 Li Li
Li Li Xiangru Li
Xiangru Li Yun Liu
Yun Liu Junda Li
Junda Li Xiaoyun Zhen1
Xiaoyun Zhen1 Jianghua Ye
Jianghua Ye
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 22 August 2024
Sec. Plant Systematics and Evolution
Volume 15 - 2024 | https://doi.org/10.3389/fpls.2024.1396389
Introduction: Among cultivated tea plants (Camellia sinensis), only four mitogenomes for C. sinensis var. assamica (CSA) have been reported so far but none for C. sinensis var. sinensis (CSS). Here, two mitogenomes of CSS (CSSDHP and CSSRG) have been sequenced and assembled.
Methods: Using a combination of Illumina and Nanopore data for the first time. Comparison between CSS and CSA mitogenomes revealed a huge heterogeneity.
Results: The number of the repetitive sequences was proportional to the mitogenome size and the repetitive sequences dominated the intracellular gene transfer segments (accounting for 88.7%- 92.8% of the total length). Predictive RNA editing analysis revealed that there might be significant editing in NADH dehydrogenase subunit transcripts. Codon preference analysis showed a tendency to favor A/T bases and T was used more frequently at the third base of the codon. ENc plots analysis showed that the natural selection play an important role in shaping the codon usage bias, and Ka/Ks ratios analysis indicated Nad1 and Sdh3 genes may have undergone positive selection. Further, phylogenetic analysis shows that six C. sinensis clustered together, with the CSA and CSS forming two distinct branches, suggesting two different evolutionary pathway.
Discussion: Altogether, this investigation provided an insight into evolution and phylogeny relationship of C. sinensis mitogenome, thereby enhancing comprehension of the evolutionary patterns within C. sinensis species.
Tea is the oldest and most popular non-alcoholic soft drink in the world, with enormous economic, cultural and scientific value (Xia et al., 2017). The main class of cultivated tea plants (Camellia sinensis) consist of C. sinensis var. sinensis (L.) O. Kuntze (Chinary type), C. sinensis var. assamica (Masters) Chang (Assamica type) and C. sinensis var. assamica subssp. Lasiocalyx Planch (Cambodia type). Of which, C. sinensis var. sinensis (CSS) and C. sinensis var. assamica (CSA) have the most obvious distinction. CSS has small leaves and is mainly grown in China and some Southeast Asian countries, while CSA has large leaves and is widely grown in India and some hot countries except southern China (Mondal, 2014; Kumarihami et al., 2016). It has long been suggested that CSS and CSA may have different origins, and that CSA is composed of two distinct lineages (Chinese Assamica type and Indian Assamica type) that were domesticated independently (Meegahakumbura et al., 2016; Wambulwa, 2018). In recent years, several studies have investigated the genetic diversity and evolution between CSS and CSA by whole genomes resequencing or complete chloroplast genomes assembly (Yengkhom et al., 2019; An et al., 2020; Li et al., 2021a). These studies confirmed that after the tea plants went through the last glacial maximum of the Quaternary, the CSA began to diverge, and the more cold-resistant CSS started to emerge. Since then, the two have evolved in parallel, forming the existing classification groups of tea plants (Zhang et al., 2021, 2023). However, the dynamic evolution of mitochondrial genomes between CSS and CSA has never been assessed until now.
Plant mitochondria are a kind of semi-autonomous organelle in eukaryotic cells. As a source of ATP energy, plant mitochondria play a variety of cellular functions during plant growth and development (Klingenberg, 2008; Liberatore et al., 2016; Niu et al., 2022). Plant mitochondrial genome (mitogenome) possess many unique features and complex dynamic structure (Kozik et al., 2019; Li et al., 2023), such as extreme variation in genome size (Petersen et al., 2020), very sparse gene distribution (Wu et al., 2022), large number of non-coding sequences (Zandueta-Criado and Bock, 2004), rich repeat sequences (Yang et al., 2022), ability to intracellular gene transfer (IGT) (Li et al., 2022), highly conservative gene sequences, and a large number of RNA editing (Fang et al., 2021). These characteristics make the plant mitogenome an important tool for studying the classification and evolution of plants (Ammiraju et al., 2007; Chen et al., 2017; Van de Paer et al., 2018; Sibbald et al., 2021). Therefore, the investigation of mitogenome is not only important for understanding mitochondrial function and cell metabolism regulation, but also provides an important theoretical basis for the study of plant species evolution and genetic diversity.
At present, only four mitogenomes of tea plants have been reported, and they all belong to Assamica type tea (C. sinensis var. assamica, CSA), three of which belong to Chinese Assamica type tea and one belongs to Indian Assamica type tea (Zhang et al., 2019; Rawal et al., 2020; Li et al., 2023). More mitogenome information of C. sinensis were needed to carry out further research. In this study, two complete mitogenomes of Chinary type tea plants (C. sinensis var. sinensis, CSS), including C. sinensis var. sinensis cv. Dahongpao (CSSDHP) and C. sinensis var. sinensis cv. Rougui (CSSRG) were sequenced and assembled using a combination of Illumina and Nanopore sequencing techniques. Both tea plants are excellent cultivars of Wuyi rock tea (Synonym: Thea bohea L.) with a long history. Of which CSSDHP is famous for being considered one of the most expensive teas in the world, more valuable by weight than gold (Rose, 2010; Li et al., 2021b), and CSSRG also is one of the highest-ranking oolong teas with the intensity of a spicy and cinnamon-like odor and a mellow and heavy taste (Xu et al., 2022). In our previous studies, the complete chloroplast genomes of CSSDHP (Accession number: MT773374) and CSSRG (Accession number: MT773375) had been assembled and could be available in the NCBI database (Li et al., 2021b; Fan et al., 2022). Here, in addition to mitogenomes of two Chinary type teas sequencing and assembly, the mitogenome structure, intracellular sequence transfer and RNA editing events have been further analyzed and compared with mitochondrial sequences of Assamica type tea. This comparative analysis would provide a more comprehensive perspective on the complexity of the mitogenome of C. sinensis and shed light on the evolution and phylogeny relationship of C. sinensis.
The cultivars of C. sinensis var. sinensis cv. Dahongpao (CSSDHP) and C. sinensis var. sinensis cv. Rougui (CSSRG) were obtained from Wuyi Mountain, Fujian Province (27°43′42.46″N, 118°0′14.40″E) in China. Young leaves were collected, mitochondria were isolated from leaves by using density gradient centrifugation and digested with DNase I (Promega, Madison, USA) to eliminate genomic DNA contamination (JRan et al., 2010). DNA were extracted using the plant DNA extraction kits (TransGene, Beijing, China), and the final DNA quality was detected by a NanoDrop spectrophotometer (Thermo Scientific, Carls-bad, CA, USA). DNA samples were preserved at −80°C at the Key Laboratory of Tea germplasm Genetic Resources of Wuyi University.
To obtain the full-length mitogenome sequence, short-read (Illumina) and long-read (Nanopore) sequencing technologies were used. The short raw reads were checked with FastQC v0.12.1 and trimmed by Trimmomatic v0.36. The long raw reads were base-called by using Albacore v2.1.7 (mean_qscore > 7) with barcode demultiplexing, and converted to fasta format with Samtools Fasta (http://www.htslib.org/doc/samtools.html). Two strategies were used to assemble the mitogenome. In the first strategy, the short clean reads were de novo assembled with GetOrganelle v1.6.4, potential mitochondrial contigs were extracted by aligning against the mitochondrial protein-coding genes from plant mitogenome database (ftp://ftp.ncbi.nlm.nih.gov/refseq/release/mitochondrion/) with BLAST v2.8.1+ (Dierckxsens et al., 2017). Then, the putative long mitochondrial reads were baited by mapping the Nanopore long reads to the potential mitochondrial contigs using BLASR v5.1 and assembled by Canu v2.1.1. In the second strategy, all Nanopore long reads were assembled de novo by using Canu directly (Koren et al., 2017). Subsequently, BWA were used to map the short clean reads to the draft contigs and improved the draft contigs with Pilon v1.22. Then, Bandage (Wick et al., 2015) was used to check whether these contigs were circular. Finally, the corrected contigs obtained from the above two assembly strategies were aligned with each other using MUMmer v3.23, and the result showed that these two contigs were identical. Examine the aligned bam files using IGV to verify the results of the assembly (Robinson et al., 2011). Based on the above assembly steps, two mitogenomes, CSSDHP and CSSRG were obtained.
Protein-coding genes and Ribosomal RNA (rRNA) were annotated by their similarity to published plant mitochondrial sequences and by using BLAST searches. The tRNA genes were annotated using tRNAscanSE (http://lowelab.ucsc.edu/tRNAscan-SE/) (Jackman et al., 2020). The position of each coding gene was determined using BLAST searches against reference mitogenome genes (OL989850 and OM809792). ORFs were predicted by ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/) with the standard genetic code and a minimal length of 102 nt, and ORFs longer than 300 bp were annotated by Blast2GO with default parameters (Conesa et al., 2005). Manual corrections of genes for start/stop codons and for intron/exon boundaries were performed in SnapGene Viewer by referencing the reference mitogenomes (OL989850 and OM809792). The mitogenome maps were drawn using the OGDRAW tool (Greiner et al., 2019).
These two complete mitogenome sequences and accompanying gene annotations had been deposited in the NCBI GenBank (Accession numbers: PP212895, CSSDHP; PP212896, CSSRG). Four reported C. sinensis mitogenomic data, including CSAOL (Accession number: OL989850), CSAOM (Accession number: OM809792), CSAMK (Accession number: MK574876 and MK574877, assembled into two rings) and CSAMH (Accession number: MH376284), was downloaded from the NCBI website. Incorrect annotation information was checked and corrected.
The BLASTN program was used for pairwise comparison of six mitogenomes of C. sinensis. Interspecies homologous regions were searched to indicate mitogenomic synteny among six C.sinensis species, fragments shorter than 100 bp was excluded from the analysis. Homologous sequences with a length of more than 500 bp were retained as conserved collinear blocks and then were collinearly visualized using AliTV 1.0.6 (https://alitvteam.github.io/AliTV/d3/AliTV.html) (Zhang et al., 2013).
The dispersed long sequence repeat across the mitogenome (Forward, Palindromic, Reverse and Complement) was detected by REPuter program (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) with the minimum repeating size set to 30 and the hamming distance set to 3 (Kurtz et al., 2001). Simple sequence repeats (SSRs) were detected by MISA program (https://webblast.ipk-gatersleben.de/misa/) with default parameters (Beier et al., 2017).
Homologous DNA fragments were discovered between the chloroplast genome and mitogenome by BLASTN (ncbi-blast-2.2.30+) with 70% identity as the threshold and the e-value of 1e-6 (Chen et al., 2015). To eliminate redundant detection, only a single IR (Inverted repeat) region of the chloroplast genome were used for analysis, and fragments with overlapping locations were combined into unique fragments. The reported chloroplast genomes of CSSDHP, CSSRG, CSAOL and CSAMK was downloaded from NCBI with accession numbers MT773374 and MT773375, OL450397and MH019307, respectively.
RNA editing events were predicted based on the online website PREPACT v3.12.0 (http://www.prepact.de/) (Lenz et al., 2018), and the setting standard are: cutoff value = 0.001. To avoid data bias, the protein-coding genes with lengths < 300 bp were excluded in codon usage calculations (Rosenberg et al., 2003). The relative synonymous codon usage (RSCU) values, the effective number of codons (ENc) value and the contents of GC at the first, second and third positions of each codon of each gene were evaluated by cusp (EMBOSS v6.6.0.0) (http://emboss.toulouse.inra.fr/cgi-bin/emboss/help/cusp). RSCU = 1 indicated that there was no preference for the use of this codon, and RSCU > 1 indicates that the codon was used preferentially by amino acids, while if RSCU < 1, the codon usage is contrary. Parity rule 2 (PR2) plot analysis were also conducted to investigate codon usage bias based on a plot of AT-bias [A3/(A3 + T3)] and GC-bias [G3/(G3 + C3)] at the third codon position of the four-codon amino acids in entire genes (Huang et al., 2022). The ENc-plot of ENc values plotted against GC3s values (ENc vs GC3s) was used to analyze the influence of base composition on the codon usage in a genome. If the points lie on or around the standard curve, the codon usage was constrained only by mutation bias. Otherwise, the codon usage pattern is influenced by other factors, such as natural selection (Wright, 1990). Ka/Ks value for each gene was calculated using the KaKs_Calculator 2.0. Ka/Ks >1 means positive selection, Ka/Ks =1 mean neutral selection, and Ka/Ks <1 mean negative selection (Wang et al., 2010). The heatmap of Ka/Ks values was drawn using ChiPlot (https://www.chiplot.online/), with a clustering method of complete and euclidean distance.
A total of 20 plant mitogenomes were downloaded from NCBI and were used to construct phylogenetic trees (Supplementary Table S10). The mitogenomes of Zea mays, Sorghum bicolor, Triticum aestivum, and Ginkgo biloba were employed as outgroups. First, MUMmer v3.23 and BLAT software were used for global and local alignment between the sample sequence and the reference genome (CSSDHP) under default parameters, and the alignment was trimmed by Gblocks_0.91b to remove low-quality regions with the parameters: -t=d -b4 = 5 -b5=h. A total of 573 SNPs was found. For each mitogenome, all SNPs were connected in the same order to obtain sequences of the same length for the construction of phylogenetic trees. Then, the genome-wide phylogenetic trees were constructed by both Maximum-likelihood (ML) and Bayesian inference (BI) methods.
The ML method was performed using PhyML v3.0 (Guindon et al., 2010). Nucleotide substitution model selection was estimated with jModelTest 2.1.10 and the Smart Model Selection feature in PhyML v3.0. The model GTR+I+G was selected for ML analyses with 1,000 bootstrap replicates for calculating the bootstrap values (BS) of the topology. MrBayes v3.2.6 (Ronquist et al., 2012) was used in BI analysis. Four chains (three heated and one cold) and two runs of 2 million generations were carried out, with each run being sampled every 100 generations. The first 10% of samples was discarded as burn-in, and the remaining trees were used to estimate Bayesian posterior probabilities (BPPs). The phylogenetic tree was visualized with iTOL v6 (Letunic and Bork, 2021).
The complete circular mitogenomes of C. sinensis var. sinensis cv. Dahongpao (CSSDHP) and C. sinensis var. sinensis cv. Rougui (CSSRG) were obtained using a combination of Illumina and Nanopore data. A total of 60,413,998 Illumina reads (about 9.1 Gb, average read length 150 bp) and 1,008,363 Nanopore reads (about 5.1 Gb, average read length 5,032 bp) were mapped to the complete mitogenome of CSSDHP. The average coverage reached 8,361× and 4,689× sequencing depth. A total of 55,973,904 Illumina reads (about 8.4 Gb, average read length 150 bp) and 652,559 Nanopore reads (about 4.9 Gb, average read length 7,608 bp) were mapped to the complete mitogenome of CSSRG. The average coverage, with 45.41× for CSSDHP and 87.61× for CSSRG, was 99.95% and 100%, respectively (Supplementary Tables S1; Supplementary Figures S1, S2). The assembly using error-corrected Nanopore reads resulted in circular genomes of 1,082,025 bp for CSSDHP and 991,788 bp for CSSRG. A total of 79 genes were identified in the mitogenome of CSSDHP, including 46 protein-coding genes (PCGs), 30 tRNA genes, and 3 rRNA genes. A total of 87 genes were identified in the mitogenome of CSSRG, including 47 PCGs, 37 tRNA genes, and 3 rRNA genes (Figure 1). When compared with the four reported C. sinensis mitogenomes, the GC content of all six mitogenomes was between 45% and 46%. However, the annotated genes and the number of genes differed significantly across the six mitogenomes (Table 1).
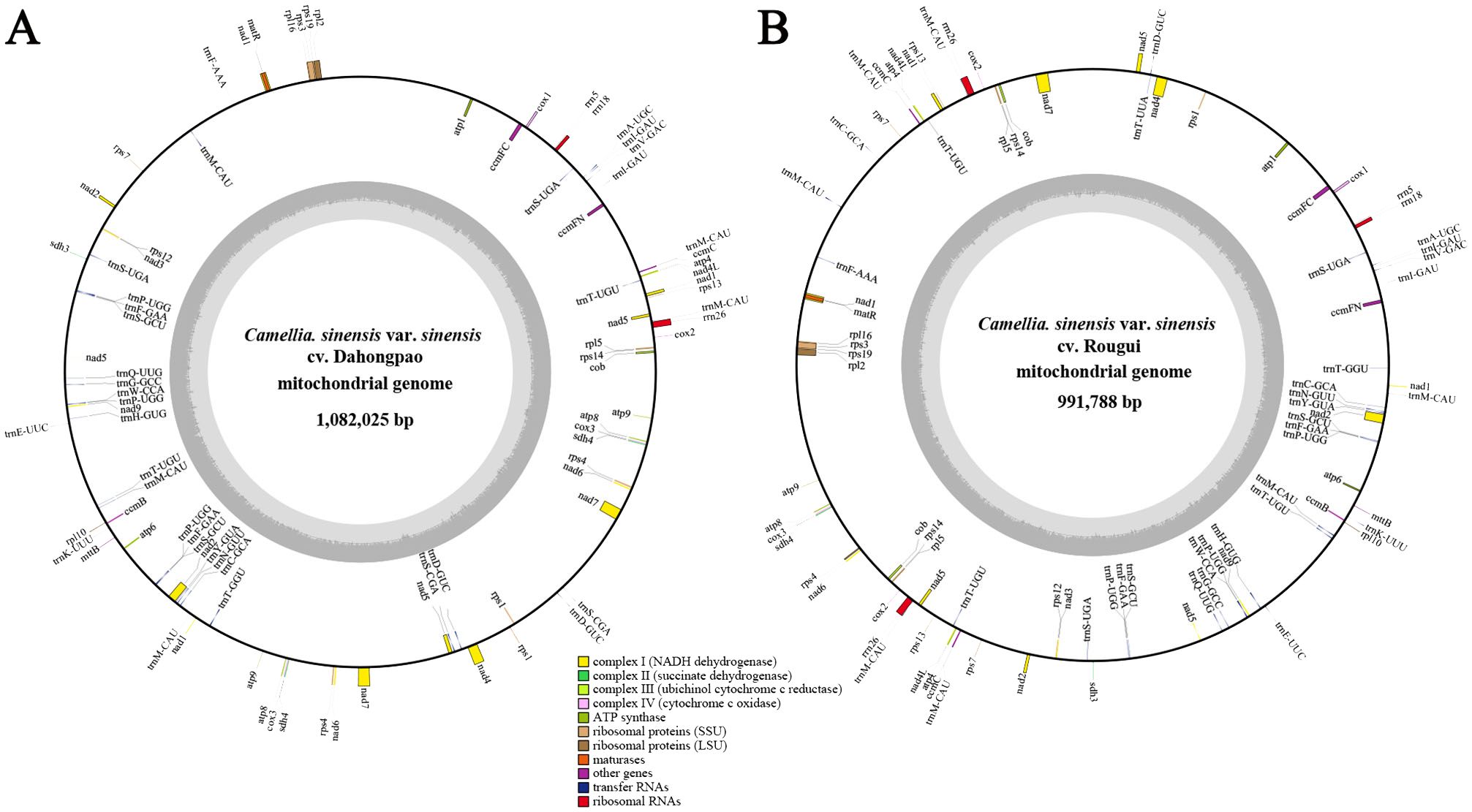
Figure 1. C. sinensis var. sinensis cv. Dahongpao (A) and C. sinensis var. sinensis cv. Rougui (B) mitochondrial genome circular map.
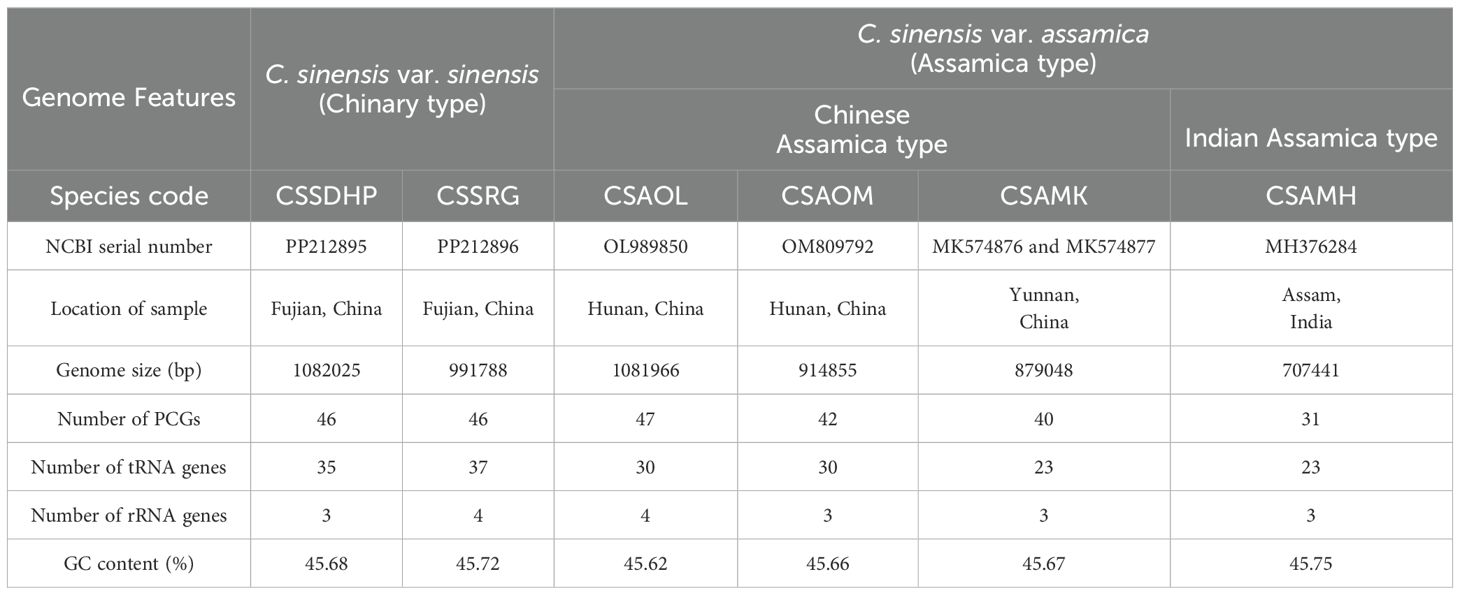
Table 1. General features of six C. sinensis mitogenomes.
CSSDHP, CSSRG, CSAOL, CSAOM and CSAMK have the complete mitogenomes while CSAMH is missing seven PCGs. A total of 46 (CSSDHP), 46 (CSSRG), 47 (CSAOL), 42 (CSAOM), 40 (CSAMK) and 31 (CSAMH) PCGs were annotated in six C. sinensis mitogenomes, respectively. Atp8, Atp9, Nad6, Nad7, Cox3, Rps1, Rps4 and Sdh4 were replicated in CSSDHP mitogenome, Atp9, Rpl2, Rpl16, Rps3 and Rps19 were replicated in CSSRG mitogenome, Atp9 (×3), Rpl2, Rpl16, Rps3 (×3) and Rps19 (×4) were replicated in CSAOL mitogenome, Atp4, Nad4L, CcmC and Rps19 were replicated in CSAOM mitogenome, Sdh3 and Rps19 were replicated in CSAMK mitogenome, and Rps19 were replicated in CSAMH mitogenome (Table 1; Supplementary Table S2). In addition, trnM-CAT(x5), trnT-TGT, trnI-GAT, trnS-TGA, trnP-TGG(x3), trnF-GAA, trnS-GCT, trnS-CGA and trnD-GTC were replicated in CSSDHP mitogenome, Rna26, trnM-CAT(x7), trnT-TGT (x3), trnI-GAT, trnS-TGA, trnP-TGG(x3), trnF-GAA, trnS-GCT and trnC-GCA were replicated in CSSRG mitogenome, Rrna18, ttrnM-CAT(x4), trnI-GAT, trnS-TGA(x3), trnP-TGG, trnS-GCT, trnN-GTT and trnC-GCA were replicated in CSAOL mitogenome, trnM-CAT (x5), trnI-GAT, trnS-TGA, trnP-TGG, trnS-GCT and trnC-GCA were replicated in CSAOM mitogenome, trnM-CAT (x4), trnP-TGG (x3), trnF-GAA, trnE-TTC, trnY-GTA and trnC-GCA were replicated in CSAMK mitogenome, and trnM-CAT(x4), trnI-GAT, trnS-TGA (x3), trnS-GCT and trnN-GTT were replicated in CSAMH mitogenome. In six C. sinensis mitogenomes, Nad4L, CcmFC, Rpl2, Rps3, trnT-TGT, trnI-GAT, trnA-TGC, trnS-TGA and trnT-GGT has one intron, Nad4 has two introns, Nad1, Nad2, Nad5 and Nad7 have four introns. One of the two Sdh3s in CSAMK mitogenome has one intron (Supplementary Table S2).
To assess the relationship between the six mitogenomes of C. sinensis species, the BLASTN program was performed to compare homologous genes and their sequence arrangement. The conserved collinearity blocks that were over 500 bp in length were identified for analysis. The results showed that while many homologous collinear blocks were detected in the six mitogenomes of C. sinensis species, the order of arrangement of collinear blocks among six mitogenomes exhibited inconsistency. The collinear cluster analysis showed that six C. sinensis mitogenomes were classified according to the degree of collinearity. CSSDHP and CSSRG, both from Fujian Province, China, are clustered together; CSAOL and CSAOM, both from Hunan Province, China, are clustered together; and CSAMK from Yunnan Province, China, are clustered together with CSAMH from Assam, India (Figure 2).
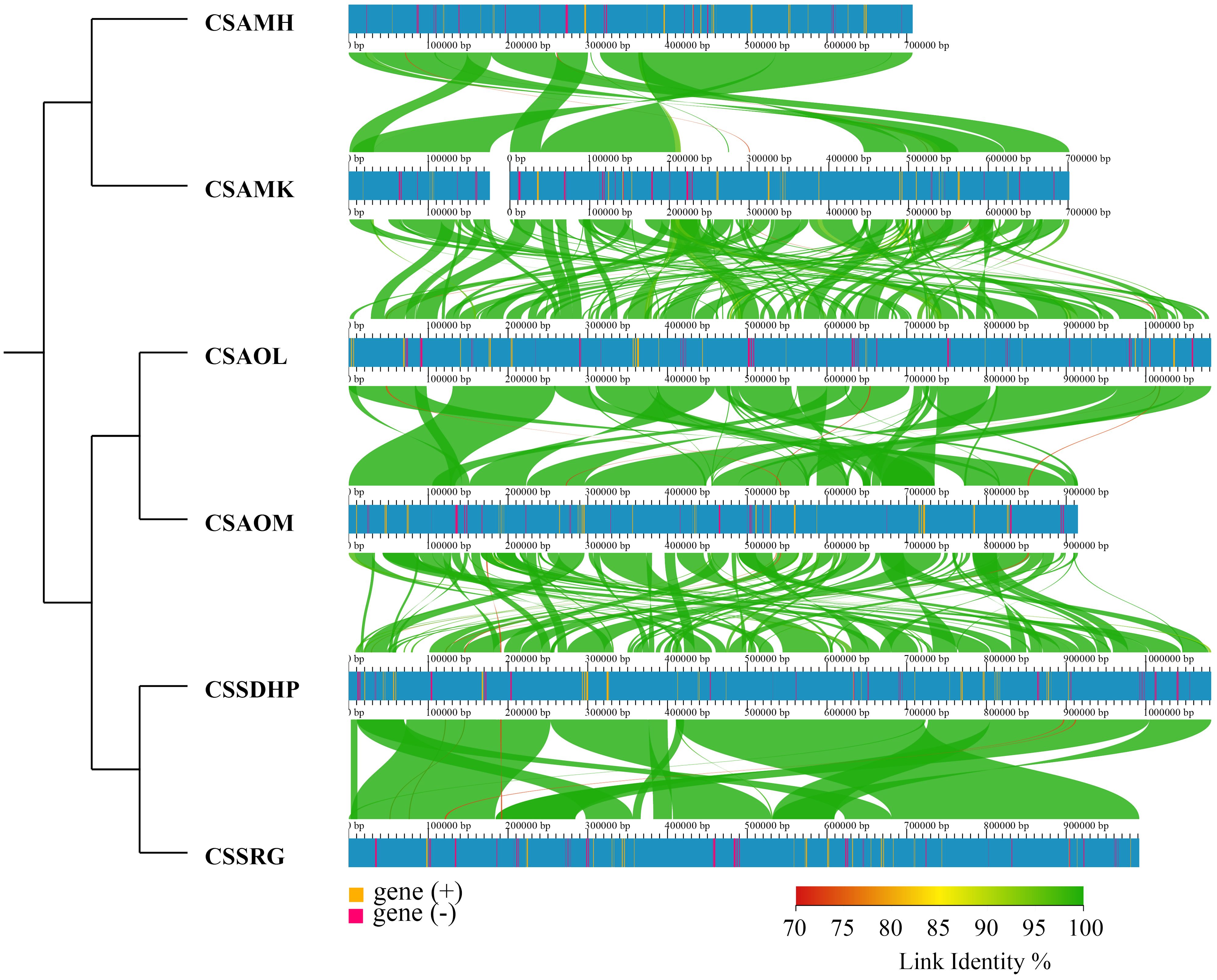
Figure 2. Collinear analysis of 6 C. sinensis species.
A large number of repetitive sequences were found in the mitogenome of tea plant. There were 828 pairs of dispersed long repeats with a length ≥ 30 bp in the CSSDHP mitogenome, including 408 pairs of forward repeats, 411 pairs of palindromic repeats, 5 pair of reverse repeats and 4 pair of complementary repeats. There were 686 pairs of dispersed long repeats with a length ≥ 30 bp in the CSSRG mitogenome, including 344 pairs of forward repeats, 339 pairs of palindromic repeats, 1 pairs of reverse repeats and 2 pairs of complementary repeats. When compared with the four reported CSA species, the number of long repeats was 828 (CSSDHP), 686 (CSSRG), 769 (CSSOL), 542 (CSSOM), 462 (CSSMK) and 349 (CSSMH), respectively. The number of the long repetitive sequences was proportional to the total length of the mitogenome (Supplementary Table S3; Figure 3A).
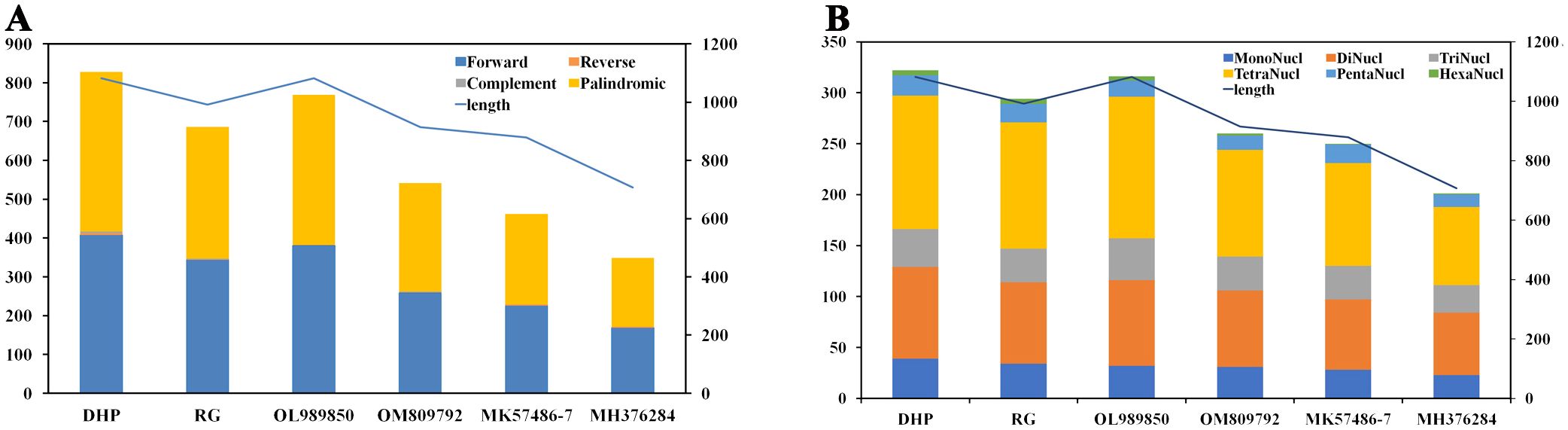
Figure 3. C. sinensis mitochondrial repeats. (A) The dispersed long sequence repeats. (B) The simple sequence repeats. Histograms display the repeat number of given lengths and the curve show the size of the mitochondrial genome.
A total of 322 and 294 SSRs were detected in CSSDHP and CSSRG mitogenomes, respectively. The number of SSRs in four reported CSA mitogenomes was 316 (CSAOL), 260 (CSAOM), 250 (CSAMK) and 201 (CSAMH), respectively. SSR tetra-nucleotide was the most abundant SSR type. The number of SSRs was proportional to the total length of the mitogenome (Supplementary Table S4; Figure 3B).
The global alignment between the chloroplast genome and mitogenome for each of 4 C. sinensis species showed a total of 43 (20,733 bp), 46 (21,607 bp), 40 (21,701 bp) and 46 (21,496 bp) mitogenomic DNA fragments homologous to the chloroplast genome, accounting for 1.91%, 2.18%, 2.01% and 2.45% of the length of the whole mitogenome in CSSDHP, CSSRG, CSAOL and CSAMK, respectively (Figure 4). The majority of homologous fragments for each species occurred in the size range of 100 - 500 bp. It was noticed that most homologous regions involved repeat sequences, including SSR sequences or Long repeat sequences (especially the IR region), which account for 90.4% (18,746 bp, CSSDHP), 91.9% (19,849 bp, CSSRG), 92.8% (20,136 bp, CSAOL), and 88.7% (19,058 bp, CSAMK) of the total homologous sequence length, respectively. Among the four mitogenomes, the longest segments homologous to the chloroplast genome were 7,722 bp (CSSDHP), 7,721 bp (CSSRG), 9572 bp (CSAOL), and 6661 bp (CSAMK), all of which were in the IR region of their respective chloroplast genomes. In addition, within these homologous mitogenomic DNA, there were ten complete genes, all of which are tRNA genes (trnH-GUG, trnD-GUC, trnM-CAU, trnW-CCA trnP-UGG, trnI-CAU, trnI-GAU, trnA-UGC, trnV-GAC and trnN-GUU) (Supplementary Table S5).
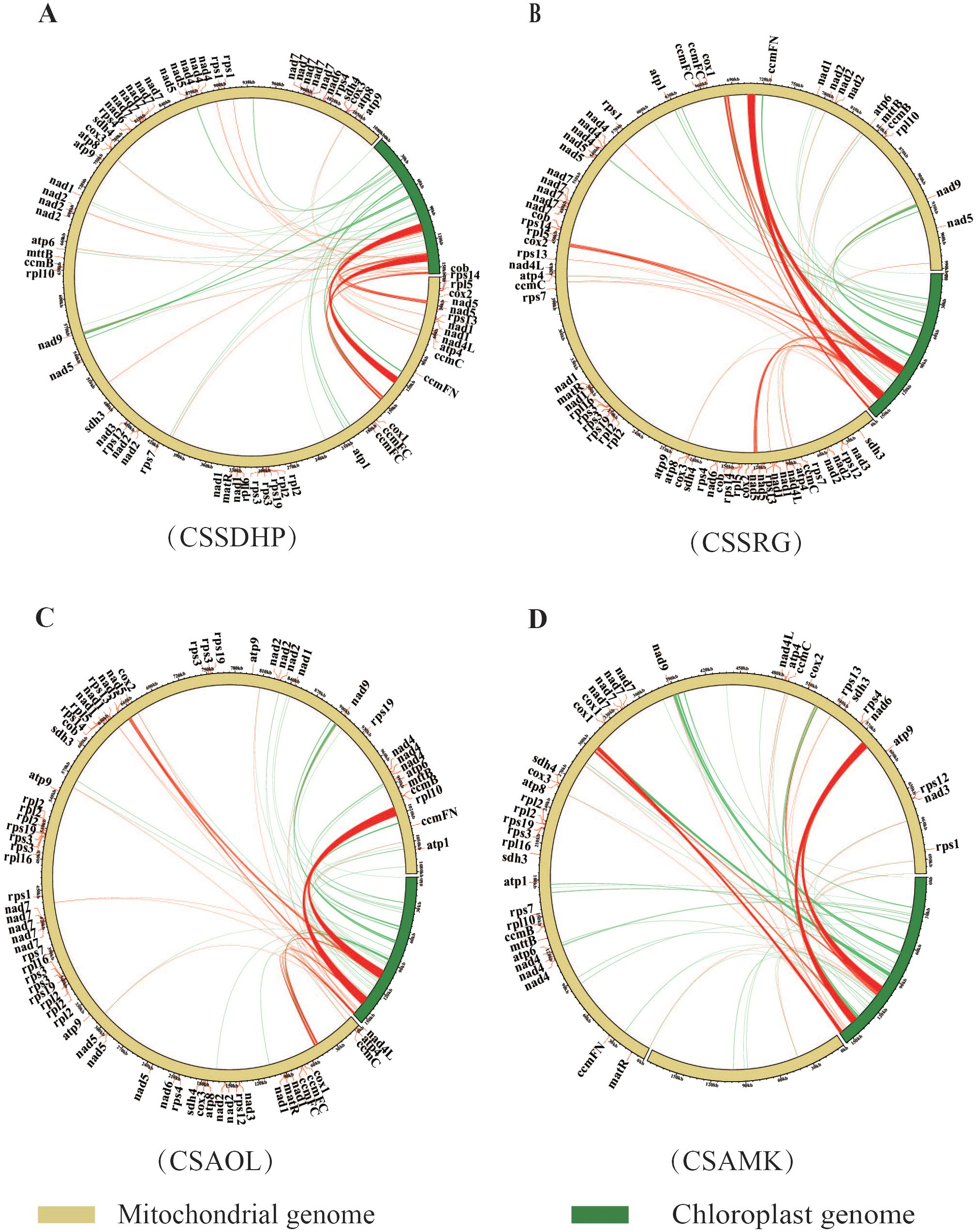
Figure 4. Schematic for the chloroplast-to-mitochondrial gene transfer. Sequence similarity between the mitochondrial and chloroplast genomes in (A) CSSDHP, (B) CSSRG, (C) CSAOL and (D) CSAMK. Homologous sequences connected by red lines are repetitive sequences, and non-repetitive sequences are connected by green lines.
RNA editing site were predicted in six mitogenomes of tea plant (Supplementary Table S6). There were a total of 720 (CSSDHP), 718 (CSSRG), 679 (CSAOL), 710 (CSAOM), 701 (CSAMK), and 546 (CSAMH) RNA editing sites identified in 37 gene types, respectively. The results showed that there was not only U-to-C RNA editing types, but also C-to-U RNA editing types. Among them, the Nad4 gene had the most predictive RNA editing sites (51-54 sites), followed by Nad5 (34-36 sites), while Rps14 had minor editing sites (only 1 site) (Figure 5A). Among these substitutions, the most amino acid changes were serine to leucine (S-to-L), proline to leucine (P-to-L), and serine to phenylalanine (S-to-F), while stop codon to Arginine (*-to-R) were the least (Figure 5B). Most amino acid changes involved the conversion of amino acid hydrophobicity, 7.0% of the RNA editing amino acids were converted from hydrophobic to hydrophobic, 17.1% from hydrophobic to hydrophilic, 2.8% from hydrophilic to hydrophilic, 72.7% from hydrophilic to hydrophobic, and only 4% involved the conversion of termination codons. In each of the six mitogenomes, an editing event from CGA (Arginine) to UGA (Termination codon) were predicted in CcmFC and Atp9 genes. In addition to CSAMH, an editing event from UGA (Termination codon) to CGA (Arginine) was predicted in Rps19 gene. In addition to CSAOL,CSAMK, and CSAMH, two editing events from ACG (Threonine) to ATG (Initiation codon) were predicted in Cob, Cox1, Nad5, Nad4L and Cox2 genes (Supplementary Table S6). In addition, Nad1, Nad2, and Nad5 genes contained both cis-spliced and trans-spliced introns, while the CcmFC, Nad4, Nad7, Rpl2, and Rps3 genes contained only cis-spliced introns (Figure 6).
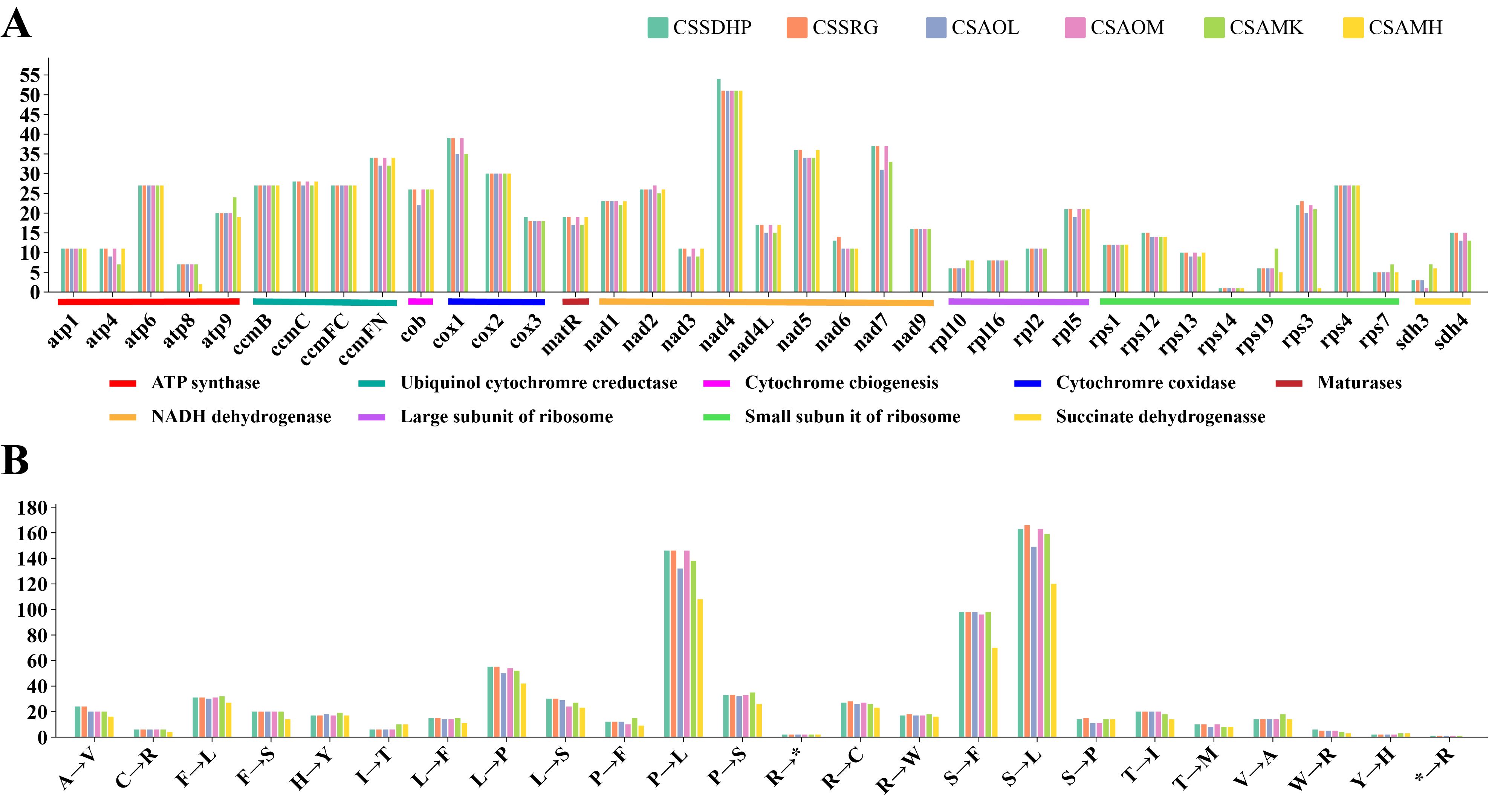
Figure 5. The Prediction of RNA Editing in six C. sinensis mitogenomes. (A) RNA editing sites in different coding genes. (B) Amino acid conversion type.
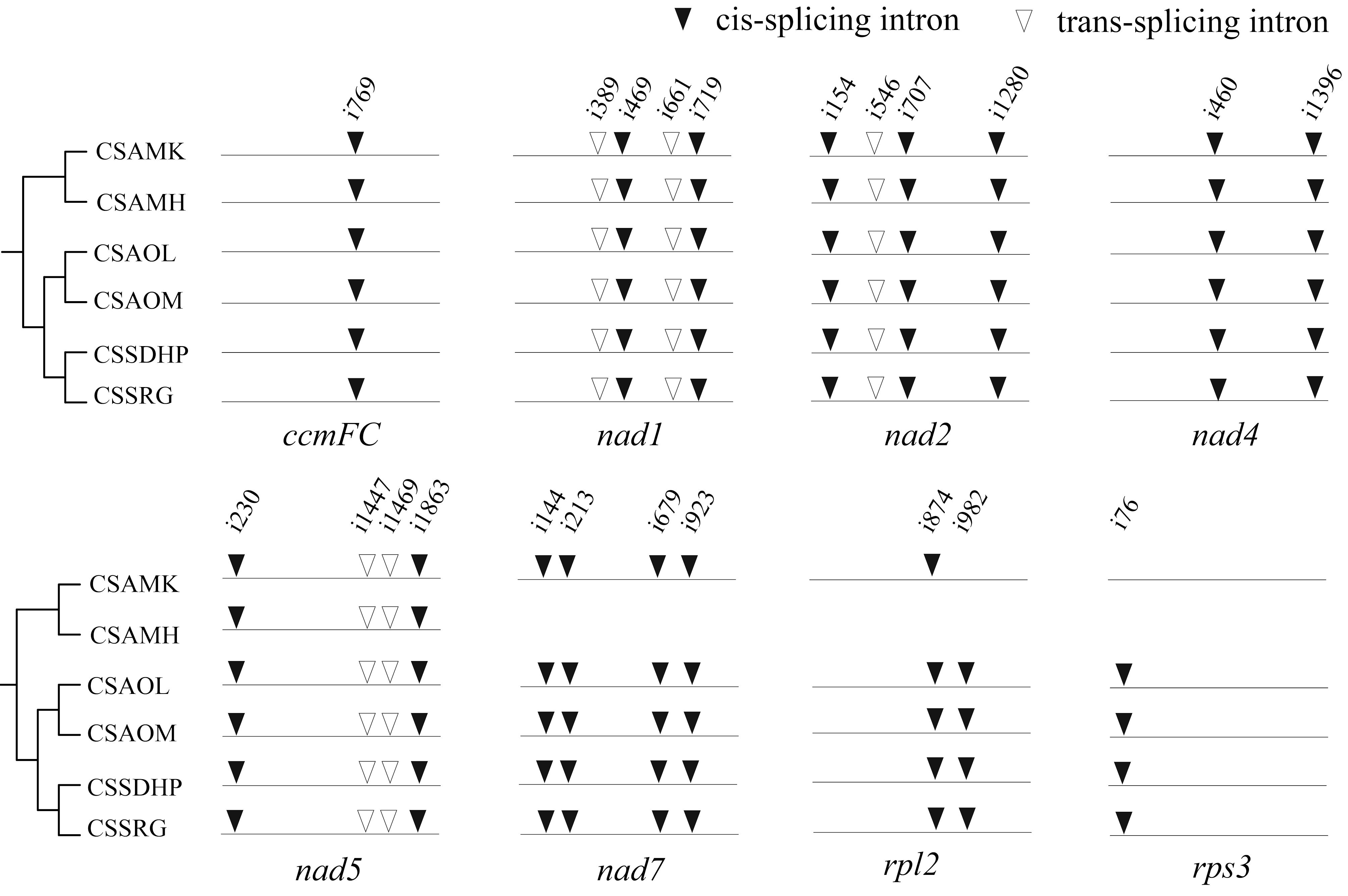
Figure 6. Comparison of mitochondrial introns among six C. sinensis. The arrowhead indicates the position of an intron insertion. Solid and hollow triangles represent cis- and trans-spliced introns, respectively.
The patterns of synonymous codon usage and the preference for G/C-ended codons were analyzed by RSCU analysis of codons. The results showed that most amino acids except methionine (AUG) and tryptophan (UGG) had bias in codon usage pattern (Figure 7). In the six mitogenomes, Alanine (Ala) had a high preference for the use of GCU, and its average RSCU value was the highest among mitogenome PCGs (RSCU: 1.5249—1.567), followed by Histidine (His) for the use of CAU (RSCU: 1.4948—1.5448), the termination codon for the use of UAA (RSCU: 1.4545—1.5429) and Tyrosine (Tyr) for the use of UAU (RSCU: 1.4961—1.5342). Tyrosine had the fewest preference for the use of UAC, with only 0.4647— 0.5039. Except for UUG, all codons with RSCU>1 end in A/T (Supplementary Table S7).

Figure 7. Relative synonymous codon usage (RSCU) of six C. sinensis mitogenomes.
The GC content was calculated for the first (GC1), second (GC2), and third (GC3) positions of the PCGs in 6 mitogenomes and results showed average GC content of these different positions (GC1, GC2, and GC3) were less than 50% (Supplementary Table S8), suggesting an AT bias. PR2 plot analysis was further conducted to assess the codon usage bias. In all six mitogenomws, most genes were distributed in the lower quadrant of the PR2-plot (Figure 8), implying that T (pyrimidines) were used more frequently than A (purines) in C. sinensis codons. ENc-plot analysis (ENc vs GC3S) was used to determine the major factors affecting codon usage bias, and the result showed only a few points lay near the curve, however, most of the genes with lower ENc values than expected values lay below the curve (Figure 9), suggesting the codon usage bias was slightly affected by the mutation pressure, but selection pressure and other factors have played an important role.
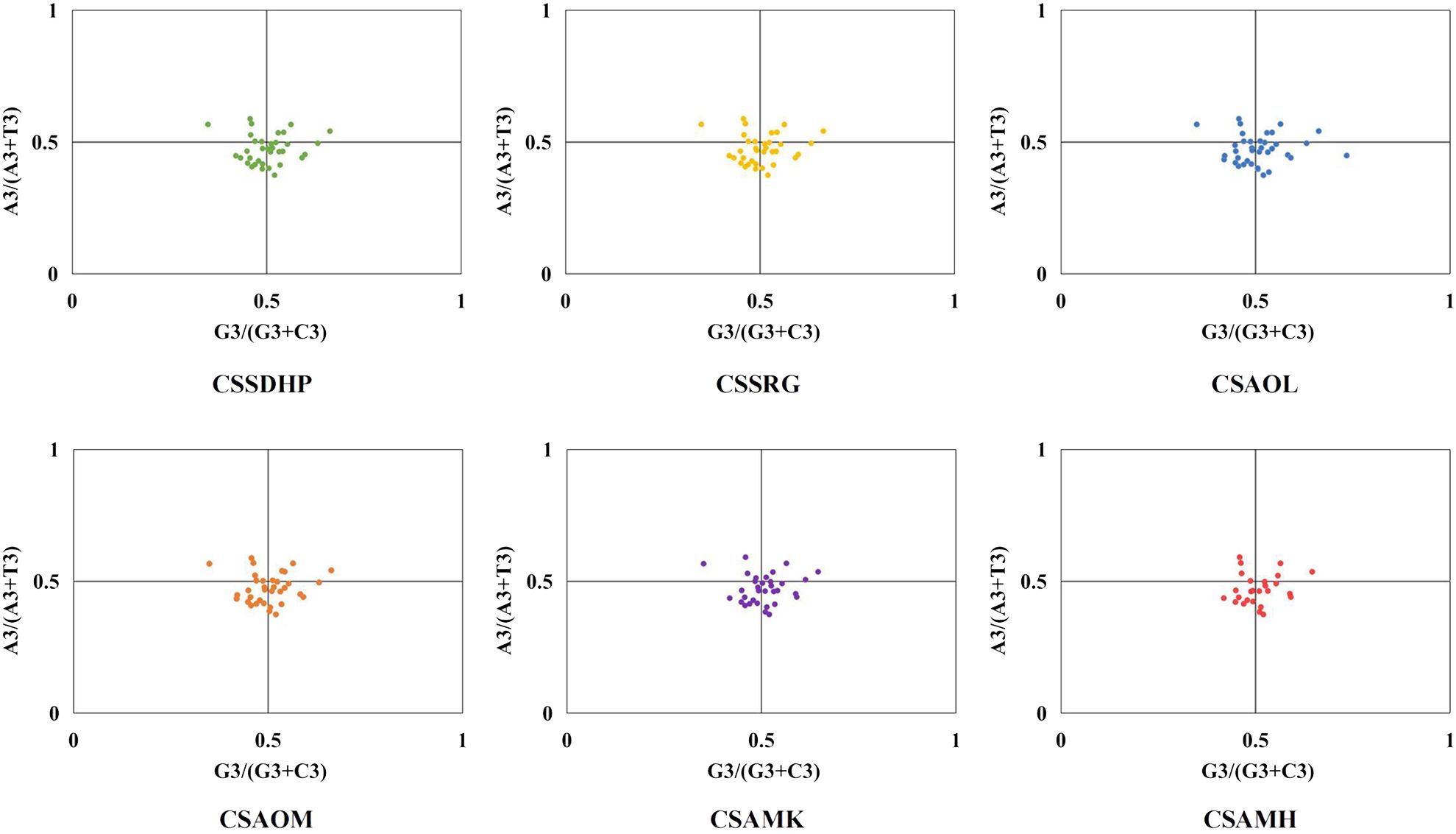
Figure 8. PR2-plot of six C. sinensis mitogenomes.
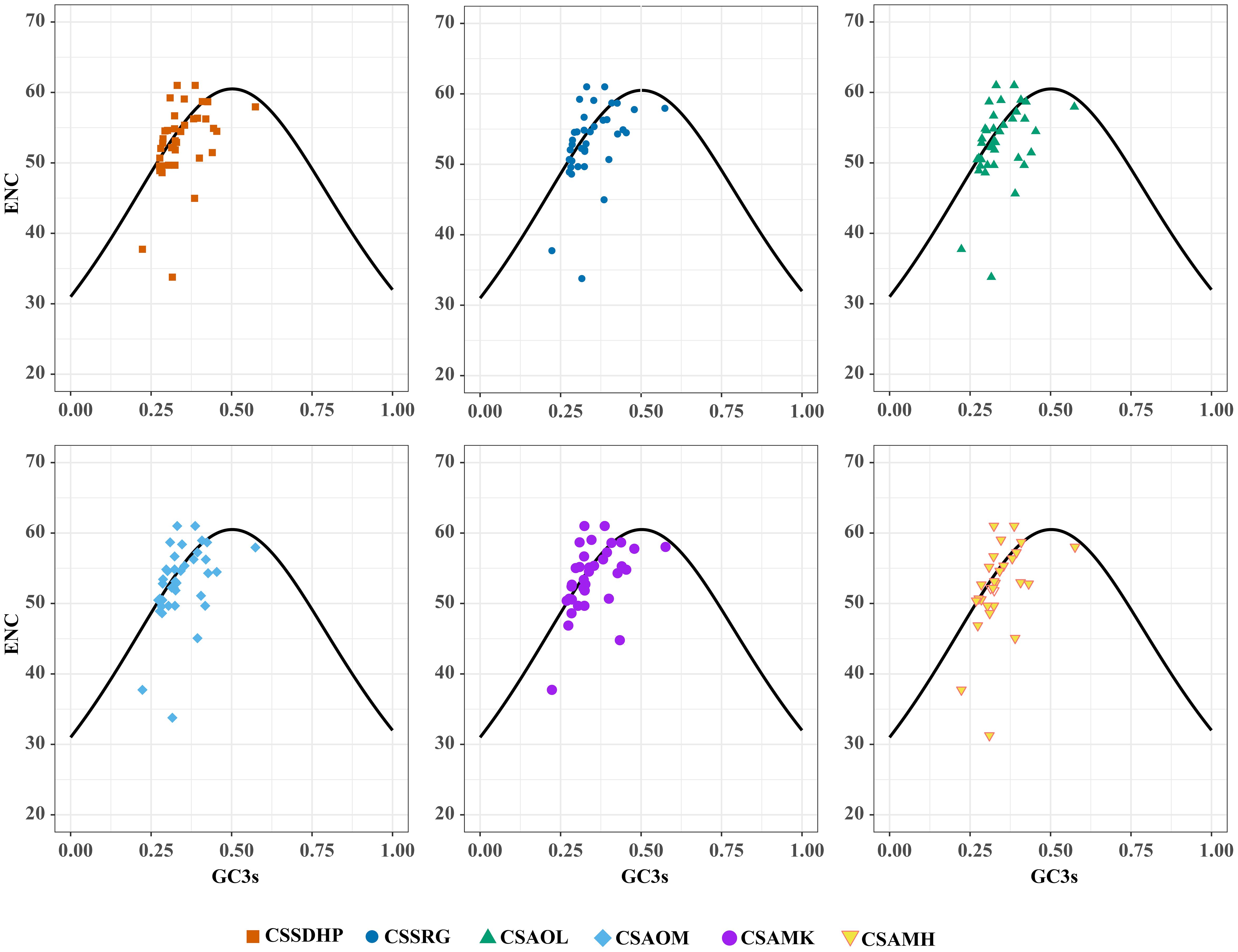
Figure 9. ENc-plot of six C. sinensis mitogenomes.
The PCGs of six mitogenomes were compared in pairs, and the Ka/Ks ratios were calculated for the shared PCGs between the pairs. The results showed that the Ka/Ks ratio of most genes were less than 0.5 or equal to 0, suggesting that those genes had undergone significant purification selection. In contrast, Ka/Ks ratios of two genes (Nad1 and Sdh3 gene) were greater than 1, indicating positive selection of these two genes in C. sinensis species. In addition, Ka/Ks ratios between two same type species (CSS vs CSS or CSA vs CSA) and two different type species (CSS vs CSA) were clearly divided into two clusters (Figure 10, Supplementary Table S9).
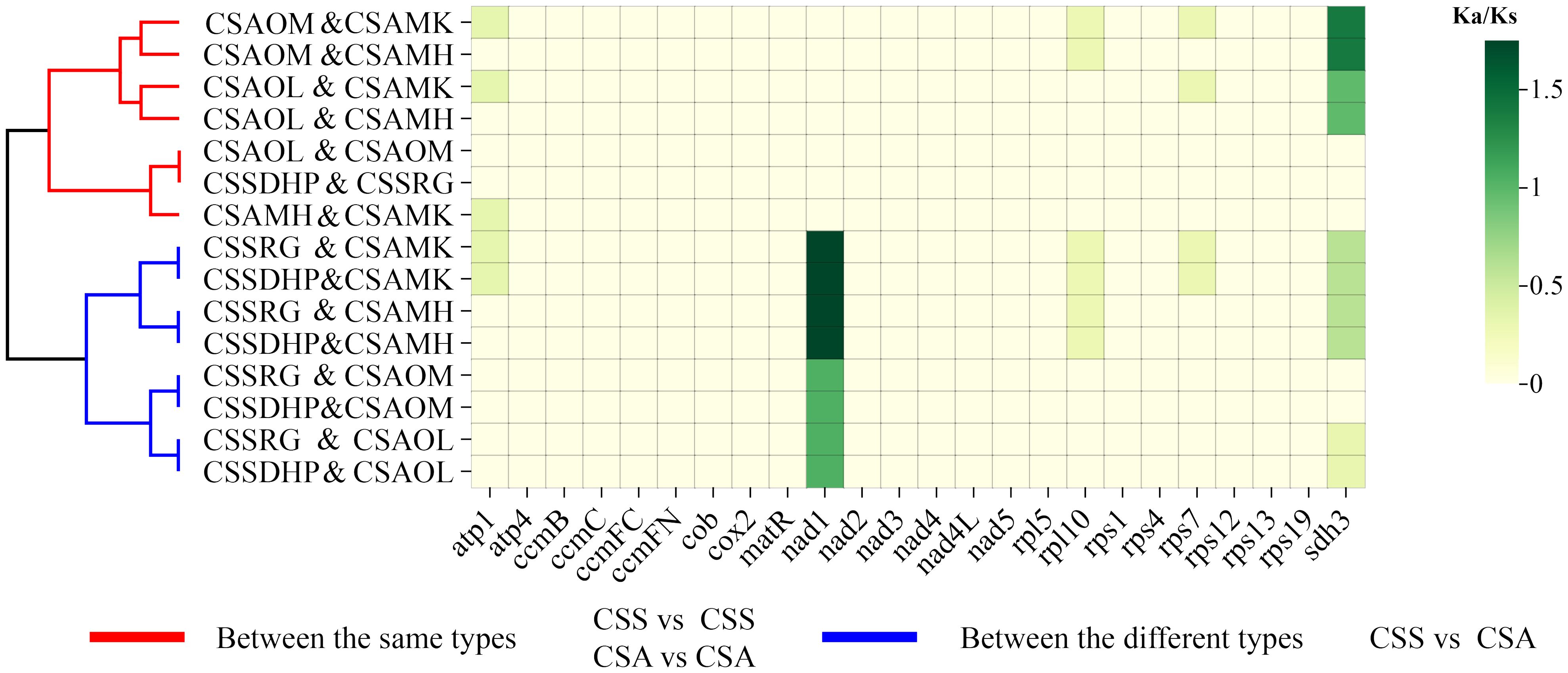
Figure 10. Analysis of Ka/Ks substitution rates among six C. sinensis mitogenomes.
To better understand the evolution of C. sinensis mitogenome, the phylogenetic trees were generated based on six C. sinensis mitogenomes and nineteen other published plant mitogenomes through a combination of the maximum likelihood method and the Bayesian method (Figure 11). The results of the classification of phylogenetic trees by the two methods were consistent with each other. Phylogenetic tree results showed that gymnosperms and dicotyledonous plants were different from monocotyledonous plants and dicotyledonous plants respectively, and the clustering of phylogenetic trees matched the family and genus relationships of these species, and Bootstrap analysis showed that all nodes had more than 80% support (BS) and 99% support (BPPs). Among 20 nodes, 17 nodes had a bootstrap value of more than 90% (BS) and BPPs =100%, which confirmed the credibility of the clustering based on mitogenome.
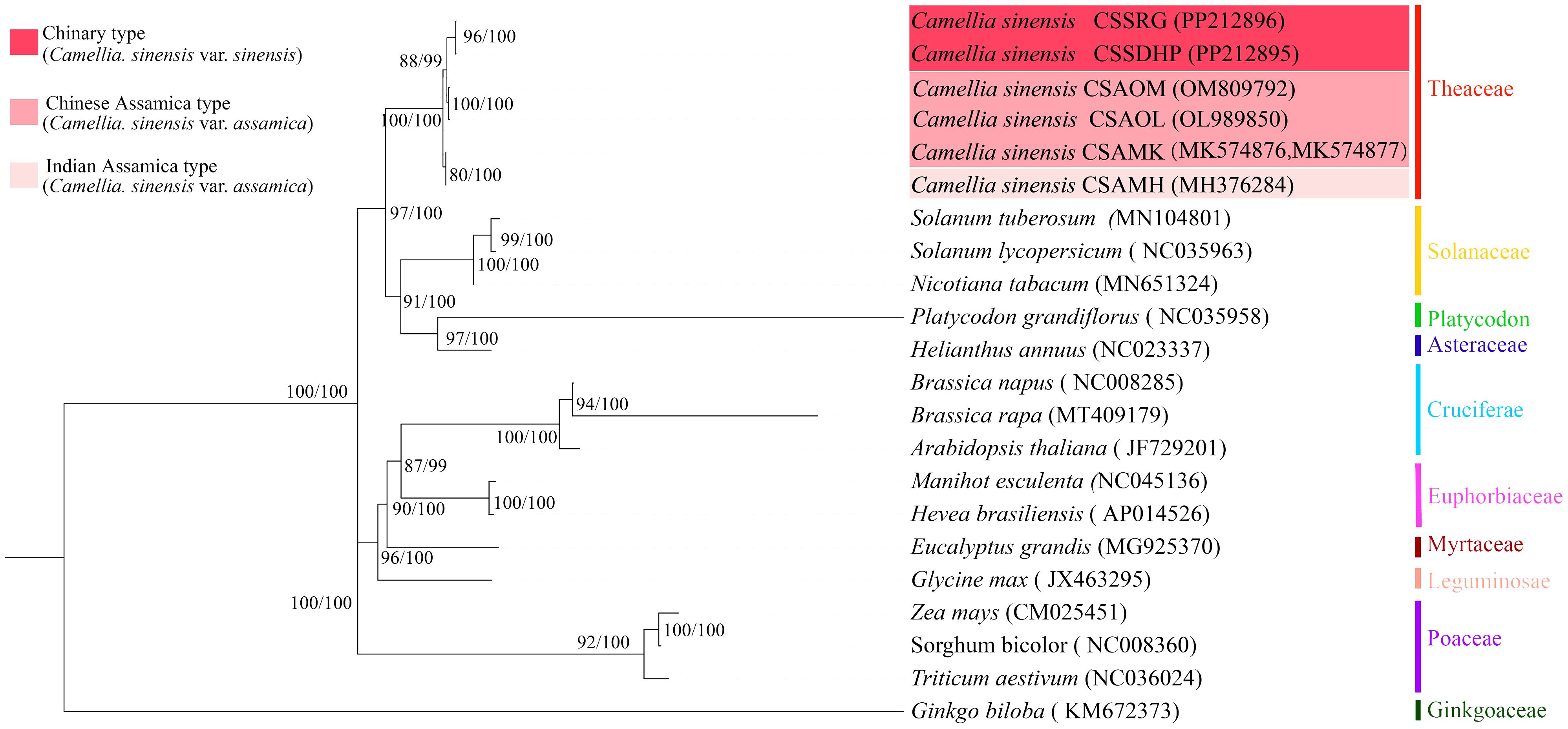
Figure 11. Maximum likelihook (ML) and Bayesian inference (BI) tree based on 20 species. Zea mays, Sorghum bicolor, Triticum aestivum, and Ginkgo biloba were classified as outgroups. Numbers beside nodes indicate bootstrap support values and Bayesian posterior probabilities.
The phylogenetic tree showed that six C. sinensis mitogenomes were clustered together and shared a common ancestor. Their ancestors evolved into two clades, one diverging into CSAMK and CSAMH, while the other continued to diverge into CSSDHP and CSSRG, CSAOL and CSAOM, respectively. The topological structure was consistent with collinear cluster (Figure 2) and intron cluster results (Figure 6).
In order to better understand the evolutionary characteristics of C. sinensis mitogenome, two CSS (C. sinensis var. sinensis cv. Dahongpao, CSSDHP and C. sinensis var. sinensis cv. Rougui, CSSRG) mitogenomes were high quality gap-free assembled based on a hybrid strategy combining Illumina and Nanopore long sequencing reads, data, and perform comprehensive comparisons with four reported CSA mitogenomes (CSAOL, CSAOM, CSAMK and CSAMH) in terms of their structure, gene content, synteny, intercellular gene transfer, and RNA editing. Mitogenome comparison showed a huge heterogeneity among the six C. sinensis species. Complex plant mitogenomes can have circular, branched, linear, or mixed forms of genomic structure (Sloan, 2013; Lai et al., 2022). Of the six genomes, five consisted of a single circular structure, whereas CSAMK mitogenome was unique and consisted of a double circular structure (Zhang et al., 2019). Except for a few transfer RNA (tRNA) genes, the gene content of five mitogenomes was consistent, while CSAMH mitogenome (Rawal et al., 2020) was missing nine genes. The loss of some genes in the mitochondrial genome during evolution had been reported in previous studies (Notsu et al., 2002; Handa, 2003), suggesting that CSAMH might have lost these genes during evolution. However, this could also be due to defects in the early sequencing assembly technology, leading to incomplete assembly. In addition, several genes were found to have copies, the number of copies was not the same in six C. sinensis mitogenomes, and the number of introns contained in the genes also varied (Supplementary Table S2). In order to better adapt to various environments, multiple copies of functional genes may appear during genome evolution (Liu et al., 2020). Therefore, these multiple copies of functional genes may confer greater stress resistance on tea plants. For example: in this study, compared to the large-leaf tea plant CSAMK, the more cold-tolerant and drought-tolerant small-leaf tea plants CSSDHP and CSSRG have more copies of the Cytochrome coxidase and NADH dehydrogenase genes, which have been reported to be involved in the plant’s defense against stressors such as drought and low temperatures (Liu et al., 2008; Møller et al., 2021; Wang et al., 2022).
Collinearity analysis revealed that six C. sinensis mitogenomes had undergone a significant amount of rearrangement, resulting in the order of genes varies greatly, which was consistent with previous studies indicating that there is little conservation of gene order in plant mitogenomes, even among close relatives (Kubo et al., 2000; Satoh et al., 2004). Despite the variability between these mitogenomes, the length of homologous sequences among different plant mitogenomes was consistent with taxonomies, and the closely related species always shared the greatest sequences, even in the non-coding regions (Li et al., 2009). Therefore, these complex mitogenomic DNA structures could also be used to trace common ancestors among diverse species (Xu et al., 2021). In collinear cluster analysis, CSSDHP was clustered with CSSRG, CSAOL was clustered with CSAOM, and CSAMK was clustered with CSAMH, suggesting that CSSDHP and CSSRG, CSAOL and CSAOM, CSAMK and CSAMH had a closer relationship respectively. It was worth mentioning that both CSSDHP and CSSRG come from Fujian Province of China, both CSAOL and CSAOM come from Hunan Province of China, and the place where CSAMK comes from (Yunnan Province of China) and the place where CSAMH comes from (Assam State of India) had similar environmental and climatic characteristics (Willson and Clifford, 1992; Hoffmann et al., 2023). So it also implied that the variation of plant mitogenomic structure might be related to environmental adaptation.
Genomic repetitive sequences had been shown to be essential for intermolecular recombination, which was important evidence of the evolution and genetic characteristics of species (Ammiraju et al., 2007; Hu et al., 2015; Dong et al., 2018). Numerous repetitive sequences, including Long repeat and SSR sequences, were found in six C. sinensis mitogenomes, which suggests that frequent intermolecular recombination had dynamically altered the structure and conformation of the mitogenome during the evolution of C. sinensis. There were obvious differences in the quantity and types of the repetitive sequence (including Long repeats and SSR) among six C. sinensis mitogenomes, which might be caused by gene duplication or variation, as well as geographical and ecological factors (Han et al., 2022). In addition, the number of repetitive sequence in both Long repeat and SSR sequences was found to be proportional to the length of the mitogenome, suggesting that the sequence repetition were closely related to the structure and size of the mitogenome.
During the evolution of plant mitogenome, DNA transfer events have occurred frequently (Timmis et al., 2004). The most common transfer direction is from the plastome to the mitogenome, and the length and sequence similarity of the migrating fragments vary among species (Wang et al., 2012; Zhao et al., 2019). Among four C. sinensis studied species, the total length of homologous fragments between chloroplast genome and mitogenomes ranged from 20,733 bp to 21,496 bp, accounting for 1.91% to 2.45% of mitogenomes, respectively. CSSDHP has the largest mitochondrial genome size (1082,025 bp), but homologous fragment with chloroplast genome was not the longest in length (20,733 bp), suggesting that integration of DNA fragments derived from the plastome contributes to limited mitogenome expansion in size. Notably, the total length of repetitive sequences (Long repeats and SSR) accounted for 88.7%- 92.8% of the total length of homologous fragments, suggesting that sequence repetition might be important driver for intracellular gene transfer. In addition, the complete genes detected in the homologous region were all tRNA genes, and tRNA genes were also detected in the homologous with region top-ranked length, which suggested that tRNA genes were more conserved in the mitogenome than protein-coding genes that are transferred, and might be involved in the integration of DNA fragments derived from the plastome. The tRNA genes play a critical role in protein synthesis. Some studies have shown that some tRNA genes in the mitochondrial genome need to be transferred from the plastid genome to maintain the stability and functionality of the mitochondria (Cheng et al., 2021; Yang et al., 2022; Lu et al., 2023).
RNA editing is a post-transcriptional process, which is closely related to the potential molecular functions and physiological processes of mitochondria in higher plants (Sloan and Wu, 2016; He et al., 2018). In this study, the number of predictive RNA editing sites in six C. sinensis mitogenome varied from 546 to 720 sites, and both C-to-U and U-to-C types of RNA editing were observed, with more of the C-to-U type. RNA editing that occurs in the first and second positions of codon can lead to changes in the properties of amino acids that affect the function of proteins (Møller et al., 2021). The predictive RNA editing events showed 72.7% of the modifications of the codons altered the amino acids from hydrophilic to hydrophobic, which might contribute to protein stability (Yi et al., 2015). The occurrence of RNA editing would result in a diversity of initiation or termination codons in protein-coding genes (Varre et al., 2019; He et al., 2021; Takenaka, 2022). In six C. sinensis mitogenomes, some editing events related to initiation or termination codons also were predicted, including editing event from CGA to UGA in CcmFC and Atp9 genes, from UGA to CGA in Rps19 gene, and from ACG to ATG in Cob, Cox1, Nad5, Nad4L and Cox2 genes. However, whether these editing events were actually activated at the starting or ending position required further transcriptome experiments to verify. In addition, Nad4 gene had the most predictive RNA editing sites, followed by Nad5 gene. Meanwhile, unlike other intron-containing genes, Nad1, Nad2, and Nad5 genes have contained not only cis-spliced introns, but also trans-spliced introns, indicating significant editing in the NADH dehydrogenase subunit transcript.
During evolution, plant mitogenomes undergo changes in genomic structure and nucleotide composition, as well as loss and transfer of protein-coding genes and tRNA genes, which are thought to be the result of a combination of natural selection, species mutation, and genetic drift (Adams et al., 2002; Liu et al., 2014; Christensen, 2018). Codon use preference play a critical role in protein function, translation accuracy and efficiency, and conducting codon preference analysis could provide insights into these evolutionary fitness of the genome (Hershberg and Petrov, 2008; Tuller et al., 2010).
In this study, the GC content at various positions and the relative synonymous codon usage (RSCU) in six C. sinensis mitogenomes were assessed. Total GC content of all six genomes were between 45.62% - 45.75%, indicating a tendency to favor A/T bases, From the RSCU values of codons, a total of 30 codons had RSCU values > 1, of which only one codon was G-ending while the rest were A/T-ending codons, indicating that the genes had little or no bias towards the G/C ending codons in six C. sinensis mitogenomes. Further, PR2-plot analysis implied that T (pyrimidines) were used more frequently than A (purines). ENc-plot analysis showed only a few points lay near the curve, and some of the genes with lower ENc values than expected values lay below the curve, which implied that the mutational pressure was not the only factor that contributed to codon use bias, and other factors such as natural selection may play an important role in all six mitogenomes (Kumar et al., 2016). These results were similar to codon preference in the chloroplast genome of C. sinensis (Yengkhom et al., 2019), and were consistent with previous studies suggesting that dicot plants exhibit a bias towards A/T-ending codons (Mower, 2020).
In genetics, Ka/Ks ratio is significant for understanding evolutionary dynamics of protein-coding genes across similar and yet diverged species (Xie et al., 2019). A pair Ka/Ks analysis was performed for six C. sinensis species, and Ka/Ks ratios of most genes were less than 0.5, indicating that these coding genes were highly conserved and did not undergo rapid evolution during the evolution process (Bi et al., 2022). In contrast, Nad1 gene and Sdh3 gene were found to have a Ka/Ks ratio > 1 in pairwise comparisons, suggesting that the two genes had undergone positive selection in C. sinensis species. Both Nad1 and Sdh3 genes had been reported to be related to plant resistance to stress such as cold tolerance (Fuentes et al., 2011; Sickmann et al., 2011; Møller et al., 2021). CSS is a slower growing shrub with smaller leaves that can withstand cooler climates, while CSA is a fast growing shrub with larger leaves that are highly sensitive to cold weather and mainly grows in warmer tropical regions (Wei et al., 2018). Thus, the positive selection of these two genes might be related to the adaptive evolution of tea plants. Cluster analysis showed that Ka/Ks ratios between the same types (CSS vs CSS or CSA vs CSA) and between the different types (CSS vs CSA) were clearly divided into two clusters, this also further indicated CSS and CSA have undergone different adaptive selections.
In this study, the result of phylogenetic tree constructed based on six C. sinensis mitogenomes and nineteen other published plant mitogenomes was consistent with the taxonomic information of species. In phylogenetic tree, six C. sinensis clustered together, with the CSA and CSS forming two distinct branches. The topological structure was consistent with collinear clustering and intron clustering (Figures 2, 10). This suggested that the CSA and CSS have undergone different evolutionary paths under long-term selective pressures.
Plant mitochondrial gene sequences are highly conserved, so far only a few genes, such as Atp1, Atp9, Cob, Nad3, eg., have been reported for phylogenetic relationship analysis (Rawal et al., 2020; Yang et al., 2023). However, due to the insufficient genetic information of a single or a few genes, the resolution of phylogenetic relationship was often unclear. In this study, phylogenetic relationships constructed using complete mitogenome SNPs were consistent with previous phylogenetic relationships based on complete chloroplast genome (Li et al., 2021a) and nuclear genome (Zhang et al., 2021; Cheng et al., 2022; Zhang et al., 2023) with high phylogenetic resolution (BS> 80%, BPPs> 99%). Therefore, it is feasible to construct plant phylogenetic relationships using complete mitogenome SNPs as super molecular markers.
This is the most detailed comparative description of the sequence, structure, and evolutionary characteristics of C. sinensis mitogenomes to date. In this study, two mitogenomes (CSSDHP and CSSRG) were successfully sequenced and assembled, supplementing the information of C. sinensis var. sinensis (CSS) mitogenome for the first time and comparing them with the C. sinensis var. assamica (CSA) mitogenomes. The mitogenome of C. sinensis exhibited a high degree of heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing, reflecting the outcomes of adaptive evolution. The phylogenetic results are consistent with those of species classification, suggesting the validity of using complete mitogenomes to construct phylogenetic relationships. This study provided an insight into evolution and phylogeny relationship of C. sinensis mitogenome, and help to deepen the understanding of the evolution of tea plant.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PP212895, https://www.ncbi.nlm.nih.gov/genbank/, PP212896.
LL: Software, Methodology, Conceptualization, Writing – original draft. XL: Methodology, Software, Data curation, Writing – review & editing. YL: Methodology, Software, Writing – review & editing. JL: Software, Validation, Writing – review & editing. XZ: Formal analysis, Validation, Writing – review & editing. YH: Formal analysis, Validation, Writing – review & editing. JY: Formal analysis, Resources, Writing – review & editing. LF: Funding acquisition, Project administration, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Natural Science Foundation of Fujian Province (No. 2021J011135), the Scientific Research Launch Fund of Wuyi University (No. YJ201902), Collaborative education mechanism mode and practice based on new ecology of industrial College (No. XGK202001), Central Leading Local Science and Technology Development Project (No. 2021L3058).
We thank Biozeron Biotechnology Co., Ltd. (Shanghai, China) for help in genome sequencing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1396389/full#supplementary-material
Supplementary Figure S1 | Sequencing Depth and Coverage Map of CSSDHP.
Supplementary Figure S2 | Sequencing Depth and Coverage Map of CSSRG.
Mitogenome, Mitochondrial genome; PCGs, Protein-coding genes; Ka/Ks: Non-synonymous/synonymous mutation ratio; RSCU, Relative synonymous codon usage; tRNA, Transfer RNA; rRNA, Ribosomal RNA; SSRs, Simple sequence repeats; ENc, Effective number of codon; GC3s, GC content at the third synonymously variable coding position; IGT, Intracellular gene transfer; ML, Maximum likelihood; BI, Bayesian inference; BS, Bootstrap values; BPPs, Bayesian posterior probabilities.
Adams, K. L., Qiu, Y. L., Stoutemyer, M., Palmer, J. D. (2002). Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. U.S.A 99, 9905–9912. doi: 10.1073/pnas.042694899
Ammiraju, J. S. S., Zuccolo, A., Yu, Y., Song, X., Piegu, B., Chevalier, F., et al. (2007). Evolutionary dynamics of an ancient retrotransposon family provides insights into evolution of genome size in the genus Oryza. Plant J. 52, 342–351. doi: 10.1111/j.1365-313X.2007.03242.x
An, Y., Mi, X., Zhao, S., Guo, R., Xia, X., Liu, S., et al. (2020). Revealing distinctions in genetic diversity and adaptive evolution between two varieties of Camellia sinensis by whole-genome resequencing. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.603819
Beier, S., Thiel, T., Muench, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.914635
Chen, Y., Ye, W., Zhang, Y., Xu, Y. (2015). High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 43, 7762–7768. doi: 10.1093/nar/gkv784
Chen, Z., Nie, H., Grover, C. E., Wang, Y., Li, P., Wang, M., et al. (2017). Entire nucleotide sequences of Gossypium raimondii and G.arboreum mitochondrial genomes revealed A-genome species as cytoplasmic donor of the allotetraploid species. Plant Biol. 19, 484–493. doi: 10.1111/plb.12536
Cheng, L., Li, M., Han, Q., Qiao, Z., Hao, Y., Balbuena, T. S., et al. (2022). Phylogenomics resolves the phylogeny of theaceae by using low-copy and multi-copy nuclear gene makers and uncovers a fast radiation event contributing to tea plants diversity. Biology 11, 1007. doi: 10.3390/biology11071007
Cheng, Y., He, X., Priyadarshani, S. V. G. N., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genomics 22, 167. doi: 10.1186/s12864-021-07490-9
Christensen, A. C. (2018). Mitochondrial DNA repair and genome evolution. Annu. Plant Rev. 50, 11–31. doi: 10.1002/9781119312994.apr0544
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Dierckxsens, N., Mardulyn, P., Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. doi: 10.1093/nar/gkw955
Dong, S., Zhao, C., Chen, F., Liu, Y., Zhang, S., Wu, H., et al. (2018). The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genomics 19, 614. doi: 10.1186/s12864-018-4991-4
Fan, L., Li, L., Hu, Y., Huang, Y., Hong, Y., Zhang, B. (2022). Complete chloroplast genomes of five classical Wuyi tea varieties (Camellia sinensis, Synonym: Thea bohea L.), the most famous Oolong tea in China. Mitochondrial DNA Part B-Resources 7, 655–657. doi: 10.1080/23802359.2022.2062263
Fang, B., Li, J., Zhao, Q., Liang, Y., Yu, J. (2021). Assembly of the complete mitochondrial genome of Chinese plum (Prunus salicina): characterization of genome recombination and RNA editing sites. Genes 12, 1970. doi: 10.3390/genes12121970
Fuentes, D., Meneses, M., Nunes-Nesi, A., Araújo, W. L., Tapia, R., Gómez, I., et al. (2011). A deficiency in the flavoprotein of Arabidopsis mitochondrial complex II results in elevated photosynthesis and better growth in nitrogen-limiting conditions. Plant Physiol. 157, 1114–1127. doi: 10.1104/pp.111.183939
Greiner, S., Lehwark, P., Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of phyML 3.0. Systematic Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Han, F., Qu, Y., Chen, Y., Xu, L., Bi, C. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1031769
Handa, H. (2003). The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 31, 5907–5916. doi: 10.1093/nar/gkg795
He, P., Xiao, G., Liu, H., Zhang, L., Zhao, L., Tang, M., et al. (2018). Two pivotal RNA editing sites in the mitochondrial atp1 mRNA are required for ATP synthase to produce sufficient ATP for cotton fiber cell elongation. New Phytol. 218, 167–182. doi: 10.1111/nph.14999
He, Z.-S., Zhu, A., Yang, J.-B., Fan, W., Li, D.-Z. (2021). Organelle genomes and transcriptomes of Nymphaea reveal the interplay between intron splicing and RNA editing. Int. J. Mol. Sci. 22, 9842. doi: 10.3390/ijms22189842
Hershberg, R., Petrov, D. A. (2008). Selection on codon bias. Annu. Rev. Genet. 42, 287–299. doi: 10.1146/annurev.genet.42.110807.091442
Hoffmann, T. D., Kurze, E., Liao, J., Hoffmann, T., Song, C., Schwab, W. (2023). Genome-wide identification of UDP-glycosyltransferases in the tea plant (Camellia sinensis) and their biochemical and physiological functions. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1191625
Hu, J., Gui, S., Zhu, Z., Wang, X., Ke, W., Ding, Y. (2015). Genome-wide identification of SSR and SNP markers based on whole-genome re-sequencing of a Thailand wild sacred lotus (Nelumbo nucifera). PLoS One 10, e0143765. doi: 10.1371/journal.pone.0143765
Huang, X., Jiao, Y., Guo, J., Wang, Y., Chu, G., Wang, M. (2022). Analysis of codon usage patterns in Haloxylon ammodendron based on genomic and transcriptomic data. Gene, 845, 146842. doi: 10.1016/j.gene.2022.146842
Jackman, S. D., Coombe, L., Warren, R. L., Kirk, H., Trinh, E., MacLeod, T., et al. (2020). Complete mitochondrial genome of a gymnosperm, Sitka spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol. 12, 1174–1179. doi: 10.1093/gbe/evaa108
Klingenberg, M. (2008). The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta (BBA)-Biomembranes 1778, 1978–2021. doi: 10.1016/j.bbamem.2008.04.011
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 15, e1008373. doi: 10.1371/journal.pgen.1008373
Kubo, T., Nishizawa, S., Sugawara, A., Itchoda, N., Estiati, A., Mikami, T. (2000). The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNA(Cys)(GCA). Nucleic Acids Res. 28, 2571–2576. doi: 10.1093/nar/28.13.2571
Kumar, N., Bera, B. C., Greenbaum, B. D., Bhatia, S., Sood, R., Selvaraj, P., et al. (2016). Revelation of influencing factors in overall codon usage bias of equine influenza viruses. PLoS One 11, e0154376. doi: 10.1371/journal.pone.0154376
Kumarihami, H. P. C., Oh, E. U., Nesumi, A., Song, K. J. (2016). Comparative study on cross-compatibility between Camellia sinensis var. sinensis (China type) and C. sinensis var. assamica (Assam type) tea. Afr. J. Agric. Res. 11, 1092–1101. doi: 10.5897/AJAR2015.9951
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lai, C., Wang, J., Kan, S., Zhang, S., Li, P., Reeve, W. G., et al. (2022). Comp arative analysis of mitochondrial genomes of Broussonetia spp. (Moraceae) reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1052151
Lenz, H., Hein, A., Knoop, V. (2018). Plant organelle RNA editing and its specificity factors: enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinf. 19, 255. doi: 10.1186/s12859-018-2244-9
Letunic, I., Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, L., Hu, Y., He, M., Zhang, B., Wu, W., Cai, P., et al. (2021a). Comparative chloroplast genomes: insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genomics 22, 138. doi: 10.1186/s12864-021-07427-2
Li, L., Hu, Y., Wu, L., Chen, R., Luo, S. (2021b). The complete chloroplast genome sequence of Camellia sinensis cv. Dahongpao: a most famous variety of Wuyi tea (Synonym: Thea bohea L.). Mitochondrial DNA Part B-Resources 6, 3–5. doi: 10.1080/23802359.2020.1844093
Li, X., Li, M., Li, W., Zhou, J., Han, Q., Lu, W., et al. (2023). Comparative Analysis of the Complete Mitochondrial Genomes of Apium graveolens and Apium leptophyllum Provide Insights into Evolution and Phylogeny Relationships. Int. J. Mol. Sci. 24, 14615. doi: 10.3390/ijms241914615
Li, J., Li, J., Ma, Y., Kou, L., Wei, J., Wang, W. (2022). The complete mitochondrial genome of okra (Abelmoschus esculentus): using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC Genomics 23, 665–678. doi: 10.1007/s00239-009-9240-7
Li, J., Tang, H., Luo, H., Tang, J., Zhong, N., Xiao, L. (2023). Complete mitochondrial genome assembly and comparison of Camellia sinensis var. Assamica cv. Duntsa. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1117002
Li, L., Wang, B., Liu, Y., Qiu, Y.-L. (2009). The complete mitochondrial genome sequence of the hornwort Megaceros aenigmaticus shows a mixed mode of conservative yet dynamic evolution in early land plant mitochondrial genomes. J. Mol. Evol. 68, 665–678. doi: 10.1007/s00239-009-9240-7
Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. (2016). The role of mitochondria in plant development and stress tolerance. Free Radical Biol. Med. 100, 238–256. doi: 10.1016/j.freeradbiomed.2016.03.033
Liu, Y., Cox, C. J., Wang, W., Goffinet, B. (2014). Mitochondrial phylogenomics of early land plants: mitigating the effects of saturation, compositional heterogeneity, and codon-usage bias. Systematic Biol. 63, 862–878. doi: 10.1093/sysbio/syu049
Liu, F., Fan, W., Yang, J. B., Xiang, C. L., Mower, J. P., Li, D. Z., et al. (2020). Episodic and guanine-cytosine-biased bursts of intragenomic and interspecific synonymous divergence in Ajugoideae (Lamiaceae) mitogenomes. New Phytol. 228, 1107–1114. doi: 10.1111/nph.16753
Liu, Y. J., Norberg, F. E., Szilágyi, A., De Paepe, R., Akerlund, H. E., Rasmusson, A. G. (2008). The mitochondrial external NADPH dehydrogenase modulates the leaf NADPH/NADP+ ratio in transgenic Nicotiana sylvestris. Plant Cell Physiol. 49, 251–263. doi: 10.1093/pcp/pcn001
Lu, G., Zhang, K., Que, Y., Li, Y. (2023). Assembly and analysis of the first complete mitochondrial genome of Punica granatum and the gene transfer from chloroplast genome. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1132551
Meegahakumbura, M. K., Wambulwa, M. C., Thapa, K. K., Li, M. M., Moller, M., Xu, J. C., et al. (2016). Indications for Three Independent Domestication Events for the Tea Plant (Camellia sinensis (L.) O. Kuntze) and New Insights into the Origin of Tea Germplasm in China and India Revealed by Nuclear Microsatellites. PLoS One 11, e0155369. doi: 10.1371/journal.pone.0155369
Møller, I. M., Rasmusson, A. G., Van Aken, O. (2021). Plant mitochondria - past, present and future. Plant J. 108, 912–959. doi: 10.1111/tpj.15495
Mondal, T. K. (2014). Breeding and biotechnology of tea and its wild species (Berlin: Springer Science & Business Media), 1–8. doi: 10.1007/978-81-322-1704-6
Mower, J. P. (2020). Variation in protein gene and intron content among land plant mitogenomes. Mitochondrion 53, 203–213. doi: 10.1016/j.mito.2020.06.002
Niu, L., Zhang, Y., Yang, C., Yang, J., Ren, W., Zhong, X., et al. (2022). Complete mitochondrial genome sequence and comparative analysis of the cultivated yellow nutsedge. Plant Genome 15, e20239. doi: 10.1002/tpg2.20239
Notsu, Y., Masood, S., Nishikawa, T., Kubo, N., Akiduki, G., Nakazono, M., et al. (2002). The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. genomics: MGG 268, 434–445. doi: 10.1007/s00438-002-0767-1
Petersen, G., Anderson, B., Braun, H.-P., Meyer, E. H., Moller, I. M. (2020). Mitochondria in parasitic plants. Mitochondrion 52, 173–182. doi: 10.1016/j.mito.2020.03.008
Ran, J. H., Gao, H., Wang, XQ. (2010). Fast evolution of the retroprocessed mitochondrial rps3 gene in Conifer II and further evidence for the Phylogeny of gymnosperms. Mol Phylogenet Evol. 54 (1), 136–149. doi: 10.1016/j.y.mpev2009.09.011
Rawal, H. C., Kumar, P. M., Bera, B., Singh, N. K., Mondal, T. K. (2020). Decoding and analysis of organelle genomes of Indian tea (Camellia assamica) for phylogenetic confirmation. Genomics 112, 659–668. doi: 10.1016/j.ygeno.2019.04.018
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rose, S. (2010). For all the tea in China: how England stole the world’s favorite drink and changed history (New York: Viking Adult), 272.
Rosenberg, M. S., Subramanian, S., Kumar, S. (2003). Patterns of transitional mutation biases within and among mammalian genomes. Mol. Biol. Evol. 20, 988–993. doi: 10.1093/molbev/msg113
Satoh, M., Kubo, T., Nishizawa, S., Estiati, A., Itchoda, N., Mikami, T. (2004). The cytoplasmic male-sterile type and normal type mitochondrial genomes of sugar beet share the same complement of genes of known function but differ in the content of expressed ORFs. Mol. Genet. Genomics 272, 247–256. doi: 10.1007/s00438-004-1058-9
Sibbald, S. J., Lawton, M., Archibald, J. M. (2021). Mitochondrial genome evolution in pelagophyte algae. Genome Biol. Evol. 13, evab018. doi: 10.1093/gbe/evab018
Sickmann, A., Rehling, P., Guiard, B., Hunte, C., Warscheid, B., der Laan, v., et al. (2011). Dual function of Sdh3 in the respiratory chain and TIM22 protein translocase of the mitochondrial inner membrane. Mol. Cell 44, 811–818. doi: 10.1016/j.molcel.2011.09.025
Sloan, D. B. (2013). One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200, 978–985. doi: 10.1111/nph.12395
Sloan, D. B., Wu, Z. (2016). Molecular evolution: the perplexing diversity of mitochondrial RNA editing systems. Curr. Biol. 26, R22–R24. doi: 10.1016/j.cub.2015.11.009
Takenaka, M. (2022). Quantification of mitochondrial RNA editing efficiency using Sanger sequencing data. Methods Mol. Biol. (Clifton N.J.) 2363, 263–278. doi: 10.1007/978
Timmis, J. N., Ayliffe, M. A., Huang, C. Y., Martin, W. (2004). Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5, 123–135. doi: 10.1038/nrg1271
Tuller, T., Waldman, Y. Y., Kupiec, M., Ruppin, E. (2010). Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. U.S.A 107, 3645–3650. doi: 10.1073/pnas.0909910107
Van de Paer, C., Bouchez, O., Besnard, G. (2018). Prospects on the evolutionary mitogenomics of plants: A case study on the olive family (Oleaceae). Mol. Ecol. Resour. 18, 407–423. doi: 10.1111/1755-0998.12742
Varre, J.-S., D'Agostino, N., Touzet, P., Gallina, S., Tamburino, R., Cantarella, C., et al. (2019). Complete sequence, multichromosomal architecture and transcriptome analysis of the Solanum tuberosum mitochondrial genome. Int. J. Mol. Sci. 20, 4788. doi: 10.3390/ijms20194788
Wambulwa, M. (2018). Preliminary investigations on the genetic relationships and origin of domestication of the tea plant (Camellia sinensis (L.) using genotyping by sequencing. Trop. Agric. Res. 29, 229–240. doi: 10.4038/tar.v29i3.8263. M, MDZ, LLM, G.
Wang, D., Rousseau-Gueutin, M., Timmis, J. N. (2012). Plastid sequences contribute to some plant mitochondrial genes. Mol. Biol. Evol. 29, 1707–1711. doi: 10.1093/molbev/mss016
Wang, J., Xu, G., Ning, Y., Wang, X., Wang, G. L. (2022). Mitochondrial functions in plant immunity. Trends Plant Sci. 27, 1063–1076. doi: 10.1016/j.tplants.2022.04.007
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinf. 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wei, C., Yang, H., Wang, S., Zhao, J., Liu, C., Gao, L., et al. (2018). Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. U.S.A 115, E4151–E4158. doi: 10.1073/pnas.1719622115
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Willson, K. C., Clifford, M. N. (1992). Tea: cultivation to consumption (Berlin: Springer Science & Business Media). doi: 10.1007/978-94-011-2326-6
Wright, F. (1990). The 'effective number of codons' used in a gene. Gene 87, 23–29. doi: 10.1016/0378-1119(90)90491-9
Wu, Z.-Q., Liao, X.-Z., Zhang, X.-N., Tembrock, L. R., Broz, A. (2022). Genomic architectural variation of plant mitochondria-A review of multichromosomal structuring. J. Systematics Evol. 60, 160–168. doi: 10.1111/jse.12655
Xia, E.-H., Zhang, H.-B., Sheng, J., Li, K., Zhang, Q.-J., Kim, C., et al. (2017). The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant 10, 866–877. doi: 10.1016/j.molp.2017.04.002
Xie, D.-F., Yu, H.-X., Price, M., Xie, C., Deng, Y.-Q., Chen, J.-P., et al. (2019). Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00460
Xu, J., Luo, H., Nie, S., Zhang, R.-G., Mao, J.-F. (2021). The complete mitochondrial and plastid genomes of Rhododendron simsii, an important parent of widely cultivated azaleas. Mitochondrial DNA Part B-Resources 6, 1197–1199. doi: 10.1080/23802359.2021.1903352
Xu, K., Tian, C., Zhou, C., Zhu, C., Weng, J., Sun, Y., et al. (2022). Non-targeted metabolomics analysis revealed the characteristic non-volatile and volatile metabolites in the Rougui wuyi rock tea (Camellia sinensis) from different culturing regions. Foods 11, 1694. doi: 10.3390/foods11121694
Yang, Z., Ni, Y., Lin, Z., Yang, L., Chen, G., Nijiati, N., et al. (2022). De novo assembly of the complete mitochondrial genome of sweet potato (Ipomoea batatas L. Lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant Biol. 22, 285. doi: 10.1186/s12870-022-03665-y
Yang, H., Ni, Y., Zhang, X., Li, J., Chen, H., Liu, C. (2023). The mitochondrial genomes of Panax notoginseng reveal recombination mediated by repeats associated with DNA replication. Int. J. Biol. macromolecules 252, 126359. doi: 10.1016/j.ijbiomac.2023.126359
Yengkhom, S., Uddin, A., Chakraborty, S. (2019). Deciphering codon usage patterns and evolutionary forces in chloroplast genes of Camellia sinensis var. assamica and Camellia sinensis var. sinensis in comparison to Camellia pubicosta. J. Integr. Agric. 18, 2771–2785. doi: 10.1016/S2095-3119(19)62716-4
Yi, Y., Wei, Y., Deng, C., Wu, S. (2015). Research progress of amino acid residues in the protein thermal stability mechanism. J. Guangxi. Uni. Sci. Technol. 26, 1–5. doi: 10.16375/j.cnki.cn45-1395/t.2015.04.001
Zandueta-Criado, A., Bock, R. (2004). Surprising features of plastid ndhD transcripts:: addition of non-encoded nucleotides and polysome association of mRNAs with an unedited start codon. Nucleic Acids Res. 32, 542–550. doi: 10.1093/nar/gkh217
Zhang, X., Chen, S., Shi, L., Gong, D., Zhang, S., Zhao, Q., et al. (2021). Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant Camellia sinensis. Nat. Genet. 53, 1250–1259. doi: 10.1038/s41588-021-00895-y
Zhang, F., Li, W., Gao, C.-w., Zhang, D., Gao, L.-z. (2019). Deciphering tea tree chloroplast and mitochondrial genomes of Camellia sinensis var. assamica. Sci. Data 6, 209. doi: 10.1038/s41597-019-0201-8
Zhang, H., Meltzer, P., Davis, S. (2013). RCircos: an R package for Circos 2D track plots. BMC Bioinf. 14, 244. doi: 10.1186/1471-2105-14-244
Zhang, Z. B., Xiong, T., Chen, J. H., Ye, F., Cao, J. J., Chen, Y. R. (2023). Understanding the origin and evolution of tea (Camellia sinensis [L.]): genomic advances in tea. J. Mol. Evol. 91, 156–168. doi: 10.1007/s00239-023-10099-z
Keywords: Camellia sinensis, mitochondrial genome, genome comparison, codon preference, positive selection, phylogenetic analysis
Citation: Li L, Li X, Liu Y, Li J, Zhen X, Huang Y, Ye J and Fan L (2024) Comparative analysis of the complete mitogenomes of Camellia sinensis var. sinensis and C. sinensis var. assamica provide insights into evolution and phylogeny relationship. Front. Plant Sci. 15:1396389. doi: 10.3389/fpls.2024.1396389
Received: 05 March 2024; Accepted: 29 July 2024;
Published: 22 August 2024.
Edited by:
Xinghui Li, Nanjing Agricultural University, ChinaReviewed by:
SeonJoo Park, Yeungnam University, Republic of KoreaCopyright © 2024 Li, Li, Liu, Li, Zhen, Huang, Ye and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Li, bGlsaUB3dXlpdS5lZHUuY24=; Li Fan, Mjc4NzAxOTEyQHFxLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.