 Zhihui Wang
Zhihui Wang Yong Lei
Yong Lei Boshou Liao
Boshou Liao- 1Key Laboratory of Biology and Genetic Improvement of Oil Crops, Ministry of Agriculture and Rural Affairs, Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences (CAAS), Wuhan, China
- 2National Key Laboratory of Crop Genetic Improvement, National Center of Crop Molecular Breeding Technology, National Center of Oil Crop Improvement (Wuhan), Huazhong Agricultural University, Wuhan, China
Peanuts (Arachis hypogaea) are an essential oilseed crop known for their unique developmental process, characterized by aerial flowering followed by subterranean fruit development. This crop is polyploid, consisting of A and B subgenomes, which complicates its genetic analysis. The advent and progression of omics technologies—encompassing genomics, transcriptomics, proteomics, epigenomics, and metabolomics—have significantly advanced our understanding of peanut biology, particularly in the context of seed development and the regulation of seed-associated traits. Following the completion of the peanut reference genome, research has utilized omics data to elucidate the quantitative trait loci (QTL) associated with seed weight, oil content, protein content, fatty acid composition, sucrose content, and seed coat color as well as the regulatory mechanisms governing seed development. This review aims to summarize the advancements in peanut seed development regulation and trait analysis based on reference genome-guided omics studies. It provides an overview of the significant progress made in understanding the molecular basis of peanut seed development, offering insights into the complex genetic and epigenetic mechanisms that influence key agronomic traits. These studies highlight the significance of omics data in profoundly elucidating the regulatory mechanisms of peanut seed development. Furthermore, they lay a foundational basis for future research on trait-related functional genes, highlighting the pivotal role of comprehensive genomic analysis in advancing our understanding of plant biology.
Introduction
As a source of edible vegetable oil and protein, peanut (Arachis hypogaea L.) is an oil and economic crop of worldwide importance. The peanut are now cultivated in more than 100 countries, mainly distributed in developing countries in Asia, Africa and South America. The global peanut production has been about 54 million tons annually, with a consistent cultivation area of approximately 31 million hectares (ha) in recent years (http://faostat.fao.org). Peanuts have a high nutritional value, as they are rich in fats (35%~60%) and proteins (22%~35%), and also provide dietary fiber, minerals, vitamins and bioactive macromolecules (Zhao et al., 2020b; Chen et al., 2021; Zhou et al., 2021). The traits of peanuts cover various characteristics such as seed weight, oil content, protein content, fatty acid composition, sucrose content, seed coat color, etc. The improvement of these traits is currently a key area of focus in in peanut genetic breeding. These traits are intricately linked to the expression and regulation of genes during seed development. Consequently, the elucidation of the peanut seed development process based on omics data has become a research hotspot in recent years, shedding light on the regulatory mechanisms governing the formation and variation of essential traits in peanut seeds.
The peanut seed development process spans from flowering to subterranean fruiting, illustrating the unique geocarpic growth habit of peanuts. In this process, the flower pollinates above ground, and then the peg, carrying the fertilized ovule, elongates and burrows into the soil to form the seed. The development process of peanut seeds is highly intricate, governed by numerous genes that regulate various seed traits such as size, weight, oil content, seed coat color, fatty acid composition, and the concentration of functional substances. Therefore, researching the regulatory genes and related molecular mechanisms involved in the peanut development process is of significant importance for the genetic improvement of peanut traits.
The omic-technology with illumina or long sequencing reads was utilized to construct the reference genome of peanut. The genome of the diploid progenitors of cultivated peanut, A. duranensis and A. ipaensis, was sequenced first (Bertioli et al., 2016; Chen et al., 2016b; Hu et al., 2018), followed by the genome of the cultivated allotetraploid peanut A. hypogaea (Bertioli et al., 2019; Zhuang et al., 2019). Since the release of the peanut reference genome, significant progress have been achieved in the investigation of quantitative trait loci (QTL) mapping, expression regulation, epigenetics, and other facets pertaining to seed-related traits (Table 1). These advancements have been facilitated through the comprehensive analysis of diverse omics datasets, such as resequencing data, transcriptome, proteome, metabolome, and epigenome, etc (Figure 1).
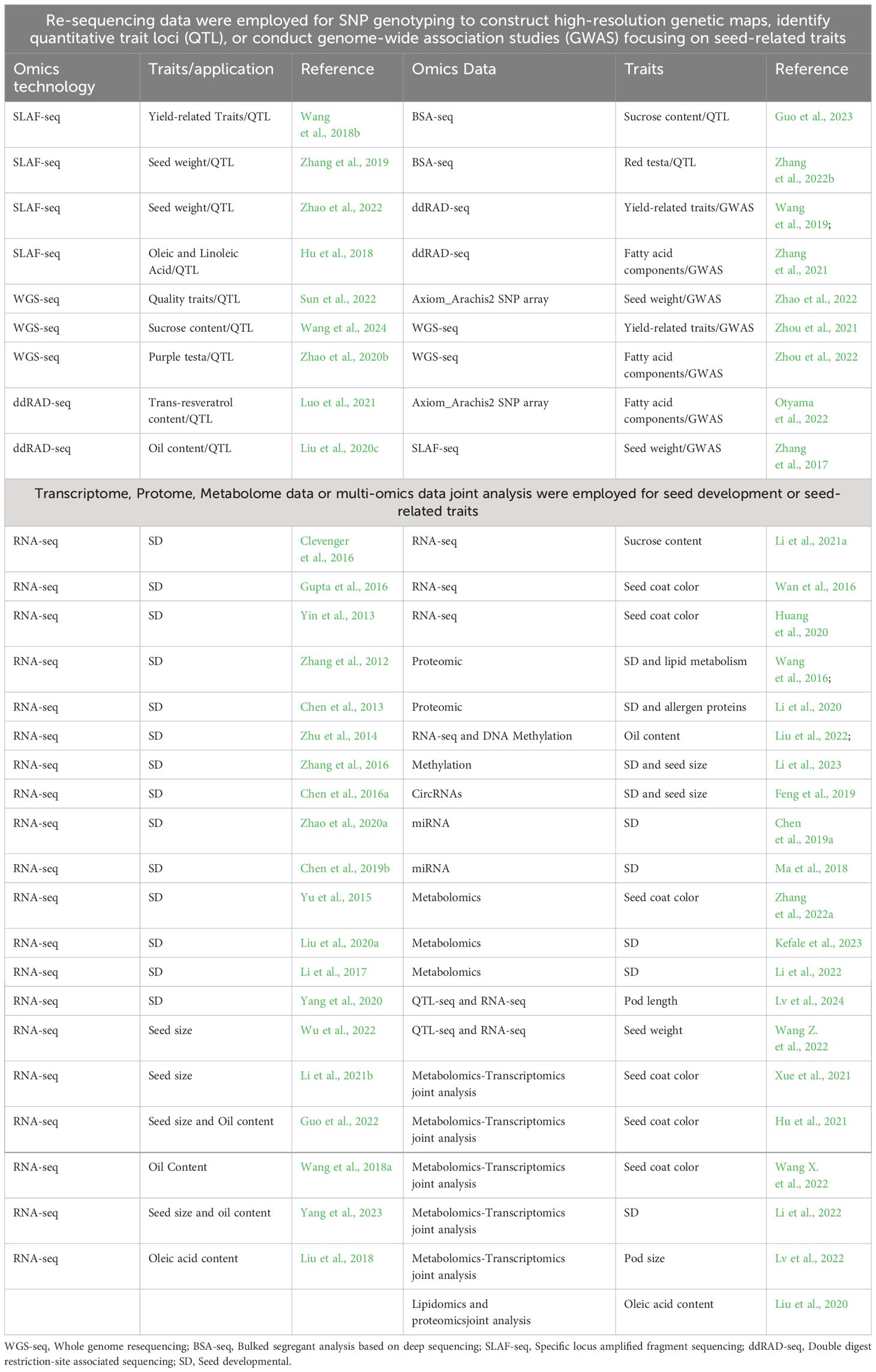
Table 1 Reference list for omics-driven research on peanut seed development.
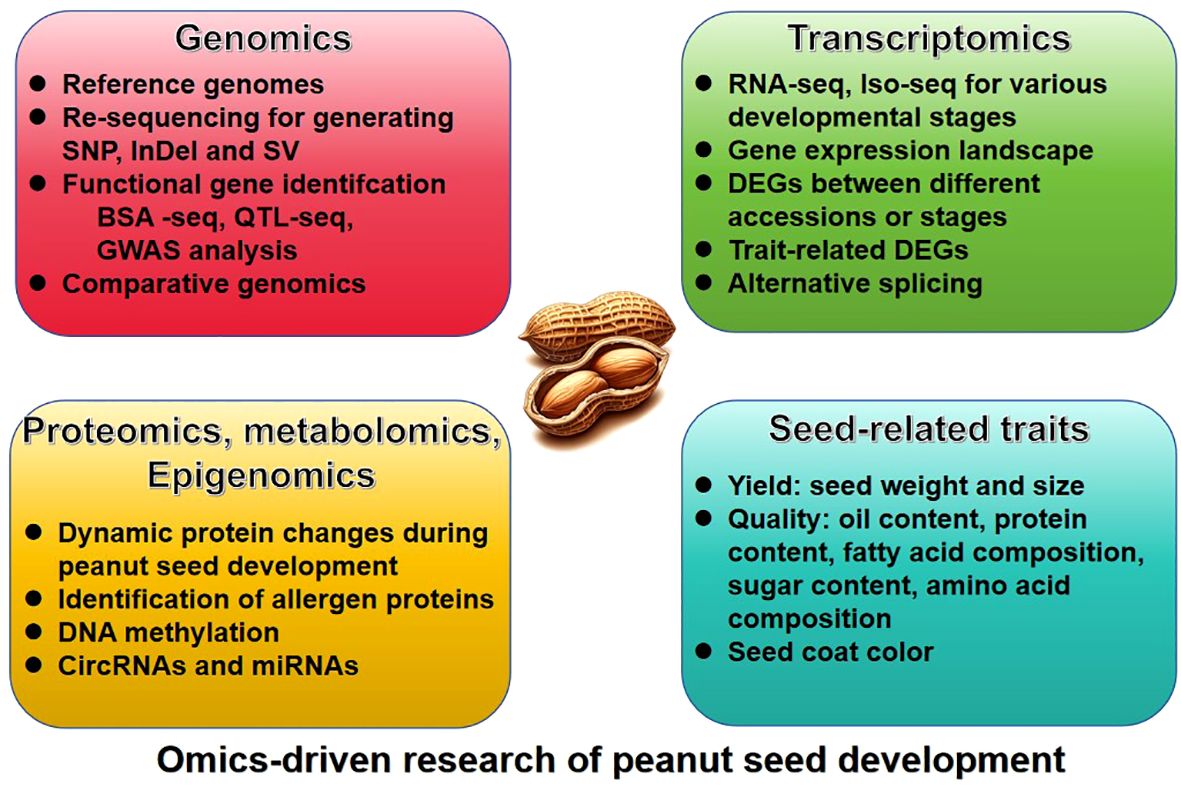
Figure 1 The overview of omics-driven research of peanut seed development.
QTL mapping and GWAS analysis of peanut seed-related traits driven by re-sequencing data
The foundation for mapping QTLs associated with seed traits in peanuts has been laid through the use of a genetic map, where the quality and precision of QTL mapping, as well as the accurate localization of QTL regions, are significantly influenced by the number and density of markers. Recent research efforts employing specific locus amplified fragment sequencing (SLAF-seq) (Wang et al., 2018b; Zhang et al., 2019; Zhao et al., 2022), genotyping-by-sequencing (GBS) (Zhang et al., 2017), and double-digest restriction-site-associated DNA sequencing (ddRAD-seq) (Wang et al., 2019; Luo et al., 2021; Zhang et al., 2021) have contributed to the generation of over 2,000 SNP markers on genetic maps, underscoring the role of omics data in enhancing genetic analysis. With the declining cost of sequencing, whole-genome re-sequencing has emerged as a powerful approach for generating large-scale SNP markers and constructing high-density genetic maps. Notably, this approach has led to the development of four high-density genetic linkage maps, each containing over 8,000 SNPs (Agarwal et al., 2018, 2019; Liu et al., 2020b; Jiang et al., 2021), making a significant advance in the ability to identify markers on a large scale, especially in the context of low genetic diversity in peanut germplasm (Jiang et al., 2011; Wang et al., 2011; Mukri et al., 2012).
In recent genetic studies, various quantitative trait loci (QTL) associated with seed-related traits have been identified, offering insights into the complex genetic foundations of these traits. Noteworthy discoveries include the identification of one stable QTL linked to seed weight on the terminal regions of chromosome B07 (Wang et al., 2018b). Further research identified additional stable QTLs influencing seed weight located on chromosomes A02 and B06 (Zhang et al., 2019). In terms of nutritional traits, a major QTL, qA05.1, was found to have a significant impact on oil, protein, and six fatty acids across diverse environments, highlighting the intricate genetic interactions shaping the nutritional composition of peanuts (Sun et al., 2022). A detailed examination conducted by Hu et al. (2018) identified QTLs related to oleic acid (C18:1), linoleic acid (C18:2), and the oleic-to-linoleic acid ratio (O/L) on chromosomes A03, A04, A09, B09, and B10, illuminating the genetic regulation of fatty acid composition. Liu et al. (2020c) discovered a stable QTL, qOCA08.1, on chromosome A08, which explained a substantial proportion of phenotypic variation in oil content. Fine-mapping of this QTL revealed a ~0.8-Mb genomic region harboring two annotated genes influencing oil synthesis, providing vital insights into the genetic determinants of oil-related traits in peanut. Employing BSA-seq technology, Guo et al. (2023) uncovered four QTLs for sucrose content on chromosomes A03 and A06, while Wang et al. (2024) further identified two homologous QTLs on chromosomes A06 and B06, providing valuable information on the genetic factors impacting this essential trait. Furthermore, studies on color traits identified key genes controlling red testa color. QTL analysis and fine-mapping identified the AhRt2 gene on chromosome 12, associated with a SNP in the third exon, as crucial for red testa color (Zhang et al., 2022b). Additionally, the AhTc1 gene, encoding an R2R3-MYB transcription factor, was found to regulate purple testa color (Zhao et al., 2020b), while AhRt1 was mapped to a region on chromosome A03, associated with a bHLH transcription factor gene, further elucidating the genetics underlying testa color in peanut (Chen et al., 2021).
Advances in genome-wide association study (GWAS) analyses have significantly contributed to the understanding of peanut seed traits. Zhao et al. (2022) identified SNP markers associated with hundred seed weight, branch number, and pod shape. In the Chinese peanut core collection, Zhou et al. (2021) uncovered two major loci exhibiting pleiotropic effects on yield-related traits, explaining about 20% of phenotypic variation. Furthermore, Zhou et al. (2022) identified three stable major associated loci, including two on chromosome A09 for oleic acid and linoleic acid and one on B06 for stearic acid. Extending the research to the USDA peanut core collection, Otyama et al. (2022) explored genetic markers tied to variations in fatty acid composition, unveiling 10 markers affecting oleic and linoleic acid contents, with the alleles having inverse impacts on these acid concentrations. Moreover, Zhang et al. (2017) uncovered 18 significant markers related to seed weight distributed across six chromosomes. Wang et al. (2019) further identified seven peak SNPs associated with yield per plant, pod weight, and seed weight. Zhang et al. (2021) identified five stable significant SNPs associated with oil content and three stable significant SNPs associated with C24:0. Collectively, these GWAS findings offer a comprehensive view of the genetic architecture underlying various peanut seed traits, facilitating targeted breeding efforts for improved cultivars.
The regulation of peanut seed development based on transcriptome data
The study of peanut seed development through transcriptome analysis has led to significant insights, with numerous publications highlighting the intricate genetic networks involved. A pivotal study by Clevenger et al. (2016) sequenced a comprehensive transcriptome map covering 22 tissue types throughout the peanut’s reproductive development, from flowering to seed maturation. This work, in conjunction with additional RNA-seq data from Yin et al. (2013); Gupta et al. (2016), and Zhang et al. (2012), has provided a detailed gene expression landscape during seed development.
Peanuts exhibit a unique botanical feature: aerial flowering followed by subterranean fruit development. The failure of peg penetration into the soil inhibits the start of pod swelling, resulting in the development of aerial pods and ultimately leading to seed abortion. Comparative transcriptomic analyses between aerial and subterranean pods have identified genes associated with early embryo abortion, including up-regulated photosynthesis-related genes and senescence-associated genes in aerial pods, which may hinder pod swelling (Chen et al., 2013; Zhu et al., 2014). Further transcriptome analyses have identified crucial genes in the embryo and basal regions of the peg, both before and after soil penetration. These genes, including MADS-box transcription factors and cellulose synthase, are vital for embryo development and pod formation (Zhang et al., 2016). Chen et al. (2016a) expanded the research to encompass two whole pod stages and nine stages of isolated pod walls, revealing a developmental gradient of gene expression and highlighting the roles of transcription factors in pod development. Similarly, MADS-box transcription factors play a pivotal role in regulating seed development in both grapevine (Grimplet et al., 2016) and Arabidopsis (Simona et al., 2011), highlighting their fundamental importance across diverse plant species. Zhao et al. (2020a) explored alternative splicing in early swelling pods, finding it mainly related to ovule development, root hair cells enlargement, root apex division, and seed germination. Chen et al. (2019b) and Yu et al. (2015) focused on oil metabolism, identifying over 2,500 genes related to lipid biosynthesis. Their expression patterns during seed development offer insights into peanut lipid biosynthesis.
Studies on the dynamic transcriptomic changes during pod filling have shed light on genotypic variations in lipid metabolism and pod filling efficiency, with the “Hanoch” genotype showing superior pod-filling capabilities (Gupta et al., 2016). Liu et al. (2020a) examined the developmental transcriptome of underground peanut pods and identified 165,689 transcripts, revealing a shift from DNA synthesis and cell division to cell expansion and storage during seed development, with photosynthetic genes active in both aerial and subterranean pods. Moreover, the role of calcium, a crucial signaling molecule, in peanut pod development has been explored. Li et al. (2017) and Yang et al. (2020) investigated the effects of calcium deficiency on gene expression related to calcium signaling and hormone regulation during pod development. Similar to findings in peanuts, studies on wheat and Chinese cabbage reveal the pivotal role of calcium in sustaining plant health, underscoring how calcium deficiency influences gene expressions linked to calcium signaling and hormonal regulation in these diverse agricultural crops (Aslam et al., 2017; Zhang et al., 2022). These studies illustrate the complex interplay between calcium signal transduction, hormone pathways, and the genetic regulation of peanut seed development, providing valuable insights for targeted breeding and genetic improvement initiatives.
The trait-related regulation of peanut seed development based on transcriptome data
Comprehensive transcriptomic analyses have shed light on the genetic mechanisms behind peanut seed development, size, and oil content. For instance, genes such as PNC, YUC, and GASA were found to influence auxin synthesis and seed size, while specific variant sites like GCP4 and RPPL1 within QTL intervals play roles in cell tissue microtubule nucleation (Wu et al., 2022). These findings were consistent with the research in other plants (Cao et al., 2020), which highlighted the importance of auxin and related genes in seed development and grain yield. RNA-seq data from cultivated peanuts and wild Arachis monticola identified genes uniquely expressed during seed development, with certain proteins potentially linked to increased seed size in cultivated varieties (Li et al., 2021b). Differences in gene expression between genotypes with varying seed size and oil content have identified pathways related to plant hormones and fatty acid biosynthesis as critical for seed related traits (Guo et al., 2022).
In terms of oil content, the analysis of 49 cultivars uncovered significant markers on chromosome A03, aiding marker-assisted selection in breeding (Wang et al., 2018a). Comparative studies between peanut varieties with different seed sizes and oil levels identified genes and networks involved in fatty acid synthesis, suggesting strategies for improving seed yield and quality (Yang et al., 2023). Similar to research conducted on peanuts, studies in other crops like safflower, soybean, and rapeseed have also explored key genes and networks involved in regulating traits such as like seed size, oil content, and fatty acid composition (Fan et al., 2023; Guan et al., 2023; Zhao et al., 2023). Research on oleic acid content between cultivars highlighted the role of FAB2 in unsaturated fatty acid biosynthesis and lipid oxidation (Liu et al., 2018), which agreed with the reports in other plants (Dar et al., 2017; He et al., 2020).
For sucrose content, comparative transcriptomics between high- and low-sucrose peanut varieties revealed genes linked to sucrose metabolism, offering targets for molecular breeding (Li et al., 2021a). Studies on seed coat color have utilized transcriptomics to identify genes and markers associated with testa color, providing valuable information for breeding peanuts with desired coat characteristics (Wan et al., 2016; Huang et al., 2020). These findings collectively enhance our understanding of peanut genomics and support targeted breeding efforts for trait improvement.
Deciphering peanut seed development based on proteomic, metabolic, and epigenetic data
The exploration of peanut seed development through integrated approaches combining proteomics, metabolomics, and epigenetics has yielded profound insights into the molecular underpinnings of key seed traits such as size, oil content, allergenicity, and amino acid composition.
Proteomic analyses have significantly advanced our understanding of the dynamic protein changes during peanut seed development. Studies have identified a diverse array of proteins involved in carbohydrate, amino acid, and lipid metabolism, highlighting their crucial roles in seed development (Wang et al., 2016; Li et al., 2020). Some of the proteins identified during the development of peanut seeds were also found in other crops such as rice (Yoon et al., 2023), soybeans (Wei et al., 2020), wheat (Dong et al., 2015), and barley (Chen et al., 2022). Notably, the identification of allergen proteins and their expression patterns offers valuable insights into allergen accumulation processes, informing breeding strategies aimed at reducing allergenicity (Li et al., 2020). Moreover, the differential expression of proteins related to lipid metabolism during seed development and post-germination stages underscores the complex regulatory mechanisms governing oil accumulation and degradation (Wang et al., 2016).
Epigenetic modifications, specifically DNA methylation, have been elucidated as pivotal regulatory mechanisms influencing peanut seed development. Comparative analyses have demonstrated global methylation changes accompanying seed development, with significant correlations between methylation levels and gene expression, particularly in pathways related to seed size and oil content (Liu et al., 2022; Li et al., 2023). These findings suggest that epigenetic regulation plays a substantial role in modulating seed trait expression.
The regulation of gene expression during seed development has also been a focus, with studies revealing the importance of circRNAs and miRNAs in this process. CircRNAs have been implicated in seed development and size regulation, pointing to their involvement in post-transcriptional regulatory networks (Feng et al., 2019). Similarly, miRNA-mediated regulatory networks have been identified as key contributors to embryo development under calcium deficiency and seed expansion, highlighting the roles of specific miRNAs in modulating gene expression related to growth and development processes (Ma et al., 2018; Chen et al., 2019a).
Metabolomics has recently been embraced in peanut research, offering novel insights into our understanding of its seed develpment. Zhang et al. (2022) employs UPLC-MS/MS to profile metabolites in the testa of four peanut germplasms with varied colors, identifying 85 metabolites and highlighting the significant diversity and differential accumulation of these compounds, including proanthocyanidins, isoflavones, flavonols, and anthocyanidins (Zhang et al., 2022a). Kefale et al. (2023) identified many differentially accumulated metabolites related to amino acid metabolism, phenylpropanoid biosynthesis, flavonoid biosynthesis, and lipid metabolism between peanut and other oil crops. Li et al. (2022) found that during the early stages of development, most amino acids were present at significantly lower levels. However, this trend shifted in the middle and late stages, where the levels of amino acids were notably higher.
Deciphering peanut seed development and seed-related traits based on joint multi-omics analysis
The joint multi-omics analysis, including genomics-transcriptomics, metabolomics-transcriptomics, proteomics-transcriptomics, serves as a powerful toolkit for elucidating the mechanisms regulating peanut seed development and controlling seed-related traits. In terms of genomics-transcriptomics joint analysis, QTL-seq and RNA-seq have been successfully applied to identify candidate genes for pod length (Lv et al., 2024) and seed weight (Wang et al., 2022). For metabolomics-transcriptomics joint analysis, three studies untilize a integrated approach to investigate the regulatory mechanisms behind testa pigmentation in peanuts. Xue et al. (2021) led this exploration, uncovering the intricate anthocyanin metabolism, highlighting the importance of petunidin 3-O-glucoside and cyanidin O-acetylhexoside in color differentiation. Their analysis identified crucial genes and transcription factors, such as CHS, DFR, MYB, bHLH, and WD40, as pivotal in regulating the distinct pigmentation of peanut testa. Hu et al. (2021) further analyzed the flavonoid biosynthesis pathway, identifying 27 significantly differentially expressed genes (DEGs) associated with testa color development, emphasizing the roles of cyanidin and delphinidin. Wang et al. (2022) broadened this investigation by profiling 133 flavonoids across four peanut cultivars, correlating specific flavonoid components with a variety of testa colors and detailing the roles of cyanidin-based anthocyanins, MYB-like transcription factors, anthocyanidin reductases (ANR), and UDP-glycosyltransferases (UGT) in color modulation (Wang et al., 2022). Additionally, transcriptomic and metabolomic analyses have shed light on the genetic and metabolic pathways involved in seed development, with the identification of genes and proteins involved in amino acid metabolism, notably arginine biosynthesis, providing avenues for enhancing the nutritional quality of peanut seeds (Li et al., 2022). Lv et al. (2022) identifies that the accumulation of p-coumaryl alcohol and its associated biosynthesis pathway, particularly the differential expression of gene LOC112771695, plays a critical role in determining peanut pod size. By integrating lipidomics and proteomics, Liu et al. (2020) unravel the complex dynamics of lipid molecular species and their association with the FAD2 mutation in high-oleic acid peanut seeds.
Perspectives
This review demonstrates the pivotal role of omics technologies and related data in achieving a comprehensive peanut reference genome and deepening our insight into the regulatory mechanisms governing peanut seed development. Extensive analysis across various developmental phases, encompassing gene expression, proteomics, metabolomics, and epigenetics, has unveiled the molecular underpinnings and identified key regulatory mechanisms and QTLs linked to seed traits. Such discoveries have substantially contributed greatly to our comprehension of peanut seed development and trait regulation. However, there is still a relative scarcity of research on the cloning and functional study of peanut functional genes. Accelerating the fine mapping of QTLs and employing multi-omics techniques to identify functional genes are essential next steps. Additionally, the application of gene-editing technologies for the improvement of seed traits and the creation of new germplasms represents a crucial research direction for the future. The genome navigation system, including platforms like RiceNavi (Wei et al., 2021), was anticipated to be developed for the purpose of QTN pyramiding and optimizing breeding routes in peanuts. These approaches will not only deepen our understanding of peanut biology but also facilitate the breeding of varieties with enhanced yield, quality, and environmental resilience.
Author contributions
ZW: Writing – original draft, Writing – review & editing. YL: Writing – original draft, Writing – review & editing. BL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No. 32201770), the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Science (CAAS-ASTIP-2021-OCRI), Natural Science Foundation of Hubei Province (2022CFB332).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agarwal, G., Clevenger, J., Kale, S. M., Wang, H., Pandey, M. K., Choudhary, D., et al. (2019). A recombination bin-map identified a major QTL for resistance to Tomato Spotted Wilt Virus in peanut (Arachis hypogaea). Sci. Rep. 9, 18246. doi: 10.1038/s41598-019-54747-1
Agarwal, G., Clevenger, J., Pandey, M. K., Wang, H., Shasidhar, Y., Chu, Y., et al. (2018). High-density genetic map using whole-genome resequencing for fine mapping and candidate gene discovery for disease resistance in peanut. Plant Biotechnol. J. 16, 1954–1967. doi: 10.1111/pbi.12930
Aslam, R., Williams, L. E., Bhatti, M. F., Virk, N. (2017). Genome-wide analysis of wheat calcium ATPases and potential role of selected ACAs and ECAs in calcium stress. BMC Plant Biol. 17, 174. doi: 10.1186/s12870-017-1112-5
Bertioli, D. J., Cannon, S. B., Froenicke, L., Huang, G. D., Farmer, A. D., Cannon, E. K., et al. (2016). The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet 48 (4), 438–46. doi: 10.1038/ng.3517
Bertioli, D. J., Jenkins, J., Clevenger, J., Dudchenko, O., Gao, D. Y., Seijo, G., et al. (2019). The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat Genet 51 (5), 877–884. doi: 10.1038/s41588-019-0405-z
Cao, J., Li, G., Qu, D., Li, X., Wang, Y. (2020). Into the seed: auxin controls seed development and grain yield. Int. J. Mol. Sci. 21, 1662. doi: 10.3390/ijms21051662
Chen, H., Chen, X. Y., Xu, R. R., Liu, W. J., Liu, N. A., Huang, L., et al. (2021). Fine-mapping and gene candidate analysis for a major dominant locus responsible for testa color in cultivated peanut. Theor. And Appl. Genet. 134, 3721–3730. doi: 10.1007/s00122-021-03924-w
Chen, X. P., Li, H. J., Pandey, M. K., Yang, Q. L., Wang, X. Y., Garg, V., et al. (2016b). Draft genome of the peanut A-genome progenitor provides insights into geocarpy, oil biosynthesis, and allergens. P Natl. Acad. Sci. U.S.A. 113, 6785–6790. doi: 10.1073/pnas.1600899113
Chen, X., Lu, Q., Liu, H., Zhang, J., Hong, Y., Lan, H., et al. (2019b). Sequencing of Cultivated Peanut, Arachis hypogaea, Yields Insights into Genome Evolution and Oil Improvement. Mol. Plant 12, 920–934. doi: 10.1016/j.molp.2019.03.005
Chen, Y., Wang, J., Yao, L., Li, B., Ma, X., Si, E., et al. (2022). Combined proteomic and metabolomic analysis of the molecular mechanism underlying the response to salt stress during seed germination in barley. Int. J. Mol. Sci. 23, 10515. doi: 10.3390/ijms231810515
Chen, H., Yang, Q., Chen, K., Zhao, S., Zhang, C., Pan, R., et al. (2019a). Integrated microRNA and transcriptome profiling reveals a miRNA-mediated regulatory network of embryo abortion under calcium deficiency in peanut (Arachis hypogaea L.). BMC Genomics 20, 392. doi: 10.1186/s12864-019-5770-6
Chen, X., Yang, Q., Li, H., Li, H., Hong, Y., Pan, L., et al. (2016a). Transcriptome-wide sequencing provides insights into geocarpy in peanut (Arachis hypogaea L.). Plant Biotechnol. J. 14, 1215–1224. doi: 10.1111/pbi.12487
Chen, X., Zhu, W., Azam, S., Li, H., Zhu, F., Li, H., et al. (2013). Deep sequencing analysis of the transcriptomes of peanut aerial and subterranean young pods identifies candidate genes related to early embryo abortion. Plant Biotechnol. J. 11, 115–127. doi: 10.1111/pbi.12018
Clevenger, J., Chu, Y., Scheffler, B., Ozias-Akins, P. (2016). A developmental transcriptome map for allotetraploid arachis hypogaea. Front. Plant Sci. 7, 1446. doi: 10.3389/fpls.2016.01446
Dar, A. A., Choudhury, A. R., Kancharla, P. K., Arumugam, N. (2017). The FAD2 Gene in plants: occurrence, regulation, and role. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01789
Dong, K., Zhen, S., Cheng, Z., Cao, H., Ge, P., Yan, Y. (2015). Proteomic analysis reveals key proteins and phosphoproteins upon seed germination of wheat (Triticum aestivum L.). Front. Plant Sci. 6. doi: 10.3389/fpls.2015.01017
Fan, K., Qin, Y., Hu, X., Xu, J., Ye, Q., Zhang, C., et al. (2023). Identification of genes associated with fatty acid biosynthesis based on 214 safflower core germplasm. BMC Genomics 24, 763. doi: 10.1186/s12864-023-09874-5
Feng, G., Zhang, M., Ma, S., Zhang, X., Ma, X., Ning, L., et al. (2019). Genome-wide identification of circular RNAs in peanut (Arachis hypogaea L.). BMC Genomics 20, 653. doi: 10.1186/s12864-019-6020-7
Grimplet, J., Martínez-Zapater, J. M., Carmona, M. J. (2016). Structural and functional annotation of the MADS-box transcription factor family in grapevine. BMC Genomics 17. doi: 10.1186/s12864-016-2398-7
Guan, M., Shi, X., Chen, S., Wan, Y., Tang, Y., Zhao, T., et al. (2023). Comparative transcriptome analysis identifies candidate genes related to seed coat color in rapeseed. Front Plant Sci 14, 1154208. doi: 10.3389/fpls.2023.1154208
Guo, J. J., Qi, F. Y., Qin, L., Zhang, M. N., Sun, Z. Q., Li, H. Y., et al. (2023). Mapping of a QTL associated with sucrose content in peanut kernels using BSA-seq. Front. In Genet. 13. doi: 10.3389/fgene.2022.1089389
Guo, F., Zhu, X., Zhao, C. (2022). Transcriptome analysis and gene expression profiling of the peanut small seed mutant identified genes involved in seed size control. Int. J. Mol. Sci. 23 (17), 9726. doi: 10.3390/ijms23179726
Gupta, K., Kayam, G., Faigenboim-Doron, A., Clevenger, J., Ozias-Akins, P., Hovav, R. (2016). Gene expression profiling during seed-filling process in peanut with emphasis on oil biosynthesis networks. Plant Sci. 248, 116–127. doi: 10.1016/j.plantsci.2016.04.014
He, M., Qin, C. X., Wang, X., Ding, N. Z. (2020). Plant unsaturated fatty acids: biosynthesis and regulation. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.00390
Hu, M., Li, J., Hou, M., Cui, S., Yang, X., Liu, L., et al. (2021). Transcriptomic and metabolomic joint analysis reveals distinct flavonoid biosynthesis regulation for variegated testa color development in peanut (Arachis hypogaea L.). Sci. Rep. 11, 10721. doi: 10.1038/s41598-021-90141-6
Hu, X. H., Zhang, S. Z., Miao, H. R., Cui, F. G., Shen, Y., Yang, W. Q., et al. (2018). High-density genetic map construction and identification of QTLs controlling oleic and linoleic acid in peanut using SLAF-seq and SSRs. Sci. Rep. 8 (1), 5479. doi: 10.1038/S41598-018-23873-7
Huang, L., Liu, X., Pandey, M. K., Ren, X., Chen, H., Xue, X., et al. (2020). Genome-wide expression quantitative trait locus analysis in a recombinant inbred line population for trait dissection in peanut. Plant Biotechnol. J. 18, 779–790. doi: 10.1111/pbi.13246
Jiang, Y., Luo, H., Yu, B., Ding, Y., Kang, Y., Huang, L., et al. (2021). High-density genetic linkage map construction using whole-genome resequencing for mapping QTLs of resistance to aspergillus flavus infection in peanut. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.745408
Jiang, C., Ramchiary, N., Ma, Y., Jin, M., Feng, J., Li, R., et al. (2011). Structural and functional comparative mapping between the Brassica A genomes in allotetraploid Brassica napus and diploid Brassica rapa. Theor. Appl. Genet. 123, 927–941. doi: 10.1007/s00122-011-1637-1
Kefale, H., Segla, K. D. S., Li, F., Jiang, N., Zhou, R., Wang, L., et al. (2023). Widely targeted metabolic profiling provides insights into variations in bioactive compounds and antioxidant activity of sesame, soybean, peanut, and perilla. Food Res. Int. 174, 113586. doi: 10.1016/j.foodres.2023.113586
Li, W., Huang, L., Liu, N., Pandey, M. K. (2021a). Key regulators of sucrose metabolism identified through comprehensive comparative transcriptome analysis in peanuts. Int. J. Mol. Sci. 22 (14), 7266. doi: 10.3390/ijms22147266
Li, H., Liang, X., Zhou, B., Chen, X., Hong, Y., Zhou, R., et al. (2020). A proteomic analysis of peanut seed at different stages of underground development to understand the changes of seed proteins. PloS One 15, e0243132. doi: 10.1371/journal.pone.0243132
Li, C., Lai, X., Luo, K., Zheng, Y., Liu, K., Wan, X. (2022). Integrated metabolomic and transcriptomic analyses of two peanut (Arachis hypogaea L.) cultivars differing in amino acid metabolism of the seeds. Plant. Physiol. Biochem. 185, 132–143. doi: 10.1016/j.plaphy.2022.05.037
Li, Z., Liu, Q., Zhao, K., Cao, D., Cao, Z., Zhao, K., et al. (2023). Dynamic DNA methylation modification in peanut seed development. iScience 26, 107062. doi: 10.1016/j.isci.2023.107062
Li, Y., Meng, J., Yang, S., Guo, F., Zhang, J., Geng, Y., et al. (2017). Transcriptome analysis of calcium- and hormone-related gene expressions during different stages of peanut pod development. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01241
Li, Z., Zhang, X., Zhao, K., Zhao, K., Qu, C., Gao, G., et al. (2021b). Comprehensive transcriptome analyses reveal candidate genes for variation in seed size/weight during peanut (Arachis hypogaea L.) domestication. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.666483
Liu, N., Guo, J. B., Zhou, X. J., Wu, B., Huang, L., Luo, H. Y., et al. (2020c). High-resolution mapping of a major and consensus quantitative trait locus for oil content to a ~ 0.8-Mb region on chromosome A08 in peanut. Theor. And Appl. Genet. 133, 37–49. doi: 10.1007/s00122-019-03438-6
Liu, H., Hong, Y., Lu, Q., Li, H., Gu, J., Ren, L., et al. (2020). Integrated analysis of comparative lipidomics and proteomics reveals the dynamic changes of lipid molecular species in high-oleic acid peanut seed. J. Agric. Food Chem. 68, 426–438. doi: 10.1021/acs.jafc.9b04179
Liu, H., Li, H., Gu, J., Deng, L., Ren, L., Hong, Y., et al. (2018). Identification of the Candidate Proteins Related to Oleic Acid Accumulation during Peanut (Arachis hypogaea L.) Seed Development through Comparative Proteome Analysis. Int. J. Mol. Sci. 19 (4), 1235. doi: 10.3390/ijms19041235
Liu, H., Liang, X., Lu, Q., Li, H., Liu, H., Li, S., et al. (2020a). Global transcriptome analysis of subterranean pod and seed in peanut (Arachis hypogaea L.) unravels the complexity of fruit development under dark condition. Sci. Rep. 10, 13050. doi: 10.1038/s41598-020-69943-7
Liu, H., Sun, Z., Zhang, X., Qin, L., Qi, F., Wang, Z., et al. (2020b). QTL mapping of web blotch resistance in peanut by high-throughput genome-wide sequencing. BMC Plant Biol. 20, 249. doi: 10.1186/s12870-020-02455-8
Liu, N., Wu, B., Pandey, M. K., Huang, L., Luo, H., Chen, Y., et al. (2022). Gene expression and DNA methylation altering lead to the high oil content in wild allotetraploid peanut (A. monticola). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1065267
Luo, H. Y., Guo, J. B., Yu, B. L., Chen, W. G., Zhang, H., Zhou, X. J., et al. (2021). Construction of ddRADseq-based high-density genetic map and identification of quantitative trait loci for trans-resveratrol content in peanut seeds. Front. In Plant Sci. 12. doi: 10.3389/fpls.2021.644402
Lv, Z., Lan, G., Bai, B., Yu, P., Wang, C., Zhang, H., et al. (2024). Identification of candidate genes associated with peanut pod length by combined analysis of QTL-seq and RNA-seq. Genomics 116, 110835. doi: 10.1016/j.ygeno.2024.110835
Lv, Z., Zhou, D., Shi, X., Ren, J., Zhang, H., Zhong, C., et al. (2022). Comparative multi-omics analysis reveals lignin accumulation affects peanut pod size. Int. J. Mol. Sci. 23, 13533. doi: 10.3390/ijms232113533
Ma, X., Zhang, X., Zhao, K., Li, F., Li, K., Ning, L., et al. (2018). Small RNA and Degradome Deep Sequencing Reveals the Roles of microRNAs in Seed Expansion in Peanut (Arachis hypogaea L.). Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00349
Mukri, G., Nadaf, H. L., Bhat, R. S., Gowda, M. V. C., Upadhyaya, H. D., Sujay, V. (2012). Phenotypic and molecular dissection of ICRISAT mini core collection of peanut (Arachis hypogaea L.) for high oleic acid. Plant Breed. 131, 418–422. doi: 10.1111/j.1439-0523.2012.01970.x
Otyama, P. I., Chamberlin, K., Ozias-Akins, P., Graham, M. A., Cannon, E. K. S., Cannon, S. B., et al. (2022). Genome-wide approaches delineate the additive, epistatic, and pleiotropic nature of variants controlling fatty acid composition in peanut (Arachis hypogaea L.). G3 (Bethesda) 12 (1), jkab382. doi: 10.1093/g3journal/jkab382
Simona, M., Lucia, C., Paul, E. G., Arp, S., Martin, M. K. (2011). The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 23, 865–872. doi: 10.1105/tpc.110.081737
Sun, Z. Q., Qi, F. Y., Liu, H., Qin, L., Xu, J., Shi, L., et al. (2022). QTL mapping of quality traits in peanut using whole-genome resequencing. Crop J. 10, 177–184. doi: 10.1016/j.cj.2021.04.008
Wan, L., Li, B., Pandey, M. K., Wu, Y., Lei, Y., Yan, L., et al. (2016). Transcriptome analysis of a new peanut seed coat mutant for the physiological regulatory mechanism involved in seed coat cracking and pigmentation. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01491
Wang, Z., Huai, D., Zhang, Z., Cheng, K., Kang, Y., Wan, L., et al. (2018b). Development of a high-density genetic map based on specific length amplified fragment sequencing and its application in quantitative trait loci analysis for yield-related traits in cultivated peanut. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00827
Wang, X., Liu, Y., Ouyang, L., Yao, R., He, D., Han, Z., et al. (2022). Metabolomics combined with transcriptomics analyses of mechanism regulating testa pigmentation in peanut. Front. Plant Sci. 16. doi: 10.3389/fpls.2022.1065049
Wang, Y., Ma, X., Zhang, X., He, X., Li, H., Cui, D., et al. (2016). ITRAQ-based proteomic analysis of the metabolic mechanisms behind lipid accumulation and degradation during peanut seed development and postgermination. J. Proteome Res. 15, 4277–4289. doi: 10.1021/acs.jproteome.6b00345
Wang, M. L., Sukumaran, S., Barkley, N. A., Chen, Z., Chen, C. Y., Guo, B., et al. (2011). Population structure and marker-trait association analysis of the US peanut (Arachis hypogaea L.) mini-core collection. Theor. Appl. Genet. 123, 1307–1317. doi: 10.1007/s00122-011-1668-7
Wang, X., Xu, P., Yin, L., Ren, Y., Li, S., Shi, Y., et al. (2018a). Genomic and transcriptomic analysis identified gene clusters and candidate genes for oil content in peanut (Arachis hypogaea L.). Plant Mol. Biol. Rep. 36, 518–529. doi: 10.1007/s11105-018-1088-9
Wang, Z., Yan, L., Chen, Y., Wang, X., Huai, D., Kang, Y., et al. (2022). Detection of a major QTL and development of KASP markers for seed weight by combining QTL-seq, QTL-mapping and RNA-seq in peanut. Theor. Appl. Genet. 135, 1779–1795. doi: 10.1007/s00122-022-04069-0
Wang, J., Yan, C. X., Li, Y., Li, C. J., Zhao, X. B., Yuan, C. L., et al. (2019). GWAS discovery of candidate genes for yield-related traits in peanut and support from earlier QTL mapping studies. Genes 10 (10), 803. doi: 10.3390/genes10100803
Wang, Z., Zhang, Y., Huai, D., Chen, Y., Wang, X., Kang, Y., et al. (2024). Detection of two homologous major QTLs and development of diagnostic molecular markers for sucrose content in peanut. Theor. Appl. Genet. 137, 61. doi: 10.1007/s00122-024-04549-5
Wei, J., Liu, X., Li, L., Zhao, H., Liu, S., Yu, X., et al. (2020). Quantitative proteomic, physiological and biochemical analysis of cotyledon, embryo, leaf and pod reveals the effects of high temperature and humidity stress on seed vigor formation in soybean. BMC Plant Biol. 20, 127. doi: 10.1186/s12870-020-02335-1
Wei, X., Qiu, J., Yong, K., Fan, J., Zhang, Q., Hua, H., et al. (2021). A quantitative genomics map of rice provides genetic insights and guides breeding. Nat. Genet. 53, 243–253. doi: 10.1038/s41588-020-00769-9
Wu, Y., Sun, Z., Qi, F., Tian, M., Wang, J., Zhao, R., et al. (2022). Comparative transcriptomics analysis of developing peanut (Arachis hypogaea L.) pods reveals candidate genes affecting peanut seed size. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.958808
Xue, Q., Zhang, X., Yang, H., Li, H., Lv, Y., Zhang, K., et al. (2021). Transcriptome and metabolome analysis unveil anthocyanin metabolism in pink and red testa of peanut (Arachis hypogaea L.). Int. J. Genomics 2021, 5883901. doi: 10.1155/2021/5883901
Yang, S., Wang, J., Tang, Z., Guo, F., Zhang, Y., Zhang, J., et al. (2020). Transcriptome of peanut kernel and shell reveals the mechanism of calcium on peanut pod development. Sci. Rep. 10, 15723. doi: 10.1038/s41598-020-72893-9
Yang, L., Yang, L., Ding, Y., Chen, Y., Liu, N., et al. (2023). Global transcriptome and co-expression network analyses revealed hub genes controlling seed size/weight and/or oil content in peanut. Plants 12 (17), 3144. doi: 10.3390/plants12173144
Yin, D., Wang, Y., Zhang, X., Li, H., Lu, X., Zhang, J., et al. (2013). De novo assembly of the peanut (Arachis hypogaea L.) seed transcriptome revealed candidate unigenes for oil accumulation pathways. PloS One 8, e73767. doi: 10.1371/journal.pone.0073767
Yoon, J., Min, C. W., Kim, J., Baek, G., Kim, D., Jang, J. W., et al. (2023). Quantitative proteomic analysis deciphers the molecular mechanism for endosperm nuclear division in early rice seed development. Plants 12, 3715. doi: 10.3390/plants12213715
Yu, M., Liu, F., Zhu, W., Sun, M., Liu, J., Li, X. (2015). New features of triacylglycerol biosynthetic pathways of peanut seeds in early developmental stages. Funct. Integr. Genomics 15, 707–716. doi: 10.1007/s10142-015-0447-y
Zhang, S., Gao, H., Wang, L., Zhang, Y., Zhou, D., Anwar, A., et al. (2022). Comparative Transcriptome and Co-Expression Network Analyses Reveal the Molecular Mechanism of Calcium-Deficiency-Triggered Tipburn in Chinese Cabbage (Brassica rapa L. ssp. Pekinensis). Plants 11, 3555. doi: 10.3390/plants11243555
Zhang, S., Hu, X., Miao, H., Chu, Y., Cui, F., Yang, W., et al. (2019). QTL identification for seed weight and size based on a high-density SLAF-seq genetic map in peanut (Arachis hypogaea L.). BMC Plant Biol. 19, 537. doi: 10.1186/s12870-019-2164-5
Zhang, J., Liang, S., Duan, J., Wang, J., Chen, S., Cheng, Z., et al. (2012). De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genomics 13, 90. doi: 10.1186/1471-2164-13-90
Zhang, K., Ma, J., Gangurde, S. S., Hou, L., Xia, H., Li, N., et al. (2022a). Targeted metabolome analysis reveals accumulation of metabolites in testa of four peanut germplasms. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.992124
Zhang, H., Wang, M. L., Dang, P., Jiang, T., Zhao, S. Z., Lamb, M., et al. (2021). Identification of potential QTLs and genes associated with seed composition traits in peanut using GWAS and RNA-Seq analysis. Gene 769, 145215. doi: 10.1016/j.gene.2020.145215
Zhang, Y., Wang, P., Xia, H., Zhao, C., Hou, L., Li, C., et al. (2016). Comparative transcriptome analysis of basal and zygote-located tip regions of peanut ovaries provides insight into the mechanism of light regulation in peanut embryo and pod development. Genes (Basel) 17, 606. doi: 10.1186/s12864-016-2857-1
Zhang, K., Yuan, M., Xia, H., He, L. Q., Ma, J., Wang, M. X., et al. (2022b). BSA-seq and genetic mapping reveals as a candidate gene responsible for red testa of peanut. Theor. And Appl. Genet. 135, 1529–1540. doi: 10.1007/s00122-022-04051-w
Zhang, X. G., Zhang, J. H., He, X. Y., Wang, Y., Ma, X. L., Yin, D. M. (2017). Genome-wide association study of major agronomic traits related to domestication in peanut. Front. In Plant Sci. 8. doi: 10.3389/fpls.2017.01611
Zhao, X., Li, C., Zhang, H., Yan, C., Sun, Q., Wang, J., et al. (2020a). Alternative splicing profiling provides insights into the molecular mechanisms of peanut peg development. BMC Plant Biol. 20, 488. doi: 10.1186/s12870-020-02702-y
Zhao, Y. H., Ma, J. J., Li, M., Deng, L., Li, G. H., Xia, H., et al. (2020b). Whole-genome resequencing-based QTL-seq identified gene encoding a R2R3-MYB transcription factor controlling peanut purple testa colour. Plant Biotechnol. J. 18, 96–105. doi: 10.1111/pbi.13175
Zhao, H. L., Tian, R. Z., Xia, H., Li, C. S., Li, G. H., Li, A. Q., et al. (2022). High-density genetic variation map reveals key candidate loci and genes associated with important agronomic traits in peanut. Front. In Genet. 13. doi: 10.3389/fgene.2022.845602
Zhao, X., Wang, J., Xia, N., Liu, Y., Qu, Y., Ming, M., et al. (2023). Combined analysis of the metabolome and transcriptome provides insight into seed oil accumulation in soybean. Biotechnol. Biofuels 16 (1), 70. doi: 10.1186/s13068-023-02321-3
Zhou, X. J., Guo, J. B., Pandey, M. K., Varshney, R. K., Huang, L., Luo, H. Y., et al. (2021). Dissection of the genetic basis of yield-related traits in the Chinese peanut mini-core collection through genome-wide association studies. Front. In Plant Sci. 12. doi: 10.3389/fpls.2021.637284
Zhou, X. J., Luo, H. Y., Yu, B. L., Huang, L., Liu, N. A., Chen, W. G., et al. (2022). Genetic dissection of fatty acid components in the Chinese peanut minicore collection under multi-environments. PloS One 17 (12), e0279650. doi: 10.1371/journal.pone.0279650
Zhu, W., Chen, X., Li, H., Zhu, F., Hong, Y., Varshney, R. K., et al. (2014). Comparative transcriptome analysis of aerial and subterranean pods development provides insights into seed abortion in peanut. Plant Mol. Biol. 85, 395–409. doi: 10.1007/s11103-014-0193-x
Keywords: omics, seed development, peanut, yield, quality
Citation: Wang Z, Lei Y and Liao B (2024) Omics-driven advances in the understanding of regulatory landscape of peanut seed development. Front. Plant Sci. 15:1393438. doi: 10.3389/fpls.2024.1393438
Received: 29 February 2024; Accepted: 18 April 2024;
Published: 03 May 2024.
Edited by:
Zhaorong Hu, China Agricultural University, ChinaReviewed by:
Xin Wei, Shanghai Normal University, ChinaCopyright © 2024 Wang, Lei and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Lei, bGVpeW9uZ0BjYWFzLmNu; Boshou Liao, bGJvc2hvdUBob3RtYWlsLmNvbQ==