 Liyuan Tang
Liyuan Tang Cunjing Liu
Cunjing Liu Sujun Zhang
Sujun Zhang Jianhong Zhang
Jianhong Zhang- Institute of Cotton, Hebei Academy of Agriculture and Forestry Sciences, Key Laboratory of Cotton Biology and Genetic Breeding in Huanghuaihai Semiarid Area, Ministry of Agriculture and Rural Affairs, Shijiazhuang, Hebei, China
High fiber strength (FS) premium cotton has significant market demand. Consequently, enhancing FS is a major objective in breeding quality cotton. However, there is a notable lack of known functionally applicable genes that can be targeted for breeding. To address this issue, our study used specific length–amplified fragment sequencing combined with bulk segregant analysis to study FS trait in an F2 population. Subsequently, we integrated these results with previous quantitative trait locus mapping results regarding fiber quality, which used simple sequence repeat markers in F2, F2:3, and recombinant inbred line populations. We identified a stable quantitative trait locus qFSA06 associated with FS located on chromosome A06 (90.74–90.83 Mb). Within this interval, we cloned a gene, GhALDH7B4_A06, which harbored a critical mutation site in coding sequences that is distinct in the two parents of the tested cotton line. In the paternal parent Ji228, the gene is normal and referred to as GhALDH7B4_A06O; however, there is a nonsense mutation in the maternal parent Ji567 that results in premature termination of protein translation, and this gene is designated as truncated GhALDH7B4_A06S. Validation using recombinant inbred lines and gene expression analysis revealed that this mutation site is correlated with cotton FS. Virus-induced gene silencing of GhALDH7B4 in cotton caused significant decreases in FS and fiber micronaire. Conversely, GhALDH7B4_A06O overexpression in Arabidopsis boosted cell wall component contents in the stem. The findings of our study provide a candidate gene for improving cotton fiber quality through molecular breeding.
Introduction
Cotton is a primary natural fiber source in the textile industry (Wen et al., 2023). Upland cotton (Gossypium hirsutum L.) is extensively cultivated because of its high yield and adaptability, and it accounts for over 95% of cultivated cotton (Yuan et al., 2019). Because of the negative correlation between cotton yield and fiber quality, relying on traditional breeding to simultaneously improve yield and quality is challenging (Wang et al., 2020). This has resulted in moderate fiber quality of commonly cultivated varieties. With the improvement of modern textile processes, the growing consumer demand for high-quality cotton products, and damage to cotton fibers caused by current mechanical harvesting, there is a substantial market demand for premium cotton with high fiber strength (FS) (Zang et al., 2022). Enhancing FS has become a major goal in cotton quality breeding.
Cotton fiber, a single-cell trichome with an extended and thickened seed surface, also serves as an excellent model for investigating cellular development processes (Haigler et al., 2012; Xu et al., 2021). Cotton fiber development progresses through four interconnected stages: fiber initiation, elongation, secondary cell wall (SCW) thickening, and desiccation maturation. Fiber length (FL; mm), fiber strength (FS; cN/tex), fiber uniformity ratio (FU), fiber micronaire (FM), and fiber elongation (FE) are the primary variables regulating the fiber’s characteristics (Liu et al., 2016). FS primarily develops during the SCW thickening stage (Zang et al., 2022). SCW thickening is an intricate biological process that is controlled by the biosynthesis-related genes responsible for cell wall primary components, cellulose, hemicellulose, and lignin, and it is regulated by numerous transcription factors, microRNAs, and phytohormones (Kumar and Turner, 2015; Zang et al., 2022; Ma et al., 2018; Huang et al., 2018; Li et al., 2018; Cao et al., 2020; Sun et al., 2020; Zhang et al., 2021; Tang et al., 2023). Furthermore, it is apparent that there are numerous genes and complex regulatory networks associated with SCW. For example, among NAC (NAM, ATAF and CUC) type transcription factors, a total of 38 GhNAC genes have been identified to be involved in cotton fiber development (Sun et al., 2018). FS value is influenced by cellulose deposition, which determines cell wall thickness, and cellulose fiber organization, which determines cellulose crystallinity, during the critical period of SCW thickening (Zang et al., 2022).
Previous researchers revealed numerous qualitative trait loci (QTLs) related to FS by employing linkage analysis or genome-wide association study using various markers such as amplified fragment length polymorphisms, restriction fragment length polymorphisms, and simple sequence repeats (SSRs) on samples from different populations (Fang et al., 2017; Ning et al., 2014). With the refinement of cotton reference genome sequencing (Li et al., 2015; Zhang et al., 2015; Hu et al., 2019; Wang et al., 2019; Ma et al., 2021), high-density single-nucleotide polymorphisms (SNPs), individually and in conjunction with SSRs and other markers, were used to identify several FS-related genes (Islam et al., 2016; Li et al., 2016; Sun et al., 2017; Zhang et al., 2017; Ma et al., 2018; Feng et al., 2020; He et al., 2021; Yang et al., 2022). For example, Ma et al. (2018) conducted a re-sequencing study on 419 core germplasm resources and detected 630 FS-related SNPs. Keerio et al. (2018) mapped six FS-related QTLs on three chromosomes using specific length–amplified fragment sequencing (SLAF-seq) of an introgression line population and its parent. These studies identified FS-related SNPs and genes, which increased the density of marker loci in intraspecific genetic maps and greatly improved candidate gene location accuracy.
Numerous FS-related candidate genes have been identified and the functions of some genes validated (Zang et al., 2021). Nonetheless, many potential genes have yet to be confirmed, and this is largely attributed to genotypic limitations and protracted transformation periods involved in establishing stable transgenic cotton plants with targeted characteristics via genetic modification (Ge et al., 2023). Virus-induced gene silencing (VIGS) is a rapid method commonly employed for preliminary gene function determination (Tian et al., 2022). VIGS studies have revealed the role of strigolactone biosynthetic genes and strigolactone-responsive transcription factor genes in modulating cotton fiber development because they affect SCW thickness and fiber elongation (Tian et al., 2022; Wen et al., 2023). Other research has used VIGS to demonstrate the involvement of genes such as GhERF41, GhLTP1, GhSTLs, and GhAPs in the formation of FL or FS, respectively (Deng et al., 2016; Guo et al., 2022; Gao et al., 2023; Zhang et al., 2024). Similarly, Arabidopsis serves as a useful model for fiber trait investigations, with studies showing that overexpression of genes such as GhMYB7, GhMYB25-like, and GhCesAs can modify cell wall composition and boost cellulose content, which impact fiber quality (Huang et al., 2016; Chen et al., 2006; Betancur et al., 2010). Sun et al. (2020) found that the cellulose and lignin contents in the stems and roots of transgenic Arabidopsis lines were reduced, which revealed that GhFSN5 is a negative regulator of SCW formation.
In our previous studies, F2, F2:3, and recombinant inbred line (RIL) (F2:9) populations were used for genetic mapping to identify QTLs associated with fiber quality traits in upland cotton. A stable FS QTL on chromosome A06 was consistently identified across multiple generations (Zhang et al., 2020). In this study, we used SLAF-seq combined with bulk segregant analysis (SLAF-BSA-seq) to map a candidate interval for FS for the F2 population. We were able to fine map a smaller genomic region, qFSA06, with which the identified QTL overlapped on chromosome A06. Cloning analysis revealed GhALDH7B4 as the potential candidate gene for qFSA06, and this was supported by a nonsense mutation and the correlation of GhALDH7B4 gene expression with FS. Functional validation experiments using VIGS in cotton and overexpression in Arabidopsis thaliana demonstrated that GhALDH7B4 positively regulates cotton FS.
Materials and methods
Plant materials
The parents (Ji567 and Ji228) of the high fiber quality hybrid cotton Jí1518 and its offspring F2 and RIL populations were employed in this study. Zhang et al. (2020) presented an extensive overview of the population’s growth process and the phenotypic evaluation of fiber quality. Ji567, the female parent, has a high yield and moderate FS. Ji228, the male parent, has high fiber quality and high FS; it has the genetic background of sea island cotton and carries the chromosomal segments from island cotton (Liu et al., 2009). RIL131 and RIL229 were selected from the RIL population because they have similar genome composition except that RIL 229 harbors the target FS QTL on chromosome A06; these RILs exhibited maintained stability of agronomic traits over the past 5 years (Tang et al., 2023).
SLAF-BSA-seq
Both parents and two bulks were chosen to perform SLAF-BSA-seq. In the F2 population, 26 plants had the highest FS (H-bulk), and 31 plants had the lowest FS (L-bulk) (Table 1). Genomic DNA from all plants in each group was combined in equal quantities to produce bulks that had a final purity level of 40 ng/µL. The SLAF library construction followed Sun et al. (2013), with 364- to 414-bp DNA strands prepared by Biomics Technologies Company (Beijing, China) for pair-end sequencing using Illumina High-seq 2500 platform (Illumina, CA, USA). The data has been deposited in the NCBI database with the accession number PRJNA1049971. After sequencing, the clean reads from the four bulks were mapped to the G. hirsutum reference genome (Zhang et al., 2015). GATK and Samtools were employed for SNP analysis (Li et al., 2009; McKenna et al., 2010). Polymorphic SNPs between bulks were used for association research. SNP-index correlation by Abe et al. (2012) and Euclidean distance (ED) by Deza et al (2009). were integrated for association mapping.
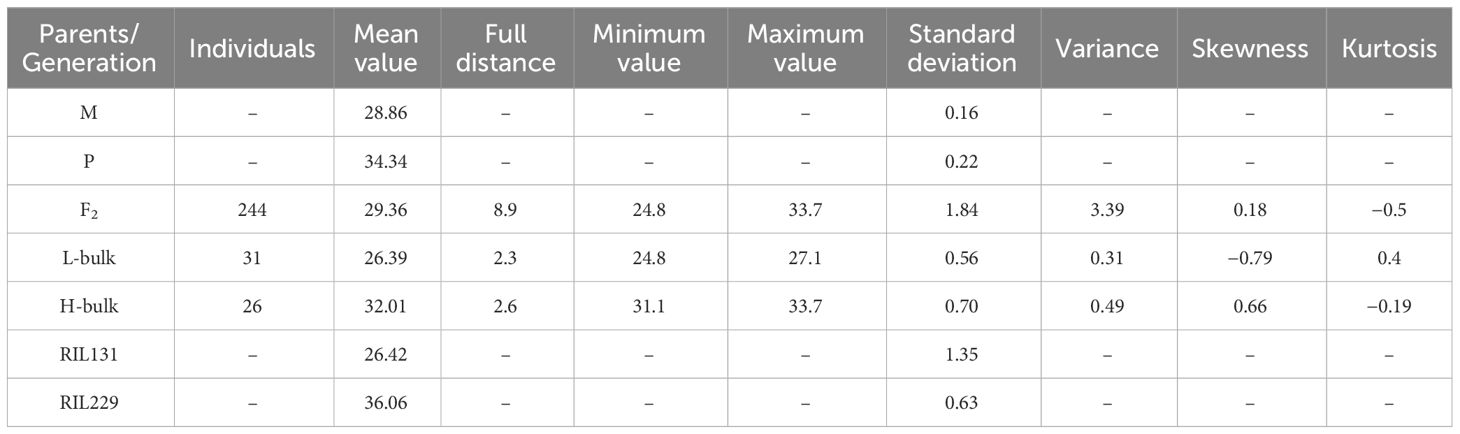
Table 1 FS characters statistics of parents, F2 population, and RIL lines.
Physical location search of SSR markers
The study employed SSR markers previously used for fiber quality QTL mapping in the population (Zhang et al., 2020) and analyzed their physical locations through literature reported (Liu et al., 2016) and the CottonGen database (https://www.cottongen.org/) (Yu et al., 2021).
Sequence alignment and analysis
Using the PrimeStar HS high-fidelity enzyme (TaKaRa, China), potential genes were amplified from fiber cDNA of the two parents. For each PCR product, a minimum of eight clones from the pEASY-Blunt cloning vector (Transgen, China) were sequenced. Sequence alignment was performed using the DNAMAN 8.0, and the protein sequence was subjected to BLAST search in the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Conserved domains of proteins were analyzed by the Pfam database (http://pfam.xfam.org/).
Verification of the association between FS and a SNP in the candidate gene GhALDH7B4_A06
Twenty RILs were arbitrarily selected to investigate the potential association between the GhALDH7B4_A06 SNP and FS. The SNP was confirmed through Sanger sequencing, and the primers used are listed in Supplementary Table 1. The FS phenotype was determined on the basis of the average over the preceding 3 years.
Gene expression analysis using quantitative real-time PCR (qRT-PCR)
Various tissues (leaf, stem, and root); ovules at 0 days post-anthesis (DPA); and fibers at 5, 10, 15, 20, and 25 DPA were retrieved from healthy plants of both parents, RIL131 and RIL229. Three biological copies were used in this research. RNA extraction, cDNA synthesis, and qRT-PCR were carried out with the RNAprep Pure Plant Kit (TIANGEN, China), the PrimeScript RT Reagent Kit (TaKaRa, China), and TB Green Premix Ex Taq II (TaKaRa, China), respectively, following the manufacturer’s instructions. As internal control, histone3 (AF024716) and TUB2 were employed for gene expression analysis in cotton and Arabidopsis, respectively. Relative expression rates were assessed using the 2−ΔΔCt method (Livak and Schmittgen, 2001). Supplementary Table 1 lists the specific primers that were designed using Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/).
VIGS of GhALDH7B4 in cotton
The 300-bp GhALDH7B4 fragment was cloned into the cotton leaf crumple virus (CLCrV) vector using SpeI and AscI restriction enzymes because recent investigations established the virus’s capacity to influence SCW synthesis during cotton fiber development (Liu et al., 2019; Tian et al., 2022). Primers used are shown in Supplementary Table 1. Agrobacterium strain LBA4404 was used to transform the VIGS vectors. The bacterial liquid involved in the test mainly included the auxiliary plasmid CLCrVB, the empty vector CLCrVA, the positive control vector CLCrVA-PDS, and the target gene vector CLCrVA-GhALDH7B4.
The transformed LBA4404 was injected into the RIL229 plants with high FS using established protocols (Gu et al., 2014; Tian et al., 2022). The silencing effect was initially evaluated by the whitening symptoms observed in the cotton plants expressing the positive control CLCrV : PDS. To extend the duration of gene silencing and maintain its efficacy during later stages, a secondary injection was administered at the leaf axil of the primary stem in the early phase of cotton squaring.
Gene silencing efficiency was determined by qRT-PCR using 25-DPA fibers of CLCrV:00 negative control and CLCrV : GhALDH7B4 plants. The cotton fiber was harvested per plant when the cotton boll opened naturally and matured. Six strains with high silencing efficiency were selected, and fiber samples from two plants were randomly combined as a biological replicate. Each set comprised three biological replicates for the assessment of fiber quality.
Overexpression of GhALDH7B4_A06 in Arabidopsis
The open reading frame of GhALDH7B4_A06O was inserted into the pCAMBIA1302 vector with 35S CaMV promoter using homologous recombination and Golden Gate seamless assembly techniques to construct the GhALDH7B4_A06O overexpression vector. The primers that were used are outlined in Supplementary Table 1. Arabidopsis thaliana transformation was conducted with the flower dipping method (Qin et al., 2017). Positive plants were screened in a medium of 1/2 Murashige and Skoog (MS) + Hygromycin B (75 mg/L) to the T3 generation. Both wild-type (WT) and transgenic Arabidopsis plants were subjected to consistent growth conditions and standard maintenance protocols. At maturity, which typically occurred after approximately 8 weeks, the first stem node of flower stems from both transgenic and WT plants, measuring approximately 10 cm in length from the base to the apex, was chosen for evaluation. The cellulose, hemicellulose, and lignin contents of samples were analyzed using specific detection kits (G0715W, G0716W, and G0708W) (Grace, China) designed for plant material.
Phenotypic detection and statistical analysis
USTER HVI1000 M700 (Uster Technologies, Switzerland) large-capacity cotton detector was used to test the characteristics of cotton fiber quality, including FL, FS, FM, FU, and FE. All experiments were independently repeated a minimum of three times. Data were analyzed with Microsoft Excel 2021 and SigmaPlot 14.0. Significant differences were determined by ANOVA.
Results
Fiber qualities of parents, recombinant lines, and populations
In this investigation, the parents and offspring of the hybrid Ji1518 were examined. Overall, there were significant variations in FS between the two parents (Ji567, 28.86 cN/tex; Ji228, 34.34 cN/tex), and the F2 population consisting of 244 individuals displayed a continuous range of FS variation (Table 1). Significant discrepant FS values were observed between the H-bulk (31.1–33.7 cN/tex) and L-bulk (24.8–27.1 cN/tex) selected from the F2 population, which can serve as representatives of high and low FS phenotypes (Table 1; Figure 1A). RIL131 and RIL229 displayed differences in the loci of the FS QTL on chromosome A06, as evidenced by genotyping indicating the presence of a SNP in the coding sequence (CDS) region of the candidate gene GhALDH7B4_A06, with RIL131 carrying the GhALDH7B4_A06S allele and RIL229 carrying the GhALDH7B4_A06O allele. RIL229 displayed comparable agronomic traits to RIL131 but demonstrated significantly higher FS, with a notable average difference of 9.64 cN/tex (Table 1).
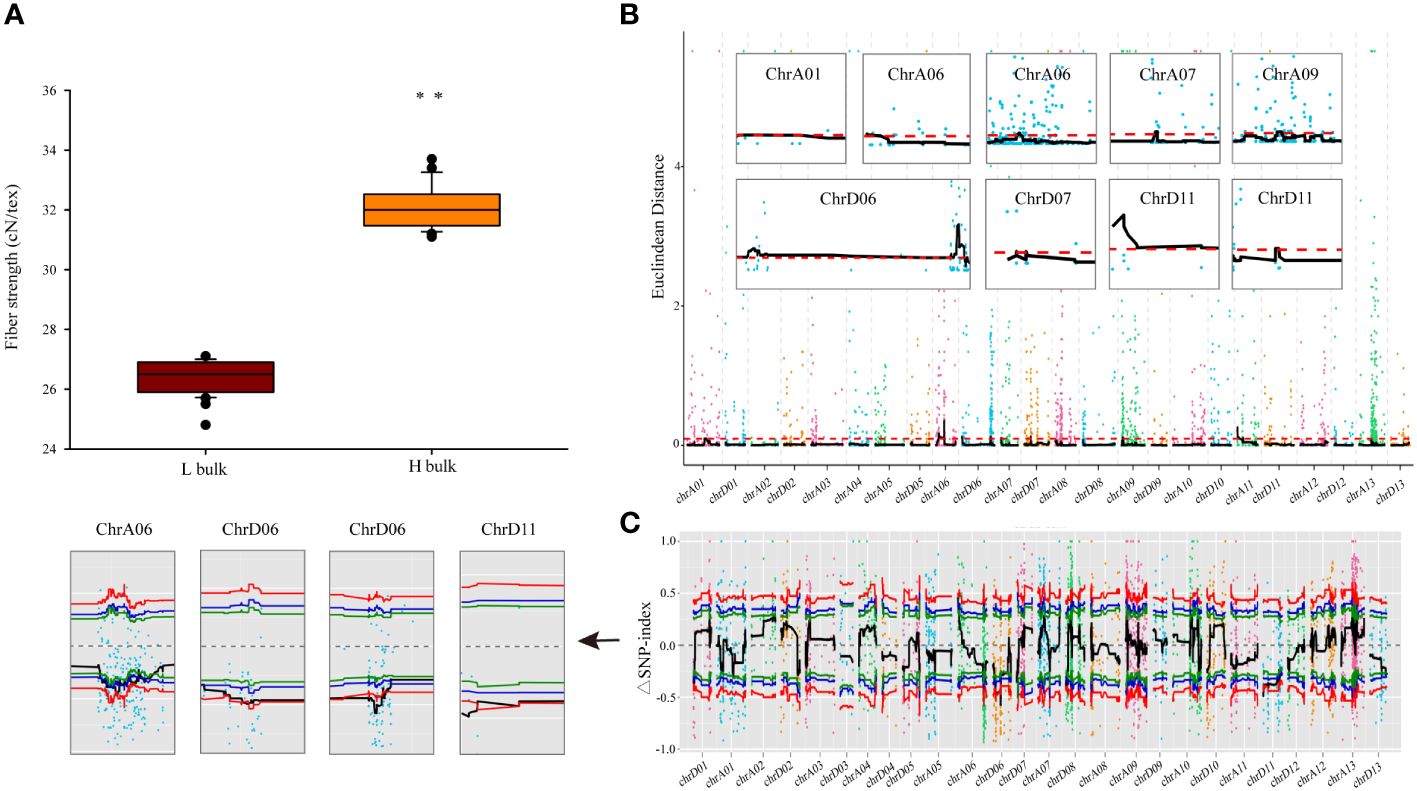
Figure 1 BSA gene bulk and genetic mapping of FS phenotypes. (A) FS values in different BSA gene bulks. Error bars represent the SD of different individuals in respective bulks, ** indicates significant difference at p < 0.01. (B, C) Statistical algorithms used to map FS candidate genes under default parameters: Euclidean distance association analysis (B) and SNP-index association analysis (C). Each colored point represents the calculated ED value or ΔSNP-index value, whereas the black lines indicate the respective fitted values. In the ED algorithm graph (B), red dashed lines indicate thresholds. In the SNP-index algorithm graph (C), the red, blue, and green lines represent thresholds with confidence levels of 0.99, 0.95, and 0.90, respectively.
FS QTL identification by SLAF-seq in (Ji567×Ji228) F2 population
To identify the major QTL governing cotton FS, SLAF-seq was performed on four libraries from two parents and two bulks from its F2 descendants (H bulk and L bulk). A total of 307,849 SLAF tags were obtained (Supplementary Table 2), with a uniform distribution across the genome’s chromosomes (Supplementary Figure 1A). The average sequencing depth of parents was 25.01× and that of gene bulk with different FS was 34.17× (Supplementary Table 3). A total of 125,969 SNPs were uniformly distributed on each chromosome (Supplementary Figure 1B). Subsequently, an association analysis was performed using 6,758 high-quality SNPs, which were obtained after rigorous filtering processes (Supplementary Table 4).
The ED and SNP-index methodologies were applied to identify QTLs linked to FS. The ED approach, with a threshold of 0.10, revealed that nine QTLs spanning a cumulative length of 26.43 Mb were identified on seven chromosomes (A01, A06, A07, A09, D06, D07, and D11) and encompassed 1,270 genes (Figure 1B; Table 2). Among these, two QTLs were located on both chromosomes A06 and D11, whereas the rest were single-locus QTLs. Additionally, employing the ΔSNP-index method with a confidence level of 0.99 revealed five QTLs that included 244 genes covering a range of 5.23 Mb on three chromosomes (A06, D06, and D11) (Figure 1C; Table 2). Integration of results from both methodologies led to the identification of four potential FS-associated QTLs distributed on chromosomes A06, D06, and D11, with a total length of 4.96 Mb and encompassing 242 genes (Table 2; Supplementary Table 5). Notably, the mapping interval on chromosome D06 exhibited the greatest size, whereas the region on chromosome D11 harbored the highest number of genes, with the smallest mapping interval and least number of genes identified on chromosome A06.
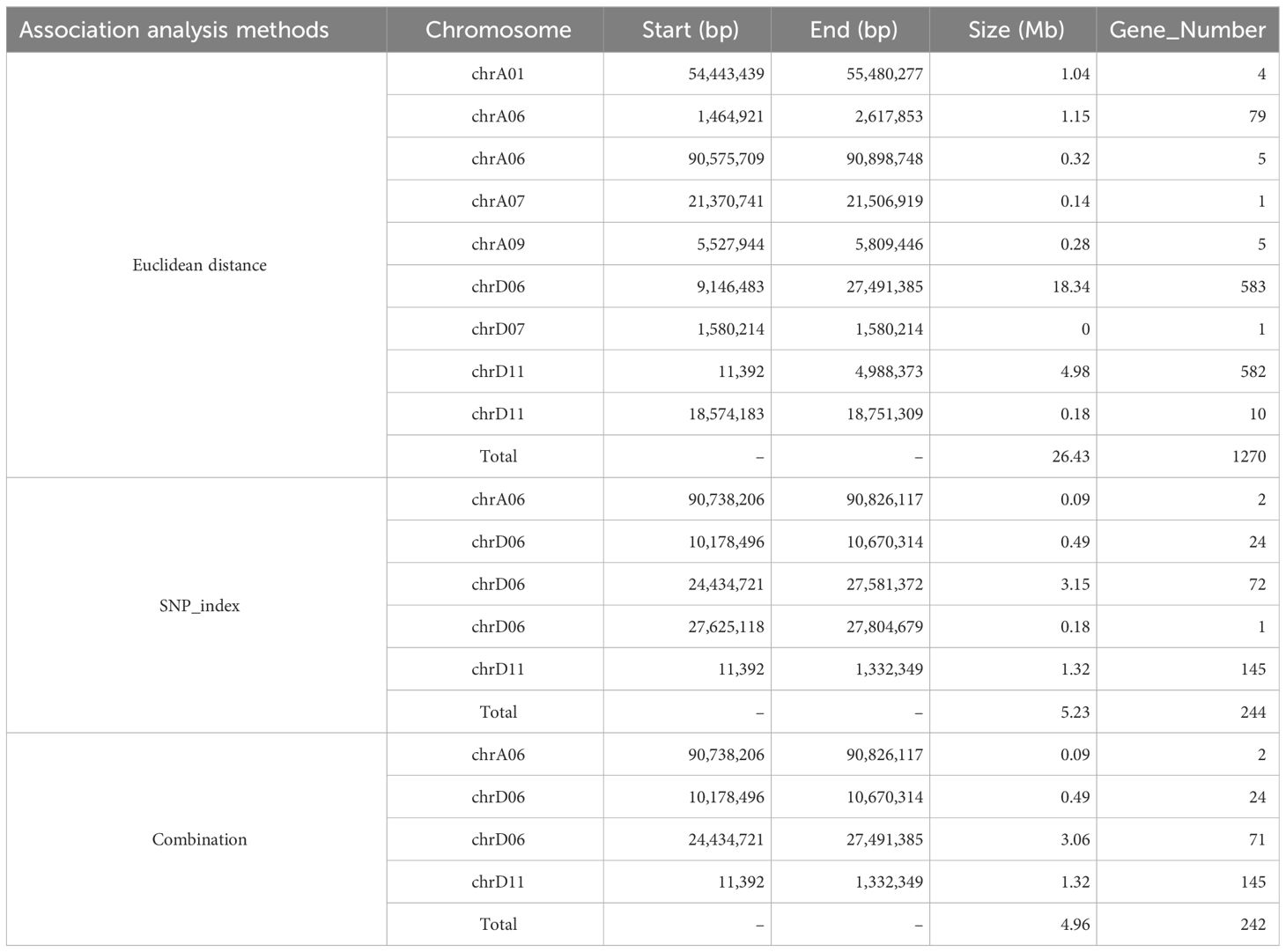
Table 2 QTL regions by the two association analysis methods.
Identification of FS-related candidate genes on chromosome A06
To narrow down the target region within the identified extensive interval, integration of previous QTL mapping results for fiber quality traits using SSR markers was conducted. Previous investigations across various populations, including F2, F2:3, and RIL populations, identified linkage groups associated with FS on chromosome A06, and two SSR markers—HAU2119 and HAU2349—consistently co-segregated within the same linkage group. In the F2/F2:3 populations, three FS QTLs were positioned between these markers, which explained the phenotypic variances ranging from 3.33% to 13.14%. However, in the RIL population, a single QTL was identified as the exclusive FS QTLs using SSR markers and was associated with 5.10% of the phenotypic variance. We tentatively hypothesize the existence of a stable major-effect QTL between the two markers on chromosome A06 based on the study population. Upon retrieving the physical positions of these markers from the CottonGen database (Supplementary Table 6), we identified that the QTL (90.74–90.83 Mb) on chromosome A06 via SLAF-BSA-seq fell within the region demarcated by HAU2119 and HAU2349 (Figure 2A), whereas the stability of the presence of QTL on chromosome D06 and D11 by SLAF-BSA-seq cannot be confirmed as no relevant QTL were previously mapped in the RIL population by SSR markers. Therefore, the region on chromosome A06 was selected for further investigation and designated as qFSA06, and it spanned an 87.9-kb interval.
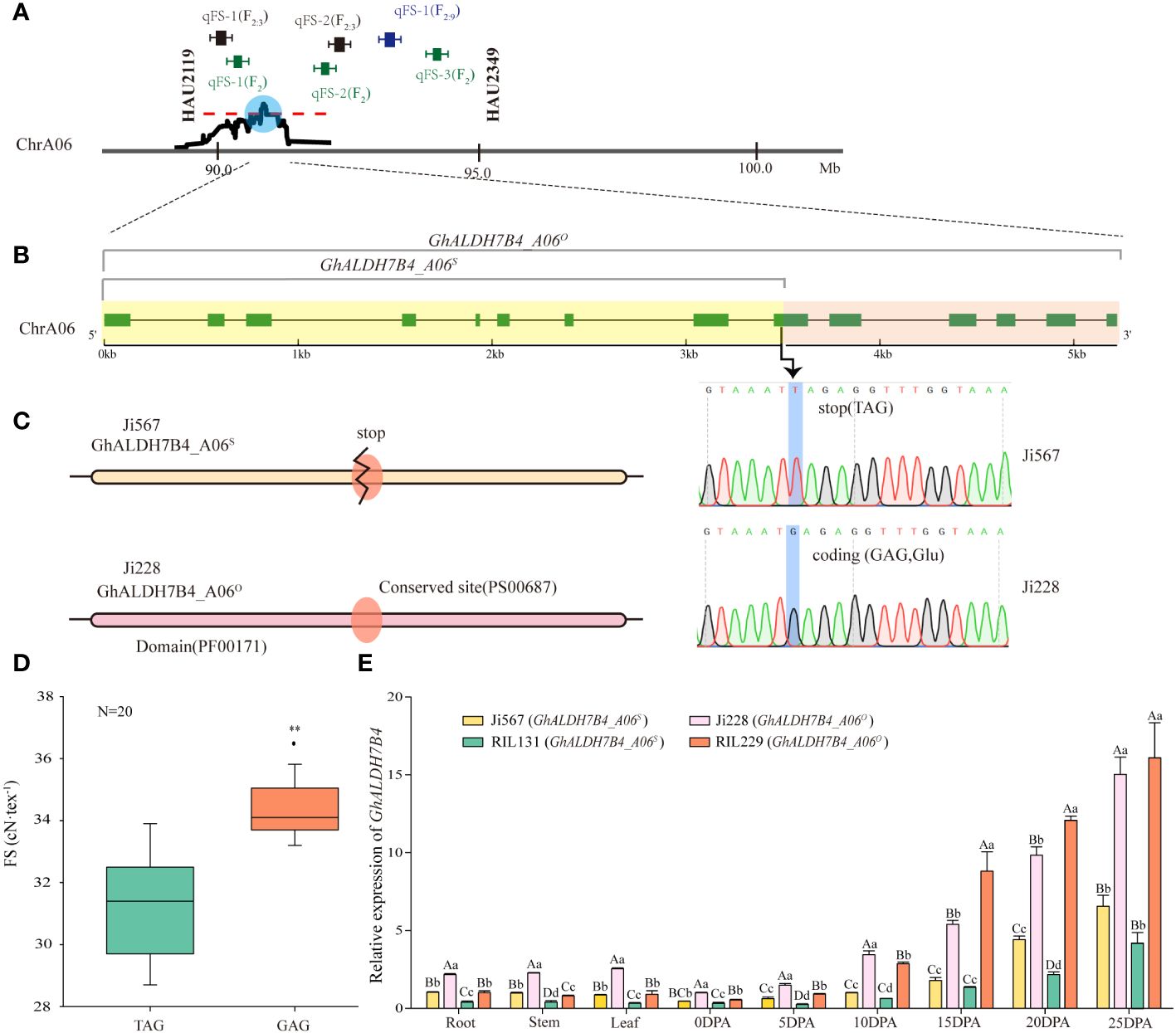
Figure 2 Designation, sequence variations, and differential expression of GhALDH7B4_A06. (A) QTL interval determination by comparing QTL mapping results based on SSR markers and SLAF-BSA-seq. (B) Allelic variations in GhALDH7B4_A06. (C) Prediction of the protein domain and conserved site of GhALDH7B4_A06. ** indicate significant differences at p < 0.01. (D) Correlation of FS and allelic variations in GhALDH7B4_A06 in 20 RILs. (E) GhALDH7B4 expression in different tissues of parents and two RILs. Lowercase and uppercase letters indicate significance at p < 0.05 and p < 0.01, respectively.
Structural and expression analyses of candidate gene GhALDH7B4 _A06
A gene located within the chromosome A06 interval was isolated from fiber cDNAs of the parental lines Ji567 and Ji228 and demonstrated a substantial genetic variation between the two lines. In Ji228, the gene’s CDS full length was determined to be 1,527 bp, whereas an SNP variant was identified at position 772 bp in Ji567 that resulted in a G-to-T substitution. This nucleotide change led to the conversion of the normal codon (GAG) to a stop codon (TAG) (Figure 2B), which caused premature termination of protein translation.
Protein domain prediction analysis indicated that the presence of stop codons resulted in incomplete structural domains and the absence of conserved glutamic acid active sites (PS00687) (Figure 2C). This alteration was discovered to be associated with FS variations in 20 RILs (Figure 2D). In particular, the gene did not exhibit any nonsense mutations in the RILs with FS exceeding 34 cN/tex. Therefore, the gene could be divided into full-length and truncated types in the study population. We tentatively assigned this gene as a candidate for qFSA06.
The candidate gene encoded a protein consisting of 508 amino acids with aldehyde dehydrogenase activity and exhibited 84.84% similarity with AtALDH7B4; thus, it was named GhALDH7B4 in cotton. This gene had one copy in each of the At and Dt sub-genomes in upland cotton, with 17 nucleotide differences in the CDS region, which led to the nomenclature of GhALDH7B4_A06 and GhALDH7B4_D06 (GenBank: PP584503) based on their chromosomal locations. The full-length variant on chromosome A06 was named GhALDH7B4_A06O (GenBank: PP210923), whereas the truncated form was named GhALDH7B4_A06s (GenBank: PP584502) (Supplementary Table 7). Cloning results indicated no differences in the GhALDH7B4_D06 CDS between the two parents (Supplementary Table 7).
qRT-PCR was employed to assess the expression pattern of GhALDH7B4 (Figure 2E). The consistent predominance of GhALDH7B4 expression in cotton fibers during 15–25 DPA, irrespective of the sample source, indicated its potential involvement in SCW formation; therefore, it likely contributes to FS development in cotton. Furthermore, the parent Ji228 and its progeny RIL229, which carry GhALDH7B4_A06O, displayed significantly higher or extremely significantly higher expression levels across diverse tissues compared with Ji567 and RIL131, which carry GhALDH7B4_A06S, indicated a possible association between gene expression levels and genotypes.
Functional analysis of GhALDH7B4 _A06 by silencing in cotton and overexpression in Arabidopsis
A VIGS experiment was performed in cotton to confirm the role of GhALDH7B4_A06 in FS formation. The findings demonstrated the persistence of whitening symptoms in various tissues, including cotton boll bracts, boll shells, and leaves, throughout boll development in the positive control group, which indicated successful silencing (Figure 3A). Notably, CLCrV : GhALDH7B4 plants exhibited a slender and fragile phenotype, although no discernible changes in external morphology were observed post-injection compared with the WT plants (Figure 3A). qRT-PCR analysis displayed a silencing efficiency of 65.16% for GhALDH7B4, with values ranging from 52.91% to 70.18% (Figure 3B). Fiber quality test results indicated a significant reduction in FS and FM in CLCrV : GhALDH7B4 cotton fiber, with reductions of 2.83cN/tex and 0.97, respectively. However, no substantial differences were observed in other fiber quality parameters compared with the negative control (Figure 3C). This VIGS experiment provided compelling evidence that supported the potential positive role of GhALDH7B4 in cotton FS development.
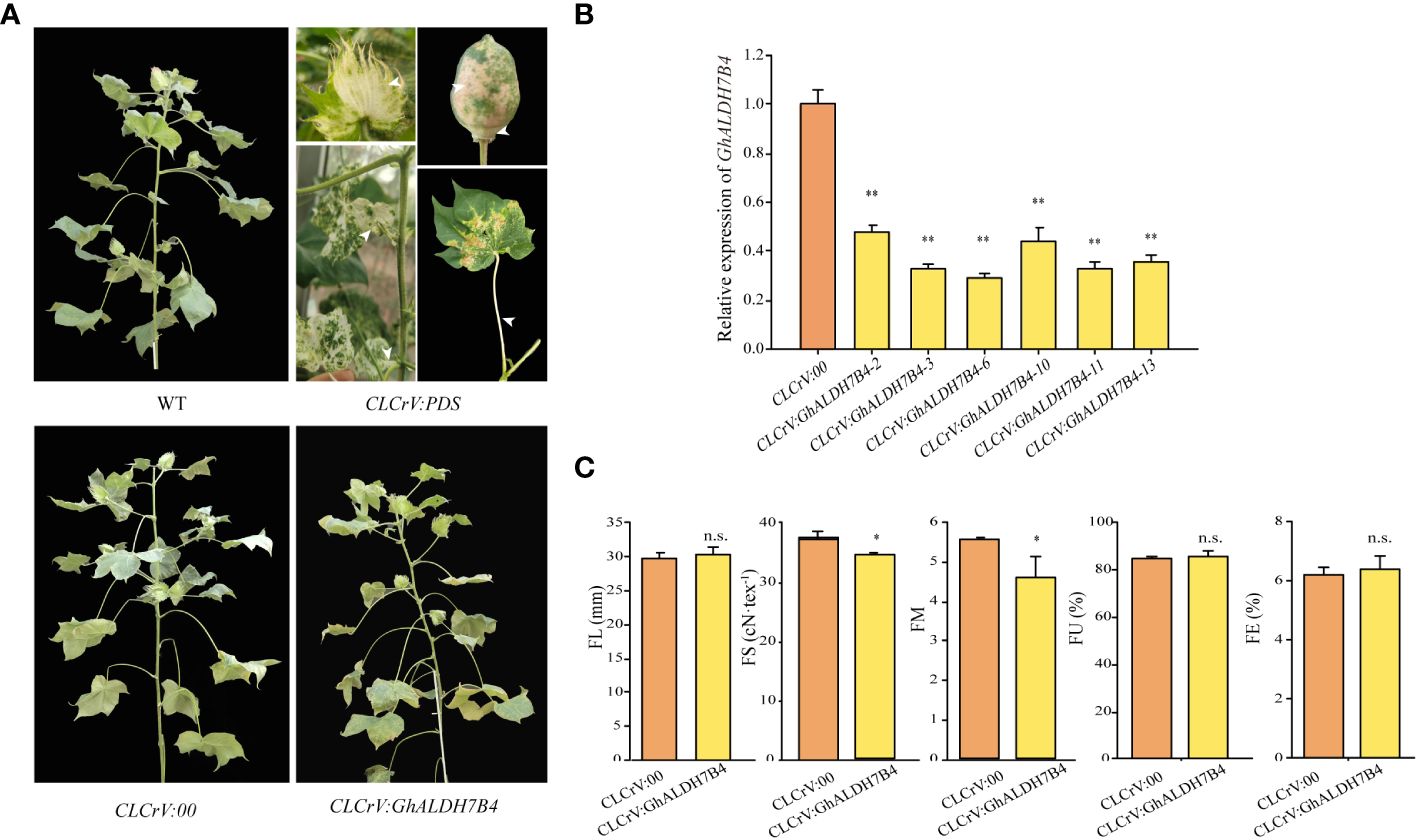
Figure 3 Phenotype, gene expression in fiber, and fiber quality traits of GhALDH7B4-silenced cotton plants. (A) Phenotypic characteristics in flowering and boll stages after the VIGS injection. (B) Gene expression analysis of GhALDH7B4 by qRT-PCR. (C) Fiber quality trait analysis. Error bars represent the SD of three replicates. * and ** indicate significant differences at p < 0.05 and p < 0.01, respectively, and n.s. indicates not significant.
For complementary functional assessment, we executed an overexpression experiment of GhALDH7B4_A06O in Arabidopsis to investigate its potential regulatory role in SCW biosynthesis. Phenotypic evaluation of the transgenic lines revealed accelerated growth rates (Figure 4A) and enhanced stem strength compared with the WT (Figure 4B). Subsequent evaluation focused on two transgenic lines (OE-3 and OE-15) characterized by elevated expression levels of GhALDH7B4_A06O (Figure 4C). Analysis of cell wall constituents indicated higher levels of cellulose, hemicellulose, and lignin in the transgenic progenies relative to the WT (Figure 4D). In particular, the most prominent disparity in cellulose content was observed in the overexpression lines compared with the WT, which demonstrated that GhALDH7B4_A06O overexpression may stimulate augmentation of compounds associated with SCW synthesis.
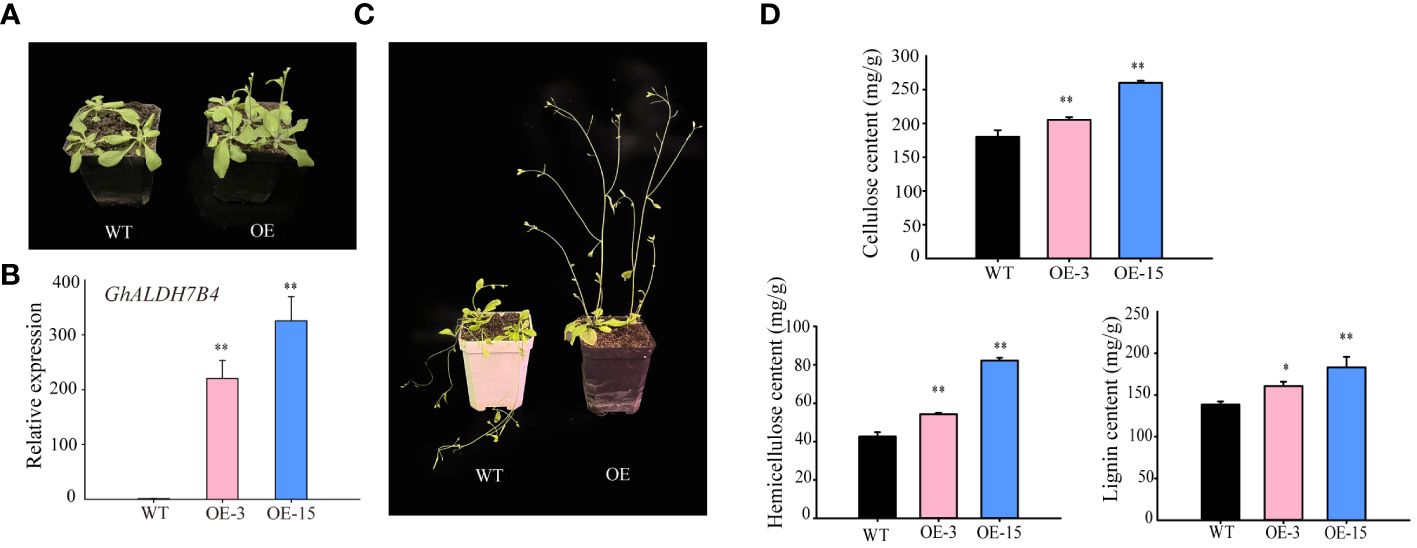
Figure 4 GhALDH7B4_A06 overexpression in Arabidopsis. (A) Phenotypic characteristics in 3-week-old Arabidopsis plants. (B) Phenotypic characteristics in 5-week-old Arabidopsis plants. (C) Gene expression analysis of GhALDH7B4 by qRT-PCR. (D) Assay of cell wall component (cellulose, hemicellulose, and lignin) contents from the stem in GhALDH7B4_A06 transgenic Arabidopsis. Cell wall component contents were determined in Arabidopsis stems older than 8 weeks of age. Error bars represent the SD of three replicates. * and ** indicate significant differences at p < 0.05 and p < 0.01, respectively. WT, wild type; OE3 and OE15, GhALDH7B4 _A06 transgenic lines.
Discussion
GhALDH7B4_A06 is a candidate gene associated with cotton FS
The combination of BSA and sequencing is effective for rapidly mapping major QTLs and is widely used in various crops (Takagi et al., 2013; Illa-Berenguer et al., 2015; Chen et al., 2021). In cotton, this approach has been applied to studies on important traits such as fiber quality, boll weight, agronomic traits, and disease resistance (Zhang et al., 2016; Cui et al., 2021; Ma et al., 2022; Jia et al., 2023; Zhang et al., 2024). In our study, an F2 population was used for SLAF-BSA-seq, and four QTLs associated with FS were mapped to three chromosomes. The large interval encompassed 242 genes. However, we focused on the small mapping region qFSA06, primarily because FS-related QTLs were detected within the linked region of chromosome A06 across the F2, F2:3, and RIL populations using SSR markers. As qFSA06 resides within this region, we hypothesize that it represents a stable major-effect QTL within these populations. Through gene cloning analysis in the parents, one gene, GhALDH7B4_A06, was identified within qFSA06, and it had a critical nonsense mutation within the CDS region. Subsequent genotyping of RILs and gene expression analysis confirmed the stable presence of this variant locus, which showed significant correlation with FS in these populations.
Liu et al. (2016) employed 6,975 F2 populations to map QTL clusters associated with FL, FS, FM, and FU on chromosome A06 of upland cotton. The QTL identified in their investigation overlapped with qFSA06 in our research, and GhALDH7B4_A06 was also considered one of candidate genes. Moreover, Liu et al. (2023) identified GhALDH7B4_A06 as a candidate gene related to SCW biosynthesis through comparative transcriptome analysis of fiber tissues between Gossypium barbadense and G. hirsutum. However, neither of those studies reported on the presence of site mutations in this gene. Our findings revealed that a nonsense mutation in GhALDH7B4_A06 determined the functional expression of the protein. On the basis of the expression of this gene reported in the above studies, we speculate that this site mutation may be prevalent in upland cotton because one parent, Ji228, is the genetic background of sea island cotton and has chromosomal segments from island cotton (Liu et al., 2009). The full-length–type protein GhALDH7B4_A06O showed 100% similarity in sequence alignment with the gene on chromosome A06 of G. barbadense (GenBank: KAB2078339.1). Such high similarity was not found in proteins from upland cotton, which indicates that the full-length GhALDH7B4_A06O likely originated from high fiber quality island cotton. In a future study, we will investigate the germplasm of upland cotton from different sources to test our hypothesis that qFSA06 represents a stable major-effect QTL. Furthermore, FS is a quantitative trait controlled by multiple genes (Zhang et al., 2017). Because of the broad mapping intervals, we will perform detailed fine mapping studies in the future to pinpoint candidate genes for the other candidate regions identified by SLAF-BSA-seq in this study.
GhALDH7B4_A06 positively regulated FS in upland cotton
FS primarily develops during the SCW thickening stage (Zang et al., 2022). On the basis of identified genes with regulatory functions during this period in cotton, Wen et al. (2023) constructed a model to enhance our understanding of the regulatory mechanisms of genes expressed during SCW development. However, there is still no comprehensive understanding of the intricate regulatory network, which necessitates further identification of FS-related genes.
GhALDH7B4 encodes an aldehyde dehydrogenase and is the sole member of the ALDH7 family in upland cotton, with one copy present in each of the At and Dt sub-genomes (Dong et al., 2017; Guo et al., 2017). ALDH7B4 has been commonly documented in plants to operate under abiotic stress conditions, including drought, abscisic acid, and salinity (Hou et al., 2022), with its role in cotton fiber development remaining largely unexplored. Our experiments involving gene silencing of GhALDH7B4_A06O in cotton and overexpression in Arabidopsis revealed that GhALDH7B4_A06O may positively regulate upland cotton FS.
A multitude of ALDHs have been identified in various crops such as A. thaliana, poplar, and maize, and it was proposed that they likely participate in SCW synthesis (Nair et al., 2004; Guillaumie et al., 2008; Bosch et al., 2011; Tian et al., 2015). It was suggested that ALDHs may have distinct roles in different stages of cell wall biosynthesis (Islam and Ghosh, 2022). However, the molecular regulatory mechanisms of ALDH genes remain unclear.
As previously mentioned, FS is a quantitative trait controlled by a complex network of multiple genes (Zhang et al., 2017). GhALDH7B4_A06 is unlikely to act independently but rather exists within this network of interactions. However, the role of GhALDH7B4_A06 in the regulatory network and its upstream substrate and downstream products remain unknown. Future investigations using transgenic lines for overexpression and gene knockout are anticipated to provide further insight into its function and metabolic pathways and to facilitate its practical application through the development of linked marker combinations.
Conclusion
This study employed forward genetics using SLAF-BSA-seq and SSR markers to locate a stable FS locus, qFSA06, and identified the candidate gene GhALDH7B4_A06. A premature stop codon within the candidate gene was associated with FS lines of RIL populations and resulted in a truncated variant, GhALDH7B4_A06S, which may lead to functional protein loss. The subsequent VIGS and overexpression experiments demonstrated that GhALDH7B4_A06 likely plays a positive regulatory role in FS of upland cotton. This discovery significantly contributes to advancing our understanding of the regulatory network associated with cotton fiber development.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
LT: Funding acquisition, Investigation, Writing – review & editing, Data curation, Software, Validation, Writing – original draft. CL: Investigation, Writing – review & editing, Formal analysis, Supervision. XL: Investigation, Writing – review & editing, Software. HW: Investigation, Writing – review & editing, Project administration. SZ: Writing – review & editing, Data curation, Software, Validation. XC: Software, Writing – review & editing, Formal analysis, Methodology, Supervision. JZ: Writing – review & editing, Funding acquisition, Investigation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Basic Research Funds of Hebei Academy of Agriculture and Forestry Sciences (HAAFS) (2021070205), HAAFS Agriculture Science and Technology Innovation Project (2022KJCXZX-MHS-1 and 2023KJCXZX-MHS-11), Hebei Key Research and Development Project (21326314D), and Hebei Agriculture Research System (HBCT2024100202).
Acknowledgments
We thank Mallory Eckstut, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the English text of a draft of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1377682/full#supplementary-material
Supplementary Figure 1 | SLAF and SNP distribution on genome.
References
Abe, A., Kosugi, S., Yoshida, K., Natsume, S., Takagi, H., Kanzaki, H., et al. (2012). Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. doi: 10.1038/nbt.2095
Betancur, L., Singh, B., Rapp, R. A., Wendel, J. F., Marks, M. D., Roberts, A. W., et al. (2010). Phylogenetically distinct cellulose synthase genes support secondary wall thickening in arabidopsis shoot trichomes and cotton fiber. J. Integr. Plant Biol. 52, 205–220. doi: 10.1111/j.1744-7909.2010.00934.x
Bosch, M., Mayer, C.-D., Cookson, A., Donnison, I. S. (2011). Identification of genes involved in cell wall biogenesis in grasses by differential gene expression profiling of elongating and non-elongating maize internodes. J. Exp. Bot. 62, 3545–3561. doi: 10.1093/jxb/err045
Cao, J., Zhao, B., Huang, C.-C., Chen, Z., Zhao, T., Liu, H.-R., et al. (2020). The miR319-targeted GhTCP4 promotes the transition from cell elongation to wall thickening in cotton fiber. Mol. Plant 13, 1063–1077. doi: 10.1016/j.molp.2020.05.006
Chen, Y., Yang, X., He, K., Liu, M., Li, J., Gao, Z., et al. (2006). The MYB transcription factor superfamily of arabidopsis: expression analysis and phylogenetic comparison with the rice MYB Family. Plant Mol. Biol. 60, 107–124. doi: 10.1007/s11103-005-2910-y
Chen, Z., Tang, D., Hu, K., Zhang, L., Yin, Y., Ni, J., et al. (2021). Combining QTL-seq and linkage mapping to uncover the genetic basis of single vs. paired spikelets in the advanced populations of two-ranked maize×teosinte. BMC Plant Biol. 21, 572. doi: 10.1186/s12870-021-03353-3
Cui, Y., Ge, Q., Zhao, P., Chen, W., Sang, X., Zhao, Y., et al. (2021). Rapid mining of candidate genes for verticillium wilt resistance in cotton based on BSA-seq analysis. Front. Plant Sci. 12, 26829. doi: 10.3389/fpls.2021.703011
Deng, T., Yao, H., Wang, J., Wang, J., Xue, H., Zuo, K. (2016). GhLTPG1, a cotton GPI-anchored lipid transfer protein, regulates the transport of phosphatidylinositol monophosphates and cotton fiber elongation. Sci. Rep. 6, 26829. doi: 10.1038/srep26829
Deza, M. M., Deza, E. (2009). Encyclopedia of Distances (Berlin: Heidelberg), 1–583. doi: 10.1007/978-3-642-00234-2_1
Dong, Y., Liu, H., Zhang, Y., Hu, J., Feng, J., Li, C., et al. (2017). Comparative genomic study of ALDH gene superfamily in Gossypium: A focus on Gossypium hirsutum under salt stress. PloS One 12, e0176733–e0176733. doi: 10.1371/journal.pone.0176733
Fang, X., Liu, X., Wang, X., Wang, W., Liu, D., Zhang, J., et al. (2017). Fine-mapping qFS07.1 controlling fiber strength in upland cotton (Gossypium hirsutum L.). Theor. Appl. Genet. 130, 795–806. doi: 10.1007/s00122-017-2852-1
Feng, L., Zhou, C., Su, Q., Xu, M., Yue, H., Zhang, S., et al. (2020). Fine-mapping and candidate gene analysis of qFS-Chr.D02, a QTL for fibre strength introgressed from a semi-wild cotton into Gossypium hirsutum. Plant Sci. 297, 110524–110524. doi: 10.1016/j.plantsci.2020.110524
Gao, Y., Cui, Y., Li, S., Feng, M., Liu, L., Xu, W., et al. (2023). GhERF41, a group IIIe ERF protein, participates in regulating cell wall synthesis in cotton fiber. Ind. Crop Prod. 192, 116001–116001. doi: 10.1016/j.indcrop.2022.116001
Ge, X., Xu, J., Yang, Z., Yang, X., Wang, Y., Chen, Y., et al. (2023). Efficient genotype-independent cotton genetic transformation and genome editing. J. Integr. Plant Biol. 65, 907–917. doi: 10.1111/jipb.13427
Gu, Z., Huang, C., Li, F., Zhou, X. (2014). A versatile system for functional analysis of genes and microRNAs in cotton. Plant Biotechnol. J. 12, 638–649. doi: 10.1111/pbi.12169
Guillaumie, S., Goffner, D., Barbier, O., Martinant, J.-P., Pichon, M., Yves Barrière (2008). Expression of cell wall related genes in basal and ear internodes of silking brown-midrib-3, caffeic acid O-methyltransferase (COMT) down-regulated, and normal maize plants. BMC Plant Biol. 8, 71. doi: 10.1186/1471-2229-8-71
Guo, Y., Chen, F., Luo, J., Qiao, M., Zeng, W., Li, J., et al. (2022). The DUF288 domain containing proteins GhSTLs participate in cotton fiber cellulose synthesis and impact on fiber elongation. Plant Sci. 316, 111168–111168. doi: 10.1016/j.plantsci.2021.111168
Guo, X., Wang, Y., Lu, H., Cai, X., Wang, X., Zhou, Z., et al. (2017). Genome-wide characterization and expression analysis of the aldehyde dehydrogenase (ALDH) gene superfamily under abiotic stresses in cotton. Gene 628, 230–245. doi: 10.1016/j.gene.2017.07.034
Haigler, C. H., Betancur, L., Stiff, M. R., Tuttle, J. R. (2012). Cotton fiber: a powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 3. doi: 10.3389/fpls.2012.00104
He, S., Sun, G., Geng, X., Gong, W., Dai, P., Jia, Y., et al. (2021). The genomic basis of geographic differentiation and fiber improvement in cultivated cotton. Nat. Genet. 53, 916–924. doi: 10.1038/s41588-021-00844-9
Hou, Q., Zhang, T., Zhao, W., Wang, L., Lu, L., Qi, Y., et al. (2022). Genetic background and cis-organization regulate ALDH7B4 gene expression in Eutrema salsugineum: a promoter analysis case study. Planta 255, 52. doi: 10.1007/s00425-022-03836-z
Hu, Y., Chen, J., Fang, L., Zhang, Z., Ma, W., Niu, Y., et al. (2019). Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 51, 739–748. doi: 10.1038/s41588-019-0371-5
Huang, J., Chen, F., Wu, S., Li, J., Xu, W. (2016). Cotton GhMYB7 is predominantly expressed in developing fibers and regulates secondary cell wall biosynthesis in transgenic Arabidopsis. Sci. China Life Sci. 59, 194–205. doi: 10.1007/s11427-015-4991-4
Huang, J., Guo, Y., Sun, Q., Zeng, W., Li, J., Li, X., et al. (2018). Genome-wide identification of R2R3-MYB transcription factors regulating secondary cell wall thickening in cotton fiber development. Plant Cell Physiol. 60, 687–701. doi: 10.1093/pcp/pcy238
Illa-Berenguer, E., Van Houten, J., Huang, Z., van der Knaap, E. (2015). Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor. Appl. Genet. 128, 1329–1342. doi: 10.1007/s00122-015-2509-x
Islam, M., Ghosh, A. (2022). Evolution, family expansion, and functional diversification of plant aldehyde dehydrogenases. Gene 829, 146522. doi: 10.1016/j.gene.2022.146522
Islam, S., Zeng, L., Thyssen, G. N., Delhom, C. D., Hee, J. K., Li, P., et al. (2016). Mapping by sequencing in cotton (Gossypium hirsutum) line MD52ne identified candidate genes for fiber strength and its related quality attributes. Theor. Appl. Genet. 129, 1071–1086. doi: 10.1007/s00122-016-2684-4
Jia, X., Wang, S., Zhao, H., Zhu, J., Li, M., Wang, G. (2023). QTL mapping and BSA-seq map a major QTL for the node of the first fruiting branch in cotton. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1113059
Keerio, A., Shen, C., Nie, Y., Ahmed, M., Zhang, X., Lin, Z. (2018). QTL mapping for fiber quality and yield traits based on introgression lines derived from Gossypium hirsutum × G. tomentosum. Int. J. Mol. Sci. 19, 243. doi: 10.3390/ijms19010243
Kumar, M., Turner, S. (2015). Plant cellulose synthesis: CESA proteins crossing kingdoms. Phytochemistry 112, 91–99. doi: 10.1016/j.phytochem.2014.07.009
Li, F., Fan, G., Lu, C., Xiao, G., Zou, C., Kohel, R. J., et al. (2015). Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530. doi: 10.1038/nbt.3208
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, X., Jin, X., Wang, H., Zhang, X., Zhang, L. (2016). Structure, evolution, and comparative genomics of tetraploid cotton based on a high-density genetic linkage map. DNA Res. 23, 283–293. doi: 10.1093/dnares/dsw016
Li, Y., Wang, N., Wang, Y., Liu, D., Tian, J., Li, L., et al. (2018). The cotton XLIM protein (GhXLIM6) is required for fiber development via maintaining dynamic F-actin cytoskeleton and modulating cellulose biosynthesis. Plant J. 96, 1269–1282. doi: 10.1111/tpj.14108
Liu, S., Geng, J., Ciu, R., Wang, Z., Liu, C., Guo, B., et al. (2009). Comprehensive analysis of traits for high quality and bollworm resistance new cotton variety Ji228. Chin. Agric. Sci. Bull. 25, 220–223.
Liu, G., Ji, L., Pei, W., Li, X., Wang, N., Ma, J., et al. (2019). Analysis of the MIR160 gene family and the role of MIR160a_A05 in regulating fiber length in cotton. Planta 250, 2147–2158. doi: 10.1007/s00425-019-03271-7
Liu, Z., Sun, Z., Ke, H., Chen, B., Gu, Q., Zhang, M., et al. (2023). Transcriptome, ectopic expression and genetic population analysis identify candidate genes for fiber quality improvement in cotton. Int. J. Mol. Sci. 24, 8293–8293. doi: 10.3390/ijms24098293
Liu, D., Zhang, J., Liu, X., Wang, W., Liu, D., Teng, Z., et al. (2016). Fine mapping and RNA-Seq unravels candidate genes for a major QTL controlling multiple fiber quality traits at the T1 region in upland cotton. BMC Genomics 17, 295. doi: 10.1186/s12864-016-2605-6
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Ma, Z., He, S., Wang, X., Sun, J., Zhang, Y., Zhang, G., et al. (2018). Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 50, 803–813. doi: 10.1038/s41588-018-0119-7
Ma, C., Rehman, A., Li, H. G, Zhao, Z. B, Sun, G., Du, X. M. (2022). Mapping of dwarfing QTL of Ari1327, a semi-dwarf mutant of upland cotton. BMC Plant Biol. 22, 5. doi: 10.1186/s12870-021-03359-x
Ma, Z., Zhang, Y., Wu, L., Zhang, G., Sun, Z., Li, Z., et al. (2021). High-quality genome assembly and resequencing of modern cotton cultivars provide resources for crop improvement. Nat. Genet. 53, 1385–1391. doi: 10.1038/s41588-021-00910-2
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Nair, R. B., Bastress, K. L., Ruegger, M. O., Denault, J. W., Chapple, C. (2004). The Arabidopsis thaliana REDUCED EPIDERMAL FLUORESCENCE1 gene encodes an aldehyde dehydrogenase involved in ferulic acid and sinapic acid biosynthesis. Plant Cell 16, 544–554. doi: 10.1105/tpc.017509
Ning, Z., Chen, H., Mei, H., Zhang, T. (2014). Molecular tagging of QTLs for fiber quality and yield in the upland cotton cultivar Acala-Prema. Euphytica 195, 143–156. doi: 10.1007/s10681-013-0990-3
Qin, X., Huang, S., Liu, Y., Bian, M., Shi, W., Zuo, Z., et al. (2017). Overexpression of A RING finger ubiquitin ligase gene AtATRF1 enhances aluminium tolerance in Arabidopsis thaliana. J. Plant Biol. 60, 66–74. doi: 10.1007/s12374-016-0903-9
Sun, H., Hu, M., Li, J., Chen, L., Li, M., Zhang, S., et al. (2018). Comprehensive analysis of NAC transcription factors uncovers their roles during fiber development and stress response in cotton. BMC Plant Biol. 18, 150. doi: 10.1186/s12870-018-1367-5
Sun, Q., Huang, J., Guo, Y., Yang, M., Guo, Y., Li, J., et al. (2020). A cotton NAC domain transcription factor, GhFSN5, negatively regulates secondary cell wall biosynthesis and anther development in transgenic Arabidopsis. Plant Physiol. Bioch. 146, 303–314. doi: 10.1016/j.plaphy.2019.11.030
Sun, X., Liu, D., Zhang, X., Li, W., Liu, H., Hong, W., et al. (2013). SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PloS One 8, e58700. doi: 10.1371/journal.pone.0058700
Sun, Z., Wang, X., Liu, Z., Gu, Q., Zhang, Y., Li, Z., et al. (2017). Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 15, 982–996. doi: 10.1111/pbi.12693
Takagi, H., Abe, A., Yoshida, K., Kosugi, S., Natsume, S., Mitsuoka, C., et al. (2013). QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. doi: 10.1111/tpj.12105
Tang, L., Cai, X., Wang, H., Li, X., Zhang, S., Liu, C., et al. (2023). Genome-wide identification of cotton FLA gene family and functional analysis of GhFLA05 in cotton fiber development. Scientia Agricultura Sin. 56, 4602–4620. doi: 10.3864/j.issn.0578-1752.2023.23.004
Tian, F., Zang, J.-L., Wang, T., Xie, Y., Zhang, J., Hu, J. (2015). Aldehyde dehydrogenase gene superfamily in Populus: organization and expression divergence between paralogous gene pairs. PloS One 10, e0124669–e0124669. doi: 10.1371/journal.pone.0124669
Tian, Z., Zhang, Y., Zhu, L., Jiang, B., Wang, H., Gao, R., et al. (2022). Strigolactones act downstream of gibberellins to regulate fiber cell elongation and cell wall thickness in cotton (Gossypium hirsutum). Plant Cell 34, 4816–4839. doi: 10.1093/plcell/koac270
Wang, M., Tu, L., Yuan, D., Zhu, D., Shen, C., Li, J., et al. (2019). Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 51, 224–229. doi: 10.1038/s41588-018-0282-x
Wang, H., Zhang, R., Shen, C., Li, X., Zhu, D., Lin, Z. (2020). Transcriptome and QTL analyses reveal candidate genes for fiber quality in Upland cotton. Crop J. 8, 98–106. doi: 10.1016/j.cj.2019.05.002
Wen, X., Chen, Z., Yang, Z., Wang, M., Jin, S., Wang, G., et al. (2023). A comprehensive overview of cotton genomics, biotechnology and molecular biological studies. Sci. China Life Sci. 66 (10), 2214–2256. doi: 10.1007/s11427-022-2278-0
Wen, Y., He, P., Bai, X., Zhang, H., Zhang, Y., Yu, J. (2023). Strigolactones modulate cotton fiber elongation and secondary cell wall thickening. J. Integr. Agr. doi: 10.1016/j.jia.2023.07.009
Xu, F., Chen, Q., Huang, L., Luo, M. (2021). Advances about the roles of membranes in cotton fiber development. Membranes 11, 471. doi: 10.3390/membranes11070471
Yang, Z., Gao, C., Zhang, Y., Yan, Q., Hu, W., Yang, L., et al. (2022). Recent progression and future perspectives in cotton genomic breeding. J. Integr. Plant Biol. 65, 548–569. doi: 10.1111/jipb.13388
Yu, J., Jung, S., Cheng, C.-H., Lee, T., Zheng, P., Buble, K., et al. (2021). CottonGen: the community database for cotton genomics, genetics, and breeding research. Plants 10, 2805. doi: 10.3390/plants10122805
Yuan, Y., Zhang, H., Wang, L., Xing, H., Mao, L., Tao, J., et al. (2019). Candidate quantitative trait loci and genes for fiber quality in Gossypium hirsutum L. detected using single- and multi-locus association mapping. Ind. Crop Prod. 134, 356–369. doi: 10.1016/j.indcrop.2019.04.010
Zang, Y., Hu, Y., Dai, F., Zhang, T. (2022). Comparative transcriptome analysis reveals the regulation network for fiber strength in cotton. Biotechnol. Lett. 44, 547–560. doi: 10.1007/s10529-022-03236-z
Zang, Y., Hu, Y., Xu, C., Wu, S.-J., Wang, Y., Ning, Z., et al. (2021). GhUBX controlling helical growth results in production of stronger cotton fiber. iScience 24, 102930–102930. doi: 10.1016/j.isci.2021.102930
Zhang, Z., Ge, Q., Liu, A., Li, J., Gong, J., Shang, H., et al. (2017). Construction of a high-density genetic map and its application to QTL identification for fiber strength in upland cotton. Crop Sci. 57, 774–788. doi: 10.2135/cropsci2016.06.0544
Zhang, T., Hu, Y., Jiang, W., Fang, L., Guan, X., Chen, J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. doi: 10.1038/nbt.3207
Zhang, S., Jiang, Z., Chen, J., Han, Z., Chi, J., Li, X., et al. (2021). The cellulose synthase (CesA) gene family in four Gossypium species: phylogenetics, sequence variation and gene expression in relation to fiber quality in Upland cotton. Mol. Genet. Genomics 296, 355–368. doi: 10.1007/s00438-020-01758-7
Zhang, Z., Shang, H., Yuzhen, S., Huang, L., Li, J., Ge, Q., et al. (2016). Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to quantitative trait loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum.). BMC Plant Biol. 16, 79. doi: 10.1186/s12870-016-0741-4
Zhang, S., Wang, H., Li, X., Tang, L., Cai, X., Liu, C., et al. (2024). Aspartyl proteases identified as candidate genes of a fiber length QTL, qFLD05, that regulates fiber length in cotton (Gossypium hirsutum L.). Theor. Appl. Genet. 137, 159. doi: 10.1007/s00122-024-04559-3
Keywords: fiber strength, bulk segregant analysis (BSA), quantitative trait locus (QTL), GhALDH7B4_A06, upland cotton (Gossypium hirsutum L.)
Citation: Tang L, Liu C, Li X, Wang H, Zhang S, Cai X and Zhang J (2024) An aldehyde dehydrogenase gene, GhALDH7B4_A06, positively regulates fiber strength in upland cotton (Gossypium hirsutum L.). Front. Plant Sci. 15:1377682. doi: 10.3389/fpls.2024.1377682
Received: 28 January 2024; Accepted: 09 April 2024;
Published: 26 April 2024.
Edited by:
Baohua Wang, Nantong University, ChinaReviewed by:
Dinesh Kumar Saini, Texas Tech University, United StatesXinlian Shen, Jiangsu Academy of Agricultural Sciences (JAAS), China
Wuwei Ye, Chinese Academy of Agricultural Sciences, China
Copyright © 2024 Tang, Liu, Li, Wang, Zhang, Cai and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianhong Zhang, bWhzempoQDEyNi5jb20=