
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 09 July 2024
Sec. Functional and Applied Plant Genomics
Volume 15 - 2024 | https://doi.org/10.3389/fpls.2024.1383135
This article is part of the Research Topic Genetics and Genomics of Emerging and Multifactorial Stresses Affecting Plant Survival and Associated Plant Microbiomes View all 14 articles
 Abu Bakar Sultan1
Abu Bakar Sultan1 Humera Nawaz2
Humera Nawaz2 Fozia Saleem1Sehar Nawaz1Muhammad Danial1
Fozia Saleem1Sehar Nawaz1Muhammad Danial1 Romana Iftikhar1Umer Maqsood3Amna Areej1Sidra Shakoor1
Romana Iftikhar1Umer Maqsood3Amna Areej1Sidra Shakoor1 Nada H. Aljarba4
Nada H. Aljarba4 Rizwan Maqbool5Muhammad Rizwan1
Rizwan Maqbool5Muhammad Rizwan1 Saad Serfraz1*
Saad Serfraz1*Within the family Fabaceae, the genus Glycine is composed of two subgenera annuals (2n=40) and perennials. This life strategy transition may have differentially affected the evolution of various gene families. Its cultivated species G. max has high level of susceptibility to major pathogens including viruses, bacteria and fungi. Understanding nucleotide-binding domain leucine-rich repeat (NLR) genes evolution in soybean is of paramount importance due to their central role in plant immunity and their potential in improving disease resistance in soybean cultivars. In this study, we investigated the significance of this annual-perennial transition on the macroevolution of NLR genes in the genus Glycine. Our results reveal a remarkable distinction between annual species such as Glycine max and Glycine soja, which exhibit an expanded NLRome compared to perennial species (G. cyrtoloba, G. stenophita, G. dolichocarpa, G. falcata, G. syndetika, G. latifolia and G. tomentella). Our evolutionary timescale analysis pinpoints recent accelerated gene duplication events for this expansion, which occurred between 0.1 and 0.5 million years ago, driven predominantly by lineage-specific and terminal duplications. In contrast, perennials initially experienced significant contraction during the diploidisation phase following the Glycine-specific whole-genome duplication event (~10 million years ago). Despite the reduction in the NLRome, perennial lineages exhibit a unique and highly diversified repertoire of NLR genes with limited interspecies synteny. The investigation of gene gain and loss ratios revealed that this diversification resulted from the birth of novel genes following individual speciation events. Among perennials, G. latifolia, a well-known resistance resource, has the highest ratio of these novel genes in the tertiary gene pool. Our study suggests evolutionary mechanisms, including recombination and transposition, as potential drivers for the emergence of these novel genes. This study also provides evidence for the unbalanced expansion of the NLRome in the Dt subgenome compared with the At subgenome in the young allopolyploid G. dolichocarpa. To the best of our knowledge, this is the first study to investigate the effect of annuality and perenniality life transition on the evolution of NLR genes in the genus Glycine to identify its genomics resources for improving the resistance of soybean crop with global importance on the economy and food security.
Soya (Glycine max), a legume, is the fourth most important agricultural commodity in the world. It is widely cultivated and used to produce biodiesel, high-quality livestock feed and the cheapest edible protein for humans (Sedivy et al., 2017). The genus Glycine comprises two subgenera (Glycine subgenus and Soja subgenus) that diverged approximately 5 million years ago (Mya) and exhibit different life strategies. The subgenus Soja consists of annual plants such as the soybean (Glycine max) and its wild ancestor, Glycine soja, which was domesticated about 6,000–9,000 years ago. These plants are indigenous to eastern Asia, including China, Japan, Korea, and some parts of Russia. In contrast, the subgenus Glycine encompasses around 30 perennial species. The majority of these species are found in Australia and inhabit a variety of environments, including deserts, sandy beaches, rocky outcrops, as well as monsoonal, temperate, and subtropical forests (Innes et al., 2008). All diploid Glycine species have a chromosome count of (2n = 4x = 38 or 40), which is different from most species in the legume tribe Phaseoleae, which usually have (2n = 20 or 22). Both Glycine subgenera basically have (2n = 40). More recently, probably within the past few tens of thousands of years, allopolyploid taxa have appeared within the subgenus Glycine. These freshly developed allopolyploids of the Glycine subgenus have been discovered as far north as Taiwan and the Ryukyu Islands (Egan and Doyle, 2010; Forrester and Ashman 2018). The progenitor of the entire Glycine genus underwent a polyploidy event approximately 5–13 million years ago. Within the last 500,000 years, an additional wave of genome duplication has occurred in the subgenus Glycine, giving rise to a vast and fully characterized polyploid complex (Sherman-Broyles et al., 2014; Kim et al., 2015). This complex has eight allopolyploid species (2n = 78, 80) and one autopolyploid species (2n = 80), which were produced from diverse combinations of diploid (2n = 38, 40) genomes. Among them, G. dolichocarpa (4n=80) is a tetraploid that consists of two subgenomes At and Dt, which arose from two progenitor species G. syndetika (A) and G. tomentella (D) (Li et al., 2014; Manzoor et al., 2019).
A reference genome for the cultivated soybean (G. max) accession ‘Williams 82’ was released in 2010 (Schmutz et al., 2010), while reference genomes for its annual relative G. soja accession IT182932 and W05 were published in 2019 (Xie et al., 2019). The genomes of Glycine species were divided into seven categories based on their potential to form fertile hybrids and the degree of pairing of meiotic chromosomes. These groups were labeled with capital letters A through G (Sherman-Broyles et al., 2014; Kim et al ., 2015). The annual wild cousin of soybean (Glycine soja) and the cultivated soybean (Glycine max) both belong to the G genome group and have the same number of chromosomes (2n = 40).
The perennial wild relatives of soybeans possess significantly more complex genomes compared to their annual counterparts, which are categorized into six distinct genome groups (A–F). Members of the perennials, subgenus Glycine have 2n = 38, 40, 78, and 80 chromosomes, in contrast to the subgenus Soja (Sherman-Broyles et al., 2014). Perennial soybeans have not had as much genomic study as their annual counterparts, despite efforts to explore their genetic properties.
Among plant resistance genes, nucleotide-binding domain leucine-rich repeat (NLR) genes are the principal component of the effector-triggered immunity (ETI) (Cui et al 2015). Through these intracellular NLR genes, resistance reactions result in infected plant tissues, typically accompanied by hypersensitivity reactions (HRs) (Pan et al., 2000; Jones et al., 2016). When invading pathogens are detected, these NLR proteins become activated, leading to changes in the conformation of the nucleotide-binding site (NBS) domain. To stop infected cells from spreading, the N-terminal domains of the exposed NBS domain start a downstream hypersensitive response (HR) that causes apoptosis to stop the proliferation and transmission of the pathogen (Andersen et al., 2018; Wang and Chai 2020). Based on the type of N-terminal domain, NLR genes are divided into four subclasses: Toll/Interleukin-1 receptor-like (TIR-NLR), RX-type coiled-coil (CC-NLR), CCR-NLR subclade with RTP8-type CC domain (CCR-NLR), and G10 subclade (CCG10-NLR), a recently proposed category with a distinct type of CC that forms a monophyletic group. Given the global dominance of soybeans, there is a growing need to enhance their genetic potential. Both domesticated (G. max) and wild (G. soja) annual species show lower levels of genetic diversity (Quershi et al ., 2023).
Globally soybean production is significantly hampered due to high susceptibility of G. max to viral, bacterial, fungal and nematode pathogens. In the US alone, an estimated loss of $95.48 billion were occurred during 1996 to 2016 highlighting the significant financial implication of these pathogens on soybean production (Bandara et al., 2020). Given the economic importance of soybean and the significant yield losses caused by various diseases, understanding the NLR genes in soybean is vital for crop improvement programs (Araújo et al., 2019). It enables the development of soybean varieties with enhanced disease resistance through traditional breeding or biotechnological approaches. For example, the identification and functional characterization of NLR genes can lead to the development of soybean lines harboring single or multiple NLR-encoding R gene receptors, potentially offering broad-spectrum resistance (Araújo et al., 2019). The perennial wild relatives of the subgenus Glycine are a valuable resource due to their disease resistance genes and adaptability to various habitats. To identify the unique mechanism of NLR gene evolution, it is necessary to perform a comprehensive characterization of NLR genes in both annual and perennial species of Glycine. The major objectives of this study to investigate the long-term evolutionary history of NLR genes in annuals and perennials species, identification of novel resistance resources in genus Glycine and effect of diploidization on their evolution. This study provides insights into the complexity and evolution of NLR genes in Glycine species, which could enhance disease resistance in Glycine crops and aid in the development of more resilient soybean cultivars.
The genome sequences of various species within the Glycine genus can be accessed through several genome database portals. For this study, we prioritized chromosomal anchored genomes and annotation files for all nine Glycine species and acquired them from two portals (NCBI and legume-info) database (Supplementary Table S1). We downloaded the genomes of G. max, G. cyrtoloba, G. stenophita, G. dolichocarpa, G. falcata, G. syndetika, G. latifolia, G. tomentella, and G. soja (Supplementary Table S1). To enable comparison of ancestral species, G. dolichocarpa, a tetraploid species, was divided into two distinct subgenomes, At and Dt. All genome files were labelled into their respective transcriptome, proteome, and gene transfer file formats. The reference proteomes of all nine Glycine genomes were processed using the NLRtracker pipeline (Jones et al., 2016; Kourelis et al., 2021). The NLRtracker produces output files containing sequences of NLR and NLR-associated sequences, as well as annotations of NLR, NBARC, deduplicated NBARC sequences, and domain sequences. These files were generated using interproscan and specified motif patterns. To ensure clarity, each NLR gene was subjected to manual curation using clustering and phylogenetic analysis, given the unclear nature of NLRtracker’s CCR-NLR annotation.
A library of NBARC domains has been produced by the PRG database, which includes reference NLR genes (Calle García et al., 2022). As previously mentioned, domain clusters were created using UCLUST’s 70% identity criteria (Edgar, 2010). The group category was assigned to each cluster using the subgroup nomenclature by (Seo et al., 2016). Seo et al. (2016) used classified clusters as seed probes to extract the NBARC domains of the Glycine genus produced by NLRtracker. These domains were then aligned with the NBARC seed probes to facilitate a comprehensive phylogenetic study of Glycine. Insights into evolution were obtained using the maximum likelihood technique of IQtree v 2.0 (Nguyen et al., 2015). The best fit model, VT + F + R10, was chosen along with 1000 bootstrap repetitions as the adjusted value.
The Interproscan tool was used to generate various output files, such as the GFF3 file, NLR fasta file, and an assembled file for gene density maps. After that, we used an adjusted bin size of 5 kb to intersect NLR gene sequences in the annotated file using BEDtools (Quinlan and Hall, 2010). After completing this procedure, count files were produced, which were subsequently modified by assigning each coordinate a bin number. Using the Rldeogram package, this generated a karyotype file for display in R (Hao et al., 2020).
As previously stated, Clustalw was used to align the deduced protein sequences of paralogs with their compatible subgroups (Li, 2003). The alignment of the respective nucleotide sequences was performed using the Perl-based pal2nal software (Suyama et al., 2006). For improved alignment, gaps and codons were removed, and Ks values were determined using the MA method and Kaks calculator. Depending on how nucleotide and protein sequences are substituted, Kaks values can be either non-synonymous (changing over time) or synonymous (changing over time).The frequency of evolution in various species may be inferred from Ks values. To prevent substitution saturation, Ks values larger than two were eliminated. NLR genes were grouped using the Orthovenn2 program to investigate orthologs. Orthovenn2 discovered common genes across all species by supplying the protein sequences of suspected NLR genes of various species with an E-value of 1e-2 as the default option (Xu et al., 2019). NLR genes were submitted to Orthofinder for a thorough examination of orthology (Emms and Kelly, 2019). Tree building was performed using the obtained orthologs. The number of gene gain and loss output was obtained sequentially by CAFE5 using orthogroup and ultrametric tree files as input. The available literature provides a thorough overview of evolutionary analysis (Areej et al., 2023).
Phylogenomic analysis was performed using Notung (Version 2.9) Command Line Interface (CLI). This software was used to calculate the lineage-specific genes (LSGs) in the genus Glycine. The gene tree and species tree were reconciled under postfix species labels. Prior to reconciliation, the species tree was converted into a binary tree. Following the phylogenomic analysis, Notung saved the output in a Homolog table. This file contained descriptions of Paralogs, Orthologs, and Xenologs. The data provided by these results were then used to generate an UpSet plot using R. This plot indicated the conserved lineages across different species within the genus Glycine.
To perform the expression analysis of identified NLR genes, we utilized available datasets from PRJNA628842 and PRJNA393826 bioprojects using the NCBI database (Supplementary Table S1). We aligned the raw read sequences using the reference genome of G. max and G. soja with HISAT (Pertea et al., 2015, 2016). Alignments were passed to StringTie for transcript assembly. Lastly, Ballgown was used to process the assembled transcripts and abundance to group the experimental conditions and identify the genes that were differently expressed between the conditions (Pertea et al., 2015, 2016).
Significant differences of NLR genes were observed among annual and perennial species. Identified NLR genes can be classified into four sub-classes CC-NLR, CCG10-NLR, TIR-NLR and CCR-NLR (Figure 1A). NLR genes are often incomplete and have undergone pseudogenization due to gene duplication, retro-transposition, non-processed inactivation, and nonsense mutations. We further performed manual filtering for intact NLR genes from each class and a similar trend was identified among Glycine species (Supplementary Table S2) and 318 and 348 NLR genes were found in G. soja and G. max, respectively. Relatively contracted NLR gene distribution was found in perennials, including G. falcata (77), G. dolichocarpa (184), G. syndetika (111), G. tomentella, (99), G. cyrtoloba (93), G. latifolia (140), and G. stenophita (78) (Figure 1B). NLR genes belonging to sub-class CC-NLR and TIR-NLR have gained substantial expansions as compared to their distribution among seven perennials’ species. Another subclass, CCR-NLR, which is a key component in network of NLRome. They remained highly conserved in all members of the family Fabaceae and their extent remained conserved, yet their expansion was also observed in annuals. Studying the factors contributing to the increased prevalence of NLR genes in annuals compared to perennial species is of immense importance. Recent phylogenomics suggest an increased number of non-redundant genes in annuals (129,006) as compared to perennials (109,827) (Zhuang et al., 2022). Expansion of NLR genes in annuals is also consistent with the super-pangenome of Glycine that provides evidence of a higher rate of non-core gene formation in annuals as compared to perennials. G. dolichocarpa subgenomes (At and Dt) have shown contraction of NLR genes upon comparison with their progenitor of G. syndetika (A) and G. tomentella (D). Significant reductions in TIR-NLR genes were observed in both At and Dt subgenomes. Interestingly G. dolichocarpa-Dt has shown increased ratio intact NLR genes as compared to its progenitor G. tomentella (D) in contrast to At which have shown considerable contraction as compared to G. syndetika (A) especially 66% TIR-NLR were lost after allopolyploidization. Overall, NLRome in both At and Dt have shown biased subgenomic fractionation as Dt have shown higher ratio of intact NLR genes as compared to At. Comparing the A, D, At, and Dt genomes revealed that the At and Dt subgenomes of the allopolyploid lost 7,351 genes, with a higher number of losses from D (4,109) than from A (3,242) (Zhuang et al., 2022). This suggests that this biased fractionation of NLR genes is asymmetrical to the total number of genes lost in each subgenome.
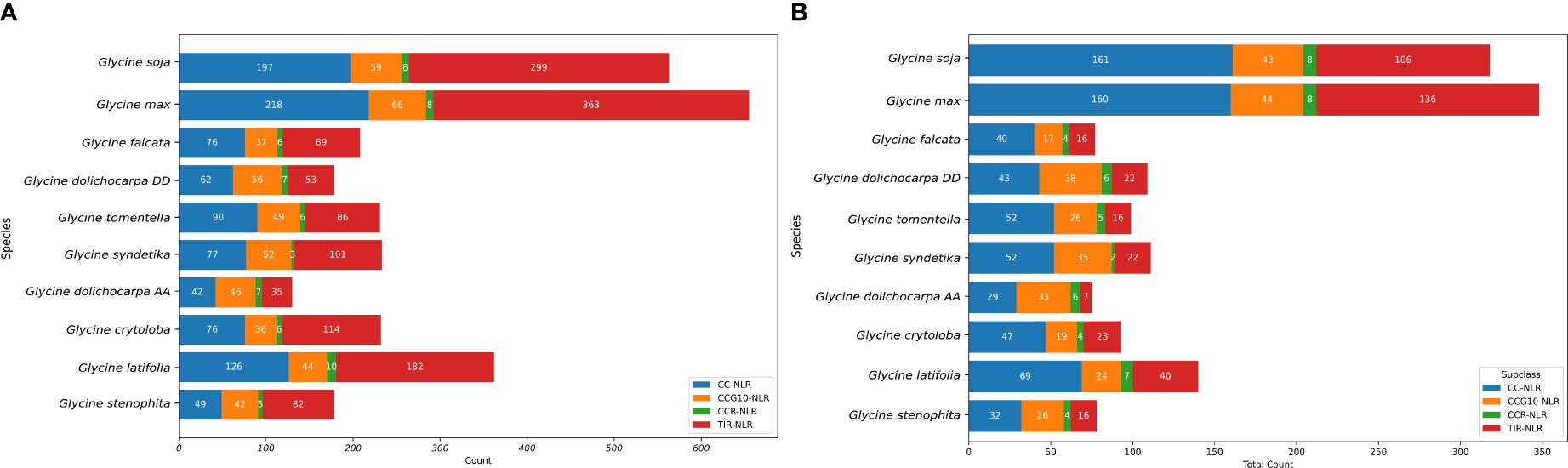
Figure 1 Comparative analysis of NLR genes and its subclasses in genus Glycine: A nucleotides domain-based perspective (A) Distribution of NLR genes in major classes across genus Glycine including partial and full length NLR genes (B) Distribution of Intact NLR Genes in Glycine Species.
We further correlated the NLRome of each species with respect to their genome size and examined the structural organization of NLR genes. All genome assemblies anchored at the chromosomal level were analyzed to construct NLR gene density maps. A significant diversity of resistant genes was established across the different members of genus Glycine. The gene density map (Figure 2; Supplementary Table S3) demonstrates that the density of NLR genes does not correlate with genome size within the genus Glycine. For instance, G. latifolia, despite having a relatively smaller genome size of 995 MB, exhibits an expanded NLRome. This contrasts with G. falcata, which has a larger genome size of 1.39GB and displays a reduced NLRome density. Similarly, G. cyrtoloba, with a genome size of 1.3GB, has a constrained NLRome. This is different from G. syndetika and G. stenophita, which, despite their smaller genome sizes of 948 and 940 MB, respectively, possess a higher NLRome density than G. cyrtoloba. Interestingly, there is a contraction of NLR gene families in both subgenome At and Dt of the allotetraploid species G. dolichocarpa when compared to its ancestors, G. tomentella and G. syndetika. The contraction of NLR gene families in G. dolichocarpa suggests that polyploidization events do not necessarily lead to an expansion of gene families but can also result in their contraction. This observation aligns with previous findings that polyploidization can either increase or decrease certain gene families, contributing to the complexity of our understanding of genome evolution (Yin et al., 2019). These findings emphasize the complex dynamics of NLR gene density and its independence from genome size, highlighting the complex interaction of genetic factors in shaping the NLRome across different species within the genus Glycine. The expanded NLRome in G. latifolia and the reduced NLRome in G. falcata, despite their contrasting genome sizes, indicate that NLR gene density is not merely a function of genome size. Instead, it may be influenced by a multitude of factors, including the species’ evolutionary history, environmental pressures, and genetic mechanisms. We further illustrated that individual gene density maps on each chromosome revealed significant variation in the distribution of NLR genes. Annual species like G. max and G. soja have more pronounced high-density regions (Supplementary Figure S1), suggesting a higher concentration of NLR genes, which could be crucial for their immune response. In contrast, perennial species show a more uniform and lower density distribution, indicating fewer NLR genes or a more even spread across their genomes.
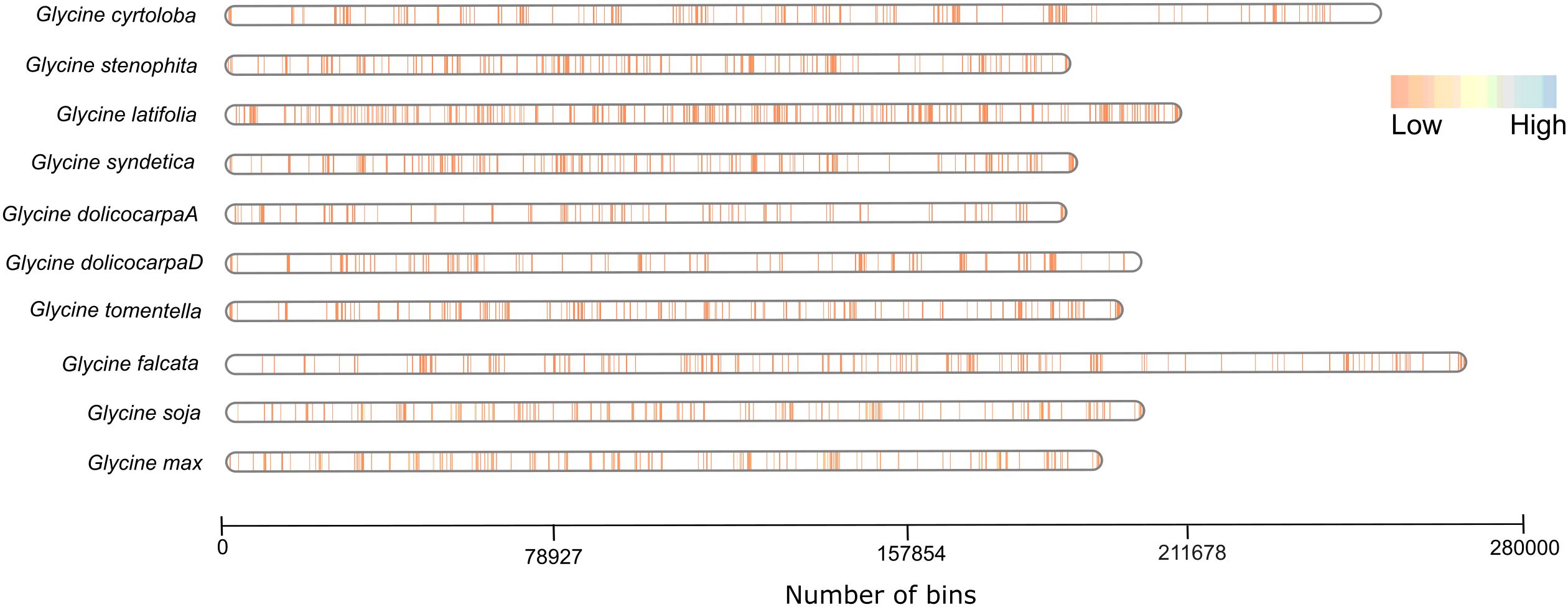
Figure 2 Gene density map: This graph displays bins on the x-axis, each representing a 5kb segment of the genome. The color gradient from brown to blue signifies the varying density of NLR genes. This graphical representation provides a quantitative correlation between NLR gene density and genomic size of different species of Glycine.
Furthermore, a comprehensive classification of NLR genes was performed across the genus Glycine. The evolutionary history of NLR genes in the genus Glycine can be traced by constructing their phylogenetic relationships. This is achieved by analyzing the amino acid sequences of the conserved NB-ARC domain. The TNL clade branched out as expected. The CNL class split into three major subclades: CCR-NLR, CC-NLR, and CCG10-NLR. Within the CC-NLR sub-clade, we identified four significant sub-groups: CNL-Un, CNL-G11, CNL-G7, and G4. The TNL clade exhibited polyphyletic traits, suggesting multiple radiations and significant diversification (Figure 3). In comparison, CCG10-CNL exhibited considerable diversity and expansion with seven radiations. CCR-NLR doesn’t diversify and remains highly conserved across all species. The complete absence of groups from G1-G8 across the genus Glycine aligns with previous findings (Asif et al., 2023, Rani et al., 2023, Areej et al., 2023). The multiple radiations of G7 and G4 also suggest their significant diversification throughout the genus Glycine. The expansion and absence of certain groups in genus Glycine directly related to the specific pathogen they detect. The encounters of Glycine species with different pathogens lead to the diversification of certain groups like we observe in G10, G4 and G7, and the absence of certain pathogens also leads to the lack of associated sub-groups, as in this case G1-G8.
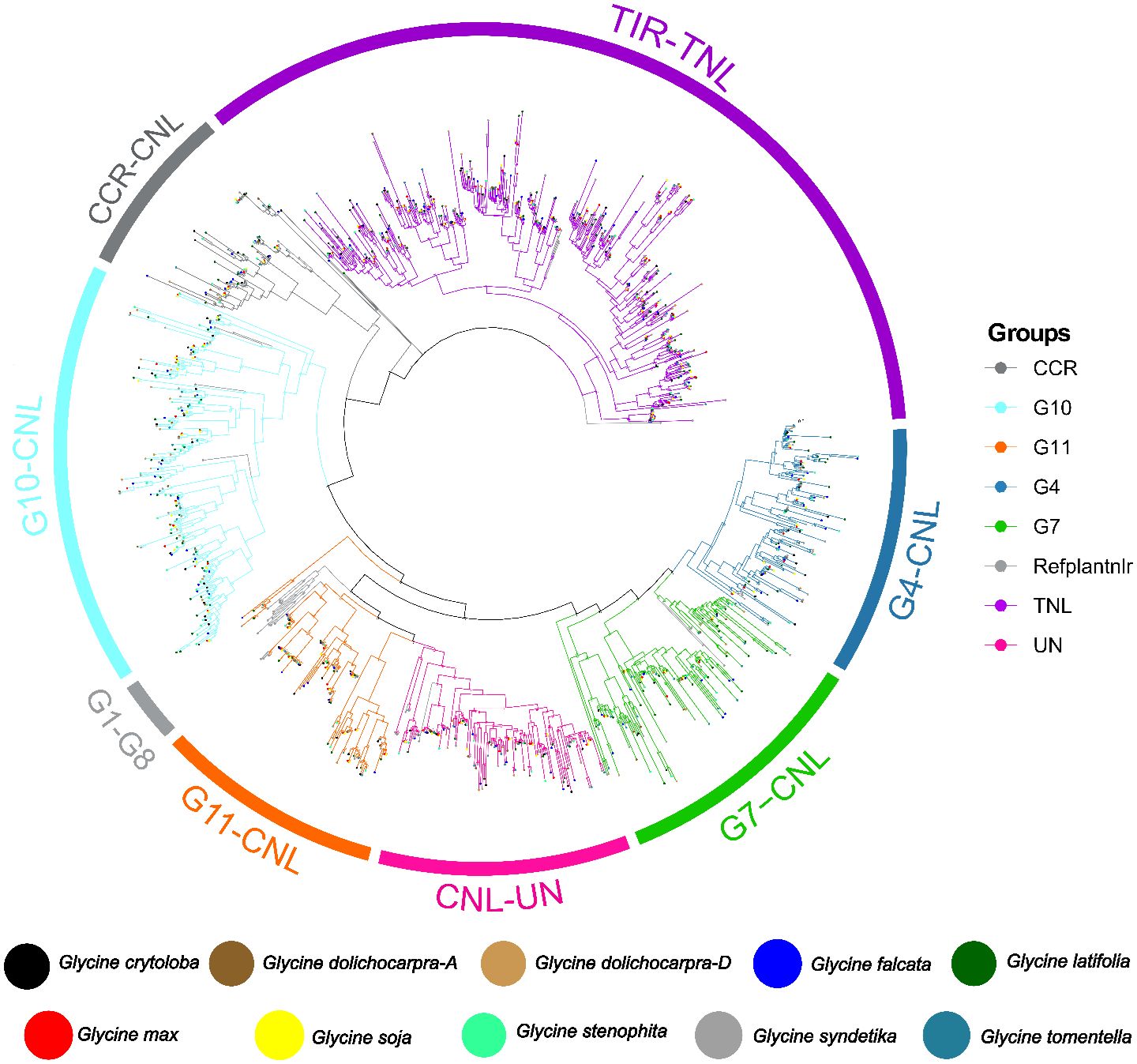
Figure 3 Phylogenetic distribution of 8 genomes and two subgenomes of genus Glycine among four major subclasses of NLR genes 1) CC-NLR 2) CCG10-NLR 3) CCR-NLR 4) TIR-NLR.
Since we have observed that the annual lineage (soja) of Glycine have an expanded NLRome as compared to the perennial lineage (wild). These expansions in the NLRome are either due to the birth of new genes or duplication of existing genes. The study of gene gain and loss of NLR genes in the genus Glycine was conducted using Orthofinder and CAFÉ analysis. Family trees for each Glycine species and its subgenome were constructed by comparing them with Phaseolus vulgaris, its closest allies in the legume tribe. Despite the unique whole genome duplication (WGD) event that occurred in the ancestor of annuals (soja) and perennials (wild), a significant loss of NLR genes was observed in the ancestral lineage (Figure 4). The contraction of NLR genes after WGD in Glycine is consistent with previous observation that the WGD event is followed by a trend of diploidization leading to contraction. Furthermore, following the divergence from perennials, the annual lineage (Soja), exhibited an expansion of NLR gene families (Figure 4). The most significant gene gain of 42 gene families were observed in the ancestral lineage of the subgenus Soja. The highest rate of terminal duplication is found in both annual species of the subgenus Soja. In contrast, the perennial subgenus G. wild demonstrates an overall decrease in the number of NLR gene families across the subgenus. The greatest gene loss was observed in G. falcata, which lost 39 gene families and gained only 6 of them. Terminal duplication also shows a downward trend across the perennials,except for G. latifolia, which gained twenty-nine gene families and lost only seven of them. Considering the evidence from gene birth and death analysis, we can hypothesize that the birth of new sub-gene families, lineage specific and terminal duplication could be the major reason for expanded NLRome in annuals species. We also compared the shared orthologs between perennials and G. max (Figures 4B, C). In total 49 shared orthologs were present between perennials diploid species (G. falcata, G. cyrtoloba and G. stenophita: 4B). Increased number of shared ortholog of up-to 59 were present between G. max and polyploid perennials (G. dolichocarpa, G. tomentella and G. syndetika:4C).
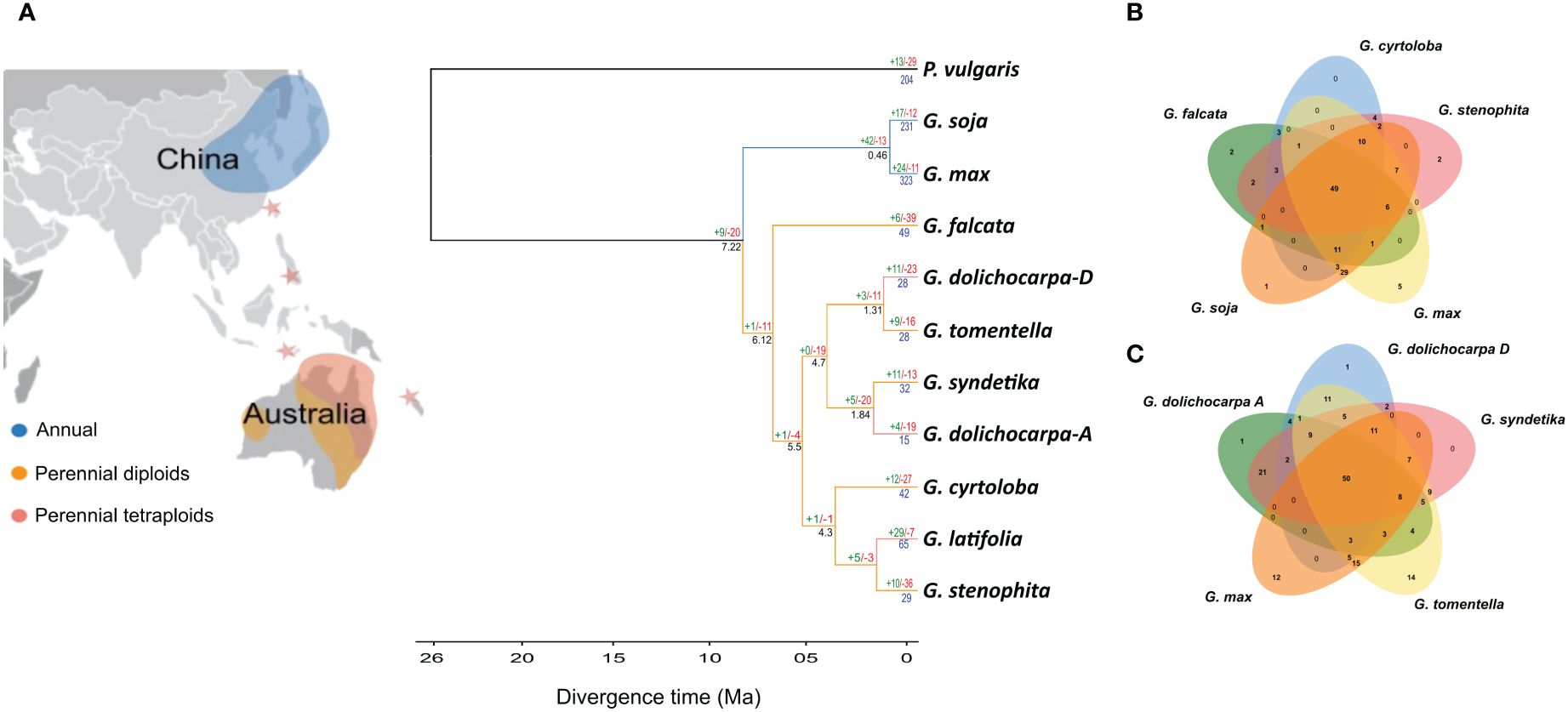
Figure 4 Unraveling gene gains and losses in Glycine species: (A) The phylogenetic tree visualizes the evolutionary trajectory of NLR genes, highlighting gene gain (green), loss (red), and duplication (blue) events across the chronological spectrum. Additionally, the branch color indicates the global distribution of each species, linking genetic evolution to geographical occurrence. (B, C) The Venn diagram illustrates the distribution of species-specific and orthologous NLR genes across the sub-genus, providing insights into the genetic diversity and evolutionary relationships among the species.
Considering the immense importance of gene duplication, as illustrated by gene gain and loss analysis. We explored the duplication history of Glycine by comparing Ks values between paralogs of each subgroup. The closest estimates suggest that divergence between annuals and perennials occurred at ~7 Mya. Collective Ks values obtained from all members of the genus Glycine have shown a common duplication curve since 26 Mya after the split from the recent common ancestor Phaseolus vulgaris. Substitution analysis further suggests that the rate of duplication increased after the Glycine-specific whole-genome duplication (WGD) event, which occurred approximately ~10 Mya (Figure 5). Despite contraction after ongoing diploidization, the perennials lineage underwent a high ratio of gene duplication between an estimated time range of 3–8 Mya (Ks= 0.05–0.01) followed by gradual reduction at the end of this marked increases in the duplication of CCG10-NLR and TIR-NLR were observed during this accelerated gene duplication cycle. However, annuals continued their gene duplications until recently, and the highest duplication rate was observed between 0.1–0.5 Mya. This suggests that annuals species of Glycine remained expanding their NLRome and the highest duplication occurred after the divergence of G. max and G. soja (annuals speciation ~ 0.47 Mya). Interestingly, both annuals’ species G. max and G. soja have showed pronounced duplications of G4-CNL subgroup that ultimately led to diversification and expansion of CC-NLR genes. In short, recent gene duplication is responsible for the expansion of the NLRome in annuals. Secondly, the duplication assay does not provide evidence of recent and accelerated gene duplication rates in subgenomes G. dolichocarpa, which is consistent with lower terminal duplication rates provided by Orthofinder analysis. It further suggests that the Dt-subgenome of G. dolichocarpa could have expanded through processes other than gene duplication, such as recombination and transposition.
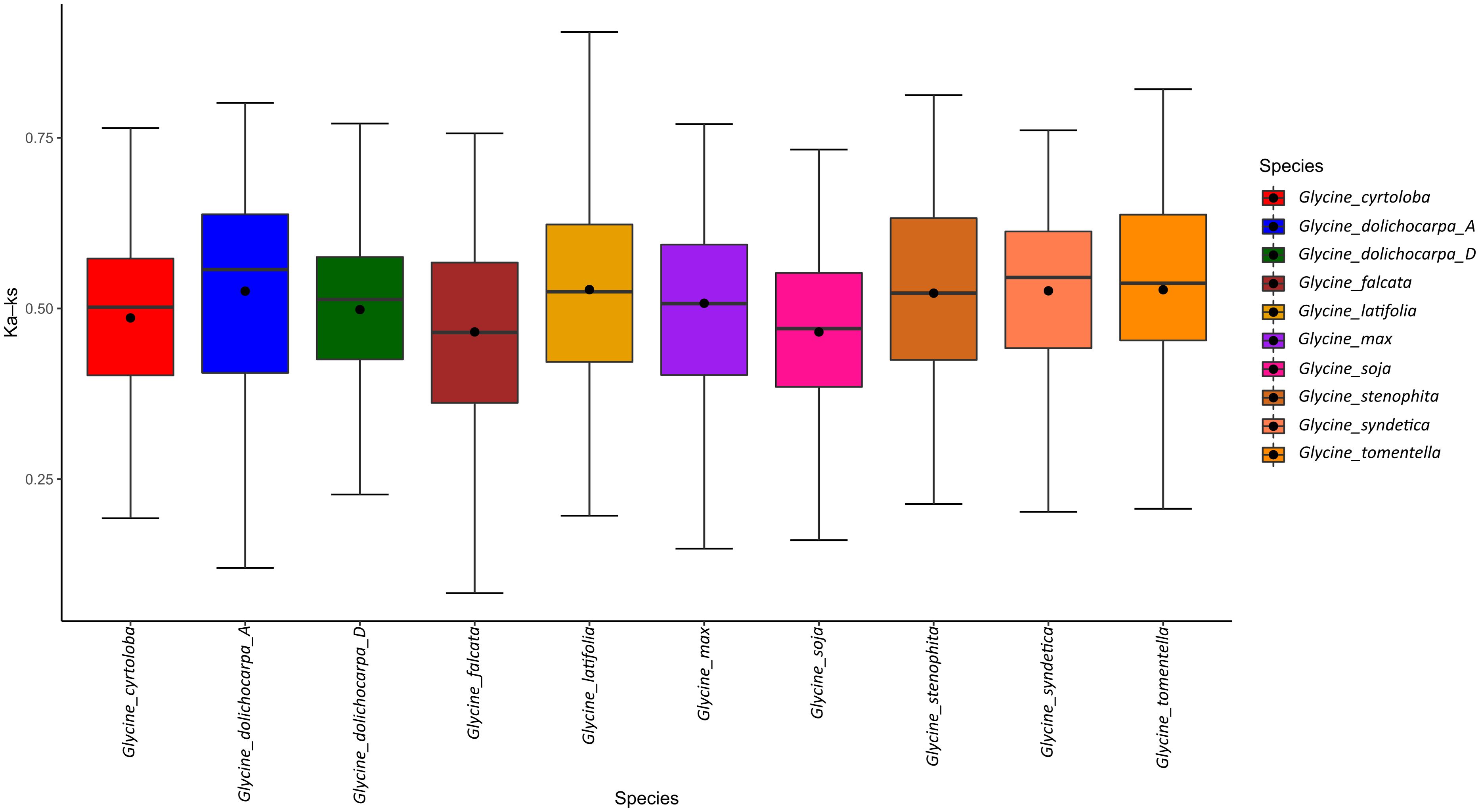
Figure 5 Evolutionary history of NLR gene duplication in Glycine species.
We further evaluated the conservation of lineage-specific gene across genus Glycine by comparing NLR gene tree and species tree using Notung tool. The highest conservation of lineage-specific genes was found among annuals (G. max and G. soja) where 64 both species shared 64 orthologs (Figure 6). Among perennials highest lineage conservation were found among G. latifolia and G. falcata which is contrast with their relatively earlier divergence (~6 Mya). Furthermore, G. falcata also showed a significant share of common orthologs with all members of perennials, suggesting the highest conservation of ancestral NLR genes. Comparison of perennials and annuals revealed ten or less than ten conserved lineages. This significant lack of conserved lineages can be explained by the geographical isolation of perennials species since most perennials are native to Australia and annuals are native to eastern China. In the case of tetraploid G. dolichocarpa, both subgenomes At and Dt have shared 24 and 26 NLR genes lineages with their progenitors G. syndetika (A) and G. tomentella (D). The highest number of species-specific lineages was observed in G. latifolia, which is consistent with the rapid birth of genes and terminal duplication discussed earlier (Figure 4). Overall, a higher ratio of species-specific genes was found in perennials and annuals, demonstrating less complex specific NLR genes repertoire.
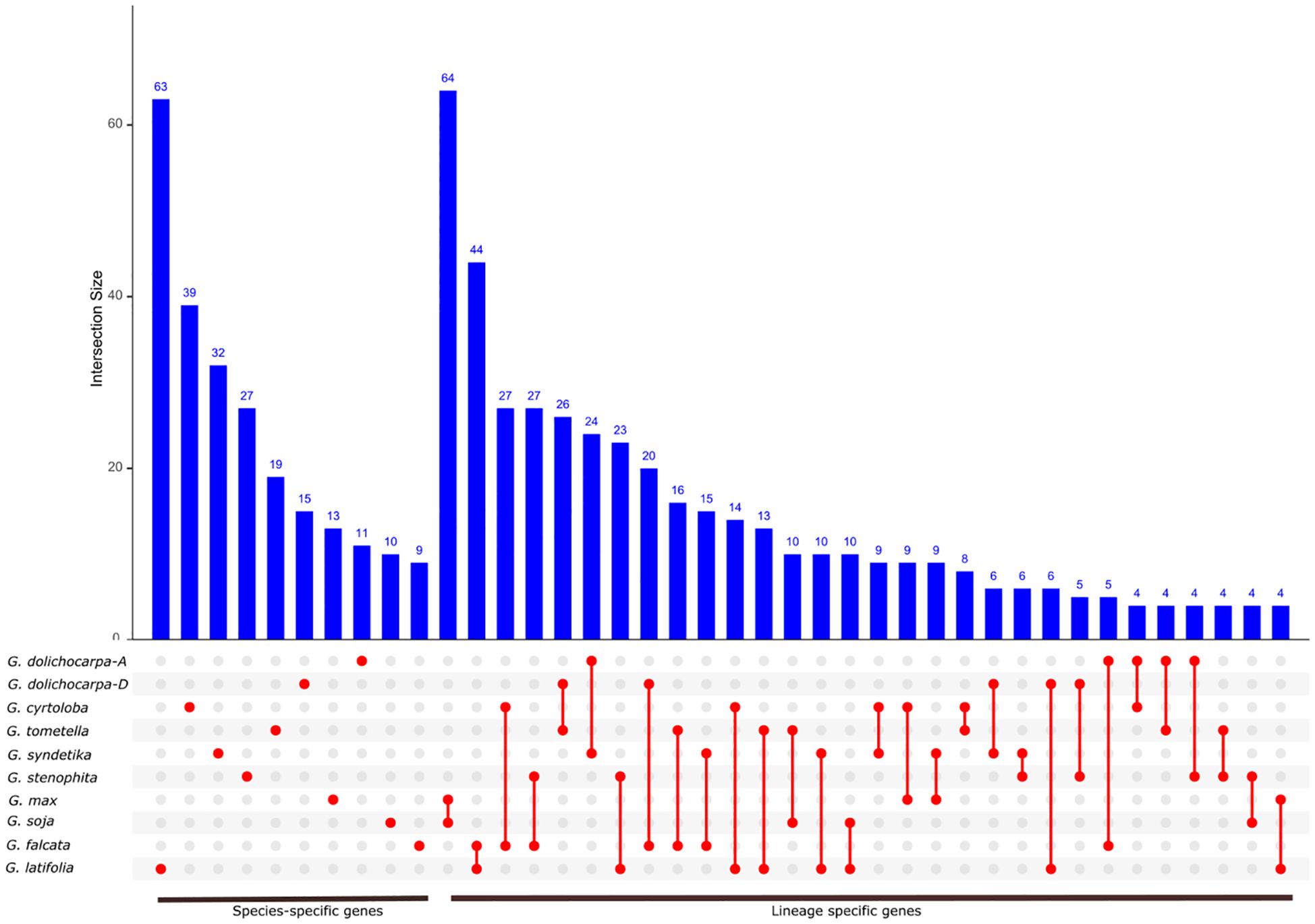
Figure 6 Identification of species and lineage specific NLR genes in genus Glycine: The blue bars denote the count of lineage-specific genes within each species. The red dots and lines below the bars highlight the lineages that are conserved across multiple species.
Considering the highest degree of lineage-specific genes among annuals and lower conservation among perennials, we further performed an in-depth comparison by using synteny analysis. It provides a deep understanding of the evolutionary connections, genomic modifications, and preserved functions across various organisms. Among annuals, a considerable amount of syntenic links were discovered among these species (Figure 3). This suggests a high level of genomic conservation between G. max and G. soja, which is expected given their close evolutionary relationship within the annuals. G. soja and G. max have shown the greatest number of ortholog clusters throughout the chromosomes (Figure 7A). We further compared annuals (G. max and G. soja) and perennial member G. latifolia. G. max and G. soja exhibit a significant degree of synteny between them, which contrasts with G. latifolia (Figure 7B). The latter demonstrates a declining trend in the syntenic relationship between orthologs within the subgenus Soja. Further, G. soja was substituted with the perennial G. dolichocarpa. Interestingly, the members of the subgenus G. wild did not exhibit as much synteny among themselves as we found within the subgenus Soja (Figure 7C). Major clusters were identified on chromosomes 1, 2, 3, 18 and 20. High syntenic relation between G. max and G. soja indicate that they both share a common ancestor, and a large part of the genome has been maintained over time, which shows high genomic stability.
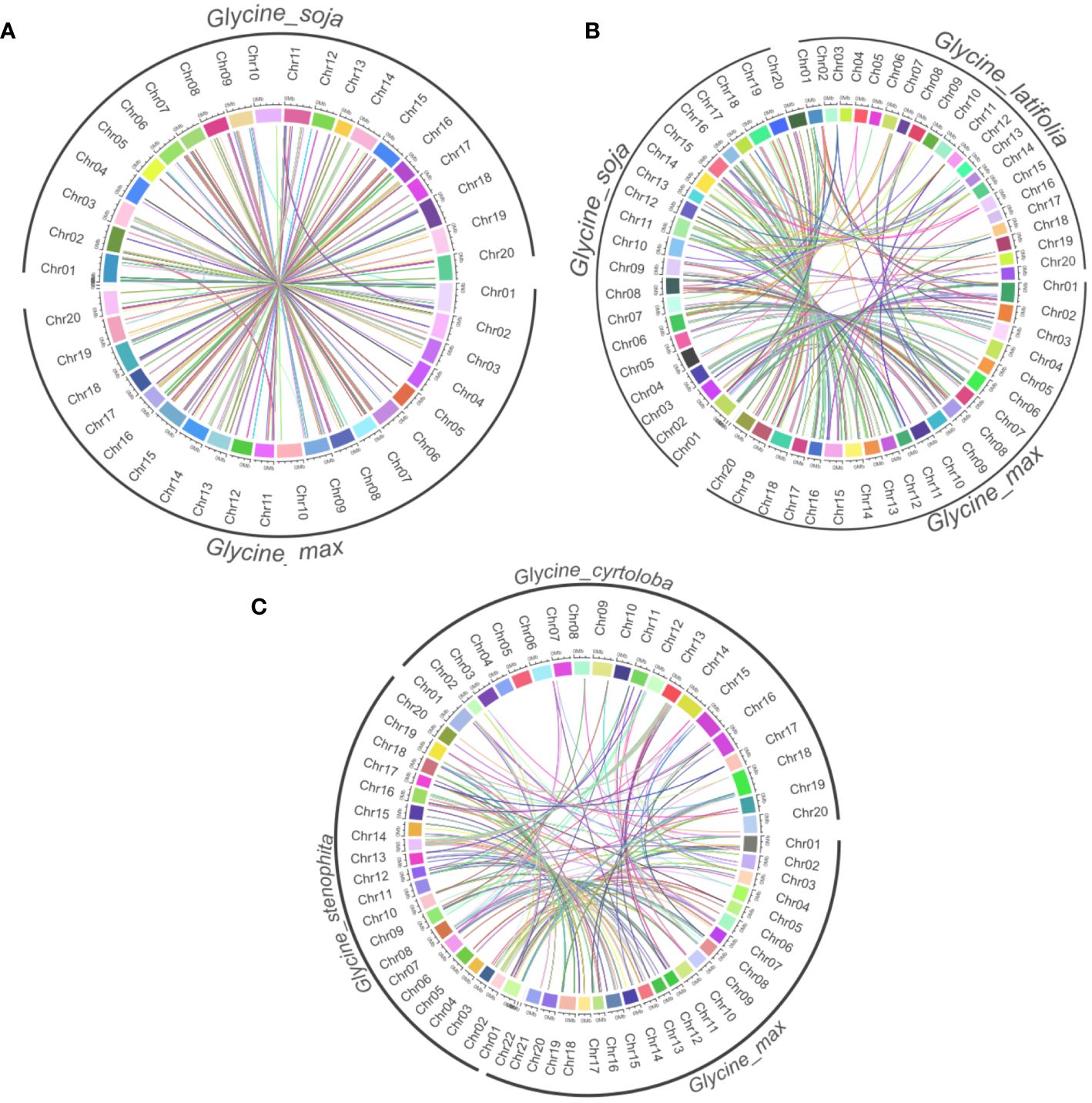
Figure 7 Synteny analysis of NLR genes among annuals and perennials species. Each panel represents syntenic relations among (A) annuals (G. max and G. soja) (B) Annuals species and G. latifolia (C) G. dolichocarpa, G. max and G. latifolia..
We further evaluated the expression identified NLR genes using available datasets. We performed a comparative transcriptomic analysis of both G. soja and G. max in response to infection by Fusarium oxysporum (Figure 8). Within the 45-day duration, a total of 45 genes were expressed in G. soja, whereas 53 genes were expressed in G. max. These genes were categorized into distinct groups: CCR, G10, G11, TNL, and CNL-UN. Notably, the expression of NLR genes was significantly higher in G. soja (wild soybean) compared to G. max (cultivated soybean). Specifically, under both infected and non-infected conditions in the wild, genes such as Glysoja.17G046920.1, Glysoja.14G038028.1, Glysoja.17G046921.1, Glysoja.05G011704.3, and Glysoja.14G038027.1 exhibited elevated expression levels in infected conditions, whereas their expression was comparatively lower in non-infected conditions. These highly expressed genes primarily belonged to the CCR-NLR and TIR-NLR groups. Contrastingly, G. max has shown opposite expression pattern. NLR gene expression was higher under non-infected conditions compared to infected conditions. Genes such as Glyma.16G215100.1, Glyma.14G079600.1, Glyma.17G245500.1, Glyma.17G245600.1, Glyma.17G179200.1, Glyma.05G082200.1, and Glyma.16G137200.1, categorized under CCR-NLR and TIR-NLR groups, demonstrated this differential expression pattern. Additionally, Glyma.14G079500.1 and Glyma.16G210600.1 exhibited notably high expression levels, specifically in non-infected conditions. Overall, the analysis indicates that the expression of NLR genes is substantially higher in wild soybean species compared to cultivated soybeans in response to Fusarium oxysporum. Furthermore, distinct expression patterns were observed between the two species under both infected and non-infected conditions, with different sets of genes displaying varied expression levels in response to the pathogen.
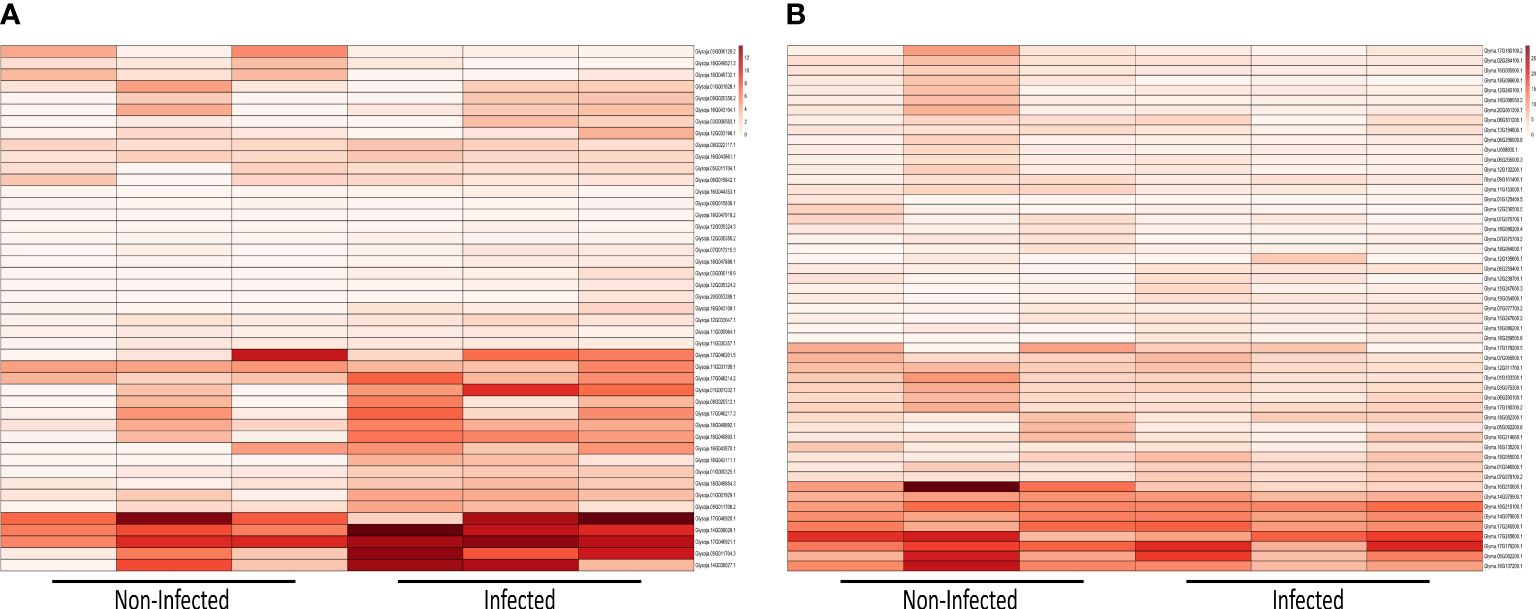
Figure 8 Expression analysis upon Fusarisum oxysporum infection (A) heatmap of NLR genes expressions in infected and non-infected G. soja (B) heatmap of NLR genes expressions in infected and non-infected G. max.
In the PRJNA628842 dataset, the seeds of two lines of G. max (LHY and WT) were grown in the Hogland Solution for two weeks and these samples were collected according to the ZT (zeitgeber time) (Figure 9). LHY is a transgenic line, that overexpresses Late Elongated Hypocotyl gene (LHY) which is responsible for the circadian movement of leaf in plants (Wang et al., 2021). We performed comparative transcriptomics between two lines of G. max LHY and WT and their response to drought stress (LHY-D and WT-D). In normal conditions relatively lower expression of 25 NLR genes were observed that can be classified into 5 groups (CCR, G10, G11, TNL and UN), highest number of genes that were expressed belonged to helper NLR (CCR-NLR). However, under drought conditions in LHY and WT, the expression rate of NLR genes were higher as compared to well-watered conditions. In total 7 genes have higher rate of expression in both LHY-D and WT-D condition belonging to CCR-NLR, TIR, and G11-CNL subgroups.
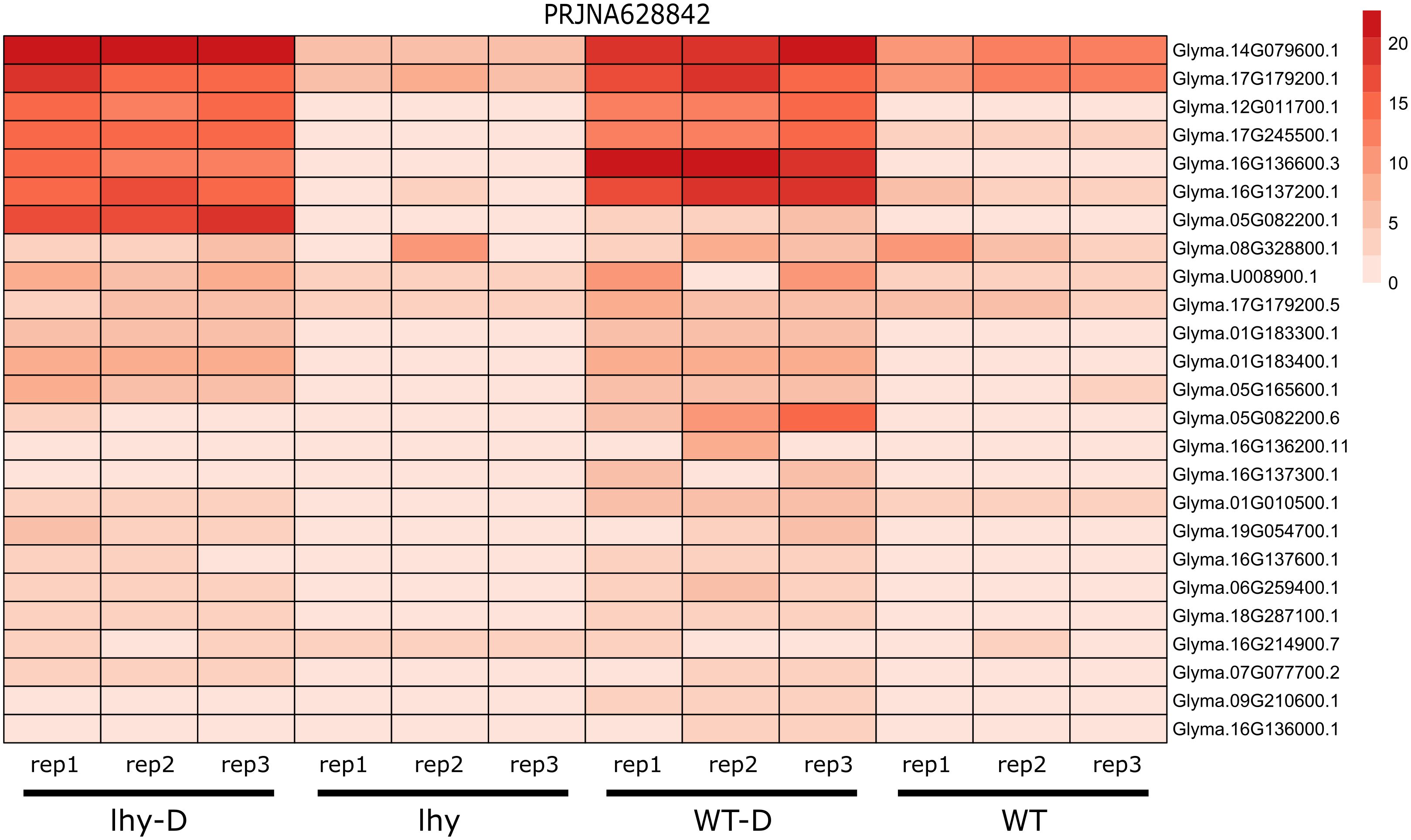
Figure 9 Expression analysis of NLR genes upon drought treatment in wild and transgenic G. max. LHY lines overexpression LHY genes illustrates higher expression of NLR genes drought conditions.
Soybean, originally from China, is now the world’s most widely grown oil and protein seed crop. Despite its agricultural importance, the primary gene pool of soybean, which includes G. max (cultivated soybean) and G. soja (wild soybean), exhibits low genetic diversity. This limited diversity is a significant constraint on the crop’s environmental resilience and yield potential. G. max is particularly susceptible to major pathogens, including viruses, bacteria, and fungi (Araújo et al., 2019; Bandara et al., 2020). Nucleotide-binding site leucine-rich repeat (NLR) genes play a crucial role in plant immunity by recognizing pathogen effectors and triggering defense responses.
Previous study for the characterization of NLR genes was limited to annual species (G. max and G. soja) and due to fragmented genome assemblies limited number of NLR gene were identified. For example, reduced NLR were identified from G. max (Zheng et al., 2016; Afzal et al., 2022). Utilization of updated reference genome and mining approaches has allowed detailed understanding of NLR gene evolution. Our study reveals that both G. max and G. soja have an expanded repertoire of NLR genes compared to their perennial relatives. This expansion is attributed to recent gene duplications occurring between 0.1 to 0.5 million years ago in their common ancestor and after their speciation. This is evidenced by a higher ratio of duplicate NLR genes to singletons and the rapid emergence of non-core genes in annuals in annual species compared to perennials (Liu et al., 2018; Zhuang et al., 2022).
Perennial species have experienced significant contraction of NLR genes following a whole-genome duplication event approximately 10 million years ago. This contraction has led to a more diversified but smaller set of NLR genes, likely due to gene duplications that occurred between 4 to 7 million years ago after diverging from the annual lineage. It is consistent with previous studies that identified multiple soybean cyst nematode (SCN) population in G. tomentella and other species G. argyria and G. pescadrensis also showed resistance to all tested SCN populations (Wen et al., 2017). Similarly perennial Glycine species (G. argyrea, G. clandestina, G. dolichocarpa, G. tomentella and G. canescens) have demonstrated resistance to soybean rust (Phakopsora pachyrhizi) (Herman et al., 2020).
The high syntenic relationship between G. max and G. soja indicates that a large part of their genome has been maintained over time, demonstrating high genomic stability in the annual lineage. Synteny analysis revealed a high degree of genomic conservation between the annual species G. max and G. soja, indicating their close evolutionary relationship and shared ancestry. This is consistent with recently constructed De-novo assembled pan genome of soybean wild relatives that provide evidence of greater genomic stability of G. max and G. soja as compared to perennial species and confirmed that large part of genome has been maintained over time (Joshi et al., 2013). In contrast, the perennial species G. latifolia exhibited a declining trend in syntenic relationship with the annuals, suggesting divergence within the subgenus Soja. Previous study has also decreased trend of syntenic relation as only 12 out of 12 G. latifolia linkage groups were identified that were colinear with G. max chromosomes (Chang et al., 2014). Within the perennial subgenus Glycine wild, the species did not exhibit as much synteny among themselves as observed within the annual subgenus Soja. Major syntenic clusters were identified on chromosomes 1, 2, 3, 18, and 20.
G. latifolia, known for its high levels of resistance to multiple soybean pathogens and pests, encodes a unique repertoire of NLR genes that are highly species-specific. This diversification might have occurred after the common duplication curve between 4–7 Mya. Despite the ongoing diploidization trend in perennial species, G. latifolia has gained 29 gene families with an accelerated terminal duplication rate relative to the closest species (G. cyrtoloba and G. stenophita). Gene gain and loss analysis provide strong evidence that the birth of new gene families and terminal duplication are significant reasons for the highly divergent evolution of NLR genes in G. latifolia. Previous studies highlighted the presence of 3,148 unique sets of genes and noted the overrepresentation of NB-ARC encoding genes (Zhuang et al., 2022). Similarly, comparative analysis with five legume species showed that genes related to defense responses were significantly overrepresented in Glycine-specific orthologous gene families (Liu et al., 2018).
The evolutionary history of NLR genes in soybean annuals and perennials plants indicates that gene duplications and losses have played a significant role in shaping their current NLR gene profiles. The study of gene gain and loss of NLR genes in the genus Glycine revealed that annual species have a higher rate of gene birth and terminal duplication compared to perennials. The most significant gene gain was observed in the ancestral lineage of the subgenus Soja, while the greatest gene loss was observed in G. falcata. G. latifolia, however, showed a significant gain of gene families, indicating a dynamic evolutionary process. Recently constructed high density linkage maps for G. latifolia and their comparison with G. max has found significant chromosomal rearrangements in perennial species and annual species G. max has undergone more frequent gene duplication contributing to their genetic diversity adaptability (Chang et al., 2014). These dynamics are crucial for understanding how plants adapt to pathogen pressures and environmental changes.
The study found the highest conservation of lineage-specific genes among annuals (G. max and G. soja), while perennials showed a higher ratio of species-specific genes. This significant lack of conserved lineages between annuals and perennials can be explained by their geographical isolation and different evolutionary pressures.
This study provides new insights into the effect of allopolyploidy on the evolution of NLR genes. The availability of the complete genome of A and D diploid progenitors allows a precise definition of its sub-genome and its genome-wide distribution of NLR genes. Comparing the A, D, At, and Dt genomes revealed that the At and Dt sub-genomes of the allopolyploid lost 7,351 genes, with a higher number of losses from D (4,109) than from A (3,242) (Zhuang et al., 2022). Conversely, NLR distribution was asymmetrical to the already described complete gene fractionation pattern. Marked expansion of NLR gene distribution was observed for Dt as compared to At. This asymmetric expansion of the NLRome in the subgenomes of G. dolichocarpa could possibly be due to homologous sequence exchanges (HSEs) and illegitimate recombination. The availability of chromosomal-anchored genome sequences of additional polyploids and their progenitors will further provide a better understanding of the role of polyploids on the evolution of NLR genes.
The findings highlight the importance of leveraging the genetic diversity within the Glycine genus to improve soybean’s disease resistance. By identifying and introgressing beneficial NLR genes from wild relatives and other sources, breeders can develop soybean varieties with enhanced resilience and yield potential. This study has significant implications for crop breeding and development of disease-resistance soybean cultivars. The phylogenetic distribution of NLR genes provides a catalog of genetic resources that can be used for crop improvement. By utilizing diverse Glycine germplasm, breeders can introduce new resistance genes into elite cultivars, enhancing genetic diversity and crop resilience. The conserved NLR genes are valuable resource for developing broad-specturm disease resistance. G4, G7, G11, CCR-CNL and CCG10-CNL perennial lineage-specific NLR genes of Glycine offer an opportunity to develop unique disease resistance traits tailored to regional challenges. These genes can be incorporated into breeding programs to create cultivars with enhanced or novel resistance traits. The use of genomic and transcriptomic resources has enabled researchers to uncover the mechanisms underlying the diversification and maintenance of NLR gene repertoires, providing valuable insights for future studies on plant immune system evolution and disease resistance. The provided results offer valuable insights into the genetic diversity, evolution, and regulation of NLR genes in related plant species, supporting the findings of the study on the genus Glycine. These results underscore the broader significance of NLR gene research in the context of plant immunity and evolutionary biology.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
AS: Data curation, Formal Analysis, Methodology, Software, Writing – original draft, Writing – review & editing. HN: Formal Analysis, Funding acquisition, Project administration, Resources, Visualization, Writing – original draft, Writing – review & editing. FS: Project administration, Resources, Software, Writing – original draft, Writing – review & editing. SN: Data curation, Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. MD: Data curation, Funding acquisition, Methodology, Resources, Software, Writing – original draft, Writing – review & editing. RI: Data curation, Formal Analysis, Methodology, Validation, Writing – original draft, Writing – review & editing. UM: Data curation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. AA: Data curation, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. NA: Data curation, Funding acquisition, Methodology, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing. RM: Data curation, Methodology, Software, Writing – original draft, Writing – review & editing. SSh: Data curation, Methodology, Writing – original draft. MR: Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. SSe: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R62), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1383135/full#supplementary-material
Supplementary Figure 1 | Gene density map with respect to each chromosome of Glycine species (ranged 1–20). The gradient color is provided to represent the gene density of each chromosome with respect to number bins present in the chromosome.
Afzal, M., Alghamdi, S. S., Nawaz, H., Migdadi, H. H., Altaf, M., El-Harty, E., et al. (2022). Genome- wide identification and expression analysis of CC-NB-ARC-LRR (NB-ARC) disease-resistant family members from soybean (Glycine max L.) reveal their response to biotic stress. J. King Saud Univ. - Sci. 34. doi: 10.1016/j.jksus.2021.101758
Andersen, E. J., Ali, S., Byamukama, E., Yen, Y., Nepal, M. P. (2018). Disease resistance mechanisms in plants. Genes 9, 339. doi: 10.3390/genes9070339
Araújo, A. C., Fonseca, F. C. D. A., Cotta, M. G., Alves, G. S. C., Miller, R. N. G. (2019). Plant NLR receptor proteins and their potential in the development of durable genetic resistance to biotic stresses. Biotechnol. Res. Innov. 3, 80–94. doi: 10.1016/j.biori.2020.01.002
Areej, A., Nawaz, H., Aslam, I., Danial, M., Qayyum, Z., Rasool, U. A., et al. (2023). Investigation of NLR genes reveals divergent evolution on NLRome in diploid and polyploid species in genus trifolium. Genes (Basel) 14, 867. doi: 10.3390/genes14040867
Asif, J., Qureshi, F., Zain, M., Nawaz, H., Naz, E., Fareed, S., et al. (2023). Investigation of resistance genes in genus vigna reveals highly variable NLRome in parallel domesticated member species. Genes (Basel) 14, 1129. doi: 10.3390/genes14061129
Bandara, A. Y., Weerasooriya, D. K., Bradley, C. A., Allen, T. W., Esker, P. D. (2020). Dissecting the economic impact of soybean diseases in the United States over two decades. PloS One 15. doi: 10.1371/journal.pone.0231141
Calle García, J., Guadagno, A., Paytuvi-Gallart, A., Saera-Vila, A., Amoroso, C. G., D’esposito, D., et al. (2022). PRGdb 4.0: an updated database dedicated to genes involved in plant disease resistance process. Nucleic Acids Res. 50, D1483–D1490. doi: 10.1093/NAR/GKAB1087
Chang, S., Thurber, C. S., Brown, P. J., Hartman, G. L., Lambert, K. N., Domier, L. L. (2014). Comparative mapping of the wild perennial Glycine latifolia and soybean (G. max) reveals extensive chromosome rearrangements in the genus Glycine. PloS One 9. doi: 10.1371/journal.pone.0099427
Cui, H., Tsuda, K., Parker, J. E. (2015). Effector-triggered immunity: from pathogen perception to robust defense. Annu. Rev. Plant Biol. 66, 487–511. doi: 10.1146/annurev-arplant-050213-040012
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Egan, A. N., Doyle, J. (2010). A comparison of global, gene-specific, and relaxed clock methods in a comparative genomics framework: dating the polyploid history of soybean (Glycine max). Syst. Biol. 59, 534–547. doi: 10.1093/sysbio/syq041
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20. doi: 10.1186/s13059-019-1832-y
Forrester, N. J., Ashman, T. L. (2018). The direct effects of plant polyploidy on the legume-rhizobia mutualism. Ann. Bot. 121, 209–220. doi: 10.1093/aob/mcx121
Hao, Z., Lv, D., Ge, Y., Shi, J., Weijers, D., Yu, G., et al. (2020). RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 6, 1–11. doi: 10.7717/peerj-cs.251
Herman, T. K., Han, J., Singh, R. J., Domier, L. L., Hartman, G. L. (2020). Evaluation of wild perennial Glycine species for resistance to soybean cyst nematode and soybean rust. Plant Breed. 139, 923–931. doi: 10.1111/pbr.12834
Innes, R. W., Ameline-Torregrosa, C., Ashfield, T., Cannon, E., Cannon, S. B., Chacko, B., et al. (2008). Genome analysis differential accumulation of retroelements and diversification of NB-LRR disease resistance genes in duplicated regions following polyploidy in the ancestor of soybean 1[W][OA]. Plant Physiol. 148, 1740–1759. doi: 10.1104/pp.108.127902
Jones, J. D. G., Vance, R. E., Dangl, J. L. (2016). Intracellular innate immune surveillance devices in plants and animals. Sci. (1979) 354. doi: 10.1126/science.aaf6395
Joshi, T., Valliyodan, B., Wu, J. H., Lee, S. H., Xu, D., Nguyen, H. T. (2013). Genomic differences between cultivated soybean, G. max and its wild relative G. soja. BMC Genomics, 14. doi: 10.1186/1471-2164-14-S1-S5
Kim, K., El Baidouri, M., Abernathy, B., Iwata-Otsubo, A., Chavarro, C., Gonzales, M., et al. (2015). A comparative epigenomic analysis of polyploidy-derived genes in soybean and common bean. Plant Physiol. 168, 1433–1447. doi: 10.1104/pp.15.00408
Kourelis, J., Sakai, T., Adachi, H., Kamoun, S. (2021). RefPlantNLR is a comprehensive collection of experimentally validated plant disease resistance proteins from the NLR family. PloS Biol. 19. doi: 10.1371/journal.pbio.3001124
Li, K.B. (2003). ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics 19, 1585–1586. doi: 10.1093/bioinformatics/btg192
Li, Y. H., Zhou, G., Ma, J., Jiang, W., Jin, L. G., Zhang, Z., et al. (2014). De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 32, 1045–1052. doi: 10.1038/nbt.2979
Liu, Q., Chang, S., Hartman, G. L., Domier, L. L. (2018). Assembly and annotation of a draft genome sequence for Glycine latifolia, a perennial wild relative of soybean. Plant J. 95, 71–85. doi: 10.1111/tpj.13931
Manzoor, A., Ahmad, T., Bashir, M. A., Hafiz, I. A., Silvestri, C. (2019). Studies on colchicine induced chromosome doubling for enhancement of quality traits in ornamental plants. Plants (Basel) 8. doi: 10.3390/plants8070194
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Pan, Q., Wendel, J., Fluhr, R. (2000). Divergent evolution of plant NBS-LRR resistance gene homologues in dicot and cereal genomes. J. Mol. Evol. 50, 203–213. doi: 10.1007/s002399910023
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Quinlan, A. R., Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Qureshi, F., Mehmood, A., Khan, S. A., Bilal, M., Urooj, F., Alyas, M., et al. (2023). Macroevolution of NLR genes in family Fabaceae provides evidence of clade specific expansion and contraction of NLRome in Vicioid clade. Plant Stress 10, 100254. doi: 10.1016/j.stress.2023.100254
Rani, S., Zahra, R., Bakar, A., Rizwan, M., Sultan, A.-B., Zain, M., et al. (2023). Dynamic Evolution of NLR Genes in Dalbergioids. Genes 14, 377. doi: 10.3390/GENES14020377
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Sedivy, E. J., Wu, F., Hanzawa, Y. (2017). Soybean domestication: the origin, genetic architecture and molecular bases. New Phytol. 214, 539–553. doi: 10.1111/nph.14418
Seo, E., Kim, S., Yeom, S. I., Choi, D. (2016). Genome-wide comparative analyses reveal the dynamic evolution of nucleotide-binding leucine-rich repeat gene family among solanaceae plants. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01205
Sherman-Broyles, S., Bombarely, A., Powell, A. F., Doyle, J. L., Egan, A. N., Coate, J. E., et al. (2014). The wild side of a major crop: Soybean’s perennial cousins from Down Under. Am. J. Bot. 101, 1651–1665. doi: 10.3732/AJB.1400121
Suyama, M., Torrents, D., Bork, P. (2006). PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34. doi: 10.1093/nar/gkl315
Wang, J., Chai, J. (2020). Molecular actions of NLR immune receptors in plants and animals. Sci. China Life Sci. 63, 1303–1316. doi: 10.1007/s11427-019-1687-6
Wang, W., Chen, L., Fengler, K., Bolar, J., Llaca, V., Wang, X., et al. (2021). A giant NLR gene confers broad-spectrum resistance to Phytophthora sojae in soybean. Nat. Commun. 12. doi: 10.1038/s41467-021-26554-8
Wen, L., Yuan, C., Herman, T. K., Hartman, G. L. (2017). Accessions of perennial Glycine species with resistance to multiple types of soybean cyst nematode (Heterodera glycines). Plant Dis. 101, 1201–1206. doi: 10.1094/PDIS-10-16-1472-RE
Xie, M., Chung, C. Y. L., Li, M. W., Wong, F. L., Wang, X., Liu, A., et al. (2019). A reference-grade wild soybean genome. Nat. Commun. 10, 1–12. doi: 10.1038/s41467-019-09142-9
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58. doi: 10.1093/nar/gkz333
Yin, D., Ji, C., Song, Q., Zhang, W., Zhang, X., Zhao, K., et al. (2019). Comparison of arachis monticola with diploid and cultivated tetraploid genomes reveals asymmetric subgenome evolution and improvement of peanut. Adv. Sci. (Weinh) 7. doi: 10.1002/ADVS.201901672
Zheng, F., Wu, H., Zhang, R., Li, S., He, W., Wong, F. L., et al. (2016). Molecular phylogeny and dynamic evolution of disease resistance genes in the legume family. BMC Genomics 17, 1–13. doi: 10.1186/s12864-016-2736-9
Keywords: glycine, perennials, annuals, NLR genes, diversification, evolution
Citation: Sultan AB, Nawaz H, Saleem F, Nawaz S, Danial M, Iftikhar R, Maqsood U, Areej A, Shakoor S, Aljarba NH, Maqbool R, Rizwan M and Serfraz S (2024) Divergent evolution of NLR genes in the genus Glycine: impacts of annuals and perennials’ life history strategies. Front. Plant Sci. 15:1383135. doi: 10.3389/fpls.2024.1383135
Received: 06 February 2024; Accepted: 12 June 2024;
Published: 09 July 2024.
Edited by:
Peng Wang, Jiangsu Province and Chinese Academy of Sciences, ChinaReviewed by:
Xuming Li, Hugo Biotechnologies Co., Ltd., ChinaCopyright © 2024 Sultan, Nawaz, Saleem, Nawaz, Danial, Iftikhar, Maqsood, Areej, Shakoor, Aljarba, Maqbool, Rizwan and Serfraz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saad Serfraz, c2VyZnJhei5zYWFkQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.