 Jiazhu Peng
Jiazhu Peng Yin Gao
Yin Gao Yanchun Qiao1*
Yanchun Qiao1* Guoping Wang
Guoping Wang- 1Vegetable Research Science Department, Guangzhou Academy of Agricultural and Rural Sciences, Guangzhou, China
- 2Vegetable Research Institute, Guangzhou Academy of Agricultural Sciences, Guangzhou, China
- 3College of Horticulture, South China Agricultural University, Guangzhou, China
Introduction: Flesh color is an essential trait in chieh-qua (Benincasa hispida Cogn. var. Chieh-qua How); however, the inheritance and molecular basis of green flesh trait remain unclear.
Methods: In the present study, two F2 populations, derived from 1742 (white flesh) × FJ3211 (green flesh) and J16 (white flesh) × FJ5 (green flesh), were used to identify the green flesh (Cqgf) locus.
Results: Genetic analysis revealed that the presence of green flesh was a quantitative trait that closely followed a normal distribution. Combining the results from QTL mapping and BSA-seq analysis, the Cqgf locus was preliminarily determined to be located on chromosome 05 and was narrowed down to a 2.55-Mb interval by linkage analysis. A large J16 × FJ5 F2 population comprising 3,180 individuals was subsequently used to screen the recombinants, and the Cqgf locus was fine-mapped to a region of 329.70 kb that harbors six genes. One of the candidate genes, Bch05G003700, the zinc-finger transcription factor LOL1 (lsd one like 1 protein; CqLOL1), was the strongest candidate gene for the Cqgf locus according to sequence variation and expression analysis. Additionally, a point mutation (A > C) in CqLOL1 resulted in the substitution of threonine (T) with proline (P) in the amino acid sequence, showing a complete relationship linked with flesh color in a panel of 45 germplasms.
Discussion: The study suggests that CqLOL1 promotes the accumulation of chlorophyll content in chieh-qua and lead to green flesh. Our findings establish a theoretical and technical foundation for breeding different flesh color lines and elucidating the underlying mechanisms of flesh color in chieh-qua.
Introduction
The color of fruit flesh is a crucial characteristic that holds significant commercial value. It is primarily determined by the content and composition of lycopene, anthocyanin, carotenoids, and chlorophyll, making it a key trait for breeding purposes (Blas et al., 2010; Wang et al., 2019; Zhao et al., 2022). Chlorophyll, composed of chlorophyll a and b, contributes predominantly to green pigmentation and plays a critical role in plant development. It serves as the site of photosynthesis and facilitates the production of other important components such as hormones, amino acids, and fatty acids. Cucurbit crops exhibit a diverse range of flesh colors, including red, yellow, orange, white, and green.
In watermelon (Citrullus lanatus), the red flesh is composed mainly of lycopene and a small amount of β-carotene (Kang et al., 2010), orange flesh is largely pigmented by β-carotene (Branham et al., 2017), and the yellow and canary yellow flesh, respectively, results from small amounts of violaxanthin and neochrome (Bang et al., 2010). The genetic basis of flesh color in watermelon is complex, and several loci/genes are known to affect the flesh color of watermelon. The C and I loci govern canary yellow and red flesh in watermelon (Poole, 1944). In addition, canary yellow (C) is dominant over red flesh (c) but can be suppressed by homozygous recessive i loci which can lead to red flesh regardless of which C allele is present (Henderson et al., 1998). The Cyf locus for canary yellow flesh color was determined to be a 79.62-kb region on chromosome 6 harboring 10 predicted genes, and Cla97C06G122050 and Cla97C06G122120 (pentatricopeptide repeat protein) were predicted to be important candidate genes (Liu et al., 2023). Compared with canary yellow flesh, white flesh is controlled by a recessive locus, and this locus (Clwf2) was determined to be located within a 132.3-kb region on chromosome 6 which included 13 candidate genes (Yi et al., 2023). A locus associated with pale yellow and white flesh color was mapped on chromosome 6 and further narrowed into a 66.8-kb region containing nine candidate genes, of which Cla007528, which has a non-synonymous mutation, was suggested to be the strongest candidate gene (Wang et al., 2021). A major locus responsible for β-carotene accumulation was determined on chromosome 1 through linkage analysis (Branham et al., 2017). Red flesh color is recessive to pale yellow color, and the locus was narrowed to a 24-kb region on chromosome 4 harboring two genes, of which the lycopene β-cyclase gene (LCYB) was considered the key gene (Wang et al., 2019). Furthermore, the downregulation of LCYB leads to a change in flesh color from pale yellow to red, while the overexpression of LCYB in red flesh lines leads to a change to orange (Zhang et al., 2020). Genome-wide association studies have also shown that the mutation of the LCYB gene from phenylalanine to valine leads to increased lycopene accumulation, and other candidate gene ClTST2 on chromosome 2 likely promotes carotenoid accumulation through ClPHT4 (Guo et al., 2019). In addition, the expression of ClPHT4 was necessary for carotenoid lycopene accumulation (Zhang et al., 2016), and ClPSY1 was positively responsible for lycopene accumulation (Guo et al., 2019). The scarlet red flesh (Yscr) was dominant over coral red flesh in watermelon and was located on chromosome 6 within a 40-kb interval which included five putative genes (Li et al., 2020). In addition, a major QTL (qfc10.1) related to pale green flesh color and chlorophyll content was found within a 519-kb region on chromosome 10 harboring 22 annotated genes (Pei et al., 2021).
In melon (Cucumis melo L.), due to the accumulation of chlorophyll and carotenoids, the flesh color mainly appears light green, white, or orange, and β-carotene is the predominant pigment (Shahwar et al., 2023). In previous studies, the genetic basis of flesh color variation was partly resolved, and two major genes, green flesh (gf) and white flesh (wf), were shown to govern the flesh color. CmOr, located on chromosome 9 and previously described as the gf locus in melon, had two haplotypes (alleles), one of which was associated with orange flesh and the other was associated with white or green flesh (Tzuri et al., 2015). The SNP within CmOr was also confirmed by complete segregation with orange flesh (Gur et al., 2017). The wf locus was likely to be CmPPR1 (MELO3C003069), which was located on chromosome 8. This gene encodes pentatricopeptide protein and participates in RNA processing in the plastid where carotenoids and chlorophyll accumulate (Galpaz et al., 2018). To decipher the wf locus, a 96-kb-overlap interval containing 11 protein coding genes was determined by QTL and GWAS, which excluded the CmPPR1 gene, but MELO3C003097 was confirmed to be a strong candidate gene (Zhao et al., 2019). In addition, CmAPRR2 was identified as a causal gene for pigment content in the rind and flesh of mature melon fruits (Oren et al., 2019).
Most of the cucumber flesh is white, while some materials have orange or green flesh because of the accumulation of β-carotene and chlorophyll. Compared with that in white flesh, the β-carotene content in the mesocarp was controlled by two recessive genes (Cuevas et al., 2010), and the β-carotene content in the endocarp was controlled by a recessive gene (Bo et al., 2012). The locus associated with orange flesh in the endocarp (CsOr) was subsequently identified on chromosome 6, which included two carotenoid-related genes, of which Csa6G452720, homolog of CmOr, was considered the causal gene of orange flesh (Kishor et al., 2021). Green flesh (Csgf) seems to be incompletely recessive to white flesh, and Csgf was controlled by two QTLs, qgf5.1 and qgf3.1 (Bo et al., 2019). The yellow flesh (yf) locus was fine-mapped into a 150-kb region on chromosome 7 harboring 21 candidate genes (Lu et al., 2015).
Chieh-qua (Benincasa hispida Cogn. var. Chieh-qua How), one of the most important cucurbit vegetables, is classified as a variety of wax gourd (Benincasa hispida (Thunb.) Cogn.) and is extensively cultivated in southern China and various Southeast Asian countries. Unlike the mature stage consumption of wax gourd, chieh-qua fruits can be harvested at both immature and mature stages, depending on market demands, and mature fruits have the advantage of long-term storage. In contrast to those of other cucurbits, the flesh color of chieh-qua can be categorized into two primary types: green and white. However, the genetic basis underlying flesh color in chieh-qua has not been determined. Since the completion of wax gourd genome sequencing (Luo et al., 2023; Xie et al., 2019), a scientific foundation has been established to unravel the molecular regulatory mechanisms governing traits, including flesh color.
This study aimed to elucidate the regulatory gene responsible for the green flesh trait in chieh-qua. To achieve this, two F2 populations with different genetic backgrounds were utilized for the genetic analysis of green flesh. QTL mapping, BSA-seq, and fine-mapping were performed to identify the Csgf locus. By combining expression and sequence variation analysis, we identified an important candidate gene Bch05G003700, namely CqLOL1, which belongs to the zinc-finger transcription factor LOL1 family. Our findings not only facilitate marker-assisted selection but also enhance our understanding of the molecular mechanisms underlying green flesh development.
Materials and methods
Plant materials and phenotypic evaluation
To generate segregating populations for analysis, two white flesh lines, 1742 and J16, were crossed with two green flesh lines, FJ3211 and FJ5. The resulting populations included 120 individuals in the 1742 × FJ3211 F2 population and 363 individuals in the J16 × FJ5 F2 population. Two distinct sets of materials were utilized to investigate the inheritance patterns of green flesh. Specifically, the 1742 × FJ3211 F2 population was employed for the construction of genetic maps and for conducting QTL mapping, while the J16 × FJ5 F2 population was utilized for BSA-seq, linkage mapping, and precise mapping of the candidate genes associated with green flesh. The flesh of J16 and FJ5 were sampled at 5, 10, 15, 20, 25, and 30 days after pollination (DAP), and the flesh color was significantly different at all stages (Supplementary Figure S1). To conduct shading experiments, light-impermeable bags were employed to completely cover the female flower of FJ5, including the ovary, after pollination. Additionally, the flesh sampled at 10 DAP under shading conditions were collected to analyze the expression of candidate genes.
In addition, 45 extreme chieh-qua materials were used to clone and compare the causal gene controlling the flesh color and used for marker-assisted selection tests. All the materials were provided by Guangzhou Academy of Agricultural Sciences and cultivated at the Nansha Experimental Base of the Guangzhou Academy of Agricultural Sciences (Guangzhou, China, 23.4 N, 113.4 E). The phenotype of flesh was evaluated by two indices at 10 DAP, flesh color grouping (FCG), and flesh chlorophyll content (FCC). FCG was categorized into four types by visual inspection, and FCC was extracted with 95% alcohol and calculated by using a previous method (Bo et al., 2019).
QTL mapping analysis
The genotypes of F2 individuals derived from 1742 × FJ3211 were obtained through SLAF-seq (Sun et al., 2013), and a high-density genetic map (unpublished) was constructed by using a method developed previously (Li et al., 2008). The map was divided into 12 linkage groups containing 5,354 markers and spanned a total of 1,997.32 cM, with an average distance of 0.37 cM between adjacent markers. QTL mapping was conducted using Highmap software via the composite interval mapping (CIM) method, and the maximum limit of detection (LOD) threshold was determined via permutations (1,000 times) (Liu et al., 2014).
BSA-seq analysis
The green flesh (GF) and white flesh (WF) DNA pools were constructed by mixing equal amounts of DNA samples from 30 green and 30 white flesh F2 individuals derived from J16 × FJ5, respectively. The two extreme bulks were subjected to whole-genome resequencing on the Illumina HiSeq platform of Biomarker Technologies (www.biomarker.com.cn). The resequencing data were filtered and aligned to the reference genome of wax gourd (GX-19; unpublished) using BWA software with default parameters. Then, the variations including InDels and SNPs were detected by using GATK software, and high-quality InDels and SNPs with GQ>50 in the two bulks were subjected to InDel and SNP index analysis. Then, the ΔInDel-index and ΔSNP-index were obtained by using the InDel and SNP index algorithms, respectively, for the two bulks by sliding window analysis with a 200-kb window size and a step length of 100 kb, and the fitted values with 99% confidence interval were used to identify the candidate region responsible for the green flesh. After overlapping the regions identified by two algorithms, we obtained the final candidate interval governing green flesh from BSA-seq.
Genotyping and linkage mapping analysis
The resequencing data of materials J16 and FJ5 were also aligned to the reference genome of wax gourd, and eight polymorphic InDel markers, Fc_1–Fc_8, within the candidate region were developed to genotype their 363 F2 individuals using Primer 3plus (https://primer3.ut.ee/). A molecular linkage map was constructed using IciMapping software (Meng et al., 2015). Polymerase chain reaction (PCR) was conducted in a 10-µL reaction mixture containing 1 µL of the DNA template (50–100 ng µL−1), 1 µL of forward primer (10 µmol L−1) and reverse primer (10 µmol L−1), 5 µL of 2× GoTaq Green Master Mix, and 2 µL of ddH2O. The PCR procedure was as follows: 95°C for 3 min; 34 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s; and 72°C for 5 min. The PCR products were subjected to 6% polyacrylamide gel eletrophoresis (PAGE). To narrow the region of Cqgf, a large J16 × FJ5 F2 population consisting of 3,810 individuals was genotyped by two flanking markers, Fc1 and Fc5, for screening of homozygous recombinants. Furthermore, five InDel markers, Fc_9–Fc_13, within the region were developed to genotype the recombinants. All InDel markers are listed in Supplementary Table S2. Then, we constructed genotype-based haplotypes for the recombinants and inferred the most likely region of Cqgf.
Sequence variation analysis and dCAPS marker development
Total RNA was extracted from the flesh using the Eastep Super Total RNA Extraction Kit (Promega, Shanghai, China), and cDNA was obtained by reverse transcription using a cDNA synthesis kit (Promega, Shanghai, China). Candidate genes responsible for the green flesh were cloned and subjected to Sanger sequencing by Beijing Tsingke Biotech Co., Ltd. Sequence alignment was performed using DNAMAN software.
Expression analysis
The expression of candidate genes was assessed in different tissues including root, stem, leaf, and flesh at six different stages (5 to 30 DAP) and the 10 DAP flesh under shading using quantitative real-time polymerase chain reaction (qRT-PCR). The expression levels of three biological and technical repeats for each sample were calculated with the ΔΔCt method (Livak and Schmittgen, 2001). The primer pairs are shown in Supplementary Table S4.
Construction of the phylogenetic tree
The homologous genes of LOL1 in different crops were searched from NCBI database to obtain the corresponding amino acid sequences. Then, all of the amino acid sequences were aligned by using MUSCLE, and a neighbor-joining phylogenetic tree was constructed using MEGA11.0 with 1,000 bootstrap replications. Finally, the phylogenetic tree and predicted motif were integrated and drawn with TBtools (Chen et al., 2023).
Results
Phenotypic analysis of green flesh
To identify the inheritance pattern of flesh color, two white flesh (1742 and J16) and two green flesh (FJ3211 and FJ5) chieh-qua inbreds were used to construct two segregating populations, 1742 × FJ3211 and J16 × FJ5 F2 population, and the flesh of parents and their F1 are shown in Figure 1A. The phenotypes of FCG were categorized based on visual observation (Figure 1B), and FCC were collected from two populations, including two panels of parents, their F1, and 93 and 363 individuals from 1742 × FJ3211 and J16 × FJ5 population, respectively. Significant differences in FCC were observed among the female parent, male parent, and F1 progeny. Notably, two male parents exhibited the highest FCC values, and their F1 progeny displayed a biased preference toward white flesh (Figure 1C). The frequency distributions of FCG and FCC from both crosses were continuous, suggesting the quantitative nature of flesh color (Figures 1D–F).
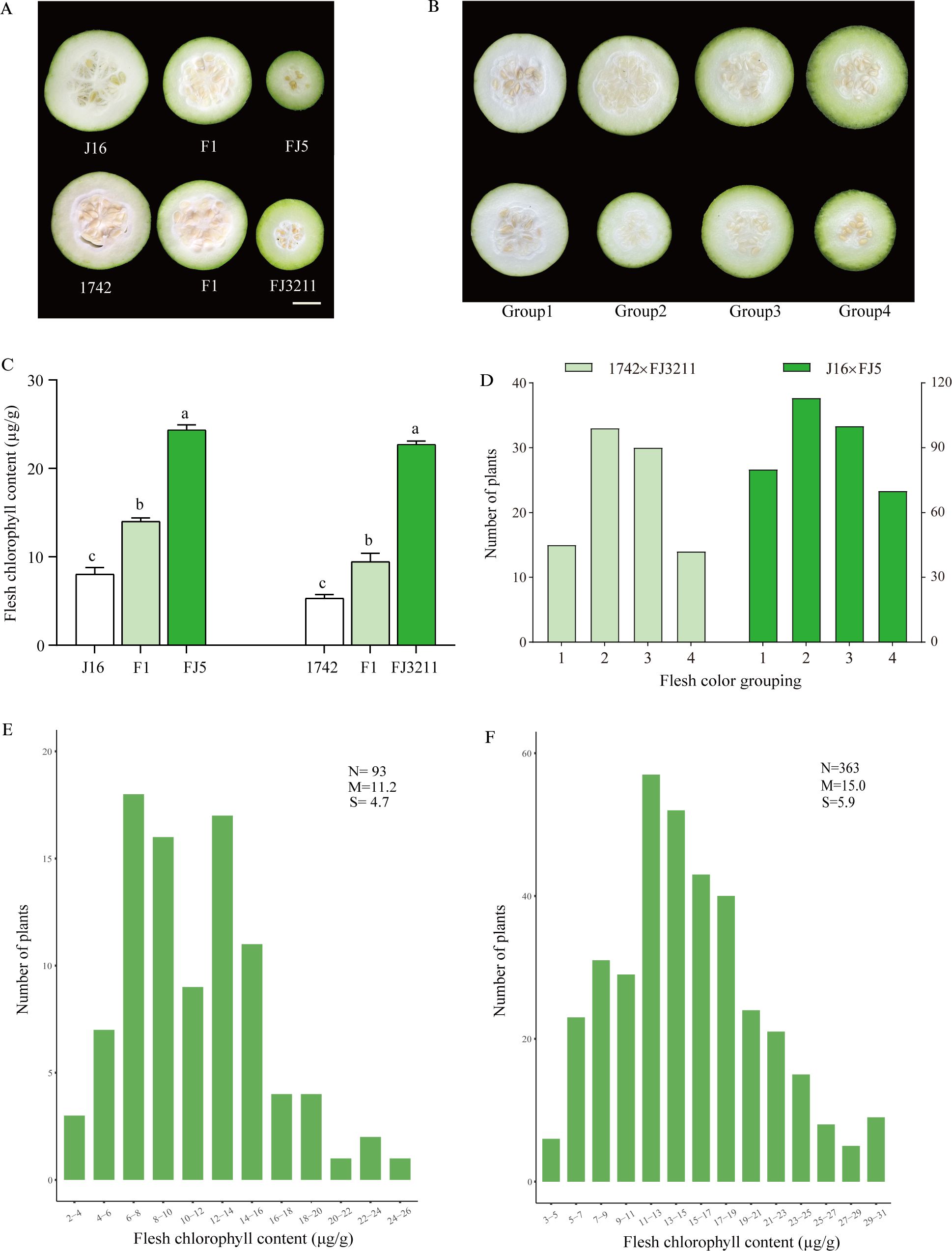
Figure 1. Flesh color and chlorophyll content of the four parents, their F1 and F2 generations. (A) Flesh color performances of four parents and their F1, scale bar = 3 cm. (B) Flesh color of the four groups in F2 population. (C) Flesh chlorophyll content of four parents and their F1. The chlorophyll contents are the mean ± SD (n = 3). (D) Flesh color grouping distributions of two F2 populations. (E) Flesh chlorophyll content distribution of the F2 population derived from 1742 × FJ3211. (F) Flesh chlorophyll content distribution of the F2 population derived from J16 × FJ5. a,b,c represent significant difference at the 0.05 level; N, the number of investigated plants; M, mean; S, standard deviation.
QTL mapping analysis
A high-density genetic map was generated using sequencing data obtained from the F2 population of 1742 × FJ3211. Subsequently, a QTL analysis was conducted using the composite interval mapping (CIM) method, employing FCG and FCC indexes. The results revealed the presence of a single peak for both FCG and FCC on chromosome 05 (Figures 2A, B). Notably, these two QTLs, designated qfcg5 and qfcc5, were consistently located at 24.67–25.51 cM and exhibited LODs of 19.84 and 11.33, respectively (Table 1). These QTLs accounted for 48.62% and 38.75% of the phenotypic variation, respectively, suggesting that a major locus governs the green flesh trait.
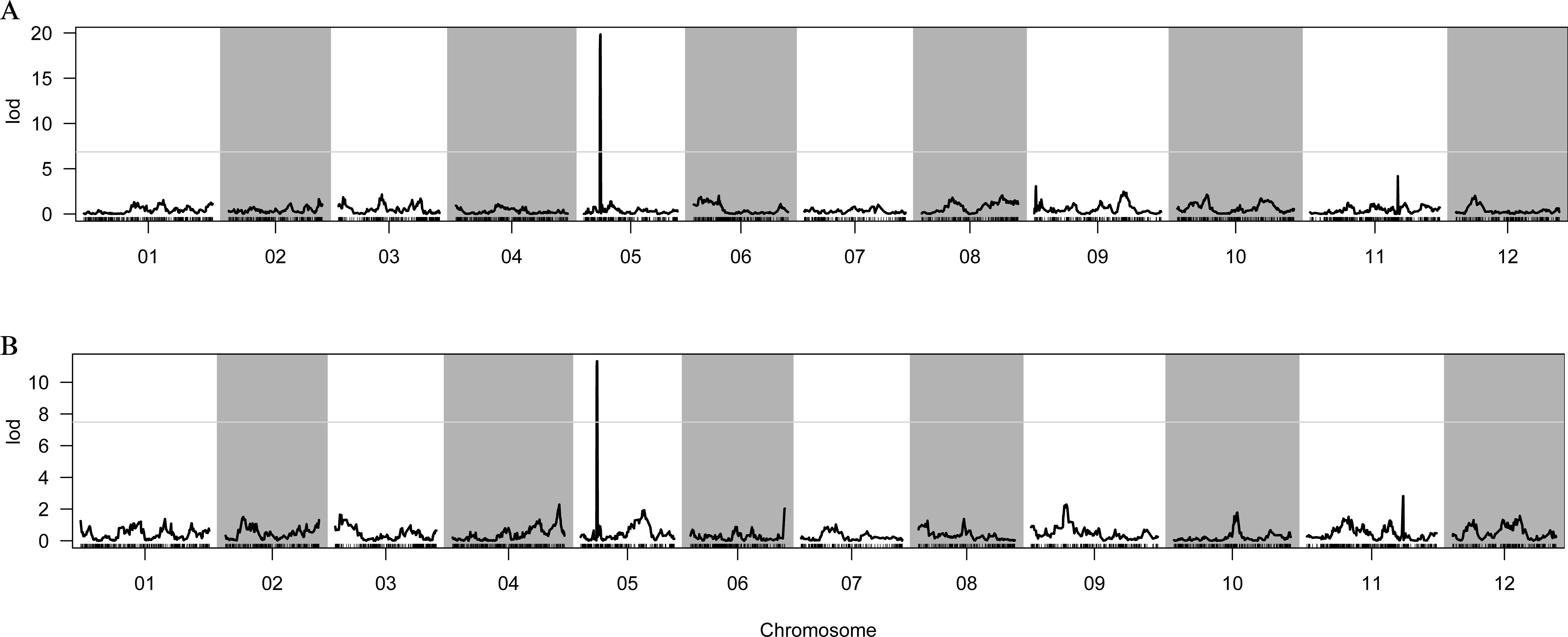
Figure 2. Genetic mapping of FCG and FCC by the 1742 × FJ3211 F2 population. (A) Distribution of LODs of FCG. (B) Distribution of LOD values of FCC. 01–12 represent the linkage groups corresponding to the chromosomes of the reference genome.
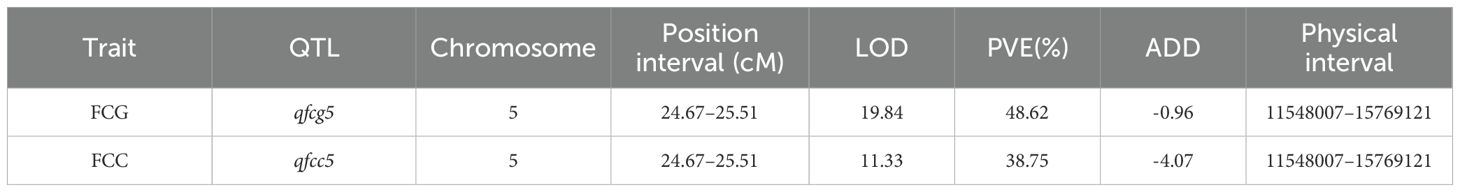
Table 1. Quantitative trait loci (QTLs) for flesh color in the F2 population.
BSA-seq identified the Cqgf locus
After filtering the raw reads from the whole-genome resequencing of the two extreme bulks (GF and WF), 2.03 billion (~30.32 Gb) and 2.12 billion (~31.67 Gb) clean reads were generated, with Q20 values of 98.95% and 98.86%, respectively (Supplementary Table S1). Then, a large number of high-quality InDels and SNPs were identified through variant calling and used to identify the Cqgf locus. The application of the InDel index algorithm to two bulks identified a prominent locus on Chr05 spanning from 8.56 to 20.06 Mb (Figure 3A). Furthermore, the employment of the SNP index algorithm with two bulks revealed three distinct loci. One of these regions was situated on Chr05, covering the genomic interval from 8.32 to 21.40 Mb (Figure 3B). The remaining two regions were located on chr06 (50.30 to 52.76 Mb) and chr10 (20.68 to 22.94 Mb). Overlapping the regions identified by two algorithms, Cqfc locus was preliminarily located on Chr05 from 8.56 to 20.06 Mb, with a physical distance of 11.60 Mb.
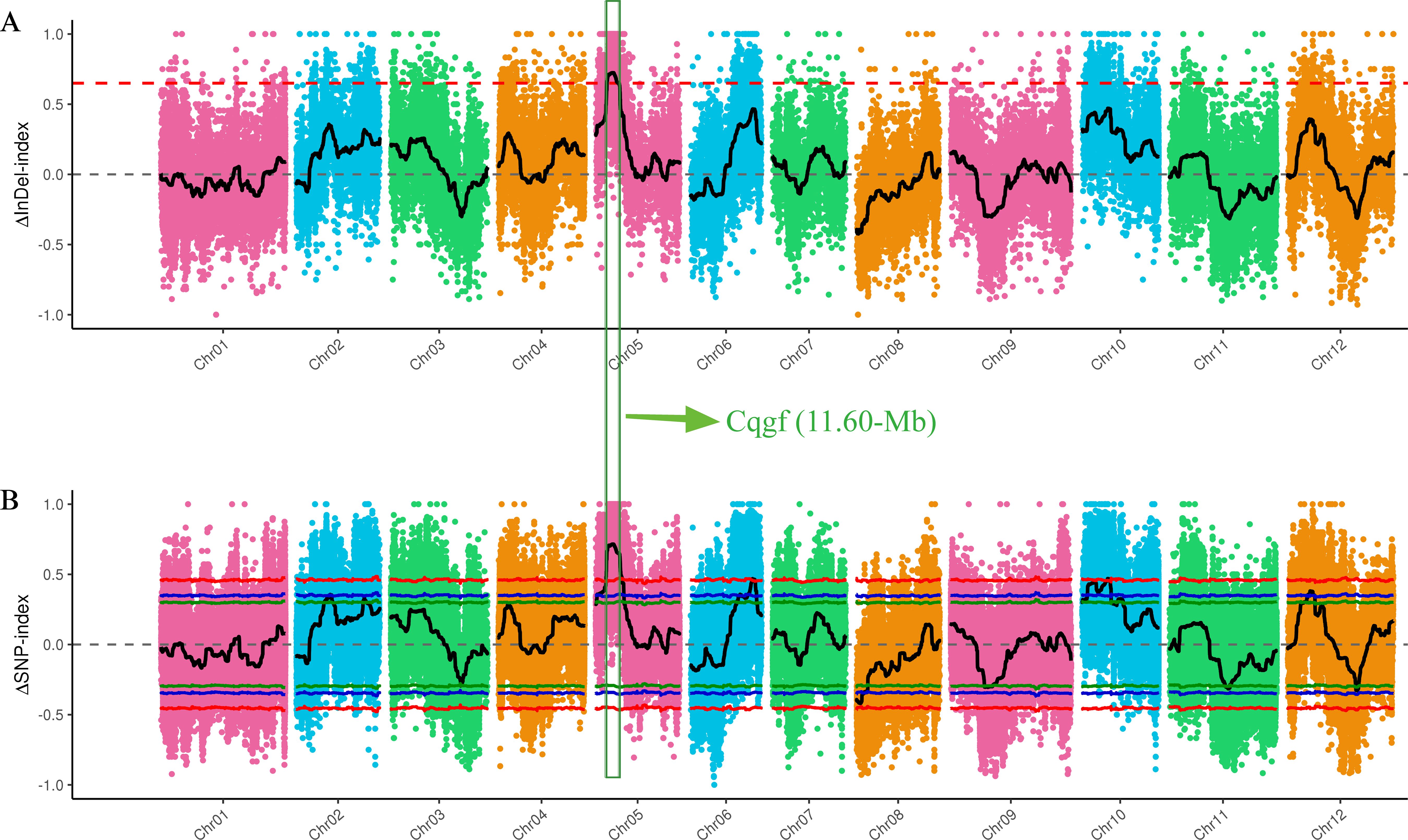
Figure 3. BSA-seq mapping of the Cqgf locus. (A) InDel index algorithm for two bulks. (B) SNP index algorithm for two bulks. The overlapping region is surrounded by the green box.
Fine-mapping of the Cqgf locus
After integrating the findings from genetic mapping and BSA-seq, the Cqgf locus was determined to be located on Chr05. Given that the Cqgf locus was previously identified through analysis of diverse segregating populations, the 8.58–20.18-Mb region, which encompasses the region identified in the genetic mapping analysis, was selected as the locus for subsequent investigations. To further verify the Cqgf locus, eight polymorphic InDel markers, Fc_1–Fc_8, distributed within the candidate region were developed and used to genotype 363 individuals from the J16 × FJ5 F2 population. Based on the genotypes and phenotypes, a molecular linkage map with a genetic distance ranging from 0 to 17.08 cM was constructed, and Cqgf was narrowed to a 2.55- Mb region from 10.40 to 12.96 Mb flanked by the InDel markers Fc_1 and Fc_5 (Figure 4A).
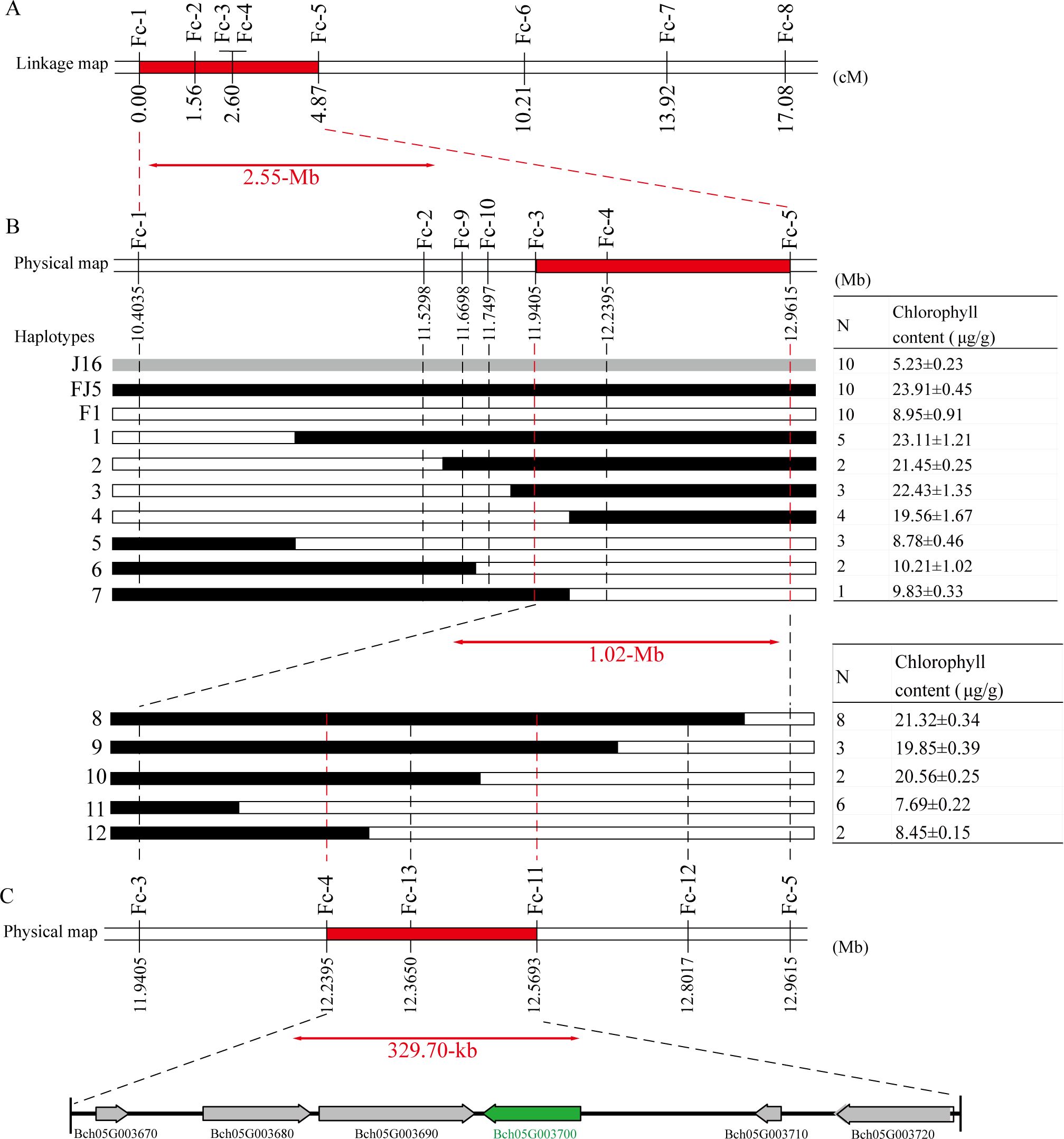
Figure 4. Fine mapping of the Cqgf locus. (A) Linkage mapping of the Cqgf locus. The number below the bars represents the genetic distance. The Cqgf locus is located in the region highlighted in red color and flanked by markers Fc_1 and Fc_5. (B) Physical map of the markers used to genotype the 20 recombinant plants. The number below the bar corresponds to the physical position of the reference genome. Seven haplotypes (1–7) representing the 20 recombinant plants screened (with markers Fc_1 and Fc_5) from 781 F2 individuals and five haplotypes (8–12) representing the 21 recombinant plants screened (with markers Fc_3 and Fc_5) from 2,399 F2 individuals. The red bar represents the Cqgf locus. (C) Physical map of the markers used to genotype the 21 recombinant plants. The arrows represent the predicted genes.
Based on genotyping with the two flanking markers (Fc_1 and Fc_5), 20 recombinant plants were identified from the 781 F2 individuals. Then, those recombinants were divided into seven haplotypes using the markers Fc_2–Fc_4 and two newly developed markers (Fc_9 and Fc_10). By utilizing the chlorophyll content and genotype data of recombinants, the Cqgf locus was further mapped into a 1.02-Mb physical interval flanked by the markers Fc_3 and Fc_5 (Figure 4B).
To determine a more precise region for the Cqgf locus, a large J16 × FJ5 F2 population consisting of 2,399 individuals was genotyped by the markers Fc_3 and Fc_5, and a total of 21 homozygous recombinants were obtained. Using the Fc_4 marker and three newly developed markers (Fc_11–Fc_13), those recombinants were divided into five haplotypes. Notably, the marker Fc_13 showed a complete linkage relationship with the chlorophyll content of 21 recombinants. Thus, the Cqgf locus was finally fine-mapped to a 329.7-kb region from 12.24 to 12.57 Mb (Figure 4C). Based on the genome of wax gourd, six genes were predicted (Table 2). The genes Bch05G003680, Bch05G003690, Bch05G003700, and Bch05G003720 were found to encode the phospholipid hydroperoxide glutathione peroxidase, 2Fe-2S ferredoxin-like protein, lsd one like 1 protein, and plant intracellular ras-group-related LRR protein 4, respectively. The remaining two genes were found to encode uncharacterized proteins.
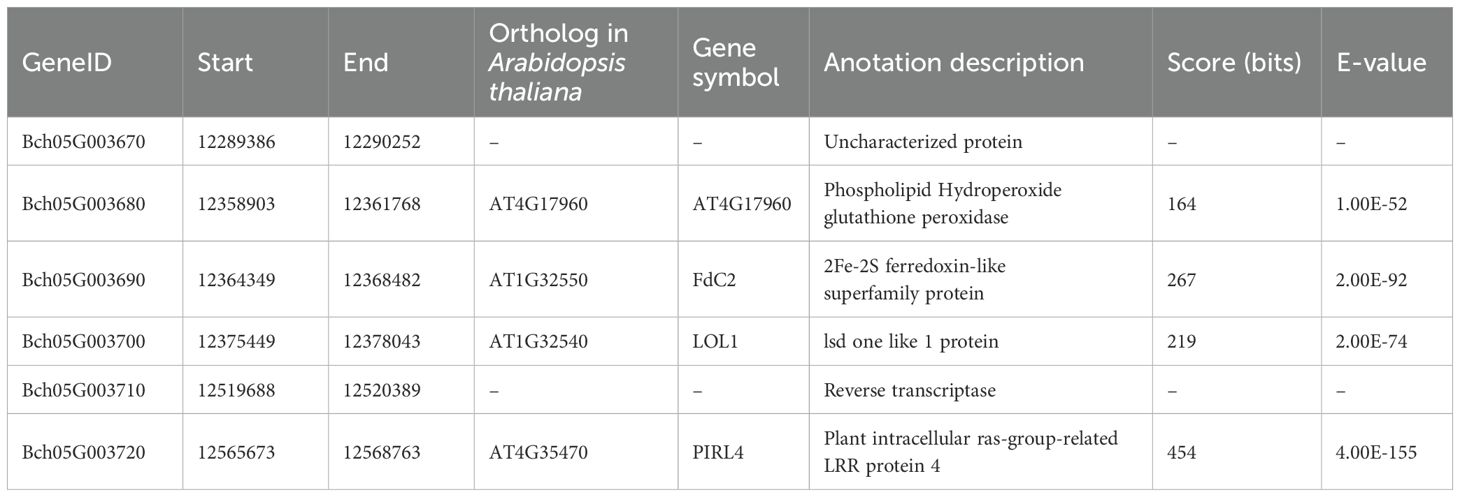
Table 2. List of six annotated genes for the Cqgf locus.
Expression analysis of candidate genes by qRT-PCR
To further determine the causal gene of flesh color, we compared the relative expression levels of the six candidate genes from 5 to 30 DAP. The results demonstrated that the expression levels of the four genes, Bch05G003670, Bch05G003680, Bch05G003710, and Bch05G003720, did not exhibit significant differences across all developmental stages (Figures 5A, B, E, F). Remarkably, the expression levels of Bch05G003700 were significantly higher than that in J16 at six stages, which corresponded to the observed difference in flesh color of the parents (Supplementary Figure S1), reaching its peak at 10 DAP and subsequently decreasing (Figure 5C). Similarly, the expression level of Bch05G003690 in FJ5 was significantly higher than that in J16 at 5, 10, and 15 DAP, whereas no significant difference was observed from 20 to 30 DAP (Figure 5D).
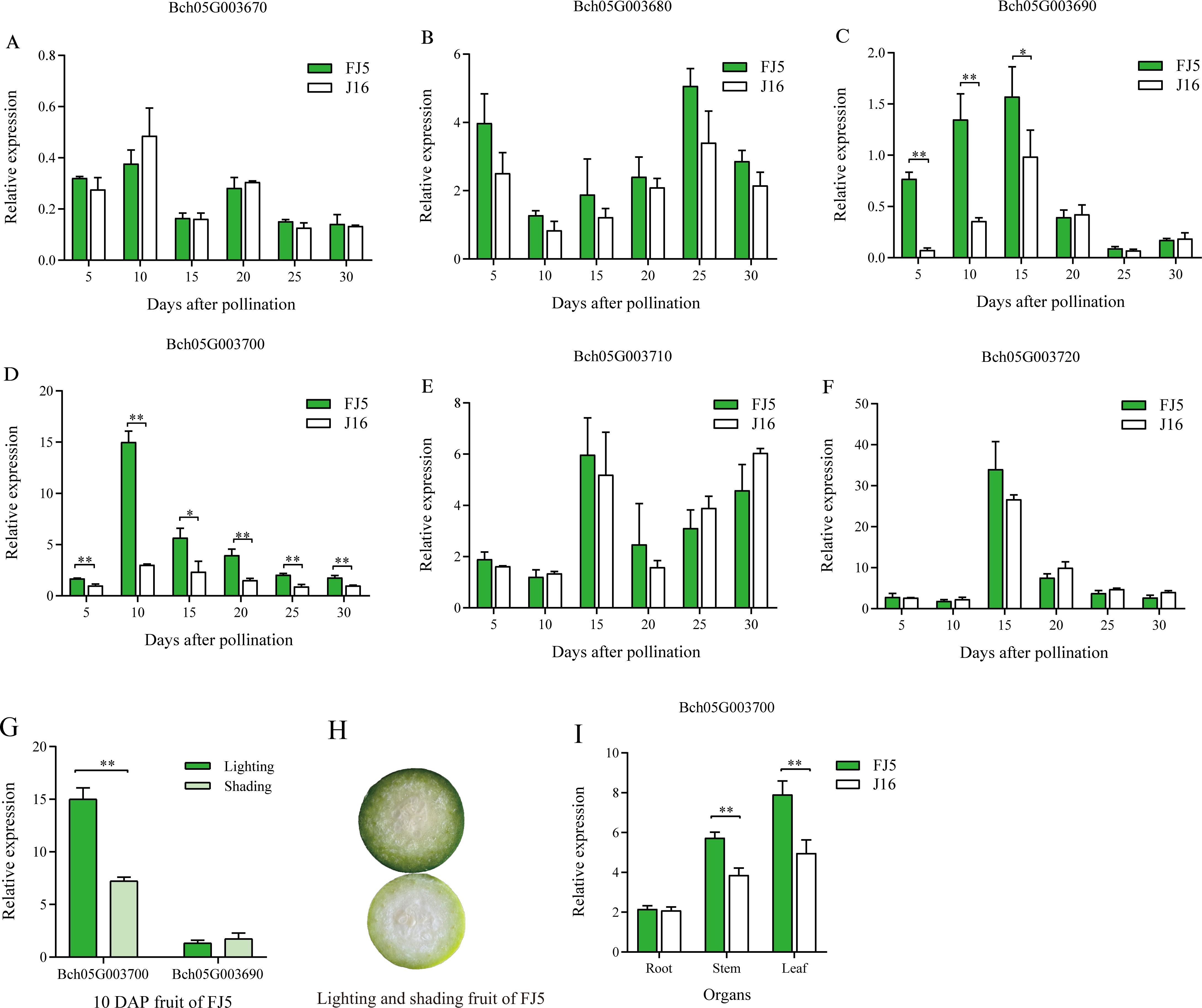
Figure 5. Expression analysis of the candidate genes between FJ5 and J16. (A) The relative expression levels of Bch05G003670 from 5 to 30 DAP. (B) Relative expression levels of Bch05G003680 from 5 to 30 DAP. (C) Relative expression levels of Bch05G003690 from 5 to 30 DAP. (D) Relative expression levels of Bch05G003700 from 5 to 30 DAP. (E) Relative expression levels of Bch05G003710 from 5 to 30 DAP. (F) Relative expression levels of Bch05G003720 from 5 to 30 DAP. (G) Expression levels of Bch05G003690 and Bch05G003700 in 10 DAP flesh of FJ5 under lighting and shading condition. (H) Phenotypes of the 10 DAP flesh in FJ5 under lighting and shading condition. The upper part of fruit was under normal lighting and the below part was under shading. (I) Expression levels of Bch05G003700 in root, stem, and leaf. The relative expression levels are mean ± SD (n = 3). * represents significance at the 0.05 level (Student’s t-test), ** represents significance at the 0.01 level (Student’s t-test).
The homologs of these two genes were associated with chlorophyll synthesis in other species. Specifically, Bch05G003690, classified as a ferredoxin-like protein, played a crucial role in electron transfer during chlorophyll biosynthesis, and Bch05G003700, a zinc-finger transcription factor LOL1, was identified as a regulator of programmed cell death in Arabidopsis and rice (Epple et al., 2003; Wang et al., 2005) and as a regulator of controlled chloroplast development in tomato and pepper (Borovsky et al., 2019). Thus, these two genes were most likely causal genes associated with green flesh in chieh-qua. Furthermore, as the green flesh color was lighter under shading conditions (Figure 5H), the 10 DAP flesh of FJ5 under shading was used to evaluate the impact of shading on the expression levels of Bch05G003690 and Bch05G003700. The results showed that there was no significant alteration in the expression level of Bch05G003690 between lighting and shading conditions (Figure 5G). In contrast, the expression level of Bch05G003700 significantly decreased under shading conditions and reached approximately half of the expression level observed under lighting conditions (Figure 5G). The reduced expression level of Bch05G003700 corresponded to the observed phenotypic difference. In addition, Bch05G003700 gene was also highly expressed in leaf and stem, indicating that Bch05G003700 was highly expressed in green tissue, and there was a significant difference in the expression of stem and leaf between two parents (Figure 5I).
Sequence characterization of the candidate genes
We then focused mainly on the variation in the coding sequences of Bch05G003690 and Bch05G003700 based on the resequencing data of J16 and FJ5. As a result, no variation was detected in the coding sequence of Bch05G003690 (Supplementary Figure S2), while Bch05G003700 had a point mutation (A>C) in the coding sequence, resulting in the conversion of the amino acid T to P (Figures 6A, C). The mutation in Bch05G003700 was also confirmed through cDNA cloning and Sanger sequencing (Figure 6A). Moreover, a perfect correlation between the mutation and flesh color was observed among 45 chieh-qua materials, which had 23 green flesh samples and 22 white flesh samples, including inbred lines and commercial F1 varieties (Figure 6B; Supplementary Table S5). Taken together, we speculate that Bch05G003700, CqLOL1, was the strongest candidate gene for the Cqgf locus, and the mutation might be responsible for the accumulation of chlorophyll content.

Figure 6. Variation of the nucleotide and amino acid sequences of Bch05G003700. (A) Gene structure and variation position between J16 and FJ5. (B) Nucleotide variation in Bch05G003700 in 45 chieh-qua germplasms. (C) Corresponding amino mutational loci. “G” and “W” represent green and white flesh color, respectively.
Furthermore, this mutation was developed into a dCAPS marker, named GF-dCAPS, by introducing a mismatch base (G) at the end of the forward primer to create a AgeI restrictive enzyme recognition site. The 45 chieh-qua materials were genotyped with the marker, and the result showed a complete correlation with flesh color (Supplementary Figure S3). In addition, the InDel marker Fc_13 was cosegregated with Cqgf in all recombinants, so Fc_13 was used to genotype the 45 chieh-qua materials, which also showed a complete accuracy (Supplementary Figure S4).
Phylogenetic analysis
To analyze the relationship between CqLOL1 and its homologous protein, we constructed a phylogenetic tree through neighbor-joining. The phylogenetic tree showed that LOL1 was conserved in different plant species, and CqLOL1 was closely related to Cucurbitaceae (Figure 7).
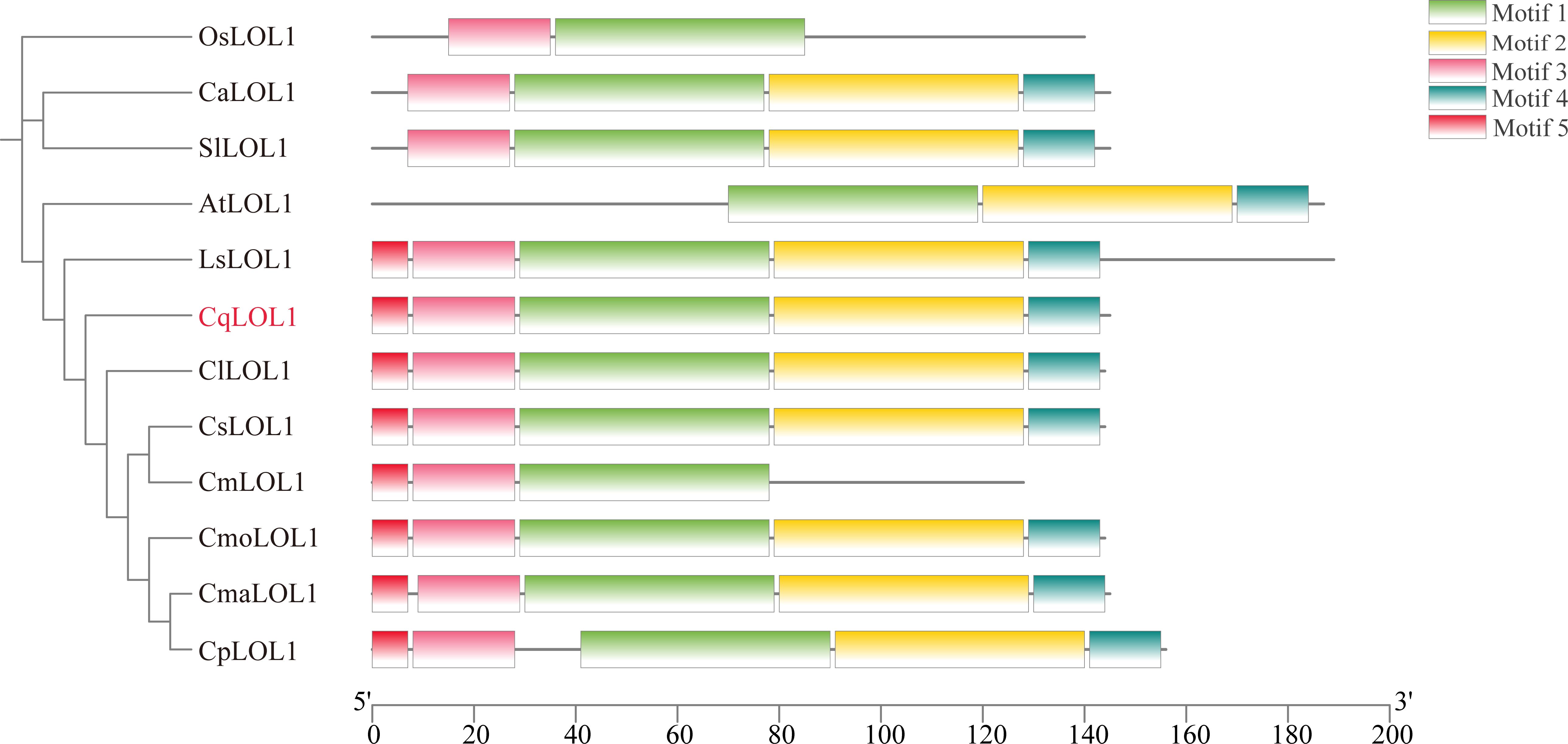
Figure 7. Phylogenetic analysis and conceptual motif information for LOL1 in chieh-qua and other species. The phylogenetic tree and motif distribution of LOL1 from Oryza sativa (Os), Arabidopsis (At), Capsicum annuum (Ca), Solanum lycopersicum (Sl), Cucumis melon (Cm), Cucumis lanatus (Cl), Lagenaria siceraria (Ls), Cucumis sativus (Cs), Cucurbita moschata (Cmo), Cucurbita maxima (Cma), and Cucurbita pepo (Cp) are shown. The green, yellow, pink, blue, and red bars represent different predicted motifs.
Discussion
Fruit flesh color, an essential feature for consumer preference, is considered a key trait for breeding. To our knowledge, chieh-qua exhibits white and green flesh, and green flesh is significantly associated with soluble sugar content (unpublished data). Genetic analysis indicated that green flesh was a quantitative trait nearly related to normal distribution. To decipher the molecular mechanism of green flesh formation, only one peak, named Cqgf, was determined on chromosome 5 through QTL mapping and BSA-seq and was consistent with the major QTL for wax gourd flesh color (fc5.1) (Su et al., 2022). Furthermore, based on the large J16 × FJ5 F2 population, Cqgf was ultimately fine-mapped into a small region (329.70 kb).
Several genes or loci related to green flesh have been identified in cucumber, watermelon, and melon. The green flesh of immature cucumber resulting from the accumulation of chlorophyll was governed by two loci (qgf5.1 and qgf3.1), of which qgf5.1 was the major-effect QTL, and the gene Csa5G021320, which encodes Sec14 cytosolic factor, was a key candidate gene (Bo et al., 2019). In watermelon, a major-effect QTL (qfc10.1) related to pale green flesh and chlorophyll content was found within a 519-kb region harboring 22 genes, and the most important candidate gene, Cla97C10G185970, encodes a plastid lipid-associated protein and participates in the photoprotection of photosystem II (Pei et al., 2021). In melon, the wf locus controls white and green flesh color, and two closely related genes, MELO3C003069 and MELO3C003097, were both reported to be causal genes. MELO3C003069 belongs to the pentatricopeptide protein family and participates in the processing of RNA in plastids (Galpaz et al., 2018), while the ortholog of MELO3C003097 in Arabidopsis (SG1) has been identified to be essential for chloroplast development and chlorophyll biosynthesis (Zhao et al., 2019). According to our results, the Cqgf region contains six protein-coding genes based on the annotation of the reference genome GX-19. Unfortunately, none of these genes were homologous to the regulatory genes related to the green color of cucurbits mentioned above. However, both the homologous genes of Bch05G003690 and Bch05G003700 have been shown to be required for chlorophyll biosynthesis in other crops. Bch05G003690 encodes ferredoxin-like proteins with a C-terminal extension (FdC2) that was involved in electron transfer, and the mutation of FdC2 resulted in decreased chlorophyll content in the leaf of Arabidopsis (Tournaire et al., 2023) and rice (Li et al., 2015). The other gene, Bch05G003700, encodes a zinc-finger transcription factor LOL1 (lsd one like 1), which is highly conserved in different plant species—for example, AtLOL1 positively regulated programmed cell death in Arabidopsis (Epple et al., 2003), and overexpression of OsLOL1 in rice increased the content of chlorophyll (Wang et al., 2005). The downregulation of CcLOL1 with a single C nucleotide insertion caused a decrease in chlorophyll content in pepper fruit, and CcLOL1 was highly expressed in green tissue (Borovsky et al., 2019). The SlLOL1 knockout mutant in tomato resulted in light green immature fruits (Borovsky et al., 2019).
We then conducted an expression analysis of six candidate genes within the fine-mapping region, especially Bch05G003690 and Bch05G003700. However, Bch05G003690 and Bch05G003700 both exhibited significantly different expression levels between J16 and FJ5. Remarkably, Bch05G003700 exhibited significance from 5 to 30 DAP, which was accordant with the phenotypic differences at six stages. In addition, the flesh color of chieh-qua is influenced by the strength of light, so we conducted shading experiments, and the green flesh clearly turned to pale green under shading (Figure 5H). Compared with that under normal lighting conditions, the expression level of Bch05G003700 under shading was lower, indicating that Bch05G003700 was induced by light, while there was no significant change in Bch05G003690 (Figure 5G). We subsequently compared the coding sequence variation of candidate genes, and there is no variation in Bch05G003690. However, there is a point mutation (A>C) in the coding sequence of Bch05G003700, and the amino acid sequence was changed from T to P. Thus, Bch05G003700 was most likely the causal gene controlling green flesh. To further reveal the association between the SNP of Bch05G003700 and green flesh, we cloned Bch05G003700 of 45 chieh-qua germplasms from different genetic backgrounds, including commercial hybrids and inbred lines. As a result, homozygous C had white flesh, while homozygous A had green flesh.
Previous studies have revealed candidate genes for several important traits in wax gourd and chieh-qua, including fruit shape (Cheng et al., 2021), fruit peel color (Ma et al., 2021), fruit cuticular wax (Yan et al., 2022), seed shape (Luo et al., 2022), seed size (Yang et al., 2023), and gyneocy (Wang et al., 2023), and molecular markers have been developed for marker-assisted breeding (Huang et al., 2022). Interestingly, the locus controlling the peel color was also located on chromosome 5, and the causal gene BhAPRR2 (Bch05G003950) is closely related to CqLOL1 (Bch05G003700) with a physical distance of 1.11 Mb, which indicates that the two genes have a certain degree of linkage. Particularly, the chieh-qua fruits with green flesh always have pale green peel—for example, FJ3211 has green flesh and light green peel, and the linkage between green flesh and light green peel was observed in the F2 population. In this study, a dCAPS marker based on a point mutation in CqLOL1 was developed to test the concordance of genotypes and phenotypes among 45 extreme materials, the result of which shows a complete correlation. The InDel marker Fc_13, close to CqLOL1, also showed an accuracy level of 100%. Thus, the two markers are useful for breeding the green flesh varieties of chieh-qua in the future.
On the other hand, the regulation of chlorophyll, including chlorophyll biosynthesis and degradation, is complex—for example, several genes involved in chlorophyll biosynthesis in rice, including OsCHLH, OsCHLD, OsCHLI, OsCHLG, OsDVR, OsPOR, and OsCAO, have been identified, and any mutations in these genes result in chlorophyll deficiency (Chen et al., 2018; Jung et al., 2003). Several YGL genes participate in chloroplast development, and these mutations lead to yellow green leaf in rice (Wu et al., 2007; Zhu et al., 2016). The key regulatory factors SGRs, PPH, and PAO that accelerate chlorophyll degradation have been identified in various species. In addition, several TFs, including NAC (Tak et al., 2018; Zou et al., 2023), HZP (Wu et al., 2024), TCP (Zheng et al., 2022), and MADS-box (Zhu et al., 2023) transcription factors, modulated SGRs, and PPH and PAO were regulated by MYB transcription factors (Wu et al., 2020). LSD1 is involved in programmed cell death, and it regulates photosynthesis-related genes, including PHANGs (photosynthesis-associated nuclear genes) and PHAPGs (photosynthesis-associated plastid genes), through its interaction with GOLDEN2-LIKE (GLK) transcription factors, thereby influencing the development and photosynthesis of chloroplasts (Lv et al., 2019; Li et al., 2022); in Capsicum chinense, the CcLOL1 controls the immature fruit color, possibly by regulating chlorophyll synthesis genes such as POR, CAO, glutamyl-tRNA reductase (HEMA1), and the chlorophyll degradation gene pheophorbide oxygenase (Borovsky et al., 2019). The above-mentioned genes may be downstream genes of CqLOL1, and further efforts are needed to demonstrate it.
In conclusion, genetic analysis revealed that green flesh was a quantitative trait closely related to a normal distribution, and the Cqgf locus was first fine-mapped to a 329.70-kb region. By combining expression and sequence variation analysis, CqLOL1 (Bch05G003700), which encodes the zinc-finger transcription factor LOL1, was proposed as the strongest candidate gene. Our findings not only provide a basis for marker-assisted selection but also help in understanding the molecular mechanism of green flesh formation.
Data availability statement
The data presented in the study are deposited in the Genome Sequence Archive in National Genomics Data Center, accession number CNP0005362.
Author contributions
JP: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal Analysis, Funding acquisition. YG: Data curation, Formal Analysis, Investigation, Writing – original draft, Writing – review & editing. YQ: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing – review & editing. GW: Conceptualization, Methodology, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Seed Industry Revitalization Project of Special Funds for Province Rural Revitalization Strategy (2022-NPY-01-004), the Guangzhou Basic and Applied Basic Research Projects (202201011716), the Guangzhou Agriculture and Rural Finance Fund Project (23107180), the Guangzhou Science and Technology Plan Projects (202002020086), and the Guangzhou Agricultural Support Fund Project (22103411).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1388115/full#supplementary-material
References
Bang, H., Davis, A. R., Kim, S., Leskovar, D. I., King, S. R. (2010). Flesh color inheritance and gene interactions among canary yellow, pale yellow, and red watermelon. J. Amer. Soc. Hortic. Sci. 135, 362–368. doi: 10.1560/IJPS.60.4.425
Blas, A. ,. L., Ming, R., Liu, Z. Y., Veatch, O. J., Paul, R. E., Moore, P. H., et al. (2010). Cloning of the papaya chromoplast-specific lycopene β-cyclase, CpCYC-b, controlling fruit flesh color reveals conserved microsynteny and a recombination hot spot. Plant Physiol. 152, 2013–2022. doi: 10.1104/pp.109.152298
Bo, K. L., Song, H., Shen, J., Qian, C. T., Staub, J. E., Simon, P. W., et al. (2012). Inheritance and mapping of the ore gene controlling the quantity of β-carotene in cucumber (Cucumis sativus L.) endocarp. Mol. Breed. 30, 335–344. doi: 10.1007/s11032-011-9624-4
Bo, K. L., Wei, S., Wang, W. P., Miao, H., Dong, S. Y., Zhang, S. P., et al. (2019). QTL mapping and genome-wide association study reveal two novel loci associated with green flesh color in cucumber. BMC Plant Bio. 19, 243. doi: 10.1186/s12870-019-1835-6
Borovsky, Y., Monsonego, N., Mohan, V., Shabtai, S., Kamara, I., Faigenboim, A., et al. (2019). The zinc-finger transcription factor CcLOL1 controls chloroplast development and immature pepper fruit color in Capsicum chinense and its function is conserved in tomato. Plant J. 99, 41–55. doi: 10.1111/tpj.14305
Branham, S., Vexler, L., Meir, A., Tzuri, G., Frieman, Z., Levi, A., et al. (2017). Genetic mapping of a major codominant QTL associated with β-carotene accumulation in watermelon. Mol. Breed. 37, 146. doi: 10.1007/s11032-017-0747-0
Chen, C. J., Wu, Y., Li, J. W., Wang, X., Zeng, Z. H., Xu, J., et al. (2023). TBtools-II: A "one for all, all for one" bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742. doi: 10.1016/j.molp.2023.09.010
Chen, P., Hu, H. T., Zhang, Y., Wang, Z. W., Dong, G. J., Cui, Y. T., et al. (2018). Genetic analysis and fine-mapping of a new rice mutant, white and lesion mimic leaf1. Plant Growth Regul. 85, 425–435. doi: 10.1007/s10725-018-0403-7
Cheng, Z. K., Liu, Z. G., Xu, Y. C., Ma, L. L., Chen, J. Y., Gou, J. Q., et al. (2021). Fine mapping and identification of the candidate gene BFS for fruit shape in wax gourd (Benincasa hispida). Theor. Appl. Genet. 134, 3983–3995. doi: 10.1007/s00122-021-03942-8
Cuevas, H. E., Song, H., Staub, J. E., Simon, P. W. (2010). Inheritance of beta-carotene-associated flesh color in cucumber (Cucumis sativus L.) fruit. Euphytica. 171, 301–311. doi: 10.1007/s10681-009-0017-2
Epple, P., Mack, A. A., Morris, V. R. F., Dangl, J. L. (2003). Antagonistic control of oxidative stress-induced cell death in Arabidopsis by two related, plant-specific zinc finger proteins. Proc. Natl. Acad. Sci. U. S. A. 100, 6831–6836. doi: 10.1073/pnas.1130421100
Galpaz, N., Gonda, I., Shem-Tov, D., Barad, O., Tzuri, G., Lev, S., et al. (2018). Deciphering genetic factors that determine melon fruit-quality traits using RNA-Seq-based high-resolution QTL and eQTL mapping. Plant J. 94, 169–191. doi: 10.1111/tpj.13838
Guo, S. G., Zhao, S. J., Sun, H. H., Wang, X., Wu, S., Lin, T., et al. (2019). Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat. Genet. 51, 1616–1623. doi: 10.1038/s41588-019-0518-4
Gur, A., Tzuri, G., Meir, A., Sa’ar, U., Portnoy, V., Katzir, N., et al. (2017). Genome-wide linkage-disequilibrium mapping to the candidate gene level in melon (Cucumis melo). Sci. Rep. 7, 9770. doi: 10.1038/s41598-017-09987-4
Henderson, W. R., Scott, G. H., Wehner, T. C. (1998). Interaction of flesh color genes in watermelon. HortScience 31, 602–602. doi: 10.21273/HORTSCI.31.4.602d
Huang, X. C., Wu, W. T., Su, L. W., Lv, H. X., Cheng, Z. K., Yang, W. R., et al. (2022). Development and application of InDel markers linked to fruit-shape and peel-colour genes in Wax Gourd. Genes 13, 1567. doi: 10.3390/genes13091567
Jung, K. H., Hur, J. H., Ryu, C. H., Choi, Y. J., Chung, Y. Y., Akio, M. Y., et al. (2003). Characterization of a rice chlorophyll-deficient mutant using the T-DNA gene-trap system. Plant Cell Physiol. 44, 463–472. doi: 10.1093/pcp/pcg064
Kang, B. S., Zhao, W. E., Hou, Y. B., Tian, P. (2010). Expression of carotenogenic genes during the development and ripening of watermelon fruit. Sci. Horti. 124, 368–375. doi: 10.1016/j.scienta.2010.01.027
Kishor, D. S., Lee, H. Y., Alavilli, H., You, C. R., Kim, J. G., Lee, S. Y. (2021). Identification of an allelic variant of the CsOr gene controlling fruit endocarp color in cucumber (Cucumis sativus L.) using genotyping-by-sequencing (GBS) and whole-genome sequencing. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.802864
Li, C. M., Hu, Y., Huang, R., Ma, X. Z., Wang, Y., Liao, T. T., et al. (2015). Mutation of FdC2 gene encoding a ferredoxin-like protein with C-terminal extension causes yellow-green leaf phenotype in rice. Plant Sci. 238, 127–134. doi: 10.1016/j.plantsci.2015.06.010
Li, M. P., Lee, K. P., Liu, T., Dogra, V., Duan, J. L., Li, M. S., et al. (2022). Antagonistic modules regulate photosynthesis-associated nuclear genes via GOLDEN2-LIKE transcription factors. Plant Physiol. 188, 2308–2324. doi: 10.1093/plphys/kiab600
Li, N., Shang, J. L., Wang, J. M., Zhou, D., Li, N. N., Ma, S. W. (2020). Discovery of the genomic region and candidate genes of the scarlet red flesh color (Yscr) locus in watermelon (Citrullus Lanatus L.). Front. Plant Sci. 11. doi: 10.3389/fpls.2020.00116
Li, R. Q., Li, Y. R., Kristiansen, K., Wang, J. (2008). SOAP: short oligonucleotide alignment program. Bioinformatics. 24, 713–714. doi: 10.1093/bioinformatics/btn025
Liu, D. Y., Ma, C. X., Hong, W. G., Huang, L., Liu, M., Liu, H., et al. (2014). Construction and analysis of high-density linkage map using high-throughput sequencing data. PloS One 9, e98855. doi: 10.1371/journal.pone.0098855
Liu, S., Liu, M. Q., Cao, Y., Xu, Y., Liu, H. Y., Zhu, Q. L., et al. (2023). Identification of chromosome region and candidate genes for canary-yellow flesh (Cyf) locus in watermelon (Citrullus lanatus). Plant Sci. 329, 111594. doi: 10.1016/j.plantsci.2023.111594
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta C(T)) method. Methods. 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, H. W., Miao, H., Tian, G. L., Wehner, T. C., Gu, X. F., Zhang, S. P. (2015). Molecular mapping and candidate gene analysis for yellow fruit flesh in cucumber. Mol. Breed. 35, 64. doi: 10.1007/s11032-015-0263-z
Luo, C., Yan, J. Q., Liu, W. R., Xu, Y. C., Sun, P. Y., Wang, M., et al. (2022). Genetic mapping and genome-wide association study identify BhYAB4 as the candidate gene regulating seed shape in wax gourd (Benincasa hispida). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.961864
Luo, W. L., Yan, J. Q., Luo, S. W., Liu, W. R., Xie, D. S., Jiang, B. (2023). A chromosome-level reference genome of the wax gourd (Benincasa hispida). Sci. data. 10, 78. doi: 10.1038/s41597-023-01986-7
Lv, R. Q., Li, Z. H., Li, M. P., Dogra, V., Lv, S. S., Liu, R. Y., et al. (2019). Uncoupled expression of nuclear and plastid photosynthesis-associated genes contributes to cell death in a lesion mimic mutant. Plant Cell. 31, 210–230. doi: 10.1105/tpc.18.00813
Ma, L. L., Liu, Z. G., Cheng, Z. K., Gou, J. Q., Chen, J. Y., Yu, W. J., et al. (2021). Identification and application of BhAPRR2 controlling peel colour in wax gourd (Benincasa hispida). Front. Plant Sci. 12. doi: 10.3389/fpls.2021.716772
Meng, L., Li, H., Zhang, L., Wang, J. K. (2015). QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 3, 269–283. doi: 10.1016/j.cj.2015.01.001
Oren, E., Tzuri, G., Vexler, L., Dafna, A., Meir, A., Faigenboim, A., et al. (2019). The multi-allelic APRR2 gene is associated with fruit pigment accumulation in melon and watermelon. J. Exp. Bot. 70, 3781–3794. doi: 10.1093/jxb/erz182
Pei, S., Liu, Z., Wang, X. Z., Luan, F. S., Dai, Z. Z., Yang, Z., et al. (2021). Quantitative trait loci and candidate genes responsible for pale green flesh colour in watermelon (Citrullus lanatus). Plant Breed. 140, 349–359. doi: 10.1111/pbr.12908
Poole, C. (1944). Genetics of cultivated cucurbits. J. Hered. 35, 122–128. doi: 10.1093/oxfordjournals.jhered.a105364
Shahwar, D., Khan, Z., Park, Y. (2023). Molecular marker-assisted mapping, candidate gene identification, and breeding in melon (Cucumis melo L.): A review. Int. J. Mol. Sci. 24, 15490. doi: 10.3390/ijms242015490
Su, L. W., Gou, J. Q., Lv, H. X., Cheng, Z. K., Ma, L. L., Huang, X. C., et al. (2022). High-density genetic map and quantitative trait loci map of fruit-related traits in wax gourd (Benincasa hispida). Euphytica. 218, 117. doi: 10.1007/s10681-022-03074-0
Sun, X. W., Liu, D. Y., Zhang, X. F., Li, W. B., Liu, H., Hong, W. G., et al. (2013). SLAF-seq: An efficient method of large-scale denovo SNP discovery and genotyping using high-throughput sequencing. PloS One 8, e58700. doi: 10.1371/journal.pone.0058700
Tak, H., Negi, S., Gupta, A., Ganapathi, T. R. (2018). A stress associated NAC transcription factor MpSNAC67 from banana (Musa x paradisiaca) is involved in regulation of chlorophyll catabolic pathway. Plant Physiol. Bio. 132, 61–71. doi: 10.1016/j.plaphy.2018.08.020
Tournaire, M. D., Scharff, L. B., Kramer, M., Goss, T., Vuorijoki, L., Rodriguez-Heredia, M., et al. (2023). Ferredoxin C2 is required for chlorophyll biosynthesis and accumulation of photosynthetic antennae in Arabidopsis. Plant Cell Environ. 46 (11), 3287–3304. doi: 10.1111/pce.14667
Tzuri, G., Zhou, X., Chayut, N., Yuan, H., Portnoy, V., Meir, A., et al. (2015). A ‘golden’ SNP in CmOr governs the fruit flesh color of melon (Cucumis melo). Plant J. 82, 267–279. doi: 10.1111/tpj.12814
Wang, C. N., Luan, F. S., Liu, H. Y., Davis, A. R., Zhang, Q. A., Dai, Z. Y., et al. (2021). Mapping and predicting a candidate gene for flesh color in watermelon. J. Integr. Agr. 20, 2100–2111. doi: 10.1016/j.plantsci.2023.111594
Wang, C. N., Qiao, A. H., Fang, X. F., Sun, L., Gao, P., Davis, A. R., et al. (2019). Fine mapping of lycopene content and flesh color related gene and development of molecular marker–assisted selection for flesh color in watermelon (Citrullus lanatus). Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01240
Wang, L. J., Pei, Z. Y., Tian, Y. C., He, C. Z. (2005). OsLSD1, a rice zinc finger protein, regulates programmed cell death and callus differentiation. Mol. Plant Microbe Interact. 18, 375–384. doi: 10.1094/MPMI-18-0375
Wang, M., Yang, S. G., Liu, W., Cao, Z. Q., Chen, L., Liu, W. R., et al. (2023). Fine mapping and candidate gene analysis of gynoecy trait in chieh-qua (Benincasa hispida Cogn. var. chieh-qua How). Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1158735
Wu, M. B., Xu, X., Hu, X. W., Liu, Y. D., Cao, H. H., Chan, H. L., et al. (2020). SlMYB72 regulates the metabolism of chlorophylls, carotenoids, and flavonoids in tomato fruit. Plant Physiol. 183, 854–868. doi: 10.1104/pp.20.00156
Wu, Y. Y., Wang, L. L., Lin, Y. L., Li, X., Liu, X. F., Xu, Z. H., et al. (2024). AcHZP45 is a repressor of chlorophyll biosynthesis and activator of chlorophyll degradation in kiwifruit. J. Exp. Bot. 75, 204–218. doi: 10.1093/jxb/erad361
Wu, Z. M., Zhang, X., He, B., Diao, L. P., Sheng, S. L., Wang, J. L., et al. (2007). A chlorophyll-deficient rice mutant with impaired chlorophyllide esterification in chlorophyll biosynthesis. Plant Physiol. 145, 29–40. doi: 10.1104/pp.107.100321
Xie, D. S., Xu, Y. C., Wang, J. P., Liu, W. R., Zhou, Q., Luo, S. B., et al. (2019). The wax gourd genomes offer insights into the genetic diversity and ancestral cucurbit karyotype. Nat. Commun. 10, 5158. doi: 10.1038/s41467-019-13185-3
Yan, J. Q., Chen, F., Sun, P. Y., Liu, W. R., Xie, D. S., Qian, Y. L., et al. (2022). Genome-wide association study and genetic mapping of BhWAX conferring mature fruit cuticular wax in wax gourd. BMC Plant Bio. 22, 539. doi: 10.1186/s12870-022-03931-z
Yang, W. R., Wang, P., Liu, T., Nong, L. F., Cheng, Z. K., Su, L. W., et al. (2023). Fine mapping of the major gene BhHLS1 controlling seed size in wax gourd (Benincasa hispida). Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1266796
Yi, L. C., Zhou, W., Zhang, Y., Chen, Z. B., Wu, N., Wang, Y. Q., et al. (2023). Genetic mapping of a single nuclear locus determines the white flesh color in watermelon (Citrullus lanatus L.). Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1090009
Zhang, J., Guo, S. G., Ren, Y., Zhang, H. Y., Gong, G. Y., Zhou, M., et al. (2016). High-level expression of a novel chromoplast phosphate transporter ClPHT4;2 is required for flesh color development in watermelon. New Phytol. 213, 1208–1221. doi: 10.1111/nph.14257
Zhang, J., Sun, H. H., Guo, S. G., Ren, Y., Li, M. Y., Wang, J. F., et al. (2020). Decreased protein abundance of lycopene β-cyclase contributes to red flesh in domesticated watermelon. Plant Physiol. 183, 1171–1183. doi: 10.1104/pp.19.01409
Zhao, B. T., Sun, M., Li, J. Y., Su, Z. W., Cai, Z. X., Shen, Z. J., et al. (2022). Carotenoid profiling of yellow-flesh peach fruit. Foods. 11, 1669. doi: 10.3390/foods11121669
Zhao, G. W., Lian, Q., Zhang, Z. H., Fu, Q. S., He, Y. H., Ma, S. W., et al. (2019). A comprehensive genome variation map of melon identifies multiple domestication events and loci influencing agronomic traits. Nat. Genet. 51, 1607–1615. doi: 10.1038/s41588-019-0522-8
Zheng, X. H., Lan, J. Q., Yu, H., Zhang, J. Z., Zhang, Y., Qin, Y. M., et al. (2022). Arabidopsis transcription factor TCP4 represses chlorophyll biosynthesis to prevent petal greening. Plant Commun. 3, 100309. doi: 10.1016/j.xplc.2022.100309
Zhu, K. J., Chen, H. Y., Mei, X. H., Lu, S. W., Xie, H. P., Liu, J. W., et al. (2023). Transcription factor CsMADS3 coordinately regulates chlorophyll and carotenoid pools in Citrus hesperidium. Plant Physiol. 193, 519–536. doi: 10.1093/plphys/kiad300
Zhu, X. Y., Guo, S., Wang, Z. W., Du, Q., Xing, Y. D., Zhang, T. Q., et al. (2016). Map-based cloning and functional analysis of YGL8, which controls leaf colour in rice (Oryza sativa). BMC Plant Bio. 16, 134. doi: 10.1186/s12870-016-0821-5
Keywords: chieh-qua, flesh color, chlorophyll content, fine-mapping, zinc-finger transcription factor LOL1
Citation: Peng J, Gao Y, Qiao Y and Wang G (2024) A point mutation in the zinc-finger transcription factor CqLOL1 controls the green flesh color in chieh-qua (Benincasa hispida Cogn. var. Chieh-qua How). Front. Plant Sci. 15:1388115. doi: 10.3389/fpls.2024.1388115
Received: 19 February 2024; Accepted: 17 September 2024;
Published: 21 October 2024.
Edited by:
Kaijing Zuo, Shanghai Jiao Tong University, ChinaReviewed by:
Shyam Gundu, West Virginia State University, United StatesShi Liu, Northeast Agricultural University, China
Kang Wei, Chinese Academy of Agricultural Sciences, China
Copyright © 2024 Peng, Gao, Qiao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanchun Qiao, cXljMTk3OTAxMjhAMTYzLmNvbQ==; Guoping Wang, Z3B3YW5nQHNjYXUuZWR1LmNu
†These authors have contributed equally to this work