 Lin Jia
Lin Jia Ning Xu
Ning Xu Bin Xia2
Bin Xia2 Qingran Meng
Qingran Meng Qiang Li
Qiang Li- 1School of Forestry, Northeast Forestry University, Harbin, China
- 2College of Landscape Architecture, Northeast Forestry University, Harbin, China
- 3School of Ecological Technology and Engineering, Shanghai Institute of Technology, Shanghai, China
- 4School of Perfume and Aroma Technology, Shanghai Institute of Technology, Shanghai, China
- 5Heilongjiang Academy of Forestry, Harbin, China
Background: Thymus mandschuricus is an aromatic and medicinal plant with notable antibacterial and antioxidant properties. However, traditional breeding methods rely on phenotypic selection due to a lack of molecular resources. A high-quality reference genome is crucial for marker-assisted breeding, genome editing, and molecular genetics.
Results: We utilized PacBio and Hi-C technologies to generate a high-quality chromosome-level reference genome for T. mandschuricus, with a size of 587.05 Mb and an N50 contig size of 8.41 Mb. The assembled genome contained 29,343 predicted protein-coding genes, and evidence of two distinct whole-genome duplications in T. mandschuricus was discovered. Comparative genomic analysis revealed rapid evolution of genes involved in phenylpropanoid biosynthesis and the CYP450 gene family in T. mandschuricus. Additionally, we reconstructed the gene families of terpenoid biosynthesis structural genes, such as TPS, BAHD, and CYP, and identified regulatory networks controlling the expression of aroma-synthesis genes by integrating transcriptome data from various organs and developmental stages. We discovered that hormones and transcription factors may collaborate in controlling aroma-synthesis gene expression.
Conclusion: This study provides the first high-quality genome sequence and gene annotation for T. mandschuricus, an indigenous thyme species unique to China. The genome assembly and the comprehension of the genetic basis of fragrance synthesis acquired from this research could potentially serve as targets for future breeding programs and functional studies.
Background
The family Labiatae is among the biggest in flowering plants, containing 236 genera and over 7,000 species (Raja, 2012). With its attractive appearance, special taste, strong scent, and therapeutic capability, Labiatae has significant ecological, economic, and cultural importance due to rich phytochemical compositions (Mulas, 2006). Because of these desirable qualities, it is extensively cultivated worldwide. T. mandschuricus, a member of the Lamiaceae family, is widely used as a spice and a traditional Chinese herb (Saroukolai et al., 2010; Kim et al., 2003; Qiao et al., 2021; Koul et al., 2008). It is often found in the northern part of China and can be applied in many occasions: traditional Chinese medicine, household cleaning, environmental safeguarding, and even in the preparation of mutton dishes. The extract of T. mandschuricus has been shown to have anti-cancer, anti-bacterial, and anti-inflammatory properties (Afonso et al., 2020; Gordo et al., 2012).
Natural antifungal agents from plant essential oils and their active components are a hot area. T. mandschuricus has strong floral and leaf aromas from which aromatic oils were extracted in industry and usually used for medical purposes (Wang et al., 2018). Previous studies have shown that thymol and carvacrol are the main components of essential oils in T. mandschuricus and have good antifungal activity (Hosseinzadeh et al., 2015). Bioactive components such as thymol, carvacrol, citral, geraniol, and nerolidol are responsible for the therapeutic benefits of T. mandschuricus as antibacterial and antioxidant agent (Nieto, 2020; Tohidi et al., 2017; Martin et al., 2003). Most of these substances belong to the terpenoid family. Examining terpene synthases provides an excellent opportunity to gain insights into the evolutionary aspects of terpenoid biosynthesis. Building blocks for terpenoids in plants come from either the cytosolic mevalonate (MVA) or plastidial methylerythritol phosphate (MEP) pathways (Tholl, 2006; Dudareva and Pichersky, 2008; Chen et al., 2020) and include isopentenyl diphosphate (C-5) and isomeric dimethylallyl diphosphate. The enzymes cytochrome P450 (CYP450), acyltransferases, 2-oxoglutarate-dependent dioxygenases, methyltransferases, and glycosyltransferases change and further diversify scaffolds, making terpenoids structurally varied.
It seems that the evolution of plant gene clusters, which resulted in the diversity of specialized terpenoids, was driven by gene duplication and neofunctionalization (Godden et al., 2019; Panchy et al., 2016). Gene duplication is common in plant genomes, with the most common mechanisms being whole-genome duplication (WGD) and tandem duplication (Chen et al., 2011). A number of plant families, including TPS (trehalose phosphate phosphatase), CYP450, and BAHD (BAHD family of acyltransferases), expand their gene repertoires through both mechanisms (Nelson and Werck-Reichhart, 2011; Weitzel and Simonsen, 2015; Stracke et al., 2020). These repetitions give useful material that may assist in the evolution of various activities, including pest resistance and stress tolerance, all of which improve plant adaptation (Xu et al., 2021; Hansen et al., 2017). There may be less chance of one-way gene loss due to recombination events and inadequate cluster expression if genes involved in terpenoid biosynthesis are clustered together. The diversification of monoterpenes and sesquiterpenes is intimately linked to the growth of the terpenoid biosynthetic gene family. Increases in the size of the TPS-b subfamily are positively connected with increases in the variety of monoterpenes found in plants (Jones et al., 2011), whereas the TPS-a subfamily is responsible for the synthesis of sesquiterpenes (Kejnovsky et al., 2009).
Using PacBio sequencing and chromatin conformation capture technologies, we have determined the reference genome sequence of T. mandschuricus. Mechanisms for polyphenol and terpenoid production in T. mandschuricus have been uncovered using comparative genomic, phylogenetic, and transcriptomic analyses as well as genome-scale sequencing data processing. This species is presumed to be haploid with a chromosome number of n = 13 (Sun et al., 2022). This study offers new insights into molecular breeding and the identification of functional genes associated with key traits of thyme. Moreover, the T. mandschuricus genome presented herein serves as a valuable resource for future investigations.
Results
Chromosome-level genome assembly and annotation of T. mandschuricus
Utilizing PacBio and Illumina HiSeq sequencing technologies, we established the complete genome sequence of T. mandschuricus. PacBio third-generation sequencing yielded 23 Gb (43.50×) of clean data, whereas sequences from Illumina sequencing were 117.26× in depth. Sequence reads were collected, trimmed, removed of contaminated data, and finally assembled. The assembled T. mandschuricus genome spans 587.05 Mb and contains a contig N50 of 8.41 Mb, which was not far from the k-mer–based estimate. The mounting rates of T. mandschuricus scaffold assembly on the 13 chromosomes using HiC assembly were all more than 90% (Figure 1A; Supplmentary Table S1). Short reads aligned to the reference genome was at a rate of above 98%. With an examination of 303 conserved core genes, BUSCO demonstrated that the T. mandschuricus genome was more than 96.8% complete; with an evaluation of 458 conserved core genes of eukaryotes, CEGMA demonstrated that the T. mandschuricus genome was more than 95.56% complete. These results indicate that our genome assembly is of high quality. The genome guanine and cytosine (GC) ratio, GC skew, unknown bases (N), and gene density for each chromosome were also calculated, and the findings showed that various chromosomes had significantly diverged in some cases (Figure 1A). We predicted 29,343 protein-coding genes, 4,474 microRNAs, 2,198 rRNAs, 856 transfer RNAs (tRNAs), and 2,106 small nuclear ribonucleic acid (snRNAs) in the assembled genome. According to the results of comprehensive functional annotation of all genes, 28,205 out of 29,343 have established roles in at least one database.
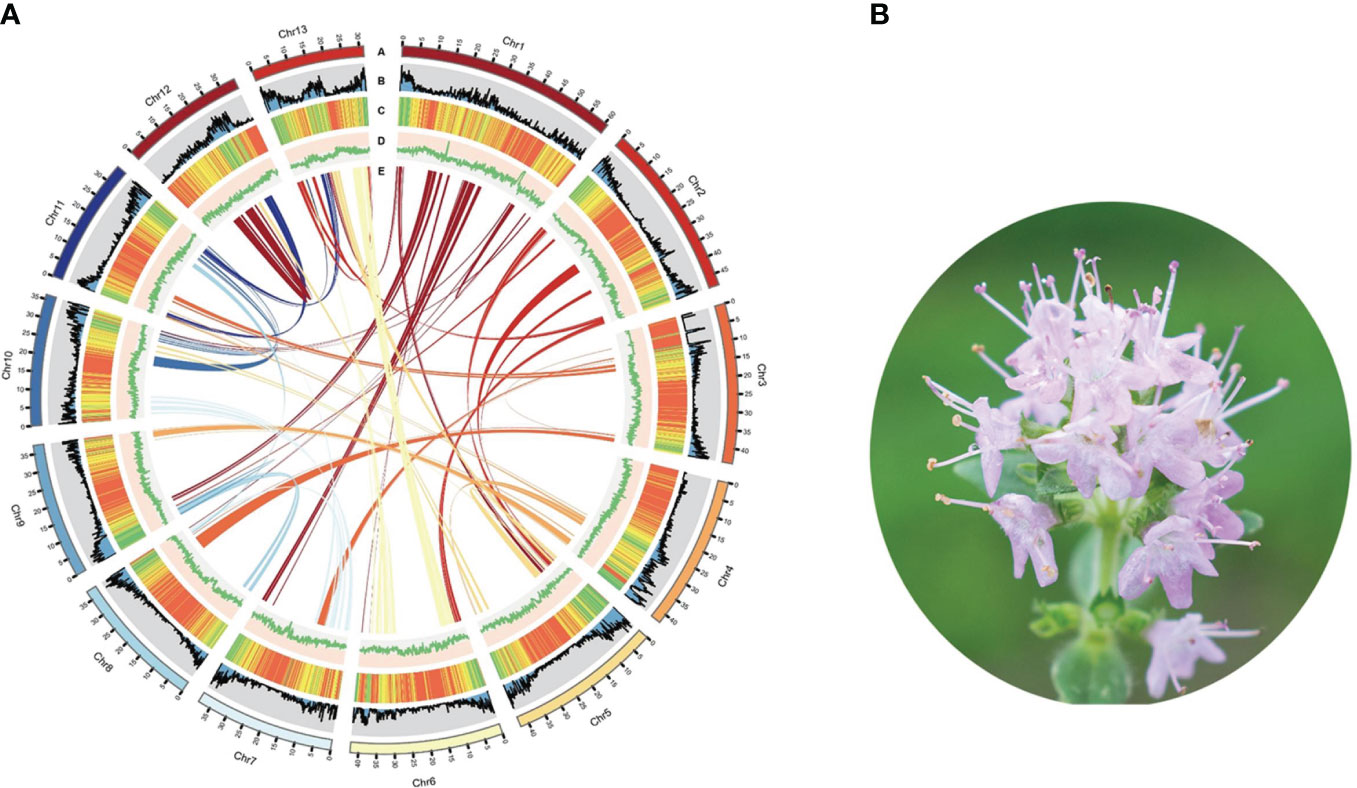
Figure 1 Summary of T. mandschuricus genome assembly. (A) Circos plot showing T. mandschuricus genomic features: A, karyotyping results; B, protein-coding gene density; C, transposable element density; D, GC content; and E schematic presentation of major interchromosomal relationships in the T. mandschuricus genome. (B) Photo of T. mandschuricus in blooming period (FS).
Furthermore, we employed structure prediction and de novo prediction to build a database of the genome’s repeat sequences. According to the findings, 637,535 repeat sequences account for 67.70% of the whole genome in T. mandschuricus. The three most prevalent forms of repetition in the genome are trithoracic repeat elements (TRF), long terminal repeats (LTR), and long interspersed nuclear elements (LINE). The distribution of major repeats was analyzed throughout the genome, and the results showed that they dispersed differently depending on chromosome and location (Figure 1A). In addition, we compared the T. mandschuricus genome with that of Salvia splendens to look for collinear regions (Supplementary Figure S1). By comparison, 4,495, 30, and 2,949 loci were found for chromosomes 1, 2, and 6, respectively. T. mandschuricus and S. splendens share many genetic similarities. However, the fact that orthologous sites in T. mandschuricus are dispersed over many chromosomes of S. splendens suggests that structural variation and replication timing have been distinct between the two species (Figure 1B). More gene cluster duplications and possibly entire genome duplications may have occurred in S. splendens.
The phylogeny and evolution of T. mandschuricus
We were able to compare the de novo–assembled T. mandschuricus genome with those of the 10 previously described species by employing protein-coding genes from all of these plants. To investigate the evolutionary history of T. mandschuricus, we analyzed the genomes of 11 selected angiosperm species. There were many variations in the number of copies of gene families between species, and a total of 69 single-copy orthologous genes were found in eleven different types of organisms (Figure 2A). Our findings suggest that T. mandschuricus, along with other members of the family Lamiaceae—Salvia splendens, Salvia bowleyana, and Salvia miltiorrhiza—all belong to the same clade. The estimated divergence time showed that T. mandschuricus, Salvia splendens, Salvia bowleyana, and Salvia miltiorrhiza may have gradually diverged from their most recent common ancestor (MRCA) around the same period at 53.21 million years ago (Mya) (Figure 2A). We examined the reduction and growth of 11 gene families in T. mandschuricus to better understand how this species adapts to the environment. A total of 6,635 genes were lost in T. mandschuricus, whereas 6,083 shrank in T. mandschuricus (from 15 gene families). Fast evolution occurred in 600 of these gene families in T. mandschuricus (Figure 2A). Analysis of the expanded gene families in T. mandschuricus revealed functional similarities to the phenylpropanoid biosynthesis, diterpenoid biosynthesis, MAPK:Mitogen-Activated Protein Kinase (MAPK) signaling pathway, ribosome biogenesis, and other pathways (Figure 2B). While this went on, T. mandschuricus may have experienced two distinct WGDs: one around 69.5 Mya and the other at 3.49 Mya (Figures 2A, C). There was a massive increase in the number of genes in the T. mandschuricus family, where OG0002816 was shown to be involved in phenylpropanoid biosynthesis (Figure 2D). One of the genetic processes of T. mandschuricus to create distinctive fragrances may be the metabolism of phenylpropanoid and diterpenoid, which is linked to the formation of polyphenols, terpenoids, and other aromas. Furthermore, PFAM clustering of rapid evolving genes in T. mandschuricus revealed that they are mostly associated with cytochrome P450, MULE transposase domain, and other activities (Figure 2E); cytochrome P450 genes (CYP) may be involved in the manufacture of terpenoids in T. mandschuricus.
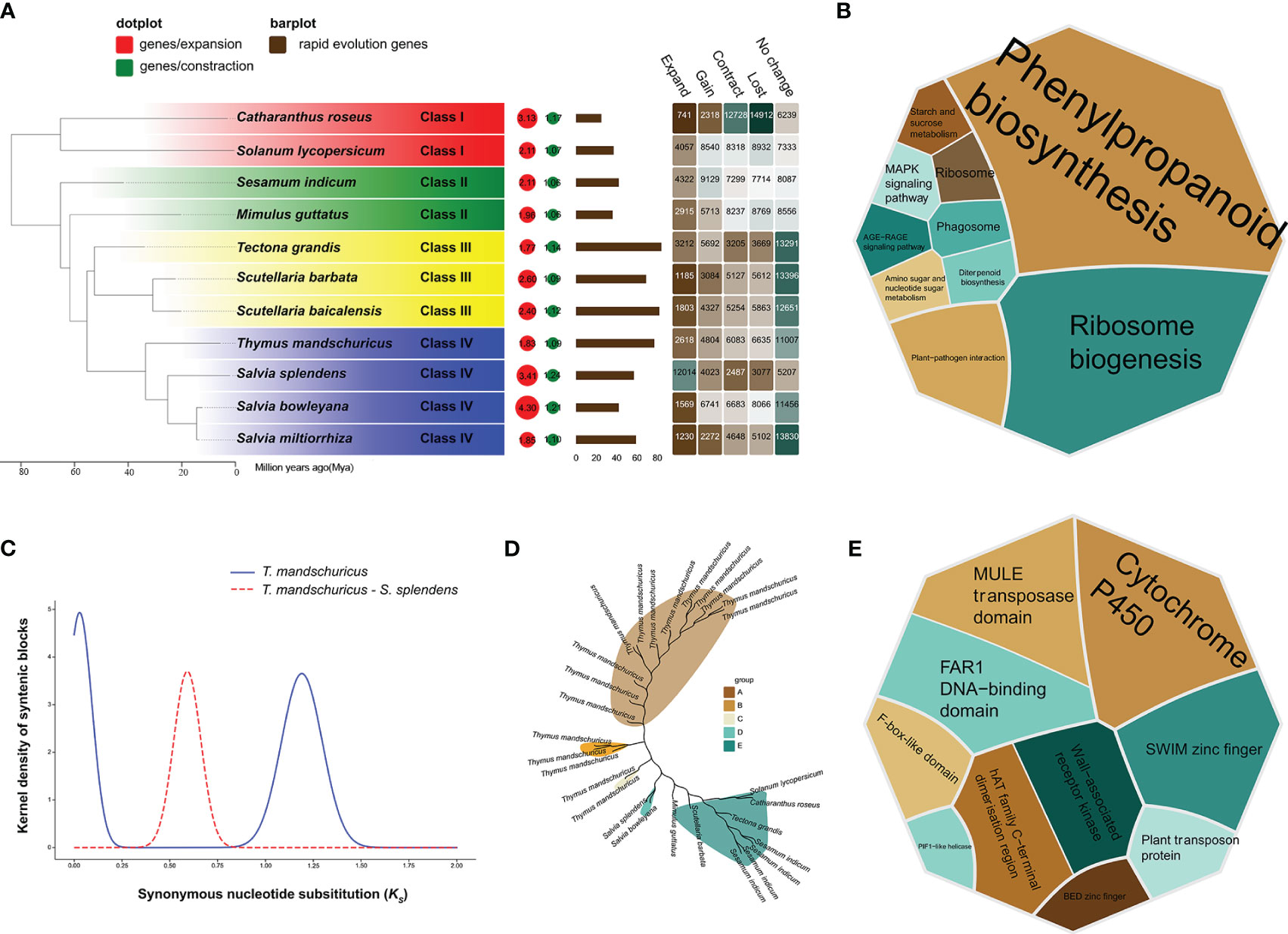
Figure 2 Phylogenetic study of T. mandschuricus with 10 distinct species, including Salvia splendens, Salvia bowleyana, Salvia miltiorrhiza, Mimulus guttatus, Sesamum indicum, Catharanthus roseus, Solanum lycopersicum, Tectona grandis, Scutellaria barbata, and Scutellaria baicalensis. (A) A phylogenetic tree of 11 different species based on single-copy genes. Bar plot indicates the number of genes in each species that have undergone fast evolution, and heatmap reflects the number of genes that have undergone expansion, contraction, loss, or gain in each species, respectively. Circles symbolize recent whole-genome duplication (WGD) occurrences. (B) A KEGG functional enrichment study of genes expanded gene family in T. mandschuricus. (C) An analysis of WGD events that have taken place in T. mandschuricus and other species. (D) Expansion of the gene family OG0002816 related to phenylpropanoid metabolism in T. mandschuricus. (E) PFAM clustering of genes in T. mandschuricus that have undergone fast evolutionary change.
Pathway and structural gene reconstruction of polyphenol synthesis
Thymol, a phenolic chemical, significantly contributes to the distinctive odor of T. mandschuricus. Previous studies have demonstrated that thyme possesses a wide array of phenolic and monoterpenoid compounds, owing to its rich volatile component content. Analysis of the genomes of T. mandschuricus and 10 other species revealed a notable expansion in the number of gene families associated with phenylpropanoid production. The shikimate pathway generates shikimic acid from the combination of phosphoenolpyruvate produced during glycolysis and erythrose-4-phosphate generated during the pentose phosphate cycle. L-phenylalanine, along with other aromatic amino acids, is created by the second route. We have identified an l-phenylalanine-dependent polyphenol production pathway (Figure 3). Structural genes such as PAL (phenylalanine ammonia lyase), C4H (cinnamate-4-hydroxylase), 4CL (coumarate CoA ligase), STS (stilbene synthase), CHS (chalcone synthase), CHI (chalcone isomerase), FNS (flavone synthase), F3H (flavanone 3-hydroxylase), and F3′H (BnF3′H-1) were identified (Figure 3).
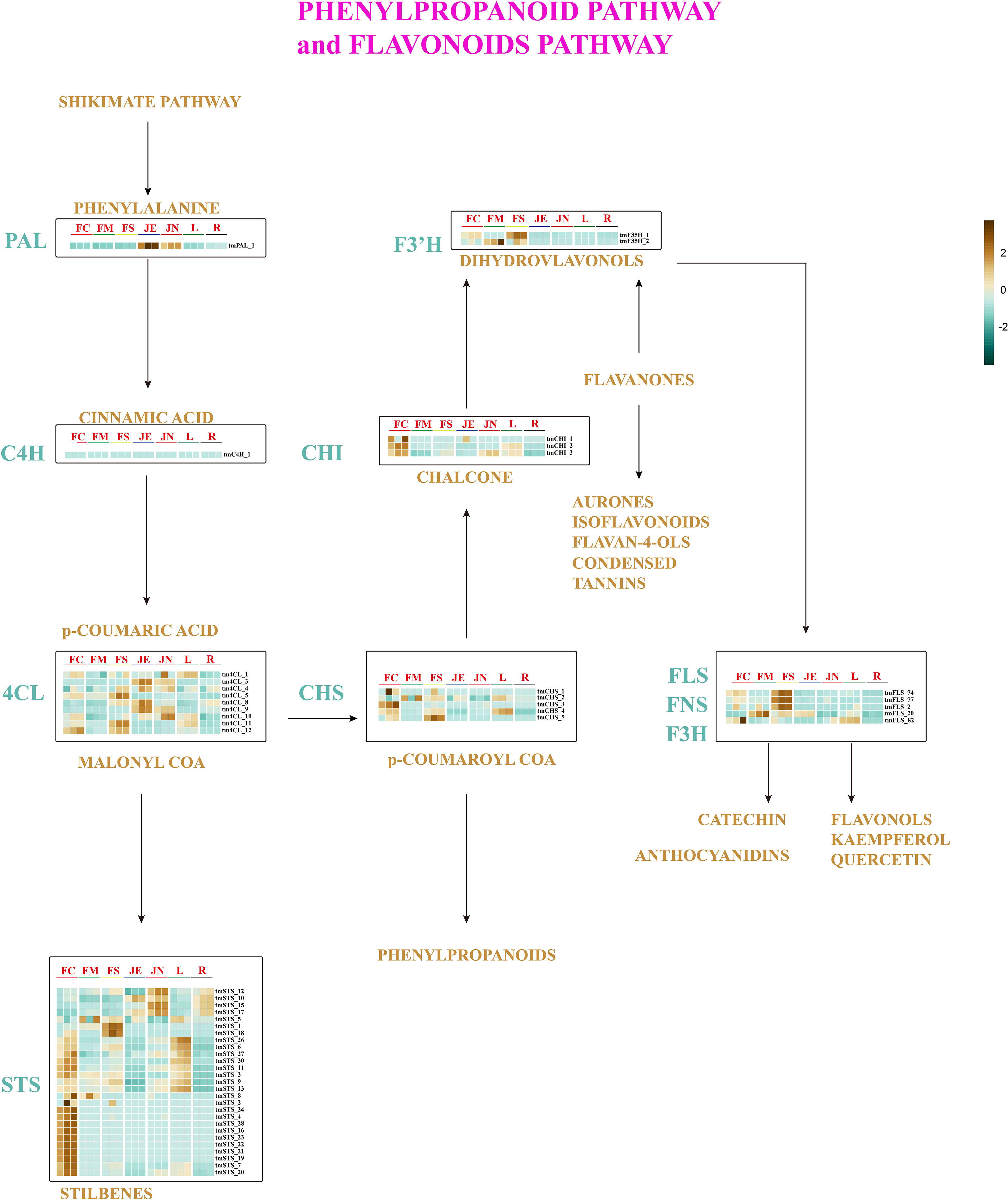
Figure 3 Pathways for phenylpropane metabolism and flavonoids metabolism in T. mandschuricus. Abbreviations for these enzymes are as follows: PAL, phenylalanine ammonia lyase; C4H, cinnamate-4-hydroxylase; 4CL, coumarate CoA ligase; STS, stilbene synthase; CHS, chalcone synthase; CHI, chalcone isomerase; FNS, flavone synthase; F3H, flavanone 3-hydroxylase; F3′H, flavonoid 3′ hydroxylase; F3′5′H, flavonoid 3′5′ hydroxylase; FLS, flavonol synthase; FC, initial flowering stage; FM, final flowering stage; FS, blooming period; JE, secondary branch; JN, tender stem; L, leaf; R, root.
To further elucidate the pathways involved in phenylpropane metabolism and flavonoid metabolism in T. mandschuricus, we analyzed RNA-seq data from the genome. All three tmCHI genes show significantly upregulated at initial flowering stage (FC). Conversely, 22 tmSTS genes, including tmSTS 9 and tmSTS 13, were significantly downregulated at secondary stem (JE) and root (R) (Figure 3). A number of tmSTS, tmF3′5′H, tmFLS, tmF3H, and tmFNS were identified to be uniquely expressed at different phases of floral development, suggesting a robust synthesis of diverse polyphenols throughout the flower development of T. mandschuricus. The specific expression of FLS (FNS and F3H) in flowers indicates that a significant portion of phenylpropanes in T. mandschuricus flowers is synthesized during the floral development stage. The key genes involved in flavonoid biosynthesis such as STS, CHS, and CHI are expressed in flowers, stems, and leaves of T. mandschuricus, providing molecular insights into the fragrance in stems and leaves of T. mandschuricus.
Identification and phylogeny of terpenoid biosynthesis-related genes
Analysis of the genomes of T. mandschuricus and 10 other species showed that the gene family responsible for diterpenoid synthesis had significantly expanded. From the 2-C-methyl-D-erythritol-4-phosphate (MEP) and mevalonate (MVA) routes, two 5-carbon “building blocks” [isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP)] are produced, which are then used in the biosynthesis pathway of terpenoids in plants. Members of 14 gene families are expressed in the terpene synthesis pathway. These includes acyl-coenzyme A:cholesterol acyltransferase (ACAT), hydroxymethylglutaryl coenzyme A synthase (HMGS), hydroxymethylglutaryl coenzyme A reductase (HMGR), mevalonate kinase (MVK), and phospho-mevalonate kinase (PMK). (Figure 4A). tmCMK 1 and tmCMK 2 exhibited a significant upregulation in the leaf (L), whereas they demonstrated a significant downregulation in the root (R) (Figure 4A). The MEP and MVA pathways were identified by transcriptome analysis. Terpene production in T. mandschuricus may include multi-organ cooperation and transport. Genes involved in the first half of the process (structural genes) were significantly expressed in leaves and secondary stems, whereas genes involved in the second half (FPPS/GPPS) were substantially expressed in flowers. The GPP and FPP reactions, catalyzed by tmTPSs, formed the C10 backbone of monoterpenes and C15 backbone of sesquiterpenes, respectively. We found 47 TPS-encoding genes that fell into five distinct groupings based on common conserved patterns (Figures 4B-D). During the blossoming and development of T. mandschuricus, we discovered 21 highly expressed tmTPSs in the flowers. The cytochrome P450 (CYP450) enzyme is responsible for the hydroxylation of monoterpenes and sesquiterpenes, whereas the BAHD family of plant acyltransferases is responsible for the esterification of these compounds. We found 338 CYP450 and 138 BAHD gene family members, approximately the same numbers as other types of spices. In addition, we found that genes in the terpenoid-synthesizing CYP450 and BAHD families are expressed at precise times throughout flower development, suggesting their importance in T. mandschuricus. We also examined the regulatory elements of transcription factors in the upstream area of the flower-expressed tmTPS promoter and found a high concentration of V-myb myeloblastosis viral oncogene 245 homolog (MYB), Myelocytomatosis transcription factors (MYC), antioxidant 246 responsive element (ARE), estrogen response element (ERE), ABA-responsive element (ABRE), stress response element (STRE) and other regulatory elements (Figures 4E, F). This may suggest that environmental influences, hormones, and endogenous transcription factors all play roles in regulating tmTPS gene transcription.
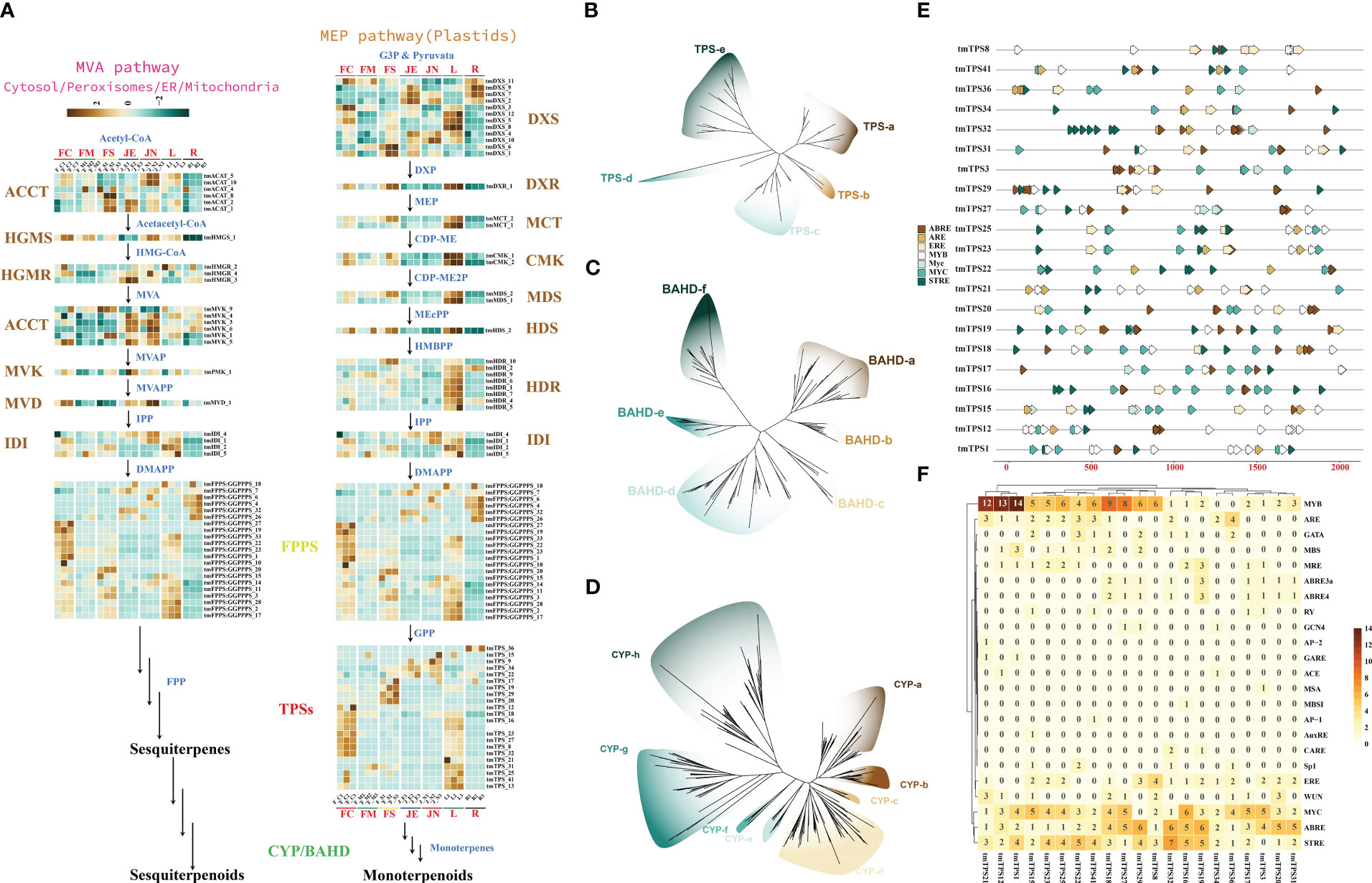
Figure 4 Reconstruction and phylogeny of the terpenoid biosynthesis pathway in T. mandschuricus. (A) Pathways of terpene biosynthesis in T. mandschuricus and relative expression of structural genes therein. These include acyl-coenzyme A:cholesterol acyltransferase (ACAT), hydroxymethylglutaryl coenzyme A synthase (HMGS), hydroxymethylglutaryl coenzyme A reductase (HMGR), mevalonate kinase (MVK), phospho-mevalonate kinase (PMK), mevalonate diphosphate decarboxylase (MVD), 1-deoxy-D-xylulose 5-phosphate synthase (DXS), 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR), 2-C-methyl-D-erythritol-4-phosphate cytidylyltransferase (MCT), 4-(cytidine-5-diphospho)-2-C-methyl-D-erythritol kinase (CMK), 2-C-methyl-D-erythritol-2,4-cyclodiphosphate synthase (MDS), (E)-4-hydroxy-3-methyl-but-2- enyl-pyrophosphate synthase (HDS), (E)-4-hydroxy-3-methyl-but-2-enyl-pyrophosphate reductase (HDR), isopentenyl diphosphate isomerase (IDI), and lavandulyl diphosphate synthase (LPPS). (B–D) Phylogenetic analysis of TPS, BAHD, and CYP gene families in T. mandschuricus, respectively. (E) The primary transcriptional regulatory components in the upstream 2k region of the TPS gene promoter are shown here in the form of a schematic diagram. (F) Prediction of transcription factor node sites in the upstream 2k region of the promoter of the TPS gene expressed in flower development of T. mandschuricus.
Regulatory analysis of tmTPS genes
Terpene synthase genes (TPSs) are critical structural genes that regulate terpene synthesis in T. mandschuricus, because they catalyze the synthesis of the essential framework for monoterpenes (C10) and sesquiterpenes (C15). Researchers found a plethora of gene regulatory elements, including MYB, MYC, ARE, ERE, ABRE, and STRE, in the promoter region of the tmTPS gene (Figure 4E). MYB binding sites were abundant in the upstream 2k regions of the promoters of many genes, including tmTPS8, tmTPS41, tmTPS32, tmTPS1, tmTPS12, and tmTPS21; other MYB binding genes were identified as tmMYB 11, tmMYB 184, tmMYB 290, and tmMYB 151. This may indicate that transcription of TPS genes is under the control of MYB transcription factors. Numerous MYB and TPS genes were expressed at the same time during the first stage of flower development in T. mandschuricus, and these MYB transcription factors were classified into five distinct subclasses (Figures 5A, C). T. mandschuricus’s MYC transcription factor family is likewise split into five groups, with subgroups B and C being particularly abundant in floral tissues (Figures 5B, D).
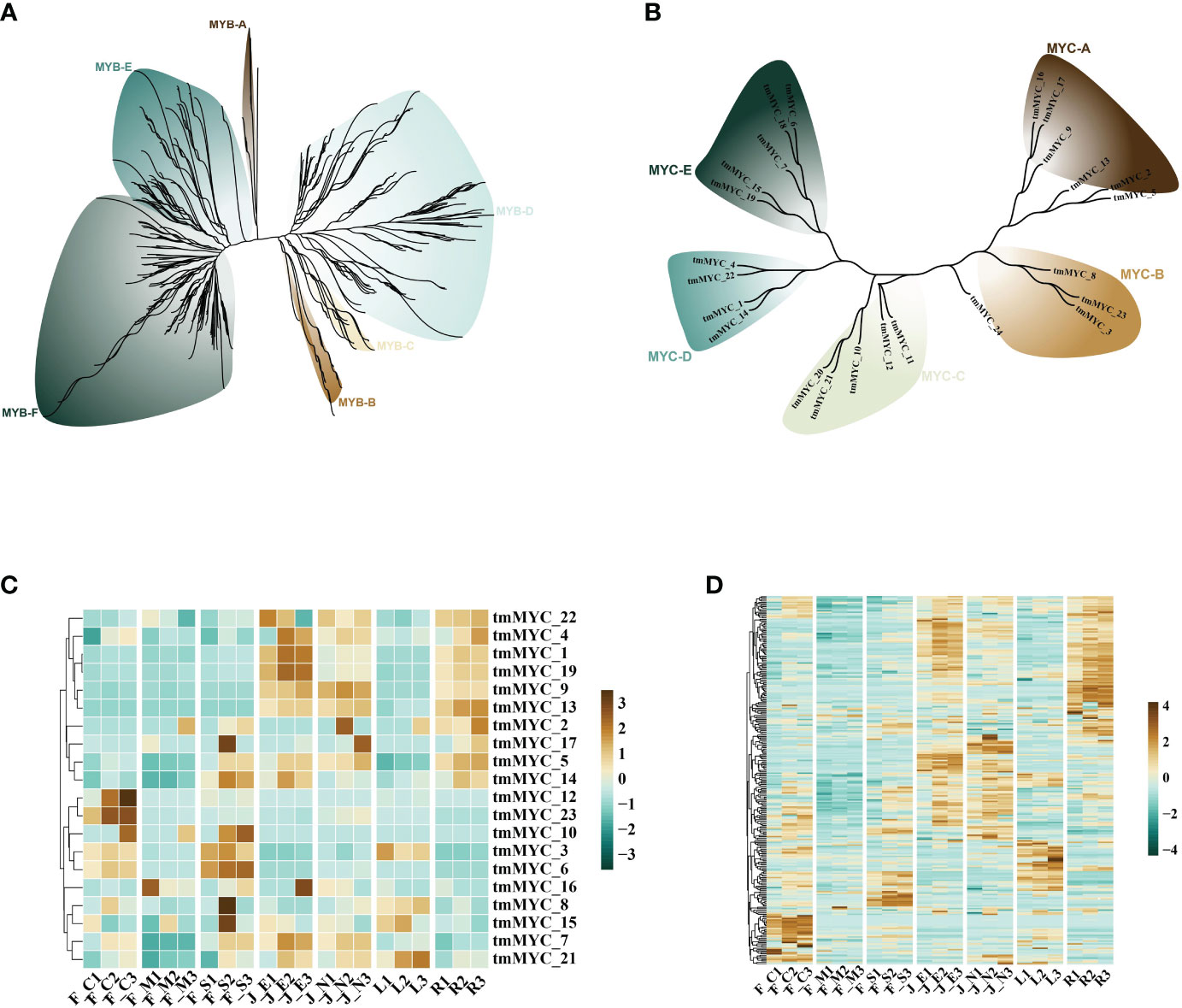
Figure 5 Analysis of regulatory systems governing the expression of TPS genes during flower development in T. mandschuricus. (A, B) Phylogenetic analyses of the MYB and MYC transcription factor families, respectively, in T. mandschuricus. (C, D) Relative expression levels of genes belonging to the MYB and MYC transcription factor families in T. mandschuricus during various stages of flower development, respectively. FC, initial flowering stage; FM, initial flowering stage; FS, blooming period; JE, secondary branch; JN, tender stem; L, tender stem; R, root.
STRE elements concentrated in the upstream 2k areas of the promoters of genes including tmTPS32, tmTPS25, tmTPS22, tmTPS19, and tmTPS16, suggesting that the transcription of these genes may be activated by biotic and abiotic stimuli. Another putative regulatory link between TPS and MYC is suggested by the high expression of the MYC transcription factor family members (tmMYC12, tmMYC23, and tmMYC10) in T. mandschuricus flowers (Figure 5D).
Discussion
Plants genomes are different from those of animals in that they are more variable, complex, and redundant (Murat et al., 2010). Plants are particularly vulnerable to environmental stress because of their immobile state (Tank et al., 2015). It is believed that genomic redundancy acts as raw materials for plant species diversification and evolutionary novelty via polyploidy/WGD events, which are often accompanied by a large number of repetitive genes, significant genome rearrangements, and increases in genetic variety (Wang et al., 2020; Jia et al., 2021). A calculation of the divergence period reveals that T. mandschuricus, Salvia splendens, Salvia bowleyana, and Salvia miltiorrhiza may have progressively diverged at 53.21 Mya from their MRCA. As time goes on, T. mandschuricus may have undergone two separate WGDs: one around 69.5 Mya and the other at 3.49 Mya. This agrees with the results from earlier investigations of extensive WGD occurrences in labiate (Lichman et al., 2020; Sun et al., 2022). Recent research has shown that the predicted time of WGD events of T. quinquecostatus is highly correlated with that of T. mandschuricus (Ntalli et al., 2010). The divergence of Thymus species may have coincided with the occurrences of these WGD episodes.
The Labiatae family of plants may be found in many different regions, including thyme, lavender, mint, basil, rosemary, marjoram, sage, and skullcap plants, which possess different therapeutic properties due to their different specialized metabolites. The Lamiaceae family produces a variety of secondary metabolites, the most prevalent of which are terpenes, phenolic acids, and flavonoids (Fraternale et al., 2014). A better understanding of the evolution of plant secondary compound biosynthetic pathways will be possible with the availability of high-quality genome sequences, which will promote the development of molecular basis studies of the diversity of specialized metabolites in various members of the Lamiaceae family.
When flowers open, volatile organic compounds (VOCs) including thymol, carvacrol, p-cymene, γ-terpinene, α-terpinene, and 1,8-cineole are found in T. mandschuricus, more in blooms than in folige. Previous research suggested that the VOCs levels of cistanche bud dropped, in some cases to as low as zero as flowers opened. The levels of VOCs in bud lavender rose initially but subsequently decreased as the flowers opened (Li et al., 2019; Rao et al., 2014). Structural genes in the MEP and MVA pathways had the highest expression in T. mandschuricus during the initial flowering stage (FC) but then declined progressively throughout the blooming period (FS) and finally were expressed at the lowest levels during the final flowering stage (FM). This might suggest that VOCs and terpenoid synthesis gene accumulation in T. mandschuricus follow a similar pattern. Higher levels of expression in CHS, STS, FLS, FNS, and essential structural genes in flavonoid metabolism were also seen during the FC and FS phases, which may further imply that terpenes are produced during these blooming stages in T. mandschuricus. Additionally, we hypothesize that the structural genes of flavonoid metabolism of T. mandschuricus have expanded with a process of exceptional paralogous gene family expansion because of a high similarity between the F3’5’H-F3’H and F3H-FLS-FNS gene families.
Conclusions
Thymus plants are often used for their taste and scent as spices, herbal teas, and insecticides. Here, we offer the genomic sequence of T. mandschuricus, an indigenous thyme species unique to China. The genome of T. mandschuricus consists of 13 pseudochromosomes, counting 587.14 Mb in size. Genomic comparisons indicated that T. mandschuricus was most closely related to sages like Salvia splendens, Salvia bowleyana, and Salvia miltiorrhiza. Ks analysis revealed two distinct WGD events in the T. mandschuricus genome. Key genes and regulatory networks involved in polyphenol and terpene production were identified by an in-depth investigation of the T. mandschuricus genome. We believe the datasets and analysis offered here will help future research in molecular breeding and functional gene discovery in Thymus.
Materials and methods
Sample collection
In this study, T. mandschuricus, an important medicinal plant, was obtained from the Maoer Mountain forest area in Shangzhi City, Heilongjiang Province, China. Young leaf samples of T. mandschuricus were used for genome sequencing, whereas T. mandschuricus collected from seven different tissues, including the initial flowering stage (FC), the final flowering stage (FM), the blooming period (FS), the tender stem (JN), the secondary branch (JE), the root (R), and the leaf (L), was used for transcriptome sequencing. It is important to note that all samples were obtained from the same strain. Three biological repeated samples were taken from each part, resulting in a total of 21 samples. The samples were immediately frozen with liquid nitrogen or carbon dioxide ice after collection and stored at −80°C. The total DNA of all samples was extracted.
DNA and RNA extraction
Genomic DNA was extracted by classic phenol–chloroform method, whereas total RNA was extracted with a TRIzol kit. The quality and quantity of extracted DNA/RNA were assessed by an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA, USA), and integrity was evaluated on agarose gel after a stain with ethidium bromide. The resulting DNA/RNA samples were stored at −80°C until subsequent library construction and genomic/transcriptomic sequencing. All samples were collected by personnel affiliated with our research group.
Library construction and sequencing
The libraries of T. Mandschuricus were constructed and sequenced on a PacBio sequencing platform (https://www.pacb.com/products-and-services/consumables/). For Hic sequencing, genomic DNA samples were pretreated according to Suhas S.P. Rao et al (Bolger et al., 2014). Under the adsorption of avidin magnetic beads, biotinized DNA was captured, and DNA fragments were end-repaired, adapter ligated, PCR-amplified, and purified in strict accordance with the Illumina Hi-C library protocol. Then, the quality of library was tested according to standard steps of library quality control. Qubit 2.0 was utilized for preliminary quantification, and the library was diluted to 1 ng/μl. Then, integrity of library DNA fragments and size of insert were detected by Agilent 2100. Then, quantitative PCR (qPCR) was employed for detecting the effective concentration of library for accurate quantification (effective concentration > 2 nM). After library inspection, different libraries were pooled according to the requirements of effective concentration and target data volume. Later, Illumina PE150 sequencing was performed according to the manufacturer’s protocols.
Quality control of sequencing data
For the next-generation sequencing data of Illumina, low-quality reads, linker sequences, and repetitive sequences were removed by trimmomatic (Gordon and Hannon, 2010) and FASTX-Toolkit (FastQC). Finally, data quality was evaluated by fastqc 39. For long PacBio reads, mean quality for each read was calculated and only reads longer than 1 kb with a mean quality of ≥7 were retained. For the Hic data, sequences containing linkers (N > 10%) were removed using HicPro.
Transcriptome sequencing
The rRNA was removed using the Ribo-Zero rRNA removal kit (Epicenter, Madison, WI, USA) with 1.5 μg of RNA from each sample as feedstock. The NEBNextR UltraTM Directional RNA Library Prep Kit (NEB, USA) and the NEBNextR UltraTM small RNA Sample Library Prep Kit (NEB, USA) were used according to the manufacturer’s recommendations. Transcriptome and small RNA libraries (Long noncoding RNAs (lncRNA), MicroRNAs (microRNA), and Circular RNAs (circRNA)) were constructed and sequenced on the Illumina HiSeq 2500/2000 platform. Three biological repeats were sequenced for each tissue sample.
RNA-seq expression analysis
Clean reads were obtained after the original data were filtered, the sequencing error rate and GC content distribution were identified, and the data were then compared with the T. mandschuricus reference genome sequence. FPKM (fragments per kilobase of transcript per million fragments) values were used to indicate transcript or gene expression levels. The original count data were analyzed by using DESeq2 v1.22.1 software. The Benjamini–Hochberg method was employed to adjust the probability (P-value) of the hypothesis test in order to obtain the false discovery rate (FDR). The differentially expressed genes were selected as screening criteria, occurring one or more times, with FDR < 0.05. Enrichment analysis was conducted using the Kyoto Encyclopedia of Genes and Genomes (KEGG), and a hypergeometric distribution test was used with pathway units.
Estimation of genomic size and genomic assembly
Genomic sizes were estimated by k-mer method with short-insert library reads (Koren et al., 2017). Here, 17-mer was selected for k-mer analysis and genomic size (Mb) estimated with the following formula: G = Knum/Kdepth, where Knum and Kdepth denoted total number and peak depth of 17-mers, respectively. De novo assembly of the long reads from the PacBio SMRT Sequencer was performed using wtdbg2 (Luo et al., 2012). The Fuzzy Bruijn Graph algorithm was used to assemble and integrate 1,024-bp sequences from the reads into vertex sequences, and, then, based on the position of the vertex sequences on the reads, the vertex sequences were concatenated to obtain the genome sequences. Subsequently, the Hi-C sequencing data were aligned to the assembled scaffolds by Burrows–Wheeler aligner-maximum exact matches (BWA-MEM) (Vasimuddin et al.,), and the scaffolds were clustered onto chromosomes with LACHESIS (Kajitani et al., 2014).
Quality assessment of genomic assemblies for T. mandschuricus was performed by BUSCO and CEGMA (Parra et al., 2007). BUSCO (Benchmarking Universal Single-Copy Orthologs) utilized a single-copy orthologous gene library along with tblastn, Augustus, HMMER, and other software tools to evaluate the integrity of assembled genomes. CEGMA (Core Eukaryotic Genes Mapping Approach) was adopted for selecting conserved genes (458 genes) in six eukaryotic model organisms and constructing a core gene library along with tblastn, genewise, and geneid software tools, which were conducted to evaluate the integrity of assembled genome.
Genomic annotation
Genome annotation comprises three main aspects: repeat sequence annotation, gene annotation, and non-coding RNA annotation. Two methods, namely, homologous sequence alignment and de novo prediction, were employed for repeat sequence labeling. The homologous sequence alignment method utilized Repeatmasker and repeatproteinmask software to identify the repeated sequences. Ab initio prediction was carried out using software such as LTR_FINDER, RepeatScout, RepeatModeler, and others. The ab repeat sequence library was established first, followed by prediction using Repeatmasker software (Chen, 2004; Flynn et al., 2020; Price et al., 2005; Xu and Wang, 2007). Protein-coding genes were annotated using a combination of de novo prediction, homology-based annotation, and transcription-based repetitive hidden genome annotation. For ab initio forecasting, Augustus (v.3.2.1) and GENSCAN (v.1.0) were used (Stanke and Morgenstern, 2005). The protein sequences of related species were downloaded from the NCBI database based on homology annotations. BLAST (Altschul et al., 1990) and GeneWise (Birney et al., 2004) were then used to predict the genetic structure. The annotation of gene sets and databases such as SwissProt (http://www.uniprot.org/), Nr (http://www.ncbi.nlm.nih.gov/protein), Pfam (http://pfam.xfam.org), KEGG (http://www.genome.jp/kegg/), InterPro (https://www.ebi.ac.uk/interpro/), and others were compared for gene function information. tRNAscan-SE software was used to locate tRNA sequences in the genome. INFERNAL software was employed to predict miRNA and snRNA sequence information in Rfam using the Rfam family covariance model.
Identification of orthologous genes and phylogenetic tree construction
Homologous species and nine closely related species were identified. The similarity relationship between the protein sequences of each species was determined using the whole-to-whole-embryo method, and the results were clustered using OrthoMCL software (Li et al., 2003). A maximum likelihood phylogenetic tree was constructed based on multiple sequence alignments using RAxML (Stamatakis, 2014).
Estimation of gene family expansion
Expansion and contraction of gene family was determined by CAFÉ software (v.3.1) (De Bie et al., 2006). Phylogenetic tree and divergence time of previous steps were imported into CAFE to infer the changes of gene family sizes using a probability model. Protein sequences that have been annotated as being involved in the terpenoid backbone biosynthesis pathway have been retrieved from all plant species using the KEGG database (Ko00900). These sequences include ACAT, HMGS, HMGR, MVK, PMK, MVD, DXR, DXS, MCT, CMK, MDS, HDS, HDR, IDI, and generate geranyl diphosphate synthase [(FPPS)/GGPPPS/FPPS]. Then, using BLASTP and setting the threshold for the E-value to 1e-5, we searched for proteins that were similar to those in the genome of T. mandschuricus. We predicted the TPS genes by using a BLAST method that was based on conserved domains (PF01397 and PF03936) and a homolog-based algorithm. Conserved domains were used as search queries inside HMMER’s hmmsearch module (Johnson et al., 2010). To identify the TPSs of T. mandschuricus, TPS protein sequences from all plant species were used as queries. For the purpose of BAHD identification, members of the BAHD family from Atha were used as queries for the BLASTP (1e-5) prediction of T. mandschuricus BAHD. We used the CYP450 protein sequences of Ath as queries to look for homologs and conserved domains in order to locate the genes that code for the CYP450 proteins (PF00067).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA929318.
Author contributions
LJ: Data curation, Formal Analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. NX: Investigation, Visualization, Writing – original draft. BX: Investigation, Visualization, Writing – review & editing. WG: Formal Analysis, Investigation, Resources, Writing – review & editing. QM: Formal Analysis, Investigation, Resources, Writing – review & editing. QL: Formal Analysis, Investigation, Resources, Writing – review & editing. YS: Formal Analysis, Investigation, Resources, Writing – review & editing. SX: Formal Analysis, Investigation, Resources, Writing – review & editing, Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Software, Supervision, Validation, Visualization. MH: Conceptualization, Supervision, Writing – review & editing. HG: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No. 31370610); the Fundamental Research Funds for the Central Universities (No. 2572017PZ05).
Acknowledgments
We thank Northeast Forestry University for providing a platform for our experiments, Lc-bio and Novogene for performing sequencing for this project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1368869/full#supplementary-material
Abbreviations
MVA, mevalonate; MEP, methylerythritol phosphate; TRF, trithoracic repeat elements; LTR, long terminal repeats; LINE, long interspersed nuclear elements; MRCA, most recent common ancestor; Mya, million years ago; WGD, whole-genome duplication; PAL, phenylalanine ammonia lyase; C4H, cinnamate-4-hydroxylase; 4CL, coumarate CoA ligase; STS, stilbene synthase; CHS, chalcone synthase; CHI, chalcone isomerase; FNS, flavone synthase; F3H, flavanone 3-hydroxylase; F3′H, BnF3′H-1; IPP, isopentenyl diphosphate; DMAPP, dimethylallyl diphosphate; ACAT, cholesterol acyltransferase; HMGS, hydroxymethylglutaryl coenzyme A synthase; HMGR, hydroxymethylglutaryl coenzyme A reductase; MVK, mevalonate kinase; PMK, phospho-mevalonate kinase; VOCs, volatile organic compounds; BAHD, BAHD family of acyltransferases; TPS, trehalose phosphate phosphatase; CYP450, the enzymes cytochrome P450; CYP, cytochrome P450 genes.
References
Afonso, A. F., Pereira, O. R., Cardoso, S. M. (2020). Health-promoting effects of Thymus phenolic-rich extracts: Antioxidant, anti-inflammatory and antitumoral properties. Antioxidants 9, 814. doi: 10.3390/antiox9090814
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Birney, E., Clamp, M., Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Chen, N. (2004). Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 5, 4–10. doi: 10.1002/0471250953.bi0410s05
Chen, H., Köllner, T. G., Li, G., Wei, G., Chen, X., Zeng, D., et al. (2020). Combinatorial evolution of a terpene synthase gene cluster explains terpene variations in Oryza. Plant Physiol. 182, 480–492. doi: 10.1104/pp.19.00948
Chen, F., Tholl, D., Bohlmann, J., Pichersky, E. (2011). The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. doi: 10.1111/j.1365-313X.2011.04520.x
De Bie, T., Cristianini, N., Demuth, J. P., Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Dudareva, N., Pichersky, E. (2008). Metabolic engineering of plant volatiles. Curr. Opin. Biotechnol. 19, 181–189. doi: 10.1016/j.copbio.2008.02.011
FastQC. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (Accessed 10 Feb 2018).
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Fraternale, D., Flamini, G., Ricci, D., Giomaro, G. (2014). Flowers volatile profile of a rare red apple tree from Marche region (Italy). J. oleo sci. 63, 1195–1201. doi: 10.5650/jos.ess14088
Godden, G. T., Kinser, T. J., Soltis, P. S., Soltis, D. E. (2019). Phylotranscriptomic analyses reveal asymmetrical gene duplication dynamics and signatures of ancient polyploidy in mints. Genome Biol. Evolution 11, 3393–3408. doi: 10.1093/gbe/evz239
Gordo, J., Máximo, P., Cabrita, E., Lourenço, A., Oliva, A., Almeida, J., et al. (2012). Thymus mastiChina: chemical constituents and their anti-cancer activity. Natural product Commun. 7, 1934578X1200701120. doi: 10.1177/1934578X1200701120
Gordon, A., Hannon, G. (2010) Fastx-toolkit. FASTQ/A short-reads pre-processing tools. Available online at: http://hannonlab.cshl.edu/fastx_toolkit.
Hansen, N. L., Heskes, A. M., Hamberger, B., Olsen, C. E., Hallström, B. M., Andersen-Ranberg, J., et al. (2017). The terpene synthase gene family in Tripterygium wilfordii harbors a labdane-type diterpene synthase among the monoterpene synthase TPS-b subfamily. Plant J. 89, 429–441. doi: 10.1111/tpj.13410
Hosseinzadeh, S., Jafarikukhdan, A., Hosseini, A., Armand, R. (2015). The application of medicinal plants in traditional and modern medicine: a review of Thymus vulgaris. Int. J. Clin. Med. 6, 635–642. doi: 10.4236/ijcm.2015.69084
Jia, K. H., Liu, H., Zhang, R. G., Xu, J., Zhou, S. S., Jiao, S. Q., et al. (2021). Chromosome-scale assembly and evolution of the tetraploid Salvia splendens (Lamiaceae) genome. Horticult. Res. 8, 177.1. doi: 10.1038/s41438-021-00614-y
Johnson, L. S., Eddy, S. R., Portugaly, E. (2010). Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinf. 11, 1–8. doi: 10.1186/1471-2105-11-431
Jones, C. G., Moniodis, J., Zulak, K. G., Scaffidi, A., Plummer, J. A., Ghisalberti, E. L., et al. (2011). Sandalwood fragrance biosynthesis involves sesquiterpene synthases of both the terpene synthase (TPS)-a and TPS-b subfamilies, including santalene synthases. J. Biol. Chem. 286, 17445–17454. doi: 10.1074/jbc.M111.231787
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Kejnovsky, E., Leitch, I. J., Leitch, A. R. (2009). Contrasting evolutionary dynamics between angiosperm and mammalian genomes. Trends Ecol. Evolution 24, 572–582. doi: 10.1016/j.tree.2009.04.010
Kim, S. I., Roh, J. Y., Kim, D. H., Lee, H. S., Ahn, Y. J. (2003). Insecticidal activities of aromatic plant extracts and essential oils against Sitophilus oryzae and Callosobruchus chinensis. J. Stored products Res. 39, 293–303. doi: 10.1016/S0022-474X(02)00017-6
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Koul, O., Walia, S., Dhaliwal, G. S. (2008). Essential oils as green pesticides: potential and constraints. Biopesticides Int. 4, 63–84. doi: 10.1109/ipdps.2019.00041
Li, H., Li, J., Dong, Y., Hao, H., Ling, Z., Bai, H., et al. (2019). Time-series transcriptome provides insights into the gene regulation network involved in the volatile terpenoid metabolism during the flower development of lavender. BMC Plant Biol. 19, 1–7. doi: 10.1186/s12870-019-1908-6
Li, L., Stoeckert, C. J., Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lichman, B. R., Godden, G. T., Buell, C. R. (2020). Gene and genome duplications in the evolution of chemodiversity: perspectives from studies of Lamiaceae. Curr. Opin. Plant Biol. 55, 74–83. doi: 10.1016/j.pbi.2020.03.005
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 2047–217X. doi: 10.1186/2047-217X-1-18
Martin, V. J., Pitera, D. J., Withers, S. T., Newman, J. D., Keasling, J. D. (2003). Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat. Biotechnol. 21, 796–802. doi: 10.1038/nbt833
Mulas, M. (2006). Traditional uses of Labiatae in the Mediterranean area. Inl: InI International Symposium on the Labiatae: Advances in Production, Biotechnology and Utilisation. 723. 25–32.
Murat, F., Xu, J. H., Tannier, E., Abrouk, M., Guilhot, N., Pont, C., et al. (2010). Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffling as a source of plant evolution. Genome Res. 20, 1545–1557. doi: 10.1101/gr.109744.110
Nelson, D., Werck-Reichhart, D. (2011). A P450-centric view of plant evolution. Plant J. 66, 194–211. doi: 10.1111/j.1365-313X.2011.04529.x
Nieto, G. (2020). A review on applications and uses of thymus in the food industry. Plants 9, 961. doi: 10.3390/plants9080961
Ntalli, N. G., Ferrari, F., Giannakou, I., Menkissoglu-Spiroudi, U. (2010). Phytochemistry and nematicidal activity of the essential oils from 8 Greek Lamiaceae aromatic plants and 13 terpene components. J. Agric. Food Chem. 58, 7856–7863. doi: 10.1021/jf100797m
Panchy, N., Lehti-Shiu, M., Shiu, S. H. (2016). Evolution of gene duplication in plants. Plant Physiol. 171, 2294–2316. doi: 10.1104/pp.16.00523
Parra, G., Bradnam, K., Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Price, A. L., Jones, N. C., Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Qiao, Y., Yu, Z., Bai, L., Li, H., Zhang, S., Liu, J., et al. (2021). Chemical composition of essential oils from Thymus mongolicus, Cinnamomum verum, and Origanum vulgare and their acaricidal effects on Haemaphysalis longicornis (Acari: Ixodidae). Ecotoxicol. Environ. Safety 224, 112672. doi: 10.1016/j.ecoenv.2021.112672
Raja, R. R. (2012). Medicinally potential plants of Labiatae (Lamiaceae) family: an overview. Res. J. medicinal Plant 6, 203–213. doi: 10.3923/rjmp.2012.203.213
Rao, S. S., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Saroukolai, A. T., Moharramipour, S., Meshkatalsadat, M. H. (2010). Insecticidal properties of Thymus persicus essential oil against Tribolium castaneum and Sitophilus oryzae. J. Pest sci. 83, 3–8. doi: 10.1007/s10340-009-0261-1
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467. doi: 10.1093/nar/gki458
Stracke, C., Meyer, B. H., Hagemann, A., Jo, E., Lee, A., Albers, S. V., et al. (2020). Salt stress response of Sulfolobus acidocaldarius involves complex trehalose metabolism utilizing a novel trehalose-6-phosphate synthase (TPS)/trehalose-6-phosphate phosphatase (TPP) pathway. Appl. Environ. Microbiol. 86, e01565–e01520. doi: 10.1128/AEM.01565-20
Sun, M., Zhang, Y., Zhu, L., Liu, N., Bai, H., Sun, G., et al. (2022). Chromosome-level assembly and analysis of the Thymus genome provide insights into glandular secretory trichome formation and monoterpenoid biosynthesis in thyme. Plant Commun. 3, 100413. doi: 10.1016/j.xplc.2022.100413
Tank, D. C., Eastman, J. M., Pennell, M. W., Soltis, P. S., Soltis, D. E., Hinchliff, C. E., et al. (2015). Nested radiations and the pulse of angiosperm diversification: increased diversification rates often follow whole genome duplications. New Phytologist 207, 454–467. doi: 10.1111/nph.13491
Tholl, D. (2006). Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr. Opin. Plant Biol. 9, 297–304. doi: 10.1016/j.pbi.2006.03.014
Tohidi, B., Rahimmalek, M., Arzani, A. (2017). Essential oil composition, total phenolic, flavonoid contents, and antioxidant activity of Thymus species collected from different regions of Iran. Food Chem. 220, 153–161. doi: 10.1016/j.foodchem.2016.09.203
Vasimuddin, M., Misra, S., Li, H., Aluru, S. (2019). “Efficient architecture-aware acceleration of BWA-MEM for multicore systems,” in 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). 314–324 (IEEE).
Wang, P., Luo, Y., Huang, J., Gao, S., Zhu, G., Dang, Z., et al. (2020). The genome evolution and domestication of tropical fruit mango. Genome Biol. 21, 1–7. doi: 10.1186/s13059-020-01959-8
Wang, H., Yang, Z., Ying, G., Yang, M., Nian, Y., Wei, F., et al. (2018). Antifungal evaluation of plant essential oils and their major components against toxigenic fungi. Ind. Crops Products 120, 180–186. doi: 10.1016/j.indcrop.2018.04.053
Weitzel, C., Simonsen, H. T. (2015). Cytochrome P450-enzymes involved in the biosynthesis of mono-and sesquiterpenes. Phytochem. Rev. 14, 7–24. doi: 10.1007/s11101-013-9280-x
Xu, Y., Tie, W., Yan, Y., Xu, B., Liu, J., Li, M., et al. (2021). Identification and expression of the BAHD family during development, ripening, and stress response in banana. Mol. Biol. Rep. 48, 1127–1138. doi: 10.1007/s11033-020-06132-9
Keywords: Thymus mandschuricus, phylogeny, CYP450, terpenoid biosynthesis, aroma production
Citation: Jia L, Xu N, Xia B, Gao W, Meng Q, Li Q, Sun Y, Xu S, He M and Gu H (2024) Chromosome-level genome of Thymus mandschuricus reveals molecular mechanism of aroma compounds biosynthesis. Front. Plant Sci. 15:1368869. doi: 10.3389/fpls.2024.1368869
Received: 11 January 2024; Accepted: 22 February 2024;
Published: 13 March 2024.
Edited by:
Shaojun Dai, Shanghai Normal University, ChinaReviewed by:
Chao Ma, China Agricultural University, ChinaZhuping Fan, Leibniz Institute of Vegetable and Ornamental Crops, Germany
Copyright © 2024 Jia, Xu, Xia, Gao, Meng, Li, Sun, Xu, He and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huiyan Gu, Z2h1aXlhbkBuZWZ1LmVkdS5jbg==; Miao He, aGVtaWFvQG5lZnUuZWR1LmNu