 Jie Wang1,2†
Jie Wang1,2† Junhu Kan2†Jie Wang2,3†
Junhu Kan2†Jie Wang2,3† Xinlin Yan2,4Yi Li2Thida Soe2
Xinlin Yan2,4Yi Li2Thida Soe2 Luke R. Tembrock5Guoming Xing1
Luke R. Tembrock5Guoming Xing1 Sen Li1*
Sen Li1* Zhiqiang Wu1,2*Minlong Jia1,2*
Zhiqiang Wu1,2*Minlong Jia1,2*- 1College of Horticulture, Shanxi Agricultural University, Jinzhong, China
- 2Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, China
- 3College of Science, Health, Engineering and Education, Murdoch University, Perth, WA, Australia
- 4College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, China
- 5Department of Agricultural Biology, Colorado State University, Fort Collins, CO, United States
Backgrounds: Prunus mume in the Rosaceae and commonly referred to as mei or Chinese plum is widely used as a traditional ornamental flowering plant and fruit tree in China. Although some population and genetic analyses have been conducted for this species, no extensive comparisons of genetic variation from plastomes have yet been investigated.
Methods: We de novo assembled a total of 322 complete P. mume plastomes in this study and did a series of comparative analyses to better resolve pan-plastomic patterns of P. mume. To determine the phylogeny and domestication history of this species, we reconstructed the phylogenetic tree of Prunus genus, and resolved the population structure of P. mume. We also examined the nucleotide variation of P. mume to find potential DNA barcodes.
Results: The assembled plastomes exhibited a typical quadripartite structure and ranged from 157,871 bp to 158,213 bp in total size with a GC content ranging from 36.73 to 36.75%. A total of 112 unique genes were identified. Single nucleotide variants (SNVs) were the most common variants found among the plastomes, followed by nucleotide insertions/deletions (InDels), and block substitutions with the intergenic spacer (IGS) regions containing the greatest number of variants. From the pan-plastome data six well-supported genetic clusters were resolved using multiple different population structure analyses. The different cultivars were unevenly distributed among multiple clades. We also reconstructed a phylogeny for multiple species of Prunus to better understand genus level diversity and history from which a complex introgressive relationship between mei and other apricots/plums was resolved.
Conclusion: This study constructed the pan-plastome of P. mume, which indicated the domestication of P. mume involved multiple genetic origins and possible matrilineal introgression from other species. The phylogenetic analysis in Prunus and the population structure of P. mume provide an important maternal history for Prunus and the groundwork for future studies on intergenomic sequence transfers, cytonuclear incompatibility, and conservation genetics.
Introduction
The Rosaceae family is extensively distributed in temperate regions and comprises ca. 120 genera and 3,300 species several of which are of major commercial importance (Zhang et al., 2017). Prunus is a large genus in the Rosaceae containing approximately 250–400 species of trees and shrubs that are found mainly in the northern temperate zone, subtropical zones, and tropical regions (Rehder, 1956), with Eastern Asia being an important center of diversity (Chin et al., 2014). Several species of Prunus are highly prized for their economic qualities as food crops, such as peach, plum, almond, and sweet cherry (Bortiri et al., 2001). In addition to their value as fruits these species also hold a significant place in gardens all over the world thanks to their beautiful vibrant fruits, leaves, and flowers (Zheng et al., 2022). These species are also considered an important source of timber and medicine (Andro & Riffaud, 1995; Lee and Wen, 2001; Wen et al., 2008). Prunus species possess various inflorescence types including racemes, corymbs, and solitary flowers, allowing them to be an ideal system to study the inflorescence evolution (Su et al., 2023). However, the taxonomy of Prunus species, particularly proper species delimitation remains unclear (Rehder, 1956; Lee and Wen, 2001; Shi et al., 2013). It has been widely accepted that Prunus comprises five subgenera, including Amygdalus, Cerasus, Lauro-cerasus, Padus, and Prunus (Rehder, 1956; Wen et al., 2008; Chin et al., 2010; Zhao et al., 2016). However, ongoing hybridization between Prunus species during evolutionary history has made it extremely difficult to resolve phylogenetic relationships (Chin et al., 2010, 2014). By employing various genomic regions, including plastid markers, nuclear ribosomal ITS, and other nuclear loci, researchers over the last 20 years have attempted to reconstruct the evolutionary relationships of Prunus (Bortiri et al., 2001, 2002, 2006; Wen et al., 2008; Chin et al., 2010; Shi et al., 2013; Chin et al., 2014; Zhao et al., 2016; Hodel et al., 2021). In recent years, several nuclear genomes from Prunus have been released, including P. mume (Zhang et al., 2018), P. avium (Wang et al., 2020), P. dulcis (Alioto et al., 2020), P. persica (Verde et al., 2013), P. yedoensis (Baek et al., 2018), P. domestica (Callahan et al., 2021), P. armeniaca, and P. salicina (Jiang et al., 2019; Liu et al., 2020), which laid a foundation to analyze numerous important characters and the phylogeny of Prunus.
Prunus mume commonly referred to as mei and Chinese plum has been widely used as a traditional ornamental flowering plant and fruit tree in China for thousands of years (Chen, 2010). The earliest cultivation of P. mume originated from the Yangtze River Basin, southern China around 4000–5000 years ago and spread throughout East Asia (Zhang, 1987). Prunus mume is a woody decorative plant of early spring that has long been regarded as a symbol of Chinese culture (Zhang et al., 2012), which is noted for colorful corollas, diverse flower morphology, and an ability to tolerate low temperatures (Chu, 1999a). Prunus mume has been classified into eleven cultivar groups based on morphological differences and include Pendulous, Single Flowered, Versicolor, Pink Double, Flavescens, Tortuosa, Green Calyx, Alboplena, Cinnabar Purple, Apricot Mei and Meiren (Chen, 2010). The history of these cultivars has involved complex gene introgression and multiple hybridization events among different cultivars of P. mume (Zhang et al., 2018).
With the continuous update and development of sequencing technology, T2T genomes of many species have also been published, which will promote the continuous in-depth research of genomes (Lan et al., 2023, 2024). Plastome data is regularly used in phylogenomic and population genomic studies now due to the reliability of assembling these genomes and the large comparative databases for analyses (Wang et al., 2023). Most plastomes have a conserved circular structure, comprising two inverted repeats (IRs) separated by a large single copy (LSC) and a small single copy (SSC) region (Jansen & Ruhlman, 2012). They usually comprise 120–130 genes that are linked to photosynthesis, transcription, and translation (Wang et al., 2024). Most plastomes are parthenogenetic, haploid, and non-recombinant, making them applicable in complicated lineages with incomplete lineage sorting and frequent introgression (Biersma et al., 2020; Jiang et al., 2023). Moreover, due to plastomes moderate mutation rate (higher than mitochondrial genes but lower than nuclear genes), they are widely utilized in comparative genomics research and evolutionary studies (Wu & Ge, 2012; Dong et al., 2020; Lin et al., 2023). Compared with traditional single plastome studies, multiple plastome datasets across a species have substantially advanced studies of agronomic plant species. Some recent examples include a study of 321 complete plastomes used to distinguish pepper (Capsicum) cultivars (Magdy et al., 2019), 343 complete plastomes within Petato section of Solanum which provided new insights into potato diversity, phylogeny, and species differentiation (Yan et al., 2022), as well as studies of pan-plastomes constructed for species such as Hemerocallis citrina (Jia et al., 2024). Nelumbo nucifera (Wang et al., 2022), Fagopyrum tataricum (Zhou et al., 2023), Oryza (He et al., 2021), and Beta vulgaris (Sielemann et al., 2022). However, no such investigation has been conducted for P. mume.
Here, we de novo assembled a large plastome dataset consisting of 322 complete P. mume plastomes to: (a) construct a reliable pan-plastome for P. mume; (b) identify the hypervariable regions of plastomes for developing potential molecular markers; (c) resolved the phylogenetic relationships and population structure of P. mume to comprehensively survey the diversity in this species; and (d) reconstructed phylogenetic relationships of Prunus to infer the evolutionary history of Prunus species especially as relates to introgression with P. mume.
Materials and methods
Plant materials, plastome assembly, and genome annotation
We de novo assembled a total of 322 complete P. mume plastomes from the whole-genome resequencing data, which were downloaded from the ENA database (https://www.ebi.ac.uk/ena/browser/; Study accession No. PRJNA352648)(Supplementary Table S1) (Zhang et al., 2018). FastQC v0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was utilized for data quality control and Trimmomatic (Bolger et al., 2014) was employed to acquire clean reads. The obtained clean raw WGS reads were aligned to a published P.mume plastome (GenBank accession: MN101214.1) to filter plastid-origin reads with bwa v0.7.17 (Li & Durbin, 2010) and SAMtools v1.9 (Li et al., 2009). Then, SPAdes v3.15.2 (Bankevich et al., 2012) and Bandage v0.8.1 (Wick et al., 2015) were used for de novo assembly of whole plastomes from resulting plastid-origin reads. MAFFT v.7 (Katoh & Standley, 2013) was used to confirm that the assembled plastomes were oriented correctly. The LSC and SSC orientations were manually adjusted to maintain collinearity in the resulting plastome sequences using MEGA7 (Kumar et al., 2016), The total length and GC content of different plastome structures, including LSC, SSC, and IR regions, were calculated using a customized Perl script to provide assembly metrics. Genome annotation of the plastomes was performed using the online program GeSeq (Tillich et al., 2017), and manually reviewed and modified as needed. Finally, the program Chloroplot (Zheng et al., 2020) was used to visualize the pan-plastome map of P.mume.
Haplotype and genetic diversity analyses
Using the corrected alignment of all plastome sequences, the haplotypes of plastomes were ascertained in DnaSP 6 (Rozas et al., 2017), with the option to eliminate gaps and missing loci. Using TCS (Clement et al., 2000) and the median-joining approach, haplotype networks were inferred and displayed in Popart v1.7 (Leigh et al., 2015) to examine the ancestry of the identified haplotypes. The output figure was further modified to be more readable in Adobe Illustrator software (Adobe Systems Incorporated, USA). The programs DnaSP 6 (Rozas et al., 2017) and MEGA7 (Kumar et al., 2016) were used to calculate the haplotype diversity (Hd) and nucleotide diversity (Π) for each genetic cluster of haplotypes, and the evolutionary distances based on the Tamura-Nei distance model was used to calculate population differentiation index (Fst), between different genetic clusters by using the plastomic SNVs. The principal coordinates analysis (PCA) was conducted in TASSEL 5.0 (Bradbury et al., 2007) and the PCA diagrams were drawn in the package ggplot2 in R v 4.2.1 (Ginestet, 2011).
Phylogenetic and population structure analyses
To resolve the phylogenetic status of P. mume, we combined 322 newly assembled P. mume plastomes and 151 additional Prunus plastomes from several species which were downloaded from NCBI database (https://www.ncbi.nlm.nih.gov/) (Supplementary Table S2). To perform the phylogenetic analysis, complete sequences of plastid genomes were aligned in MAFFT 7 (Katoh and Standley, 2013) and manually adjusted in MEGA7 (Kumar et al., 2016). Subsequently, maximum likelihood (ML) trees were constructed by IQtree v.2.1 (Nguyen et al., 2015). The visualization of phylogenetic trees was performed by FigTree v.1.4.2 (http://beast.bio.ed.ac.uk/FigTree). SNV-sites were used to derive a SNV dataset from the entire-plastome alignment (Page et al., 2016). Based on the SNV data matrix and with K values ranging from 1 to 12, population structure analysis was carried out using Structure v.2.3.4, with 10 replicates for each K value (Hubisz et al., 2009). CLUMPP (Jakobsson & Rosenberg, 2007) was utilized to calculate the best K value, and the ggplot2 package of R v 4.2.1 (Ginestet, 2011) was used to display the results.
Nucleotide variant analysis across the Prunus mume pan-plastome
Using the complete plastome alignments, nucleotide variants were identified for P. mume plastomes, using the full plastome alignments. Single nucleotide variants (SNVs), block substitutions (two or more consecutive nucleotide variants), nucleotide insertions or deletions (InDels), and mixed sites (which comprise two or more of the preceding three variants at a given site) are the four categories into which nucleotide variants were separated. Graphs produced by variant analyses were created using R v4.2.1’s ggplot2 package.
Results
Phylogenetic relationships of Prunus
To reveal the evolutionary relationship among Prunus, an ML tree was constructed based on our 322 newly assembled P.mume plastomes combined with complete plastomes from an additional 151 Prunus species. Neillia gracilis and Prinsepia uniflora were used as outgroups. As shown in Figure 1, Prunus was divided into six main clades. According to the previous classification of Prunus (Rehder, 1956; Wen et al., 2008; Chin et al., 2010), there were five subgenera in this genus, referred to as Laurocerasus, Padus, Cerasus, Amygdalus and Prunus (Zhao et al., 2016) (Figure 1). Since our phylogenetic tree indicated that Laurocerasus and Padus are not monophyletic, we further grouped them together into a clade, called the Laurocerasus & Padus clade. Therefore, six major clades were resolved, corresponding to Laurocerasus & Padus clade, Cerasus clade, Amygdalus clade A, Amygdalus clade B, Prunus clade A and Prunus clade B. Laurocerasus & Padus clade including species from Laurocerasus, Padus and Maddenia. The Cerasus clade included Lobopetalum, Pseudocerasus, Eucerasus and Phyllomahaleb. The Amygdalus clade included Amygdalus the flowering plums and peaches. The Prunus clade including Armeniaca, Salicina, Microcerasus and P. mume. Due to the large sampling of P. mume accessions contained in this ML tree, we found that some P. armeniaca and P. mume clustered together. Moreover, we also observed that a subset of Lobopetalum did not cluster with Cerasus, but with Amygdalus. To better understand the evolutionary relationship in Prunus, apart from the molecular data, we classified it based on the type of inflorescence, including racemes, corymbs, and solitary flowers (Figure 1). The species in Laurocerasus & Padus clade only belonged to the racemose group, while the Cerasus clade contained three inflorescence types. We also found subg. Amygdalus was divided into the solitary-flower group. The results also shown that subg. Prunus mostly belonged to the solitary-flower group, with only P. salicina divided into the corymbose group.
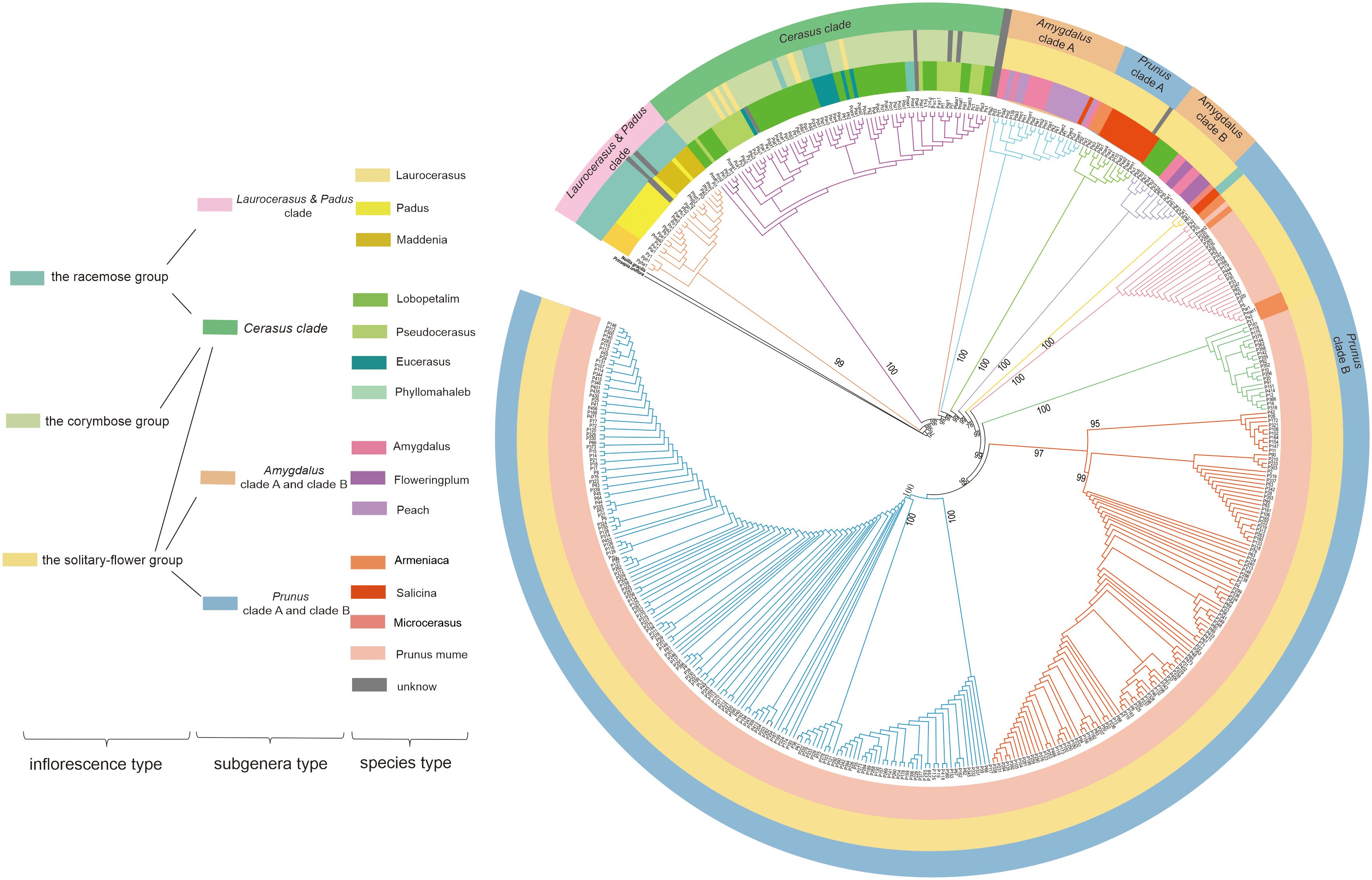
Figure 1 ML tree of section Prunus based on whole plastid genome dataset. To the left of the tree is a color-coded legend indicating the different categories into which the samples were placed. The first column refers to the inflorescence type, the second column refers to the subgenera type and the third column refers to the species type.
Phylogenetic relationships and haplotype analysis of Prunus mume
To explore the phylogenetic relationships of 322 different accessions of P. mume, we constructed evolutionary trees based on the complete plastomes of all accessions, with P. padus and P. triloba used as outgroups. 322 P. mume accessions were classified into 16 groups, including 11 cultivar groups (Pendulous, Single Flowered, Versicolor, Pink Double, Flavescens, Tortuosa, Green Calyx, Alboplena, Cinnabar Purple, Apricot Mei and Meiren), Old Mei, Wild Mei, Japanese apricot, Ornamental value and fruit production Mei, the accessions that could not be reasonably classified were labelled as unknown breed group (Chu, 1999b; Chen, 2010). Six clades were clearly resolved, which was consistent with the results of haplotype analysis (Figure 2A). Except for clade II, which only contains the Single Flowered cultivar group, the other clades contain different types of cultivar groups. Similarly, it cannot be ruled out that there are Single Flowered or other cultivar groups within the unknown breed group included in clade II. As the most abundant variety, the Pink Double group (101) was widely distributed in clades I, III, V, and VI, among which clade VI was the most abundant (38, 37.60%). Alboplena cultivars (11) were also distributed in clades I, III, V, and VI, with the most in clade VI (6, 54.50%). Cinnabar Purple cultivars (45) were distributed in clades I, III, V, and VI, with the most in clade III (25, 55.60%). Apricot Mei cultivars (12) were mainly distributed in clade I, with smaller distributions in clade III and IV. Green Calyx cultivars (13) were distributed in clade III, V, and VI with the same abundance in clade III and clade VI, but only one in clade V. Versicolor cultivars (5) were distributed in clade III and clade VI. Pendulous cultivars (30) were distributed in clades I, III, and VI. The majority of Pendulous cultivars were found in clade III (25, 83.30%), followed by clade VI (5, 16.70%), while clade I contained only one. Tortuosa cultivars were solely distributed within clade VI, which contains only one accession. The Flavescens and Meiren cultivars were also only distributed in clade VI. Single Flowered cultivars (24) were distributed among all clades, with the greatest number in clade VI and only one each in clades I, II, and IV. Old Mei cultivars (7) were distributed in clades I, II, III and VI, with most distributed in clade II (4, 57.14%). Wild Mei cultivars (12) were distributed in clades II, III, IV, V, and VI, although mainly in clade II (5, 41.70%). Ornamental value and fruit production Mei cultivars were only distributed in clade II which contained two accessions. Japanese apricot cultivars (14) were distributed in clades I, II, III, and VI, with most in clade III (8, 57.14%) and the fewest in clade VI (1, 7.14%).

Figure 2 (A) ML tree of 322 P. mume accessions. (B) Haplotype network of P. mume accessions. The size of pie chart is proportional to the number of accessions with the same haplotype. The colors in the pie chart indicate the percentage of accessions from different cultivars.
By utilizing an alignment matrix encompassing 322 P. mume plastomes, we conducted a SNV-based haplotype analysis, resulting in the identification of 46 haplotypes. The two haplotypes (h1-h2) represented outgroups, which did not belong to P. mume and appeared to show a large number of nucleotide variations. The h1 stands for P. padus, the h2 stands for P. triloba. Prunus mume accessions were clearly divided into six genetic clusters, which agreed with the phylogenetic tree (Figure 2B). There were two haplotypes in clade I, 12 in clade II, 11 in clade III, two in clade IV, one in clade V, and 17 in clade VI. The two haplotypes (h3-h4) may be old haplotypes.
Population structure and genetic diversity of P. mume
To gain a deeper understanding of the matrilineal history of P. mume, population structure was inferred by STRUCTURE with K = 5 based on an SNV-only input matrix being the best supported partition (Figure 3C). Our results showed that different K values indicate difference in group stability of P. mume lineages. At all K values the first three clades were resolved in the bar plot in all cases. Similarly, mixed membership was observed at all K values in some of the clades beyond the earliest diverging three. (Figures 3A–C). Previous studies have shown introgression between P. mume cultivars and other Prunus species [17]. Shared membership in the STRUCTURE barplots using plastome data also suggest that introgression may have occurred during P. mume domestication but because plastomes are uniparentally inherited this might also be the result of incomplete lineage sorting. (Figures 3A, B). Principal component analysis (PCA) was conducted to further investigate the genetic diversity based on all 322 accessions with the SNV dataset (Figure 3E). The results of PCA using whole plastome alignments were similar to those seen in other population analyses conducted herein. Nucleotide and haplotype diversity among the six clades of P. mume showed minor variations. Nucleotide diversity (Pi) differed among different clades (Figure 3D). The highest nucleotide diversity (Pi) was in clade II (Pi=0.00013), followed by clade VI (Pi=0.00004), clade IV (Pi=0.00001) clade I (Pi=0.00001), and clade III (Pi=0.00000) and clade V (Pi=0.00000). The highest haplotype diversity (Hd) was in clade II (Hd = 0.924), followed by clade VI (Hd = 0.564) and clade III (Hd = 0.254), clade IV (Hd=0.200), and clade V (Hd=0.000). Our analyses of genetic divergence between 322 P. mume accessions were found in Fst values. Among all clades, clade III had the highest level of divergence (III to I Fst = 0.996; III to II Fst = 0.896; III to IV Fst = 0.970; III to V Fst = 0.992; and III to VI Fst = 0.899). Clade II also exhibited high levels of divergence. Clade III and clade V showed lower levels of divergence compared to the other clades.

Figure 3 Phylogenetic and population structure analyses of P. mume. (A) The structure of phylogenetic topology in Bayesian inference. (B) a bar-plot of all samples showing the percentage assignment to a given group across different values of K (C) Delta K across a range of K values from 3−9. (D) Genetic diversity and differentiation of six clades of P. mume. Pairwise Fst between the corresponding genetic clusters is represented by the numbers above the lines joining two bubbles. (E) PCA analysis including outgroup (larger panel) and excluding outgroup (inset panel) showing the first two components in both cases.
Plastome structure and organization of Prunus mume
All P. mume plastomes exhibited a typical quadripartite and circular organization. The size of the complete plastomes ranged from 157,871 bp to 158,213 bp (Med = 157,910 bp). Among the different plastome regions, the LSC size ranged from 86,102 bp to 86,412 bp (Med = 86,115 bp), the SSC size ranged from 18,963 bp to 19,011bp (Med = 19,011 bp) and the IR size ranged from 26,387 bp to 26,400 bp (Med = 26,390 bp). The total GC content (%) of the complete plastomes ranged from 36.73 to 36.75 (Med = 37.74), with 34.53 to 34.59 (Median = 34.57) for the LSC, 30.36 to 30.45 (Median = 30.38) for the SSC, and 42.56 to 42.57 (Median = 42.56) for the IRs. The GC content of the IR regions was higher than that of the LSC and SSC (Supplementary Table S3, Figure 4).
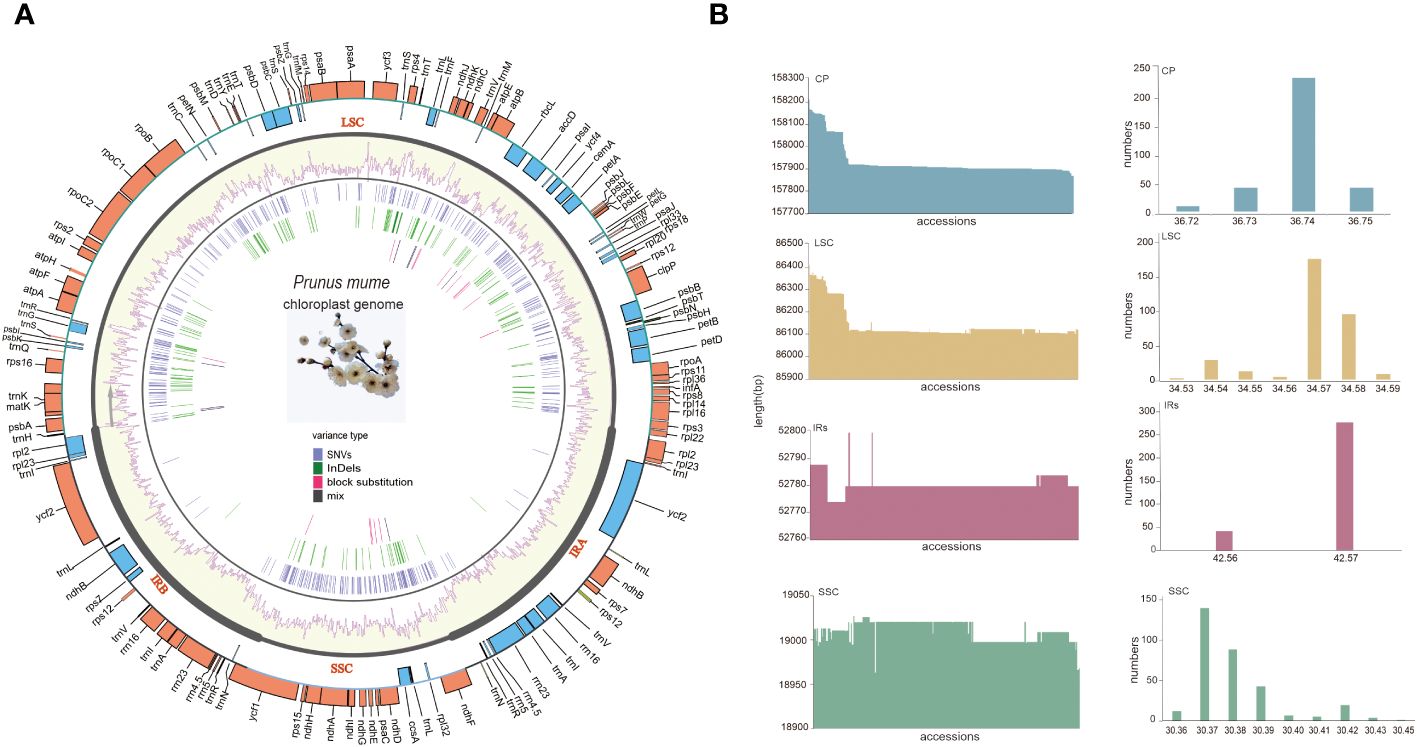
Figure 4 The pan-plastome of P. mume. (A) The general plastome structure P. mume. The outer circle shows gene placement and annotation across the genome, the transcribed genes that run clockwise and counterclockwise are indicated by the light blue and orange bars respectively. The GC content is displayed as yellow bars inside of the tetrad divisions (LSC, SSC, IRA, and IRB) in the second outer circle. SNVs, InDels, block substitutions and mixed variants are represented within purple, green, red and black lines, respectively. Single nucleotide variants (SNVs), block substitutions (BS, two or more consecutive nucleotide variants), nucleotide insertions or deletions (InDels), and mixed sites (which comprise two or more of the preceding three variants at a particular site) are the four categories into which variants are divided. (B) Sequence length and the GC content barplots of the total plastome, LSC, SSC and IRs.
A total of 112 unique genes were annotated and grouped into functional categories as follows: 78 protein-coding genes (PCGs), 30 transfer RNA (tRNA) genes, and 4 ribosomal RNA (rRNA) genes (Figure 4A). The distribution of these identified genes in the LSC, SSC, and IRs was as follows: a total of 83 genes in the LSC, including 62 PCGs and 21 tRNA genes; 13 in the SSC, including 12 PCGs and 1 tRNA gene, and 16 duplicated in the IRs, including 5 PCGs, 7 tRNA genes, and 4 rRNA genes (all genes in the IRs were counted once). Among these genes 18 (13 located in LSC,1 in SSC and 4 in the IRs) including 12 PCGs (atpF, clpP, ndhA, ndhB, petB, petD, rpl2, rpl16, rpoC1, rps12, rps16, and ycf3) and 6 tRNA (trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC) contained introns. In particular, clpP, ycf3, and trans-spliced rps12 (characterized by the first exon locating in LSC and the other two in the IRs) contained two introns (Supplementary Table S4, Figure 4B).
Analysis of nucleotide variation in Prunus mume plastomes
To gain a deeper understanding of nucleotide variation of the P. mume pan-plastome, 322 newly assembled plastomes were aligned for following analyses. A total of 682 variants were identified among coding sequences (CDS) and intergenic spacers (IGS) of 322 plastomes. Variants were classified as SNVs, InDels, block substitutions and mixed variants. Among these variants, SNVs had the largest number and proportion (455, 66.72%), followed by InDels (201, 29.50%), block substitutions (18, 2.64%) and mixed variants (8, 1.17%) (Figure 5, Supplementary Table S5). Variants were dispersed unevenly across the pan-plastome. The LSC (474, 69.50%) included the greatest number of variants, followed by the SSC (161, 23.61%), and IRs (47, 6.90%) contained the fewest variants. When analyzing the variants by genic position, the IGS regions had the highest number of variants (381, 55.87%), followed by CDS (220, 32.26%).
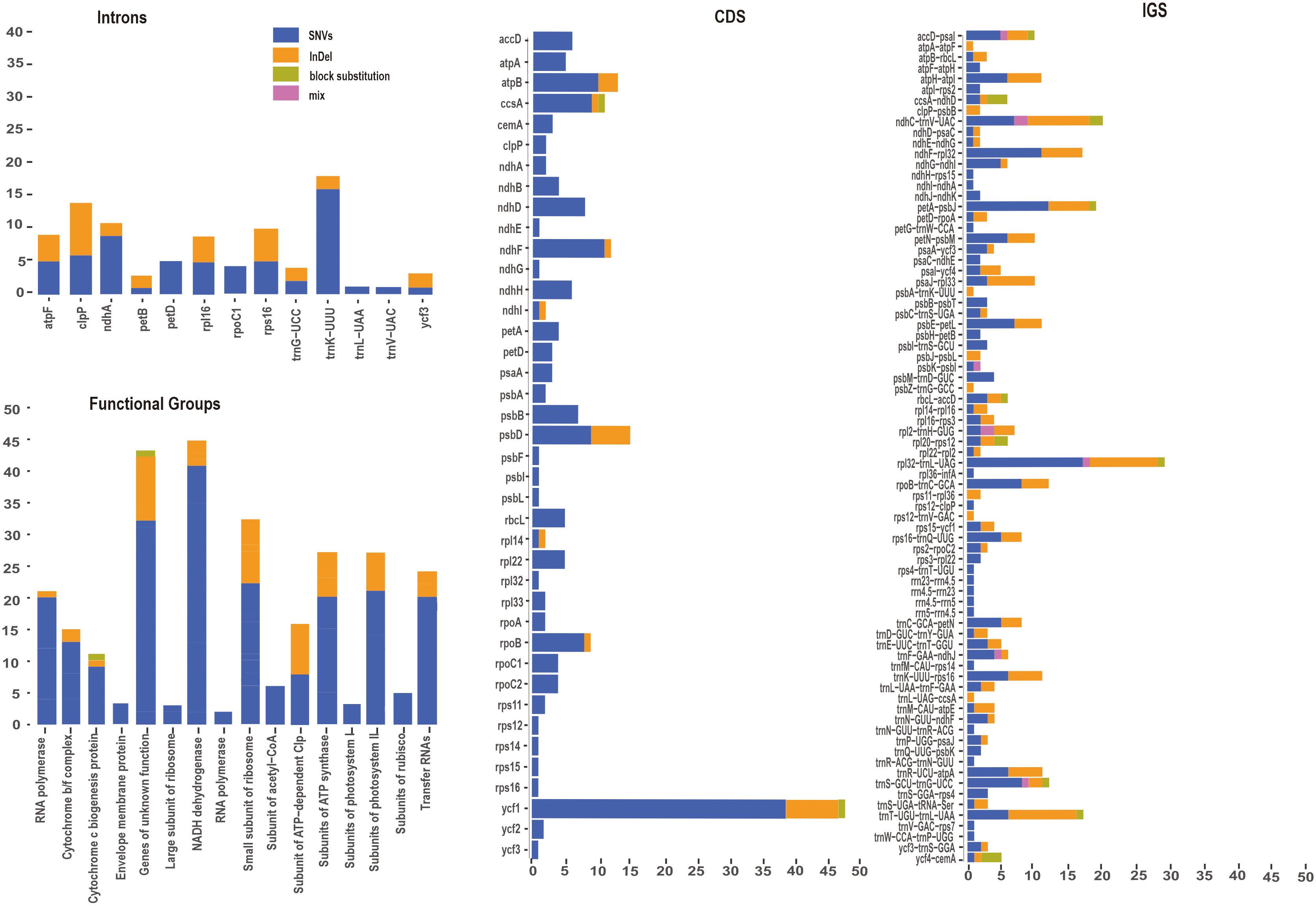
Figure 5 Variant locations categorized by genic position (CDS, Introns and IGS) and functional group.
Among the CDS, ycf1 (48 total variants) had the most variants, including SNVs (38), InDels (7) and block substitutions (1). ccsA (11) also had three types of variations, including SNVs (9), InDels (1) and block substitutions (1). Eleven genes including ycf3, rps12, rps14, rps15, rps16, rpl32, psbF, psbL, psbI, ndhG, and ndhE had only one SNV each (Figure 5). Seven genes including psbA, rpl33, clpP, rpoA, rps11, ndhB and ndhA had two SNVs, rpl14 and ndhl each included one SNV and one InDel The genes rpoB included eight SNVs and one InDels, psbD included nine SNVs and six InDels.
Hypervariable regions across the Prunus mume pan-plastome
Numerous nucleotide mutations were observed throughout the P. mume plastome, despite structural and gene conservation. These nucleotide variations have the potential to be employed in the development of molecular markers. According to the evolutionary tree, we found several potential loci to distinguish each clade (Supplementary Table S4). Clade 1 contained the most hypervariable regions, followed by clade II. Here, we selected genes located in the boundary region to show the sequence differences for each clade (Figure 6). We found that ycf1 had a large number of variant loci, which contained unique SNVs to clade I, II and III. There were also some SNVs that clade I and II exclusively contained. We also found some loci that contain unique InDels, such as rpl2, which contained the unique InDels to clade II and clade V. We also found loci which have unique SNVs in the IGS, such as trnL-CAA-ycf2 containing unique SNVs to clade VI. The high variability in IGSs could improve resolution of intraspecific studies.
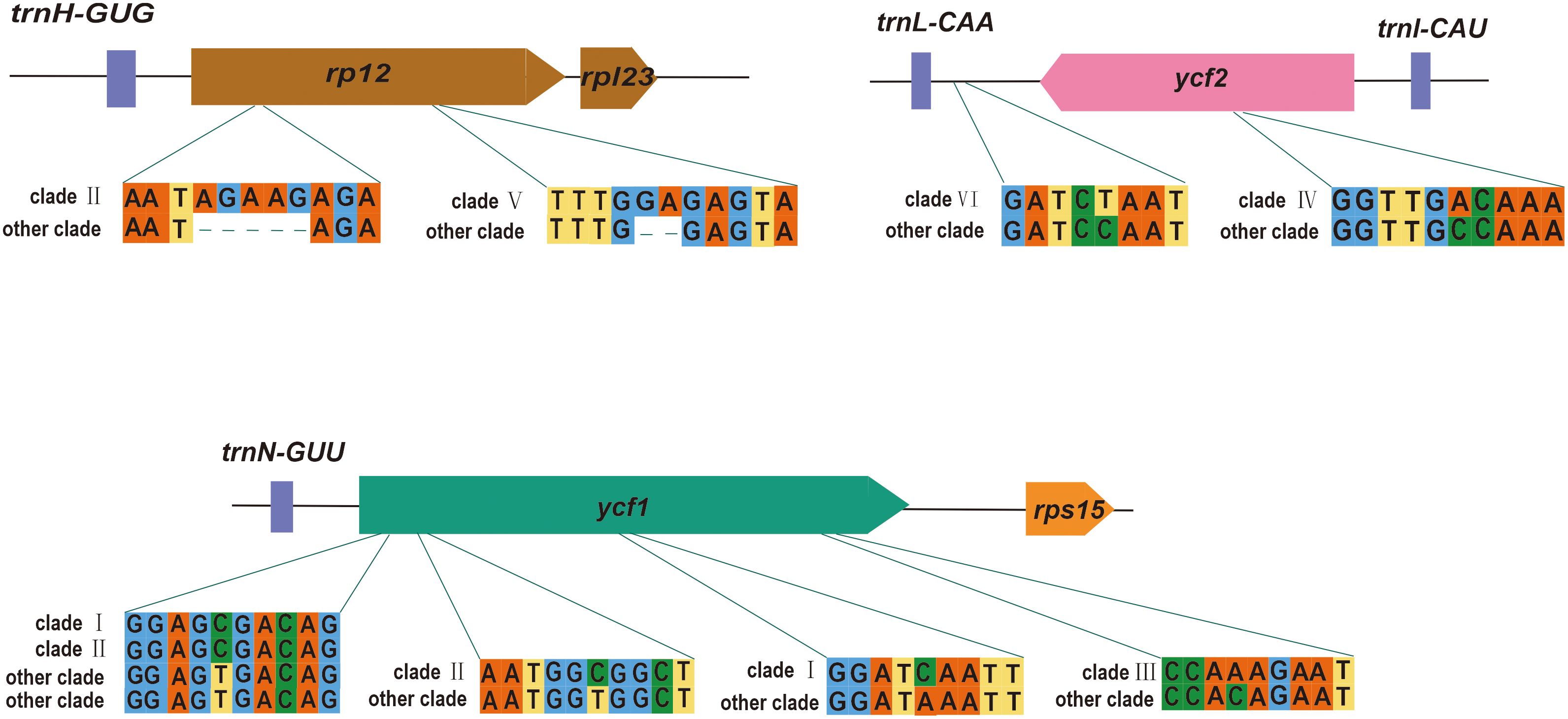
Figure 6 Examples of variable sites.
Discussion
Phylogenetic relationships in Prunus
Our research combined plastome data of Prunus to resolve evolutionary relationships in the genus more broadly to better understand the evolution of inflorescences and possible past maternal introgression. Previous studies have indicated that the racemose, solitary-flowering, and corymbose inflorescences may provide signal for delimiting Prunus lineages. Past plastomic studies, have determined that the Prunus species in corymbose group had a close relationship with the solitary-flower group and the corymbose group was resolved as sister to the racemose group, which was the ancestral state of inflorescence in Prunus (Chin et al., 2010, 2014; Zhao et al., 2016). Our reconstructed phylogenetic tree in Prunus also exhibited a similar pattern as these results although some polyphyly in the inflorescence type mapped onto our plastome tree suggests a complex history and evolution among Prunus species. We also suggest that Padus and Lauroploidus may have originated from allopolyploidy and hybridization events for both are not monophyletic and clustered into a clade. Moreover, our study also provided strong evidence for supporting the placement of P. maackii in subg. Padus (Zhao et al., 2016; Su et al., 2023). Some studies have suggested that two species from Microcerasus should be divided into the solitary-flower group instead of the corymbose group (Bortiri et al., 2001; Lee and Wen, 2001; Chin et al., 2014). Genetic analyses based on different types of molecular data also supported this opinion (Bortiri et al., 2002, 2006; Liu et al., 2012; Shi et al., 2013; Zhao et al., 2016). Our reconstructed phylogenetic tree was congruent with these previous studies and also did not support the inclusion of Microcerasus into the Cerasus group (Kataoka et al., 1988). All species in the corymbose group were from the subgenus Cerasus. The Microcerasus species were recorded scattering in the solitary flower group (Shi et al., 2013), and our results also support this conclusion. Furthermore, it was noted that Microcerasus species bred with solitary-flower group species more frequently than with Cerasus group species (Mowrey and Werner, 1990). According to morphological and genetic data, the classification of Microcerasus into the solitary-flower group was reasonable (Baack and Rieseberg, 2007). To further understand the origin (s) and phylogenetic status of Microcerasus species, future research should sample these species as well as closely related taxa more intensively. The phylogenetic relationships among P. mume, P. armeniaca, and P. salicina are still a matter of debate, due to frequent hybridization, apomixis and a complex history of cultivation (Baack and Rieseberg, 2007; Xue et al., 2019; Numaguchi et al., 2020). We found that some apricots mei and P. salicina clustered together, suggesting that past introgression was still retained in P. armeniaca and P. mume. Based on the maternal inheritance of plastome and biparental inheritance of nuclear genome, apricots, Mei, and P. salicina may be originated from the same paternal parent and several maternal parents through past introgression.
Hypervariable regions in the P. mume pan-plastome
In this study, we employed 322 newly assembled plastomes to construct the pan-plastome of P. mume. Based on these plastome data, we conducted comprehensive analyses, such as structural feature and nucleotide variation. Similar to other research, our study revealed a significant conservation of quadripartite structure, sequence length, gene order and GC content in P. mume plastomes (Daniell et al., 2016). The IR regions exhibited higher GC contents than the LSC and SSC regions, which could contribute to their greater conservation (Khakhlova and Bock, 2006; Fan et al., 2018). Despite the conserved characteristics of plastome sequences, there are still abundant polymorphisms among plastomes for effective phylogenetic analysis and species identification (Gitzendanner et al., 2018; Xue et al., 2019). By comparing the plastomes in a specific genus or species, one can identify potential DNA barcodes for genus/species identification. These DNA barcodes often exhibit higher accuracy and discriminatory power in plant identification compared to universal barcodes (Dong et al., 2021). In the nucleotide variation analysis of plastomes of P. mume, ycf1 had the highest number of variants in the CDS region. Additionally, psbD and atpB were found to have a greater number of variants than other genes and they could be employed as potential DNA barcodes to distinguish different accessions of P. mume. The IGS regions were determined to be more variable than the CDS regions in plastomes from other studies (Song et al., 2020). Likewise, InDels are more common beyond CDS regions, and the P. mume pan-plastome also exhibited a similar pattern of variant abundance and type as other plastomes (Wang et al., 2022). As more research on pan-plastomes is completed, intronic regions and hypervariable regions from IGSs are also regarded as valuable resources for species identification at the intraspecific level, with higher resolution compared to some common DNA barcode from CDS regions, such as rbcL and matK. This study proved that some IGS regions: rpl32-trnL-UAG and ndhC-trnV-UAC were especially rich in informative markers. Similar to pan-plastomes of Brassica napus, Nelumbo nucifera, our pan-plastome research revealed that SNVs are by far the most prevalent form of mutation (Song et al., 2020; Tao et al., 2021; Wang et al., 2022). Nowadays, the plastomes’ crucial role in evolution is more deeply understood by researchers and further pan-plastome research from a variety of cultivated taxa will no doubt clarify the history of domestication in P. mume. Further pan-plastome research from a variety of cultivated and wild taxa will allow for improved descriptions of patterns unique to domesticated lineages. Such work will improve the development of elite varieties.
Phylogenetic relationships and divergence among clades of Prunus mume
Numerous processes, such as genetic drift, reproductive isolation, local adaptation, demographic fluctuations, mode of reproduction, artificial selection, and human domestication, are known to have an impact on plant population structure and genetic diversity (Lovette, 2004). It is commonly believed that plastomes lack any genes related to domestication. The nucleotide diversity of plastomes is expected to be reduced due to severe artificial selection during domestication (Haudry et al., 2007; Gross and Olsen, 2010; Avni et al., 2017; He et al., 2021) however this may not be the case in long-lived woody species. According to early records, Chinese plum landraces were selected from a wild lineage of P. mume, the true mume branch. In addition to the true mume lineage, two hybrid lineages are also known between species P. mume and P. armeniaca as well as P. mume and P. cerasifera (Zhang et al., 2018). From our phylogeny the nearly all P. mume samples appeared to be from P. mume maternal origins although a few samples within the P. mume lineage were identified as P. armeniaca suggesting past introgression. Such imbalance in P. mume plastomes despite known instances of interspecific introgression may suggest that unidirectional nucleocytoplasmic incompatibly (Guo et al., 2019; Wang et al., 2021) has occurred in the hybridization history of P. mume but more sampling needs to be done before this can be concluded with confidence. If such a pattern of nucelocytoplasmic incompatibly occurred in P. mume domestication this will be valuable to know in future breeding projects to avoid mismatching parents.
From our reconstructed phylogenetic tree based on pan-plastomes of P. mume, six well-supported clades were identified and each clade differed in terms of haplotypic diversity and the cultivar group membership (Figure 2). The morphological characteristics demonstrated significant variations among different cultivar groups within P. mume. We also found multiple cultivar groups instead of any single one distributed in the same clade. Although, most wild Mei and old Mei were distributed in clade II, and a majority of cultivar accessions of P. mume were distributed in clades I, III, IV, V and VI. There were a large number of haplotypes located in clade II and most haplotypes contained only one accession. We speculate that clade II was a wild lineage due to this disruption pattern. These discoveries could help clarify the patterns of introgression during P. mume domestication. These findings also support the reliability of whole plastome data in studying plant diversity at multiple taxonomic levels, especially at the intraspecific level. In summary, these pan-plastome resources will not only provide new insights into the natural and domestication history of Prunus, but also advance the progress of research on organelles.
Conclusions
Our understanding of the evolution and phylogeny of section Prunus have been greatly aided by numerous studies (Zhao et al., 2016; Xue et al., 2019; Ding et al., 2022; Su et al., 2023). However, a lack of pan-plastomic studies have hindered our understanding of matrilineal relationships within this important genus of plants. The current study where we have de novo assembled 322 different P. mume plastomes and 151 other Prunus accessions plastomes to reconstruct the Prunus evolutionary tree is an important contribution in understanding natural plastome evolution as well as plastome evolution under artificial selection within the cultivated P. mume lineage. For instance, we found that six well-supported plastome lineages could be resolved within P. mume and that most named cultivar groups were selected from two of these clades. Furthermore, we resolved polyphyly between P. mume and P. armeniaca indicating past introgression in the domestication of these apricots. Lastly our comprehensive description of nucleotide variants in the mei pan-plastome will aid future researchers and breeders in understanding the molecular evolution of the plastome in this important fruit species and utilize this knowledge in trait improvement and preservation of wild and cultivated germplasm.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
JW(1st author): Writing – original draft. JK: Writing – review & editing, Methodology. JW(3rd author): Writing – review & editing, Methodology. XY: Data curation, Writing – review & editing. YL: Writing – review & editing, Formal analysis. TS: Data curation, Writing – review & editing. LT: Writing – review & editing. GX: Writing – review & editing. ZW: Writing – review & editing, Funding acquisition. SL: Writing – review & editing, Methodology. MJ: Writing – review & editing, Methodology.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors have been supported by several grants, including the Guangdong Pearl River Talent Program (grants 2021QN02N792), the Science Technology and Innovation Commission of Shenzhen Municipality (grants RCYX20200714114538196); Shanxi Agricultural University's "Special" and "Excellent" Agricultural High Quality Development Science and Technology Support Project and (grants TYGC24-59); the Chinese Academy of Agricultural Sciences Elite Youth Program (grant 110243160001007).
Acknowledgments
The authors gratefully acknowledge helpful comments from reviewers on earlier versions of this manuscript. We sincerely thank members of Wu Lab for their help in preparing this paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1404071/full#supplementary-material
References
Alioto, T., Alexiou, K. G., Bardil, A., Barteri, F., Castanera, R., Cruz, F., et al. (2020). Transposons played a major role in the diversification between the closely related almond and peach genomes: results from the almond genome sequence. Plant J. 101, 455–472. doi: 10.1111/tpj.14538
Andro, M.-C., Riffaud, J.-P. (1995). Pygeum africanum extract for the treatment of patients with benign prostatic hyperplasia: A review of 25 years of published experience. Curr. Ther. Res. 56, 796–817. doi: 10.1016/0011-393X(95)85063-5
Avni, R., Nave, M., Barad, O., Baruch, K., Twardziok, S., Gundlach, H., et al. (2017). Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357, 93–97. doi: 10.1126/science.aan0032
Baack, E. J., Rieseberg, L. H. (2007). A genomic view of introgression and hybrid speciation. Curr. Opin. Genet. Dev. 17, 513–518. doi: 10.1016/j.gde.2007.09.001
Baek, S., Choi, K., Kim, G. B., Yu, H. J., Cho, A., Jang, H., et al. (2018). Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol. 19, 127. doi: 10.1186/s13059-018-1497-y
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Biersma, E. M., Torres-Díaz, C., Molina-Montenegro, M. A., Newsham, K. K., Vidal, M. A., Collado, G. A., et al. (2020). Multiple late-Pleistocene colonization events of the Antarctic pearlwort Colobanthus quitensis(Caryophyllaceae) reveal the recent arrival of native Antarctic vascular flora. J. Biogeogr. 47, 1663–1673. doi: 10.1111/jbi.13843
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bortiri, E., Heuvel, B. V., Potter, D. (2006). Phylogenetic analysis of morphology in Prunus reveals extensive homoplasy. Plant Syst. Evol. 259, 53–71. doi: 10.1007/s00606-006-0427-8
Bortiri, E., Oh, S.-H., Fang-You, G., Dan, P. (2002). The phylogenetic utility of nucleotide sequences of sorbitol 6-phosphate dehydrogenase in prunus (Rosaceae). Am. J. Bot. 89, 1697–1708. doi: 10.3732/ajb.89.10.1697
Bortiri, E., Oh, S. H., Jiang, J., Baggett, S., Granger, A., Weeks, C., et al. (2001). Phylogeny and systematics of Prunus (Rosaceae) as determined by sequence analysis of ITS and the chloroplast trnL-trnF spacer DNA. Systematic Bot. 26, 797–807. doi: 10.1043/0363–6445-26.4.797
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Callahan, A. M., Zhebentyayeva, T. N., Humann, J. L., Saski, C. A., Galimba, K. D., Georgi, L. L., et al. (2021). Defining the 'HoneySweet' insertion event utilizing NextGen sequencing and a de novo genome assembly of plum (Prunus domestica). Hortic. Res. 8, 8. doi: 10.1038/s41438-020-00438-2
Chen, J. (2010). China mei flower (Prunus mume) cultivars in color (Beijing: China Forestry Publishing House).
Chin, S. W., Shaw, J., Haberle, R., Wen, J., Potter, D. (2014). Diversification of almonds, peaches, plums and cherries - molecular systematics and biogeographic history of Prunus (Rosaceae). Mol. Phylogenet Evol. 76, 34–48. doi: 10.1016/j.ympev.2014.02.024
Chin, S. W., Wen, J., Johnson, G., Potter, D. (2010). Merging Maddenia with the morphologically diverse Prunus (Rosaceae). Bot. J. Linn. Soc 164, 236–245. doi: 10.1111/j.1095-8339.2010.01083.x
Clement, M., Posada, D., Crandall, K. A. (2000). TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 1–29. doi: 10.1186/s13059-016-1004-2
Ding, A., Bao, F., Ding, A., Zhang, Q. (2022). Cold hardiness of prunus mume ‘Xiang ruibai’ and its parents based on biological indexes and physical parameters. Forests 13, 2163. doi: 10.3390/f13122163
Dong, W. P., Sun, J. H., Liu, Y. L., Xu, C., Wang, Y. H., Suo, Z. L., et al. (2021). Phylogenomic relationships and species identification of the olive genus Olea (Oleaceae). J. Syst. Evol. 60, 1263–1280. doi: 10.1111/jse.12802
Dong, W., Xu, C., Wen, J., Zhou, S. (2020). Evolutionary directions of single nucleotide substitutions and structural mutations in the chloroplast genomes of the family Calycanthaceae. BMC Evol. Biol. 20, 96. doi: 10.1186/s12862-020-01661-0
Fan, W. B., Wu, Y., Yang, J., Shahzad, K., Li, Z. H. (2018). Comparative chloroplast genomics of dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00689
Ginestet, C. (2011). ggplot2: elegant graphics for data analysis. Stat Society. 174, 245–246. doi: 10.1111/j.1467-985X.2010.00676_9.x
Gitzendanner, M. A., Soltis, P. S., Yi, T. S., Li, D. Z., Soltis, D. E. (2018). Plastome phylogenetics: 30 years of inferences into plant evolution. ScienceDirect 85, 293–313. doi: 10.1016/bs.abr.2017.11.016
Gross, B. L., Olsen, K. M. (2010). Genetic perspectives on crop domestication. Trends Plant Sci. 15, 529–537. doi: 10.1016/j.tplants.2010.05.008
Guo, C., Guo, X. H., Li, D. Z. (2019). Phylogenomic analyses reveal intractable evolutionary history of a temperate bamboo genus (Poaceae: Bambusoideae). Plant Diversity. 41, 213–219. doi: 10.1016/j.pld.2019.05.003
Haudry, A., Cenci, A., Ravel, C., Bataillon, T., Brunel, D., Poncet, C., et al. (2007). Grinding up wheat: A massive loss of nucleotide diversity since domestication. Mol. Biol. Evol. 24, 1506–1517. doi: 10.1093/molbev/msm077
He, W., Chen, C., Xiang, K., Wang, J., Zheng, P., Tembrock, L. R., et al. (2021). The history and diversity of rice domestication as resolved from 1464 complete plastid genomes. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.781793
Hodel, R. G. J., Zimmer, E., Wen, J. (2021). A phylogenomic approach resolves the backbone of Prunus (Rosaceae) and identifies signals of hybridization and allopolyploidy. Mol. Phylogen. Evol. 160, 107118. doi: 10.1016/j.ympev.2021.107118
Hubisz, M. J., Falush, D., Stephens, M., Pritchard, J. K. (2009). Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332. doi: 10.1111/j.1755-0998.2009.02591.x
Jakobsson, M., Rosenberg, N. A. (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. doi: 10.1093/bioinformatics/btm233
Jansen, R. K., Ruhlman, T. A. (2012). “Plastid genomes of seed plants. genomics of chloroplasts and mitochondria,” in Advances in Photosynthesis and Respiration, vol 35. (Springer, Dordrecht), 103–126. Available at: https://doi.org/10.1007/978-94-007-2920-9_5.
Jia, M., Wang, J., Cao, D., Jiang, C., Li, W., Tembrock, L. R., et al. (2024). The pan-plastome of Hemerocallis citrina reveals new insights into the genetic diversity and cultivation history of an economically important food plant. BMC Plant Biol. 24, 44. doi: 10.1186/s12870-023-04668-z
Jiang, D., Cai, X., Gong, M., Xia, M., Xing, H., Dong, S., et al. (2023). Complete chloroplast genomes provide insights into evolution and phylogeny of Zingiber (Zingiberaceae). BMC Genomics 24, 30. doi: 10.1186/s12864–023-09115–9
Jiang, F., Zhang, J., Wang, S., Yang, L., Luo, Y., Gao, S., et al. (2019). The apricot (Prunus Armeniaca L.) genome elucidates Rosaceae evolution and beta-carotenoid synthesis. Hortic.Res. 6, 128. doi: 10.1038/s41438–019-0215–6
Kataoka, I., Sugiura, A., Tomana, T. (1988). Interspecific Hybridization between Microcerasus and other Prunus spp. J. Jpn Soc. Hortic. Sci. 56, 398–407. doi: 10.2503/jjshs.56.398
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khakhlova, O., Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lan, L., Leng, L., Liu, W., Ren, Y., Reeve, W., Fu, X., et al. (2024). The haplotype-resolved telomere-to-telomere carnation (Dianthus caryophyllus) genome reveals the correlation between genome architecture and gene expression. Hortic. Res. 11, uhad244. doi: 10.1093/hr/uhad244
Lan, L., Zhao, H., Xu, S., Kan, S., Zhang, X., Liu, W., et al. (2023). A high-quality Bougainvillea genome provides new insights into evolutionary history and pigment biosynthetic pathways in the Caryophyllales. Hortic. Res. 10, uhad124. doi: 10.1093/hr/uhad124
Lee, S., Wen, J. (2001). A phylogenetic analysis of prunus and the amygdaloideae (Rosaceae) using ITS sequences of nuclear ribosomal DNA. Am. J. Bot. 88, 150–160. doi: 10.2307/2657135
Leigh, J. W., Bryant, D., Nakagawa, S. (2015). popart: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210X.12410
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lin, N., Liu, R., Wang, Y., Guo, P., Wang, Y., Liu, Y., et al. (2023). The complete chloroplast genome of Ulmus mianzhuensis with insights into structural variations, adaptive evolution, and phylogenetic relationships of Ulmus (Ulmaceae). BMC Genomics 24, 366. doi: 10.1186/s12864-023-09430-1
Liu, C., Feng, C., Peng, W., Hao, J., Wang, J., Pan, J., et al. (2020). Chromosome-level draft genome of a diploid plum (Prunus salicina). Gigascience 9 (12), giaa130. doi: 10.1093/gigascience/giaa130
Liu, X.-L., Wen, J., Nie, Z.-L., Johnson, G., Liang, Z.-S., Yang, C. (2012). Polyphyly of the Padus group of Prunus (Rosaceae) and the evolution of biogeographic disjunctions between eastern Asia and eastern North America. J. Plant Res. 126, 351–361. doi: 10.1007/s10265–012-0535–1
Lovette, I. J. (2004). Molecular markers, natural history, and evolution. 2nd edition. The Auk. 121, 1298–1299. doi: 10.1093/auk/121.4.1298
Magdy, M., Ou, L., Yu, H., Chen, R., Zhou, Y., Hassan, H., et al. (2019). Pan-plastome approach empowers the assessment of genetic variation in cultivated Capsicum species. Hortic. Res. 6, 108. doi: 10.1038/s41438-019-0191-x
Mowrey, B. D., Werner, D. J. (1990). Phylogenetic relationships among species of Prunus as inferred by isozyme markers. Theor. Appl. Genet. 80, 129–133. doi: 10.1007/BF00224026
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Numaguchi, K., Akagi, T., Kitamura, Y., Ishikawa, R., Ishii, T. (2020). Interspecific introgression and natural selection in the evolution of Japanese apricot (Prunus mume). Plant J. 104, 1551–1567. doi: 10.1111/tpj.15020
Page, A. J., Taylor, B., Delaney, A. J., Soares, J., Seemann, T., Keane, J. A., et al. (2016). SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2, e000056. doi: 10.1099/mgen.0.000056
Rehder, A. (1956). Manual of Cultivated Trees and Shrubs Hardy in North America: exclusive of the subtropical and warmer temperate regions (New York: Macmillan).
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Shi, S., Li, J., Jiahui, S., Yu, J., Zhou, S. (2013). Phylogeny and Classification of Prunus sensu lato (Rosaceae). J. Integr. Plant Biol. 55, 1069–1073. doi: 10.1111/jipb.12095
Sielemann, K., Pucker, B., Schmidt, N., Viehover, P., Weisshaar, B., Heitkam, T., et al. (2022). Complete pan-plastome sequences enable high resolution phylogenetic classification of sugar beet and closely related crop wild relatives. BMC Genomics 23, 113. doi: 10.1186/s12864-022-08336-8
Song, J. M., Guan, Z., Hu, J., Guo, C., Yang, Z., Wang, S., et al. (2020). Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants. 6, 34–45. doi: 10.1038/s41477-019-0577-7
Su, N., Hodel, R. G. J., Wang, X., Wang, J. R., Xie, S. Y., Gui, C. X., et al. (2023). Molecular phylogeny and inflorescence evolution of Prunus (Rosaceae) based on RAD-seq and genome skimming analyses. Plant Divers. 45, 397–408. doi: 10.1016/j.pld.2023.03.013
Tao, Y., Luo, H., Xu, J., Cruickshank, A., Zhao, X., Teng, F., et al. (2021). Extensive variation within the pan-genome of cultivated and wild sorghum. Nat. Plants 7, 766–773. doi: 10.1038/s41477-021-00925-x
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Verde, I., Abbott, A. G., Scalabrin, S., Jung, S., Shu, S., Marroni, F., et al. (2013). The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494. doi: 10.1038/ng.2586
Wang, J., Kan, S., Liao, X., Zhou, J., Tembrock, L. R., Daniell, H., et al. (2024). Plant organellar genomes: much done, much more to do. Trends Plant Sci. doi: 10.1016/j.tplants.2023.12.014
Wang, J., Liao, X., Gu, C., Xiang, K., Wang, J., Li, S., et al. (2022). The Asian lotus (Nelumbo nucifera) pan-plastome: diversity and divergence in a living fossil grown for seed, rhizome, and aesthetics. Ornamental Plant Res. 2, 1–10. doi: 10.48130/OPR-2022-0010
Wang, J., Liao, X., Li, Y., Ye, Y., Xing, G., Kan, S., et al. (2023). Comparative Plastomes of Curcuma alismatifolia (Zingiberaceae) Reveal Diversified Patterns among 56 Different Cut-Flower Cultivars. Genes 14, 1360–1385. doi: 10.3390/genes14091743
Wang, J., Liu, W., Zhu, D., Hong, P., Zhang, S., Xiao, S., et al. (2020). Chromosome-scale genome assembly of sweet cherry (Prunus avium L.) cv. Tieton obtained using long-read and Hi-C sequencing. Hortic. Res. 7, 122. doi: 10.1038/s41438–020-00343–8
Wen, J., Berggren, S., Lee, C.-H., Ickert-Bond, S., Yi, T., Yoo, K.-O., et al. (2008). Phylogenetic inferences in Prunus (Rosaceae) using chloroplast ndhF and ribosomal ITS sequences. J. Syst. Evol. 46, 322–332. doi: 10.3724/SP.J.1002.2008.08050
Wang, J. H., Moore, M. J., Wang, H., Zhu, Z. X., Wang, H. F. (2021). Plastome evolution and phylogenetic relationships among Malvaceae subfamilies. Gene. 765, 145103. doi: 10.1016/j.gene.2020.145103
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z. Q., Ge, S. (2012). The phylogeny of the BEP clade in grasses revisited: evidence from the whole-genome sequences of chloroplasts. Mol. Phylogenet Evol. 62, 573–578. doi: 10.1016/j.ympev.2011.10.019
Xue, S., Shi, T., Luo, W., Ni, X., Iqbal, S., Ni, Z., et al. (2019). Comparative analysis of the complete chloroplast genome among Prunus mume, P. Armeniaca, and P. salicina. Hortic. Res. 6, 89. doi: 10.1038/s41438–019-0171–1
Yan, L. J., Zhu, Z. G., Wang, P., Fu, C. N., Guan, X. J., Kear, P., et al. (2022). Comparative analysis of 343 plastid genomes of Solanum section Petota: Insights into potato diversity, phylogeny, and species discrimination. J. Syst. Evol. 61, 599–612. doi: 10.1111/jse.12898
Zhang, Q. (1987). The interspecific crossing of mei flower and cold hardiness breeding. J. Beijing Forestry Univ. 9, 69–79.
Zhang, Q., Chen, W., Sun, L., Zhao, F., Huang, B., Yang, W., et al. (2012). The genome of Prunus mume. Nat. Commun. 3, 1318. doi: 10.1038/ncomms2290
Zhang, S. D., Jin, J. J., Chen, S. Y., Chase, M. W., Soltis, D. E., Li, H. T., et al. (2017). Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol. 214, 1355–1367. doi: 10.1111/nph.14461
Zhang, Q., Zhang, H., Sun, L., Fan, G., Ye, M., Jiang, L., et al. (2018). The genetic architecture of floral traits in the woody plant Prunus mume. Nat. Commun. 9, 1702. doi: 10.1038/s41467-018-04093-z
Zhao, L., Jiang, X. W., Zuo, Y. J., Liu, X. L., Chin, S. W., Haberle, R., et al. (2016). Multiple events of allopolyploidy in the evolution of the racemose lineages in prunus (Rosaceae) based on integrated evidence from nuclear and plastid data. PloS One 11, e0157123. doi: 10.1371/journal.pone.0157123
Zheng, T., Li, P., Zhuo, X., Liu, W., Qiu, L., Li, L., et al. (2022). The chromosome-level genome provides insight into the molecular mechanism underlying the tortuous-branch phenotype of Prunus mume. New Phytol. 235, 141–156. doi: 10.1111/nph.17894
Zheng, S., Poczai, P., Hyvonen, J., Tang, J., Amiryousefi, A. (2020). Chloroplot: an online program for the versatile plotting of organelle genomes. Front. Genet. 11. doi: 10.3389/fgene.2020.576124
Keywords: pan-plastome, phylogeny, population structure, Prunus, P. mume
Citation: Wang J, Kan J, Wang J, Yan X, Li Y, Soe T, Tembrock LR, Xing G, Li S, Wu Z and Jia M (2024) The pan-plastome of Prunus mume: insights into Prunus diversity, phylogeny, and domestication history. Front. Plant Sci. 15:1404071. doi: 10.3389/fpls.2024.1404071
Received: 20 March 2024; Accepted: 29 April 2024;
Published: 03 June 2024.
Edited by:
Wenpan Dong, Beijing Forestry University, ChinaCopyright © 2024 Wang, Kan, Wang, Yan, Li, Soe, Tembrock, Xing, Li, Wu and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sen Li, c2F1bGlzZW5AMTYzLmNvbQ==; Zhiqiang Wu, d3V6aGlxaWFuZ0BjYWFzLmNu; Minlong Jia, eXlzam1sQDE2My5jb20=
†These authors have contributed equally to this work