 Shenghui Su
Shenghui Su Min Ji2
Min Ji2 Chenxia Cheng
Chenxia Cheng- 1College of Horticulture, Qingdao Agricultural University, Qingdao, Shandong, China
- 2Engineering Laboratory of Genetic Improvement of Horticultural Crops of Shandong Province, Qingdao Agricultural University, Qingdao, China
- 3National Technology Centre for Whole Process Quality Control of FSEN Horticultural Products (Qingdao), Qingdao Agricultural University, Qingdao, China
- 4Laboratory of Quality & Safety Risk Assessment for Fruit (Qingdao), Ministry of Agriculture and Rural Affairs, Qingdao Agricultural University, Qingdao, China
- 5Qingdao Key Lab of Modern Agricultural Quality and Safety Engineering, Qingdao Agricultural University, Qingdao, China
- 6Academy of Dongying Efficient Agricultural Technology and Industry on Saline and Alkaline Land in Collaboration with Qingdao Agricultural University, Dongying, China
Histone methylation is an important type of histone modification that regulates gene expression in plants. In this study, we identified 14 arginine methylation-related genes (Protein Arginine Methyltransferase, MdPRMT) and 32 demethylation-related genes (JmjC Domain-Containing Family, MdJMJ) in apple. Furthermore, we investigated the phylogenetic relationship, chromosome distribution, gene structure, motif analysis, promoter sequence analysis, and expression patterns of MdPRMT and MdJMJ genes. Homology analysis showed a high degree of conservation and homology between PRMT and JMJ genes in Arabidopsis and apple. We identified the types of duplicated genes in the MdJMJ and MdPRMT gene families, found a large number of whole-genome duplicates (WGD) gene pairs and a small number of tandem duplicates (TD) pairs, transposed duplication (TRD) gene pairs as well as proximal duplicates (PD) pairs, and discussed the possible evolutionary pathways of the gene families from the perspective of duplicated genes. Homology analysis showed a high degree of conservation and homology between PRMT and JMJ genes in Arabidopsis and apple. In addition, the promoter regions of MdPRMT and MdJMJ contain numerous cis-acting elements involved in plant growth and development, hormone response, and stress responses. Based on the transcriptional profiles of MdPRMT and MdJMJ in different tissues and developmental stages, it was found that MdPRMT and MdJMJ may play multiple roles in apple growth and development, for example, MdJMJ21 may be involved in the regulation of apple endosperm formation. MdPRMT and MdJMJ exhibit different expression patterns in response to hormone signaling in apple, MdJMJ3, MdJMJ18, MdJMJ30, MdPRMT2, MdPRMT13, and MdPRMT14 may play roles in apple response to drought stress, while the expression of MdJMJ13, MdPRMT3, MdPRMT4, and MdPRMT6 is affected by cold stress. Our study provides a foundation for determining the functional roles of MdPRMT and MdJMJ genes in apple.
1 Introduction
In eukaryotes, the basic structural unit of chromatin is the nucleosome (Kouzarides, 2007). Generally, nucleosomes are composed of octamers consisting of two copies each of four histone subunits, H2B, H2A, H3, and H4 (Thomas Jenuwein, 2001; Liu et al., 2010). The N-terminal amino acid residues of histones are susceptible to post-translational modifications, including acetylation, methylation, phosphorylation, ubiquitination, and other histone modifications (Kouzarides, 2007). In recent years, with the advancement in detection techniques for histone modifications and further research, it has been discovered that the middle and C-terminal regions of histones can also be specifically modified. These modifications affect the compactness and accessibility of chromatin in different ways, thereby influencing gene expression and ultimately affecting various physiological and developmental processes in organisms. They are one of the most important epigenetic regulatory mechanisms of gene expression in eukaryotes (Lawrence et al., 2016). Histone modifications are reversible covalent modifications. The occurrence, removal, and functional roles of these covalent modifications are mainly regulated by histone-modifying enzymes and their corresponding cofactors, including Writers, Erasers, and Reader/Effectors (Liu et al., 2010). Writers are enzymes that catalyze the addition of chemical groups to histones for modification, such as histone acetyltransferases (HATs), histone methyltransferases (HMTs), kinases, and ubiquitinases. Erasers are enzymes that remove these modifications from histones, such as histone deacetylases (HDACs), histone demethylases (HDMs), phosphatases, and deubiquitinases. Readers are proteins or protein complexes that recognize and specifically bind to substrates with specific post-translational modifications (Liu et al., 2010). Among them, histone methylation, as one of the main types of histone modifications, primarily affects the binding of histones to Reader proteins, leading to changes in chromatin structure, and thus transcriptional repression or activation (Genschik et al., 2014). Histone methylation can be divided into lysine methylation and arginine methylation based on their occurrence sites (Alvarez-Venegas, 2005), so HMTs can also be classified into HKMTs and PRMTs. The removal of histone methylation modifications is mainly accomplished by HDMs (Luo et al., 2014).
Protein arginine methylation is catalyzed by type I and II PRMTs, with type I PRMTs catalyzing asymmetric dimethylation of arginine residues and type II PRMTs catalyzing symmetric dimethylation of arginine residues (Bedford and Richard, 2005). Studies have shown that Arabidopsis protein arginine methyltransferase 5 (AtPRMT5), a homolog of human PRMT5, is an enzyme capable of catalyzing symmetric dimethylation of arginine residues, and it plays a role in Arabidopsis growth and development, especially in promoting flowering (Wang et al., 2001; Pei et al., 2007). In addition, other type I PRMTs in Arabidopsis that can catalyze asymmetric dimethylation of arginine residues (AtPRMT10, AtPRMT4a, and AtPRMT4b) have also been shown to promote flowering (Niu et al., 2007, 2008). Overall, existing evidence suggests that protein arginine methyltransferases play important roles in plant growth and development. HDMs are key factors regulating the steady state of histone methylation. HDMs can be divided into two types based on their mechanisms: lysine-specific demethylase KDM1/LSD1 and demethylases containing Jumonji C (JmjC) domains. KDM1/LSD1 performs demethylation of mono- and dimethylated lysine residues through FAD-dependent amine oxidation reactions, while JmjC proteins catalyze demethylation reactions that are dependent on iron (II) and α-ketoglutarate, and they play important roles in histone demethylation. They can remove methylation modifications at H3K4, H3K9, H3K27, and H3K36 (Anand and Marmorstein, 2007). Arabidopsis contains 21 JmjC domain proteins (JMJs), which are divided into five subfamilies (KDM5/JARID1, KDM4/JHDM3, KDM3/JHDM2, JMJD6, and JmjC domain-only) based on protein sequence similarity (Luo et al., 2014). In rice, 20 JmjC domain proteins have been identified, among which OsJMJ706 specific to H3K9 and OsJMJ703 specific to H3K4 have been shown to be involved in the regulation of rice flower organ development and transposon silencing, respectively (Sun and Zhou, 2008; Copenhaver et al., 2013; Cui et al., 2013). These findings collectively indicate that PRMTs and HDMs play important roles in plant growth and development.
Apple (Malus domestica) is one of the most important fruits in the world (Perini et al., 2014). Previous studies have shown that histone lysine methylation in apples is primarily mediated by a class of proteins containing SET domains (SDGs). The apple genome contains a total of 67 SDG genes, which play important roles in apple development and stress responses (Li et al., 2021). However, research on protein arginine methyltransferases and histone demethylases in apples is very limited, so it is necessary to reveal their functional characteristics in apples.
In this study, a total of 32 JMJ members and 14 PRMT members were identified in the apple genome, and they were subjected to detailed analysis in terms of systematic evolution, homology relationships, conserved domains, gene structure, and cis-acting elements. In addition, the expression profiles of MdPRMTs and MdMJMs in different organs of apple at different developmental stages, under biotic and abiotic stresses, and in response to hormones were also studied. In conclusion, this study provides a comprehensive analysis of JMJ and PRMT genes in apple, laying a foundation for further exploration of their regulatory roles in apple development, stress responses, and hormone signaling.
2 Materials and methods
2.1 Identification and classification of histone methylation modification genes
Following previous research, the Arabidopsis Information Resource (TAIR, https://www.arabidopsis.org/) was used to obtain the Arabidopsis thaliana histone methylation modification genes protein sequences. The whole genome data of apples (Malus domestica) was obtained from the Apple Genome Database (http://bioinformatics.cau.edu.cn/AppleMDO/) (Da et al., 2019). And the gene identifier (ID) can be found in the Supplementary Table S1.
Candidate histone methylation modification genes (PRMTs and JMJs) in apple were identified using; two basic local alignment search tool (BLAST) methods implemented in TBtools (Chen et al., 2020). The Arabidopsis PRMTs and JMJs protein sequences were used as queries for BLAST analysis. Additionally, relevant hidden Markov models were obtained from the Pfam database (http://pfam.xfam.org/), and employed to search the apple protein sequence data using the HMMER 3.0 software with a stringent threshold of E ≤ 10–20. After removing redundancy and duplicate sequences, a preliminary set of candidate sequences was determined. The intersection of the gene family candidate members was obtained by combining the results obtained from the above-mentioned methods. Subsequently, the conserved protein domains of the target gene family in apples were analyzed using the Web CD-Search tool on the NCBI website (https://www.ncbi.nlm.nih.gov/Structure/bwrb/bwrpsb.cgi/). This analysis aimed to determine whether the conserved domain related to the target gene family protein was present in each candidate sequence. Only the candidate sequences containing complete domains were retained for further analysis. In the case of those sequences that contained different or incomplete domains, their sequence integrity was assessed by submitting them to the SoftBerry website (http://linux1.softberry.com/) before subjecting them to the Batch Web CD-Search tool for comparing the similarity between their domains and those of Arabidopsis PRMTs and JMJs sequences.
The protein physicochemical properties of gene family members were calculated using the ExPASy online software ProtParam (https://web.expasy.org/protparam/). These properties encompass various parameters such as the number of amino acids, molecular weight, theoretical pI, instability index, aliphatic index, and grand average of hydropathicity. The chromosome location of each confirmed histone methylation modification gene was retrieved from the GFF3 file of apple genome, and then visualized on apple chromosomes using TBtools (Chen et al., 2020).
2.2 Phylogenetic analysis of histone methylation modification proteins in M. domestica and A. thaliana
The amino acid sequences of PRMTs, and JMJs in Arabidopsis and apple were obtained individually from the TAIR and AppleMDO databases. These sequences were aligned using MUSCLE in MEGA 11 software. Then, the optimal model was determined using MEGA 11 software, and the phylogenetic tree was constructed using the maximum likelihood method(ML) (Tamura et al., 2021). The bootstrap consensus tree inferred from 1000 replicates is taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). The phylogenetic tree was visualized and optimized by TBtools.
2.3 Chromosomal distribution and synteny analysis
The gene location analysis was performed using the Amazing Gene Location tool in TBtools, which mapped the PRMTs and JMJs to their respective chromosomes based on the information extracted from the GTF/GFF files. To visualize the synteny blocks within the apple genome and between the genomes of apple, Arabidopsis, rice and maize, the Dual Synteny Plotter tool in TBtools was employed. These synteny blocks were generated using the One Step MCScanX-Super Fast method. Rice and maize genome data were both downloaded from the EnsemblPlants database (https://plants.ensembl.org/index.html). The Simple Ka/Ks calculator (NG) in TBtools was utilized to calculate the synonymous (Ks) and non-synonymous (Ka) values for duplicated genes.
2.4 Identification of duplicated gene types
We employed the DupGen_finder tool (https://github.com/qiao-xin/DupGen_finder) to classify the types of duplicated genes. DupGen_finder_unique categorizes plant genome duplicated genes into five categories based on a specific algorithm: whole-genome duplication (WGD), proximal duplication (PD), transposed duplication (TRD), dispersed duplication (DSD), and tandem duplication (TD) (Qiao et al., 2019). Using Arabidopsis as an outgroup for apple, we followed the recommended workflow of DupGen_finder for our analysis and parameter configuration as previously described (Kenchanmane Raju et al., 2023).
2.5 Sequence analysis
The conserved features of PRMTs and JMJs sequences were analyzed and visualized based on motifs using the Multiple Em for Motif Elicitation (MEME) suite 5.4.1 (https://meme-suite.org/meme/tools/meme) (Bailey et al., 2015) and TBtools. The gene structure of PRMT and JMJ genes were assessed using Gene Structure Shower in TBtools, utilizing information from the apple genome GFF3 file. To identify the cis-elements in the promoters of PRMT and JMJ genes, PlantCare (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was employed (Lescot et al., 2002).
2.6 Expression profiling of PRMT and JMJ genes in apple
The expression pattern of PRMT and JMJ genes following abiotic, hormone and biotic stress treatment were obtained from SRA database in NCBI website or National Genomics Data Center. The study IDs were as follows: for abiotic stress treatment: salt treatment (SRP229388), cold stress (CRA002596) and drought treatment (SRP347250); for hormone treatment: 1-Methylcyclopropene (1-MCP) treatment (SRP334206), Gibberellin Acid 3 (GA3) and 1-Naphthaleneacetic acid (NAA) treatment (SRP185711) and Indole-3-butyric acid (IBA) treatment (SRP330812); for biotic stress treatment: Alternaria alternata (SRP091754), Pythium ultimum (SRP048684), Fusarium solani (SRP239526) and ring rot (SRP153065). Kallisto, an RNA-seq quantification program were used to calculate the expression of gene (Bray et al., 2016). In this study, the gene expression was estimated using the fragment number per kilobase per million mapped exons (FPKM) and transcripts per kilobase of exon model per million mapped reads (TPM) method.
2.7 Quantitative reverse transcription polymerase chain reaction
Apple (‘GL3’) tissue culture seedlings were transferred to MS medium plus 200 mM sodium chloride (NaCl), 10% polyethylene glycol (PEG) 6000. Leaves were sampled at 0, 6, 12 and 24h after treatment for RNA isolation. Total RNA was extracted from apple leaves using the hot borate method as previously described (Ma et al., 2006). For reverse transcription-PCR, first-strand cDNA was synthesized from 1 μg of total RNA using Reverse Transcriptase M-MLV (RNase H-) (Takara Biomedical Technology Co., Ltd., Shiga, Japan). qRT-PCR was conducted with Takara SYBR Premix Ex Taq II (Takara Biomedical Technology Co., Ltd.) using a Light Cycler 480 instrument (Roche, Basel, Switzerland). MdEF-1α were used as internal controls. All primers used are listed in Supplementary Table S8.
3 Results
3.1 Identification of PRMTs and JMJs in apple
To identify the PRMTs and JMJs gene families in the apple genome, a comprehensive investigation was carried out using the protein sequences of the corresponding genes from Arabidopsis as search queries. After eliminating redundant and duplicate sequences, we identified 14 and 32 genes in PRMT and JMJ families, respectively, in the apple genome (Table 1). The predicted protein sequence lengths of the PRMT and JMJ families ranged from 232 (MdPRMT11) to 722 (MdPRMT3) and 187 (MdJMJ2) to 1843 (MdJMJ14) amino acids, respectively. The molecular weight (MW) of the PRMT and KDM families ranged from 25.45 (MdPRMT11) to 81.12 (MdPRMT3) and 21.43 (MdJMJ2) to 210 (MdJMJ14) kDa, respectively. The isoelectric point (pI) values for the PRMT and JMJ families varied from 25.45 (MdPRMT11) to 81.12 (MdPRMT3) and 5.05 (MdJMJ8) to 9.03 (MdJMJ28), respectively. The complete information about the genes, including gene locus ID, chromosomal position, coding sequence length, and protein sequence length, was shown in Table 1. The nomenclature of the genes followed a sequential order based on their arrangement on the chromosome.
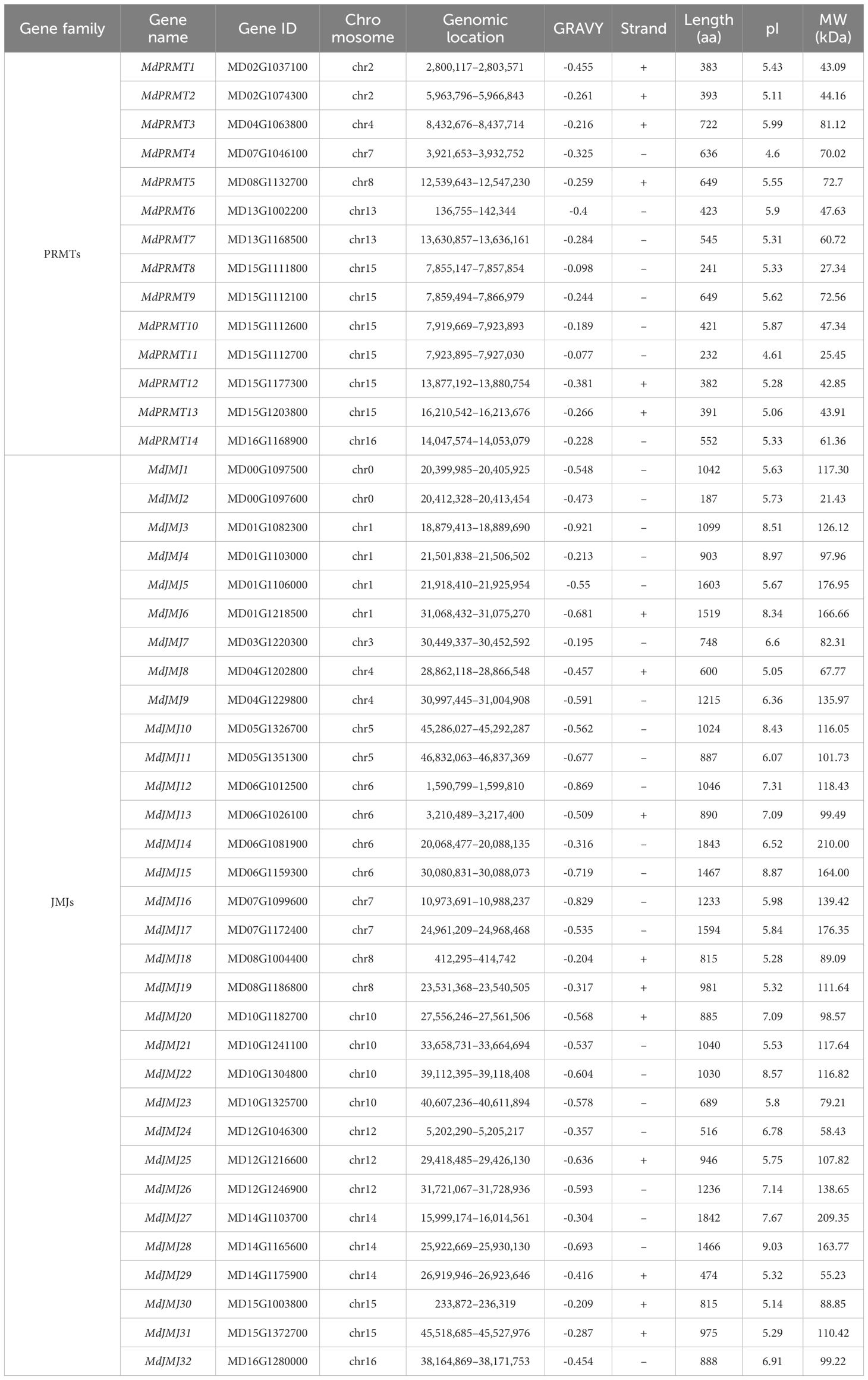
Table 1 PRMT and JMJ genes identified in apple.
3.2 Analysis of chromosomal distribution and evolutionary relationships of MdJMJ and MdPRMT genes
In Figure 1A, the chromosomal distribution of PRMTs, and JMJs in apple was shown. The analysis revealed that the distribution of these genes is irregular. Particularly, the MdJMJ genes were distributed on all chromosomes apart from chromosomes 2, 9, 11, and 17. The chromosomal distribution of MdPRMT genes was comparatively more restricted, limited to chromosomes 2, 4, 7, 8, 13, 15, and 16. Notably, chromosome 15 showed the most concentrated distribution with a total of 6 genes (MdPRMT8, MdPRMT9, MdPRMT10, MdPRMT11, MdPRMT12 and MdPRMT13).
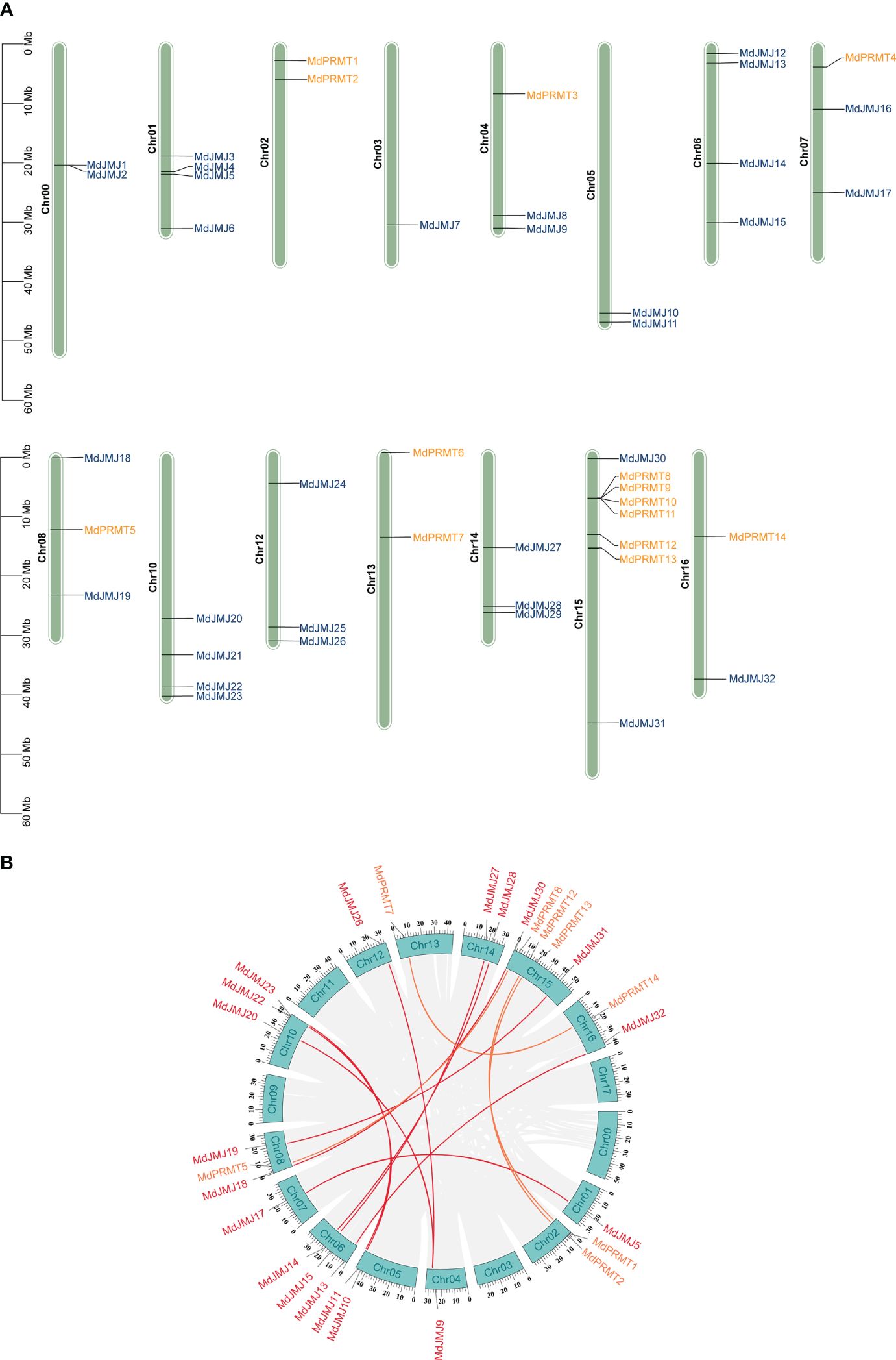
Figure 1 Chromosomal distribution and collinearity analysis of JMJ and PRMT genes between apple and A. thaliana. (A) Chromosomal distribution and duplication analysis of JMJs and PRMTs in apple. (B) Collinearity analysis of JMJ and PRMT genes family in apple.
Gene duplication events are a common occurrence across all species, playing a crucial role in generating novel functional genes and driving species evolution (Xu et al., 2012). In order to explore gene duplication events within the JMJ and PRMT gene families in apple, we conducted genome synteny analysis. The analysis revealed the presence of 10 potential duplicated genes in the MdJMJ gene family and 4 in the MdPRMT gene family (Figure 1B). This finding suggests that the expansion of the MdJMJ gene family is primarily driven by segmental duplication, as indicated by the high number of potential duplicated genes. Notably, there is a significantly gene cluster located on chromosome 15 for the MdPRMT gene family (Figure 1A).
Using Arabidopsis as an outgroup for apple, we followed the recommended workflow of DupGen_finder and found that after the divergence of the two species, the MdJMJ family contained 10 WGD-pairs, 1 TD-pairs (MdJMJ1-MdJMJ2), and 3 TRD-pairs (MdJMJ3-MdJMJ6, MdJMJ4-MdJMJ7, MdJMJ11-MdJMJ12). No DSD-pairs and PD-pairs were found in the MdJMJ family. The MdPRMT family had 4 WGD-pairs (MdPRMT7- MdPRMT14, MdPRMT5-MdPRMT8, MdPRMT2-MdPRMT13, MdPRMT1-MdPRMT12), 2 PD-pairs (MdPRMT11-MdPRMT9, MdPRMT9-MdPRMT10) and 1 TRD- pairs (MdPRMT6-MdPRMT13). No DSD-pairs and TD-pairs were found in the MdPRMT family. Detailed analytical results can be found in Supplementary Table S9. The analysis outcomes indicate that the primary mode of evolution for the MdJMJ and MdPRMT gene families is whole-genome duplication.
To elucidate the evolutionary relationships of the PRMT and JMJ gene families, a maximum likelihood (ML) phylogenetic tree was constructed using the PRMT and JMJ proteins from apple and Arabidopsis (Figure 2). The classification of the gene families was based on the established Arabidopsis JMJ gene families. The JMJ subfamily includes KDM5, KDM4, KDM3, KDM1, JMJC, and JMJD6, with 8, 6, 10, 4, 1, and 3 MdJMJ proteins, respectively (Figure 2A).
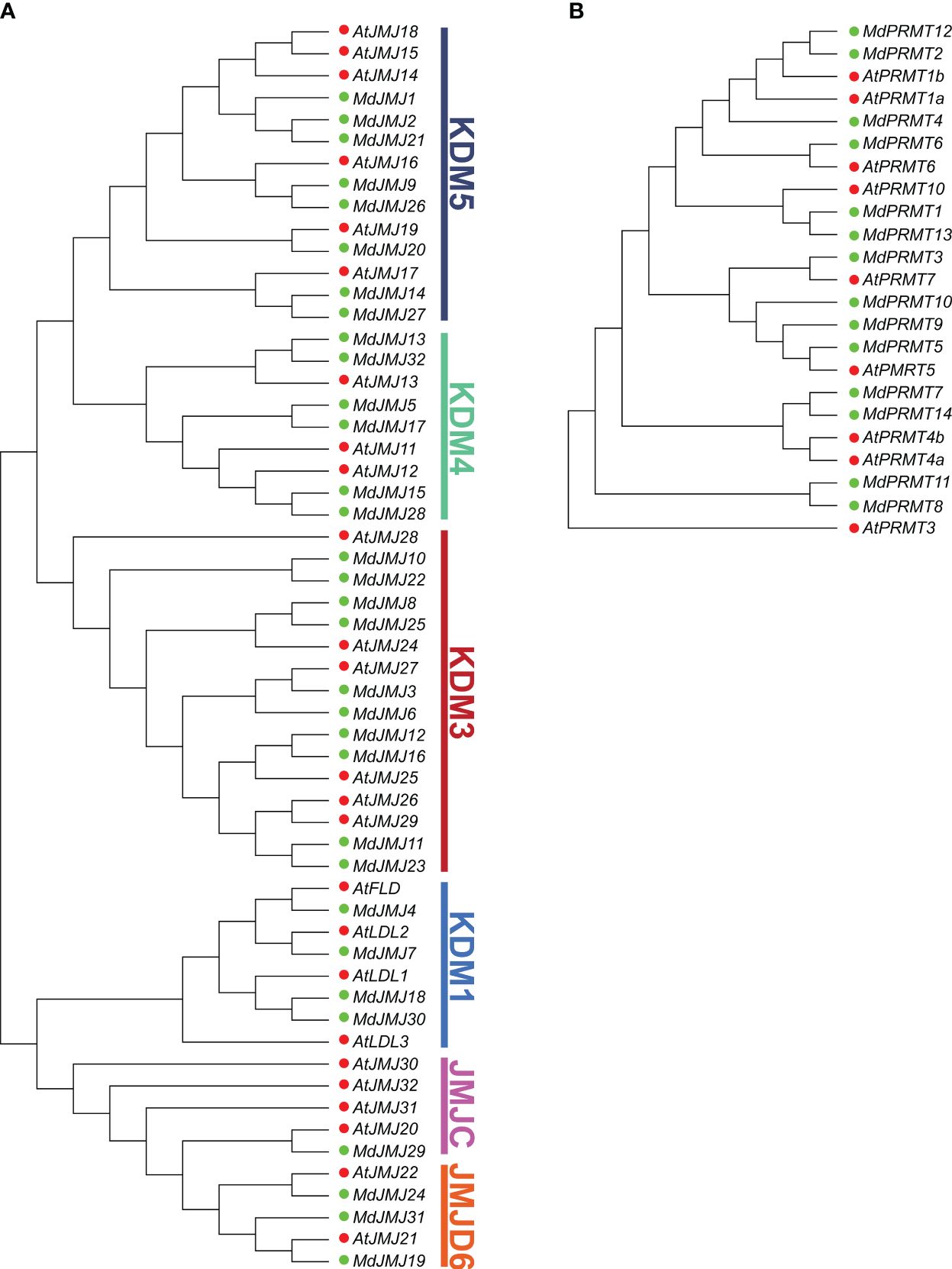
Figure 2 Phylogenetic relationship of JMJ (A) and PRMT (B) proteins from apple and Arabidopsis. The bootstrap consensus of tree was inferred from 1000 replicates, and the phylogenetic tree was constructed using the maximum likelihood method. In the figure, red dots represent Arabidopsis proteins, while green dots represent apple proteins.
To gain deeper insights into the origin, evolutionary history, and functional characteristics of PRMT and JMJ genes, we performed a comparative synteny analysis between the genomes of apple, Arabidopsis, rice and maize (Figure 3). The apple genome contained 19 JMJ genes with high synteny to the Arabidopsis genome, forming a total of 21 syntenic gene pairs, as shown in Figure 3A. These syntenic gene pairs include MdJMJ10-AtJMJ16, MdJMJ11-AtJMJ17, MdJMJ11-AtJMJ18, MdJMJ14-AtJMJ19, MdJMJ15-AtJMJ20, MdJMJ17-AtJMJ21, MdJMJ19-AtJMJ22, MdJMJ21-AtJMJ23, MdJMJ22-AtJMJ24, MdJMJ23-AtJMJ25, MdJMJ23-AtJMJ26, MdJMJ24-AtJMJ27, MdJMJ25-AtJMJ28, MdJMJ27-AtJMJ29, MdJMJ28-AtJMJ30, MdJMJ29-AtJMJ31, MdJMJ3-AtJMJ32, MdJMJ31-AtJMJ33, MdJMJ4-AtJMJ34, MdJMJ5-AtJMJ35, and MdJMJ9-AtJMJ36 (Figure 3A). 7 syntenic gene pairs were found between apples and maize, only one ortholog pair (AtJMJ21-OsFBO14) were found in Arabidopsis and rice. In addition, there were 7 PRMT genes in the apple genome that showed synteny with the Arabidopsis genome, forming a total of 11 syntenic gene pairs. These syntenic gene pairs include MdPRMT13-AtPRMT1a, MdPRMT13-AtPRMT1b, MdPRMT14-AtPRMT4b, MdPRMT14-AtPRMT4a, MdPRMT2-AtPRMT1a, MdPRMT2-AtPRMT1b, MdPRMT3-AtPRMT7, MdPRMT5-AtPRMT5, MdPRMT7-AtPRMT4b, MdPRMT7-AtPRMT4a, and MdPRMT8-AtPRMT5 (Figure 3B). Five pairs of co-linear genes (ZmPRMT6-MdPRMT6, ZmPRMT3-MdPRMT7, ZmPRMT3-MdPRMT14, ZmPRMT4-MdPRMT7, and ZmPRMT5-MdPRMT6) were identified between maize and apple, while no co-linear genes were found between Arabidopsis and rice. These results indicate a high conservation of JMJ and PRMT genes between the apple and Arabidopsis genomes.

Figure 3 Synteny analysis of JMJs (A) and PRMTs (B) of apple, Arabidopsis, maize and rice.
For evolutionary studies, Ka (non-synonymous substitution) and Ks (synonymous substitution) values can be used to predict the selective pressure on duplicated genes (Zhao et al., 2016). When the Ka/Ks ratio falls below 1, it signifies that the genes have undergone purifying selection, indicating the absence of significant functional differentiation among these genes during the evolutionary process (Hurst, 2002). We utilized the Simple Ka/Ks CalculatorNG program in TBtools to analyze 14 pairs of collinear genes in MdJMJ and MdPRMT to further investigate the evolutionary relationships among these collinear genes. The results showed that all the collinear gene pairs had a Ka/Ks ratio less than 1, indicating that these genes have undergone purifying selection and have not experienced functional differentiation (Table 2). However, it is important to note that a Ka/Ks ratio less than 1 does not imply that these collinear genes have not undergone functional differentiation or evolution. It simply suggests that, within the observed time scale, the functional differences among these genes have had a relatively minor impact on the evolutionary process.

Table 2 Calculation of synonymous (Ks) and non-synonymous (Ka) for duplicated genes in apple.
3.3 Sequence and structure analysis of JMJs and PRMTs
A phylogenetic tree for the MdJMJ and MdPRMT gene families was constructed using the Maximum Likelihood method in MEGA11.0 software (Figures 4A, E). It is evident that the genes clustered together in both gene families exhibit similar conserved domains. MdJMJ4 and MdJMJ31 contained only motif 1, while MdJMJ17 lacked any motifs. Additionally, motif 15, motif 19, and motif 20 were unique to MdJMJ19, MdJMJ29, MdJMJ28, and MdJMJ15, respectively (Figures 4B). The motif analysis was further supported by conserved domain analysis further supported the similar domain structures within gene cluster, which showed MdJMJ19, MdJMJ29, MdJMJ28, and MdJMJ15 possessed the ZZ superfamily domain, PLN02529 superfamily domain, and PLN02328 superfamily domain, respectively (Figures 4C). These three conserved domains were specific to these four genes, consistent with the motif analysis. Gene structure analysis revealed significant variation in the structure of MdJMJ genes, with clustering genes showing similar structures (Figure 4D). The number of introns ranged from 0 to 32, with MdJMJ28 and MdJMJ15 lacking introns, while MdJMJ3 and MdJMJ16 had the highest number of 32 introns.
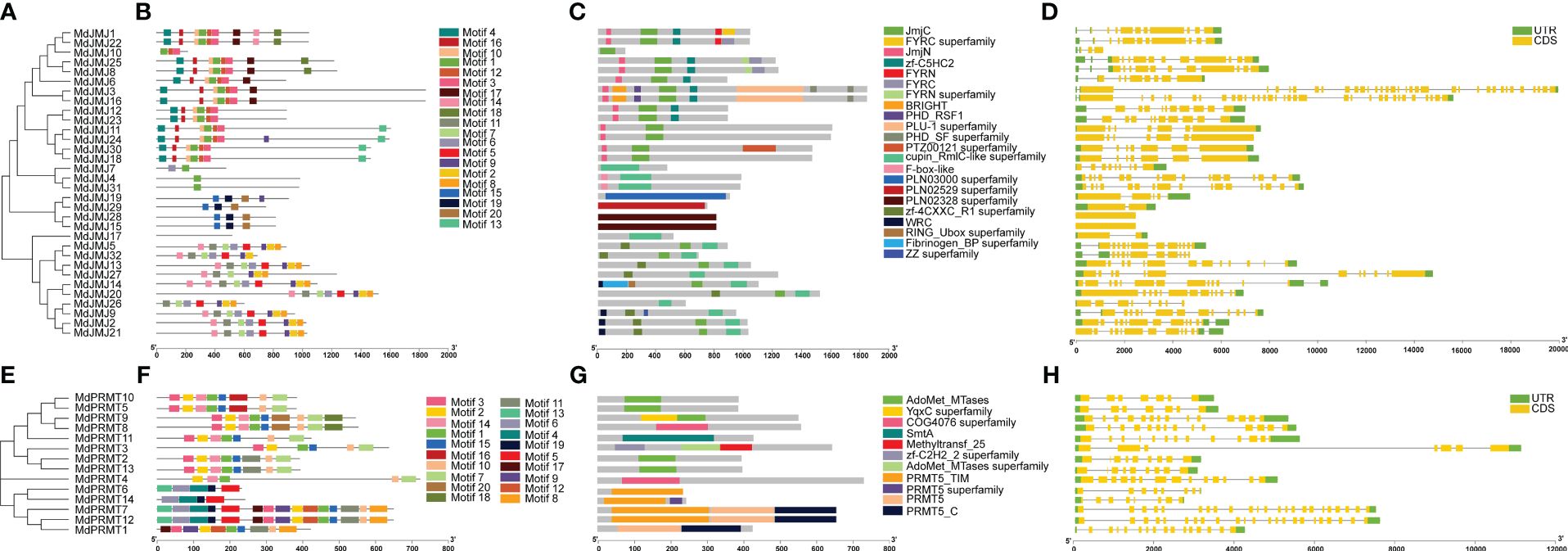
Figure 4 Phylogenetic analysis, motif composition, conserved domains of MdJMJ, and MdPRMT proteins and exon-intron structures of MdJMJ and MdPRMT genes. (A–C) The phylogenetic tree (A), motif composition (B) and the distribution of conserved domains (C) of MdJMJs. (D) Gene structures of the MdJMJ genes. (E–G) The phylogenetic tree (E), motif composition (F) and the distribution of conserved domains (G) of MdPRMTs. (H) Gene structures of the MdPRMT genes. Green rectangles represent untranslated regions (UTRs); yellow rectangles represent coding sequence (CDS) or exons; grey lines represent introns.
For the MdPRMT gene family, which had a smaller number of members, clustering of genes based on conserved motifs revealed similar motif structures within the clusters (Figures 4F). Conserved domain analysis clearly reveals that MdPRMT10, MdPRMT5, MdPRMT2, and MdPRMT13 genes only contain the AdoMet_MTases conserved domain (Figures 4G). MdPRMT4 and MdPRMT8 genes only possess the COG4076 superfamily conserved domain. The PRMT5_C and PRMT5 conserved domains are present only in MdPRMT7, MdPRMT12, and MdPRMT1 genes. All the members of the MdPRMT gene family have gene structures with introns, ranging from 6 (MdPRMT10 and MdPRMT5) to 22 (MdPRMT7 and MdPRMT12), indicating variations in gene structure within this gene family (Figures 4H).
The specific amino acid sequences of motifs of MdJMJ, MdPRMT and the detailed information of the MEME sites analysis were shown in the Supplementary Tables S2 and S3, respectively.
3.4 Cis-elements in the promoter of MdPRMTs and MdJMJs
An analysis was conducted on the promoter fragments (-2000 bp) of all MdPRMT and MdJMJ genes (Figures 5A, C). A large number of cis-regulatory elements were identified in their promoter regions, mainly including light response, cell cycle, low temperature, circadian rhythm, stress defense, growth and development (endosperm, seed-specific, meristem, cell differentiation), hormone signaling (MeJA, ABA, GA, Auxin, and SA), anoxic specific inducibility, and some transcription factors (TF), including MYB and ATBP. Additionally, we also found a wound responsive element in each of the MdJMJ. By counting the number of cis-regulatory elements (Figures 5B, D), it was found that light-responsive cis-elements were present in the promoter regions of all members of the MdPRMT and MdJMJ. MeJA, ABRE, and ARE elements were also widely distributed. Therefore, it can be inferred that MdPRMTs and MdJMJs play important roles in apple growth and development as well as hormone responses. On the other hand, less abundant cis-regulatory elements should not be overlooked. For example, the low-temperature elements were present in a significant number of MdJMJs and MdPRMTs, with 18 and 6 genes involved, respectively. Stress defense elements were found in 11, and 3 genes, while MYB-drought elements were present in 20 and 8 genes of MdJMJ and MdPRMT, respectively. These genes may play crucial roles in stress tolerance in apple. Additionally, one gene in MdJMJs contained an Endosperm-specific negative expression element and a Wound responsive element, while none of the MdPRMTs genes contained these two elements.
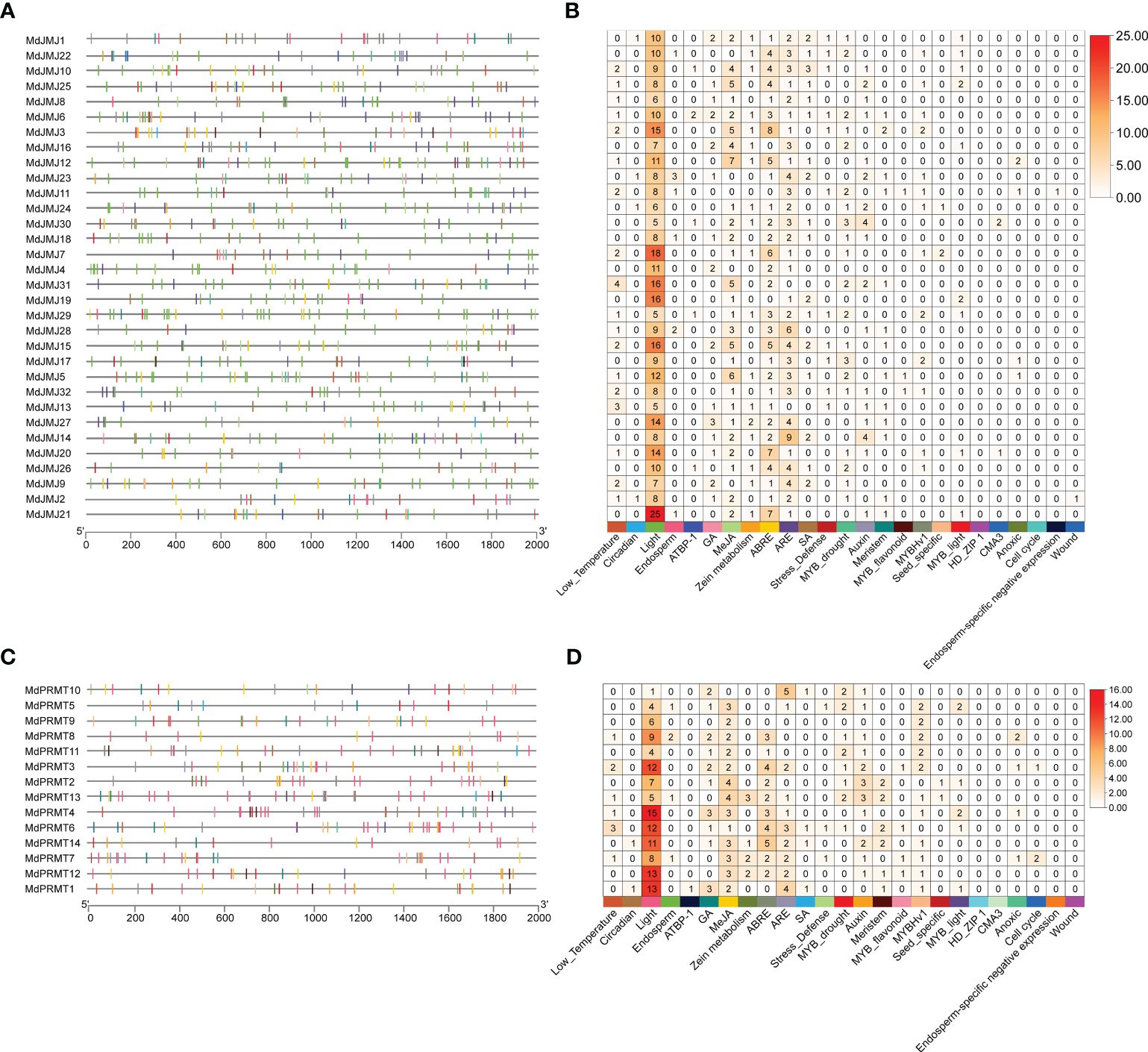
Figure 5 The cis-elements analysis of MdJMJ and MdPRMT promoters. (A, B) The distribution (A) and number (B) of cis-elements in the promoter of each MdJMJ genes. (C, D) The distribution (C) and number (D) of cis-elements in the promoter of each MdPRMT genes.
Similarly, to compare the differences in cis-regulatory elements between apple and Arabidopsis, the same method was used to construct a map of cis-regulatory elements in Arabidopsis promoters and categorize them according to the phylogenetic tree (Supplementary Figure S2). The results indicate that, on average, Arabidopsis has a fewer variety of cis-regulatory elements compared to apple. Like apple, Arabidopsis AtJMJ and AtPRMT genes also contain a large number of light-responsive and stress-response elements, which can provide some assistance for the function and evolution of the apple gene families.
3.5 Expression profiles of the apple MdPRMT and MdJMJ in various organs, tissues and developmental stage
In this study, the expression profiles of MdJMJs and MdPRMTs in different tissues and developmental stages of apple were analyzed using publicly available transcriptome data (Figure 6). According to the results, the majority of histone modification-related genes showed relatively low expression levels during fruit maturation (SRP034165). It is worth noting that 11 histone modification-related genes (MdPRMT1, MdPRMT2, MdPRMT4, MdPRMT5, MdPRMT6, MdPRMT7, MdPRMT12, MdPRMT13, MdPRMT14, MdJMJ6, and MdJMJ19) exhibited higher expression levels in the early stage of fruit maturation, and their expression levels decreased to varying degrees as the fruit matured. Among them, the MdPRMT gene family had the largest number of genes, with approximately 64% of the genes showing decreased expression levels during fruit maturation. To delineate the expression patterns of histone modification-related genes in major organs of apple, transcriptome data from seedling and tree shoot apex, dormant buds and bud break, and different parts of apple flowers were analyzed. In shoot apex tissues at different stages, members of each gene family exhibited different expression patterns (SRP050139). For example, most members of the MdPRMT gene family showed varying degrees of down-regulation during the transition from the seedling stage to the mature stage, which was consistent with the down-regulated genes during fruit maturation. This suggests that the MdPRMTs may play an important role in the growth and development of apple. Furthermore, during apple plant development, 6 down-regulated genes were identified in the MdJMJs. Thus, it can be inferred that they collectively play a role in apple plant development.
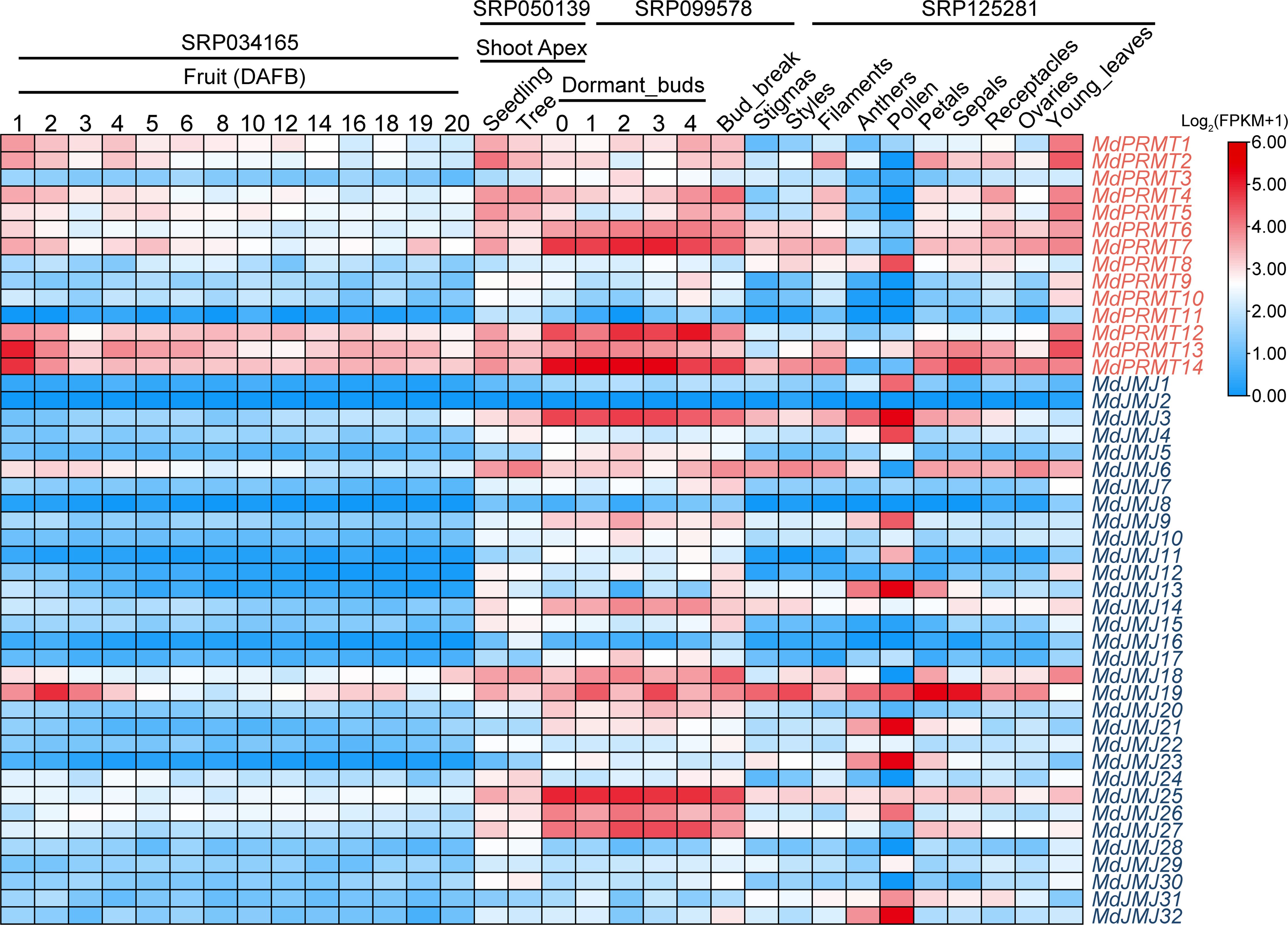
Figure 6 Expression profiles of the apple MdPRMT and MdJMJ genes in various organs, tissues and developmental stages. Different shades of red and blue denote the extent of the expression values according to the color bar provided [log2(FPKM+1)].
Dormant buds play a crucial role in the growth cycle of plants and serve as a survival strategy for apple trees under adverse conditions such as winter or drought (Rohde and Bhalerao, 2007). They are also an important means of reproduction for apple trees. We analyzed the expression patterns of histone modification-related genes in apple dormant buds during dormancy and bud break (SPR099578). The results showed that MdPRMTs and MdJMJs had 5 and 6 genes, respectively, with higher expression levels. Among them, MdPRMT7, MdJMJ3 and MdJMJ25 showed relatively minor down-regulation, while the majority of genes exhibited lower expression levels or irregular trends.
Flowers are important reproductive organs of apple. We analyzed the transcriptome data from different parts of apple flowers. The results showed that histone modification-related genes had significantly higher average expression levels in pollen compared to other parts. MdPRMTs, and MdJMJs had 1 (MdPRMT8) and 5 (MdJMJ3, MdJMJ13, MdJMJ21, MdJMJ23, MdJMJ32) genes with significantly higher expression levels in pollen, respectively. Furthermore, MdJMJ19 was expressed in all parts of the flower, with significantly higher expression levels in petals and sepals compared to other parts. Finally, it is noteworthy that MdJMJ2 showed either no expression or relatively low expression levels in fruit development, shoot apex, bud, different parts of flowers, and young leaves.
3.6 Expression profiles of apple MdPRMTs and MdJMJs under different biotic and abiotic stresses
In East Asia, Alternaria alternata apple pathotype (AAAP) is one of the main pathogens causing apple Alternaria blotch disease, significantly affecting the growth and development of apple trees and fruit yield (Abe et al., 2010). Some MdPRMTs and MdJMJs showed decreased expression levels after infection, such as MdJMJ6, MdJMJ15, and MdJMJ28. Some genes showed an upregulation, such as MdPRMT1, MdPRMT12 and MdJMJ25. In addition, certain genes demonstrated fluctuating expression levels, such as MdPRMT14 and MdPRMT7, which increased in expression level from 0 to 18 hours after infection and then decreased below the expression level at 0 hours of infection. On the other hand, MdJMJ27 and MdJMJ3 showed a decrease in expression level at the early stage of infection and then an increase in expression level after 18 hours of infection. It is worth noting that the expression level of MdJMJ13 was significantly higher at 18 and 72 hours after infection compared to other time points.
Pythium ultimum is one of the main pathogens causing Apple Replant Disease (ARD), which leads to growth inhibition and even death of apple seedlings when planted in orchard soil previously used for apple (or closely related species) cultivation (Zhu and Saltzgiver, 2020). The expression levels of MdJMJ and MdPRMT genes generally do not show significant changes after infection. However, it should be noted that MdJMJ13 expression is higher than the control after 4 hours of infection, while MdJMJ18 expression is lower than the control.
ARD causes severe growth and developmental obstacles in apple trees, and previous studies have determined Fusarium solani as the main pathogen causing ARD (Liu et al., 2022). There was a significant downregulation of MdJMJ3, MdJMJ18, MdJMJ25, MdJMJ27, MdJMJ29 and MdPRMT8, while MdPRMT2 showed an upregulation after F.solani infection. These results suggest that the above genes may play a role in apple resistance against F. solani infection.
Apple fruit ring rot (FRR) is a highly destructive disease caused by Botryosphaeria dothidea, which severely affects the apple industry’s development in the Asian region (Shen et al., 2019). The results revealed that, regardless of susceptible or resistant varieties, the expression levels of MdPRMTs and MdJMJs showed no significant changes compared to the corresponding mock after Botryosphaeria dothidea infection (Figure 7A).
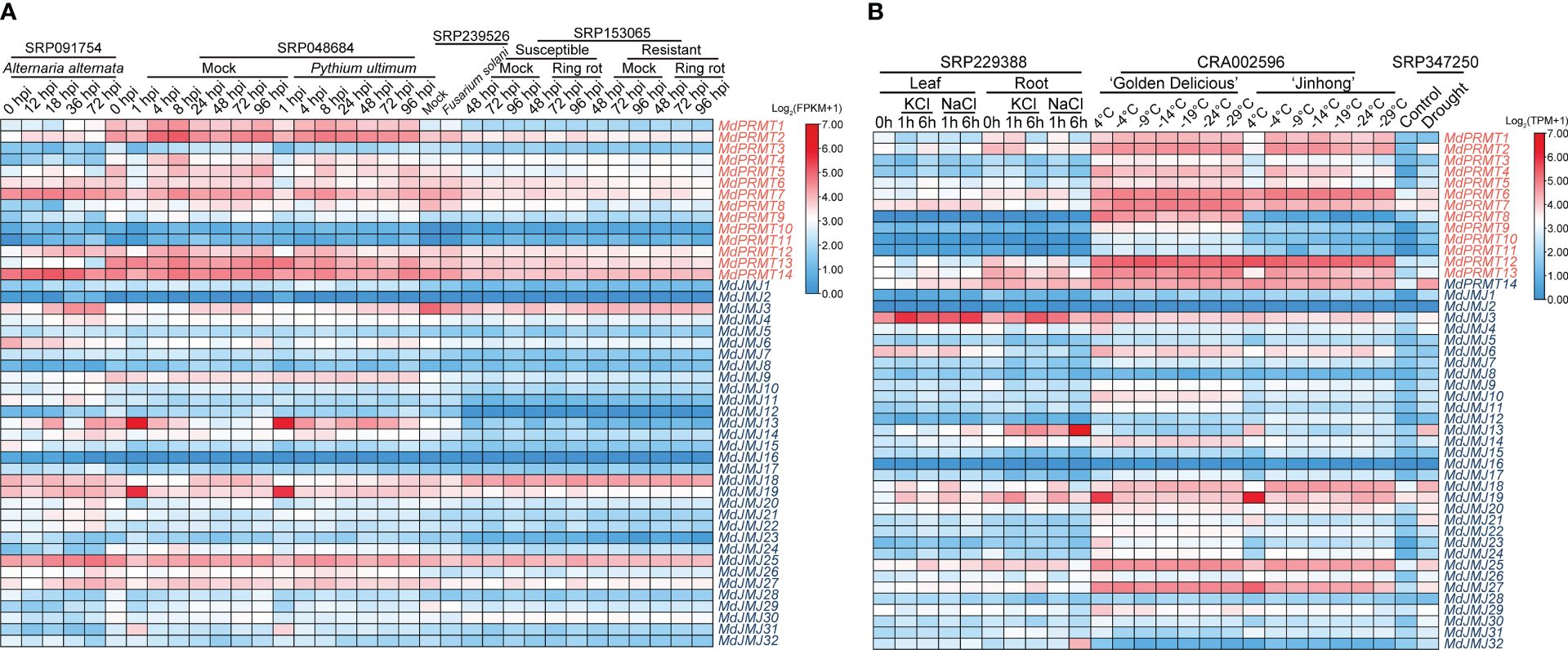
Figure 7 Expression profiles of the apple MdPRMT and MdJMJ genes under stress treatment. (A) Expression profiles of MdPRMT and MdJMJ genes at different time points under biotic stress (Alternaria alternata, Pythium ultimum, Fusarium solani, Ring rot) (SRP091754, SRP048684, SRP239526, SRP153065). (B) Expression profiles of MdPRMT and MdJMJ genes in leaves and roots at different time points under KCl and NaCl salt treatment (SRP229388), RNA-seq transcriptomes of barks (epidermis, phloem, and cambium) from one-year-old branches of two apple cultivars (‘Golden Delicious’ and ‘Jinhong’) under chilling and freezing treatments (CRA002596), and ‘GL3’ under drought stress (SRP347250). Different shades of red and blue denote the extent of the expression values according to the color bar provided [log2(FPKM+1) and log2(TPM+1)].
First, in terms of abiotic stress, we focused on salt stress (SRP229388) (Figure 6B). Transcriptional data from apple seedlings subjected to two types of salt stress, KCl and NaCl, showed that the expression levels of most MdPRMTs and MdJMJs were relatively low in the leaves. It is worth noting that MdJMJ6 showed a downregulation trend in the leaves under both types of salt stress. Conversely, MdJMJ3, MdJMJ13, MdJMJ19, and MdPRMT7 exhibited significant upregulation under both KCl and NaCl salt stress, with MdJMJ3 showing higher expression levels during the initial 1 hour of KCl salt stress, followed by a significant decrease after 6 hours, while the opposite trend was observed under NaCl salt stress. In the root system, MdJMJ13 displayed an obvious upregulation trend under both KCl and NaCl salt stress, with a significant increase in expression levels after 6 hours of NaCl stress, much higher than the expression levels under the same period of KCl stress. MdPRMT12 showed a downregulation trend, and MdJMJ19 exhibited an initial upregulation followed by a decrease in expression levels. A comparison between the leaf and root systems also revealed that the expression levels of MdJMJ13, MdPRMT2, MdPRMT13, and MdPRMT14 under both KCl and NaCl salt stress were significantly higher in the root system than in the leaf system, whereas the opposite was observed in MdJMJ6 and MdPRMT7. Neither MdJMJ16 nor MdJMJ2 showed expression.
In the northern region of China, low-temperature freeze injury is one of the common diseases in apple during winter and spring (Yang et al., 2011). We analyzed the RNA-seq transcriptomes of barks (epidermis, phloem, and cambium) from one-year-old branches of two apple cultivars, ‘Golden Delicious’ (non-cold-tolerant varieties) and ‘Jinhong’ (cold-tolerant varieties) under chilling and freezing treatments (CRA002596) (Figure 7B). The results showed that the expression level of MdPRMT8 was significantly higher in ‘Golden Delicious’ than in ‘Jinhong,’ and with the continuous decrease in temperature, its expression level showed a significant downregulation. MdPRMT9, MdPRMT10, MdPRMT11, and MdJMJ23 had higher average expression levels in ‘Golden Delicious’ than in ‘Jinhong’, but their expression levels did not show an obvious pattern of change. It is worth noting that MdJMJ19 had very high expression levels at 4°C in both apple cultivars, and as the temperature decreased, its expression levels were significantly downregulated. In particular, MdJMJ19 in ‘Golden Delicious’ showed a stable expression level at -9°C and even lower temperatures, while in ‘Jinhong’, a slight upregulation was observed starting from -4°C, and although the expression levels decreased at -14°C and -29°C, the overall trend was still upregulation. In ‘Jinhong’, the expression level of MdJMJ15 was significantly higher at -4°C and lower temperatures than at 4°C, while in ‘Golden Delicious’, the change in expression levels was not significant. MdPRMT2 and MdPRMT13 had significantly higher expression levels at -4°C in ‘Jinhong’ than at 4°C, and their expression levels were downregulated at lower temperatures, which was not observed in ‘Golden Delicious’. MdJMJ18 showed an upregulation of expression levels between -4°C and -14°C in both cultivars, and its expression levels decreased at even lower temperatures, indicating that this gene may play a role in apple’s resistance to low temperature.
Drought is also one of the main types of stress faced during apple cultivation, especially in drought-prone areas of China (Berger et al., 2016). We analyzed the RNA-seq data of ‘GL3’ under drought treatment (SRP347250) (Figure 7B). The analysis results showed that compared to the control group, most MdPRMTs and MdJMJs showed varying degrees of upregulation under drought stress, with MdPRMT2, MdPRMT6, MdPRMT14, MdJMJ18, MdJMJ13, MdJMJ20, MdJMJ21, MdJMJ25 and MdJMJ27 exhibiting significant upregulation. Therefore, it can be inferred that these genes may be closely related to apple’s resistance to drought.
We selected five MdJMJ and MdPRMT genes that were significantly upregulated under drought stress for qRT-PCR analysis. The results showed a significant and varying upregulation in all five genes upon PEG (as drought) treatment (Figure 8A). When two MdJMJ genes (MdJMJ13 and MdJMJ32) were subjected to salt stress, both exhibited a decrease in expression after 6 h, followed by an increase at 12 h (Figure 8B). Similarly, the relative expression levels of the four genes under 4°C cold stress displayed a similar pattern, with an initial increase at 6 h and subsequent decline (Figure 8C).
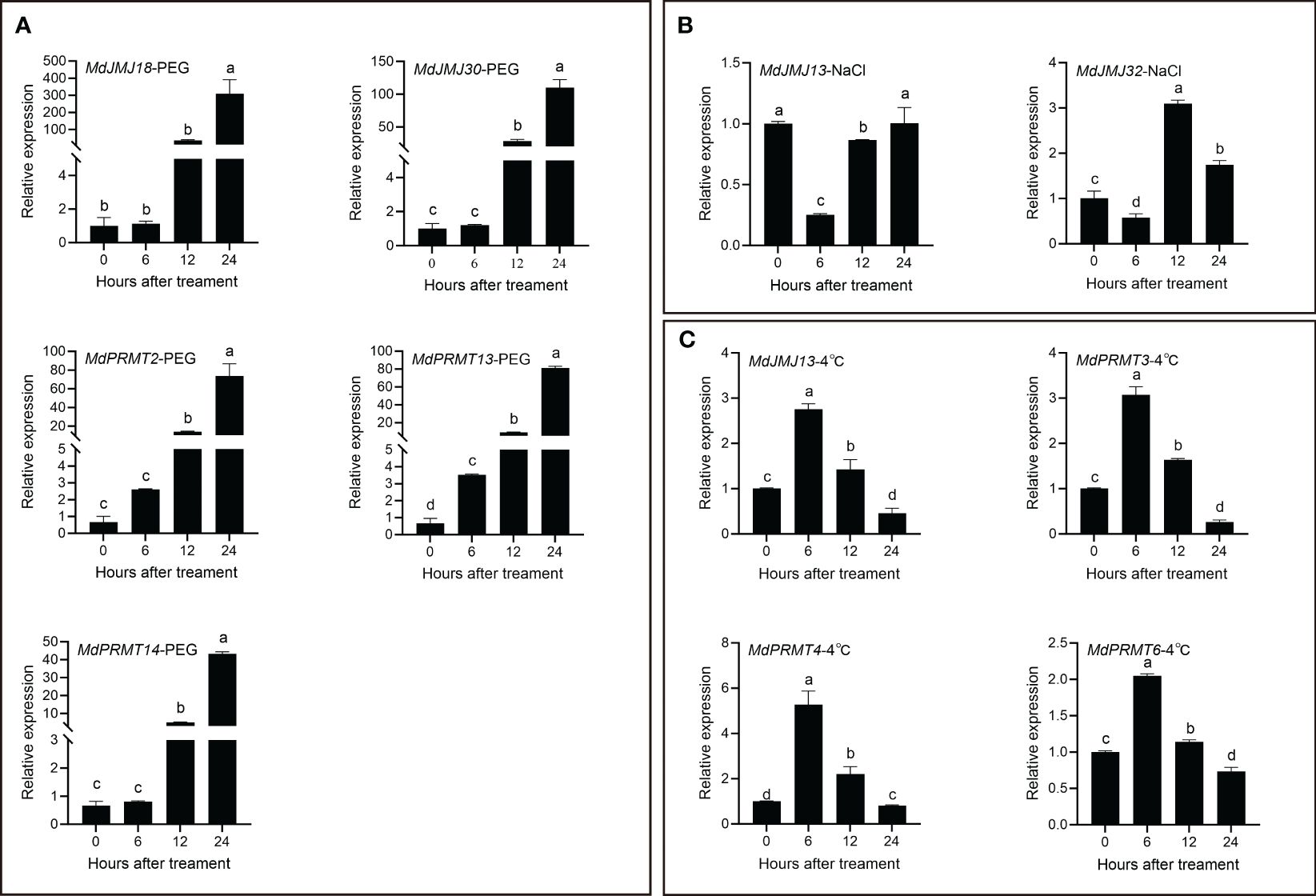
Figure 8 qRT-PCR analysis of selected MdJMJ and MdPRMT genes expression under PEG (A), NaCl (B) and 4°C (C) treatment. 25 d-old leaves were treated with PEG, NaCl and 4°C for 0 h, 6 h, 12 h and 24 h The mean values ± SEM are shown for three biological replicates. Different letters above the bars indicate significant differences according to Duncan-test (P < 0.05).
3.7 Expression profiles of the apple MdPRMTs and MdJMJs in response to hormone treatment
Plant hormones play a crucial role in the growth and development of plants. 1-MCP (1-Methylcyclopropene) is commonly used for preserving ethylene-producing or ethylene-sensitive fruits, effectively delaying fruit senescence (Berger et al., 2016). We analyzed the RNA-seq data (SRP334206) of three apple fruit varieties (‘Golden Delicious’, ‘Granny Smith’, and ‘Fuji’) treated with 1-MCP (Figure 9). The analysis revealed that MdPRMT2, MdPRMT7, MdPRMT12, MdPRMT13, and MdPRMT14 showed significantly upregulated expression in the 1-MCP treatment group compared to the control group in all three apple varieties. Additionally, the expression of MdPRMT8 in ‘Fuji’ was significantly lower than the other two varieties. Similarly, the expression of MdJMJ3, MdJMJ18, MdJMJ25, and MdJMJ27 was significantly higher in the 1-MCP treatment group, with MdJMJ3 showing the most noticeable upregulation at 60 d after 1-MCP treatment. This suggests that the aforementioned genes may be important for regulating the response of apple fruits to 1-MCP.
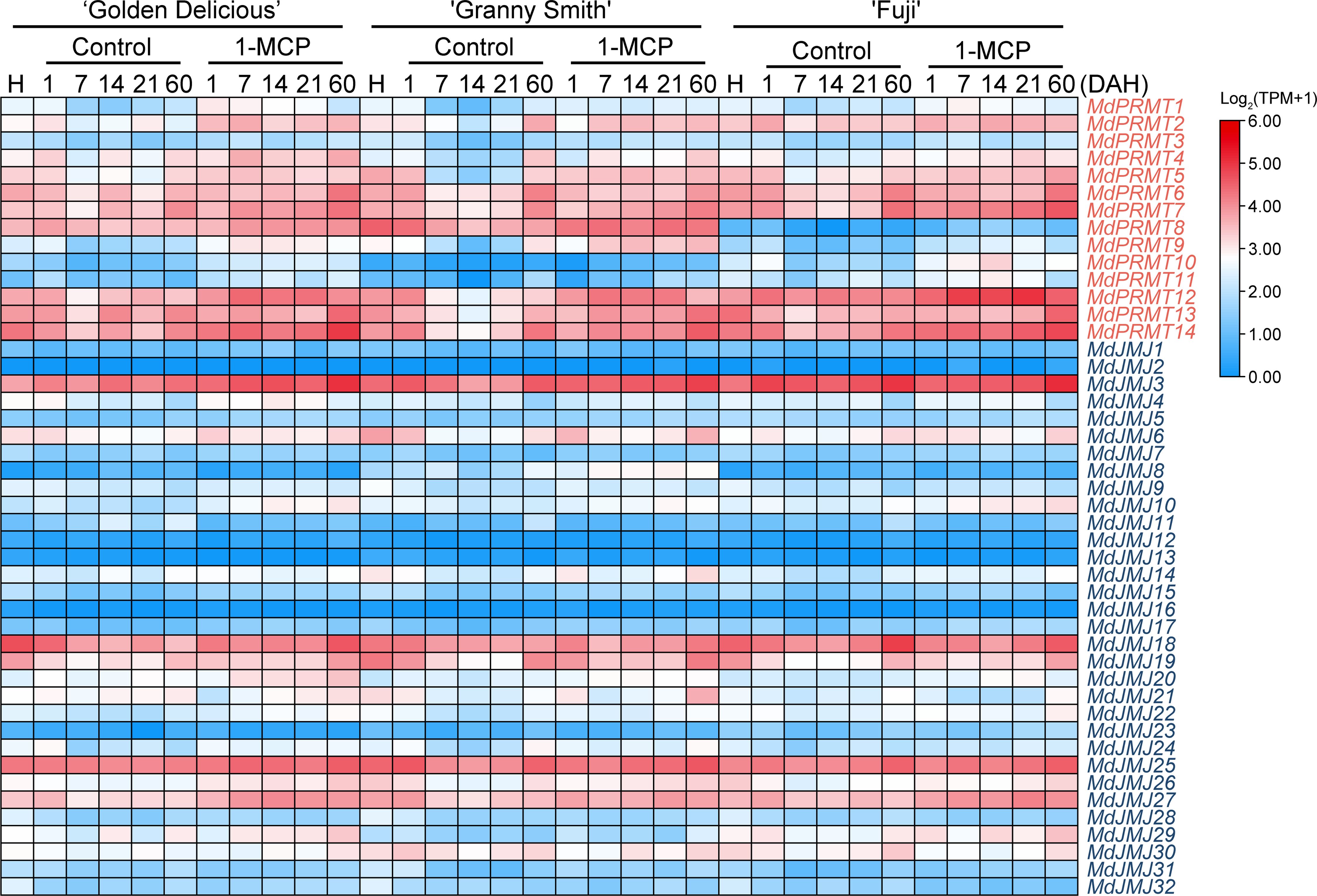
Figure 9 Expression profiles of the apple MdPRMT and MdJMJ genes during fruit response to 1-MCP treatment (SRP334206). Different shades of red and blue denote the extent of the expression values according to the color bar provided [log2(TPM+1)].
GA3 and NAA are important plant hormones involved in various plant growth and development processes (Hennerty and Forshey, 1972; Pharis and King, 1985). We analyzed the RNA-seq data (SRP185711) of the main floral organs of ‘Honeycrisp’ apples after 18 and 132 days of NAA and GA3 treatment. The analysis showed that the expression of MdJMJ6 in the hypanthium, ovary, and ovule was significantly lower after 18 days of NAA and GA3 treatment compared to the hand-pollinated control (HP). Additionally, after 132 days of GA3 treatment, all three organs showed lower expression levels of MdJMJ6. MdPRMT8 and MdJMJ19 exhibited significant upregulation in expression after 18 days of NAA and GA3 treatment compared to HP. Furthermore, the expression levels of MdPRMTs and MdJMJs in all organs were significantly lower after 132 days of GA3 treatment compared to 18 days (Figure 10).
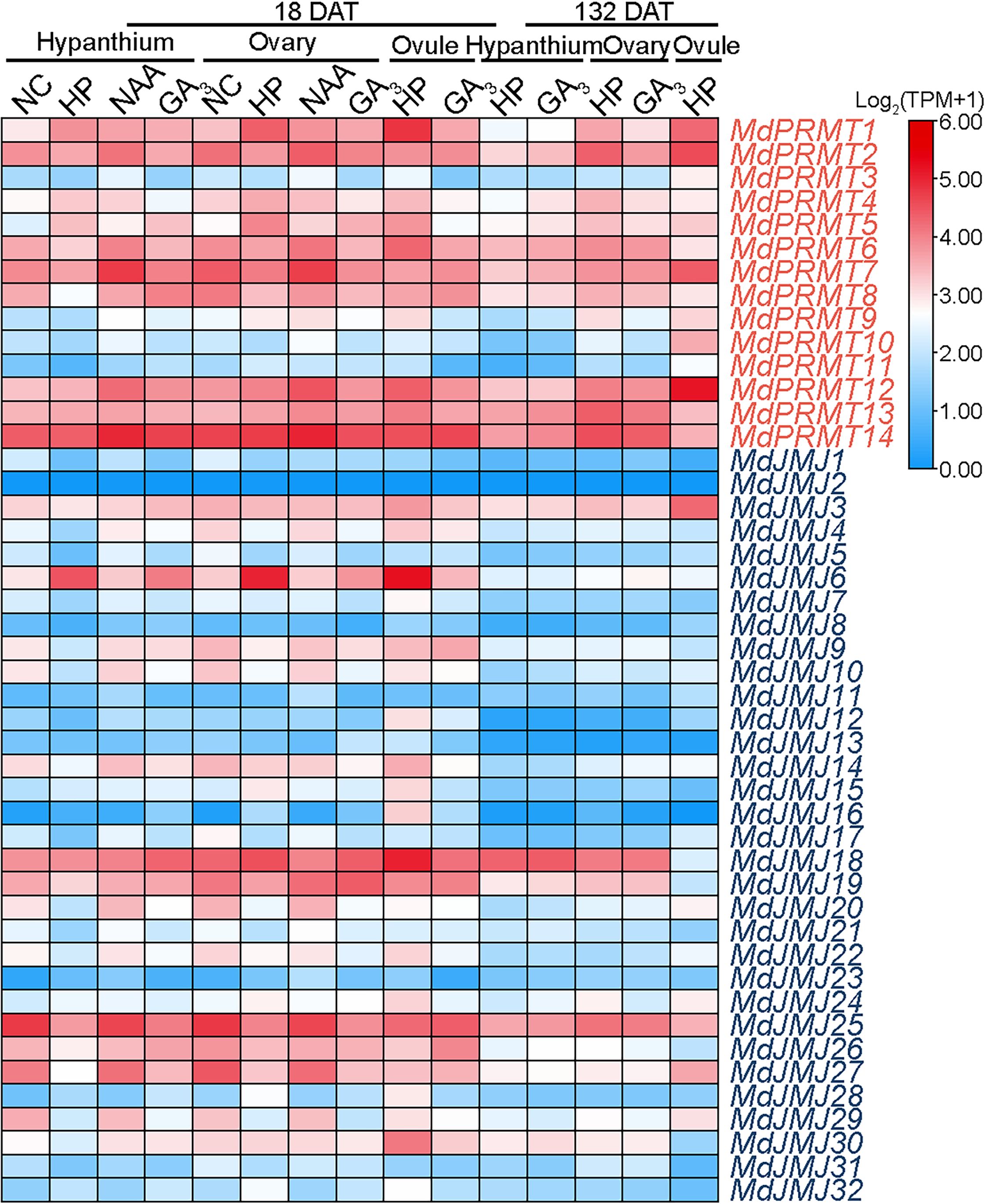
Figure 10 Expression profiles of the MdPRMT and MdJMJ genes in response to flower treatments with NAA and GA3 in apple (SRP185711). Different shades of red and blue denote the extent of the expression values according to the color bar provided [log2(TPM+1)].
Finally, we also investigated the response of apple plants at different growth and development stages to Indole-3-butyric acid (IBA), which promotes adventitious root formation (Supplementary Figure S1). The ability of apple plants to generate adventitious roots varies at different stages of growth and development. We analyzed the RNA-seq data (SRP330812) of adult and juvenile apple plants treated with IBA. Most of the MdPRMTs and MdJMJs showed no significant sensitivity to IBA treatment. However, it should be noted that in adult plants, the expression of MdJMJ19 was upregulated after 6 and 12 hours of IBA treatment compared to mock, followed by a decrease with longer treatment time. In juvenile plants, except for the 12-hour treatment, the expression of MdJMJ19 was higher than mock.
4 Discussion
Epigenetics involves stable heritable variations in organisms without changes in DNA sequences, including histone modifications (Berger et al., 2009). Histone modifications play roles in maintaining genome stability, gene regulation, and cellular processes. Research on histone modifications in important crops like rice (Ahmad et al., 2011) and tomato (Li et al., 2020) has shown their impact on gene expression and cellular activities. In this study, MdJMJ and MdPRMT families in apples were identified, characterized, and analyzed for potential roles in stress response, hormone regulation, and tissue development (Figure 11).

Figure 11 The role of the MdJMJ and MdPRMT genes in apple.
In order to better understand the phylogenetic relationship between the JMJ and PRMT gene families in apple and Arabidopsis, we constructed a phylogenetic tree. Based on the results of related studies in Arabidopsis, we classified the JMJ gene family into corresponding subfamilies, and found that the JMJ genes clustered in a logical manner. Similarly, the PRMT genes also showed reasonable clustering. Gene duplication plays a crucial role in species evolution (Panchy et al., 2016). In the current apple reference genome, we observed that most of the MdJMJ and MdPRMT genes have undergone gene duplication. In this study, we used the Arabidopsis genome as an outgroup and employed Dupgen_finder to identify the types of gene duplication within the MdJMJ and MdPRMT gene families. The results indicated that apple has recently experienced a whole-genome duplication event (Qiao et al., 2019), the predominant types of gene duplications in the MdJMJ and MdPRMT families following whole-genome duplication were WGD, with a smaller proportion of PD, TRD and TD types. Generally, different categories of gene duplication exhibit distinct patterns of functional evolution. The Ka/Ks values for most homologous gene pairs in the MdJMJ and MdPRMT gene families were less than 1, suggesting they have undergone purifying selection, eliminating deleterious mutations. Notably, a cluster of MdPRMT genes was identified on apple chromosome 15. Analysis showed that these genes were mainly involved in proximal repeats. In the promoter regions, significant differences in the types of cis-regulatory elements were observed between the PD-pair (MdPRMT9-MdPRMT11, MdPRMT9-MdPRMT10). In terms of expression profiles, MdPRMT9 exhibited higher expression levels in most parts of apple flowers compared to MdPRMT10 and MdPRMT11, under various treatments including biotic stress, cold stress and 1-MCP treatment. This suggests that TD-pairs retain higher conservation in their structural homogeneity, without affecting their functional differentiation (Qiao et al., 2019), while the WGD duplication serves as the main driver for the expansion of the MdJMJ and MdPRMT gene families, contributing to the diversity in family member structures (Feng et al., 2024).
We particularly focused on the differences in cis-regulatory elements between TD and PD repeats in the MdJMJ and MdPRMT gene families. For example, between the TD-pair (MdJMJ1-MdJMJ2), 14 types of cis-acting elements were identified, but only 7 of these were shared by both genes. A similar situation was also observed between the two PD-pairs in the MdPRMT family. This suggests that tandem duplication and proximal duplication play a significant role in the evolution of novel functions in the MdJMJ and MdPRMT gene families (Cannon et al., 2004). Furthermore, a complementary pattern of cis-regulatory elements is observed in the promoter regions of WGD gene pairs. For example, MdJMJ20 and MdJMJ26 form a WGD duplicate pair with MdJMJ9, and among the predicted 14 cis-elements, only four (Light, MYB_drought, ABRE, and ARE) are shared by all three genes. The above results can be explained by the duplication-degeneration-complementation (DDC) model. The DDC model suggests that following a gene duplication event, two alleles at two different loci experience degeneration mutations at different times, leading to a pair of duplicate genes sharing the ancestral gene’s functions and completing the ancestral gene’s functions in a complementary manner (Hahn, 2009). Comparative analysis of gene expression in various apple tissues revealed significant tissue-specific expression differences among most duplicate gene pairs (Figure 6), suggesting that these genes have evolved by differentiating ancestral gene functions or expression patterns under different tissue conditions. Additionally, we focused on comparing cis-regulatory elements in the promoters of apples (MdJMJ and MdPRMT families) with those of Arabidopsis (AtJMJ and AtPRMT families) (Supplementary Figure S2). The average number of cis-elements in apples is 9.28 for MdJMJ and 9.57 for MdPRMT, compared to 7.76 for AtJMJ and 8.22 for AtPRMT in Arabidopsis. The differences in element types can be attributed to both the high species specificity of cis-elements and the result of different selection pressures and environmental conditions acting on the ancestral genes after duplication events, leading to the acquisition of new cis-regulatory elements in their promoters (Zhang, 2003; Lan and Pritchard, 2016). The genes in model organisms generally have more comprehensive functional studies. For example, the homologous gene of MdJMJ21, AtJMJ25, has been confirmed to be involved in the formation of the very early embryo and endosperm in Arabidopsis (Day et al., 2008). We found that there is an endosperm-related cis-acting element in the promoter region of both MdJMJ21 and AtJMJ25 (Supplementary Figure S2), suggesting that MdJMJ21 may be involved in the regulation of apple endosperm formation. The Arabidopsis gene AtLDL1 has been confirmed to participate in physiological (defense) and developmental (flowering time) processes (Dutta et al., 2017), possessing ARE, ABRE, and Stress_defense-related regulatory elements. Homologous to AtLDL1, MdJMJ3 has eight ABRE and one ARE regulatory elements and shows up-regulated expression under drought stress treatment, and it is significantly expressed in anthers and pollen. Therefore, MdJMJ3 may similarly participate in the regulation of apple abiotic stress and flower development processes. Further homologous analysis of some genes in the MdJMJ and MdPRMT gene families with model organisms can reveal their potential functions.
Multiple models have been proposed to elucidate the retention and evolution of gene duplications, including gene dosage balance (Birchler et al., 2005), subfunctionalization (SF), neofunctionalization (NF) (Sandve et al., 2018), expression specialization, and pseudogenization (Qiao et al., 2019). Based on expression profiles, we studied the expression differences between duplicated genes and explored the mechanisms of gene retention. Interestingly, at different stages of fruit development, many gene pairs showed complementary expression patterns, which may indicate subfunctionalization. For instance, the gene pair MdPRMT1 and MdPRMT12, where one gene copy had higher expression levels at several stages while the other gene copy had higher expression levels at the remaining stages. Some gene pairs exhibited parallel expression patterns, suggesting that gene dosage balance is imposed in the evolution of duplicated gene pairs to maintain the total expression level of ancestral genes. For example, duplicated gene pairs like MdPRMT9-MdPRMT10 and MdJMJ1-MdJMJ2 showed similar expression patterns at different stages of fruit development. Additionally, some gene pairs showed expression specialization and nonfunctionalization, such as MdJMJ19-MdJMJ31 and MdJMJ18-MdJMJ30, where one gene copy was highly expressed at almost all stages of dormant buds, while the other gene copy showed low or no expression. We primarily focused on the performance of MdJMJ and MdPRMT under abiotic stress. It was found that some duplication gene pairs exhibited similar expression patterns under abiotic stress treatments, such as MdJMJ18-MdJMJ30 and MdJMJ13-MdJMJ32, where qPT-PCR and transcriptome analyses confirmed their consistent expression patterns under drought stress and salt stress, and a certain number of related cis-regulatory elements were identified in their promoter regions, possibly involved in regulating gene expression under stress conditions. These results demonstrate the complexity of the expansion and evolution of the MdJMJ and MdPRMT gene families, laying the foundation for further elucidating their more specific regulatory mechanisms.
5 Conclusions
In this study, a total of 14 MdPRMT genes and 32 MdJMJ genes were identified in apples. Co-linearity analysis showed that both gene families exhibited high conservation between Arabidopsis and apple. Promoter analysis indicated that MdPRMTs and MdJMJs may play important roles in plant growth and development, light response, hormone response, and stress response. Based on the analysis of transcription levels of MdPRMTs and MdJMJs in different tissues and developmental stages, we found that MdPRMTs and MdJMJs may have multiple functions in the growth and development process of apples. Especially, MdPRMT13, MdPRMT14, and MdJMJ19 showed a downtrend during fruit ripening and aging process. MdPRMT7, MdPRMT12, MdJMJ3, MdJMJ13, MdJMJ21, MdJMJ23, MdJMJ25, MdJMJ26, MdJMJ27, and MdJMJ32 exhibited high expression levels in specific organs and tissues of apples. MdJMJ25, MdPRMT7, MdPRMT14, MdJMJ9, MdPRMT2, MdJMJ3, MdJMJ18, MdJMJ27, and MdPRMT8 were involved in response to biotic stress. MdPRMT2, MdPRMT7, MdPRMT12, MdPRMT13, and MdPRMT14 showed a significant upregulation in the 1-MCP-treated group of three apple varieties. MdPRMT8 and MdJMJ19 exhibited significant upregulation within 18 days after NAA and GA3 treatment. MdJMJ19 may be associated with apple response to IBA.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SRP034165, SRP050139, SRP0999578, SRP125281, SRP334206, SRP185711, SRP229388, SRP347250, SRP091754, SRP048684, SRP239526, SRP153065, SRP330812, https://ngdc.cncb.ac.cn/, CRA002596.
Author contributions
SS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. MJ: Data curation, Formal analysis, Investigation, Software, Validation, Writing – review & editing. JC: Formal analysis, Investigation, Validation, Writing – review & editing. MZ: Investigation, Software, Validation, Writing – review & editing. XX: Conceptualization, Funding acquisition, Resources, Writing – review & editing. CC: Conceptualization, Formal analysis, Project administration, Resources, Software, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by National Natural Science Foundation of China (grant numbers 32172523); The fruit innovation team of Shandong modern agricultural industry technology system (SDAIT-06-05), and the Shandong Key R&D Plan (Agricultural Variety Project) (No. 2022LZGCQY008).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1381753/full#supplementary-material
Supplementary Table 4 | Orthologous gene pairs between apple, Arabidopsis, rice and zea mays JMJ and PRMT Genes.
References
Abe, K., Iwanami, H., Kotoda, N., Moriya, S., Takahashi, S. (2010). Evaluation of apple genotypes and Malus species for resistance to Alternaria blotch caused by Alternaria alternata apple pathotype using detached-leaf method. Plant Breed. 129, 208–218. doi: 10.1111/j.1439-0523.2009.01672.x
Ahmad, A., Dong, Y., Cao, X. (2011). Characterization of the PRMT gene family in rice reveals conservation of arginine methylation. PloS One 6, e22664. doi: 10.1371/journal.pone.0022664
Alvarez-Venegas, R. (2005). Methylation patterns of histone H3 Lys 4, Lys 9 and Lys 27 in transcriptionally active and inactive Arabidopsis genes and in atx1 mutants. Nucleic Acids Res. 33, 5199–5207. doi: 10.1093/nar/gki830
Anand, R., Marmorstein, R. (2007). Structure and mechanism of lysine-specific demethylase enzymes. J. Biol. Chem. 282, 35425–35429. doi: 10.1074/jbc.R700027200
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Bedford, M. T., Richard, S. (2005). Arginine methylation:an emerging regulator of protein function. Mol. Cell 18, 263–272. doi: 10.1016/j.molcel.2005.04.003
Berger, S. L., Kouzarides, T., Shiekhattar, R., Shilatifard, A. (2009). An operational definition of epigenetics. Genes Dev. 23, 781–783. doi: 10.1101/gad.1787609
Berger, J., Palta, J., Vadez, V. (2016). Review: An integrated framework for crop adaptation to dry environments: responses to transient and terminal drought. Plant Sci. 253, 58–67. doi: 10.1016/j.plantsci.2016.09.007
Birchler, J. A., Riddle, N. C., Auger, D. L., Veitia, R. A. (2005). Dosage balance in gene regulation: biological implications. Trends Genet. 21, 219–226. doi: 10.1016/j.tig.2005.02.010
Bray, N. L., Pimentel, H., Melsted, P., Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4, 10. doi: 10.1186/1471-2229-4-10
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Copenhaver, G. P., Chen, Q., Chen, X., Wang, Q., Zhang, F., Lou, Z., et al. (2013). Structural basis of a histone H3 lysine 4 demethylase required for stem elongation in rice. PloS Genet. 9, e1003239. doi: 10.1371/journal.pgen.1003239
Cui, X., Jin, P., Cui, X., Gu, L., Lu, Z., Xue, Y., et al. (2013). Control of transposon activity by a histone H3K4 demethylase in rice. Proc. Natl. Acad. Sci. 110, 1953–1958. doi: 10.1073/pnas.1217020110
Da, L., Liu, Y., Yang, J., Tian, T., She, J., Ma, X., et al. (2019). AppleMDO: A multi-dimensional omics database for apple co-expression networks and chromatin states. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01333
Day, R. C., Herridge, R. P., Ambrose, B. A., Macknight, R. C. (2008). Transcriptome analysis of proliferating Arabidopsis endosperm reveals biological implications for the control of syncytial division, cytokinin signaling, and gene expression regulation. Plant Physiol. 148, 1964–1984. doi: 10.1104/pp.108.128108
Dutta, A., Choudhary, P., Caruana, J., Raina, R. (2017). JMJ27, an Arabidopsis H3K9 histone demethylase, modulates defense against pseudomonas syringe and flowering time. Plant J. 91, 1015–1028. doi: 10.1111/tpj.13623
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.2307/2408678
Feng, X., Chen, Q., Wu, W., Wang, J., Li, G., Xu, S., et al. (2024). Genomic evidence for rediploidization and adaptive evolution following the whole-genome triplication. Nat. Commun. 15, 1635. doi: 10.1038/s41467-024-46080-7
Genschik, P., Marrocco, K., Bach, L., Noir, S., Criqui, M.-C. (2014). Selective protein degradation: a rheostat to modulate cell-cycle phase transitions. J. Exp. Bot. 65, 2603–2615. doi: 10.1093/jxb/ert426
Hahn, M. W. (2009). Distinguishing among evolutionary models for the maintenance of gene duplicates. J. Hered. 100, 605–617. doi: 10.1093/jhered/esp047
Hennerty, M. J., Forshey, C. G. (1972). Tree physiological condition as a source of variation in chemical thinning of apple fruits. HortScience 7, 259–260. doi: 10.21273/HORTSCI.7.3.259
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486. doi: 10.1016/S0168-9525(02)02722-1
Kenchanmane Raju, S. K., Ledford, M., Niederhuth, C. E. (2023). DNA methylation signatures of duplicate gene evolution in angiosperms. Plant Physiol. 192, 2883–2901. doi: 10.1093/plphys/kiad220
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Lan, X., Pritchard, J. K. (2016). Coregulation of tandem duplicate genes slows evolution of subfunctionalization in mammals. Science 352, 1009–1013. doi: 10.1126/science.aad8411
Lawrence, M., Daujat, S., Schneider, R. (2016). Lateral thinking: how histone modifications regulate gene expression. Trends Genet. 32, 42–56. doi: 10.1016/j.tig.2015.10.007
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Li, Z., Jiang, G., Liu, X., Ding, X., Zhang, D., Wang, X., et al. (2020). Histone demethylase SlJMJ6 promotes fruit ripening by removing H3K27 methylation of ripening-related genes in tomato. New Phytol. 227, 1138–1156. doi: 10.1111/nph.16590
Li, W., Yan, J., Wang, S., Wang, Q., Wang, C., Li, Z., et al. (2021). Genome-wide analysis of SET-domain group histone methyl transferases in apple reveals their role in development and stress responses. BMC Genomics 22, 283. doi: 10.1186/s12864–021-07596–0
Liu, Y., Liu, Q., Li, X., Tang, Z., Zhang, Z., Gao, H., et al. (2022). Exogenous dopamine and MdTyDC over expression enhance apple resistance to fusarium solani. Phytopathology 112, 2503–2513. doi: 10.1094/PHYTO-04-22-0142-R
Liu, C., Lu, F., Cui, X., Cao, X. (2010). Histone methylation in higher plants. Annu. Rev. Plant Biol. 61, 395–420. doi: 10.1146/annurev.arplant.043008.091939
Luo, M., Hung, F.-Y., Yang, S., Liu, X., Wu, K. (2014). Histone lysine demethylases and their functions in plants. Plant Mol. Biol. Rep. 32, 558–565. doi: 10.1007/s11105-013-0673-1
Ma, N., Tan, H., Liu, X., Xue, J., Li, Y., Gao, J. (2006). Transcriptional regulation of ethylene receptor and CTR genes involved in ethylene-induced flower opening in cut rose (Rosa hybrida) cv. Samantha. J. Exp. Bot. 57, 2763–2773. doi: 10.1093/jxb/erl033
Niu, L., Lu, F., Pei, Y., Liu, C., Cao, X. (2007). Regulation of flowering time by the protein arginine methyl transferase AtPRMT10. EMBO Rep. 8, 1190–1195. doi: 10.1038/sj.embor.7401111
Niu, L., Zhang, Y., Pei, Y., Liu, C., Cao, X. (2008). Redundant requirement for a pair of PROTEIN ARGININE METHYLTRANSFERASE4 homologs for the proper regulation of Arabidopsis flowering time. Plant Physiol. 148, 490–503. doi: 10.1104/pp.108.124727
Panchy, N., Lehti-Shiu, M., Shiu, S. H. (2016). Evolution of gene duplication in plants. Plant Physiol. 171, 2294–2316. doi: 10.1104/pp.16.00523
Pei, Y., Niu, L., Lu, F., Liu, C., Zhai, J., Kong, X., et al. (2007). Mutations in the type II protein arginine methyltransferase AtPRMT5 result in pleiotropic developmental defects in Arabidopsis. Plant Physiol. 144, 1913–1923. doi: 10.1104/pp.107.099531
Perini, P., Pasquali, G., Margis-Pinheiro, M., de Oliviera, P. R. D., Revers, L. F. (2014). Reference genes for transcriptional analysis of flowering and fruit ripening stages in apple (Malus × domestica Borkh.). Mol. Breed. 34, 829–842. doi: 10.1007/s11032-014-0078-3
Pharis, R. P., King, R. W. (1985). Gibberellins and reproductive development in seed plants. Annu. Rev. Plant Biol. 36, 517–568. doi: 10.1146/annurev.pp.36.060185.002505
Qiao, X., Li, Q., Yin, H., Qi, K., Li, L., Wang, R., et al. (2019). Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 20, 38. doi: 10.1186/s13059-019-1650-2
Rohde, A., Bhalerao, R. P. (2007). Plant dormancy in the perennial context. Trends Plant Sci. 12, 217–223. doi: 10.1016/j.tplants.2007.03.012
Sandve, S. R., Rohlfs, R. V., Hvidsten, T. R. (2018). Subfunctionalization versus neofunctionalization after whole-genome duplication. Nat. Genet. 50, 908–909. doi: 10.1038/s41588-018-0162-4
Shen, F., Huang, Z., Zhang, B., Wang, Y., Zhang, X., Wu, T., et al. (2019). Mapping gene markers for apple fruit ring rot disease resistance using a multi-omics approach. G3-Genes Genomes Genet. 9, 1663–1678. doi: 10.1534/g3.119.400167
Sun, Q., Zhou, D.-X. (2008). Rice jmjC domain-containing gene JMJ706 encodes H3K9 demethylase required for floral organ development. Proc. Natl. Acad. Sci. United States America 105, 13679–13684. doi: 10.1073/pnas.0805901105
Tamura, K., Stecher, G., Kumar, S., Battistuzzi, F. U. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Thomas Jenuwein, C. D. A. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Wang, H., Huang, Z.-Q., Xia, L., Feng, Q., Erdjument-Bromage, H., Strahl, B. D., et al. (2001). Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Am. Assoc. Advance. Sci. 293, 853–857. doi: 10.1126/science.1060781
Xu, G., Guo, C., Shan, H., Kong, H. (2012). Divergence of duplicate genes in exon–intron structure. Proc. Natl. Acad. Sci. 109, 1187–1192. doi: 10.1073/pnas.1109047109
Yang, W., Liu, X. D., Chi, X. J., Wu, C. A., Li, Y. Z., Song, L. L., et al. (2011). Dwarf apple MbDREB1 enhances plant tolerance to low temperature, drought, and salt stress via both ABA-dependent and ABA-independent pathways. Planta 233, 219–229. doi: 10.1007/s00425-010-1279-6
Zhang, J. (2003). Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 292–298. doi: 10.1016/S0169-5347(03)00033-8
Zhao, J., Guo, R., Guo, C., Hou, H., Wang, X., Gao, H. (2016). Evolutionary and expression analyses of the apple basic leucine zipper transcription factor family. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00376
Keywords: Prmt, JMJ, apple, gene expression, gene family
Citation: Su S, Ji M, Chen J, Zhang M, Xu X and Cheng C (2024) Genome-wide identification and expression analysis of protein arginine methyltransferase and JmjC domain-containing family in apple. Front. Plant Sci. 15:1381753. doi: 10.3389/fpls.2024.1381753
Received: 22 February 2024; Accepted: 13 May 2024;
Published: 28 May 2024.
Edited by:
Somashekhar M. Punnuri, Fort Valley State University, United StatesReviewed by:
Sunil K. Kenchanmane Raju, Michigan State University, United StatesZihao Zheng, Syngenta, United States
Copyright © 2024 Su, Ji, Chen, Zhang, Xu and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaozhao Xu, MjAxODAxMDA2QHFhdS5lZHUuY24=; Chenxia Cheng, Y2hlbmdjaGVueGlhQHFhdS5lZHUuY24=