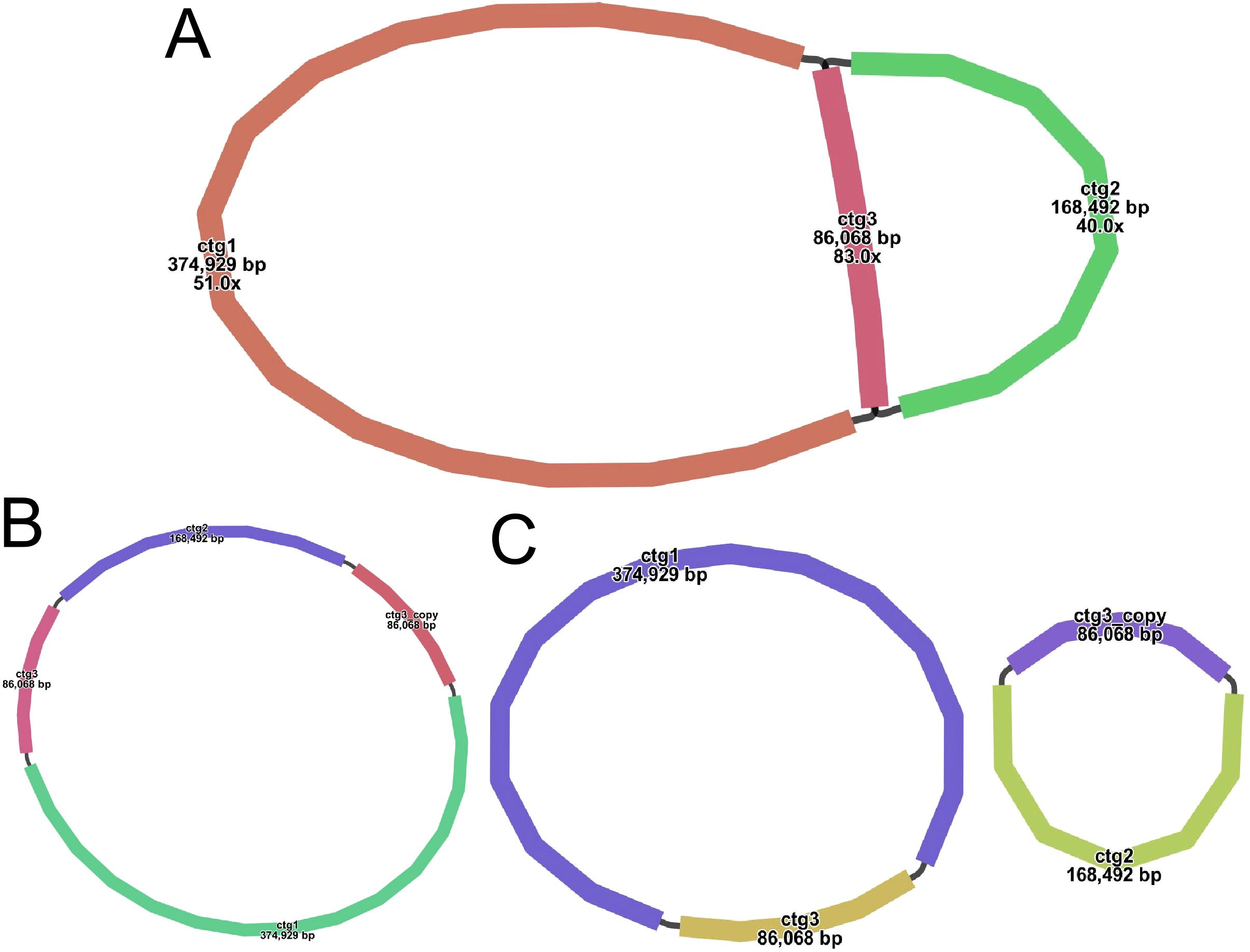
Figure 2. Various graphical mitochondrial genome structure maps of S. psammophila. (A) Sketch map of the mitochondrial genome assembly (node ID marked in the image). (B) The main configuration of the mitochondrial genome. (C) The minor configuration of the mitochondrial genome.
Codon preference of the mitochondrial genome
Codon preference analysis was performed on 33 unique PCGs of S. psammophila mitochondria, and the codon usage of individual amino acids is shown in Supplementary Table S3. All amino acids except Met (AUG) and Trp (UGG) consist of multiple codons. Codons with relative synonymous codon use (RSCU) greater than 1 are considered to be used preferentially by amino acids. As shown in Supplementary Figure S1, codon usage preferences are also prevalent in mitochondrial PCGs, except for the start codons AUG and Trp (UGG), which have an RSCU value of 1. For example, the termination codon has a high codon usage preference for UAA with an RSCU value of 1.69, which is the highest among mitochondrial PCGs. This is followed by alanine (Ala), which has a high codon usage preference for GCU with an RSCU value of 1.64.
Sequence repeat analysis
Repeat sequence analysis showed that a total of 232 SSRs were identified in the mitochondrial genome of the S. psammophila, with 86, 36, 24, 74, 9, and 3 monomers, dimers, trimers, tetramers, pentamers, and hexamers, respectively (Figure 3A). Among them, monomeric and dimeric SSRs accounted for 52.59% of the total SSRs. Thymine (T) monomeric repeat sequences accounted for 55.81% (48) of the 86 monomeric SSRs. Repeat sequences are categorized into tandem repeat sequences and dispersed repeat sequences. Tandem repeat sequences, also known as satellite DNA, are core repetitive units of approximately 7 to 200 bases that are repeated many times in tandem and are widely found in prokaryotic and eukaryotic genomes. There were 19 tandem repeats in the mitochondrial genes with greater than 85% match and lengths ranging from 12 to 28 bp, and 362 pairs of dispersed repeats with lengths greater than or equal to 30 bp. The dispersed repeats contained 180 pairs of palindromic repeats, 179 pairs of forward repeats and 2 pairs of complementary repeats (Figure 3B). Of these, the longest palindromic repeat is 260 bp and the longest forward repeat is 86,068 bp.
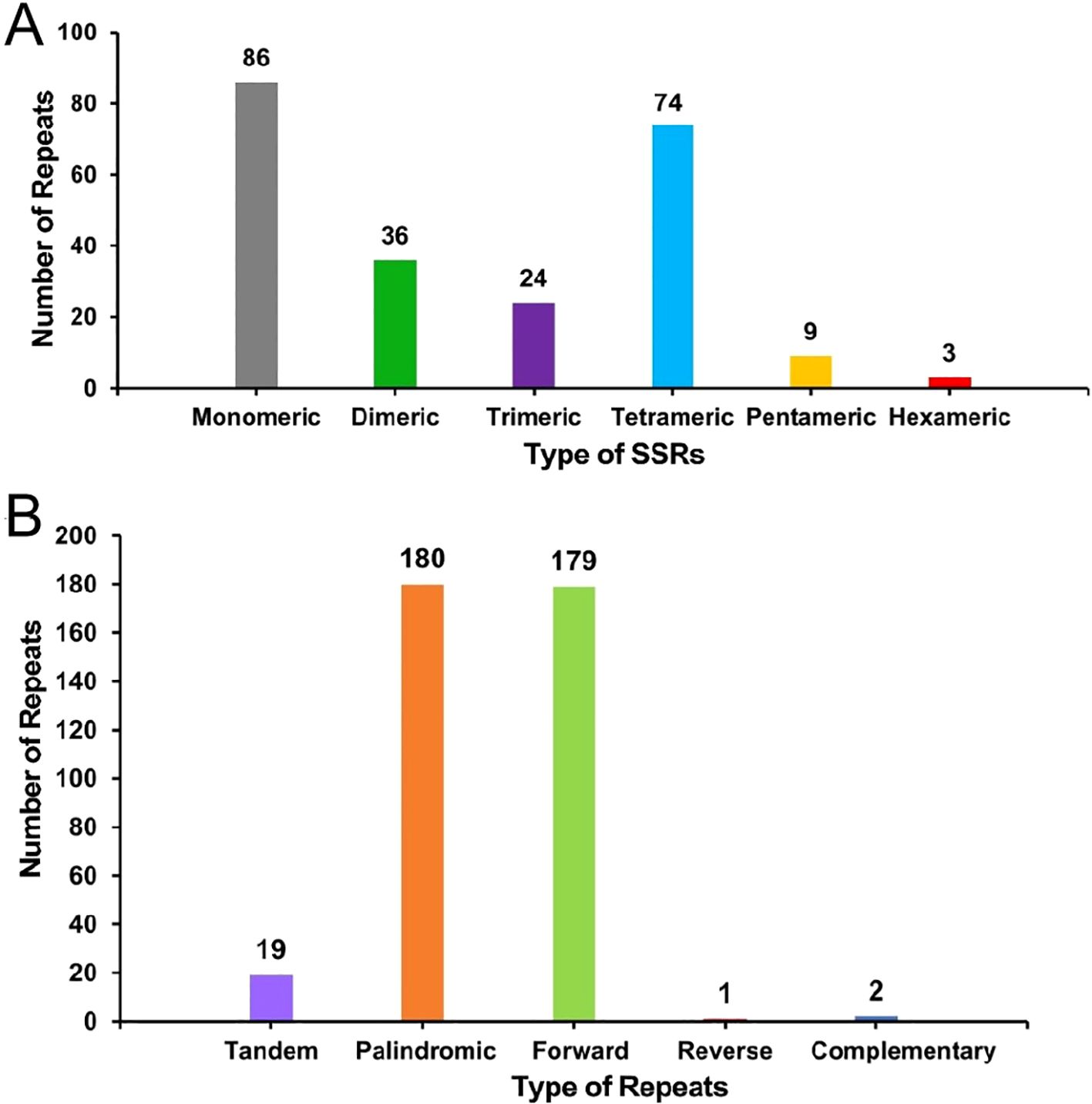
Figure 3. The histogram of the repeat sequence analysis. (A) Horizontal coordinates represent the type of SSRs, and vertical coordinates represent the number of repetitive fragments. The gray legend represents monomeric SSRs, the green legend represents dimeric SSRs, the purple legend represents trimeric SSRs, the blue legend represents tetrameric SSRs, the yellow legend represents hexameric SSRs, and the red legend represents hexameric SSRs. (B) The horizontal coordinates represent the type of repetitive sequence, and the vertical coordinates represent the number of repetitive fragments. The purple legend represents tandem repeats, the orange legend represents palindromic repeats, the green legend indicates forward repeats, the red legend represents reverse repeats, and the blue legend represents complementary repeats.
Sequence transfer analysis
During mitochondrial evolution, some fragments migrate from chloroplasts to mitochondrial genes, and the length and sequence similarity of the migrated fragments change from species to species. Based on the sequence similarity, a total of 38 fragments from the chloroplast genome were found in the mitochondrial genome of S. psammophila, with a total length of 16,536 bp, which accounted for 2.31% of the total length of the mitochondrial genome (Figure 4). Among these homologous fragments, MTPT 16 was the longest, with a length of 2,579 bp, and MTPT 8 and MTPT 9 were the shortest, both at 33 bp. Annotating these homologous sequences, we identified eight complete genes, namely, one PCG (atpE) and seven tRNA genes (trnD-GUC, trnM-CAU, trnN-GUU, trnP-UGG, trnS-GGA, trnV-UAC, and trnW-CCA).
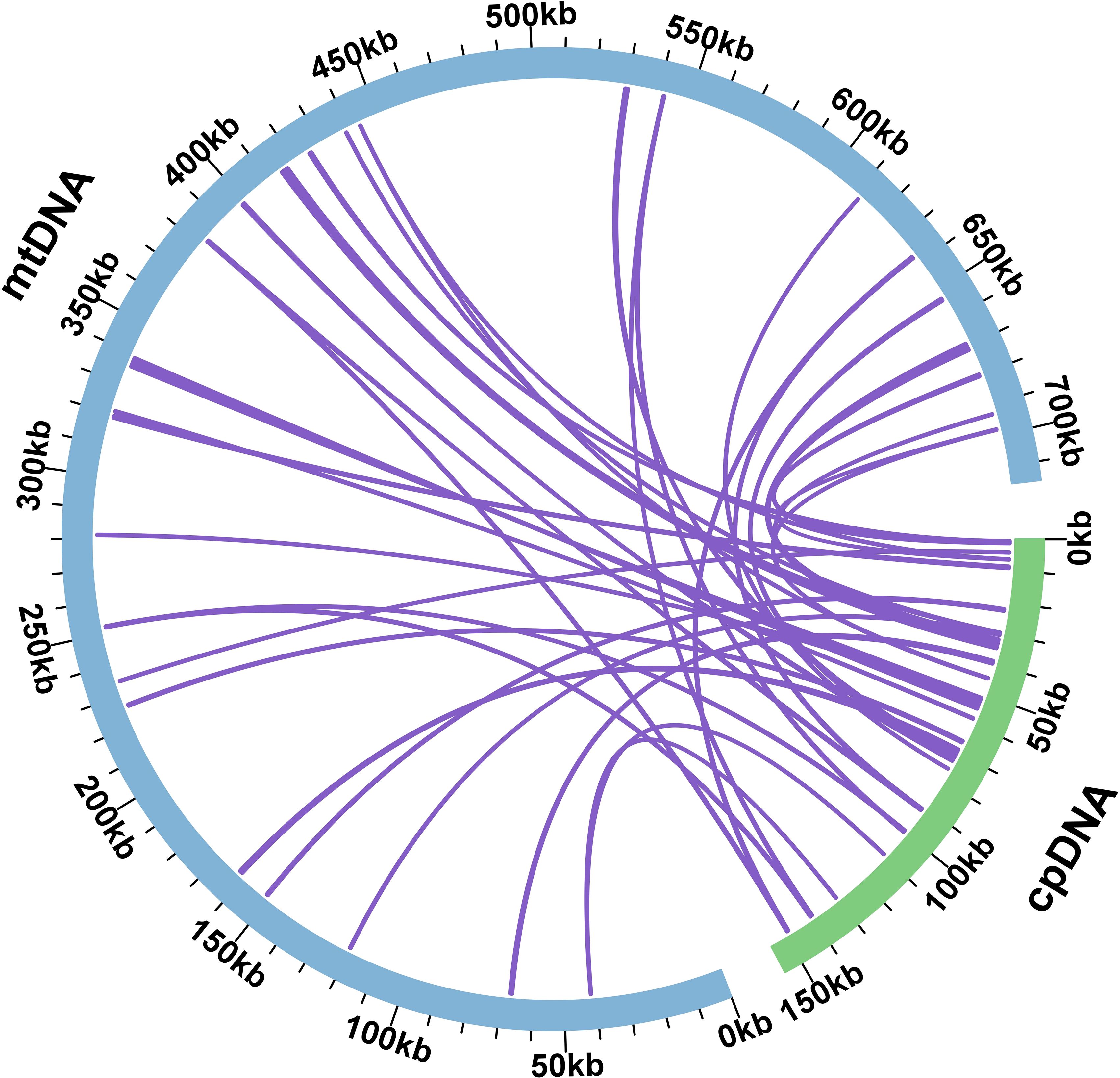
Figure 4. Homological sequences between mitochondrial and chloroplast genomes. The blue circular segment in the figure represents the mitochondrial genome, the green circular segment represents the chloroplast genome, and the purple line between the circular segments corresponds to a homologous fragment of the genome fragments.
Prediction of RNA editing sites
RNA editing events were predicted for 33 unique PCGs of the mitochondrial genome by setting the following criterion: cutoff value = 0.9. A total of 324 potential RNA editing sites were identified, all of which were C-to-U editing and basically appeared in the second or third position, with a few occurring in the third position (Supplementary Table S8). As shown in Figure 5, among these mitochondrial genes, the most edited gene was the nad4 gene, with 38 RNA editing sites identified. The least edited genes were atp1, cox2, rpl10, and rps4, with 1 RNA editing site identified.
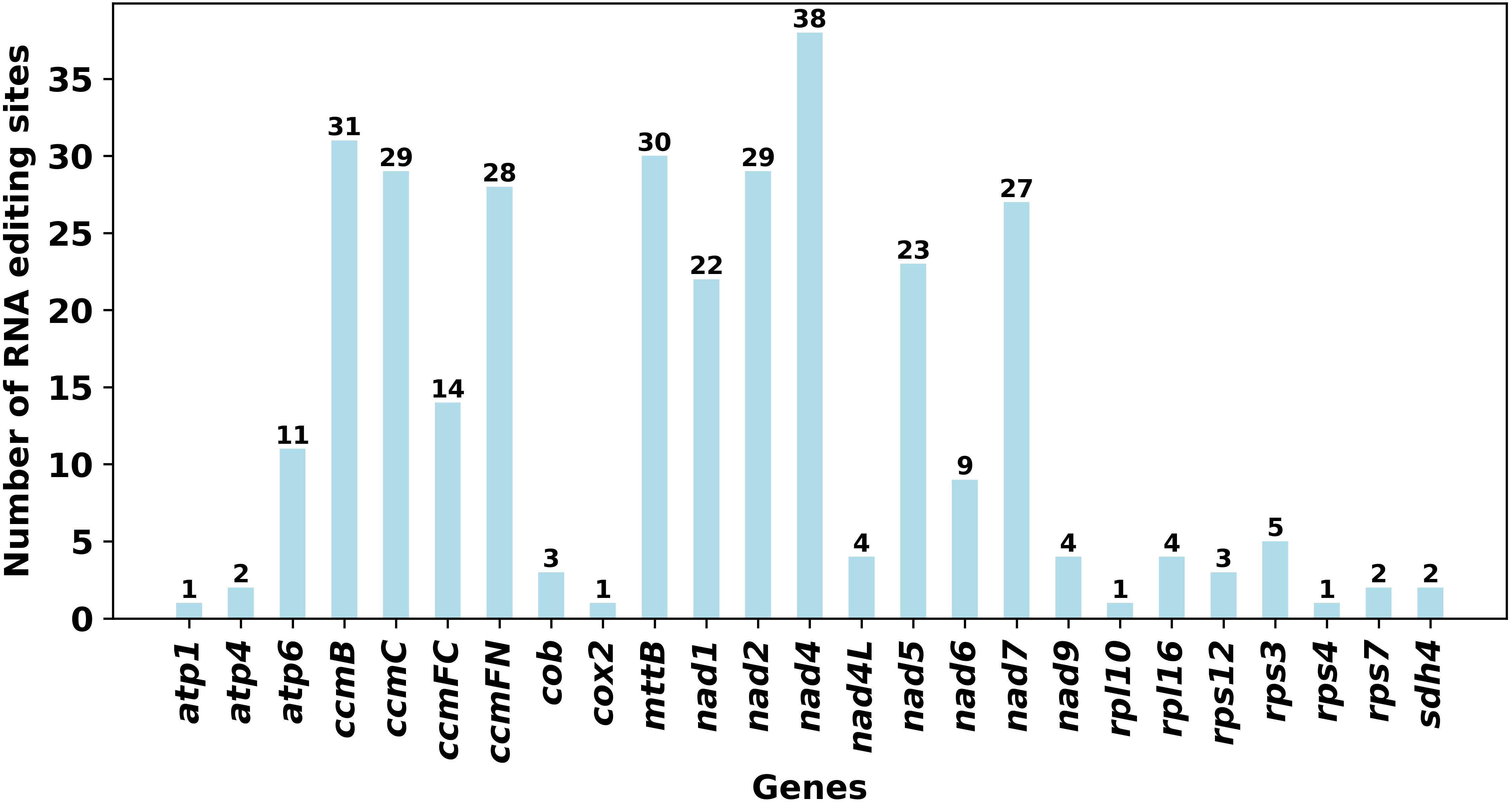
Figure 5. Number of RNA editing sites in all the PCGs.
Phylogenetic analysis
To further explore the evolutionary relationships of the mitochondria of S. psammophila, phylogenetic trees were constructed for 34 species of plants from three families, namely, Salicaceae, Euphorbiaceae, and Rhizophoraceae, based on the DNA sequences of 24 conserved mitochondrial PCGs, in which Bruguiera sexangula and Kandelia obovata of the Rhizophoraceae family were set up as the outgroups (Figure 6). The PCGs shared by these species are atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, ccmFC, ccmFN, cob, cox1, cox2, cox3, matR, mttB, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9. The topology of the mitochondrial DNA phylogeny coincides with the latest classification of the Angiosperm Phylogeny Group (APG). S. psammophila belongs to the family Salicaceae in the order Salicales and is most closely related to S. polaris.
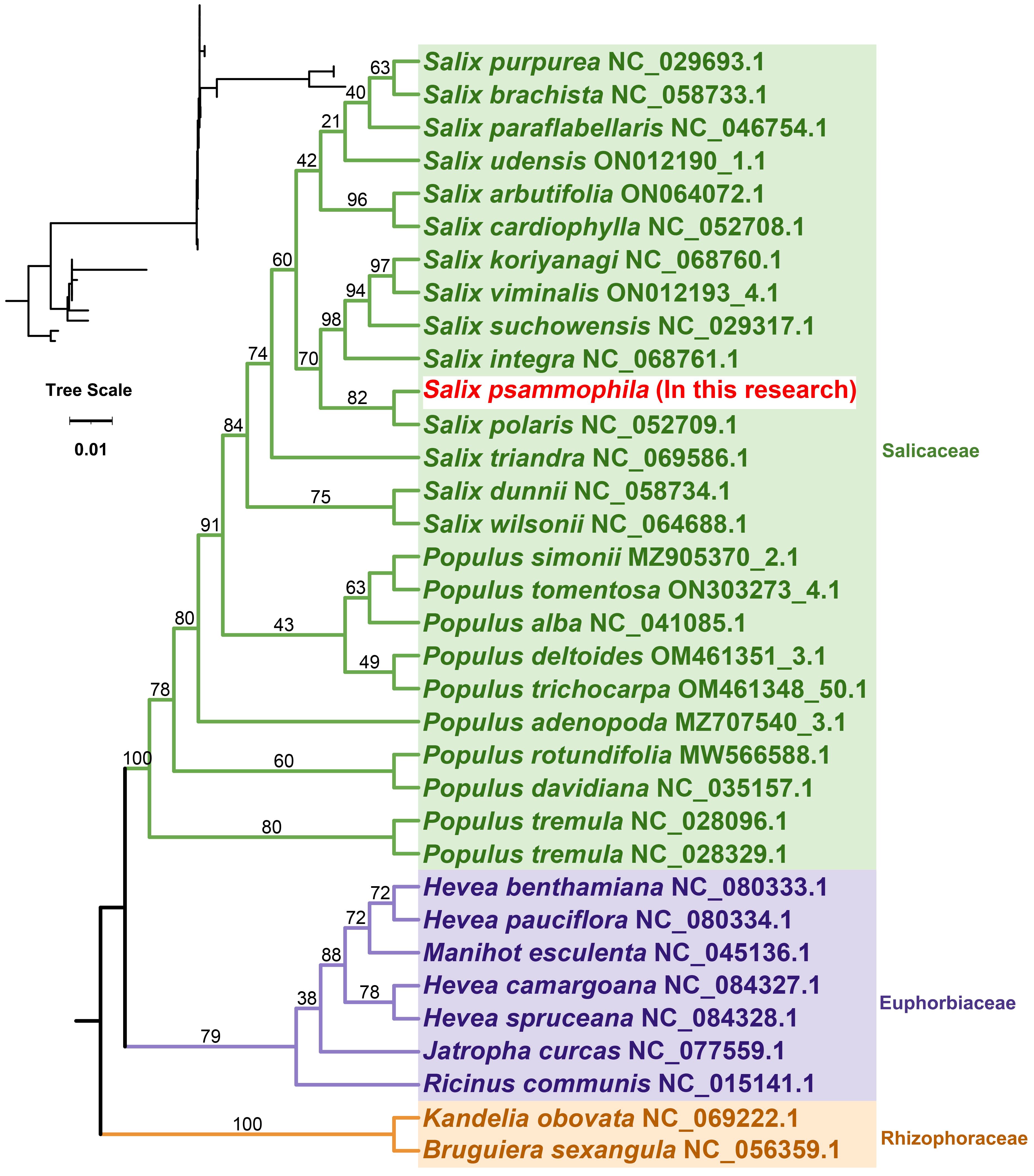
Figure 6. Phylogenetic relationships of S. psammophila.
Synteny analysis
S. koriyanagi, S. viminalis, S. suchowensis, S. integra, S. psammophila, and S. polaris were selected for the synteny analysis. As shown in Figure 7, there are a large number of homologous collinear blocks between the mitochondrial genomes of S. psammophila and the other five willow species. In addition, there are some gap regions where sequences are species-specific and have no homology with the rest of the species. The results showed that the mitochondrial genome of S. psammophila underwent genomic rearrangement with S. integra but not with S. polaris.
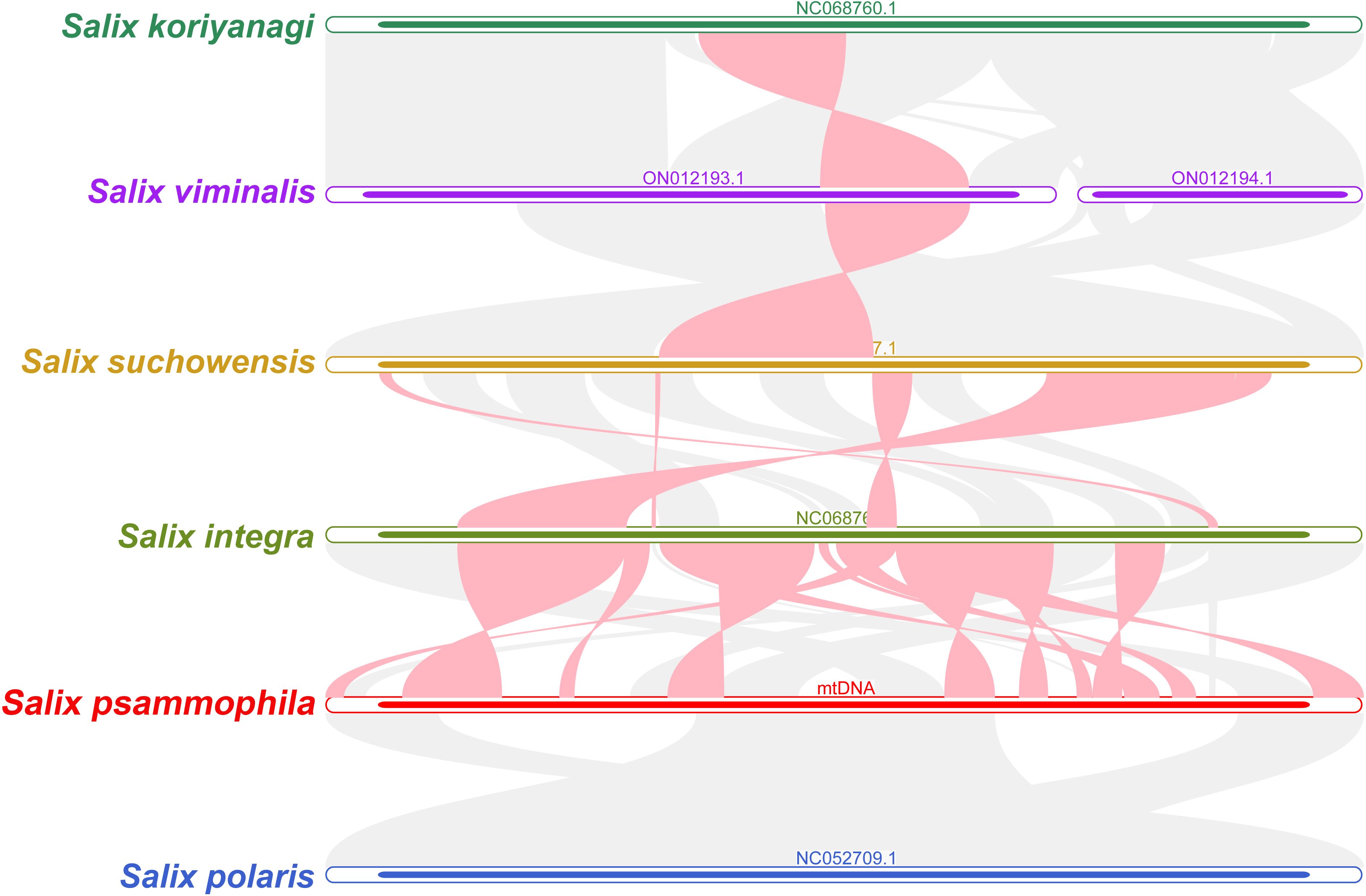
Figure 7. Synteny analysis of six different species in the genus Salix. The red curved regions indicate regions where inversion occurs, and the gray regions indicate regions of good homology.
Discussion
Complexity and variability mitochondrial genome size and structure in plants
The plant mitochondrial genome is an evolutionary dynamic entity with great plant diversity in terms of size, structure, gene content, etc (Petersen et al., 2020). S. psammophila mitochondrial genome is a circular structure with a total length of 715,555 bp and a GC content of 44.89%. The mitochondrial genome structure and GC content were similar to those of the known P. deltoides, P. simonii, S. wilsonii, and S. suchowensis, all of which were cyclic in structure, and the GC content was approximately 44.80%. However, there are also some differences in the genome structure and size of the Salicaceae plants. The mitochondrial genomes of both P. deltoides and P. simonii are three circulars, the P. tremula, P. tremula × P. alba, P. alba, and P. davidiana are the typical single circular sequences, as are the S. suchowensis, S. wilsonii, and S. purpurea (Kersten et al., 2016; Choi et al., 2017; Ye et al., 2017; Brenner et al., 2019; Chen et al., 2020; Bi et al., 2022; Han et al., 2022; Qu et al., 2022). In the genus Populus, the mitochondrial genome size is very conserved, and poplars such as P. simonii (781,478 bp), P. deltoides (802,637 bp), P. tremula (783,442 bp), and P. alba (838,420 bp) are essentially similar in mitochondrial genome size (Kersten et al., 2016; Brenner et al., 2019; Bi et al., 2022; Qu et al., 2022). Numerous studies have shown that a large number of repetitive units, the incorporation of foreign sequences, and the acquisition or loss of large genetic fragments of their own are the main causes of large differences in plant mitochondrial genomes (Wu et al., 2022). In the mitochondrial genome of the genus Populus, there are no repetitive sequences >350 bp, which may be the reason for the small differences in size and structure of the genus Populus mitochondrial genome (Bi et al., 2022). In contrast, the mitochondrial genome size of the genus Salix is highly variable, ranging from 599 kb (S. purpurea) to 735 kb (S. cardiophylla) (Wei et al., 2016; Chen et al., 2020). The size of plant mitochondrial genomes varies dramatically between families and even between different species of the same genus. For example, the mitochondrial genome size difference between woody cotton (Gossypium arboreum) and upland cotton (Gossypium hirsutum) is 65.6 kb (Ala et al., 2023). Furthermore, there was no positive correlation between genome size and gene number; P. deltoides mitochondrial genomes had the largest size but not the largest number of genes, with the largest number of genes being found in S. brachista, and the difference in the number of genes between their mitochondrial genomes was small (Supplementary Table S11). The mitochondrial genome of Welwitschia mirabilis is more than twice the size of Cycas taitungensis, but had fewer genes than Cycas taitungensis (Guo et al., 2016), Meanwhile, the sizes of the S. psammophila and S. wilsonii mitochondrial genomes are similar, and there are significant differences in the types and copy numbers of tRNA, suggesting that different species within the genus Salix possess their unique biological functions. Previous studies have demonstrated that the phylogeny of the mitogenome in Populus can effectively distinguish between different groups of poplar trees (Qu et al., 2022). Similarly, species within the genus Salix can also be categorized using phylogenetic trees. For instance, S. wilsonii and S. dunnii are classified under Sect. Wilsonianae, while S. brachista and S. paraflabellaris are grouped under Sect. Lindleyanae.
Repeat sequences in the mitochondrial genome of Salicaceae plants
A large number of repetitive sequences are distributed in the plant mitochondrial genome, and their size and arrangement are closely related to the differences and rearrangements among the plant mitochondrial genomes, which play an important role in the evolution of plant mitochondria (Tanaka et al., 2014; Wynn and Christensen, 2019; Yang et al., 2021). Repetitive sequences in plant mitochondrial genomes can be categorized into two groups, dispersed repeats and tandem repeats (Wu et al., 2019). Simple sequence repeat (SSR) is a highly variable tandem repeat sequence of 1–6 bp, commonly used as a molecular marker for species identification and genetic diversity studies (Ma et al., 2017; Li and Ye, 2020). A total of 232 SSRs were detected in the S. psammophila mitochondrial genome, and the analysis revealed that 95% of the mononucleotide SSRs were A/T, which was similar to the mitochondrial genomes of other genus Salix, with the highest proportion of A/T repeats among the mononucleotide repeats (Ye et al., 2017; Han et al., 2022). In addition to tandem repeat sequences, plant mitochondrial genomes are characterized by the presence of a large number of dispersed repeat sequences, including palindromic repeat sequences, forward repeat sequences, reverse repeat sequences, and complementary repeat sequences. There are 362 dispersed repeat sequences in the S. psammophila mitochondrial genome, with palindromic and forward repeats being the most abundant. As shown in Supplementary Table S6, a total of 236 (65.19%) dispersed repeats range from 30 to 39 bp, 108 (29.83%) repeats are between 40 and 99 bp in length, 4.7% of the repeats are within the range of 100 to 300 bp in length, and one of the repeats is 86,068 bp, which is a forward repeat. Larger repetitive sequences are critical in genomic structural changes, and paired forward and reverse repeat sequences (>1 kb) can produce subgenomic or isomeric conformations (Bi et al., 2020). In addition to S. psammophila, S. cardiophylla, S. paraflabellaris, and S. suchowensis mitochondrial genomes also contain repetitive sequences greater than 1 kb in length, and their impact on the structure and size expansion of mitochondrial genomes is of interest (Chang et al., 2013; Dong et al., 2018; Chen et al., 2020).
The rps14 function during the evolution of the genome of Salicaceae plants
The PCG analysis based on the mitochondrial genome of S. psammophila and S. suchowensis, S. wilsonii, P. simonii, and P. deltoides revealed that the mitochondrial genome of Salicaceae plants, like other plants, is relatively conserved. It has been shown that the number of PCGs gradually decreases during the evolution of species, and that the genes lost in most cases are RP genes and succinate dehydrogenase genes (Adams and Palmer, 2003). Through comparative analysis of the mitochondrial genomes of several Salicaceae plants, it was found that the rps14 gene was lost in S. psammophila, S. suchowensis, S. wilsonii, and P. simonii (Ye et al., 2017; Bi et al., 2022; Han et al., 2022; Qu et al., 2022). RPs are proteins in ribosomes that play a crucial role in ribosome biogenesis, protein synthesis, cell growth, development, and apoptosis (Ramakrishnan and White, 1998; Naora and Naora, 1999; Maguire and Zimmermann, 2001; Wang et al., 2013b). S. psammophila and P. simonii play important roles in resisting wind damage and fixing sand dunes and have excellent characteristics such as drought resistance, tolerance to barrenness, and strong adaptability. They are the main tree species in the Northwest and North China regions (Jia et al., 2019; Wu et al., 2020; Yang et al., 2022). S. suchowensis is mainly distributed in central and eastern China, and has the function of windbreak and sand fixation (Jia et al., 2020; Wang et al., 2024). The original ecological area of P. deltoides is from the southeastern United States to southern Canada and plays an important role in riparian ecosystems (Fahrenkrog et al., 2017). Based on their different geographical distribution and physiological characteristics, we speculate that the loss of rps14 in the evolution of Salicaceae plants may lead to their sensitivity to adversities such as drought (Supplementary Table S12). RPs are important for the translation of diverse proteins and are involved in overall cellular adaptation under stress conditions (Vemanna et al., 2019). It has been shown that 46 RPs are downregulated for expression under drought conditions (Narsai et al., 2013). Bi et al. found that rps14 and the ccmB, ccmFC, rps1, and rps10 genes may play an important role in the evolutionary process by analyzing selection pressures on PCGs of the Phaseolus vulgaris mitochondrial genome (Bi et al., 2020).
Conservation of RNA editing events in plant mitochondrial genomes
The oxidative phosphorylation system in mitochondria consists of five complexes (I–V), the normal assembly of which requires standard processing of pre-mRNAs, involving RNA editing and intron shearing, and is important for the maintenance of mitochondrial functions (Dudkina et al., 2006; Li et al., 2014; Sun et al., 2018). RNA editing is a phenomenon in which genetic information is altered through the insertion of nucleotides, or nucleotide deletions and transitions, into the messenger RNA of a functional gene (Keller et al., 1999). RNA editing can occur in a variety of forms, including C-to-U, U-to-C, and A-to-I conversions; U insertions and deletions; and G insertions (Small et al., 2020). In plants, RNA editing events occur mainly in plant mitochondria and chloroplasts, and take place mainly in the form of a C-to-U transition (Hao et al., 2021). We analyzed the 33 PCGs within the mitochondrial genome of S. psammophila and identified a total of 324 RNA editing sites. All of these sites exhibited C to U transitions, which is consistent with findings in S. suchowensis and P. deltoides (Ye et al., 2017; Qu et al., 2022). In addition, we found the highest number of edits to nad4 in S. psammophila and S. suchowensis mitochondria. It has also been found in plants such as Corydalis saxicola, with the most editing of the nad4 in the mitochondrial genome (Maldonado et al., 2022; Li et al., 2024). The nad4 is one of the components of complex I, which is the largest complex related to respiration in the mitochondria of land plants and is important for maintaining plant growth and development (Møller et al., 2021). Thus, extensive editing of the nad4 may be important for the functioning of complex I in plant mitochondria.
Conclusions
In this study, we successfully assembled and annotated the mitochondrial genome of S. psammophila for the first time, identifying it as a single circular structure with a total length of 715,555 bp and a GC content of 44.89%. Comparative analysis with mitochondrial genomes of S. wilsonii, S. suchowensis, P. simonii, and P. deltoides revealed that PCGs within the Salicaceae family are highly conserved, with significant variations primarily observed in RPs. Notably, rps14 is absent in S. psammophila, a species well-adapted to arid environments. This absence might indicate a role in enhancing tolerance to environmental stresses such as drought. These findings contribute valuable insights into the phylogenetic and genetic research of Salicaceae plants.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
HQ: Data curation, Formal Analysis, Writing – original draft, Resources, Software, Methodology, Validation, Visualization. YC: Data curation, Formal Analysis, Software, Writing – original draft. RW: Data curation, Resources, Writing – original draft. WZ: Data curation, Formal Analysis, Investigation, Writing – original draft. ZZ: Writing – original draft, Data curation, Formal Analysis, Validation. FY: Data curation, Resources, Writing – original draft. HY: Funding acquisition, Investigation, Methodology, Project administration, Writing – review & editing. GL: Project administration, Software, Visualization, Writing – review & editing, Conceptualization, Funding acquisition. JZ: Funding acquisition, Investigation, Project administration, Validation, Writing – original draft, Writing – review & editing, Conceptualization, Methodology, Supervision, Visualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Beijing Academy of Agriculture and Forestry Sciences Science Foundation (Nos. KJCX20230203 and JJP2024-027), the National Natural Science Foundation of China (32360401), and the Inner Mongolia Natural Science Foundation (2023MS03050).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1411289/full#supplementary-material
Supplementary Figure 1 | The codon preference analysis of the mitochondrial genome. Different colors of the same amino acid represent the RSCU values of different codons editing this amino acid.
References
Adams, K., Palmer, D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/S1055-7903(03)00194-5
Ala, K. G., Zhao, Z., Ni, L., Wang, Z. (2023). Comparative analysis of mitochondrial genomes of two alpine medicinal plants of Gentiana (Gentianaceae). PloS One 18, e0281134. doi: 10.1371/journal.pone.0281134
Bao, Y., Zhang, G. (2012). Study of adsorption characteristics of methylene blue onto activated carbon made by Salix Psammophila. Energy Proc. 16, 1141–1146. doi: 10.1016/j.egypro.2012.01.182
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C., Lu, N., Xu, Y., He, C., Lu, Z. (2020). Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by Comparative Genomic Approaches. Int. J. Mol. Sci. 21, 3778. doi: 10.3390/ijms21113778
Bi, C., Paterson, A. H., Wang, X., Xu, Y., Wu, D., Qu, Y., et al. (2016). Analysis of the complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. BioMed. Res. Int. 2016, 1–18. doi: 10.1155/2016/5040598
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.914635
Brenner, W. G., Mader, M., Müller, N. A., Hoenicka, H., Schroeder, H., Zorn, I., et al. (2019). High level of conservation of mitochondrial RNA editing sites among Four Populus Species. G3 (Bethesda) 9, 709–717. doi: 10.1534/g3.118.200763
Chang, S., Wang, Y., Lu, J., Gai, J., Li, J., Chu, P., et al. (2013). The mitochondrial genome of soybean reveals complex genome structures and gene evolution at intercellular and phylogenetic levels. PloS One 8, e56502. doi: 10.1371/journal.pone.0056502
Chen, Y., Ye, W., Zhang, Y., Xu, Y. (2015). High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 43, 7762–7768. doi: 10.1093/nar/gkv784
Chen, X., Zhang, L., Huang, Y., Zhao, F. (2020). Mitochondrial genome of Salix cardiophylla and its implications for infrageneric division of the genus of Salix. Mitochondrial DNA B Resour. 5, 3485–3486. doi: 10.1080/23802359.2020.1827065
Chen, Z., Zhao, N., Li, S., Grover, C. E., Nie, H., Wendel, J. F., et al. (2017). Plant mitochondrial genome evolution and cytoplasmic male sterility. CRC Crit. Rev. Plant Sci. 36, 55–69. doi: 10.1080/07352689.2017.1327762
Cheng, Y., He, X., Priyadarshani, S. V. G. N., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genomics 22, 167. doi: 10.1186/s12864-021-07490-9
Choi, M. N., Han, M., Lee, H., Park, H.-S., Kim, M.-Y., Kim, J.-S., et al. (2017). The complete mitochondrial genome sequence of Populus davidiana dode. Mitochondrial DNA B Resour. 2, 113–114. doi: 10.1080/23802359.2017.1289346
Dong, S., Zhao, C., Chen, F., Liu, Y., Zhang, S., Wu, H., et al. (2018). The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genomics 19, 614. doi: 10.1186/s12864-018-4991-4
Dudkina, N. V., Heinemeyer, J., Sunderhaus, S., Boekema, E. J., Braun, H.-P. (2006). Respiratory chain supercomplexes in the plant mitochondrial membrane. Trends Plant Sci. 11, 232–240. doi: 10.1016/j.tplants.2006.03.007
Edera, A. A., Small, I., Milone, D. H., Sanchez-Puerta, M. V. (2021). Deepred-Mt: deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 136, 104682. doi: 10.1016/j.compbiomed.2021.104682
Fahrenkrog, A. M., Neves, L. G., Resende, M. F. R., Dervinis, C., Davenport, R., Barbazuk, W. B., et al. (2017). Population genomics of the eastern cottonwood ( Populus deltoides ). Ecol. Evol. 7, 9426–9440. doi: 10.1002/ece3.3466
Fakih, Z., Plourde, M. B., Germain, H. (2023). Differential participation of plant ribosomal proteins from the small ribosomal subunit in protein translation under stress. Biomolecules 13, 1160. doi: 10.3390/biom13071160
Gray, M. W. (2012). Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 4, a011403–a011403. doi: 10.1101/cshperspect.a011403
Guo, W., Grewe, F., Fan, W., Young, G. J., Knoop, V., Palmer, J. D., et al. (2016). Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 33, 1448–1460. doi: 10.1093/molbev/msw024
Han, F., Qu, Y., Chen, Y., Xu, L., Bi, C. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1031769
Hao, W., Liu, G., Wang, W., Shen, W., Zhao, Y., Sun, J., et al. (2021). RNA editing and its roles in plant organelles. Front. Genet. 12. doi: 10.3389/fgene.2021.757109
Jia, H., Li, J., Zhang, J., Sun, P., Lu, M., Hu, J. (2019). The Salix psammophila SpRLCK1 involved in drought and salt tolerance. Plant Physiol. Biochem. 144, 222–233. doi: 10.1016/j.plaphy.2019.09.042
Jia, H., Wang, L., Li, J., Sun, P., Lu, M., Hu, J. (2020). Physiological and metabolic responses of Salix sinopurpurea and Salix suchowensis to drought stress. Trees 34, 563–577. doi: 10.1007/s00468-019-01937-z
Jin, J.-J., Yu, W.-B., Yang, J.-B., Song, Y., dePamphilis, C. W., Yi, T.-S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Jo, Y. D., Park, J., Kim, J., Song, W., Hur, C.-G., Lee, Y.-H., et al. (2011). Complete sequencing and comparative analyses of the pepper (Capsicum annuum L.) plastome revealed high frequency of tandem repeats and large insertion/deletions on pepper plastome. Plant Cell Rep. 30, 217–229. doi: 10.1007/s00299-010-0929-2
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keller, W., Wolf, J., Gerber, A. (1999). Editing of messenger RNA precursors and of tRNAs by adenosine to inosine conversion. FEBS Lett. 452, 71–76. doi: 10.1016/S0014-5793(99)00590-6
Kersten, B., Faivre Rampant, P., Mader, M., Le Paslier, M.-C., Bounon, R., Berard, A., et al. (2016). Genome sequences of Populus tremula chloroplast and mitochondrion: implications for holistic poplar breeding. PloS One 11, e0147209. doi: 10.1371/journal.pone.0147209
Kolmogorov, M., Yuan, J., Lin, Y., Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Letunic, I., Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Lewis, S., Searle, S., Harris, N., Gibson, M., Iyer, V., Richter, J., et al. (2002). Apollo: a sequence annotation editor. Genome Biol. 3, research0082.1. doi: 10.1186/gb-2002-3-12-research0082
Li, Z.-J. (2009). Methods for the identification of horizontal gene transfer (HGT) events and progress in related fields: methods for the identification of horizontal gene transfer (HGT) events and progress in related fields. Hereditas (Beijing) 30, 1108–1114. doi: 10.3724/SP.J.1005.2008.01108
Li, C., Liu, H., Qin, M., Tan, Y., Ou, X., Chen, X., et al. (2024). RNA editing events and expression profiles of mitochondrial protein-coding genes in the endemic and endangered medicinal plant, Corydalis saxicola. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1332460
Li, J., Ye, C. (2020). Genome-wide analysis of microsatellite and sex-linked marker identification in Gleditsia sinensis. BMC Plant Biol. 20, 338. doi: 10.1186/s12870-020-02551-9
Li, X., Zhang, Y., Hou, M., Sun, F., Shen, Y., Xiu, Z., et al. (2014). Small kernel 1 encodes a pentatricopeptide repeat protein required for mitochondrial nad7 transcript editing and seed development in maize (Zea mays) and rice (Oryza sativa). Plant J. 79, 797–809. doi: 10.1111/tpj.12584
Liu, S., Ni, Y., Li, J., Zhang, X., Yang, H., Chen, H., et al. (2023). CPGView: a package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 23, 694–704. doi: 10.1111/1755-0998.13729
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Lu, D., Hao, L., Huang, H., Zhang, G. (2019). The complete chloroplast genome of Salix psamaphila, a desert shrub in northwest China. Mitochondrial DNA B Resour. 4, 3432–3433. doi: 10.1080/23802359.2019.1675485
Ma, Q., Li, S., Bi, C., Hao, Z., Sun, C., Ye, N. (2017). Complete chloroplast genome sequence of a major economic species, Ziziphus jujuba (Rhamnaceae). Curr. Genet. 63, 117–129. doi: 10.1007/s00294-016-0612-4
Maguire, B. A., Zimmermann, R. A. (2001). The ribosome in focus. Cell 104, 813–816. doi: 10.1016/S0092-8674(01)00278-1
Maldonado, M., Abe, K. M., Letts, J. A. (2022). A structural perspective on the RNA editing of plant respiratory complexes. Int. J. Mol. Sci. 23, 684. doi: 10.3390/ijms23020684
Møller, I. M., Rasmusson, A. G., Van Aken, O. (2021). Plant mitochondria – past, present and future. Plant J. 108, 912–959. doi: 10.1111/tpj.15495
Naora, H., Naora, H. (1999). Involvement of ribosomal proteins in regulating cell growth and apoptosis: translational modulation or recruitment for extraribosomal activity? Immunol. Cell Biol. 77, 197–205. doi: 10.1046/j.1440-1711.1999.00816.x
Narsai, R., Wang, C., Chen, J., Wu, J., Shou, H., Whelan, J. (2013). Antagonistic, overlapping and distinct responses to biotic stress in rice (Oryza sativa) and interactions with abiotic stress. BMC Genomics 14, 93. doi: 10.1186/1471-2164-14-93
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Petersen, G., Anderson, B., Braun, H.-P., Meyer, E. H., Møller, I. M. (2020). Mitochondria in parasitic plants. Mitochondrion 52, 173–182. doi: 10.1016/j.mito.2020.03.008
Petersen, G., Cuenca, A., Møller, I. M., Seberg, O. (2015). Massive gene loss in mistletoe (Viscum, Viscaceae) mitochondria. Sci. Rep. 5, 17588. doi: 10.1038/srep17588
Petibon, C., Malik Ghulam, M., Catala, M., Abou Elela, S. (2020). Regulation of ribosomal protein genes: an ordered anarchy. Wiley Interdiscip. Rev. RNA 12, e1632. doi: 10.1002/wrna.1632
Qu, Y., Zhou, P., Tong, C., Bi, C., Xu, L. (2022). Assembly and analysis of the Populus deltoides mitochondrial genome: the first report of a multicircular mitochondrial conformation for the genus Populus. J. For Res. (Harbin) 34, 717–733. doi: 10.1007/s11676-022-01511-3
Ramakrishnan, V., White, S. W. (1998). Ribosomal protein structures: insights into the architecture, machinery and evolution of the ribosome. Trends Biochem. Sci. 23, 208–212. doi: 10.1016/S0968-0004(98)01214-6
Richardson, A. O., Palmer, J. D. (2006). Horizontal gene transfer in plants. J. Exp. Bot. 58, 1–9. doi: 10.1093/jxb/erl148
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. (2015). Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. U.S.A. 112, E3515–E 3524. doi: 10.1073/pnas.1504491112
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PloS Biol. 10, e1001241. doi: 10.1371/journal.pbio.1001241
Small, I. D., Schallenberg-Rüdinger, M., Takenaka, M., Mireau, H., Ostersetzer-Biran, O. (2020). Plant organellar RNA editing: what 30 years of research has revealed. Plant J. 101, 1040–1056. doi: 10.1111/tpj.14578
Smith, D. R. (2011). Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol. Evol. 3, 743–748. doi: 10.1093/gbe/evr068
Sun, F., Zhang, X., Shen, Y., Wang, H., Liu, R., Wang, X., et al. (2018). The pentatricopeptide repeat protein EMPTY PERICARP 8 is required for the splicing of three mitochondrial introns and seed development in maize. Plant J. 95, 919–932. doi: 10.1111/tpj.14030
Tanaka, Y., Tsuda, M., Yasumoto, K., Terachi, T., Yamagishi, H. (2014). The complete mitochondrial genome sequence of Brassica oleracea and analysis of coexisting mitotypes. Curr. Genet. 60, 277–284. doi: 10.1007/s00294-014-0433-2
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Vemanna, R. S., Bakade, R., Bharti, P., Kumar, M. K. P., Sreeman, S. M., Senthil-Kumar, M., et al. (2019). Cross-talk signaling in rice during combined drought and bacterial blight stress. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00193
Wang, Z., Hou, J., Lu, L., Qi, Z., Sun, J., Gao, W., et al. (2013b). Small ribosomal protein subunit S7 suppresses ovarian tumorigenesis through regulation of the PI3K/AKT and MAPK pathways. PloS One 8, e79117. doi: 10.1371/journal.pone.0079117
Wang, J., Lan, P., Gao, H., Zheng, L., Li, W., Schmidt, W. (2013a). Expression changes of ribosomal proteins in phosphate- and iron-deficient Arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genomics 14, 783. doi: 10.1186/1471-2164-14-783
Wang, Y., Wang, H., Yu, C., Yan, X., Chu, J., Jiang, B., et al. (2024). Comprehensive bioinformation analysis of homeodomain-leucine zipper gene family and expression pattern of HD-Zip I under abiotic stress in Salix suchowensis. BMC Genomics 25, 182. doi: 10.1186/s12864-024-10067-x
Wei, S., Wang, X., Bi, C., Xu, Y., Wu, D., Ye, N. (2016). Assembly and analysis of the complete Salix purpurea L. (Salicaceae) mitochondrial genome sequence. Springerplus 5, 1894. doi: 10.1186/s40064-016-3521-6
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z., Hu, K., Yan, M., Song, L., Wen, J., Ma, C., et al. (2019). Mitochondrial genome and transcriptome analysis of five alloplasmic male-sterile lines in Brassica juncea. BMC Genomics 20, 348. doi: 10.1186/s12864-019-5721-2
Wu, Z., Liao, X., Zhang, X., Tembrock, L. R., Broz, A. (2022). Genomic architectural variation of plant mitochondria-a review of multichromosomal structuring. J. Syst. Evol. 60, 160–168. doi: 10.1111/jse.12655
Wu, H., Yao, D., Chen, Y., Yang, W., Zhao, W., Gao, H., et al. (2020). De Novo genome assembly of Populus simonii further supports that Populus simonii and Populus trichocarpa belong to different sections. G3 (Bethesda) 10, 455–466. doi: 10.1534/g3.119.400913
Wynn, E. L., Christensen, A. C. (2019). Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3 (Bethesda) 9, 549–559. doi: 10.1534/g3.118.200948
Yang, H., Fan, L., Yu, X., Zhang, X., Hao, P., Wei, D., et al. (2022). Analysis of the NAC gene family in Salix and the identification of SpsNAC005 gene contributing to salt and drought tolerance. Forests 13, 971. doi: 10.3390/f13070971
Yang, H., Li, W., Yu, X., Zhang, X., Zhang, Z., Liu, Y., et al. (2021). Insights into molecular structure, genome evolution and phylogenetic implication through mitochondrial genome sequence of Gleditsia sinensis. Sci. Rep. 11, 14850. doi: 10.1038/s41598-021-93480-6
Ye, N., Wang, X., Li, J., Bi, C., Xu, Y., Wu, D., et al. (2017). Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. PeerJ 5, e3148. doi: 10.7717/peerj.3148
Zhang, T., Fang, Y., Wang, X., Deng, X., Zhang, X., Hu, S., et al. (2012). The complete chloroplast and mitochondrial genome sequences of Boea hygrometrica: insights into the evolution of plant organellar genomes. PloS One 7, e30531. doi: 10.1371/journal.pone.0030531
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, J., Liu, G., Wei, J. (2024). Assembly and comparative analysis of the first complete mitochondrial genome of Setaria italica. Planta 260, 23. doi: 10.1007/s00425-024-04386-2
Keywords: Salix psammophila, Salicaceae, mitochondrial genome, Pacbio HiFi, comparative analysis
Citation: Qiao H, Chen Y, Wang R, Zhang W, Zhang Z, Yu F, Yang H, Liu G and Zhang J (2024) Assembly and comparative analysis of the first complete mitochondrial genome of Salix psammophila, a good windbreak and sand fixation shrub. Front. Plant Sci. 15:1411289. doi: 10.3389/fpls.2024.1411289
Received: 02 April 2024; Accepted: 02 September 2024;
Published: 02 October 2024.
Edited by:
Junhua Peng, Spring Valley Agriscience Co., Ltd, ChinaReviewed by:
Yoshinori Fukasawa, Utsunomiya University, JapanYanshu Qu, Nanjing Forestry University, China
Copyright © 2024 Qiao, Chen, Wang, Zhang, Zhang, Yu, Yang, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiewei Zhang, emhhbmdqaWV3ZWlAYmFhZnMubmV0LmNu; and6aGFuZzkxOUAxNjMuY29t; Guiming Liu, bWluZ2d1aWxpdUBhbGl5dW4uY29t; Haifeng Yang, aGFpZmVuZ0BpbWF1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship