 Cui Li1,2,3†
Cui Li1,2,3† Han Liu1,2,3†Mei Qin1,2,3Yao-jing Tan4Xia-lian Ou1,2,3Xiao-ying Chen1,2,3Ying Wei1,5Zhan-jiang Zhang1,5*
Han Liu1,2,3†Mei Qin1,2,3Yao-jing Tan4Xia-lian Ou1,2,3Xiao-ying Chen1,2,3Ying Wei1,5Zhan-jiang Zhang1,5* Ming Lei1,2,3*
Ming Lei1,2,3*- 1National Center for Traditional Chinese Medicine (TCM) Inheritance and Innovation, Guangxi Botanical Garden of Medicinal Plants, Nanning, China
- 2Guangxi Key Laboratory of Medicinal Resources Protection and Genetic Improvement, Guangxi Botanical Garden of Medicinal Plants, Nanning, China
- 3Guangxi Engineering Research Center of Traditional Chinese Medicine (TCM) Resource Intelligent Creation, Guangxi Botanical Garden of Medicinal Plants, Nanning, China
- 4School of Basic Medical Sciences, Guangxi Medical University, Nanning, China
- 5Guangxi Key Laboratory for High-Quality Formation and Utilization of Dao-di Herbs, Guangxi Botanical Garden of Medicinal Plants, Nanning, China
Corydalis saxicola, an endangered medicinal plant endemic to karst habitats, is widely used in Traditional Chinese Medicine to treat hepatitis, abdominal pain, bleeding hemorrhoids and other conditions. However, to date, the mitochondrial (mt) genome of C. saxicola has not been reported, which limits our understanding of the genetic and biological mechanisms of C. saxicola. Here, the mt genome of C. saxicola was assembled by combining the Nanopore and Illumina reads. The mt genome of C. saxicola is represented by a circular chromosome which is 587,939 bp in length, with an overall GC content of 46.50%. 40 unique protein-coding genes (PCGs), 22 tRNA genes and three rRNA genes were identified. Codon usage of the PCGs was investigated and 167 simple sequence repeats were identified. Twelve homologous fragments were identified between the mt and ct genomes of C. saxicola, accounting for 1.04% of the entire mt genome. Phylogenetic examination of the mt genomes of C. saxicola and 30 other taxa provided an understanding of their evolutionary relationships. We also predicted 779 RNA editing sites in 40 C. saxicola mt PCGs and successfully validated 506 (65%) of these using PCR amplification and Sanger sequencing. In addition, we transcriptionally profiled 24 core mt PCGs in C. saxicola roots treated with different concentrations of CaCl2, as well as in other organs. These investigations will be useful for effective utilization and molecular breeding, and will also provide a reference for further studies of the genus Corydalis.
1 Introduction
Corydalis DC., which contains approximately 530 species divided into 42 sections, is the largest genus within the Papaveraceae family (Zhang et al., 2008; Xu et al., 2022a; Chen et al., 2023b). Corydalis species are found across northern temperate regions, as well as the subarctic, subtropical Indochina and in the East Africa montane forests. Notably, the Corydalis species are particularly abundant around the Qinghai–Tibet Plateau and the Hengduan Mountains of China (Wu et al., 1999; Zhang et al., 2008). Corydalis species are adapted to wildly varied habitats, including riversides, forests, shrublands, grasslands, screes, cliffsides and high elevations (up to 6000 m). Such morphological diversity has made studying the taxonomy, ecology, evolutionary history and utilization of the genus challenging (Ohara and Higashi, 1994; Ohkawara et al., 1997; Kudo et al., 2001; Ehlers and Olesen, 2004; Niu et al., 2014; Zhang et al., 2014; Niu et al., 2017; Zhu et al., 2018; Chen et al., 2023b). Many Corydalis species possess brilliantly-colored flowers and are prized by horticulturalists and hobby gardeners (Lidén and Zetterlund, 1997). In addition, several Corydalis species are used as medicines to treat hepatitis, cancer, cardiovascular diseases and pain. These include C. yanhusuo W.T.Wang ex Z.Y.Su & C.Y.Wu, C. decumbens (Thunb.) Pers, C. saxicola Bunting, C. ophiocarpa, C. cava (L.) Schweigg. & Koerte and C. tomentella Franch, among others (Wu et al., 1999; Orhan et al., 2007; Kim et al., 2010; Novak et al., 2012; Li et al., 2013; Kim et al., 2014; Xu et al., 2022b; Feng et al., 2023; Qin et al., 2023b).
Among the medicinal Corydalis species, C. saxicola is widely used in Traditional Chinese Medicine due to its antibacterial, antiviral and anticancer activities (Orhan et al., 2007; Kim et al., 2010; Li et al., 2013; Kim et al., 2014; Tian et al., 2016; Zhang et al., 2016; Liu et al., 2019; Feng et al., 2023; Qin et al., 2023b). Studies suggest that these pharmacological activities are due to the presence of unique benzylisoquinoline alkaloid compounds known as cavidines (e.g., cavidine, apocavidine, dehydrocavidine and dehydroapocavidine) (Bhakuni and Chaturvedi, 1983; Qin et al., 2023a). As a calcicole plant, C. saxicola is found exclusively in and around the rocky crevices of calcium-rich karst landscapes. Unfortunately, increasing demand and a limited range have driven C. saxicola to become endangered. In order to safeguard C. saxicola as a medicinal resource, studies should be conducted to comprehensively evaluate the molecular biology and genetics of this endangered plant species.
Both mitochondria (mt) and chloroplasts (cp) have originated as prokaryotic endosymbionts, and these possess semi-autonomous genetic systems (Jiang et al., 2023a). The crucial physiological processes regulated by chloroplasts and mitochondria are photosynthesis and respiration, respectively (Raven, 2015). In the majority of seed plants, the mt and cp genomes are maternally inherited and are critical to growth and development (Pogson et al., 2015; Liberatore et al., 2016; Wang et al., 2022). In contrast to animal mt genomes, plant mt genomes have undergone numerous changes in the architecture, leading to a broad distribution in genome size, ranging from ~66 kb to ~12 Mb (Liu et al., 2014; Oldenburg and Bendich, 2015; Cole et al., 2018; Putintseva et al., 2020; Wu et al., 2022). In addition, plant mt genomes exhibit marked interspecies differences in gene order, gene length, gene content and migration of sequences from other organelles (Liu et al., 2014; Oldenburg and Bendich, 2015; Cole et al., 2018; Wu et al., 2022; Wang et al., 2024). Typically, mt genomes are circular, double-linked molecules. However, some plant mt genomes can exist as linear, multi-branched and polycyclic structures (Sloan, 2013; Gualberto et al., 2014; Kozik et al., 2019; Jackman et al., 2020). In comparison to cp genomes, these characteristics have made the assembly and exploration of plant mt genomes relatively difficult (Shan et al., 2023). To date (as of April 5, 2023), the National Center for Biotechnology Information (NCBI) database contains 602 published mt genomes, representing only approximately 5.7% of all cp genomes (10,479) (https://www.ncbi.nlm.nih.gov/genome/browse/#!/organelles/). Recently, the complete cp genomes of several Corydalis species, including C. saxicola, have been sequenced and published (Ren et al., 2021; Xu and Wang, 2021; Xu et al., 2022a). However, to date, only one Corydalis mt genome has been published (C. pauciovulata, Genbank: OR100522.1). Fortunately, the development of long-read PacBio and Nanopore sequencing techniques have made the sequencing of organellar genomes considerably simpler and faster.
In this study, the mt genome of C. saxicola was sequenced and annotated based on data obtained from Oxford Nanopore and Illumina. The relative synonymous codon usage (RSCU), sequence repeats and mitochondrial plastid DNAs (MTPTs) were analyzed. A phylogenetic analysis was performed between C. saxicola and 30 other angiosperm species based on 23 conserved mt protein-coding genes (PCGs). In addition, by combining PCR amplification and Sanger sequencing, we conducted an in-depth analysis of the RNA editing sites within the mt PCGs. Finally, we investigated the relative expression of 24 core mt PCGs in roots and other organs of C. saxicola. In addition, PCG expression was measured after treating the roots with exogenous CaCl2. These data will be invaluable to further research on the taxonomic classification, molecular evolution and breeding of C. saxicola.
2 Materials and methods
2.1 Plant materials, DNA and RNA extraction and sequencing
C. saxicola was identified by Drs Zhan-jiang Zhang and Cui Li of the Guangxi Botanical Garden of Medicinal Plants (GBGMP) (Nanning, Guangxi Zhuang Autonomous Region, China). Voucher specimens (reference number: 450123130505040LY) were deposited in the GBGMP Herbarium.
Fresh and healthy C. saxicola tissues were collected from the greenhouse located in the GBGMP experimental area. One gram of C. saxicola leaves were used four total DNA extraction according to the 2 × cetyltrimethylammonium bromide method (Porebski et al., 1997). The same qualified DNA sample was used for Illumina sequencing, Oxford Nanopore sequencing, and PCR amplification.
Thirty mg of C. saxicola roots, stems, branches, mature leaves and young leaves were used for total RNA extraction according to the manual of RNA Isolater Total RNA Extraction Reagent (Vazyme, China). The first-strand cDNA was synthesized using a HiScript III 1st Strand cDNA Synthesis Kit (+gDNA wiper) (Vazyme, China). Qualified extracted RNA was used as a template, and random hexamers and oligo(dT)20VN were used as primers. PCR was performed using 2 × Phanta Max Master Mix (Dye Plus) (Vazyme, China) and conducted on a T100 Thermal Cycler (BIO-RAD, USA).
2.2 Assembly and annotation of mt genomes
Flye (v.2.9.1-b1780) (Kolmogorov et al., 2019) was used to assemble the long reads of C. saxicola, with default parameters. The assembled contigs were then used to build a library using makeblastdb. BLASTn (Chen et al., 2015) was utilized to identify mt genome fragments, with the conserved mitochondrial genes of Arabidopsis thaliana were used as query sequences. With the help of BWA (v0.7.17) (Li and Durbin, 2009), the short reads and long reads were mapped to the contigs and all the mapped reads were retained. Finally, Unicycler (v0.5.0) (Wick et al., 2017) was employed to combine all mapped short and long reads for hybrid assembly, with default parameters. Unicycler-generated GFA format files were visualized with the help of Bandage (Wick et al., 2015).
Referring to previously published literature (Richardson et al., 2013), we found that the mt genome of Liriodendron tulipifera had the most complete mt gene content among angiosperms. As a result, in this study, the mt genome was annotated using GeSeq (v2.03) (Michael et al., 2017), with L. tulipifera (NC_021152.1) as the reference. IPMGA (http://www.1kmpg.cn/ipmga/) was also employed for annotation and integration of the results from both tools. Forty PCGs resulted from our annotation. For genes that were present in the L. tulipifera mt genome were not annotated in our newly sequenced species, and we further queried these using the BLASTn (v.2.13.0) (Chen et al., 2015) with parameters ‘-evalue 1e-10 -outfmt 6 -num_threads 10 -max_hsps 10’. tRNAscan-SE (v.2.0.11) (Lowe and Eddy, 1997) was also utilized to annotate the tRNA genes. Apollo (v1.11.8) (Lewis et al., 2002) was employed to manually adjust and rectify any annotation mistakes found in each mt genome.
2.3 Analysis of codon usage and repeated sequences
Protein-encoding sequences were extracted with PhyloSuite (v1.1.16) (Zhang et al., 2020). Codon preference analysis and RSCU value calculation were performed with Mega (v7.0) (Kumar et al., 2016). MISA (v2.1) (https://webblast.ipk-gatersleben.de/misa/; accessed on 5 August, 2023) (Beier et al., 2017), TRF (v4.09) (https://tandem.bu.edu/trf/trf.unix.help.html; accessed on 6 August, 2023) (Benson, 1999) and REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer/; accessed on 8 August, 2023) (Kurtz et al., 2001) were employed to detect the repetitive sequences. These included microsatellite sequence repeats, tandem repeats (TR) and dispersed repeats. The results were visualized using Excel (2021) and the Circos package (v0.69-9) (Zhang et al., 2013).
2.4 Identification of MTPTs, and RNA editing events
The cp genome of C. saxicola was assembled using GetOrganelle (v1.7.7.0) (Jin et al., 2020) and annotated using CPGAVAS2 (Shi et al., 2019). CPGView (Liu et al., 2023) was employed to rectify annotation mistakes in the cp genomes. BLASTn (v.2.13.0) (Chen et al., 2015) was utilized to identify homologous fragments in the mt and cp genomes, and the results were visualized using the Circos package (v0.69-9) (Zhang et al., 2013). Putative RNA editing sites were predicted using Deepred-mt (Edera et al., 2021), with a cutoff value of 0.9.
2.5 Phylogenetic and synteny analyses
The mt genome sequences of closely related plant species were obtained from NCBI (https://www.ncbi.nlm.nih.gov; accessed on 4 August, 2023), and PhyloSuite (v1.1.16) (Zhang et al., 2020) was then employed to extract common genes. To conduct a phylogenetic analysis, the nucleotide sequences of shared genes were aligned using MAFFT (v7.505) (Katoh and Standley, 2013), and the aligned sequences were concatenated to form a data matrix. Based on this data matrix, the maximum likelihood analysis was performed to construct the phylogenetic tree using IQ-TREE (v1.6.12) (Nguyen et al., 2015), with a bootstrap value of 1000, and then visualized using ITOL (v6) (Letunic and Bork, 2019).
To investigate the collinearity of the mt genomes, BLASTn (Chen et al., 2015) was used to identify conserved homologous sequences between C. saxicola and other closely related plant species, with the following parameters: -evalue 1e-5, -word_size 9, -gapopen 5, -gapextend 2, -reward 2, and –penalty 3. Only collinear blocks of at least 500 bp in length were selected for further analysis. MCscanX (Wang Y. et al., 2012) was then used to generate a multiple synteny plot.
2.6 RT-qPCR and heatmap generation
The first-strand reaction products were diluted with sterilized distilled H2O three times, and the diluted products (1 µL) were used for qPCR (20 µL). qPCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme, China) and conducted on a QuantStudio 3 system (Thermo Fisher Scientific, USA). Three biological replicates and three technical replicates were performed. The relative expression levels of specific genes were calculated using the 2−ΔΔCt method, with glyceraldehyde-3-phosphate dehydrogenase 8 (CsGAPDH8) used as the internal control (Livak and Schmittgen, 2001). All primers used for qPCR are listed in Supplementary Table S1. The relative expression levels were visualized using tbtools (v2.008) (Chen et al., 2023a).
2.7 CaCl2 treatment of C. saxicola
To test CaCl2 response, one-month-old of C. saxicola plants grown in pots containing a matrix with a 3:1 mixture ratio of peat soil and vermiculite in the greenhouse (23 °C, 16 h light, a photo flux density of 120 µmol m-2 s-1) were treated with equal volume of CaCl2 at 4 mmol/L, 30 mmol/L, 100 mmol/L, 200 mmol/L, 300 mmol/L and 400 mmol/L for 25 d. The same batch of plants treated with the same quantity of water were set as the control (CK). The organs were then cleaned and physically isolated and immediately frozen or dried for further research.
2.8 Measurement of dehydrocaverdine content
Agilent 1260 Infinity II (Agilent Co. Ltd, NY, USA) was used to determine the relative content of dehydrocaverdine. 200.0 mg of dried plant tissue was weighed in a 15-mL tube and 10 mL of methanol was added. The samples were ultra-sonicated for 60 min, and then centrifuged at 13,000 g for 15 min. The supernatant was transferred to a new 15-mL tube and dried using nitrogen pressure reduction method at room temperature. The dried sample was then dissolved with 1-mL of mobile phase (acetonitrile: 0.01% of K2HPO4 aqueous solution = 21:79 (v/v)) and passed through a 0.22 μm filter for measurement. Dehydrocaverdine was eluted under the following conditions: Agilent XDB C18 chromatographic column (150 mm × 4.6 mm, 5 µm) (Agilent Co. Ltd, NY, USA); mobile phase (acetonitrile: 0.01% of K2HPO4 = 21:79 (v/v)); 30 °C; 347 nm of detection wavelength; 1.0 mL/min of volume flow rate. A standard sample of dehydrocaverdine was purchased from Shanghai Yuanye Co. Ltd (Shanghai, China).
3 Results
3.1 General features of C. saxicola mt genome
The C. saxicola mt genome was assembled using a hybrid assembly of both long and short reads. Finally, a graphical mt genome comprising 3 nodes (contig1, contig2 and contig3) was constructed by resolving the repeat regions (Supplementary Figure S1). Contig3 was predicted to be repeats, as its depth was approximately twice when compared with contig1 and contig2 (Supplementary Figure S1; Supplementary Table S2). Finally, we derived a single circular molecule for the C. saxicola mt genome (Genbank: OR625126) (Figure 1).
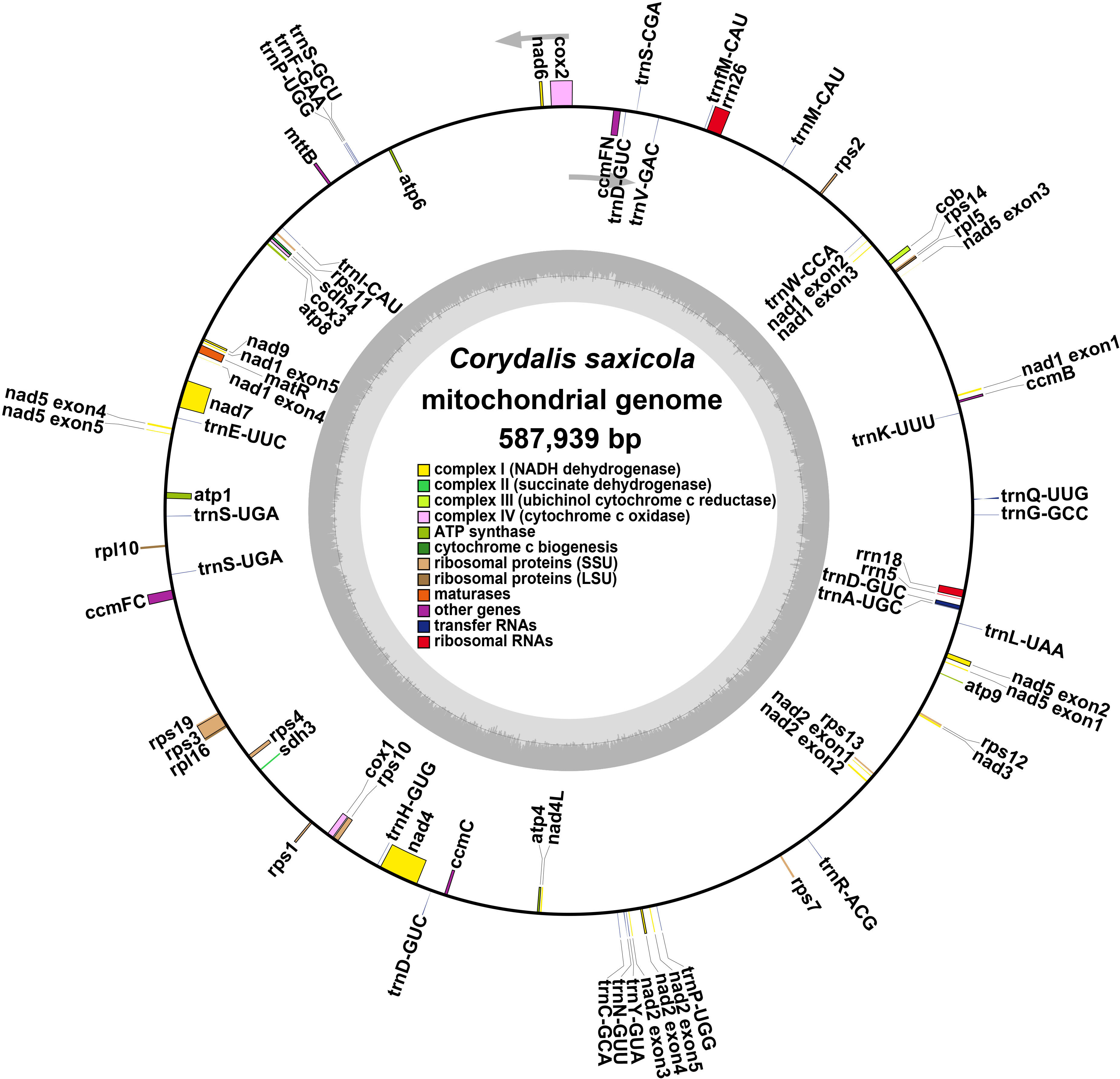
Figure 1 A map of the C. saxicola mt genome. The genes drawn inside and outside the circle are transcribed clockwise and counterclockwise, respectively.
The C. saxicola mt genome is 587,939 bp in length, with an overall GC content of 46.50%. A total of 40 unique PCGs were identified, including 24 core genes and 16 variable genes (Table 1). In addition, 22 tRNA genes and three rRNA genes were annotated. All the PCGs and rRNA genes, and most of the tRNA genes, occurred as single copies. However, two tRNA genes (trnP-UGG and trnS-UGA) occurred as double copies, and one tRNA gene (trnD-GUC) occurred as a triple copy (Table 1). Among the 65 annotated genes, 9 genes contained more than one exon. Of these, 3 (ccmFC, rps3 and rps10), 1 (cox2), 1 (nad4) and 4 (nad1, nad2, nad5 and nad7) contained 2, 3, 4 and 5 exons, respectively (Supplementary Table S3).
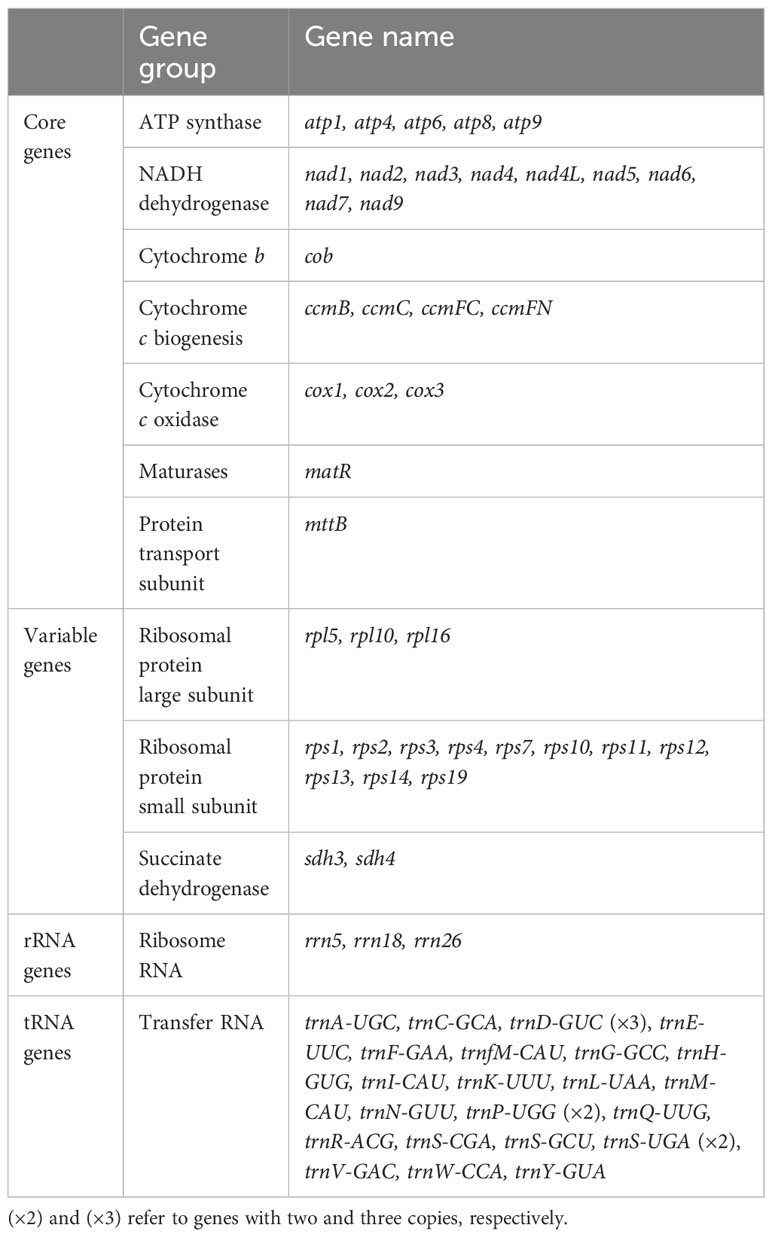
Table 1 A list of genes identified in the C. saxicola mt genome.
In addition, we also produced the complete C. saxicola cp genome (Genbank: OR625127). The cp genome was 190,564 bp in length and contained a pair of inverted repeat (IR) regions (47,692 bp each), a large single-copy (LSC) region (96,266 bp), and a small single-copy (SSC) region (8,890 bp) (Supplementary Figure S2). A total of 110 genes were identified, including 77 PCGs, 29 rRNA genes and 4 tRNA genes (Supplementary Table S4). This cp genome was subsequently used for comparative analysis with the C. saxicola mt genome in order to identify the MTPTs.
3.2 Codon usage of PCGs
The 64 codons present in eukaryotic genomes are capable of encoding 20 different amino acids, among which Met and Trp are the only two that are not encoded by multiple codons. To investigate codon usage preference, we performed codon usage analysis on the 40 PCGs identified in the C. saxicola mt genome (Figure 2, Supplementary Table S5). With the exception of the start codon (AUG) and tryptophan (Trp, UGG), both of which had a RSCU of one, we observed a shared preference for codon usage in C. saxicola mt PCGs. Specifically, alanine (Ala) was preferentially encoded by GCU, with a RSCU of 1.60. The stop codon was preferentially encoded by UAA, with a RSCU of 1.54. Interestingly, phenylalanine (Phe) had a RSCU of 1.2, showing no clear codon usage preference.
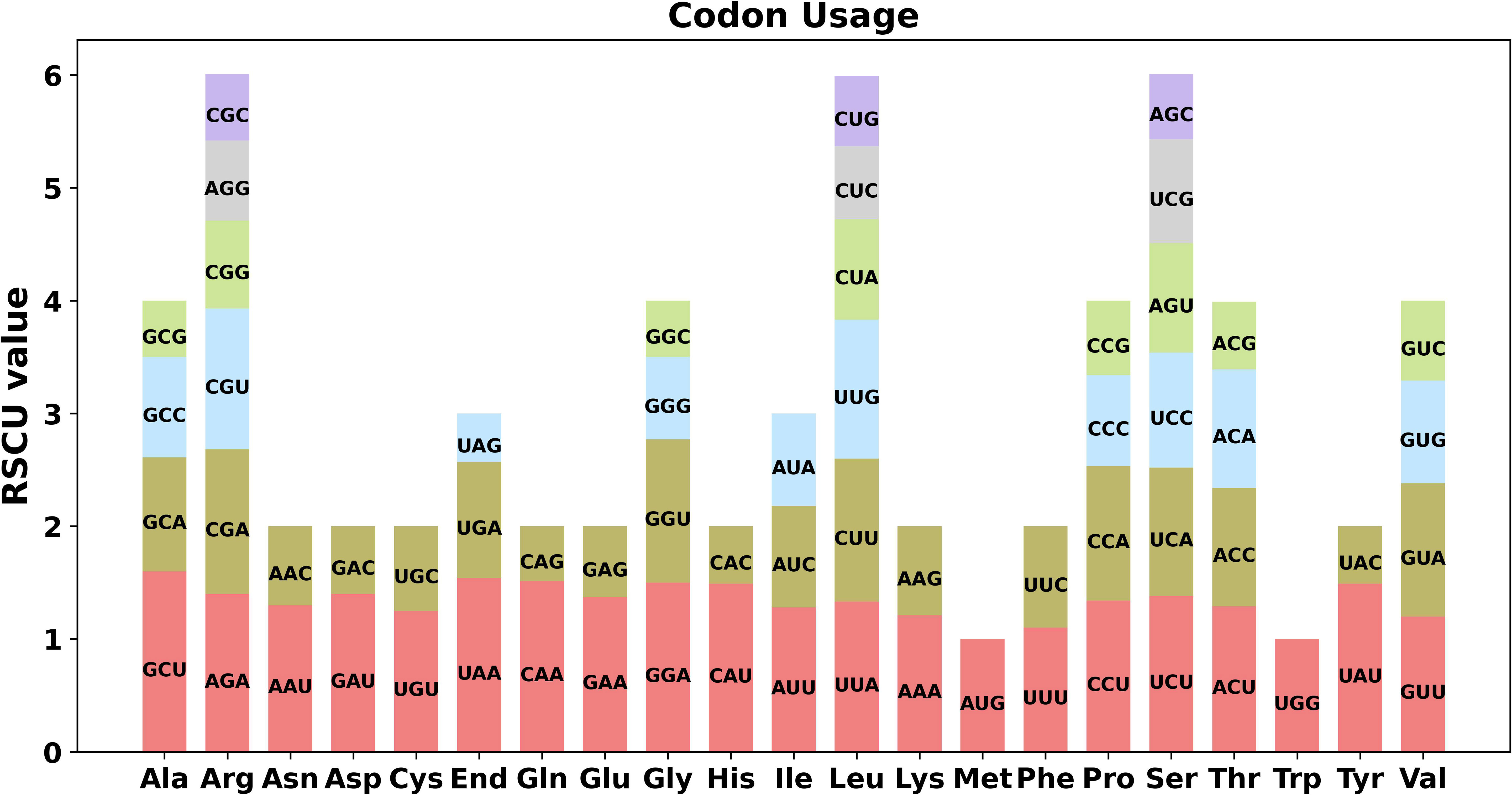
Figure 2 Codon usage of 20 amino acids and stop codons across all C. saxicola mt PCGs. The codons are represented by different colors in the histogram.
3.3 Repeated sequences
We identified 167 SSRs across the C. saxicola mt genome, including 41 mononucleotide, 42 dinucleotide, 17 trinucleotide, 61 tetranucleotide, five pentanucleotide and one hexanucleotide SSRs (Supplementary Figure S3A; Supplementary Table S6). Together, monomeric and dimeric SSRs made up 49.70% of all SSRs (Supplementary Figure S3A). Of the 13 monomeric SSRs, 53.85% (7) were adenine (A) monomeric repeats, and of the dimeric SSRs, 29.03% were TR (Supplementary Figure S3A). Tetrameric SSRs were the most common, representing 36.53% (61) of all SSRs (Supplementary Figure S3A; Supplementary Table S6). In addition, 17 TR, ranging from 12 to 42 bp in length, with a matching degree greater than 69%, were identified in the C. saxicola mt genome (Supplementary Figure S3B; Supplementary Table S7). Moreover, 797 pairs of dispersed repeats of at least 30 bases in length were identified, including 388 pairs of palindromic repeats, 408 pairs of forward repeats and one pair of reverse repeats (Supplementary Figure S3B; Supplementary Table S8). No complementary repeats were detected. The longest pairs of palindromic and forward repeats were 1559 bp and 4268 bp, respectively (Supplementary Table S8).
3.4 Identification of MTPTs
According to sequence similarity analysis, 12 homologous fragments were identified between the C. saxicola mt and ct genomes (Figure 3). The homologous fragments were 6135 bp in length, accounting for 1.04% of the entire mt genome (Supplementary Table S9). Among them, three fragments exceeded 1000 bp in length (Supplementary Table S9). These fragments were sourced from PCGs, tRNA genes and intergenic regions of the plastid genome (Supplementary Table S9). Specifically, 7 fragments were associated with either 13 partial or complete plastid PCGs, including psaB (partial), psbC (partial), psbD (partial), psbJ (partial), rpl14 (partial), rpoC2 (partial), petG, petL, psbE, psbF, psbL and psbZ (Supplementary Table S8). In addition, 7 homologous fragments encompassed 7 complete tRNA genes, including trnD-GUC, trnH-GUG, trnN-GUU, trnM-CAU, trnP-UGG, trnS-UGA and trnW-CCA (Supplementary Table S9).
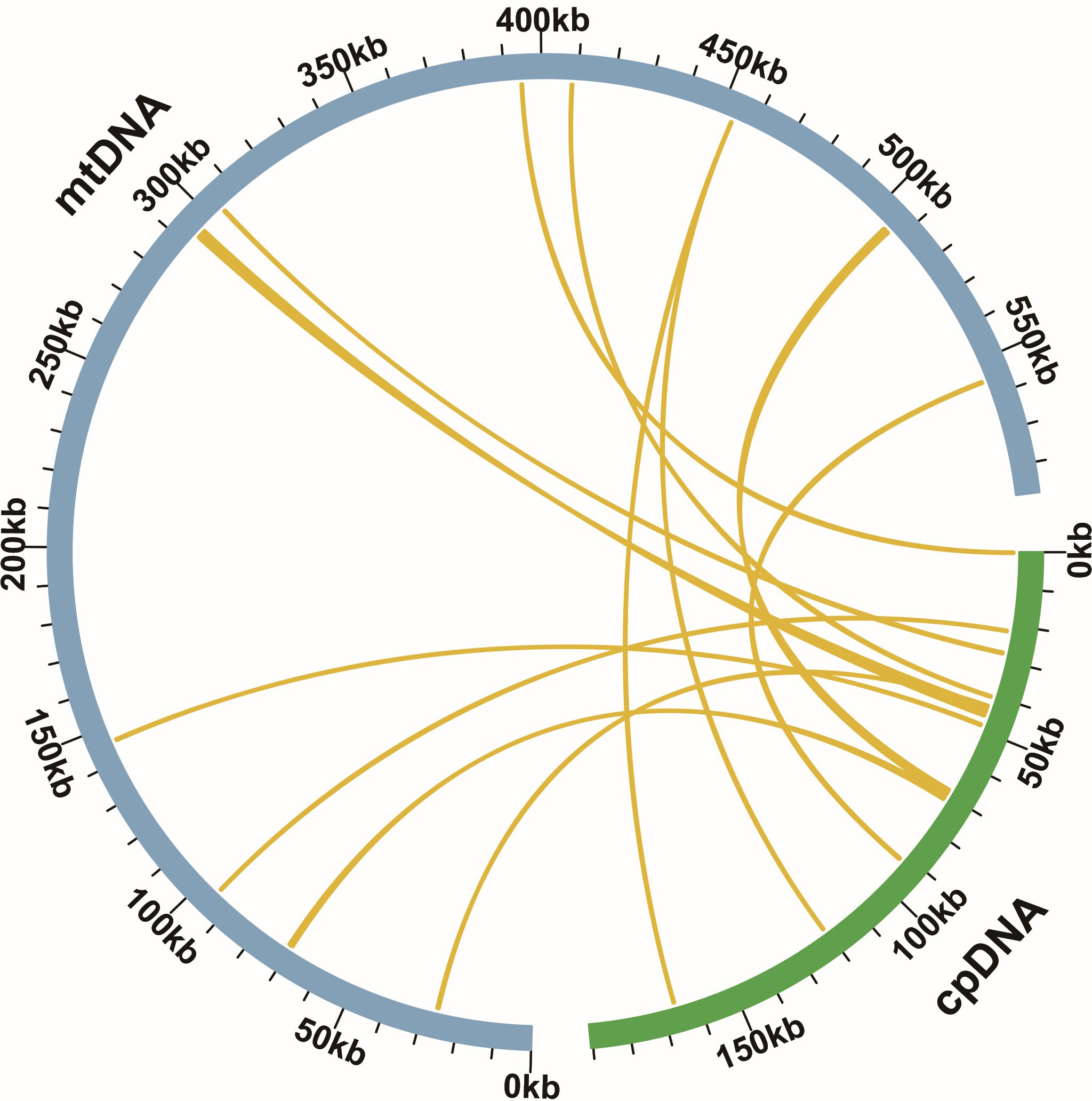
Figure 3 Homologous fragment analysis of C. saxicola cp and mt genomes. The blue and green arcs represent the mt genome and cp genome, respectively. The yellow lines correspond to homologous genomic fragments.
By analyzing sequence similarities, we created a multiple synteny plot of C. saxicola with 7 other related species. Several homologous, co-linear blocks were identified between C. saxicola and its relatives, particularly C. pauciovulata (Figure 4). Additionally, several regions that are exclusive to C. saxicola were discovered and these do not share similarities with other species. Moreover, we discovered that the co-linear blocks were not in the same order among the 8 species, indicating that extensive genomic rearrangements had occurred.

Figure 4 C. saxicola mt genome synteny. The bars signify the different mt genomes, with ribbons representing the homologous sequences between adjacent species. The red areas indicate where inversions have occurred, while the gray areas indicate regions of high homology. Blocks of less than 0.5 kb shared between species are not shown. Regions lacking a common block signify that they are exclusive to the species.
3.5 Phylogenetic and synteny analyses
To explore the evolutionary history of C. saxicola, we constructed a phylogenetic tree using the nucleotide sequences of 23 conserved core mt PCGs (atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, ccmFC, ccmFN, cob, cox2, cox3, matR, nad1, nad2, nad3, nad4L, nad5, nad6, nad7, nad9, rps3 and rps12) from 31 species representing four orders (Ranunculales, Proteales, Caryophyllales and Alismatales) (Figure 5, Supplementary Table S10). The mt genomes of Stratiotes aloides and Butomus umbellatus were selected as outgroups. Overall, the phylogenetic topology obtained from mt DNA corresponded to the most recent Angiosperm Phylogenetic Group (APG) classification (Figure 5). Unsurprisingly, C. saxicola (Papaveraceae family, Ranunculales order) was most closely related to C. pauciovoulata (Figure 5).
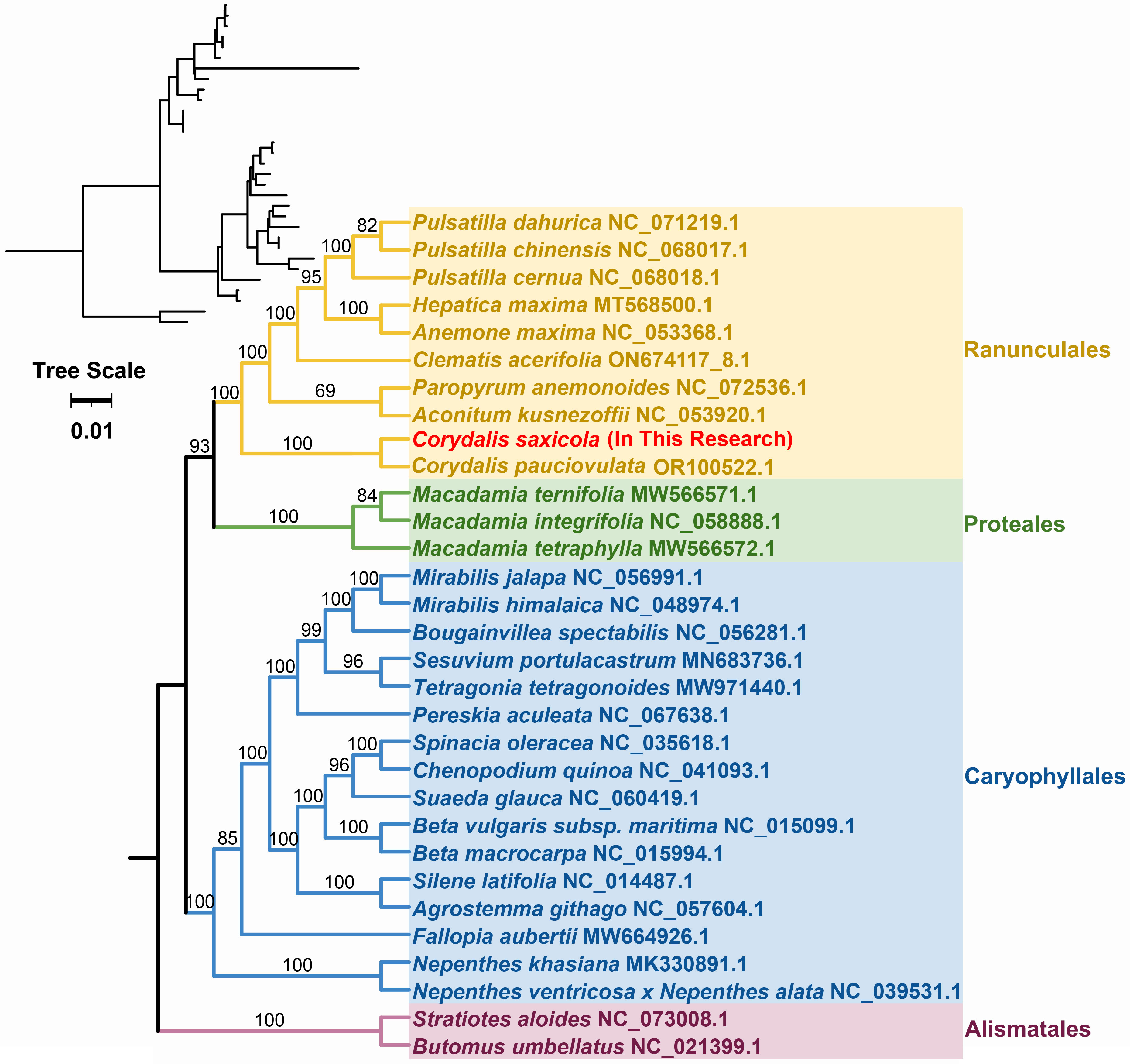
Figure 5 Phylogenetic analysis of C. saxicola and 30 other species based on the nucleotide sequences of 23 conserved core mt PCGs. S. aloides and B. umbellatus were selected as outgroups. The number at each node represents the bootstrap probability.
3.6 RNA editing events in C. saxicola
To identify potential RNA editing sites amongst the 40 C. saxicola mt PCGs, we carried out PCR amplification and Sanger sequencing of the genomic DNA (gDNA) and complementary DNA (cDNA) of all 40 PCGs. Primers were designed for flanking sequences of at least 40 bp upstream from either the start codon or downstream from the stop codon. If the gDNA contained one or more exons, primers were designed for flanking sequences upstream and downstream of each exon. The PCR amplification results are shown in Supplementary Figure S4, and the detailed primer information is listed in Supplementary Table S11. In general, the positions which varied between the gDNA and cDNA amplification products from the same gene indicated the presence of RNA editing sites. In all, we identified 546 ‘C to U’ RNA editing sites (Supplementary Tables S12, S13, Supplementary File A). Validation for 19 RNA editing sites within the cox3 gene is shown in Figure 6. Among the RNA editing sites, 25 (4.6%) were synonymous substitution sites and 516 were non-synonymous substitution sites (Supplementary Table S12). Two of RNA editing sites introduced termination codons into atp9 and rps10 (Supplementary Table S12). Among all the mt PCGs, nad4 contained the greatest number of RNA editing sites (54), while mttB and ccmB contained 46 and 43 editing events, respectively (Supplementary Tables S12, S13). Interestingly, no RNA editing sites were identified in rps11 and rps12 (Supplementary Tables S12, S13). Eight types of synonymous substitutions were identified, including Asp to Asp (1), Gly to Gly (1), Ile to Ile (6), Leu to Leu (4), Phe to Phe (6), Pro to Pro (3), Ser to Ser (3) and Tyr to Tyr (1) (Supplementary Table S12). Additionally, 14 types of non-synonymous substitutions were identified, including Ala to Val (4), Arg to Cys (43), Arg to stop codon (2), Arg to Trp (32), His to Tyr(22), Leu to Phe (19), Phe to Leu (1), Pro to Leu (119), Pro to Phe (12), Pro to Ser (48), Ser to Leu (117), Ser to Phe (81), Thr to Ile (10) and Thr to Met (7) (Supplementary Table S12).

Figure 6 Validation of 19 RNA editing sites within cox3. Each chromatogram displays the sequences before and after editing at the RNA editing site. The position of each site is indicated at the top and framed by grey dashed rectangles.
We further compared the validated RNA editing sites with those predicted by Deepred-mt (Edera et al., 2021), using a cutoff value of 0.9. A total of 779 RNA editing sites were predicted, and all were C-U (Figure 7; Supplementary Table S12). Among these predicted RNA editing sites, 506 (65.0%) were validated. Notably, 38 other RNA editing sites were validated but not predicted (Supplementary Table S12).
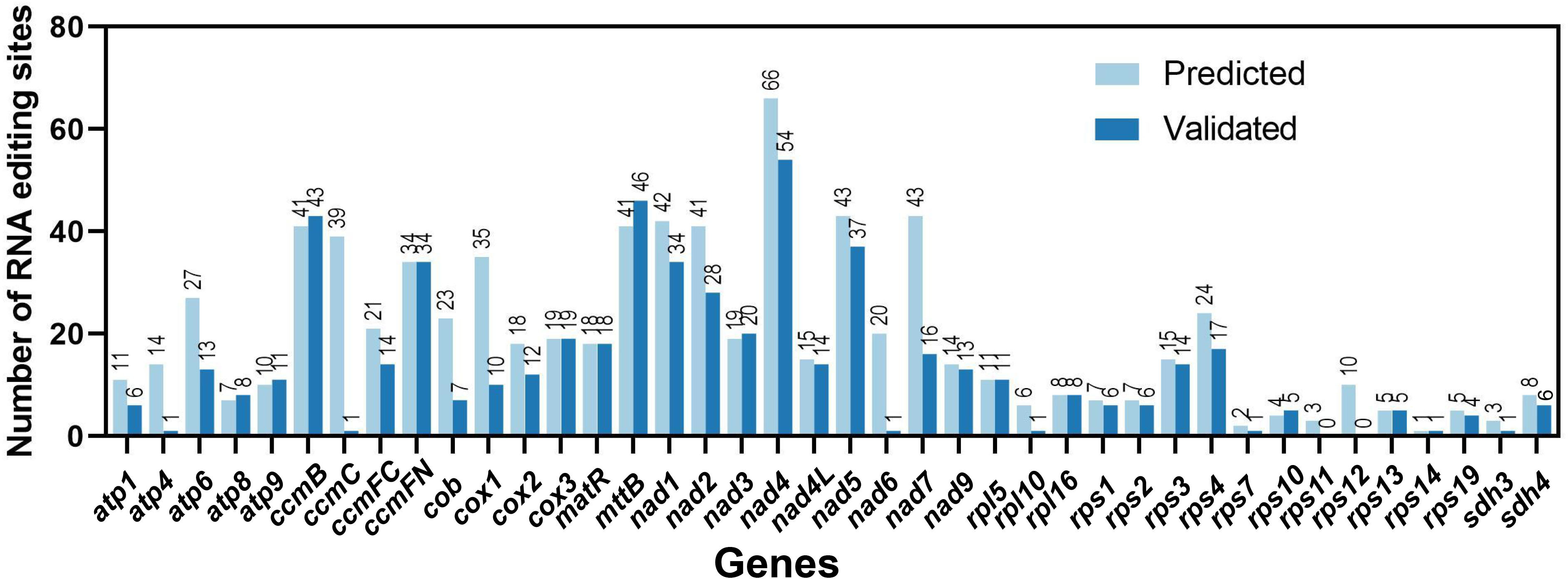
Figure 7 Prediction and validation of RNA editing sites among the 40 C. saxicola mt PCGs.
3.7 Transcript profiles of 24 core mt PCGs
To functionally characterize 24 core C. saxicola mt PCGs, we studied their expression profiles in various tissues via reverse transcription followed by quantitative real-time PCR (RT-qPCR). While transcriptional expression levels differed between the genes, the majority were minimally expressed in roots and flowers (Figure 8).
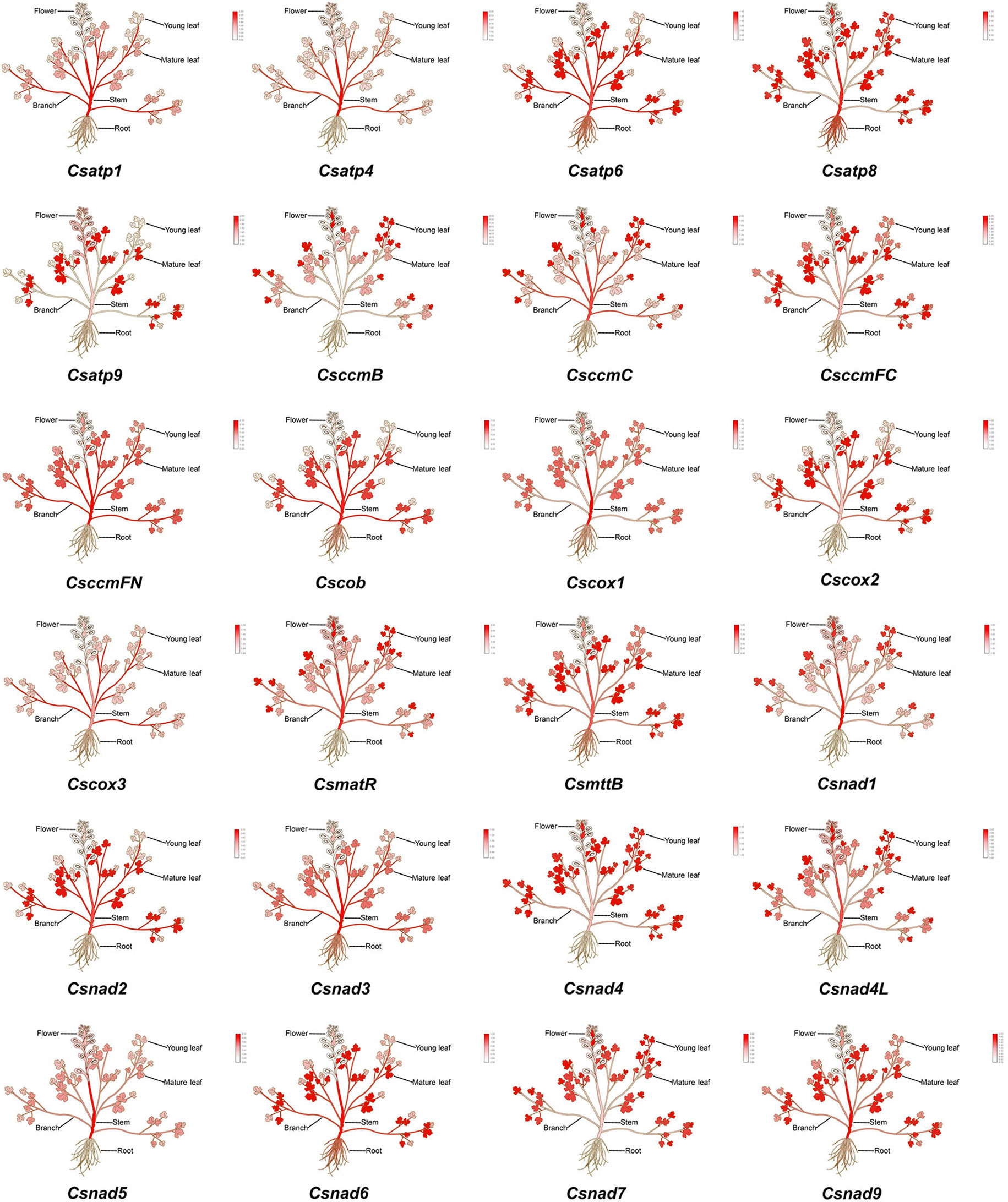
Figure 8 A heatmap of the relative expression levels of 24 core mt PCGs in the roots, stems, branches, mature leaves, young leaves and flowers of C. saxicola, as determined by RT-qPCR.
Karst habitats are distinguished by their calcium-rich limestone background, and this environment is home to many calcicole plants (Xue et al., 2018). As a plant native to karst regions, C. saxicola is particularly resistant to high concentrations of calcium. To investigate whether any of the C. saxicola mt PCGs are responsive to exogenous CaCl2, the expression levels of 24 core mt PCG transcripts were analyzed in C. saxicola roots by RT-qPCR. Interestingly, exogenous calcium resulted in the marked upregulation of the majority of the tested genes, with the exception of atp6, atp9, cox1, cox2 and cox3 (Figure 9). Compared to that in CK, the expression levels of atp6 were significantly lower in 100, 200, and 400 mmol/L CaCl2 treatments, while those of atp9 and cox1 were only significantly lower in 400 mmol/L CaCl2 treatments. However, the quantities of cox2 or cox3 transcripts in CaCl2 treatments were non-significantly different from CK.
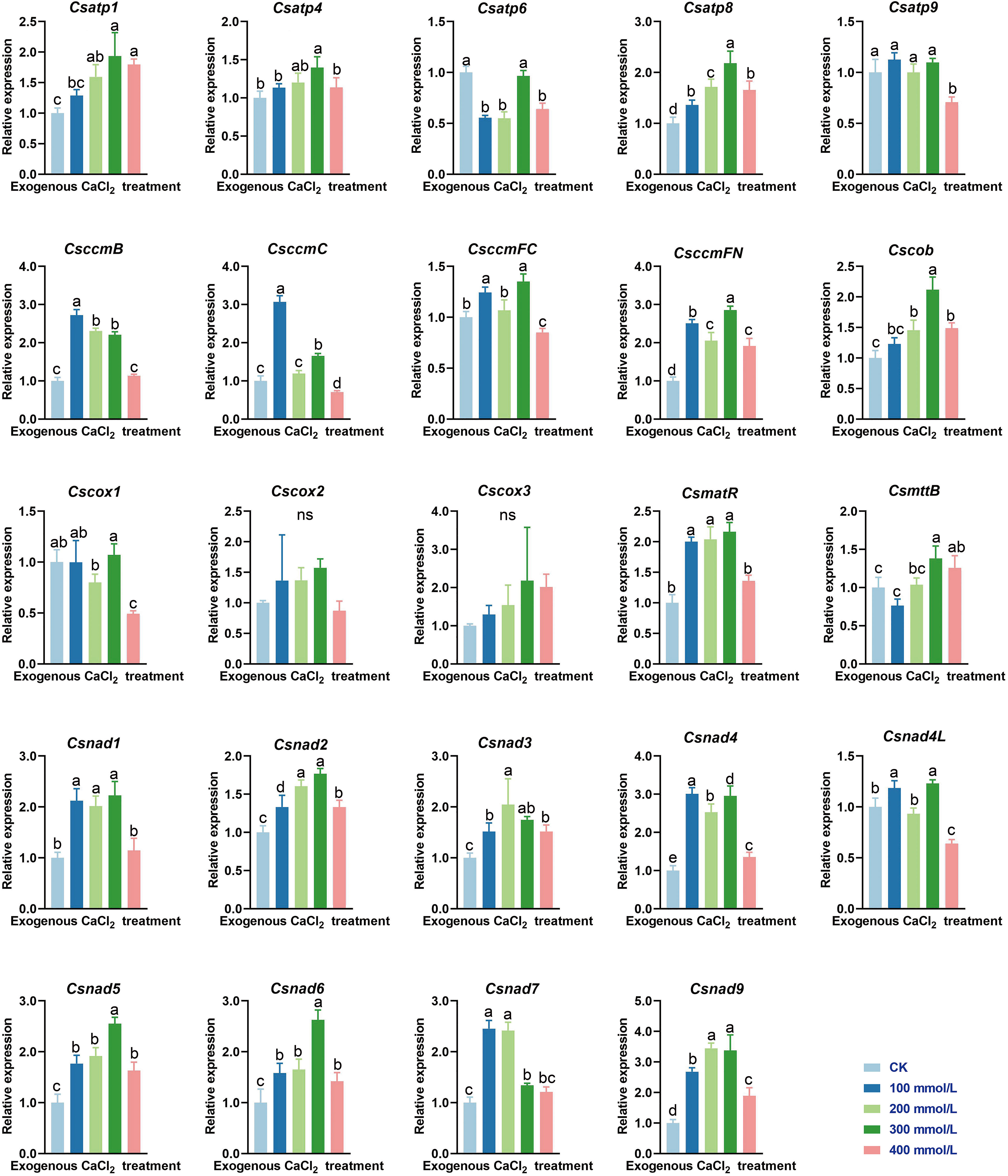
Figure 9 The relative expression levels of 24 core mt PCGs in C. saxicola roots grown under CaCl2 stress. The data are shown as the means ± SD of three replicates. Different lowercase letters indicate significant differences among treatments (ANOVA); differences were considered significant at p < 0.05.
4 Discussion
The study of mitochondria started in earnest around 1950 with the first isolation of mitochondria from animal and plant tissues (Møller et al., 2021). Mitochondria are essential for cellular functioning and are responsible for generating energy and mediating interactions between other organelles (Wang et al., 2022). Besides their role in respiration, plant mitochondria are actively engaged in a range of other cellular activities, such as photosynthesis as well as Crassulacean acid and C4 metabolism, heat generation, temperature regulation, immunity, biotic and abiotic stress resistance, programmed cell death and genomic evolution (Møller et al., 2021; Welchen et al., 2021; Le et al., 2022; Wang et al., 2022). As is the case with other eukaryotes, plant mitochondria are encased by two membranes and contain their own genetic material, the mt genome (Robles and Quesada, 2021). Similar to the cp genome, that of the mt exhibits a distinct organization and pattern of inheritance which is unlike that of the nuclear genome (Robles and Quesada, 2021). In addition, the mitochondria of animals and plants differ in several ways. First, many animal cells possess more than one mt genome copies per mitochondrion. However, plant cells contain mtDNA at a much lower rate than one copy per mitochondrion (Zhang et al., 2023). For example, direct observations via super-resolution microscopy suggest that human fibroblast cells contain at least 1.4 copies of the mt genome per mitochondrion (Kukata et al., 2011), while only one-third of A. thaliana leaf mitochondria contain mtDNA (Wang et al., 2010). Second, plant mt genomes range from ~66 kb to ~12 Mb, while animal mt genomes are far less variable in length (Wang et al., 2010). For example, mammalian mt genomes are generally 15-17 kb in length (Wang et al., 2010). Third, plant mitochondria often contain only partial mt genomes (Preuten et al., 2010; Wang et al., 2010; Rose, 2021). Finally, plant mt genomes exhibit diverse structures, including circular double-linked molecules as well as linear, multi-branched and polycyclic structures (Sloan, 2013; Gualberto et al., 2014; Kozik et al., 2019; Jackman et al., 2020). Such complexity has limited the comprehensive study of plant mt genomes.
4.1 Overview of the C. saxicola mt genome
Fortunately, the development of PacBio and Oxford Nanopore long-read sequencing techniques has made the sequencing of organellar genomes simpler and faster. However, assembling plant mt genomes often remains challenging due to their structural variety and numerous repetitive sequences. Here, we successfully assembled the mt genome of C. saxicola based on Oxford Nanopore and Illumina data. The C. saxicola mt genome is represented by a single-circle chromosome, 587,939 bp in length, and includes 65 genes (Figure 1; Table 1). These characteristics are similar to the closely related C. pauciovulata, which is the first mt genome to be published for any of the Corydalis species and it was also a single-circle structure, 675,483 bp in length and included 66 genes. To date, the largest plant mt genome, ~11.7 Mbp, has been recorded from Siberian larch (Larix sibirica Ledeb.) (Putintseva et al., 2020), which is approximately 21 times of the C. saxicola mt genome in size. Phylogenetic analysis indicated that C. saxicola is most closely related to C. pauciovoulata (Figure 5), and they share many homologous and co-linear blocks (Figure 4). These results indicate that our assembly and annotation of C. saxicola mt genome were reliable, and that genome structure, genome arrangement and gene number may be conserved to some extent among the Corydalis species, at least in C. saxicola and C. pauciovoulata. However, more mt genomes of Corydalis or Papaveraceae species need to be sequenced to analyze their variations and evolution.
Approximately 10% of the plant mt genomes that have been sequenced contain multiple chromosome structures (Wu et al., 2022). Recombination events are known to alter the abundance of sub-genomic molecules (Preuten et al., 2010). In the draft map of the C. saxicola mt genome, the coverage of contig 3 was nearly double that of either contig 1 or contig 2 (Supplementary Figure S1; Supplementary Table S2), indicating that the recombination configurations such as two smaller circles might be present in the C. saxicola mitochondria. In contrast to animal mt genomes, which exhibit very few rearrangements, continuous rearrangements are known to occur in plant mt genomes (Gualberto and Newton, 2017). These may be caused directly by continuous fusion and fission of plant mitochondria, resulting in the reunification of both damaged and undamaged mtDNA (Lannera and Connor, 2001; Gualberto and Newton, 2017). This would significantly decrease the number of point mutations, and potentially increase the longevity of certain plants (Lannera and Connor, 2001; Gualberto and Newton, 2017). Frequent intermolecular rearrangements of mtDNA might also have occurred in C. saxicola mitochondria, as evidenced by the number of SSRs, TR and dispersed repeats identified in the C. saxicola mt genome (Supplementary Figure S3; Supplementary Tables S6-S8). To date, whether these repetitive elements redundant or functional are largely unknown, but they determine the mt genome size to some extent (Putintseva et al., 2020; Wu et al., 2022). Additionally, whether and when multiple conformations exist and what functions they carry out also require further investigations.
In plants, mt genomes not only rearrange with each other, but also communicate with plastids, nuclei and even other species (Mower et al., 2012). Here, we identified a total of 12 homologous fragments between the mt and ct genomes of C. saxicola (Figure 3), accounting for 1.04% of the entire mt genome (Supplementary Table S9). These fragments contained PCGs, tRNA genes and intergenic regions of the plastid genome (Supplementary Table S9). Compared to many other species, the proportion of MTPTs was relatively low between the mt and cp genomes of C. saxicola (Jiang et al., 2023a; Jiang et al., 2023b; Shan et al., 2023). It concluded that plastid-related sequences are not responsible for the mt genome size (Putintseva et al., 2020), however, MTPTs and nuclear mitochondrial transferred fragments are crucial for mt genome evolution (Wang D. et al., 2012; Jiang et al., 2023b). Interestingly, all the tRNA genes contained in MTPT fragments were complete (Jiang et al., 2023a; Jiang et al., 2023b; Shan et al., 2023), indicating that they may still be functional. However, some of the transferred PCGs were incomplete (Supplementary Table S9), and this phenomenon has also been observed in other species (Jiang et al., 2023a). Whether these PCGs are still functional is an open question.
4.2 RNA editing events in the mt C. Saxicola genome
RNA editing, resulting in the post-translational insertion, deletion and conversion of nucleotides into coding and non-coding regions of RNA molecules, is known to occur in the nucleus, cytoplasm and DNA-containing organelles (Covello and Gray, 1989; Hiesel et al., 1989; Maier et al., 1996; Maldonado et al., 2022). Since its discovery in 1986 (Benne et al., 1986), RNA editing has been identified in animals, plants, lycophytes, ferns, protozoa and some viruses, with most organisms exhibiting variable preferred types (Hao et al., 2021). In particular, the mitochondria and chloroplasts of land plants exhibit extensive RNA editing, with C/U editing being the most common (Small et al., 2020). Furthermore, mitochondria contain approximately 20 times more RNA editing sites than plastids (Giegé and Brennicke, 1999). In both mitochondria and chloroplasts, RNA editing is involved in male sterility, seed development, environmental adaptation and pathogen resistance (García-Andrade et al., 2013; Yan et al., 2018; Yang et al., 2020). Although several methods have been developed for the detection of RNA editing sites and levels, Sanger sequencing is still the most reliable.
Here, a total of 779 ‘C to U’ RNA editing sites were predicted in the 40 C. saxicola mt PCGs by Deepred-mt (Figure 7; Supplementary Table S12) (Edera et al., 2021). Among these predicted RNA editing sites, 506 (65.0%) were validated using a combination of PCR amplification and Sanger sequencing (Supplementary Figure S4). Additionally, 38 other RNA editing sites were validated but not predicted (Supplementary Table S12). Several reasons may be responsible for these discrepancies: (1) the deep RNA sequencing data of 21 plant mt genomes, selected for the training dataset of Deepred-mt, might be not as comprehensive as possible to predict all ‘C to U’ RNA editing sites in variable plant mt genomes; (2) although Deepred-mt exhibits higher precision in the prediction of RNA editing sites compared to other two state-of-the-art predictive methods, PREP-Mt (Mower, 2009) and PREPACT (Lenz et al., 2018), the accuracy is not 100%, and the deviation might be due to efficient but still imperfect learning strategies or other factors used in computer simulation (Edera et al., 2021). In all, 546 ‘C to U’ RNA editing sites were validated, 25 (4.6%) of which were synonymous substitution sites (Supplementary Table S12). This proportion is similar to the mt genome of Taraxacum mongolicum, in which validated synonymous conversions accounted for 4.69% (10/216) (Jiang et al., 2023a). Among the 25 synonymous substitution sites, 21 occurred on the third bases of triplet codons, and the rest occurred on the first bases of triplet codons. These four synonymous conversions were identified in atp9 (base position 209), mttB (base position 37), nad1 (base position 823) and rps10 (base position 7), and all represented ‘Leu to Leu’ substitutions (Supplementary Table S12). The majority of RNA editing sites occurred in the first and second positions of triplet codons, resulting in synonymous substitutions (Supplementary Table S12). This phenomenon has also been observed in A. thaliana (Giegé and Brennicke, 1999). Among the mt PCGs, nad4 contained the greatest number of RNA editing sites (54), while mttB and ccmB contained 46 and 43 editing sites, respectively (Supplementary Tables S12, S13). Nad4 and the other eight mt nad proteins (nad1, nad2, nad3, nad4L, nad5, nad6, nad7 and nad9) are components of complex I, which are the largest of the respiratory complexes found in the mitochondria of nearly all land plants (Møller et al., 2021). Multiple RNA editing sites were validated among the nad genes (Supplementary Tables S12, S13), indicating that post-transcriptional modifications like RNA editing may be crucial to the functioning of complex I. Interestingly, analysis of the RNA-editing sites from 17 plant species also showed that nad4 contained the greatest number, consistent with our results (Figure 7, Supplementary Tables S12, S13) (Maldonado et al., 2022). Notably, the number of RNA editing sites appears to be disproportionate to the length of the gene (Supplementary Table S11) (Maldonado et al., 2022; Jiang et al., 2023a; Li et al., 2023).
Furthermore, two stop codons and one traditional start codon (AUG) were found to be introduced by RNA editing events (Supplementary Table S12). One of the stop codons occurred in atp9 and another occurred in rps10. Both events were CGA to UGA, resulting in the conversation of Arg into a stop codon (Supplementary Table S12). The validated conversion of ACG to AUG introduced a traditional start codon into nad4L (Supplementary Table S12). It should be noted that unedited ACG may also serve as a start codon in plant plastids and mitochondria (Dong et al., 1998; Zandueta-Criado and Bock, 2004). Studies have suggested that C-to-U editing results in a reduced rate of TT-dimers in DNA, increasing the UV resistance of land plants (Binder and Brennicke, 2003; Fujii and Small, 2011). Whether the conversion of ACG to AUG influences translation efficiency requires further study.
4.3 Transcript profiles of 24 core C. saxicola mt PCGs
Most mitochondrial genes are combined with other unrelated genes into poly-cistronic primary transcripts, and endo- and exo-nucleolytic reactions process the transcripts (Møller et al., 2021). RT-qPCR indicated that the expression of 24 core C. saxicola mt PCGs differed among the different organs (Figure 8). Notably, the majority exhibit relatively higher expression in aerial tissues, including stems, branches, and mature and young leaves, but minimal expression in the roots and flowers (Figure 8). Interestingly, cavidines, especially dehydrocavidine, are known to be present mostly in the roots of Corydalis species (Xu et al., 2022b). Our studies suggest that the content of dehydrocavidine is highest in flowers, followed by the roots (data not shown). Whether these results represent correlation or coincidence requires further investigation.
In plants, Ca2+ not only provides structural stability, but also acts as a dynamic intracellular messenger involved in stress response and development (He et al., 2021; Pirayesh et al., 2021). Ca2+ was also demonstrated to be involved in the biosynthesis of secondary metabolites (Martins et al., 2021; Ahmad et al., 2022). As a plant native to karst regions, C. saxicola is particularly resistant to calcium. In addition, our work indicates that high Ca2+ concentrations result in a significant increase in the dehydrocaverdine content of the roots of C. saxicola (Supplementary Figure S5). Research has revealed a physiological relationship between Ca2+ and mitochondrial function (Pirayesh et al., 2021). To investigate whether and to what extent the C. saxicola mt PCGs are responsive to exogenous CaCl2 treatment, we performed RT-qPCR to analyze the relative expression of 24 core mt PCGs in C. saxicola roots. Interestingly, the majority of these genes exhibited upregulated expression in response to exogenous CaCl2 treatment (Figure 9). These results demonstrate that high concentrations of exogenous Ca2+ influence the mitochondrial function in C. saxicola, as all 24 core mt genes were components of the mitochondrial electron transport chain (Møller et al., 2021). It has been reported that the concentration of [Ca2+] mito is approximately double that of [Ca2+]cyt under stress, and that entry of these cations into the mitochondria is critical to prevent its accumulation in the cytosol (He et al., 2021; Pirayesh et al., 2021). However, whether the concentration of [Ca2+] mito in C. saxicola roots is higher than that of [Ca2+]cyt still needs to be investigated. Furthermore, the expression of nuclear-encoded components should be analyzed to validate the functional elevation of respiratory complexes. Additionally, whether the upregulated expression of these mt PCGs in response to exogenous CaCl2 resulted in increased dehydrocaverdin biosynthesis requires further study. One possible explanation is that O-methyltransferases, the key enzymes involved in the biosynthesis of dehydrocaverdin, require ATP to transfer the methyl group from S-adenosylmethionine to its specific substrates (Xu et al., 2022b). Recently, highly efficient mt genome-editing techniques have been developed (Kang et al., 2021; Nakazato et al., 2022; Forner et al., 2023), which will facilitate the functional validation of mt genes.
5 Conclusions
In this study, we mapped out the mt genome of C. saxicola, an endangered medicinal plant endemic to karst habitats. Variable aspects of the mt genome have been investigated, including the basic composition, codon preference, repeat sequences, MTPTs and genome comparisons with those of closely related species. These discoveries can be employed to create molecular markers and assist scientists in comprehending the evolution and how to classify the plants within the Corydalis species.
We performed an extensive analysis of the RNA editing events in 40 C. saxicola mt PCGs and validated 70% (65% predicted ones and 5% were unpredicted) of these by using PCR amplification and Sanger sequencing. Additionally, we transcriptionally profiled 24 core mt PCGs in different tissues of C. saxicola. The results showed the majority were minimally expressed in roots and flowers. Interestingly, most of these genes exhibited upregulated expression in response to exogenous CaCl2 treatment. These investigations will be helpful in understanding the biological functions of the mt PCGs towards variable biotic and abiotic stresses.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Fresh and healthy C. saxicola tissues were collected for this study. C. saxicola was identified by Z-JZ and CL of GBGMP (Nanning, Guangxi Zhuang Autonomous Region, China). Voucher specimens (reference number: 450123130505040LY) were deposited in the GBGMP Herbarium. This study, including plant sample collection, complies with relevant institutional, national and international guidelines and legislation. No specific permits were required for plant collection.
Author contributions
CL: Writing – original draft, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Software. HL: Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – original draft. MQ: Validation, Writing – review & editing. Y-JT: Validation, Writing – review & editing. X-LO: Validation, Writing – review & editing. X-YC: Formal Analysis, Writing – original draft. YW: Formal Analysis, Resources, Writing – review & editing. Z-JZ: Conceptualization, Investigation, Writing – review & editing. ML: Conceptualization, Data curation, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Natural Science Foundation of Guangxi (2020GXNSFAA159151; 2023GXNSFAA026509); Guangxi Key R&D Program (Guike AB23026092); Guangxi Key Laboratory of Medicinal Resources Protection and Genetic Improvement (KL2022ZZ04, KL2023ZZ09); Guangxi Appropriate Technology Development and Promotion Project of Traditional Chinese Medicine (GZSY22-02) and Key Techniques Research and Promotion of Guangxi Medicinal Materials Varieties (GZKJ2314). The funders were not involved in the study design, data collection, analysis, publication decision and manuscript preparation.
Acknowledgments
The authors sincerely thank the experimental personnel and bioinformatics analysis at MitoRun research group participated in this project. We would also give our special thanks to Associate Professor Cheng-jie Chen from South China Agricultural University and Associate Research Fellow Ai-ping Luan from Chinese Academy of Tropical Agricultural Sciences for their kindly aid supported in the heat map illustration. Lastly, we appreciate the linguistic assistance provided by TopEdit (www.topeditsci.com) during the preparation of this manuscript and we thank Dr Dev Sooranna of Imperial College in London for editing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1332460/full#supplementary-material
Supplementary Figure 1 | The uniting graph of the mt genome of C. saxicola obtained from the de novo assembly of Illumina reads. Three contigs (ctg1, ctg2 and ctg3) were found and these are depicted in different colors in the graph.
Supplementary Figure 2 | The cp genome map of C. saxicola. Genes drawn inside and outside the circle are transcribed clockwise and counterclockwise, respectively. Genes belonging to different functional groups are color-coded. The light and darker gray in the inner circle correspond to Adenine-Thymine (AT) and GC content, respectively. LSC, large single-copy; SSC, small single-copy; IR, inverted repeat.
Supplementary Figure 3 | Repeated sequences in the C. saxicola mt genome. (A) Distribution of SSRs in the C. saxicola mt genome. (B) Distribution of tandem and dispersed repeats in the C. saxicola mt genome.
Supplementary Figure 4 | Agarose gel electrophoresis of gDNA and cDNA PCR products of 40 C. saxicola mt PCGs. The first well was loaded with 5 µL of marker, and the rest of the wells were loaded with 10 µL of PCR products for each gene. M: marker; I, II, III, IV, and V: exon-contained gene fragments.
Supplementary Figure 5 | The content of dehydrocavidine in variable organs of C. saxicola treated with different concentrations of CaCl2. Different lowercase letters indicate significant differences among treatments (ANOVA); differences were considered significant at p < 0.05.
References
Ahmad, M. A., Deng, X., Adeel, M., Rizwan, M., Shakoor, N., Yang, Y., et al. (2022). Influence of calcium and magnesium elimination on plant biomass and secondary metabolites of Stevia rebaudiana Bertoni. Biotechnol. Appl. Biochem. 69 (5), 2008–2016. doi: 10.1002/bab.2263
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Benne, R., Van Den Burg, J., Brakenhoff, J. P., Sloof, P., Van Boom, J. H., Tromp, M. C. (1986). Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 46 (6), 819–826. doi: 10.1016/0092-8674(86)90063-2
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi: 10.1093/nar/27.2.573
Bhakuni, D. S., Chaturvedi, R. (1983). The alkaloids of Corydalis meifolia. J. Nat. Prod. 46, 320–324. doi: 10.1021/np50027a004
Binder, S., Brennicke, A. (2003). Gene expression in plant mitochondria: transcriptional and post-transcriptional control. Philos. Trans. R. Soc Lond. B. Biol. Sci. 358 (1429), 181–188. doi: 10.1098/rstb.2002.1179
Chen, C., Wu, Y., Li, J., Wang, X., Zeng, Z., Xu, J., et al. (2023a). TBtools-II: a “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16 (11), 1733–1742. doi: 10.1016/j.molp.2023.09.010
Chen, J. T., Liden, M., Huang, X. H., Zhang, L., Zhang, X. J., Kuang, T. H., et al. (2023b). An updated classification for the hyper-diverse genus Corydalis (Papaveraceae: fumarioideae) based on phylogenomic and morphological evidence. J. Integr. Plant Biol. 65 (9), 2138–2156. doi: 10.1111/jipb.13499
Chen, Y., Ye, W., Zhang, Y., Xu, Y. (2015). High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 43, 7762–7768. doi: 10.1093/nar/gkv784
Cole, L. W., Guo, W., Mower, J. P., Palmer, J. D. (2018). High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evol. 35, 2773–2785. doi: 10.1093/molbev/msy176
Covello, P. S., Gray, M. W. (1989). RNA editing in plant mitochondria. Nature 341 (6243), 662–666. doi: 10.1038/341662a0
Dong, F. G., Wilson, K. G., Makaroff, C. A. (1998). The radish (Raphanus sativus L.) mitochondrial cox2 gene contains an ACG at the predicted translation initiation site. Curr. Genet. 34 (2), 79–87. doi: 10.1007/s002940050369
Edera, A. A., Small, I., Milone, D. H., Sanchez-Puerta, M. V. (2021). Deepred-Mt: deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 136, 104682. doi: 10.1016/j.compbiomed.2021.104682
Ehlers, B. K., Olesen, J. M. (2004). Flower production in relation to individual plant age and leaf production among different patches of Corydalis intermedia. Plant Ecol. 174, 71–78. doi: 10.1023/B:VEGE.0000046060.77491.b9
Feng, C., Yong, X., Jiang, Q., Su, Z., Liu, Z., Wu, T., et al. (2023). Inhibitory effects of Corydalis saxicola Bunting total alkaloids on macrophage pyroptosis. Chem. Biodiversity 20 (8), e202201255. doi: 10.1002/cbdv.202201255
Forner, J., Kleinschmidt, D., Meyer, E. H., Gremmels, J., Morbitzer, R., Lahaye, T., et al. (2023). Targeted knock-out of a conserved plant mitochondrial gene by genome editing. Nat. Plants 9, 1818–1831. doi: 10.1038/S41477-023-01538-2
Fujii, S., Small, I. (2011). The evolution of RNA editing and pentatricopeptide repeat genes. New Phytol. 191 (1), 37–47. doi: 10.1111/j.1469-8137.2011.03746.x
García-Andrade, J., Ramírez, V., López, A., Vera, P. (2013). Mediated plastid RNA editing in plant immunity. PloS Pathog. 9 (10), e1003713. doi: 10.1371/journal.ppat.1003713
Giegé, P., Brennicke, A. (1999). RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc. Natl. Acad. Sci. U.S.A. 96 (26), 15324–15329. doi: 10.1073/pnas.96.26.15324
Gualberto, J. M., Mileshina, D., Wallet, C., Niazi, A. K., Weber-Lotfi, F., Dietrich, A. (2014). The plant mitochondrial genome: dynamics and maintenance. Biochimie. 100, 107–120. doi: 10.1016/j.biochi.2013.09.016
Gualberto, J. M., Newton, K. J. (2017). Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 68, 225–252. doi: 10.1146/annurev-arplant-043015-112232
Hao, W., Liu, G., Wang, W., Shen, W., Zhao, W., Sun, J., et al. (2021). RNA editing and its roles in plant organelles. Front. Genet. 12. doi: 10.3389/fgene.2021.757109
He, J., Rössner, N., Hoang, M. T. T., Alejandro, S., Peiter, E. (2021). Transport, functions, and interaction of calcium and manganese in plant organellar compartments. Plant Physiol. 187 (4), 1940–1972. doi: 10.1093/plphys/kiab122
Hiesel, R., Wissinger, B., Schuster, W., Brennicke, A. (1989). RNA editing in plant mitochondria. Science 246 (4937), 1632–1634. doi: 10.1126/science.2480644
Jackman, S. D., Coombe, L., Warren, R. L., Kirk, H., Trinh, E., MacLeod, T., et al. (2020). Complete mitochondrial genome of a Gymnosperm, sitka spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol. 12, 1174–1179. doi: 10.1093/gbe/evaa108
Jiang, M., Ni, Y., Li, J., Liu, C. (2023a). Characterisation of the complete mitochondrial genome of Taraxacum mongolicum revealed five repeat-mediated recombinations. Plant Cell Rep. 42 (4), 775–789. doi: 10.1007/s00299-023-02994-y
Jiang, M., Ni, Y., Zhang, J., Li, J., Liu, C. (2023b). Complete mitochondrial genome of Mentha spicata L. reveals multiple chromosomal configurations and RNA editing events. Int. J. Biol. Macromol. 251, 126257. doi: 10.1016/j.ijbiomac.2023.126257
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., Li, D. Z. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21 (1), 241. doi: 10.1186/s13059-020-02154-5
Kang, B.-C., Bae, S.-J., Lee, S., Lee, J. S., Kim, A., Lee, H., et al. (2021). Chloroplast and mitochondrial DNA editing in plants. Nat. Plants 7 (7), 899–905. doi: 10.1038/s41477-021-00943-9
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kim, Y. A., Kong, C. S., Yea, S. S., Seo, Y. (2010). Constituents of Corydalis heterocarpa and their anti-proliferative effects on human cancer cells. Food Chem. Toxicol. 48, 722–728. doi: 10.1016/j.fct.2009.12.002
Kim, J. H., Ryu, Y. B., Lee, W. S., Kim, Y. H. (2014). Neuraminidase inhibitory activities of quaternary isoquinoline alkaloids from Corydalis turtschaninovii rhizome. Bioorg. Med. Chem. 22, 6047–6052. doi: 10.1016/j.bmc.2014.09.004
Kolmogorov, M., Yuan, J., Lin, Y., Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: one ring does not rule them all. PloS Genet. 15, e1008373. doi: 10.1371/journal.pgen.1008373
Kudo, G., Maeda, T., Narita, K. (2001). Variation in floral sex allocation and reproductive success within inflorescences of Corydalis ambigua (Fumariaceae): pollination efficiency or resource limitation? J. Ecol. 89, 48–56. doi: 10.1046/j.1365-2745.2001.00512.x
Kukata, C., Wurm, C. A., Spåhra, H., Falkenbergd, M., Larssona, N.-G., Jakobs, S. (2011). Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. U.S.A. 108 (33), 13534–13539. doi: 10.1073/pnas.1109263108
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 (22), 4633–4642. doi: 10.1093/nar/29.22.4633
Lannera, R. M., Connor, K. F. (2001). Does bristlecone pine senesce? Exp. Gerontol. 36 (4-6), 675–685. doi: 10.1016/s0531-5565(00)00234-5
Le, X. H., Lee, C. P., Monachello, D., Millar, A. H. (2022). Metabolic evidence for distinct pyruvate pools inside plant mitochondria. Nat. Plants. 8 (6), 694–705. doi: 10.1038/s41477-022-01165-3
Lenz, H., Hein, A., Knoop, V. (2018). Plant organelle RNA editing and its specificity factors: enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinf. 19, 255. doi: 10.1186/s12859-018-2244-9
Letunic, I., Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47 (W1), W256–W259. doi: 10.1093/nar/gkz239
Lewis, S. E., Searle, S. M. J., Harris, N., Gibson, M., Iyer, V., Richter, J., et al. (2002). Apollo: a sequence annotation editor. Genome Biol. 3 (12), research0082.1. doi: 10.1186/gb-2002-3-12-research0082
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, W., Huang, H., Zhang, Y., Fan, T., Liu, X., Xing, W., et al. (2013). Anti-inflammatory effect of tetrahydrocoptisine from Corydalis impatiens is a function of possible inhibition of TNF-alpha, IL-6 and NO production in lipopolysaccharide-stimulated peritoneal macrophages through inhibiting NF-kappa B activation and MAPK pathway. Eur. J. Pharmacol. 715, 62–71. doi: 10.1016/j.ejphar.2013.06.017
Li, J., Tang, H., Luo, H., Tang, J., Zhong, N., Xiao, L. (2023). Complete mitochondrial genome assembly and comparison of Camellia sinensis var. Assamica cv. Duntsa. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1117002
Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. (2016). The role of mitochondria in plant development and stress tolerance. Free Radic. Biol. Med. 100, 238–256. doi: 10.1016/j.freeradbiomed.2016.03.033
Lidén, M., Zetterlund, H. (1997). Corydalis: a gardener’s guide and a monograph of the tuberous species Vol. 140 (Worcestershire: Alpine Garden Society).
Liu, Y., Medina, R., Goffinet, B. (2014). 350 my of mitochondrial genome stasis in mosses, an early land plant lineage. Mol. Biol. Evol. 31, 2586–2591. doi: 10.1093/molbev/msu199
Liu, S., Ni, Y., Li, J., Zhang, X., Yang, H., Chen, H., et al. (2023). CPGView: a package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 23 (3), 694–704. doi: 10.1111/1755-0998.13729
Liu, X., Zheng, H., Lu, R., Huang, H., Zhu, H., Yin, C., et al. (2019). Intervening effects of total alkaloids of Corydalis saxicola Bunting on rats with antibiotic-induced gut microbiota dysbiosis based on 16S rRNA gene sequencing and untargeted metabolomics analyses. Front. Microb. 10, 1151. doi: 10.3389/fmicb.2019.01151
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25 (4), 402–408. doi: 10.1006/meth.2001.1262
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/gkab688
Maier, R. M., Zeltz, P., Kössel, H., Bonnard, G., Gualberto, J. M., Grienenberger, J. M. (1996). RNA editing in plant mitochondria and chloroplasts. Plant Mol. Biol. 32 (1-2), 343–365. doi: 10.1007/BF00039390
Maldonado, M., Abe, K. M., Letts, J. A. (2022). A structural perspective on the RNA editing of plant respiratory complexes. Int. J. Mol. Sci. 23, 684. doi: 10.3390/ijms23020684
Martins, V., Unlubayir, M., Teixeira, A., Gerós, H., Lanoue, A. (2021). Calcium and methyl jasmonate cross-talk in the secondary metabolism of grape cells. Plant Physiol. Biochem. 165, 228–238. doi: 10.1016/j.plaphy.2021.05.034
Michael, T., Pascal, L., Tommaso, P., Ulbricht-Jones, E. S., Axel, F., Ralph, B., et al. (2017). GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Møller, I. M., Rasmusson, A. G., Van Aken, O. (2021). Plant mitochondria - past, present and future. Plant J. 108 (4), 912–959. doi: 10.1111/tpj.15495
Mower, J. P. (2009). The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 37, W253–W259. doi: 10.1093/nar/gkp337
Mower, J. P., Case, A. L., Floro, E. R., Willis, J. H. (2012). Evidence against equimolarity of large repeat arrangements and a predominant master circle structure of the mitochondrial genome from a monkeyflower (Mimulus guttatus) lineage with cryptic CMS. Genome Biol. Evol. 4 (5), 670–686. doi: 10.1093/gbe/evs042
Nakazato, I., Okuno, M., Zhou, C., Itoh, T., Tsutsumi, N., Takenaka, M., et al. (2022). Targeted base editing in the mitochondrial genome of Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 119 (20), e2121177119. doi: 10.1073/pnas.2121177119
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Niu, Y., Chen, G., Peng, D. L., Song, B., Yang, Y., Li, Z. M., et al. (2014). Grey leaves in an alpine plant: a cryptic colouration to avoid attack? New Phytol. 203, 953–963. doi: 10.1111/nph.12834
Niu, Y., Chen, Z., Stevens, M., Sun, H. (2017). Divergence in cryptic leaf colour provides local camouflage in an alpine plant. Proc. R. Soc B. 284, 20171654. doi: 10.1098/rspb.2017.1654
Novak, Z., Chlebek, J., Opletal, L., Jiros, P., Macakova, K., Kunes, J., et al. (2012). Corylucinine, a new alkaloid from Corydalis cava (Fumariaceae), and its cholinesterase activity. Nat. Prod. Commun. 7, 859–860. doi: 10.1177/1934578X1200700712
Ohara, M., Higashi, S. (1994). Effects of inflorescence size on visits from pollinators and seed set of Corydali ambigua (Papaveraceae). Oecologia 98, 25–30. doi: 10.1007/BF00326086
Ohkawara, K., Ohara, M., Higashi, S. (1997). The evolution of ant-disperal in a spring-ephemeral Corydali ambigua (Papaveraceae): timing of seed-fall and effects of ants and ground beetles. Ecography 20, 217–223. doi: 10.1111/J.1600-0587.1997.TB00364.X
Oldenburg, D. J., Bendich, A. J. (2015). DNA maintenance in plastids and mitochondria of plants. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00883
Orhan, I., Özcelik, B., Karaoğlu, T., Sener, B. (2007). Antiviral and antimicrobial profiles of selected isoquinoline alkaloids from Fumaria and Corydalis species. Z. Naturforsch. C. J. Biosci. 62 (1-2), 19–26. doi: 10.1515/znc-2007-1-204
Pirayesh, N., Giridhar, M., Ben Khedher, A., Vothknecht, U. C., Chigri, F. (2021). Organellar calcium signaling in plants: an update. Biochim. Biophys. Acta Mol. Cell Res. 1868 (4), 118948. doi: 10.1016/j.bbamcr.2021.118948
Pogson, B. J., Ganguly, D., Albrecht-Borth, V. (2015). Insights into chloroplast biogenesis and development. Biochim. Biophys. Acta 1847, 1017–1024. doi: 10.1016/j.bbabio.2015.02.003
Porebski, S., Bailey, L. G., Baum, B. R. (1997). Modification of a CTAB DNA extraction protocol for plants containing high poly-saccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15. doi: 10.1007/BF02772108
Preuten, T., Cincu, E., Fuchs, J., Zoschke, R., Liere, K., Börner, T. (2010). Fewer genes than organelles: extremely low and variable gene copy numbers in mitochondria of somatic plant cells. Plant J. 64 (6), 948–959. doi: 10.1111/j.1365-313X.2010.04389.x
Putintseva, Y. A., Bondar, E. I., Simonov, E. P., Sharov, V. V., Oreshkova, N. V., Kuzmin, D. A. (2020). Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genomics 21, 654. doi: 10.1186/s12864-020-07061-4
Qin, F., Chen, Y., Wang, F. F., Tang, S. Q., Fang, Y. L. (2023a). Corydalis saxicola Bunting: a review of its traditional uses, phytochemistry, pharmacology, and clinical applications. Int. J. Mol. Sci. 24, 1626. doi: 10.3390/ijms24021626
Qin, J., Luo, Z., Wang, Q., Tang, C., Meng, M., Huang, Z., et al. (2023b). Integrating metabonomics and metagenomics sequencing to study the anti-liver fibrosis effects of palmatine in Corydalis saxicola Bunting. J. Ethnopharmacol. 315, 116666. doi: 10.1016/j.jep.2023.116666
Raven, J. A. (2015). Implications of mutation of organelle genomes for organelle function and evolution. J. Exp. Bot. 66, 5639–5650. doi: 10.1093/jxb/erv298
Ren, F. M., Wang, L. Q., Li, Y., Zhuo, W., Xu, Z. C., Guo, H. J., et al. (2021). Highly variable chloroplast genome from two endangered Papaveraceae lithophytes Corydalis tomentella and Corydalis saxicola. Ecol. Evol. 11, 4158–4171. doi: 10.1002/ece3.7312
Richardson, A. O., Rice, D. W., Young, G. Y., Alverson, A. J., Palmer, J. D. (2013). The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11, 29. doi: 10.1186/1741-7007-11-29
Robles, P., Quesada, V. (2021). Organelle genetics in plants. Int. J. Mol. Sci. 22 (4), 2104. doi: 10.3390/ijms22042104
Rose, R. J. (2021). Contribution of massive mitochondrial fusion and subsequent fission in the plant life cycle to the integrity of the mitochondrion and its genome. Int. J. Mol. Sci. 22 (11), 5429. doi: 10.3390/ijms22115429
Shan, Y., Li, J., Zhang, X., Yu, J. (2023). The complete mitochondrial genome of Amorphophallus albus and development of molecular markers for five Amorphophallus species based on mitochondrial DNA. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1180417
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47 (W1), W65–W73. doi: 10.1093/nar/gkz345
Sloan, D. B. (2013). One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 200, 978–985. doi: 10.1111/nph.12395
Small, I. D., Schallenberg-Rüdinger, M., Takenaka, M., Mireau, H., Ostersetzer-Biran, O. (2020). Plant organellar RNA editing: what 30 years of research has revealed. Plant J. 101 (5), 1040–1056. doi: 10.1111/tpj.14578
Tian, B., Tian, M., Huang, S. M. (2016). Advances in phytochemical and modern pharmacological research of Rhizoma Corydalis. Pharm. Microbiol. 58, 265–275. doi: 10.1080/13880209.2020.1741651
Wang, J., Kan, S., Liao, X., Zhou, J., Tembrock, L. R., Daniell, H. (2024). Plant organellar genomes: much done, much more to do. Trends Plant Sci. doi: 10.1016/j.tplants.2023.12.014
Wang, D., Rousseau-Gueutin, M., Timmis, J. N. (2012). Plastid sequences contribute to some plant mitochondrial genes. Mol. Biol. Evol. 29 (7), 1707–1711. doi: 10.1093/molbev/mss016
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49. doi: 10.1093/nar/gkr1293
Wang, J., Xu, G., Ning, Y., Wang, X., Wang, G. L. (2022). Mitochondrial functions in plant immunity. Trends Plant Sci. 27 (10), 1063–1076. doi: 10.1016/j.tplants.2022.04.007
Wang, D. Y., Zhang, Q., Liu, Y., Lin, Z. F., Zhang, S. X., Sun, M. X., et al. (2010). The levels of male gametic mitochondrial DNA are highly regulated in Angiosperms with regard to mitochondrial inheritance. Plant Cell 22 (7), 2402–2416. doi: 10.1105/tpc.109.071902
Welchen, E., Canal, M. V., Gras, D. E., Gonzalez, D. H. (2021). Cross-talk between mitochondrial function, growth, and stress signalling pathways in plants. J. Exp. Bot. 72 (11), 4102–4118. doi: 10.1093/jxb/eraa608
Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13, e1005595. doi: 10.1371/journal.pcbi.1005595
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z., Liao, X., Zhang, X., Tembrock, L. R., Broz, A. (2022). Genomic architectural variation of plant mitochondria—a review of multichromosomal structuring. J. Syst. Evol. 60 (1), 160–168. doi: 10.1111/jse.12655
Wu, Z. Y., Zhuang, X., Su, Z. Y. (1999). “Corydalis DC,” in Flora Reipublicae Popularis Sinicae, vol. 32 . Ed. Delecti Flora Reipublicae Popularis Sinicae Agendae Academiae Sinicae (Beijing: Science Press), 106–479.
Xu, Z., Li, Z., Ren, F., Gao, R., Wang, Z., Zhang, J., et al. (2022b). The genome of Corydalis reveals the evolution of benzylisoquinoline alkaloid biosynthesis in Ranunculales. Plant J. 111, 217–230. doi: 10.1111/tpj.15788
Xu, X. D., Li, X. X., Wang, D. (2022a). New insights into the backbone phylogeny and character evolution of Corydalis (Papaveraceae) based on plastome data. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.926574
Xu, X., Wang, D. (2021). Comparative chloroplast genomics of Corydalis species (Papaveraceae): evolutionary perspectives on their unusual large scale rearrangements. Front. Plant Sci. 11, 600354. doi: 10.3389/fpls.2020.600354
Xue, L., Ren, H., Long, W., Leng, X., Wang, J., Yao, X., et al. (2018). Ecophysiological responses of calcicole Cyclobalanopsis glauca (Thunb.) Oerst. to drought stress and calcium supply. Forests 9, 667. doi: 10.3390/f9110667
Yan, J., Zhang, Q., Yin, P. (2018). RNA editing machinery in plant organelles. Sci. China Life Sci. 61 (2), 162–169. doi: 10.1007/s11427-017-9170-3
Yang, Y., Fan, G., Zhao, Y., Wen, Q., Wu, P., Meng, Y., et al. (2020). Cytidine-to-Uridine RNA editing factor NbMORF8 negatively regulates plant immunity to phytophthora pathogens. Plant Physiol. 184 (4), 2182–2198. doi: 10.1104/pp.20.00458
Zandueta-Criado, A., Bock, R. (2004). Surprising features of plastid ndhD transcripts: addition of non-encoded nucleotides and polysome association of mRNAs with an unedited start codon. Nucleic Acids Res. 32 (2), 542–550. doi: 10.1093/nar/gkh217
Zhang, D., Gao, F., Li, W. X., Jakovlíc, I., Zou, H., Zhang, J., et al. (2020). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, B., Huang, R., Hua, J., Liang, H., Pan, Y., Dai, L., et al. (2016). Antitumor lignanamides from the aerial parts of Corydalis saxicola. Phytomedicine 23, 1599–1609. doi: 10.1016/j.phymed.2016.09.006
Zhang, L., Ma, J., Shen, Z., Wang, B., Jiang, Q., Ma, F., et al. (2023). Low copy numbers for mitochondrial DNA moderates the strength of nuclear–cytoplasmic incompatibility in plants. J. Integr. Plant Biol. 65 (3), 739–754. doi: 10.1111/jipb.13400
Zhang, H., Meltzer, P., Davis, S. (2013). RCircos: an R package for Circos 2D track plots. BMC Bioinf. 14, 244. doi: 10.1186/1471-2105-14-244
Zhang, M. L., Su, Z. Y., Lidén, M. (2008). “Corydalis DC,” in Flora of China (Beijing: Science Press), 295–427.
Zhang, Y. W., Zhao, J. M., Inouye, D. W. (2014). Nectar thieves influence reproductive fitness by altering behaviour of nectar robbers and legitimate pollinators in Corydalis ambigua (Fumariaceae). J. Ecol. 102, 229–237. doi: 10.1111/1365-2745.12166
Keywords: Corydalis saxicola, mitochondrial genome, RNA editing, mitochondrial plastid DNAs, expression profile
Citation: Li C, Liu H, Qin M, Tan Y-j, Ou X-l, Chen X-y, Wei Y, Zhang Z-j and Lei M (2024) RNA editing events and expression profiles of mitochondrial protein-coding genes in the endemic and endangered medicinal plant, Corydalis saxicola. Front. Plant Sci. 15:1332460. doi: 10.3389/fpls.2024.1332460
Received: 03 November 2023; Accepted: 23 January 2024;
Published: 06 February 2024.
Edited by:
Shaojun Dai, Shanghai Normal University, ChinaReviewed by:
Georgi Bonchev, Bulgarian Academy of Sciences (BAS), BulgariaZhiqiang Wu, Kunpeng Institute of Modern Agriculture at Foshan, China
Copyright © 2024 Li, Liu, Qin, Tan, Ou, Chen, Wei, Zhang and Lei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhan-jiang Zhang, enpqMTgxMUAxNjMuY29t; Ming Lei, bGVpbWluZ0BneHl5end5LmNvbQ==
†These authors have contributed equally to this work