 Tiexin Zeng
Tiexin Zeng Yang Ni
Yang Ni Jingling Li
Jingling Li Haimei Chen
Haimei Chen Mei Jiang
Mei Jiang Lijia Xu
Lijia Xu Chang Liu
Chang Liu Peigen Xiao
Peigen Xiao- Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
Rehmannia glutinosa is an economically significant medicinal plant. Yet, the structure and sequence of its mitochondrial genome has not been published, which plays a crucial role in evolutionary analysis and regulating respiratory-related macromolecule synthesis. In this study, the R. glutinosa mitogenome was sequenced employing a combination of Illumina short reads and Nanopore long reads, with subsequent assembly using a hybrid strategy. We found that the predominant configuration of the R. glutinosa mitogenome comprises two circular chromosomes. The primary structure of the mitogenome encompasses two mitochondrial chromosomes corresponding to the two major configurations, Mac1-1 and Mac1-2. The R. glutinosa mitogenome encoded an angiosperm-typical set of 24 core genes, nine variable genes, three rRNA genes, and 15 tRNA genes. A phylogenetic analysis using the 16 shared protein-coding genes (PCG) yielded a tree consistent with the phylogeny of Lamiales species and two outgroup taxa. Mapping RNA-seq data to the coding sequences (CDS) of the PCGs revealed 507 C-to-U RNA editing sites across 31 PCGs of the R. glutinosa mitogenome. Furthermore, one start codon (nad4L) and two stop codons (rpl10 and atp6) were identified as products of RNA editing events in the R. glutinosa mitogenome.
1 Introduction
Rehmannia glutinosa (Gaertn.) DC. (http://www.theplantlist.org/), a member of the Scrophulariaceae family has been widely used in traditional Chinese medicine (TCM) and is commonly known as “DiHuang” in China (Li et al., 2022). With a medicinal history spanning over two millennia, R. glutinosa is a vital industrial crop first documented in Shennong’s Classic of Materia Medica (Qin and Han Dynasties, 100 BC) (Li et al., 2022). The plant is processed into various forms, including fresh rehmannia root (Xian DiHuang), rehmannia dried rhizome (Sheng DiHuang), and prepared rehmannia root (Shu DiHuang) (Meng et al., 2017; Li et al., 2022). In its fresh form, R. glutinosa root possesses many therapeutic benefits, including antipyretic, salivary secretion enhancement, hematothermal regulation, anti-coagulative, detoxifying, and analgesic properties. It is commonly employed in the treatment of various medical conditions such as fevers, yin imbalances, glossal abnormalities, polydipsia, cutaneous eruptions, hematemesis, epistaxis, and pharyngitis (Liu et al., 2017; Meng et al., 2017; Li et al., 2022). Moreover, the Liuwei Dihuang Pill, a quintessential formulation in Traditional Chinese Medicine (TCM), features R. glutinosa as its principal component, demonstrating substantial efficacy in ameliorating diabetes and its associated complications (Zheng et al., 2020; Chen et al., 2021; Lu et al., 2022). R. glutinosa holds significant research and development value due to its extensive medicinal history and efficacy. However, cultivation is often challenged by root rot and high-stress resistance (Kim et al., 2020; Wang R et al., 2018). Traditional artificial domestication is time-consuming, and the ability of direct introduction of superior wild variety genes is limited (Fernie and Yang, 2019). Next-generation sequencing technology has enabled the integration of bioinformatics with genetic engineering, offering new possibilities for breeding R. glutinosa (Koenig et al., 2013; Meyer and Purugganan, 2013).
Mitochondria, biomacromolecules as essential cellular organelles, play a critical role in various metabolic processes, including the tricarboxylic acid (TCA) cycle, urea cycle, heme biosynthesis, calcium homeostasis, iron/sulfur cluster formation, gluconeogenesis, amino acid metabolism, and apoptosis (Osellame et al., 2012). Moreover, mitochondria are involved in synthesizing and folding essential biological macromolecules such as proteins, lipids, and nucleic acids, which are fundamental components of cellular structures and processes (Blomain and McMahon, 2012). Contrasting nuclear DNA (nDNA), mitochondrial DNA (mtDNA) is more susceptible to exogenous and endogenous stress due to its proximity to oxidative phosphorylation sites and the absence of protective histones in mitochondria. Although nucleoid structures offer some protection, mtDNA damage frequently occurs within mitochondria (Liao et al., 2022; Palozzi et al., 2022). The current research has demonstrated that the incidence of mtDNA damage in cells significantly surpasses that of nDNA damage (Liao et al., 2022; Palozzi et al., 2022; Roy et al., 2022). The mtDNA damage and repair mechanisms including Non-homologous end joining (NEHJ) often lead to homologous recombination in mitochondrial genomes, potentially mediated by repeat sequences (Dahal et al., 2018; Chevigny et al., 2020). Damage to mitochondria can also result in delusions, which are implicated in cytoplasmic male sterility (CMS) (Hu et al., 2014). Several CMS-related genes have been identified and characterized across various species, such as RT98-CMS rice and RT102-CMS rice (Igarashi et al., 2013; Okazaki et al., 2013). Given the central role of mitochondria in synthesizing and maintaining the presence of biological macromolecules, investigating the mitochondria of industrial crops holds substantial importance for cultivating high-quality crops. Genome research on R. glutinosa may yield valuable insights into the relationship between mitochondria and macromolecules, furthering our understanding of these complex interactions and their implications for crop improvement.
RNA editing events are prevalent in mitochondrial genomes and have far-reaching implications for protein function. These events often result in alterations to the amino acids specified by the genomic sequence. Such modifications not only enhance the conservation of the overall amino acid sequence but also affect the physicochemical attributes of the protein, even influencing its folding dynamics (Takenaka et al., 2008; Small et al., 2020; Kang et al., 2021). These observations underscore the pivotal role of RNA editing sites in maintaining the proper functionality of proteins. Additionally, RNA editing events appear to be intricately linked with the mechanisms of natural selection. Some researchers (Chateigner-Boutin and Small, 2010; Ichinose and Sugita, 2017) have investigated RNA editing events within the mitochondria of 17 angiosperm species. Remarkably, the nonsynonymous editing sites exhibit high conservation across these species, with approximately 80% conservation observed.
Additionally, the efficiency of the editing process is notably high, achieving an editing extent of around 80% across all examined plant species. This high level of conservation and efficiency suggests a crucial functional role for these editing events in plant mitochondrial biology. After reverse transcription into cDNA, some edited transcripts integrate into the genome through homologous recombination and are subsequently preserved. Most RNA editing sites in plant mitochondria are predominantly at the second codon position. The most frequent form of editing involves the conversion of cytosine (C) to uracil (U). This specific nucleotide alteration is thought to be correlated with an overall increase in the hydrophobicity of the resultant protein. Approximately 55% of amino acid substitutions resulting from RNA editing events exhibit a transition from hydrophilic to hydrophobic properties. This trend suggests a substantive impact on the edited protein’s physicochemical characteristics, potentially affecting its function and interaction within cellular environments (Sun et al., 2016; Mohammed et al., 2022). Additionally, the premature emergence of stop codons caused by RNA editing may result from erroneous editing, leading to the premature termination of gene translation, reducing the amino acid sequence of the encoded protein, and affecting protein function.
We successfully assembled and characterized the mitochondrial genome of R. glutinosa’s dual mitochondrial chromosomes in the present study. We validated its secondary structure through the lens of homologous recombination mediated by direct repeats. Additionally, we identified 507 RNA editing sites within the protein-coding regions, all of which involved the conversion of cytosine (C) to uracil (U). Our analysis also revealed the presence of two modified stop codons in the CDs of rps10 and atp6 and one altered start codon of nad4L, resulting from RNA editing events. These findings offer novel insights into the complexity and functional implications of RNA editing in the mitochondrial genome of R. glutinosa.
2 Materials and methods
2.1 Plant materials, DNA and RNA extraction, and sequencing
Fresh leaves of R. glutinosa plants (IMPLAD Accession Number: 202205002) were harvested at the Institute of Medicinal Plant Development (IMPLAD, Longitude: 116.267500° E, Latitude: 40.033056° N). After cleansing with deionized distilled water (ddH2O), the specimens were cryopreserved at −80°C. The leaf samples were partitioned into two sets designated for DNA sequencing (DNA-seq) and RNA sequencing (RNA-seq).
Genomic DNA was isolated employing the Magnetic Plant Genomic DNA Kit (Catalog No. DP342; Tiangen, China). Total RNA was extracted utilizing the RNAprep Pure Plant Plus Kit (Catalog No. DP441; Tiangen, China). For RNA-seq analysis, mRNA was selectively enriched from the total RNA pool using targeted probes to remove ribosomal RNA (rRNA). Fragmentation was executed using divalent cations in a high-temperature environment provided by the First Strand Synthesis Reaction Buffer (5X). Subsequently, after the adenylation of the 3’ ends of DNA fragments, NEBNext Adaptors featuring a hairpin loop structure were ligated, setting the stage for subsequent hybridization. The cDNA fragments with a predominant length range of 370 to 420 base pairs were isolated to construct the fragmented library via the AMPure XP system (Beckman Coulter, Beverly, USA). The sequencing library was constructed using the TIANSeq Fast DNA Library Kit (Illumina; Catalog No. NG102), and sequencing was performed on an Illumina NovaSeq 6000 platform (Illumina, Inc.; San Diego, CA, USA).
For Oxford Nanopore sequencing, high molecular weight (HMW) DNA was isolated using the NEB Monarch HMW DNA Extraction Kit (Catalog No. T3060L; New England Biolabs, England). Mechanical shearing of the genomic DNA to an average fragment size of approximately 10 kb was accomplished using the Covaris g-TUBE (Thermo Fisher, USA). The DNA library was assembled using the DNA Library Kit (Catalog No. SQK-LSK110) and sequenced on a PromethION platform (Novogene Co., Ltd., Beijing, China).
2.2 Genome assembly and annotation
Illumina short-read sequences were processed using Trimmomatic software, employing the default settings (Bolger et al., 2014). Nanopore long-read sequences were filtered using Guppy software, also with default settings (Wick 2017). A hybrid assembly approach was implemented for the assembly of organelle genomes. For the assembly of the plastid genome (plastome), we utilized GetOrganelle software (Jin et al., 2020) to extract plastid-specific reads from the Illumina dataset, applying parameters “-R 15 -k 21,45,65,85,105 -F embplant_pt”. These reads were assembled into a unitig graph, and bifurcation structures corresponding to inverted repeat regions were resolved by aligning the Nanopore reads to these structures via the Unicycler software (Wick et al., 2017). The orientation of the resulting assembled genome was subsequently refined using Novowrap (Wu et al., 2021). For the mitochondrial genome (mitogenome), a similar hybrid assembly strategy was employed. Initially, GetOrganelle was used to isolate mitochondrial reads from the raw data, employing parameters “-R 50 -k 21,45,65,85,105 -P 1000000 -F embplant_mt”. These reads were then assembled into a unitig graph, and bifurcation structures were resolved through nanopore read alignment via Unicycler software (Wick et al., 2017).
Annotation of the plastome was performed using both CPGAVAS2 (Shi et al., 2019) and CPGView (Liu et al., 2023). Protein-coding genes (PCGs) and ribosomal RNA (rRNA) sequences within the mitogenome were annotated using Geseq (Tillich et al., 2017). Transfer RNA (tRNA) molecules were identified using tRNA-scan (Lowe and Eddy, 1997), version 1.4. A graphical representation of the mitogenome was generated using OGdraw (Greiner et al., 2019). All organelle genome annotations underwent meticulous review and were manually corrected as needed using the Apollo software suite (Misra and Harris, 2006).
2.3 Repeat elements, mitochondrial plastid DNAs, and mitochondrial nuclear DNAs analysis
Microsatellite sequence repeats (SSRs) were detected using the MISA tool with parameters specified as “1-10 2-6 3-5 4-5 5-5 6-5.” Tandem repeats were ascertained using the Tandem Repeats Finder (TRF) with parameters set at ‘2 7 7 80 10 50 500 -f -d -m’.
Mitochondrial Plastid Sequences (MTPTs) were discerned through a reciprocal comparison strategy, employing BLASTn (version 2.2.30+) with its default parameters. The plastid genome (plastome) was assembled utilizing Illumina sequence reads via the GetOrganelle software. Comparative analysis between the plastome and the mitochondrial genome (mitogenome) was performed using BLASTn, employing specific parameters: e-value set to 1e-6 and word size configured at 7 (Chen et al., 2015). BLASTn hits shorter than 100 base pairs were excluded from the analysis. Subsequently, MTPT gene clusters within the mitogenome were delineated and defined as contiguous gene assemblies in the plastome devoid of intervening mitochondrial genes. These MTPT gene clusters were visually represented in a circular map generated using TBtools (version 1.076).
To identify putative Nuclear Mitochondrial DNA segments (NUMTs), the nuclear genome of R. glutinosa (GenBank accession JABTTQ000000000.1) was compared against the mitogenome using BLASTn. Specific BLASTn parameters were as follows: e-value of 1e-5, word size of 9, gap opening cost of 5, gap extension cost of 2, match reward of 2, mismatch penalty of -3, and turning off the dust filter. The BLASTn output was visualized using TBtools (Chen et al., 2020). Segments identified as potential NUMTs were further annotated using GeSeq software. Additionally, the nuclear genomes of R. glutinosa were similarly probed for putative NUMTs.
2.4 Identification and validation of repeat mediated recombination
To investigate the influence of repeat sequences on both intermolecular and intramolecular recombination events within the mitochondrial genome of R. glutinosa, we employed BLASTn analysis. The search parameters were meticulously chosen, incorporating an Expectation value (E-value) threshold of 1E-6 and a word size setting of 7 to identify relevant repeat sequences rigorously (Chen et al., 2015). Sequence segments of 500 base pairs (bp) in length surrounding the repeats were extracted to assess potential recombination products near the repeats based on anticipated sequences preceding and succeeding recombination. Subsequently, Nanopore long reads were mapped to the extracted sequence segments of the four configurations, and the repeat-spanning reads were enumerated.
To investigate putative recombination products identified through mapping PacBio long reads, polymerase chain reaction (PCR) primers were designed at the junction of repetitive sequences and recombination fragments using the Primer 3 web service (Untergasser et al., 2012). PCR reactions were performed in 50 μL volumes, consisting of 23 μL water, 25 μL 2 × Taq PCR Master Mix, 1 μL of each primer, and 1 μL DNA. The reactions were performed on a Pro-Flex PCR system (Applied Biosystems, Waltham, MA, USA). Subsequently, the PCR products were separated and visualized on 1.0% agarose gels. Finally, the PCR amplicons were sequenced using the Sanger method to confirm the recombination events.
2.5 Phylogenetic analysis
To construct a phylogenetic tree, we downloaded 21 Lamiales mitogenome sequences, including the original version of R. glutinosa (OM397952.2), from the National Center for Biotechnology Information (NCBI) database. The common genes from 21 mitochondrial genomes were extracted and concatenated using Phylosuite (Zhang D et al., 2020). Subsequently, the DNA sequences of the 16 protein-coding genes (PCGs) shared among these ten mitogenomes were extracted (Table 1). These sequences were aligned with MAFFT (v7.450) (Rozewicki et al., 2019), and a phylogenetic tree was constructed using Phylosuite with the maximum likelihood (ML) method based on the alignment. The credibility of the phylogenetic tree was assessed by performing bootstrap testing with 1,000 replications. Finally, the resulting maximum-likelihood tree was visualized using iTOL (https://itol.embl.de/) (Letunic and Bork, 2021).
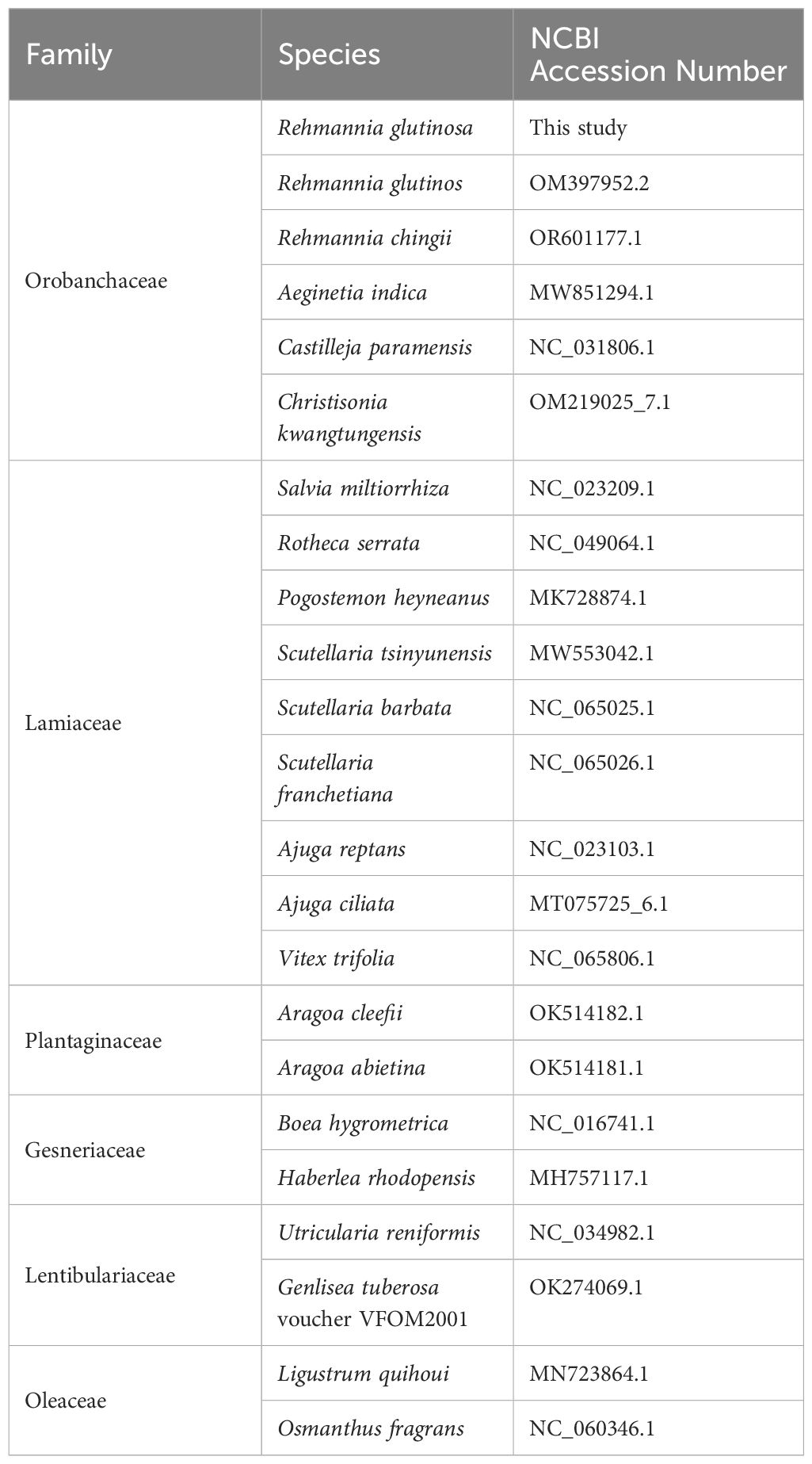
Table 1 Lamiales mitogenome sequences for the construction of the phylogenetic tree.
2.6 Identification and validation of RNA editing sites
To delineate both RNA editing sites and Single Nucleotide Polymorphism (SNP) loci, we initially extracted the coding domains (CDs) of each protein-coding gene (PCG), flanked by 100 base pair (bp) regions to serve as reference sequences. To detect SNP loci, genomic DNA sequencing reads were aligned to the reference above sequences using the Burrows-Wheeler Aligner (BWA; version 0.7.12-r1039) (Li and Durbin, 2010), with all parameters set to default. SNP loci were subsequently identified using REDItools (version 2.0), adopting identical parameters for RNA editing site identification: a minimum coverage of 5 reads and a frequency threshold of ≥ 0.1. Following this, RNA editing sites were ascertained utilizing REDItools (version 2.0) (Picardi and Pesole, 2013), with the criteria set at a coverage threshold of ≥ 5 reads and a frequency threshold of ≥ 0.1 (Wu et al., 2017). The resultant mapping data, specifically at the RNA editing loci with a minor variant frequency of ≥ 0.1, were visualized via the Integrative Genomics Viewer (IGV; version 2.15.1) (Milne et al., 2010).
3 Results
3.1 General feature of the R. glutinosa mitochondrial genome
The mitochondrial genome of R. glutinosa represented the first published genome of the genus Rehmannia and the fifth mitochondrial genome within the Orobaceae family. Genomic assembly was performed using Illumina and Nanopore sequencing technologies, generating 10.2 GB and 19.8 GB reads, respectively. The coverage depth of the long and short reads mapped to the R. glutinosa mitogenome sequences was obtained using samtools (v1.3.1) (Li et al., 2009) (Supplementary Figures S1, 2). De novo assembly of Illumina short reads was performed using the GetOrganelle software. Repeated sequences were resolved by mapping the Nanopore long sequences. Subsequently, Unicycler software was used to extract 29 contigs to construct unitig graphs, including ten double-bifurcating structures (DBS) (Figure 1A). The abundance of each configuration of DBS were calculated by mapping Nanopore long reads to the reference sequences using Unicycler. These configurations were further used for final assembly, and the results of the Unicycler analysis were subsequently loaded into bandage software with the “Merge all possible nodes” module. As a result, two chromosomes of the mitotic genome of R. glutinosa were obtained (Figure 1B).
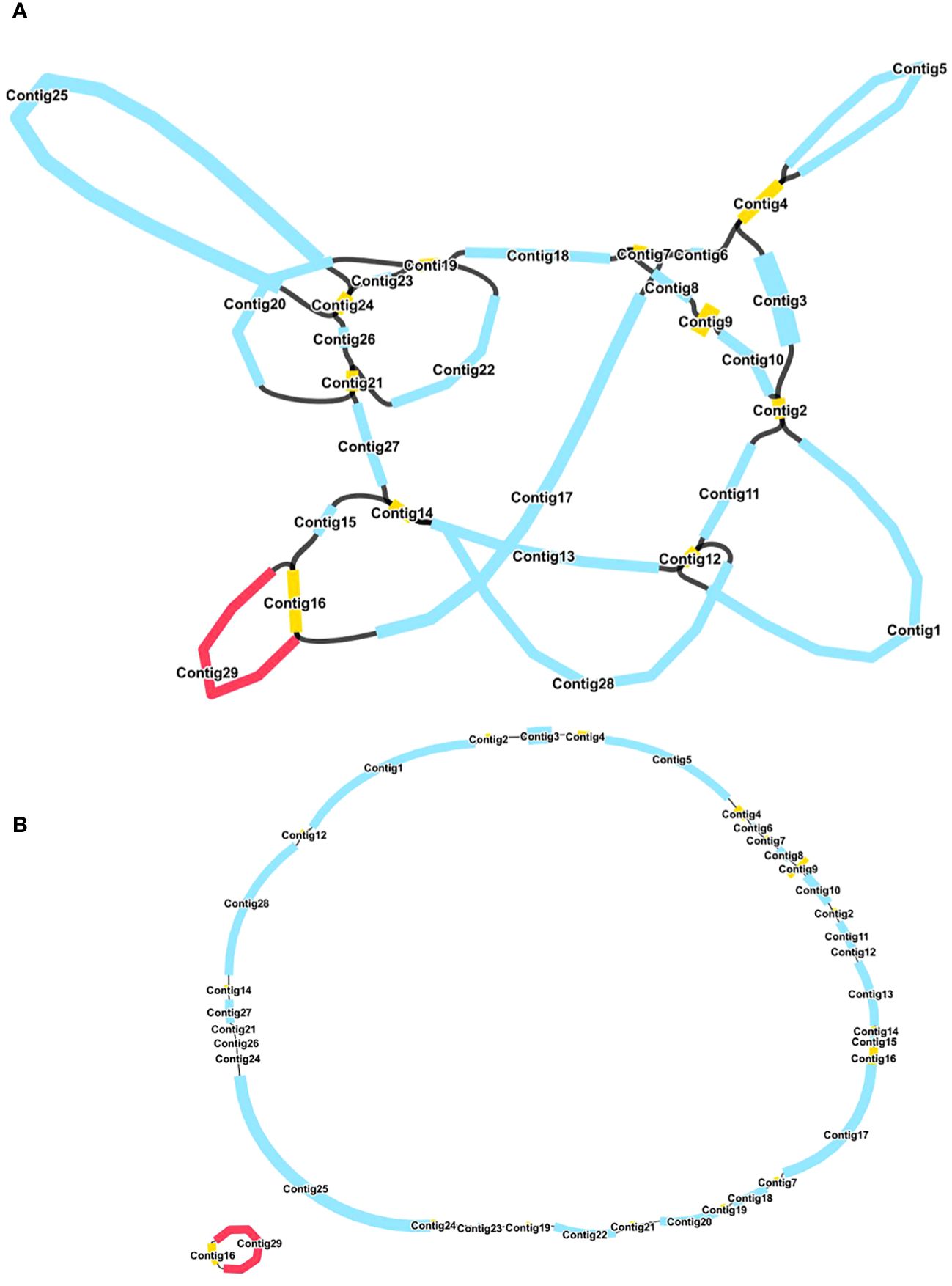
Figure 1 A schematic representation of the assembly process for the R. glutinosa mitogenome is provided. (A) A unitig graph for the R. glutinosa mitogenome was generated through de novo assembly of Illumina reads using Unicycler. This unitig graph consisted of seven contigs (depicted in yellow) that formed double bifurcating structures (DBSs). Each DBS exhibited two secondary configurations based on the Nanopore long reads. (B) A schematic diagram of the mitochondrial chromosome 1 (MC1, represented by a blue circle) and mitochondrial chromosome 2 (MC2, represented by a red circle) of R. glutinosa following the resolution of DBSs using long reads is presented. The contigs illustrated in blue and red correspond to chromosomes 1 and 2, respectively.
The R. glutinosa mitogenome had two chromosomes of 545,523 bp (chromosome 1 with 497,303bp, chromosome 2 with 48,220bp), and its entire GC was 45% (T 27.6%, C 22.5%, A 27.4%, G 22.5%). The GC content of R. glutinosa and its relative species ranged from 43.27% to 45.62%, and the genome length ranged from 225,612 bp-1,860,774 bp (Table 2). We annotated the mitochondrial genome, and the categorization of genes is shown in Table 3. The core genes consisted of five ATP synthase genes, nine NADH dehydrogenase genes, three cytochrome C biogenesis genes, three cytochrome C oxidase genes, ubiquinol cytochrome c reductase, a transport membrane protein, a maturase. The variable genes consisted of 4 large subunits of ribosome proteins (rpl2, rpl5, rpl10, and rpl16), seven small subunits of ribosome proteins (rps3, rps4, rps7, rps10, rps12, rps13 and rps14), three rRNA genes (rrn5, rrn18, and rrn26), and two respiratory genes (sdh3 and sdh4). A total of 15 unique tRNA genes were identified based on tRNAscan-SE. The schematic genome is presented in Figure 2.
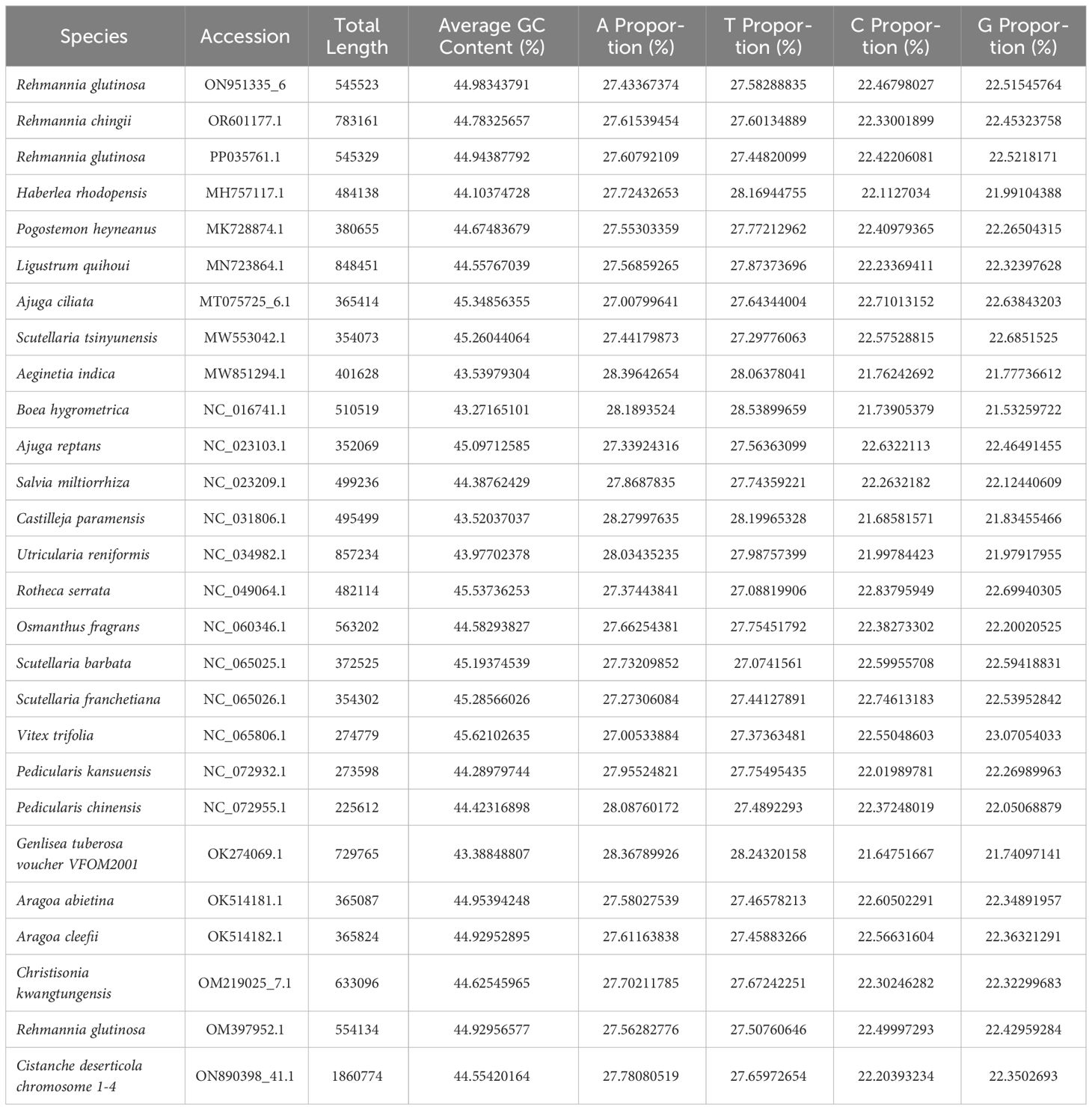
Table 2 Comparative genomic analysis of Lamiales mitogenome sequences.
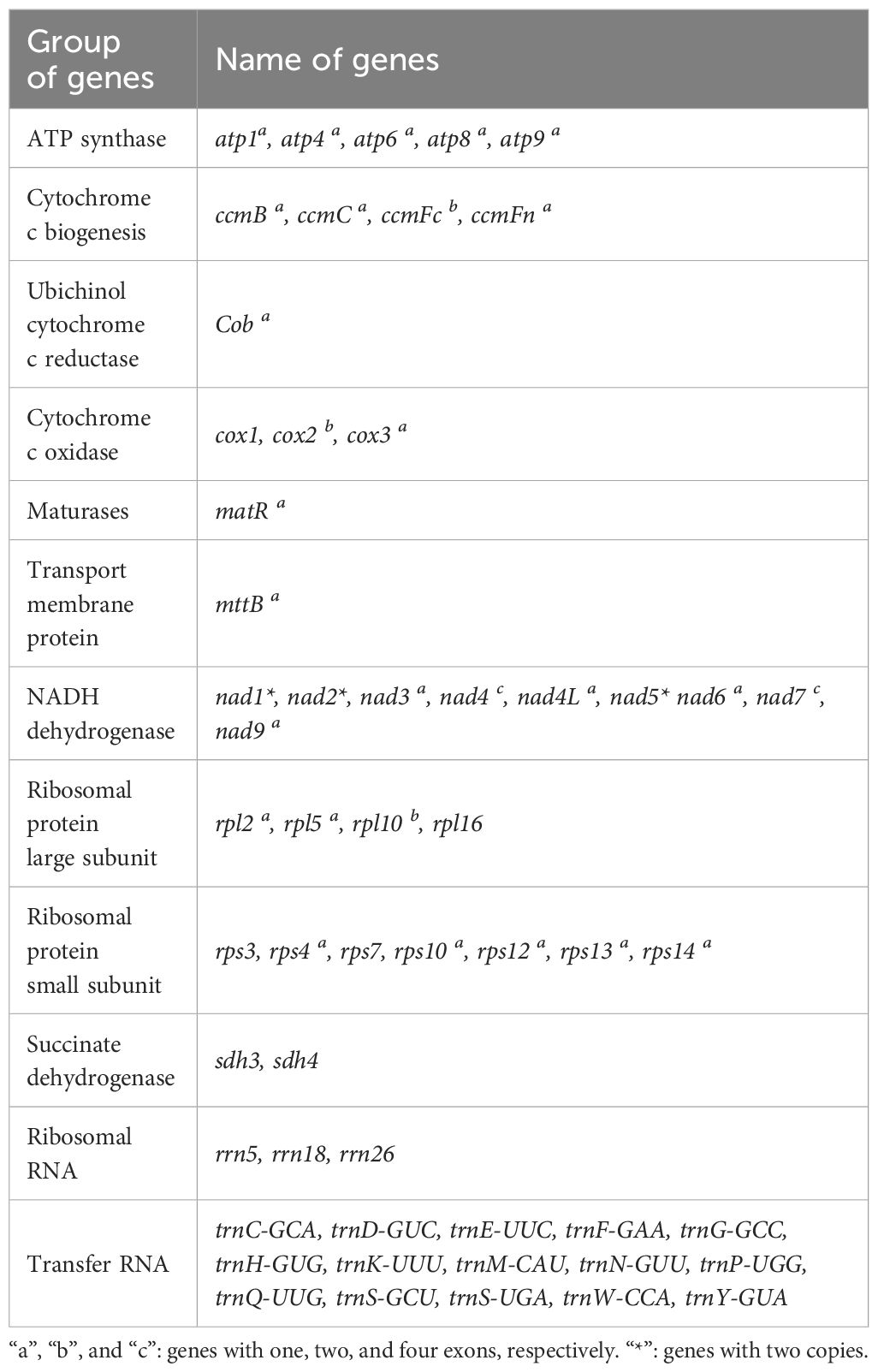
Table 3 Gene contents in the mitogenome of R. glutinosa.
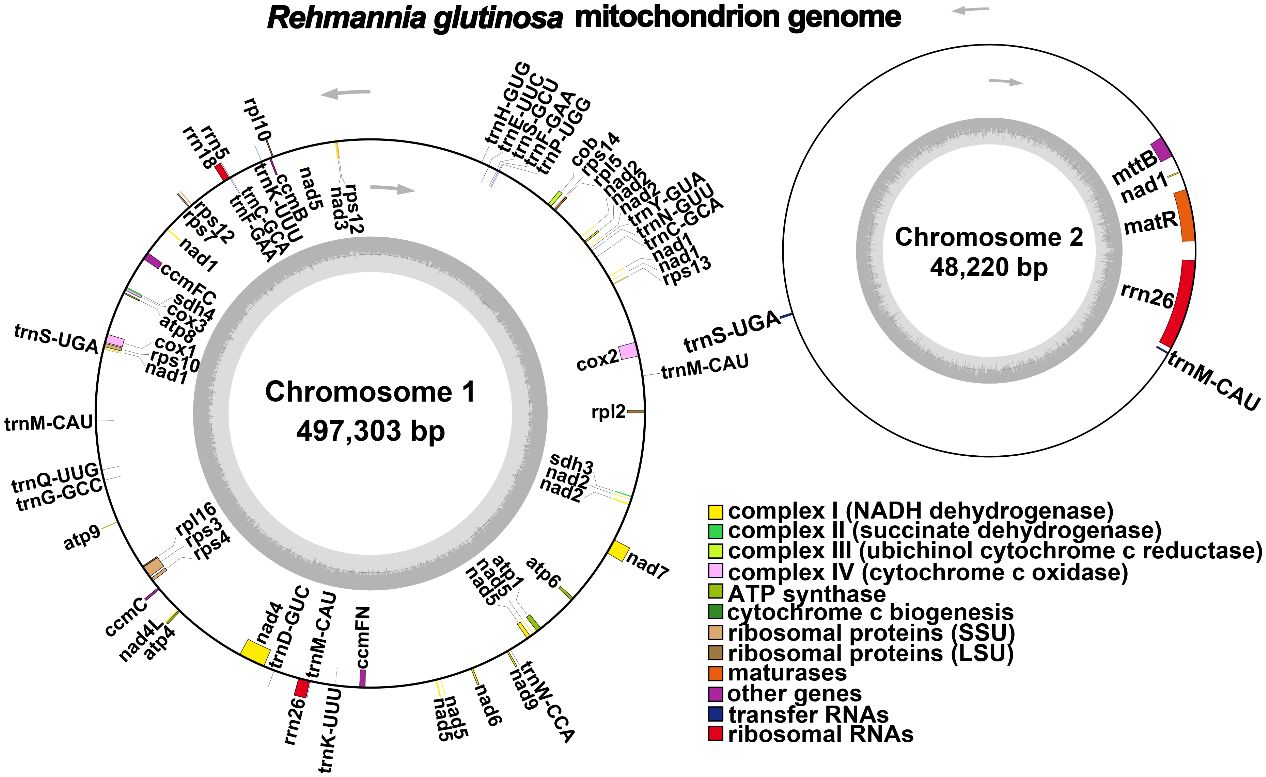
Figure 2 A schematic representation of the circular mitochondrial chromosome 1 and mitochondrial chromosome 2 of Rehmannia glutinosa is provided. Genes depicted on the inner side correspond to the negative strand, while those on the outer side represent the positive strand. Genes containing introns are marked with an asterisk (*). The gray circle illustrates the GC content, with an inner circle within the GC content graph denoting the 50% threshold. Different functional categories are indicated by the colors shown in the accompanying legend.
In this study, we analyzed the mitochondrial genome of R. glutinosa in this research and compared it with the publicly available genome sequence OM397952.2 (Supplementary Figure S1). Our findings indicate a strong collinearity between the two genomes, which is consistent from 1 bp up to 352,181 bp. Notably, there are repeated fragments spanning from 352,182 bp to 361,101 bp, and an extensive inverted repeat sequence can be observed from 419,346 bp to 547,032 bp.
3.2 Repeat elements analysis
Microsatellites, also known as simple sequence repeats (SSRs), are short repetitive DNA units composed of mononucleotide, dinucleotide, trinucleotide, tetranucleotide, or pentanucleotide motifs that are predominantly present in eukaryotic genomes [14]. In the mitochondrial genome of R. glutinosa, 100 and 16 SSR markers were identified in the major and secondary chromosomal molecules, respectively (refer to Figure 3; Supplementary Tables S2, 3). All six types of SSRs were detected in the mitochondrial genome, with 30, 17, 9, 41, and 3 SSRs having mono-, di-, tri-, tetra-, penta- or hexanucleotide repeat units in the major chromosomal molecule, and 3, 3, 1, 8, and 1 SSRs having mono-, di-, tri-, tetra-, or pentanucleotide repeat units in the chromosome 2, respectively. The most commonly occurring SSRs in the mitochondrial genome of R. glutinosa had a four-nucleotide repeat unit, accounting for 42.2% of all repeats. These microsatellite markers have the potential to serve as identification markers of R. glutinosa.
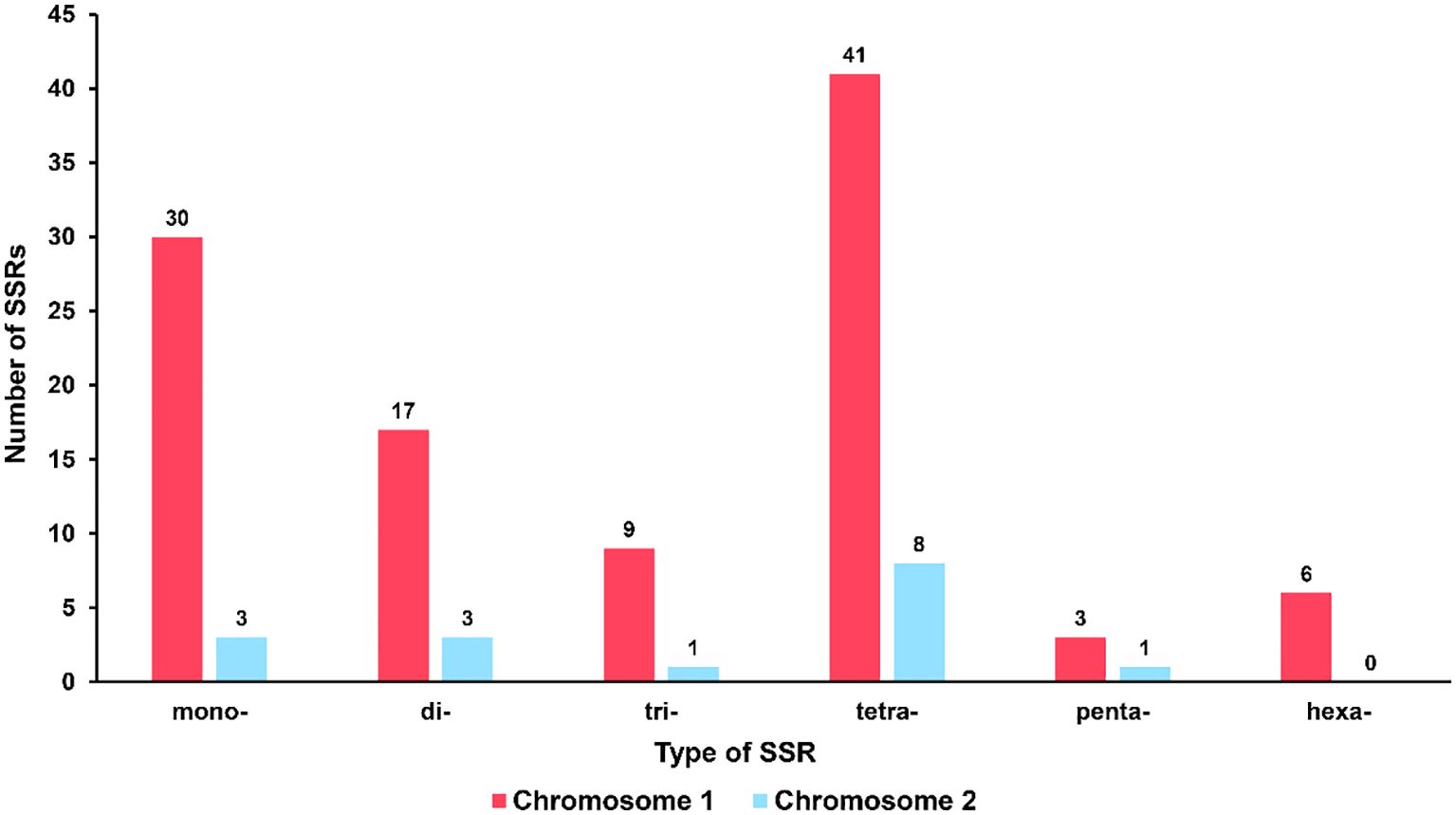
Figure 3 Type and quantity of SSR. The red column represents chromosome 1, and the blue column represents chromosome 2.
Tandemly repeated DNA sequences are characterized by a unit length greater than six base pairs and are highly variable components of the genome [15]. These repeats are commonly found in intergenic regions, although some can be located within coding sequences or pseudogenes. Six tandem repeat sequences were detected within chromosome 1 of the R. glutinosa mitogenome, with lengths ranging from 14 to 23 base pairs (Supplementary Tables S3).
3.3 Recombination mediated by repeat sequences
The mitochondrial genome of plants could not be fully represented by a single cyclic molecule, as rearrangement mediated by repeated sequences may occur to varying degrees. To investigate the possible homologous recombination in the mitochondrial genome of R. glutinosa, we detected 87 pairs of repetitive sequences in the mitochondrial genome of R. glutinosa using BLASTN with 1E-5. Based on Nanopore long reads, we carefully examined each pair of repetitive sequences for their support with long reads, and found that three pairs might support homologous recombination (Table 4; Supplementary Table S4). The length of these repeats is between 2,795 and 7,933 bp.
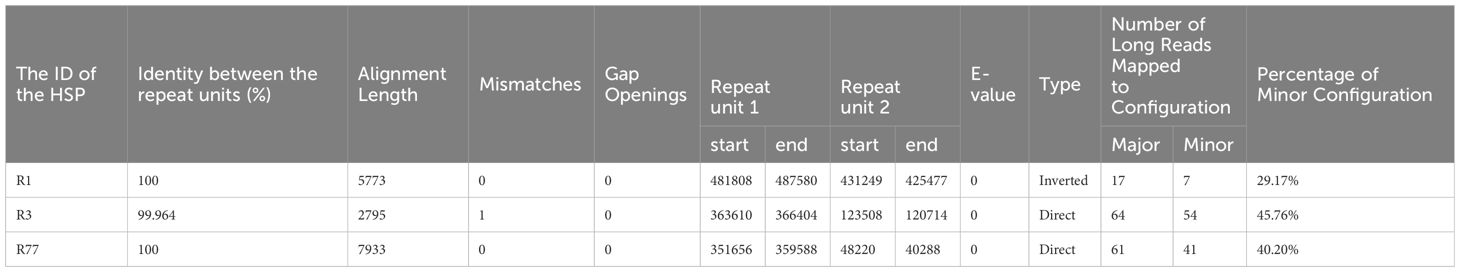
Table 4 The details of three direct repeats.
Primers were designed at each end of the repeat sequences further to investigate the recombination and potential configurations of R. glutinosa. Since the size of these repeat sequences exceeded 1,000 bp in length, specific primers were designed at the junction of each pair of repeats present on the primary single circular molecule (Figure 4A; Supplementary Table S5). In a recombinant configuration, PCR products (junctions 1-4) were shown in Figure 4B. The alignment of the Sanger sequencing results of the PCR products and the genomic sequences are shown in Supplementary Figures S4–15. We predicted the various configuration of the mitochondrial genome in Figure 4C. Three recombination events mediated by repeat sequences were confirmed (R1, R3, and R77, with R77 representing a pair of direct repeat sequences). All three sets of repeat sequences were found to generate secondary configurations, which is in accordance with according to the findings obtained through our long-read analysis (Figure 5; Supplementary Figure S16).
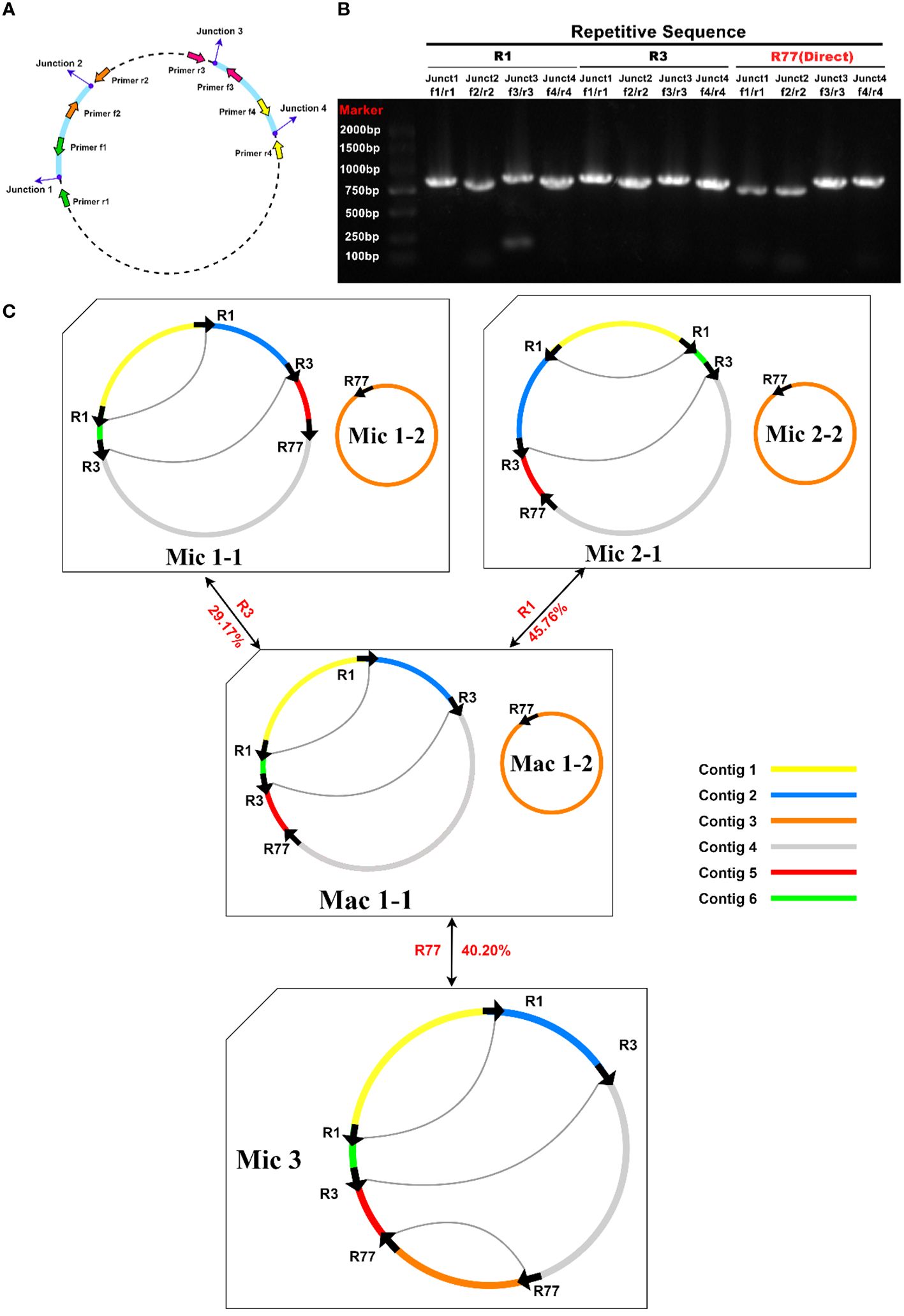
Figure 4 PCR validation of recombination products associated with repetitive sequence-mediated secondary configurations. (A) Schematic illustration of junctions related to each repetitive sequence. The corresponding primers are depicted as purple dots. F1-4: forward primers; R1-4: reverse primers. (B) Electrophoretic gel image of PCR products amplified using various forward and reverse primer combinations to amplify the DNA molecules corresponding to junctions 1-4. The name of the repetitive sequence, combinations of forward and reverse primers, expected junctions to be amplified, and lane numbers are displayed above the gel image. Each PCR product’s expected size encompasses those of the repetitive sequence and its 200-1000 bp long flanking sequences. The PCR product lengths are a rough evaluation of the successful amplification of fragments representing recombination products. (C) Hypothetical products of homologous recombination mediated by repetitive sequences R1, R3, and R77. Arrows indicate the repeat units of R1, R3, and R77. Arcs connect two repeat units if they are located on the same chromosome. Sequences surrounding the repeat units are displayed in distinct colors. Circles represent circular chromosomes. The genomic configuration is denoted by “C” followed by the configuration and chromosome numbers. Double-headed arrows indicate the source circular chromosomes, the repetitive elements, and the product circular chromosomes. The genomic configuration name is prefixed with “Ma,” representing “major” if it is the most abundant configuration; otherwise, the genomic configuration name is prefixed with “Mi,” representing “minor.” Mac is the genomic configuration containing chromosomes Mac1-1 and Mac1-2. Mac1-1 can undergo recombination mediated by R1 or R3 to form a circular chromosome Mic1-1 or Mic2-1. Mic3 only contains one circular chromosome, and it can undergo recombination mediated by R77 to form two circular chromosomes: Mac1-1 and Mac1-2.
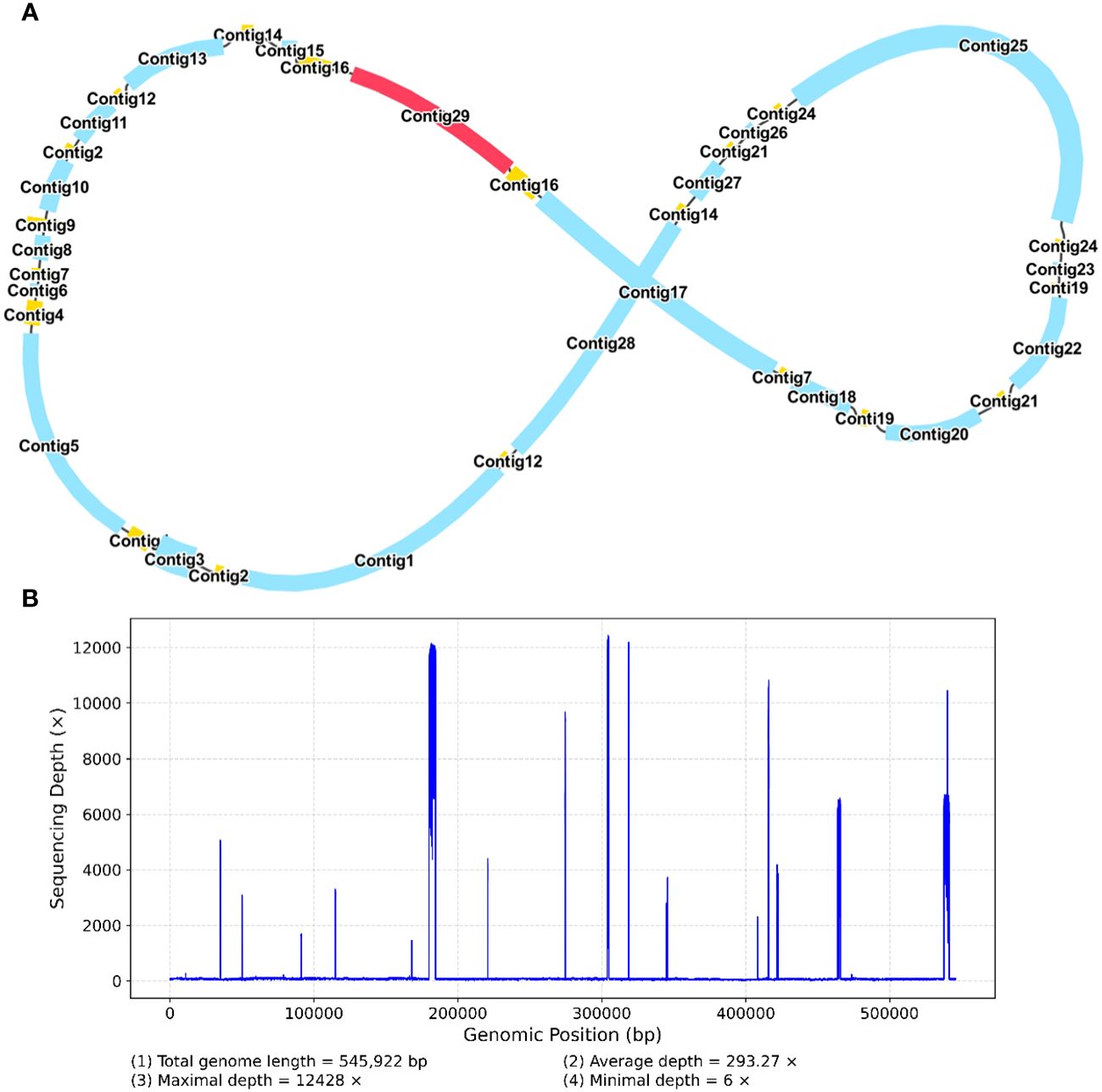
Figure 5 A schematic representation of the secondary configuration for the R. glutinosa mitogenome is provided. (A) A unitig graph for the R. glutinosa mitogenome was generated through de novo assembly of Illumina reads using Unicycler. This unitig graph consisted of seven contigs (depicted in yellow) that formed double bifurcating structures (DBSs) (B) The coverage depth of the Illumina short reads mapped to the R. glutinosa mitogenome sequences of the secondary configuration.
3.4 Mitochondrial plastid DNAs and mitochondrial nuclear DNAs analysis
Homologous sequence transfer refers to the process in which a part of the chloroplast genome sequence that was integrated into the mitochondrial genome during the evolution (Brigulla and Wackernagel, 2010; Zhang G. J. et al., 2020). In the mitochondrial genome of R. glutinosa, we found 24 homologous DNA fragments, including six fragments from the chloroplast genome IR regions. The total length of these fragments was 13,685 bp, accounting for 2.51% of the whole mitochondrial genome, of which the longest fragment was 4,513 bp. We annotated these fragments and found that they contained a part of chloroplast genes (Figure 6; Supplementary Figures S17–19, Supplementary Table S6), including 12 complete genes (ndhB, rps7, psbJ, psbL, psbF, psbE, rpl23, trnI-CAU, trnS-GGA, trnD-GUC, trnH-GUG and trnN-GUU) and 11 partial genes (trnL-CAA, rpoC1, rpoB, rpl2, trnI-GAU, psbA, psaB, trnK-UUU, rps4, ndhA and ndhB).
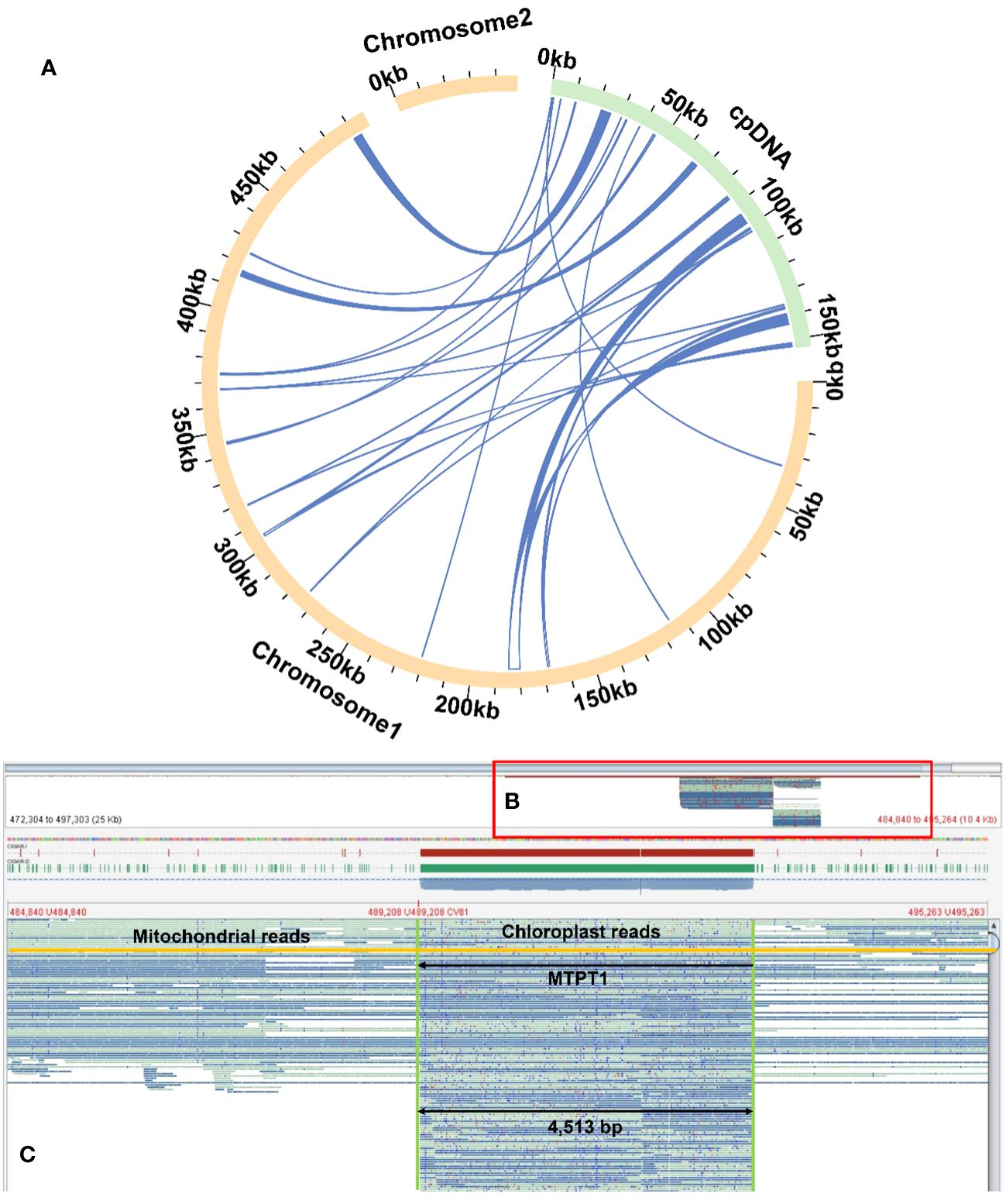
Figure 6 Examplar homologos sequences bewteen the mitogenome and chloroplastome. (A) Similar sequences are shared between the mitogenome and chloroplastome. The yellow and green arcs represent the mitogenome and chloroplastome genome (labeled as cpDNA), respectively. The inner circle arcs represent the MTPT fragments. (B) A bird’s eye view of MTPT, and the red box represents the enlarged part. (C) Mapping of long reads onto MTPT1 on chromosome 1. The MTPT sequence is highlighted in a green box. The encompassed regions illustrate upstream (mitoDNA) - MTPT - downstream (mitoDNA) sequences. A mitochondrial read is highlighted in yellow, bordered by mitoDNA sequences with MTPT sequence in the middle.
Besides chloroplasts, there are homologous sequences between the mitochondria and nuclear genomes (Supplementary Table S7). Compared with the published whole genome sequence of R. glutinosa, we found that there were 4,395 fragments of nuclear DNAs with a total of 5,073,866bp length, which were similar. Among them, 3,694 fragments, with a total of 4,742,687 bp, were homologous to mitochondrial chromosome 1, with the longest fragment being 78,947 bp and the shortest being 36 bp. There are 1,701 fragments (331,179bp) homologous to mitochondrial chromosome 2 (the longest sequence had 32,990bp, and the shortest sequence was only 34bp). The total length of homologous fragments on these nuclear DNAs far exceeded the total length of the whole mitochondrial genome (545,523 bp), which might be related to the multiple migration of mitochondrial genes (Brigulla and Wackernagel, 2010; McFarlane and Humphrey, 2010; Knoll et al., 2014).
3.5 The phylogenomic analysis of R. glutinosa
We conducted a phylogenetic analysis of the mitochondrial genomes of 21 Lamiales plants (Rehmannia glutinosa in our research: ON951335.1 – ON951336.1, Rehmannia glutinosa OM397952.2, Rehmannia chingii OR601177.1, Castilleja paramensis NC_031806.1, Aeginetia indica MW851294.1, Christisonia kwangtungensis OM219025_7.1, Salvia miltiorrhiza NC_023209.1, Rotheca serrata NC_049064.1, Pogostemon heyneanus MK728874.1, Scutellaria tsinyunensis MW553042.1, Scutellaria barbata NC_065025.1, Scutellaria franchetiana NC_065026.1, Ajuga reptans NC_023103.1, Ajuga ciliata MT075725_6.1, Vitex trifolia NC_065806.1, Aragoa cleefii OK514182.1, Aragoa abietina OK514181.1, Boea hygrometrica NC_016741.1, Haberlea rhodopensis MH757117.1, Utricularia reniformis NC_034982.1, Genlisea tuberosa voucher VFOM2001 OK274069.1, Ligustrum quihoui MN723864.1 and Osmanthus fragrans NC_060346.1). Two Oleaceae species (Ligustrum quihoui and Osmanthus fragrans) were selected as the outgroups. In total, the nucleotide sequences of 16 common genes (atp1, atp4, ccmB, ccmC, ccmFC, ccmFN, cob, cox2, cox3, matR, nad1, nad2, nad3, nad5, nad6 and rps13) were used for phylogenetic analysis. As shown in Figure 7. The R. glutinosa mitogenome assembled by us and the mitogenome released on GenBank are grouped together, which can be identified as the same species.
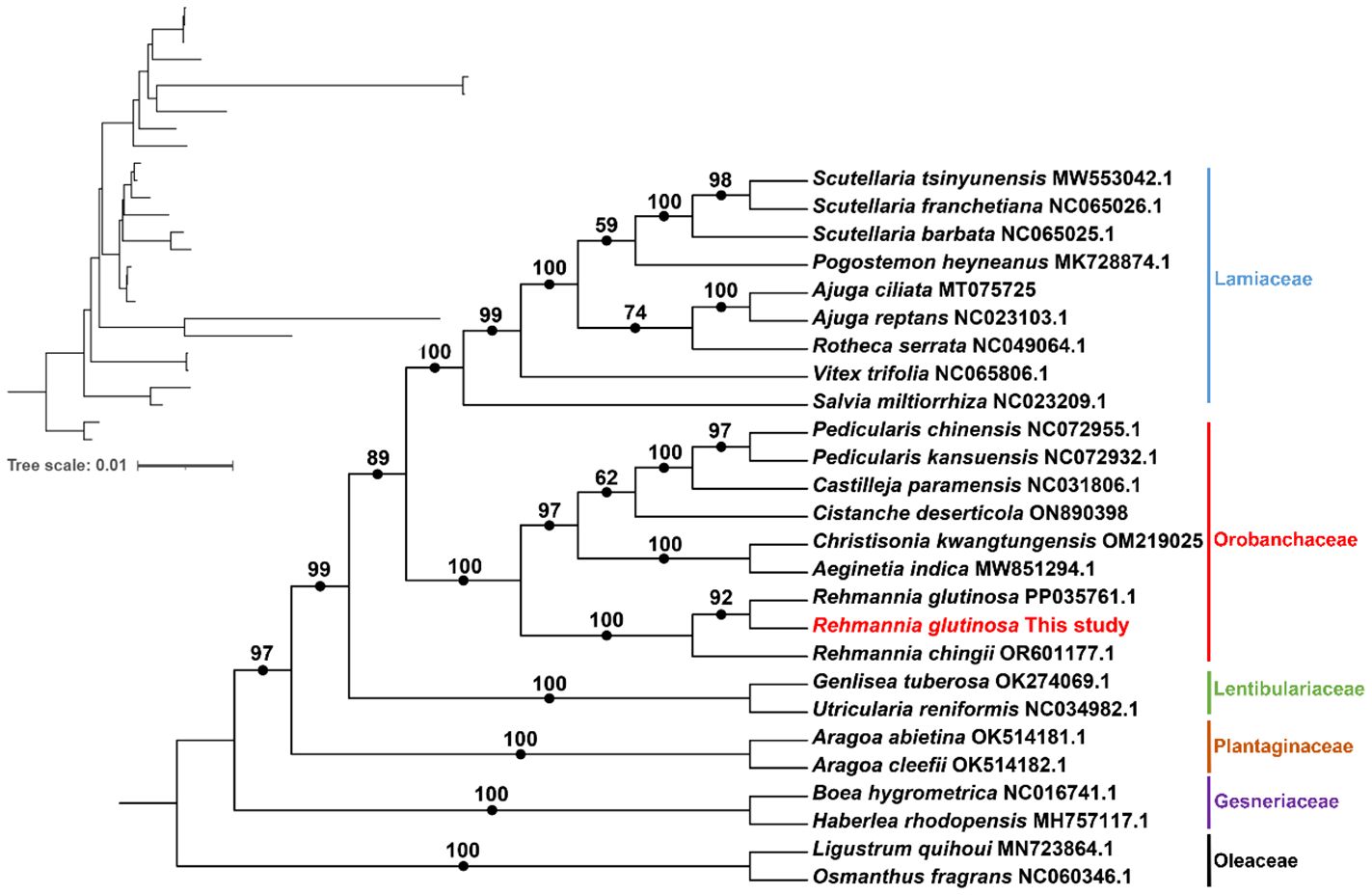
Figure 7 Molecular phylogenomic analysis of mitogenomes in Lamiales. The tree was constructed using concatenated conserved protein sequences from the mitogenomes of 21 species through maximum likelihood (ML) methods. Bootstrap scores were obtained using 1,000 replicates, and the ML bootstrap support values were indicated at the respective nodes. The tree in the upper left corner initially displays the original branch lengths. Two species from Oleaceae (Ligustrum quihoui and Osmanthus fragrans) were used as outgroups.
3.6 The RNA editing sites in the mitogenome of R. glutinosa
RNA editing has been observed in the plant mitochondrial genomes extensively [18]. By mapping the transcriptome data to the reference mitogenome, we identified 507 RNA editing sites in the protein-coding regions (Figure 8; Supplementary Table S8). These genes include the genes nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9, cob, cox1, cox2, cox3, rpl2, rpl5, rpl10, rpl16, rps10, rps3, rps4, rps12, rps13, rps14, atp4, atp6, atp8, atp9, ccmB, ccmC, ccmFC, ccmFN, matR, mttB, sdh3 and sdh4. The types of all the editing sites were from C to U. Among them, the amino acid changes caused by non-synonymous substitution (15 of a total of 24 amino acid changes) may lead to the structural changes of the final synthesized protein. We detected RNA editing of two stop codons (rps10 and atp6) and one start codon (nad4L) (Supplementary Figures S20–22), and the expression of related genes may be affected.
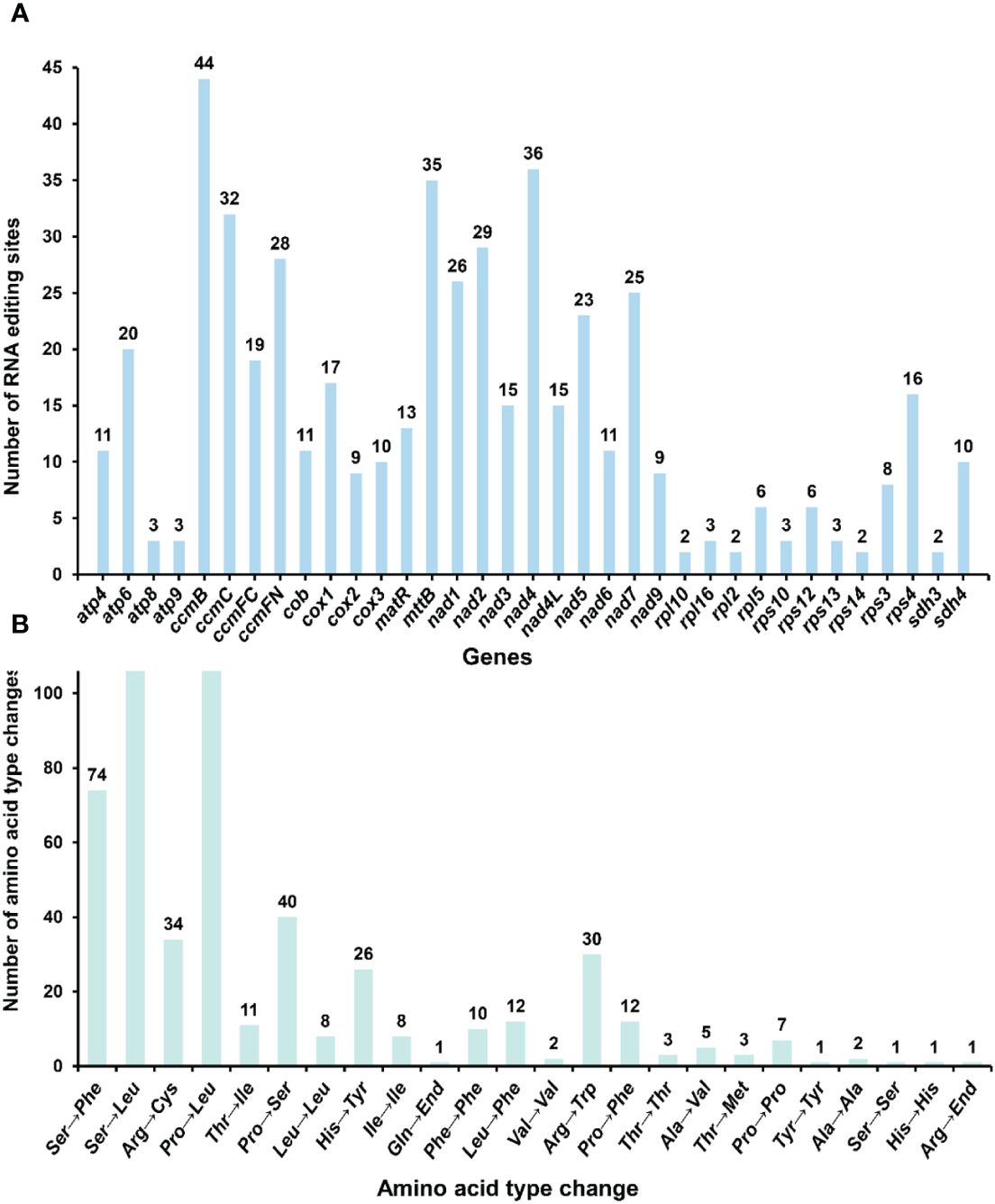
Figure 8 Statistics on the type and quantity of RNA editing events. (A) The number of RNA editing events for each gene. (B) The quantity of various amino acid changes.
4 Discussion
4.1 Graph-based method for mitochondrial genome assembly
Early investigations into plant mitochondrial genomes postulated a single master circle configuration akin to chloroplast genomes (Wu et al., 2022). However, subsequent studies had revealed that a solitary reference genome is inadequate for representing the full extent of genetic variation between individuals, particularly in plant mitochondrial genomes (Backert et al., 1996; Gonzalez et al., 1999). Graph-based genomic representations have proven more effective in capturing configurational and structural variations, as demonstrated by the soybean pan-genome comprising 26 plant materials (Tian et al., 2012). Plant mitochondrial genomes exhibit substantial differences in complexity, size, and structure, and a single circular representation fails to encompass all potential configurations. Recent publications have presented graph-based plant mitochondrial genomes of Lamiales species, such as Salvia miltiorrhiza and Scutellaria tsinyunensis (Li et al., 2021; Yang H et al., 2022). These genomes comprise two chromosomal molecules, with nine configurations reported for Salvia miltiorrhiza. More graph-based assembly tools have been published, including Master graph and PMAT (He et al., 2023; Bi et al., 2024). Additionally, there is an increasing discovery of multi-conformation mitochondrial genomes. This study provides a graph-based mitochondrial genome consisting of 29 contigs, including ten repeat regions (DBS structure), from which additional minor configurations can emerge.
4.2 Multiple chromosome configurations and homologous recombination
Traditionally, plant mitochondrial genomes were considered single circular molecules, similar to chloroplast genomes, primarily because Illumina short-read sequencing technologies struggled to resolve complex bifurcated structures. Consequently, plant mitochondrial assemblies that failed to form a circle were often considered assembly errors (Sloan, 2013). However, Nanopore long-read sequencing and graph-based genome assembly approaches have provided new solutions for mitochondrial genome assembly, and the homologous recombination of mitochondrial genomes mediated by direct repeat sequences had been confirmed (Wang et al., 2024). These advancements have also facilitated better prediction the potential complex structures of the mitochondrial genomes.
The presence of minor configurations in mitochondrial genomes had been confirmed in various plant species, including Scutellaria tsinyunensis, Ipomoea batatas, Saposhnikovia divaricata, Salvia miltiorrhiza, Cistanche deserticola, and Aeginetia indica (Li et al., 2021; Miao et al., 2022; Ni et al., 2022; Yang H. et al., 2022; Yang Z. et al., 2022; Zhong et al., 2022). Among these, Salvia miltiorrhiza displayed two distinct mitochondrial configurations, which were ascribed to an increased occurrence of homologous recombination events (Yang H. et al., 2022). Our current investigation identified three minor configurations within the R. glutinosa mitochondrial genome, leveraging long-read sequencing technology. While we confirmed the junction sites through polymerase chain reaction (PCR) amplification and Sanger sequencing, dditional experimental evidence is necessary to validate this phenomenon further.
4.3 MTPT & NUMT
4.3.1 NUMT
Mitochondria are thought to have originated from endosymbiotic α-proteobacteria, which subsequently experienced gene loss or transfer to the nucleus (Martin et al., 2015; Roger et al., 2017). The mitochondrial genome of flowering plants contains up to 40 known protein-coding genes, with the number of non-core genes varying significantly among species, apart from the 24 core protein-coding genes (Adams et al., 2002). One primary factor accounting for the variability in gene content within mitochondrial genomes is the transfer of mitochondrial genes to the nucleus during eukaryotic evolution (Brigulla and Wackernagel, 2010; McFarlane and Humphrey, 2010; Knoll et al., 2014). This functional gene transfer contributes to the co-evolution of mitochondria and the nucleus (Levin et al., 2014), has been found in mice and humans, an ongoing evolutionary process in land plants and some green algae. Due to the differing evolutionary and migration rates among plant species from various flora, gene migration frequencies also vary. For instance, the rps1 gene has been lost from the mitochondria of most Lamiales plants, a finding consistent with our study results.
For successful activation and expression of genes in the nucleus following physical transfer from mitochondria (Gualberto and Newton, 2017), these newly transferred genes must acquire promoters and other regulatory elements. If a protein lacks the necessary targeting information, it must obtain sequences of protein products targeting mitochondria. Several transferred genes have acquired mitochondrial target pre-sequences, which are removed from the protein following their introduction into mitochondria. Some genes have obtained mitochondrial pre-sequences from pre-existing mitochondrial protein genes (Liu et al., 2009; Gualberto and Newton, 2017). Once the transferred nuclear copy is activated, both this and the mitochondrial copies can be co-expressed for a period, at least at the transcript level, as demonstrated in the case of cox2 in some legumes, rpl5 in wheat, and sdh4 in poplar. The activation of transferred genes appears to be related to positive selection, but the existence of a nuclear screening mechanism for transferred genes remains uncertain. It is clear, however, that the transfer between mitochondria and the nucleus occurs frequently. In our study, we found that the nuclear DNA of R. glutinosa contained 4,329 fragments, with a total length of 4,880,380 bp, exhibiting similarity to mitochondrial sequences.
4.3.2 MTPT
DNA transfer is prevalent in flowering plants, with DNA sequences being exchanged between the nuclear genome and the mitogenome (Wang X. C. et al., 2018). The most ancient mitochondrial-to-plastid DNA transfer (MTPT) events occurred approximately 300 million years ago, before the divergence of gymnosperms and angiosperms. Although most MTPTs are non-functional, some notable exceptions have been discovered, such as contributions to the replacement of tRNA genes, the creation of promoter regions and codons, and involvement in post-transcriptional RNA processing. Small DNA plastid fragments typically migrate to mitochondria, while larger fragments are exchanged between the nucleus and mitochondria.
In this study, the longest potential transfer fragment from the chloroplast of R. glutinosa was 4,513 bp, whereas the longest from mitochondria was 78,947 bp. The imperfect repair mechanism of mitochondria may facilitate the insertion of foreign sequences, and following the integration of nuclear organelle DNA, this DNA may undergo rearrangement, mutation, elimination, breakage, and proliferation. This process may represent one of the mechanisms driving species evolution.
4.4 RNA editing sites
RNA editing events typically occur during the post-transcriptional process in mitochondria, with specific RNA positions affected by RNA editing and their corresponding DNA positions referred to as editing sites (Edera et al., 2018). Early diverging lineages exhibit the highest number of editing sites among angiosperms, with approximately 400 editing sites reported in Arabidopsis (Edera et al., 2018). Our study verified 507 RNA editing events within the protein-coding region of R. glutinosa, all of which involved C-to-U conversions. The amino acid changes induced by this type of RNA editing may be statistically correlated with alterations in protein hydrophobicity. Cytoplasmic male sterility (CMS) is also associated with reduced, deleted, or incorrect RNA editing of mitochondrial gene transcripts, which modifies gene expression patterns and the functional properties of translation products, ultimately leading to CMS (Hu et al., 2014). For example, in male-sterile lines of Sorghum, the frequency of RNA editing within the atp6 transcript is notably reduced. Additionally, two specific RNA editing sites within the atp9 maintainer transcript in rice alter arginine codons to termination codons. Intriguingly, these amino acid changes result in alterations in the expression of three genes including two stop codons (rps10 and atp6) and one start codon (nad4L). This finding provides insights for future molecular breeding of R. glutinosa.
Double-stranded DNA breaks (DSBs) are repaired primarily via two mechanisms: non-homologous end-joining (NHEJ) and homologous recombination (HR) (Roy et al., 2022). RNA can directly repair DSBs in an HR-dependent (RAD51-dependent) process, inhibited by RNases H1 and H2, known to degrade RNA-DNA hybrids (Mishra et al., 2018). Compared to other terrestrial plants, angiosperms have undergone extensive loss of editing sites through the substitution of editable cytidines with thymidines in their genomes. The homologous recombination of cDNA produced by reverse transcription of edited RNA appears to be one of the molecular mechanisms responsible for the loss of editing sites. While RNA editing is essential for DNA damage repair and genetic selection, its specific mechanism requires further investigation. The prediction and identification of these RNA editing sites offer valuable insights into inferring gene function through the introduction of novel codons. Furthermore, these findings highlight the crucial role of RNA editing in regulating mitochondrial gene expression in plants, particularly its impact on protein synthesis and functionality which subsequently influences plant growth and developmental processes.
5 Conclusion
In conclusion, this study offers a comprehensive analysis of the R. glutinosa mitochondrial genome, focusing on graph-based genome representation and identifying multiple chromosome configurations. Our findings reveal the presence of three minor configurations of the R. glutinosa mitochondrial genome, which were confirmed through PCR amplification and Sanger sequencing. Additionally, we observed the transfer of mitochondrial and chloroplast sequences to the nuclear genome, highlighting the complex interplay between organelle genomes and the nucleus. The research presented here contributes valuable insights into plant mitochondrial genomes’ intricate structure and dynamics, which can inform future molecular breeding efforts for R. glutinosa and other plant species. However, further experimental evidence is needed to fully understand the specific mechanisms of RNA editing and the potential nuclear screening processes for transferred genes. By expanding our understanding of plant mitochondrial genomes, we can better elucidate the factors that drive species evolution and develop targeted strategies for plant improvement.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GenBank (https://www.ncbi.nlm.nih.gov/) with accession numbers: OR030048.1 and ON951335.1 - ON951336.1, respectively. The associated BioProject, BioSample, and SRA numbers are PRJNA905540, SAMN37344158, and SRR26039035 for the Illumina sequencing reads and SRR26039034 for the Nanopore sequencing reads.
Author contributions
TZ: Data curation, Formal analysis, Validation, Visualization, Writing – original draft. YN: Data curation, Methodology, Software, Writing – original draft. JL: Data curation, Methodology, Validation, Visualization, Writing – original draft. HC: Formal analysis, Writing – original draft. QL: Data curation, Validation, Writing – original draft. MJ: Data curation, Writing – original draft. LX: Funding acquisition, Supervision, Writing – review & editing. CL: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. PX: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-022), the National Science & Technology Fundamental Resources Investigation Program of China [2018FY100705], National Natural Science Foundation of China [81872966], The National Science & Technology Fundamental Resources Investigation Program of China (2018FY100700), Guangxi Science and Technology base and talent project (NO. AD22080012). The funders were not involved in the study design, data collection and analysis, publication decision, or manuscript preparation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1326387/full#supplementary-material
References
Adams, K. L., Qiu, Y. L., Stoutemyer, M., Palmer, J. D. (2002). Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. United States America 99, 9905–9912. doi: 10.1073/pnas.042694899
Backert, S., Dorfel, P., Lurz, R., Borner, T. (1996). Rolling-circle replication of mitochondrial DNA in the higher plant Chenopodium album (L). Mol. Cell. Biol. 16, 6285–6294. doi: 10.1128/MCB.16.11.6285
Bi, C., Shen, F., Han, F., Qu, Y., Hou, J., Xu, K., et al. (2024). PMAT: an efficient plant mitogenome assembly toolkit using low coverage HiFi sequencing data. Horticulture Res., 11(3), uhae023. doi: 10.1093/hr/uhae023
Blomain, E. S., McMahon, S. B. (2012). Dynamic regulation of mitochondrial transcription as a mechanism of cellular adaptation. Biochim. Et Biophys. Acta-Gene Regul. Mech. 1819, 1075–1079. doi: 10.1016/j.bbagrm.2012.06.004
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinf. (Oxford England) 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brigulla, M., Wackernagel, W. (2010). Molecular aspects of gene transfer and foreign DNA acquisition in prokaryotes with regard to safety issues. Appl. Microbiol. Biotechnol. 86, 1027–1041. doi: 10.1007/s00253-010-2489-3
Chateigner-Boutin, A.-L., Small, I. (2010). Plant RNA editing. RNA Biol. 7, 213–219. doi: 10.4161/rna.7.2.11343
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, J., Teng, D., Wu, Z., Li, W., Feng, Y., Tang, Y., et al. (2021). Insights into the Molecular Mechanisms of Liuwei Dihuang Decoction via Network Pharmacology. Chem. Res. Toxicol. 34, 91–102. doi: 10.1021/acs.chemrestox.0c00359
Chen, Y., Ye, W., Zhang, Y., Xu, Y. (2015). High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 43, 7762–7768. doi: 10.1093/nar/gkv784
Chevigny, N., Schatz-Daas, D., Lotfi, F., Gualberto, J. M. (2020). DNA repair and the stability of the plant mitochondrial genome. Int. J. Mol. Sci. 21. doi: 10.3390/ijms21010328
Dahal, S., Dubey, S., Raghavan, S. C. (2018). Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cell. Mol. Life Sci. 75, 1641–1655. doi: 10.1007/s00018-017-2702-y
Edera, A. A., Gandini, C. L., Virginia Sanchez-Puerta, M. (2018). Towards a comprehensive picture of C-to-U RNA editing sites in angiosperm mitochondria. Plant Mol. Biol. 97, 215–231. doi: 10.1007/s11103-018-0734-9
Fernie, A. R., Yang, J. (2019). De novo domestication: an alternative route toward new crops for the future. Mol. Plant 12, 615–631. doi: 10.1016/j.molp.2019.03.016
Gonzalez, P., Barroso, G., Labarere, J. (1999). Molecular gene organisation and secondary structure of the mitochondrial large subunit ribosomal RNA from the cultivated Basidiomycota Agrocybe aegerita: a 13 kb gene possessing six unusual nucleotide extensions and eight introns. Nucleic Acids Res. 27, 1754–1761. doi: 10.1093/nar/27.7.1754
Greiner, S., Lehwark, P., Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64.
Gualberto, J. M., Newton, K. J. (2017). Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 68, 225–252. doi: 10.1146/annurev-arplant-043015-112232
He, W., Xiang, K., Chen, C., Wang, J., Wu, Z. (2023). Master graph: an essential integrated assembly model for the plant mitogenome based on a graph-based framework. Briefings Bioinf. 24, bbac522. doi: 10.1093/bib/bbac522
Hu, J., Huang, W., Huang, Q., Qin, X., Yu, C., Wang, L., et al. (2014). Mitochondria and cytoplasmic male sterility in plants. Mitochondrion 19, 282–288. doi: 10.1016/j.mito.2014.02.008
Ichinose, M., Sugita, M. (2017). RNA editing and its molecular mechanism in plant organelles. Genes 8.
Igarashi, K., Kazama, T., Motomura, K., Toriyama, K. (2013). Whole genomic sequencing of RT98 mitochondria derived from oryza rufipogon and northern blot analysis to uncover a cytoplasmic male sterility-associated gene. Plant Cell Physiol. 54, 237–243. doi: 10.1093/pcp/pcs177
Jin, J.-J., Yu, W.-B., Yang, J.-B., Song, Y., dePamphilis, C. W., Yi, T.-S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21. doi: 10.1186/s13059-020-02154-5
Kang, B.-C., Bae, S.-J., Lee, S., Lee, J. S., Kim, A., Lee, H., et al. (2021). Chloroplast and mitochondrial DNA editing in plants. Nat. Plants 7, 899–905. doi: 10.1038/s41477-021-00943-9
Kim, Y.-G., Komakech, R., Jeong, D. H., Park, Y. M., Lee, T. K., Kim, K. H., et al. (2020). Verification of the field productivity of rehmannia glutinosa (Gaertn.) DC. Developed through optimized in vitro culture method. Plants-Basel 9.
Knoll, A., Fauser, F., Puchta, H. (2014). DNA recombination in somatic plant cells: mechanisms and evolutionary consequences. Chromosome Res. 22, 191–201. doi: 10.1007/s10577-014-9415-y
Koenig, D., Jimenez-Gomez, J. M., Kimura, S., Fulop, D., Chitwood, D. H., Headland, L. R., et al. (2013). Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. Proc. Natl. Acad. Sci. United States America 110, E2655–E2662. doi: 10.1073/pnas.1309606110
Letunic, I., Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Levin, L., Blumberg, A., Barshad, G., Mishmar, D. (2014). Mito-nuclear co-evolution: the positive and negative sides of functional ancient mutations. Front. Genet. 5, 448. doi: 10.3389/fgene.2014.00448
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinf. (Oxford England) 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinf. (Oxford England) 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, J., Xu, Y., Shan, Y., Pei, X., Yong, S., Liu, C., et al. (2021). Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 254. doi: 10.1007/s00425-021-03684-3
Li, M., Jiang, H., Hao, Y., Du, K., Du, H., Ma, C., et al. (2022). A systematic review on botany, processing, application, phytochemistry and pharmacological action of Radix Rehmnniae. J. Ethnopharmacol 285. doi: 10.1016/j.jep.2021.114820
Liao, S., Chen, L., Song, Z., He, H. (2022). The fate of damaged mitochondrial DNA in the cell. Biochim. Et Biophys. Acta-Molecular Cell Res. 1869. doi: 10.1016/j.bbamcr.2022.119233
Liu, C., Ma, R., Wang, L., Zhu, R., Liu, H., Guo, Y., et al. (2017). Rehmanniae Radix in osteoporosis: A review of traditional Chinese medicinal uses, phytochemistry, pharmacokinetics and pharmacology. J. Ethnopharmacol 198, 351–362. doi: 10.1016/j.jep.2017.01.021
Liu, S., Ni, Y., Li, J., Zhang, X., Yang, H., Chen, H., et al. (2023). CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. doi: 10.1111/1755-0998.13729
Liu, S.-L., Zhuang, Y., Zhang, P., Adams, K. L. (2009). Comparative analysis of structural diversity and sequence evolution in plant mitochondrial genes transferred to the nucleus. Mol. Biol. Evol. 26, 875–891. doi: 10.1093/molbev/msp011
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Lu, Z., Huang, M., Lin, H., Wang, G., Li, H. (2022). Network pharmacology and molecular docking approach to elucidate the mechanisms of Liuwei Dihuang pill in diabetic osteoporosis. J. Orthopaedic Surg. Res. 17. doi: 10.1186/s13018-022-03194-2
Martin, W. F., Garg, S., Zimorski, V. (2015). Endosymbiotic theories for eukaryote origin. Philos. Trans. R. Soc. B-Biological Sci. 370. doi: 10.1098/rstb.2014.0330
McFarlane, R. J., Humphrey, T. C. (2010). A role for recombination in centromere function. Trends Genet. 26, 209–213. doi: 10.1016/j.tig.2010.02.005
Meng, X., He, M., Guo, R., Duan, R., Huo, F., Lv, C., et al. (2017). Investigation of the effect of the degree of processing of radix rehmanniae preparata (Shu dihuang) on shu dihuangtan carbonization preparation technology. Molecules 22. doi: 10.3390/molecules22071193
Meyer, R. S., Purugganan, M. D. (2013). Evolution of crop species: genetics of domestication and diversification. Nat. Rev. Genet. 14, 840–852. doi: 10.1038/nrg3605
Miao, Y., Chen, H., Xu, W., Liu, C., Huang, L. (2022). Cistanche species mitogenomes suggest diversity and complexity in lamiales-order mitogenomes. Genes 13. doi: 10.3390/genes13101791
Milne, I., Bayer, M., Cardle, L., Shaw, P., Stephen, G., Wright, F., et al. (2010). Tablet-next generation sequence assembly visualization. Bioinf. (Oxford England) 26, 401–402. doi: 10.1093/bioinformatics/btp666
Mishra, A., Saxena, S., Kaushal, A., Nagaraju, G. (2018). RAD51C/XRCC3 facilitates mitochondrial DNA replication and maintains integrity of the mitochondrial genome. Mol. Cell. Biol. 38. doi: 10.1128/MCB.00489-17
Misra, S., Harris, N. (2006). Using Apollo to browse and edit genome annotations. Curr. Protoc. Bioinf.
Mohammed, T., Firoz, A., Ramadan, A. M. M. (2022). RNA editing in chloroplast: advancements and opportunities. Curr. Issues Mol. Biol. 44, 5593–5604. doi: 10.3390/cimb44110379
Ni, Y., Li, J., Chen, H., Yue, J., Chen, P., Liu, C. (2022). Comparative analysis of the chloroplast and mitochondrial genomes of Saposhnikovia divaricata revealed the possible transfer of plastome repeat regions into the mitogenome. BMC Genomics 23. doi: 10.1186/s12864-022-08821-0
Okazaki, M., Kazama, T., Murata, H., Motomura, K., Toriyama, K. (2013). Whole mitochondrial genome sequencing and transcriptional analysis to uncover an RT102-type cytoplasmic male sterility-associated candidate gene derived from oryza rufipogon. Plant Cell Physiol. 54, 1560–1568. doi: 10.1093/pcp/pct102
Osellame, L. D., Blacker, T. S., Duchen, M. R. (2012). Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 26, 711–723.
Palozzi, J. M., Jeedigunta, S. P., Minenkova, A., Monteiro, V. L., Thompson, Z. S., Lieber, T., et al. (2022). Mitochondrial DNA quality control in the female germline requires a unique programmed mitophagy. Cell Metab. 34, 1809. doi: 10.1016/j.cmet.2022.10.005
Picardi, E., Pesole, G. (2013). REDItools: high-throughput RNA editing detection made easy. Bioinf. (Oxford England) 29, 1813–1814. doi: 10.1093/bioinformatics/btt287
Roger, A. J., Munoz-Gomez, S. A., Kamikawa, R. (2017). The origin and diversification of mitochondria. Curr. Biol. 27, R1177–R1192. doi: 10.1016/j.cub.2017.09.015
Roy, A., Kandettu, A., Ray, S., Chakrabarty, S. (2022). Mitochondrial DNA replication and repair defects: Clinical phenotypes and therapeutic interventions. Biochim. Et Biophys. Acta-Bioenergetics 1863. doi: 10.1016/j.bbabio.2022.148554
Rozewicki, J., Li, S., Amada, K. M., Standley, D. M., Katoh, K. (2019). MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47, W5–W10. doi: 10.1093/nar/gkz342
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Sloan, D. B. (2013). One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’model of plant mitochondrial DNA structure. New Phytol. 200, 978–985. doi: 10.1111/nph.12395
Small, I. D., Schallenberg-Ruedinger, M., Takenaka, M., Mireau, H., Ostersetzer-Biran, O. (2020). Plant organellar RNA editing: what 30 years of research has revealed. Plant J. 101, 1040–1056. doi: 10.1111/tpj.14578
Sun, T., Bentolila, S., Hanson, M. R. (2016). The unexpected diversity of plant organelle RNA editosomes. Trends Plant Sci. 21, 962–973. doi: 10.1016/j.tplants.2016.07.005
Takenaka, M., Verbitskly, D., van der Merwe, J. A., Zehrmann, A., Brennicke, A. (2008). The process of RNA editing in plant mitochondria. Mitochondrion 8, 35–46. doi: 10.1016/j.mito.2007.09.004
Tian, C. F., Zhou, Y. J., Zhang, Y. M., Li, Q. Q., Zhang, Y. Z., Li, D. F., et al. (2012). Comparative genomics of rhizobia nodulating soybean suggests extensive recruitment of lineage-specific genes in adaptations. Proc. Natl. Acad. Sci. United States America 109, 8629–8634.
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3-new capabilities and interfaces. Nucleic Acids Res. 40. doi: 10.1093/nar/gks596
Wang, X.-C., Chen, H., Yang, D., Liu, C. (2018). Diversity of mitochondrial plastid DNAs (MTPTs) in seed plants. Mitochondrial DNA Part A 29, 635–642. doi: 10.1080/24701394.2017.1334772
Wang, R., Wang, Y., Yang, Q., Kang, C., Li, M. (2018). Unraveling the characteristics of the microbial community and potential pathogens in the rhizosphere soil of Rehmannia glutinosa with root rot disease. Appl. Soil Ecol. 130, 271–279. doi: 10.1016/j.apsoil.2018.07.001
Wang, J., Kan, S., Liao, X., Zhou, J., Tembrock, L. R., Daniell, H., et al. (2024). Plant organellar genomes: Much done, much more to do. Trends Plant Sci. doi: 10.1016/j.tplants.2023.12.014
Wick, R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 3 (10), e000132. doi: 10.1099/mgen.0.000132
Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13. doi: 10.1371/journal.pcbi.1005595
Wu, B., Chen, H. M., Shao, J. J., Zhang, H., Wu, K., Liu, C. (2017). Identification of symmetrical RNA editing events in the mitochondria of salvia miltiorrhiza by strand-specific RNA sequencing. Sci. Rep. 7, 11. doi: 10.1038/s41598-017-00052-8
Wu, P., Chen, H., Xu, C., Yang, J., Zhang, X. C., Zhou, S. L. (2021). NOVOWrap: An automated solution for plastid genome assembly and structure standardization. Mol. Ecol. Resour. 21, 2177–2186. doi: 10.1111/1755-0998.13410
Wu, Z.-Q., Liao, X.-Z., Zhang, X.-N., Tembrock, L. R., Broz, A. (2022). Genomic architectural variation of plant mitochondria-A review of multichromosomal structuring. J. Syst. Evol. 60, 160–168. doi: 10.1111/jse.12655
Yang, H., Chen, H., Ni, Y., Li, J., Cai, Y., Ma, B., et al. (2022). De novo hybrid assembly of the salvia miltiorrhiza mitochondrial genome provides the first evidence of the multi-chromosomal mitochondrial DNA structure of salvia species. Int. J. Mol. Sci. 23. doi: 10.3390/ijms232214267
Yang, Z., Ni, Y., Lin, Z., Yang, L., Chen, G., Nijiati, N., et al. (2022). De novo assembly of the complete mitochondrial genome of sweet potato (Ipomoea batatas L. Lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant Biol. 22.
Zhang, G.-J., Dong, R., Lan, L.-N., Li, S.-F., Gao, W.-J., Niu, H.-X. (2020). Nuclear integrants of organellar DNA contribute to genome structure and evolution in plants. Int. J. Mol. Sci. 21. doi: 10.3390/ijms21030707
Zhang, D., Gao, F., Jakovlic, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zheng, W., Wang, G., Zhang, Z., Wang, Z., Ma, K. (2020). Research progress on classical traditional Chinese medicine formula Liuwei Dihuang pills in the treatment of type 2 diabetes. Biomedicine Pharmacotherapy 121. doi: 10.1016/j.biopha.2019.109564
Keywords: mitochondrial genome, repeat-mediated recombination, mitochondrial plastid DNAs, RNA editing, homologous recombination
Citation: Zeng T, Ni Y, Li J, Chen H, Lu Q, Jiang M, Xu L, Liu C and Xiao P (2024) Comprehensive analysis of the mitochondrial genome of Rehmannia glutinosa: insights into repeat-mediated recombinations and RNA editing-induced stop codon acquisition. Front. Plant Sci. 15:1326387. doi: 10.3389/fpls.2024.1326387
Received: 23 October 2023; Accepted: 24 April 2024;
Published: 14 May 2024.
Edited by:
Michael James Wilkinson, Aberystwyth University, United KingdomReviewed by:
Kun Zhang, Yangzhou University, ChinaZhiqiang Wu, Chinese Academy of Agricultural Sciences, China
Qiang Li, Chengdu University, China
Copyright © 2024 Zeng, Ni, Li, Chen, Lu, Jiang, Xu, Liu and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijia Xu, bGp4dUBpbXBsYWQuYWMuY24=; Chang Liu, Y2xpdTY2ODhAaG90bWFpbC5jb20=; Y2xpdUBpbXBsYWQuYWMuY24=
†These authors have contributed equally to this work