 Ana López-Malvar1,2
Ana López-Malvar1,2 Zoila Reséndiz-Ramirez3
Zoila Reséndiz-Ramirez3 Ana Butrón4
Ana Butrón4 Jose Cruz Jiménez-Galindo5
Jose Cruz Jiménez-Galindo5 Pedro Revilla4
Pedro Revilla4 Rosa Ana Malvar4*
Rosa Ana Malvar4*- 1Facultad, de Biología, Departamento de Biología Vegetal Y Ciencias del Suelo, Universidad de Vigo, Vigo, Spain
- 2Agrobiología Ambiental, Calidad de Suelos Y Plantas (UVIGO), Unidad Asociada a La MBG (CSIC), Vigo, Spain
- 3Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarios, Río Bravo, Tamaulipas, Mexico
- 4Misión Biológica de Galicia (CSIC), Pontevedra, Spain
- 5National Institute of Forestry Agriculture and Livestock Research (INIFAP), Cuauhtémoc, Chihuahua, Mexico
Introduction: Validations of previously detected quantitative trait loci (QTLs) to assess their reliability are crucial before implementing breeding programs. The objective of this study was to determine the reliability and practical usefulness of previously reported QTLs for resistance to stem tunneling by the Mediterranean stem borer (MSB) and yield. These authors used approximately 600 recombinant inbred lines (RILs) from a multiparent advanced generation intercross (MAGIC) population to map QTL using a genome-wide association study (GWAS) approach.
Methods: We identified RILs situated at the extremes of resistance and yield distributions within the whole MAGIC, and those QTLs were evaluated per se and crossed to a tester (A638) using lattice designs. In each set, a significant single-nucleotide polymorphism (SNP) was considered validated if (1) the same SNP was associated with the trait with a p-value < 0.02, or (2) within a ±2-Mbp interval, an SNP associated with the trait exhibited a p-value < 0.02 and demonstrated linkage disequilibrium (r2 > 0.2) with the SNPs previously reported.
Results and discussion: The novel QTL validation approach was implemented using improved experimental designs that led to higher heritability estimates for both traits compared to those estimated with the whole MAGIC population. The procedure used allowed us to jointly validate several QTL and to ascertain their possible contribution to hybrid improvement. Specifically, nearly three-quarters of the QTLs for tunnel length were confirmed. Notably, QTLs located in the genomic region 6.05–6.07 were consistently validated across different sets and have been previously linked to resistance against stalk tunneling in various mapping populations. For grain yield, approximately 10 out of 16 QTLs were validated. The validation rate for yield was lower than for tunnel length, likely due to the influence of dominance and/or epistatic effects. Overall, 9 out of 21 QTLs for tunnel length and 6 out of 17 QTLs for grain yield identified in our previous research were validated across both validation sets, indicating a moderate genetic correlation between per se and testcross performance of inbred lines. These findings offer insights into the reliability of QTL and genomic predictions, both derived from assessments conducted on the entire MAGIC population. Genomic predictions for tunnel length based on inbred line evaluations could be useful to develop more resistant hybrids; meanwhile, genomic prediction for yield could only be valid in a homozygous background.
Introduction
The primary pest affecting maize in Europe is the Mediterranean corn borer (MCB) Sesamia nonagrioides (Lef.). Stem corn borer larvae feed on the stem pith of maize producing tunnels that cause yield losses of approximately 30%, equivalent to worldwide losses of 311.3 million tons every year (Meissle et al., 2010). Tunnels produced in the stalk pith interfere with assimilate movement of nutrients toward the developing ear and increase lodging rate (López et al., 2001). One approach to pest control involves the cultivation of resistant and/or tolerant varieties, particularly in regions where the cultivation of transgenic maize is prohibited or heavily regulated, such as in Europe (Farinós et al., 2004). Genetic improvement of pest resistance relies on attaining varieties that experience reduced damage produced by insects. However, the most resistant varieties could not be the most productive in terms of grain yield. Even more, certain materials have shown a negative correlation between resistance to stem borer attack and grain yield (Sandoya et al., 2008; Butrón et al., 2012). In those cases, selection of varieties that are able to produce higher grain yields under high insect pressure could be a better alternative (Butrón et al., 1999; Ordas et al., 2012). However, at a particular site, the pest infestation level is influenced by multiple factors, exhibiting erratic behavior across years, and differences among varieties for grain yield could be highly affected by the infestation rate. Therefore, it is important to assess varieties’ performance across different scenarios concerning insect attack intensity in order to select varieties with high stable yield.
In order to optimize the development of varieties with reduced damage by insect attack and/or high yield across different scenarios, it is imperative to comprehend the inheritance of the traits involved and to study the regions of the genome or quantitative trait loci (QTLs) associated with resistance using different mapping populations in order to advance in the complete QTLome knowledge for these highly relevant traits (Salvi and Tuberosa, 2015). Accordingly, we can devise the optimal selection method (whether genomic or phenotypic), determine the feasibility of managing any QTL through marker-assisted selection (MAS), or even investigate genes involved in the inheritance of insect resistance. In the specific case of resistance to corn borers, QTLs have been widely sought using biparental populations and linkage-based QTL mapping (Ordas et al., 2009; Samayoa et al., 2014; Samayoa et al., 2015a, 2017; Jiménez-Galindo et al., 2017). However, the use of high-density molecular markers is now widespread due to its low cost, and genome-wide association studies (GWAS) are being conducted for a wide range of traits, including maize resistance to insects (Butrón et al., 2018). GWAS for resistance to the MCB have been conducted using diversity panels of inbred lines (Samayoa et al., 2015b) and multiparent advanced generation intercross (MAGIC) populations (Jiménez-Galindo et al., 2019).
Although many QTLs for resistance to stem corn borers have been found, and molecular markers significantly associated to resistance have been identified, MAS (Flint-Garcia et al., 2003; Samayoa et al., 2019) and genomic selection (Gesteiro et al., 2023) have been scarcely used to improve maize resistance to insects. External validations of detected QTL to assess QTL reliability are advisable before implementing MAS. QTL reliability can be affected by a significant QTL × environment interaction but also by low phenotyping precision.
Phenotyping genetically wide panels of inbred lines or RILs is challenging due to the difficulty of controlling the high experimental error associated with large trials; mostly when evaluations are often done using incomplete block or augmented designs in which most genotypes are assessed only once in each trial. Moreover, assessing plant traits related to performance against pest attack involves managing pest populations that could show variability for survival, voracity, or any other trait that would affect plant performance. Therefore, the high experimental errors associated with these evaluations could greatly interfere with QTL identification.
In addition, reliable additive-effect QTL could not be useful to discriminate among heterozygous genotypes if heterotic effects can counterbalance additive effects. Therefore, QTL validation should be done using inbred lines as well as hybrids to really ascertain the practical value of detected QTLs. Hence, the objective of this study was to determine the reliability and practical usefulness of QTL for resistance and yield reported by Jiménez-Galindo et al. (2019). Identification of QTL was based on a GWAS approach applied to a maize MAGIC population evaluated using augmented designs in a 2-year evaluation. For validation purposes, we selected inbred line sets at the tails of distributions for resistance and yield in the whole MAGIC to maintain genetic variability for both traits. These inbreds were evaluated per se and crossed to a tester (A638) using lattice designs. The results obtained in this study will contribute to expanding the maize QTLome knowledge for traits highly relevant for maize breeding as the they are related to pest tolerance and resistance.
Materials and methods
Field experiments
The Maize Genetics and Breeding group of the Misión Biológica de Galicia has developed an eight-way MAGIC population of 608 RILs. This MAGIC population was evaluated for resistance to corn borer attack and grain yield for 2 years (2014 and 2015) using augmented designs (Jiménez-Galindo et al., 2019). Data compiled by these authors are provided in a Supplementary Table.
Based on those evaluations, we selected a group of lines presenting either the longest or shortest tunnels made by MCB attack and/or exhibiting the highest or lowest yield. A collection of 56 RILs [inbred line validation set (IVS)], representing the widest variability for these two traits, was then established as the basis for this study. This set of RILs was crossed with A638 to obtain hybrids to validate QTL in a heterozygous background [hybrid validation set (HVS)]. The tester was chosen to avoid interferences on differences among hybrids due to divergences in heterosis as the inbred A638 belongs to the “Reid” group while RILs are characterized by the lack of Reid material in their pedigrees.
The IVS was evaluated along with the eight inbred checks used in previous evaluations (Jiménez-Galindo et al., 2019). Five of the checks were founders of the MAGIC population (A509, EP125, EP17, EP86, and F473), which are partially resistant to corn borer attack. The other three inbred checks were susceptible inbreds, EP42, EP47, and EP80 (Butrón et al., 2006). The 64 lines were evaluated in Pontevedra, in 2017 and 2018, following simple 8 × 8 lattice designs in two adjacent trials, one treated with insecticide (INSECT 5G, Chlorpyrifos 5%) (Control) and the other under artificial infestation with Sesamia nonagrioides eggs. The first insecticide treatment was applied between the sixth and seventh week after sowing, and treatment was repeated every 3 weeks until harvest.
Before flowering, in the trial under artificial infestation, pest infestation was ensured by placing ~80–120 MCB eggs between the sheath and the stem in one internode below the main ear on five plants per plot. Eggs were obtained from insect rearing in the MBG-CSIC laboratory following the methodology of Eizaguirre and Albajes (1992). In all inbred trials, the elementary plot consisted of 15 plants sown at a density of 70,000 plants/ha.
Fifty-two hybrids (HVS), obtained by crossing 52 out of the 56 selected inbreds with A638, along with four hybrid checks were also evaluated. Evaluations were made in 2017 and 2018 in Pontevedra under insecticide treatment and infestation with MCB eggs, in trials adjacent to those described for inbreds, and in Barrantes (42°29′N; 8°45′O, 115 m above sea level) and Ponte Caldelas (42°23′N 8°30′O, 291 m above sea level) in 2017 under natural infestation conditions. Natural infestation was high in both locations. The experimental design employed was a simple 8 × 7 lattice, with each elementary plot consisting of 25 plants and a density of 70,000 plants per hectare.
At harvest, grain yield per plant and plot grain yield were determined and expressed in g plant−1 and Mg ha−1 at 14% grain moisture content in IVS and HVS, respectively. In the artificial infestation trials, the five infested plants per plot were collected; the stalks were split lengthwise, and the lengths of the tunnels made by the larvae were measured. A similar procedure was used to measure damage by larvae on naturally infested hybrid trials, although it was done on five random plants per plot.
Statistical analysis
Inbred lines
A combined analysis of variance across years and condition (control and infested) was carried out for grain yield and across years at infestation conditions for tunnel length. The inbred lines were considered as fixed factors while the rest of the main factors and interactions were considered as random factors. Comparison of means was done using Fisher’s protected LSD.
The best linear unbiased estimators (BLUEs) across environments were calculated for each RIL using the Proc Mixed procedure of SAS, with genotype being considered as a fixed factor and year and block as random factors. Heritabilities on a family mean basis were calculated following Holland et al. (2003). To estimate genotypic, residual, and genotype × environment variance, we implemented the VARCOMP procedure in SAS considering all factors as random. The phenotypic variance was calculated as the sum of genotypic, residual, and genotype × environment variance.
Hybrids
A combined analysis across years and conditions was performed for grain yield and tunnel length (excluding protected trial data). Hybrid means were compared using Fisher’s protected LSD and BLUEs across environments were estimated for each RIL. Heritabilities and variance components were computed as explained above. All analyses were carried out with SAS software (SAS/STAT, 2007).
Association mapping
GWAS, based on a mixed linear model that includes a genotype–phenotype matrix, was completed with Tassel 5 (Bradbury et al., 2007) on inbred lines and hybrids, separately. Restricted maximum likelihood estimates of variance components were obtained using the “compressed MLM” and “previously determined population parameters” (P3D) methods described by Zhang et al. (2010). The BLUEs comprised the phenotypic matrices while the genotypic matrix consisted of 224,363 single-nucleotide polymorphisms (SNPs), the same matrix employed by Jiménez-Galindo et al. (2019).
In order to validate the SNPs described as significantly associated with grain yield and tunnel length by Jiménez-Galindo et al. (2019), in the current study, the linkage disequilibrium of significant (p-value < 0.02) marker–phenotype associations with published significant SNPs was studied. In the respective set (inbreds and hybrids), a published significant SNP (from Jiménez-Galindo et al., 2019) was considered validated if (1) the same SNP was associated with the trait with a p-value < 0.02, or (2) within a ±2-Mbp interval, there were SNPs associated with the trait at p-value < 0.02 and in linkage disequilibrium (r2 > 0.2) with the SNPs reported by Jiménez-Galindo et al. (2019).
Results
Means, heritability, and variance components
We observed significant differences among inbred checks and among RILs in IVS and HVS (data not shown). BLUE estimates for inbreds and hybrids are shown in Supplementary Tables 2 and 3, respectively. Regarding heritability estimates, we observed an increase in the values obtained using the set of lines showing extreme values for both traits compared to the values obtained using the whole MAGIC population (Jiménez-Galindo et al., 2019) (Table 1). We studied the decomposition of phenotypic variance in tunnel length and grain yield. Genotype × environment interaction variance is minimal in both traits, in both magnitude and proportion, though significant in IVS. In tunnel length, genetic variance prevails in IVS, less so in HVS and the entire MAGIC population. In grain yield, genetic variance remains consistent in IVS and HVS but decreases in the whole MAGIC population.
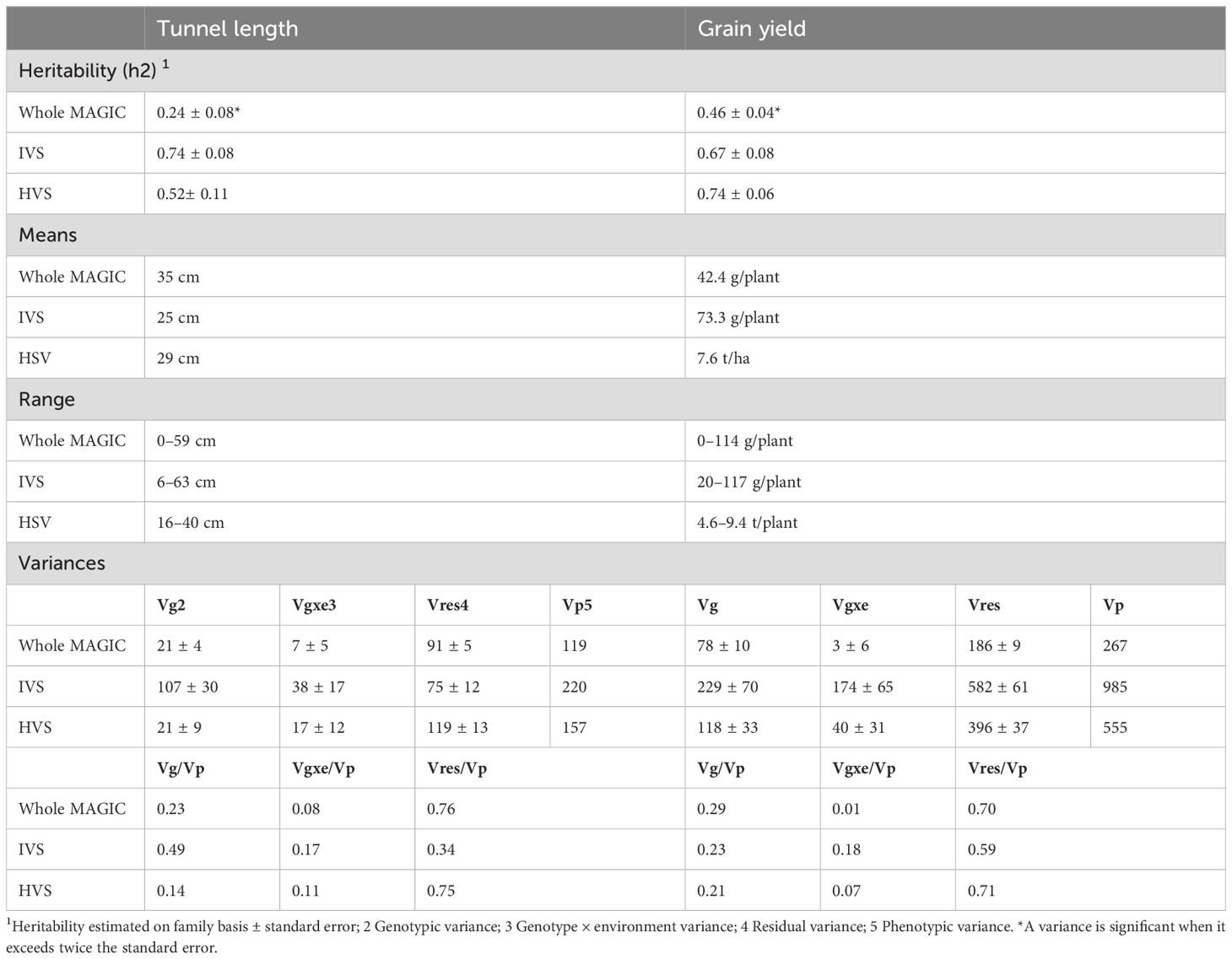
Table 1. Heritability, means, range, and variance components calculated with the whole MAGIC population, the inbred validation set (IVS), and the hybrid validation set (HVS).
Validation of QTL for tunnel length produced by Sesamia nonagrioides larvae
None of the SNPs that showed significant associations with tunnel length in the whole MAGIC (Jiménez-Galindo et al., 2019) showed significant associations (p < 0.02) with tunnel length in the IVS and only one in the HVS. However, within both sets, we have pinpointed SNPs that show a significant association with tunnel length falling within the confidence intervals of SNPs significantly linked to the trait across the entire MAGIC population.
Using the IVS, all of the QTLs detected with the whole MAGIC on chromosomes 5, 6, and 7 were validated. In addition, half of the QTLs on chromosomes 1, 2, and 8 were validated along with one out of three QTLs on chromosome 3. None of the six QTLs on chromosome 4 has been validated in IVS.
Moreover, using HVS, the same SNPs were validated as with the IVS on chromosomes 1, 5, 6, and 7. On chromosome 2, the two QTLs detected in the full set were confirmed; meanwhile, 2/3 and 2/6 were validated on chromosomes 3 and 4, respectively, although three of them were not validated with the IVS (Tables 2, 3). In summary, out of the 21 QTLs associated with tunnel length in the whole MAGIC population, 14 were validated. Nine were validated using both IVS and HVS, QTL_8_2 with IVS only, and QTL_2_2, QTL_3_3, QTL_4_2, and QTL_4_3 with HVS only (Tables 2, 3).
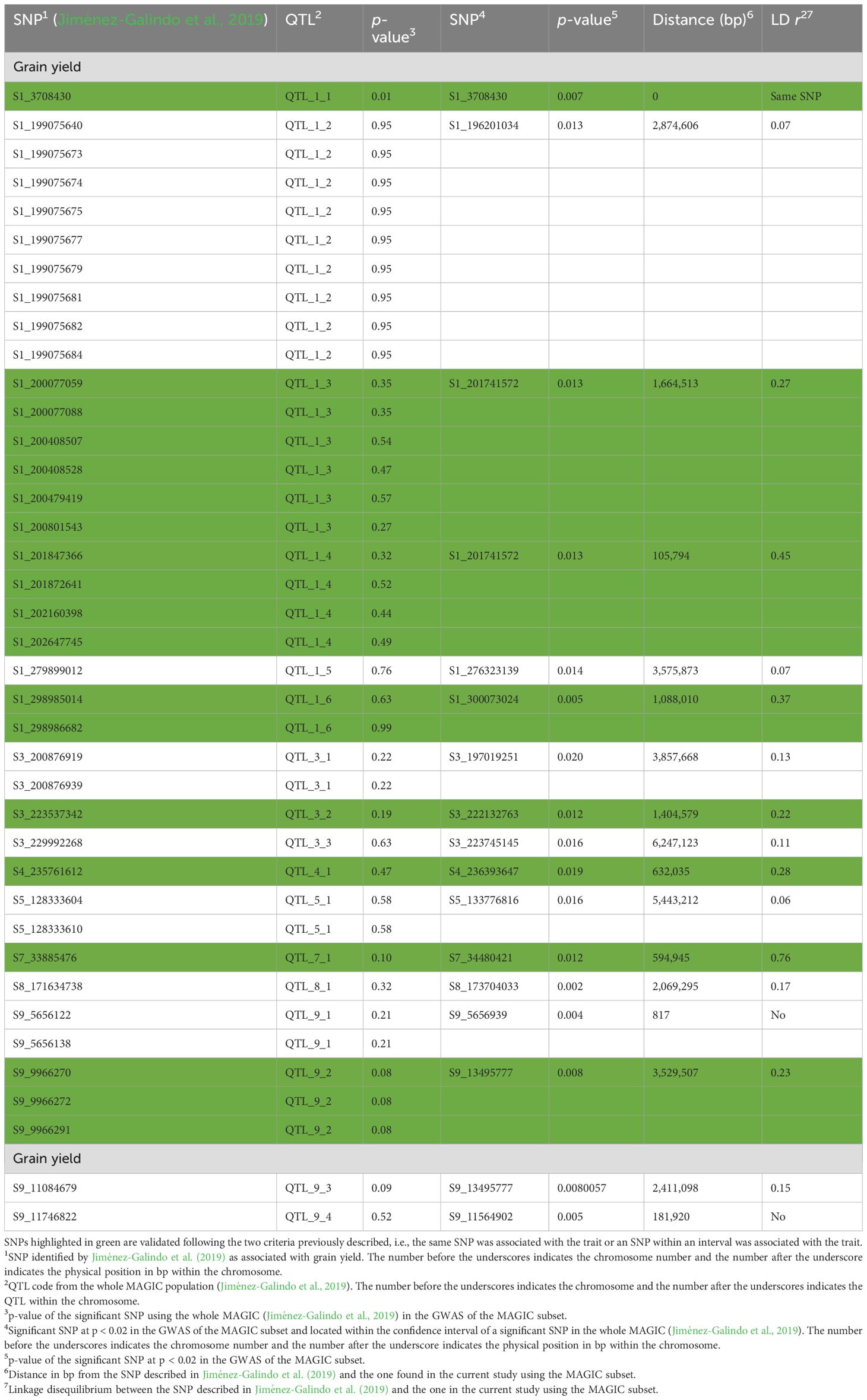
Table 2. Validation of QTL found for tunnel length in the whole MAGIC population (Jiménez-Galindo et al., 2019) using a 56-inbred subset of the MAGIC population (MAGIC subset).
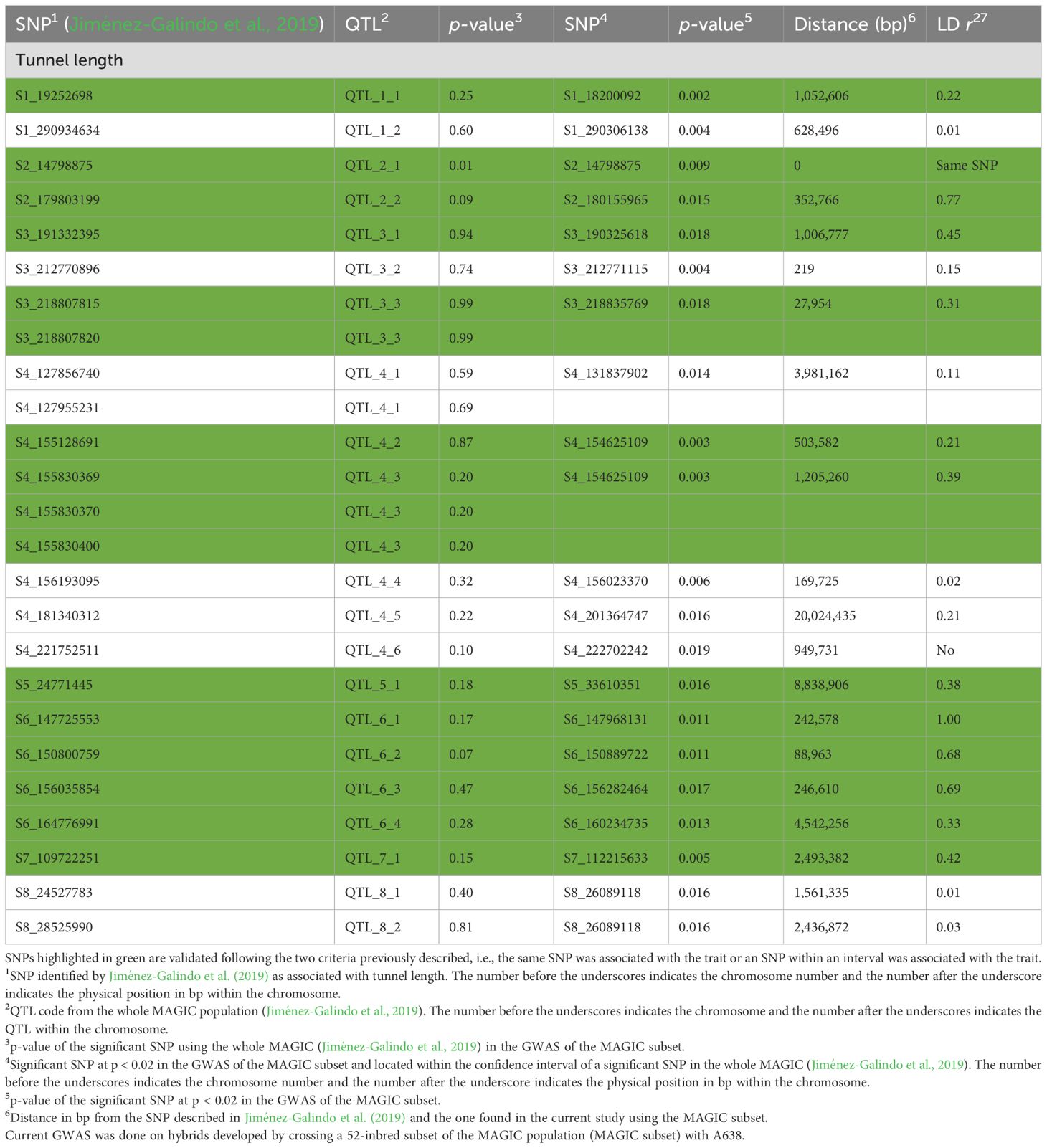
Table 3. Validation of significant QTL found for tunnel length in the whole MAGIC population (Jiménez-Galindo et al. 2019) in a hybrid background.
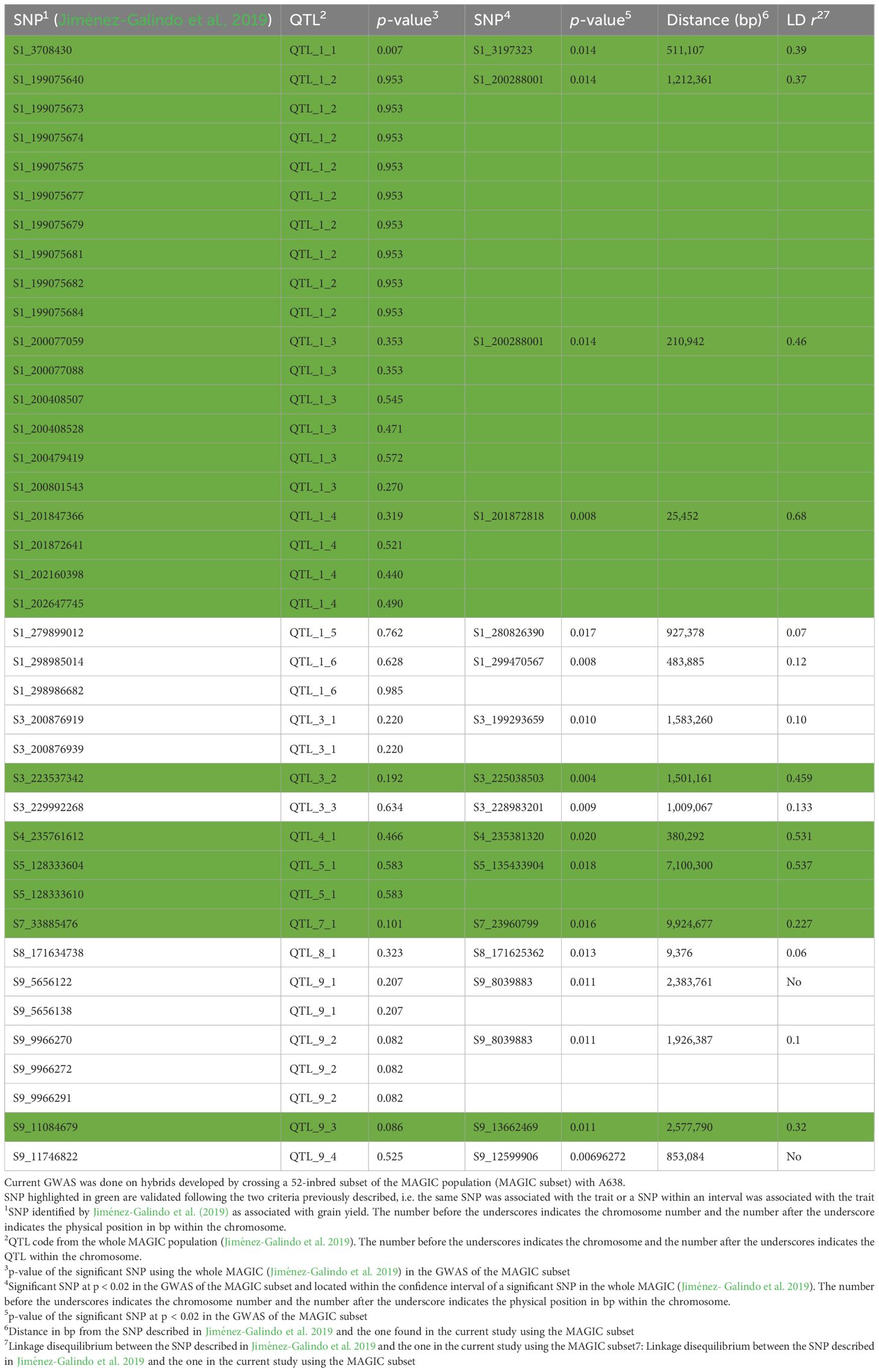
Table 4. Validation of QTL found for grain yield in the whole MAGIC population (Jiménez-Galindo et al. 2019) using a 56-inbred subset of the MAGIC population (MAGIC subset).
Validation of QTL for grain yield
The SNP S1_3708430 that was described as significantly associated with grain yield in the complete set of RILs (Jiménez-Galindo et al., 2019) also showed a significant association (p < 0.02) with yield in the current study using the IVS. In addition, three out of the six QTLs for yield found on chromosome 1 using the complete MAGIC population (Jiménez-Galindo et al., 2019) were validated using both validation sets, IVS and HVS (QTL_1_1; QTL_1_3 and QTL_1_4). QTL_1_6 was only validated in the IVS and QTL_1_2 was only validated using the HVS (Tables 4, 5). Notably, QTL_1_2 and QTL_1_3 exhibited a high degree of linkage disequilibrium (r2 = 0.80) and are situated at a distance of 1,001,419 base pairs (Figure 1). Similarly, the linkage between QTL_1_3 and QTL_1_4 is substantial (r2 = 0.72), with a genomic separation of 1,770,307 base pairs. This observation suggests the potential categorization of these adjacent QTLs as a single QTL encompassing the QTL_1_2, QTL_1_3, and QTL_1_4 reported by Jiménez-Galindo et al. (2019).
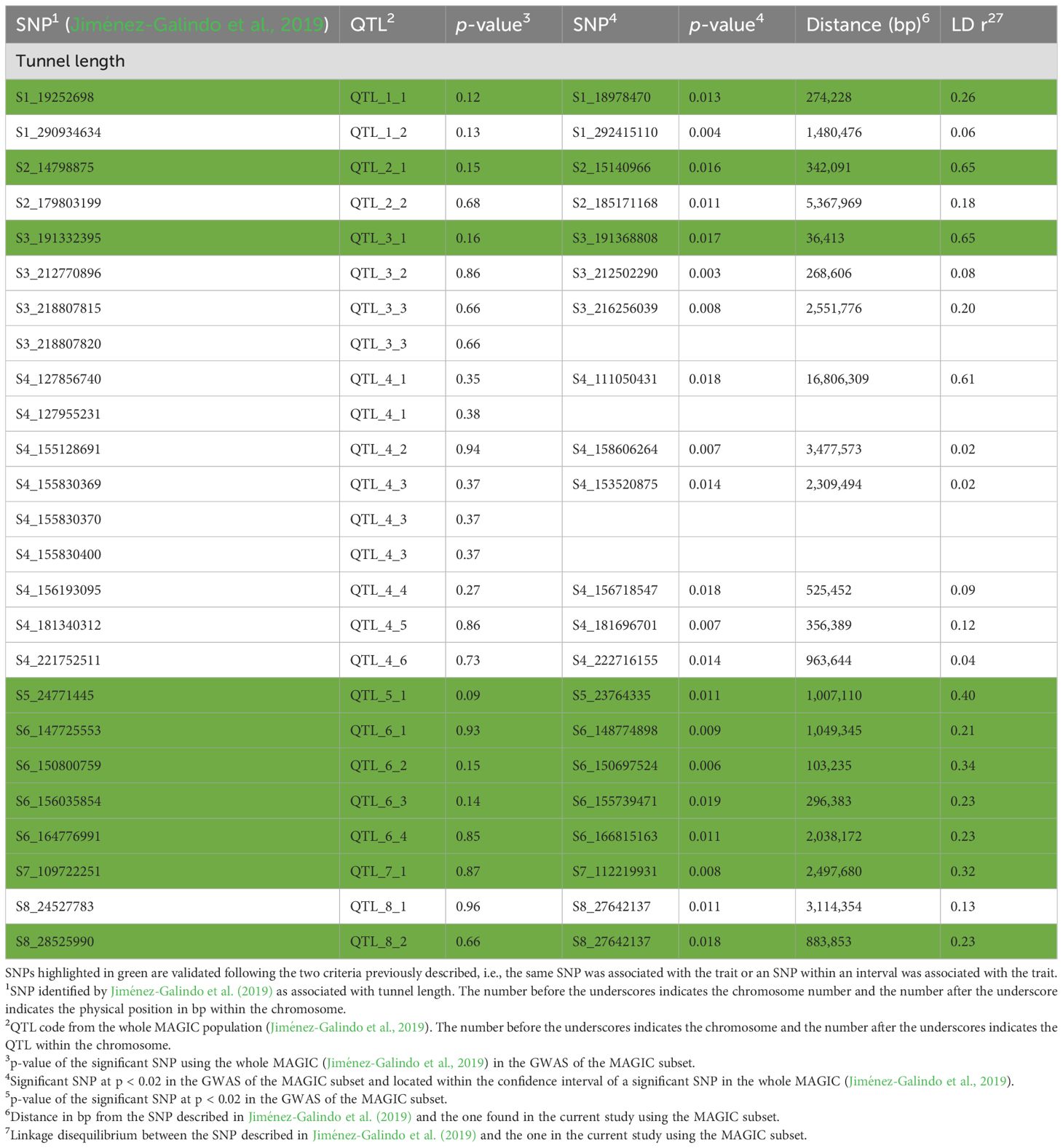
Table 5. Validation of significant QTL found for grain yield in the whole MAGIC population (MAGIC) and published in Jiménez-Galindo et al. (2019) in a hybrid background.
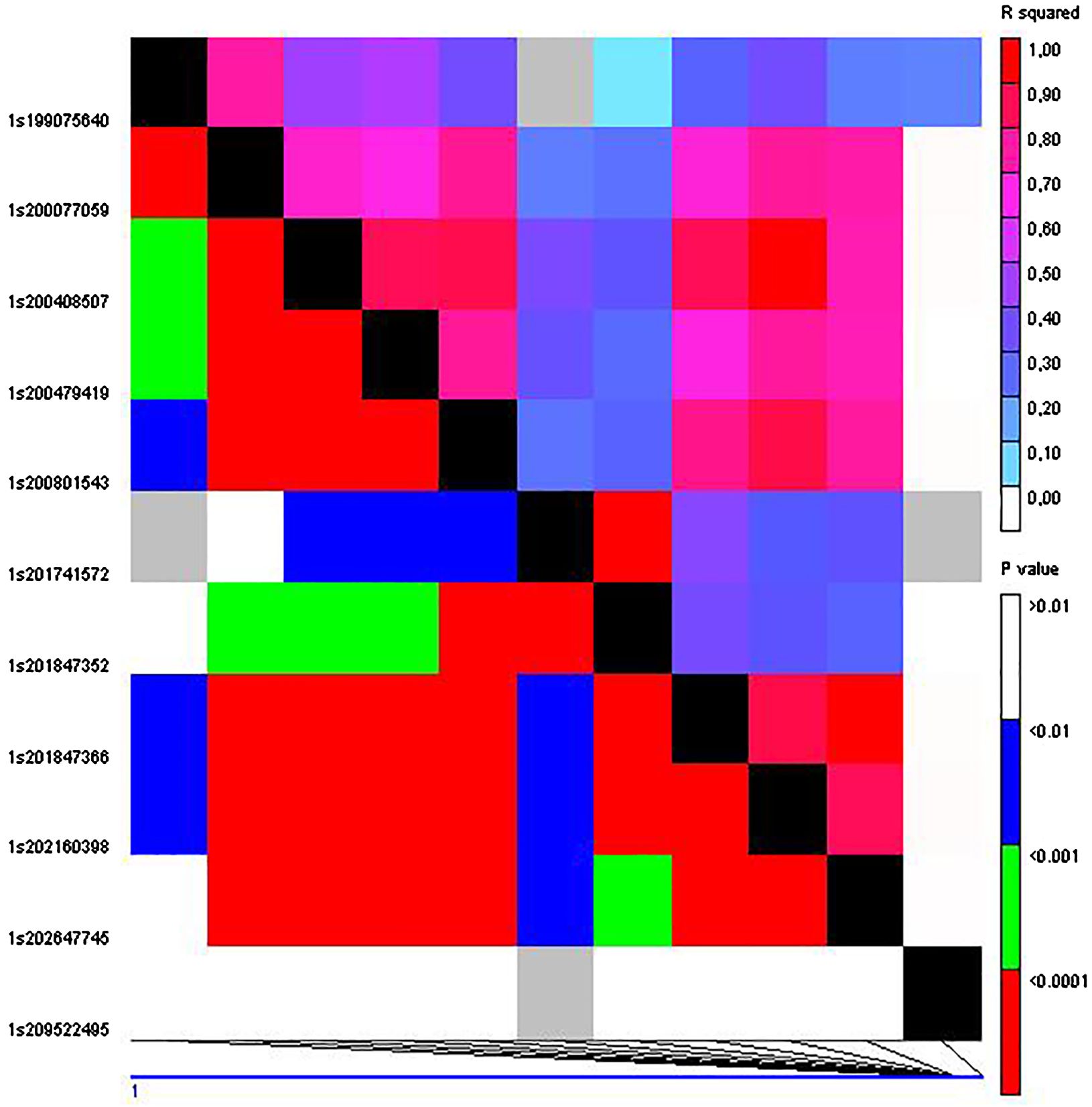
Figure 1. Linkage disequilibrium among the 23 SNPs on chromosome 1 significantly associated with grain yield using the whole MAGIC population (Jiménez-Galindo et al., 2019).
One out of the three QTLs for yield was found on chromosome 3 using the whole MAGIC, and the QTL on chromosome 7 and the QTL on chromosome 4 were validated using both validation sets. The QTL on chromosome 5 and one out of four QTLs on chromosome 9 were validated in HVS. Finally, one out of four QTLs on chromosome 9 was validated in the IVS. To summarize, out of the 39 SNPs, corresponding to 17 QTLs, associated with grain yield in the whole MAGIC population, 11 were validated. Six were validated using IVS and HVS, two with IVS only, and three with HVS only (Tables 4, 5).
Discussion
The experimental design used in Jiménez-Galindo et al., 2019 was an augmented design, originally conceived to evaluate a large number of genotypes with the objective of discarding those that presented poor performances. In this study, we have selected a reduced set of inbred lines (IVS) at the tails of distributions for resistance and yield in the whole MAGIC population in order to maintain trait variability but limiting the number of individuals. This allowed us to improve the phenotyping reliability as the new set could be evaluated using an α-lattice design with two replications instead of an augmented design. The α-lattice experimental design provides enhanced control over experimental variability among replications and blocks, improving phenotyping precision and repeatability and increasing the genotypic/phenotypic variation ratio. As a result of the improved experimental design and more accurate phenotyping, we observed a gain in heritability values in both validations sets, inbred lines and hybrids, compared to heritability estimates in the whole MAGIG population (Jiménez et al., 2019). The increased inheritance estimates for tunnel length and yield in the IVS seemed to be a consequence of higher observed genetic variability; meanwhile, observed genetic variance was not increased in the HVS but the higher number of evaluations reduced the variability of hybrid means. As expected, owing to the reduced importance of additive genetic effects in a heterozygous background, we found that hybrids presented half or less than a half of the genotypic variation of the per se inbreds agreeing with (Michel et al., 2022). The reduction of genetic variance in the HVS compared to IVS was significant only for tunnel length because additive effects are the only significant genetic effects for this trait and, consequently, dominance and/or epistasis effects would not contribute to increase genotypic variability among hybrids as it could happen for yield (Butrón et al., 1999, Cartea et al., 1999).
The QTL validation approach used in the current study greatly differed from approaches used in other works, as those studies usually restringed genetic variation to a particular genomic region (single QTL) using near-isogenic inbred lines (NIL) or heterogeneous inbred families (HIF) (Huo et al., 2016; Jiménez-Galindo et al., 2018; Zhao et al., 2022; Yang et al., 2017). Evaluation of NIL and HIF genotypes is very valuable to advance in the isolation and cloning of the genes involved in the inheritance of a particular trait, but is not useful to validate the genetic weight of a particular QTL in a more real scenario in which variation for the trait is not limited. Conversely, results derived from the current validations could provide information of the usefulness of QTL and genomic predictions, both based on the evaluation of the whole MAGIC population, for implementing marker-assisted or/and genomic selection. In addition, the validation of additive-effect QTL detected by Jiménez-Galindo et al. (2019) has been done in homozygous and heterozygous backgrounds, which would allow us to assess the value of these QTLs for improved hybrid development. For validation in a heterozygous background, IVS inbreds were crossed to the Reid inbred line A638. It has been previously shown that using testers from complementary heterotic groups to the heterotic group or groups of the evaluated inbreds is superior for detecting/validating additive-effect QTLs than testers more genetically related to the inbreds (Frascaroli et al., 2009).
In the present study, almost three-fourths of QTLs found as associated to tunnel length by Jiménez-Galindo et al. (2019) were validated. However, the p-values of current associations were high, excluding the suitability of these QTLs for performing MAS but emphasizing that genomic predictions based on genotype–phenotype associations in the whole MAGIC population could be valuable for genomic selection. QTLs in the region 6.05–6.07 were validated in both sets, and this genomic region has also been associated with resistance to stalk tunneling in different mapping populations and environments. In addition, this region encompasses several QTLs for cell wall digestibility (Ralph et al., 2004) and, consequently, deserves to be deeply studied to identify the genes behind those associations. Ten out of 16 (approximately ⅗) QTLs for grain yield found in the whole MAGIC population were validated. The QTL validation ratio was inferior for yield than for tunnel length as expected because dominance and/or epistatic effects are equally or more important than additive effects for yield and could mask additive effects (Butrón et al., 2009). Interestingly, the additive-effect QTL on the long arm of chromosome 8 detected by Jiménez-Galindo et al. (2019) could not be validated in any validation set and, in the same chromosome arm, several authors have found significant QTLs for mid-parent heterosis for yield (Schön et al., 2010.; Tang et al., 2010; Larièpe et al., 2012; Samayoa et al., 2017). Again, p-values of associations found in the current study between markers linked to QTLs detected by Jiménez-Galindo et al. (2019) and grain yield were high, suggesting that genomic prediction based on the evaluation of the whole MAGIC population could be suitable to improve yield. Gesteiro et al. (2023) have already shown that genotypic selection for yield surpassed phenotypic selection, although genotypic and phenotypic selection for reducing tunnel lengths could be preferred to improve yield and resistance simultaneously.
Nine out of 21 and 6 out of 17 QTLs identified for tunnel length and grain yield, respectively, by Jiménez-Galindo et al. (2019) were validated across both validation sets, suggesting a moderate genetic correlation between per se and testcross performance of inbred lines. However, Zhang et al. (2011), although having estimated moderate to high genetic correlations between per se and testcross evaluations for plant and ear heights, showed that few QTLs were detected in both genetic backgrounds. In any case, the validation of a moderate proportion of QTL for tunnel length in both validation sets suggested that genomic predictions for tunnel length based on inbred line evaluations could be useful to develop improved hybrids (Ma et al., 2022). Michel et al. (2022) already estimated promising genomic value predictive abilities between r = 0.49 and r = 0.55 for models predicting maize hybrid traits that were trained with per se data.
Conclusions
In this study, we have used a novel validation QTL approach together with improved experimental designs that led to higher heritability estimates for both traits compared to the whole MAGIC population. A substantial proportion of QTLs associated with tunnel length and grain yield were validated across both sets. The current validations offer insights for guiding the implementation of marker-assisted or genomic selection strategies. Moreover, the study indicates the potential value of utilizing genomic predictions based on inbred line assessments to enhance the development of superior hybrid varieties.
Phenotyping genetically-wide panels of inbred lines or RILs is challenging due to the difficulty of controlling the high experimental error associated with large trials. Therefore, the high experimental errors associated with these evaluations could greatly interfere with QTL identification making necessary to validate those QTL before implementing any maker-assisted or genomic selection program. In such scenario, a novel validation approach has been used that allows to ascertain the reliability of QTL as well as its practical value to contribute to hybrid improvement.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
AL-M: Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. ZR-R: Data curation, Investigation, Methodology, Writing – review & editing. AB: Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing. JJ-G: Data curation, Investigation, Methodology, Writing – review & editing. PR: Investigation, Resources, Supervision, Writing – review & editing. RM: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by subsequent projects financed by MCIU/AEI/FEDER, UE (RTI2018-096776-B-C21 and PID2021-122196OB-C21).
Acknowledgments
We thank the technical staff for their support in the experimental trials. AL-M thanks her postdoctoral contract granted by Xunta de Galicia. JJ-G thanks a PhD scholarship #314685 from the National Council for Science and Technology (CONACYT), Mexico. ZR-R thanks a Scholarship from the National Council of Science and Technology (CONACYT) to carry out Postdoctoral studies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1404881/full#supplementary-material
References
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/BIOINFORMATICS/BTM308
Butrón, A., Malvar, R. A., Velasco, P., Vales, M. I., Ordás, A. (1999). Combining abilities for maize stem antibiosis, yield loss, and yield under infestation and non infestation with pink stem borer. Crop Sci. 39, 691–696. doi: 10.2135/cropsci1999.0011183X003900020015x
Butrón, A., Romay, M. C., Peña-Asín, J., Alvarez, A., Malvar, R. A. (2012). Genetic relationship between maize resistance to corn borer attack and yield. Crop Sci. 52, 1176–1180. doi: 10.2135/cropsci2011.11.0584
Butrón, A., Samayoa, L. F., Santiago, R., Ordás, B., Malvar, R. A. (2018). “Genomics of insect resistance,” in The Maize Genome. Eds. T. R. Bennetzen, J., Flint-Garcia, S., Hirsch, C. (Springer Nature, Switzerland), 163–183.
Butrón, A., Sandoya, G., Revilla, P., Malvar, R. A. (2009). Genetics of resistance to the pink stem borer (Sesamia nonagrioides) in maize (Zea mays). Ann. Appl. Biol. 154, 205–217. doi: 10.1111/j.1744-7348.2008.00284.x
Butrón, A., Sandoya, G., Santiago, R., Ordás, A., Rial, A., Malvar, R. A. (2006). Searching for new sources of pink stem borer resistance in maize (Zea mays L.). Genetic Resources Crop Evol. 53, 1455–1462.
Cartea, M. E., Malvar, R. A., Butrón, A., Vales, M. I., Ordás, A. (1999). Inheritance of antibiosis to Sesamia nonagrioides (Lepidoptera: Noctuidae) in maize. J. Economic Entomol. 92 (4), 994–998.
Eizaguirre, M., Albajes, R. (1992). Diapause induction in the stem corn borer, Sesamia nonagrioides (Lepidoptera: Noctuidae). Entomologia Generalis 17 (4), 277–283. doi: 10.1127/entom.gen/17/1992/277
Farinós, G. P., de la Poza, M., Hernández-Crespo, P., Ortego, F., Castañera, P. (2004). Resistance monitoring of field populations of the corn borers Sesamia nonagrioides and Ostrinia nubilalis after 5 years of Bt maize cultivation in Spain. Entomologia Experimentalis et Applicata, 110 (1), 23–30. doi: 10.1111/j.0013-8703.2004.00116.x
Flint-Garcia, S. A., Jampatong, C., Darrah, L. L., McMullen, M. D. (2003). Quantitative trait locus analysis of stalk strength in four maize populations. Crop Sci. 43, 13–22. doi: 10.2135/cropsci2003.0013
Frascaroli, E., Canè, M. A., Pè, M. E., Pea, G., Morgante, M., Landi, P. (2009). QTL detection in maize testcross progenies as affected by related and unrelated testers. Theor. Appl. Genet. 118, 993–1004. doi: 10.1007/s00122-008-0956-3
Gesteiro, N., Ordás, B., Butrón, A., de la Fuente, M., Jiménez-Galindo, J. C., Samayoa, L. F., et al. (2023). Genomic versus phenotypic selection to improve corn borer resistance and grain yield in maize. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1162440
Holland, J. B., Nyquist, W. E., Cervantes-Martínez, C. T. (2003). Estimated and interpreting heritability for plant breeding: An update. Plant Breed Rev. 22, 9–122. doi: 10.1002/9780470650202
Huo, D., Ning, Q., Shen, X., Liu, L., Zhang, Z. (2016). QTL mapping of kernel number-related traits and validation of one major QTL for ear length in maize. PloS One 11 (5), e0155506. doi: 10.1371/journal.pone.0155506
Jiménez-Galindo, J. C., Malvar, R. A., Butrón, A., Caicedo, M., Ordás, B. (2018). Fine analysis of a genomic region involved in resistance to Mediterranean corn borer. BMC Plant Biol. 18, 1–13. doi: 10.1186/s12870-018-1385-3
Jiménez-Galindo, J. C., Malvar, R. A., Butrón, A., Santiago, R., Samayoa, L. F., Caicedo, M., et al. (2019). Mapping of resistance to corn borers in a MAGIC population of maize. BMC Plant Biol. 19, 1–17. doi: 10.1186/s12870-019-2052-z
Jiménez-Galindo, J. C., Ordás, B., Butrón, A., Samayoa, L. F., Malvar, R. A. (2017). QTL mapping for yield and resistance against Mediterranean corn borer in maize. Front. Plant Sci. 8, 698.
Larièpe, A., Mangin, B., Jasson, S., Combes, V., Dumas, F., Jamin, P., et al. (2012). The genetic basis of heterosis: multiparental quantitative trait loci mapping reveals contrasted levels of apparent overdominance among traits of agronomical interest in maize (Zea mays L.). Genetics. 190 (2), 795–811. doi: 10.1534/genetics.111.133447
López, C., Sans, A., Asin, L., EizaGuirre, M. (2001). Phenological model for sesamia nonagrioides (Lepidoptera: noctuidae). Environ. Entomol 30, 23–30. doi: 10.1603/0046-225X-30.1.23
Ma, Y., Li, D., Xu, Z., Gu, R., Wang, P., Fu, J., et al. (2022). Dissection of the genetic basis of yield traits in line per se and testcross populations and identification of candidate genes for hybrid performance in maize. Int. J. Mol. Sci. 23 (9), 5074. doi: 10.3390/ijms23095074
Meissle, M., Mouron, P., Musa, T., Bigler, F., Pons, X., Vasileiadis, V. P., et al. (2010). Pests, pesticide use and alternative options in European maize production: Current status and future prospects. J. Appl. Entomology 134, 357–375. doi: 10.1111/j.1439-0418.2009.01491.x
Michel, K. J., Lima, D. C., Hundley, H., Singan, V., Yoshinaga, Y., Daum, C., et al. (2022). Genetic mapping and prediction of flowering time and plant height in a maize Stiff Stalk MAGIC population. Genetics. 221 (2), iyac063. doi: 10.1093/genetics/iyac063
Ordas, B., Butron, A., Alvarez, A., Revilla, P., Malvar, R. A. (2012). Comparison of two methods of reciprocal recurrent selection in maize (Zea mays L.). Theor. Appl. Genet. 124, 1183–1191. doi: 10.1007/S00122-011-1778-2/TABLES/5
Ordas, B., Malvar, R. A., Santiago, R., Sandoya, G., Romay, M. C., Butron, A. (2009). Mapping of QTL for resistance to the Mediterranean corn borer attack using the intermated B73 × Mo17 (IBM) population of maize. Theor. Appl. Genet. 119, 1451–1459. doi: 10.1007/s00122-009-1147-6
Ralph, J., Guillaumie, S., Grabber, J. H., Lapierre, C., Barrière, Y. (2004). Genetic and molecular basis of grass cell-wall biosynthesis and degradability. III. Towards a forage grass ideotype. C R Biol. 327, 467–479. doi: 10.1016/j.crvi.2004.03.004
Salvi, S., Tuberosa, R. (2015). The crop QTLome comes of age. Curr. Opin. Biotechnol. 32, 179–185. doi: 10.1016/j.copbio.2015.01.001
Samayoa, L. F., Butron, A., Malvar, R. A. (2014). QTL mapping for maize resistance and yield under infestation with Sesamia nonagrioides. Mol. Breed. 34, 1331–1344. doi: 10.1007/s11032-014-0119-y
Samayoa, L. F., Butrón, A., Malvar, R. A. (2019). Usefulness of marker-assisted selection to improve maize for increased resistance to Sesamia nonagrioides attack with no detrimental effect on yield. Annals Applied Biol. 174 (2), 219–222. doi: 10.1111/aab.12480
Samayoa, L. F., Malvar, R. A., Butrón, A. (2017). QTL for maize midparent heterosis in the heterotic pattern American dent × European flint under corn borerpressure. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00573
Samayoa, L. F., Malvar, R. A., McMullen, M. D., Butrón, A. (2015a). Identification of QTL for resistance to Mediterranean corn borer in a maize tropical line to improve temperate germplasm. BMC Plant Biol. 15. doi: 10.1186/s12870-015-0652-9
Samayoa, L. F., Malvar, R. A., Olukolu, B. A., Holland, J. B., Butrón, A. (2015b). Genome-wide association study reveals a set of genes associated with resistance to the Mediterranean corn borer (Sesamia nonagrioides L.) in a maize diversity panel. BMC Plant Biol. 15, 1–15. doi: 10.1186/s12870-014-0403-3
Sandoya, G., Butrón, A., Alvarez, A., Ordás, A., Malvar, R. A. (2008). Direct response of a maize synthetic to recurrent selection for resistance to stem borers. Crop Sci. 48, 113–118. doi: 10.2135/CROPSCI2007.02.0084
Schön, C. C., Baldev, D. S., Friedrich Utz, H., Melchinger, A. E. (2010). High congruency of QTL positions for heterosis of grain yield in three crosses of maize. doi: 10.1007/s00122-009-1209-9
Tang, J., Yan, J., Ma, X., Teng, W., Wu, W., Dai, J., et al. (2010). Dissection of the genetic basis of heterosis in an elite maize hybrid by QTL mapping in an immortalized F 2 population. doi: 10.1007/s00122-009-1213-0
Yang, Z., Li, X., Zhang, N., Wang, X., Zhang, Y., Ding, Y., et al. (2017). Mapping and validation of the quantitative trait loci for leaf stay-green-associated parameters in maize. Plant Breed. 136, 188–196. doi: 10.1111/pbr.12451
Zhang, Y., Li, Y., Wang, Y., Peng, B., Liu, C., Liu, Z., et al. (2011). Correlations and QTL detection in maize family per se and testcross progenies for plant height and ear height. Plant Breed. 130, 617–624. doi: 10.1111/J.1439-0523.2011.01878.X
Zhang, Z., Ersoz, E., Lai, C. Q., Todhunter, R. J., Tiwari, H. K., Gore, M. A., et al. (2010). Mixed linear model approach adapted for genome-wide association studies. Nature Genetics. 42 (4), 355–360.
Zhao, Y., Ma, X., Zhou, M., Wang, J., Wang, G., Su, C. (2022). validating a major quantitative trait locus and predicting candidate genes associated with kernel width through qtl mapping and RNA-sequencing technology using near-isogenic lines in maize. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.935654
Keywords: Mediterranean corn borer, QTL validation, maize breeding, MAGIC population, grain yield, recombinant inbred lines
Citation: López-Malvar A, Reséndiz-Ramirez Z, Butrón A, Jiménez-Galindo JC, Revilla P and Malvar RA (2024) Validation of QTLs associated with corn borer resistance and grain yield: implications in maize breeding. Front. Plant Sci. 15:1404881. doi: 10.3389/fpls.2024.1404881
Received: 21 March 2024; Accepted: 16 September 2024;
Published: 22 October 2024.
Edited by:
Gustavo A. Slafer, Catalan Institution for Research and Advanced Studies (ICREA), SpainReviewed by:
Roberto Tuberosa, University of Bologna, ItalyRenan Santos Uhdre, Washington State University, United States
Copyright © 2024 López-Malvar, Reséndiz-Ramirez, Butrón, Jiménez-Galindo, Revilla and Malvar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa Ana Malvar, cm1hbHZhckBtYmcuY3NpYy5lcw==