 Shamshad Alam1
Shamshad Alam1 Krishna Tesman Sundaram1
Krishna Tesman Sundaram1 Uma Maheshwar Singh2
Uma Maheshwar Singh2 Madamshetty Srinivas Prasad3
Madamshetty Srinivas Prasad3 Gouri Sankar Laha3
Gouri Sankar Laha3 Pallavi Sinha1*
Pallavi Sinha1* Vikas Kumar Singh1*
Vikas Kumar Singh1*- 1Rice Breeding Innovation, International Rice Research Institute (IRRI) South Asia Hub, Hyderabad, India
- 2Rice Breeding Innovation, International Rice Research Institute-South Asia Regional Centre (ISARC), Varanasi, Uttar Pradesh, India
- 3Crop Protection, Indian Council of Agriculture Research-Indian Institute of Rice Research (ICAR-IIRR), Hyderabad, India
Rice blast and bacterial leaf blight, are major disease, significantly threatens rice yield in all rice growing regions under favorable conditions and identification of resistance genes and their superior haplotypes is a potential strategy for effectively managing and controlling this devastating disease. In this study, we conducted a genome-wide association study (GWAS) using a diverse set of 147 rice accessions for blast and bacterial blight diseases in replications. Results revealed 23 (9 for blast and 14 for BLB) significant marker-trait associations (MTAs) that corresponded to 107 and 210 candidate genes for blast and BLB, respectively. The haplo-pheno analysis of the candidate genes led to the identification of eight superior haplotypes for blast, with an average SES score ranging from 0.00 to 1.33, and five superior haplotypes for BLB, with scores ranging from 1.52cm to 4.86cm superior haplotypes. Among these, superior haplotypes LOC_OS12G39700-H4 and LOC_Os06g30440-H33 were identified with the lowest average blast scores of 0.00-0.67, and superior haplotype LOC_Os02g12660-H39 exhibited the lowest average lesion length (1.88 - 2.06cm) for BLB. A total of ten accessions for blast and eight accessions for BLB were identified carrying superior haplotypes were identified. These haplotypes belong to aus and indx subpopulations of five countries (Bangladesh, Brazil, India, Myanmar, and Pakistan). For BLB resistance, eight accessions from six countries (Bangladesh, China, India, Myanmar, Pakistan, and Sri Lanka) and four subpopulations (aus, ind1A, ind2, and ind3) were identified carrying superior haplotypes. Interestingly, four candidate genes, LOC_Os06g21040, LOC_Os04g23960, LOC_Os12g39700, and LOC_Os01g24640 encoding transposon and retrotransposon proteins were among those with superior haplotypes known to play a crucial role in plant defense responses. These identified superior haplotypes have the potential to be combined into a single genetic background through haplotype-based breeding for a broader resistance spectrum against blast and bacterial blight diseases.
1 Introduction
Rice is a crucial global crop, with over 90% of its production and consumption concentrated in the Asian region. To meet the continuous demand for rice and feed the growing population, it is projected that we need to produce 25% more rice by the year 2030. However, the potential yield of the rice crop is significantly impacted by various pests and diseases. According to Savary et al. (2000), pests and diseases are responsible for causing yield losses of up to 41% annually in rice crops across Asia. The economic impact of these plant diseases is staggering, with over $220 billion lost due to diseases and at least $70 billion lost to invasive insects every year (FAO, 2019). Moreover, climate change is emerging as a major challenge for sustainable rice production. Shifting weather patterns brought about by climate change are significantly influencing the prevalence and severity of various plant diseases. Rising temperatures and increased carbon dioxide concentration are leading to physiological changes in plants, which in turn escalate the intensity of crop diseases. Disturbances in rainfall patterns and temperature are also affecting the life cycles, reproduction, and geographical distribution of many plant pathogens, further complicating disease management strategies. In light of these pressing challenges, the development of disease-resistant rice varieties that can withstand changing climate conditions becomes paramount.
Rice plants face severe attacks from various pathogens throughout their growth stages, significantly reducing yield and quality. Among these pathogens, Blast and Bacterial leaf blight (BLB), caused by Magnaporthe oryzae and Xanthomonas oryzae, respectively, are major diseases posing a substantial threat due to their wide distribution and potential damage under favorable conditions. Blast fungus infects rice at different growth stages, leading to an annual yield loss of approximately 10-30% (Devanna et al., 2022). It is particularly devastating in lowland rice in temperate and subtropical Asia and upland rice in tropical Asia, Latin America, and Africa. On the other hand, Bacterial blight causes systemic infection during the maximum tillering stage, resulting in 20-40% yield loss (Yang et al., 2020). Susceptible cultivars can suffer yield losses of up to 100%, depending on factors such as weather conditions, variety, nitrogen fertilizer application, and early-stage infection (Yang et al., 2020). The resistance against these diseases is governed by either vertical or horizontal resistance. Vertical resistance is conferred by major genes that provide race-specific resistance, while horizontal resistance is governed by multiple genes offering partial resistance with delayed and reduced disease lesion development. Horizontal resistance provides broad-spectrum and race-non-specific resistance. Extensive research on the genetics of resistance has led to the identification of more than 100 R-genes and 500 QTLs for blast, along with over 45 BLB R-genes (Lin et al., 2018; Zhang et al., 2017a: Neelam et al., 2020). Among these, 31 blast resistance and eleven BLB genes have been cloned and characterized at the molecular level (Lin et al., 2018). However, relying solely on race-specific resistance can lead to the evolution of pathogens, posing a challenge for breeders to regularly identify new genes/QTLs and develop durable and broad-spectrum resistant rice varieties.
For decades, blast and BLB have been devastating diseases that result in significant yield penalties in rice. Consequently, the urgent need to breed rice varieties with enhanced resistance has been recognized (Khush, 2005). Recently, several resistant genes against blast and BLB have been utilized in breeding programs to improve both elite rice varieties and parental lines of hybrids through Marker-Assisted Selection (MAS) (Dixit et al., 2020; Singh et al., 2022; Janaki Ramayya et al., 2021). However, deploying a single resistance gene often leads to rapid breakdowns. In the Philippines, for instance, deploying Xa7 and other genes (xa5 and Xa21) individually resulted in resistance lasting only about three years (Vera Cruz et al., 2000; Webb et al., 2010). To achieve broad-spectrum and durable resistance, the practice of pyramiding more than one resistance gene has been recommended (Khanna et al., 2015; Xiao et al., 2016). In various breeding programs, combinations of 2-4 R genes have been extensively utilized to achieve durable resistance against prevalent pathotypes of blast and BLB (Yugander et al., 2017). A few reported gene pyramids include Pi54 + Pi1 (Jamaloddin et al., 2020), Piz5 + Pi54 (Singh et al., 2013), Pi9 + Pita (Khanna et al., 2015), Pi9 + Pi2 + Piz (Chukwu et al., 2019), Xa21+ xa13 (Jamaloddin et al., 2020), xa5+ xa13+ Xa21 (Pradhan et al., 2015), Xa4+ xa5+ Xa21 (Suh et al., 2015), xa13+ Xa21 (Swathi et al., 2019), and Xa4+ xa5+ xa13+ Xa21 (Dixit et al., 2020). These combinations have demonstrated effective and durable resistance against both blast and BLB, showcasing their potential for enhancing resilience to these devastating diseases.
The emergence of new races of pathogens in the field poses a challenge, as they can overcome resistant rice varieties within a few years after their existence (Chen et al., 2009). Therefore, it is essential to regularly identify new donors with novel or superior alleles/haplotypes and incorporate them into resistance breeding programs. Global germplasm collections, such as the 3000-rice genome project (3,000 rice genomes project, 2014) , hold a wealth of natural genetic diversity for various biotic and abiotic stresses. Exploring such diverse genetic resources enables the identification of new donors with genes/QTLs and their superior haplotypes. Genome-wide association studies (GWAS) using high-density single nucleotide polymorphisms (SNPs) and haplotype analysis have proven valuable in detecting genetic variants and novel alleles associated with traits of interest, directly benefiting genomic-assisted breeding to improve commercial cultivars (McCouch et al., 2016; Varshney et al., 2005; Varshney et al., 2009). In recent studies, association mapping has been employed to dissect the genetic architecture of blast (Kang et al., 2016; Frontini et al., 2021) and BLB resistance (Dilla-Ermita et al, 2017; Jiang et al., 2021). For instance, Lu et al. (2021) identified 11 QTLs for BLB resistance in a sub-set of 340 lines from the 3K RG panel. Raboin et al. (2016) identified three loci associated with blast resistance in Japonica and Indica panels. Additionally, Li et al. (2019) identified 56 QTLs for blast resistance in the rice diversity panel 1, and Volante et al. (2020) found 14 Marker Trait Associations (MTAs) associated with blast resistance, with 11 accessions exhibiting high resistance levels through field and growth chamber screenings.
Considering the above facts, the primary objectives of the present study are to identify significant MTAs and their underlying candidate genes conferring resistance to blast and BLB, to discover superior haplotypes (SH), and develop markers to track these SH. Furthermore, the study aims to identify novel genetic donors possessing high levels of resistance against blast and BLB, contributing to the ongoing efforts to develop resilient rice varieties against these two devastating diseases.
2 Materials and methods
2.1 Selection of a core sub-set of 3K RGP
A meticulous process was employed to select representative genotypes from 33 diverse countries, encompassing various subgroups within the 3K rice genome dataset to develop a core sub-set from 3K-RGP. This process involved curating a set of 147 accessions (28 aus, 28 ind1A, 26 ind1B, 35 ind2, 15 ind3, 55 indx, 2 trop, 1 aro and 1 admix) that collectively represents all the subpopulation of 3K rice genome panel. These accessions were chosen to facilitate the evaluation of their resistance against blast and bacterial blight diseases. To measure the genetic diversity, an analysis was conducted utilizing 0.5 million SNPs. This involved employing the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) algorithm, which computed genetic distances based on molecular markers. The outcomes of the analysis were utilized to construct a UPGMA tree, helping in the careful selection of genotypes for subsequent screening. The genetic diversity assessment confirmed the variability within the genotypes, reinforcing the suitability of the chosen core sub-set for further analysis. A core sub-set of 147 accessions of 3K RG Panel from 33 different countries which include 28 aus, 28 ind1A, 26 ind1B, 35 ind2, 15 ind3, 55 indx, 2 trop, 1 aro and 1 admix were used in this study for phenotyping blast and bacterial blight resistance.
2.2 Phenotyping for blast and bacterial blight resistance
2.2.1 Phenotyping for blast resistance
All the genotypes of sub-set 3K RG Panel along with resistant (IR64 and Tetep) and susceptible (HR12) check were screened for their reaction to blast under control conditions in three replications at CERF (Control Environment Research Facility) IRRI-South Asia Hub, ICRISAT Campus, Hyderabad during wet season 2021 (Alam et al., 2017). A highly virulent M. oryzae isolate called SPI-40 collected from IIRR, Hyderabad, Telangana was used for screening (Madhan Mohan., 2011). 8-10 Seeds of each line were sown in portray following random complete block design (RCBD). Resistant and susceptible checks were also shown in each portray. 21 days old seedlings were inoculated with blast spores using a hand sprayer. Inoculated seedlings were incubated in the dark chamber with relative humidity >90% for 24hr at 26°C. After 24 hr. of incubation, all the seedlings were grown at 28/26°C Day/night temperature and 16/8-hr light/dark photoperiod for disease development. Observation of each line was recorded on 0–5 SES scale (IRRI (International Rice Research Institute), 1996) 7 days after inoculation. Scores 0–2 were considered resistant (R), 3 as moderately resistant (MR), and 4-5 as susceptible (S). The average blast score was used for the genome-wide association mapping. An average of three replications were used for the analysis.
2.2.2 Phenotyping of bacterial blight resistance
To evaluate accession of sub-set of 3k RG panel for bacterial blight resistance, a highly virulent isolate of Xanthomonas oryzae pv. oryzae (Xoo) DX-020, belongs to pathotype-4 (Yugander et al., 2017) was used. Eight plants of each accession of sub-set of 3K RG panel along with checks were screened for their reaction to BLB under controlled conditions at the maximum tillering stage during wet season 2021 in three replications. The rice plants were inoculated with freshly prepared inoculum following the clip inoculation method with sterilized scissors (Kauffman et al., 1973). Five plants and five fully expanded leaves per plant were inoculated (2-3cm from the tip). The disease assessment was done three weeks after inoculation by measuring the lesion length in centimetres from the cut tip of inoculated leave. The mean of two fully expanded leaf from three replicate plants for each accession were taken for determining the reaction type. A plant was classified as resistant if the average lesion length was shorter than 5 cm, moderately resistant if the lesion was >5-10 cm, moderately susceptible if the lesion was >10-15 cm, and susceptible if the lesion was >15 cm (Korinsak et al., 2021)
2.3 Genetic diversity and population structure analysis
Genetic diversity and population structure was done using genome-wide 0.5 million SNP data points of all the accessions. The tree was constructed using neighbor-joining method and the calculated dissimilarity index. Structure admixture analysis was analyzed through admixture software with k=3 as cross-validation error shown low. A Neighbor-joining tree was constructed using TASSEL Software version 5.0. The Phylogenetic tree is visualized through an unrooted tree using treeio R package. The Principal Component Analysis is done using SNPRelate R Package and Genetic Diversity and Analysis of Molecular Variance (AMOVA) is done using poppr R package.
2.4 GWAS, favourable allele, haplotype analysis and haplo−pheno analysis
Genome wide association study was performed using more than 0.5 million SNPs and the average SES score of three replications for blast and the average lesion length of five BLB clipped leaves. The analysis was conducted using five different multilocus GWAS models, including mrMLM (Wang et al., 2016), FASTmrMLM, FASTmrEMMA (Tamba and Zhang, 2018 ), ISIS EM-BLASSO, and pLARmEB (Zhang et al., 2017b), using an R-based methodology (https://CRAN.R-project.org/). The criterion for significance to find peak associations (MTAs) with the target trait is a LOD score of ≥5. The phenotypic allele effect (ai) was determined by the formula given by Zhang et al., 2013 and the favorable alleles of each trait.
To identify the candidate genes underlying the MTAs, all annotated genes contained within 50kbp LD from the peak MTA of the Oryza sativa reference sequence (Os-Nipponbare-Reference-IRGSP-1.0; RAP database: http://rapdb.dna.affrc.go.jp/download/irgsp1.html) were taken into consideration. Additionally, known blast R genes or literature-based QTLs were compared to the sites of the significant MTAs (Sharma et al., 2012; Ashkani et al., 2016) and recent literature (Kang et al., 2016; Ma et al., 2015, 2015; Zhao et al., 2011; Xu et al., 2014; Su et al., 2015; Mgonja et al., 2016; Zhu et al., 2016; Jiang et al., 2020; Yang et al., 2022).
The total candidate genes were taken into the haplo-pheno analysis for the identification of superior haplotypes. Haplotype analysis was carried using in-house scripts in R programming. Then the analysis included One-way ANOVA with haplotype as a fixed factor. Subsequently, Duncan’s multiple range test (DMRT) and ANOVA were used to test the statistical significance among the mean of haplotype groups using the Agricolae package in R (De Mendiburu, 2018). All reported P values underwent adjustment using either the Benjamini-Hochberg FDR or the Bonferroni method when multiple comparisons were performed, and a statistically significant threshold of adjusted p < 0.05 was considered.
For the whole investigation, we used the ‘3K filtered’ SNP set from the SNP seek database. The following filtering criteria were used to generate the filtered from the Base SNP set: (1) alternative allele frequency at least 0.01, (2) fraction of missed calls per SNP at most 0.292, and this SNP set was previously present in the SNP seek database, which was directly used in this work (http://snp-seek.irri.org/download.zul). Only nonsynonymous SNPs and indels in the exon region that cause an amino acid change were taken into account in the haplotype analysis for the candidate locus. The SNP-seek database (http://snp-seek.irri.org/_download.zul) was used to gather data on haplotypes and their diversity in order to identify the best haplotype by classifying haplotypes according to the genotype trait means for the related genes. The R-package (https://CRAN.R-project.org/) was used to create boxplots with a significance threshold of p <= 0.05.
3 Results
3.1 Insights into genetic diversity and population structure of the panel
The SNP-based diversity analysis provided valuable insights into the diversity exhibited by the panel (Supplementary Figure 1). The admixture analysis showed a notable distinction at k=2, distinctly separating the indica and aus groups. The optimal population structure emerged at k=3, revealing population divergence within aus, ind1A, and ind1B (Supplementary Figure 1A). The structure analysis revealed that indx has a blending of all subpopulations, signifying a historical interplay of domestication and selection between indx and other clusters within the population. Remarkably, while the admixture analysis highlighted the optimal k value as 3, it couldn’t distinguish the subpopulations of ind1B and ind3. However, the cladogram analysis provided confirmed relationships and divergences within these populations (Supplementary Figure 1B). The cladogram, illustrating the genetic relationships between samples, suggested the findings of identity-by-descent and neighbor-joining tree construction methods. The cladogram unequivocally reinforced the admixture results, underlining how indx’s presence is a consequence of controlled selection and domestication of various varieties. Notably, indx accessions demonstrated a heightened relatedness between ind3 and ind1B, elucidating the dispersion of their unique diversity and their positioning within the broader subgroups of ind2 and ind1B.
Employing principal component analysis (PCA) on our population, we extracted 15% of the total variation, effectively representing two distinct clusters. These clusters shed light on the contributions of aus and indica lines to the population’s genetic makeup. Despite the relatively modest variance captured by the PCA analysis, the second cluster, attributed to indica lines, elucidates the divergence in allele composition in a blended manner (Supplementary Figures 1C, D). To further delve into genetic diversity, the poppr r package was utilized, as displayed in Supplementary Table 1. Among the subpopulations, ind1A displayed the highest Nei’s genetic distance at 0.34, while indx exhibited the lowest at 0.23. As for the Shannon diversity index, indx emerged with the highest value of 3.9, in contrast to ind1A, which recorded the lowest at 2.6. An examination of molecular variance unveiled that 87.74% of the variance exists within samples, while the remaining 12.25% of the variance is accounted for between the distinct population groups.
3.2 Evaluation of disease resistance in the sub-set of 3K-RGP
Each of the accessions within the 3K-RGP sub-set underwent comprehensive screening for resistance against blast and BLB in multiple replications. Disease reaction showed minimal variation across replications, so the average score was used for the subsequent analysis (Supplementary Figure 2). In terms of blast severity, the spectrum of severity scores spanned from 0, indicating high resistance (e.g., LALSAITA::IRGC 43915-1), to 5, indicative of high susceptibility (e.g., RTS 16::IRGC 8235-1). Among all accessions, 12.92% showed resistance with a severity score (SES) of 0-2, another 12.92% demonstrated moderate resistance with SES scores above 2 to 3, while the majority, comprising 72.78% of genotypes, exhibited susceptibility with SES scores above 3 to 5. The reference susceptible check HR12 recorded a score of 5, while the resistant check Tetep scored 0. Evidently, the phenotypic variation in blast severity was considerable among the genotypes within the panel, revealing a pronounced divergence (Supplementary Figures 2A–D).
In the case of BLB screening, all accessions were inoculated during the maximum tillering stage. Lesion length averages across the panel ranged from 1.16cm (e.g., SUWEON 311: IRGC 61890-1) to 27.2cm (e.g., BPI 76 NON SENSITIVE (GREEN)::IRGC 9790-1). The susceptible check TN1 exhibited a lesion length of 21.5 cm, while the resistant check IRBB60 displayed a length of 0.3 cm. Among the accessions, 17.07% displayed resistance with mean lesion lengths varying from 1.13 to 3.00cm. Additionally, 42% were moderately resistant, characterized by mean lesion lengths spanning 3.08 to 6.00cm. Around 14.58% were moderately susceptible, with mean lesion lengths ranging from 6.02 to 8.92cm, and 25% of genotypes showed susceptibility, their mean lesion lengths varying from 9.02 to 27.2 cm (Supplementary Figures 2B, E, F).
3.3 Unveiling significant MTAs through multi-locus GWAS
We used GWAS analysis using 0.5 million SNPs from the selected accessions and their average scores against blast and BLB to unveil meaningful MTAs. Employing various multilocus GWAS methods (mrMLM, FASTmrMLM, and ISIS EM-BLASSO), we sought to identify significant associations by considering a LOD value ≥5 along with the phenotypic variance (PVE) as a threshold for significance. A total of 23 significant MTAs identified from the analysis, comprising nine for blast resistance and 14 for BLB resistance (Table 1). The soundness of the model is evident from the well-fitted Manhattan and Q-Q plots (Figure 1). The MTAs linked to blast resistance were dispersed across chromosomes 1, 4, 6, 7, 8, 11, and 12, with PVE ranging from 2.60% to 11.96%. Correspondingly, the BLB resistance revealed 14 MTAs, distributed across chromosomes 1, 2, 5, 6, 8, 11, and 12, encompassing PVE values ranging from 3.4% to 19.13%. These associations were identified through two distinct multi-locus GWAS models.
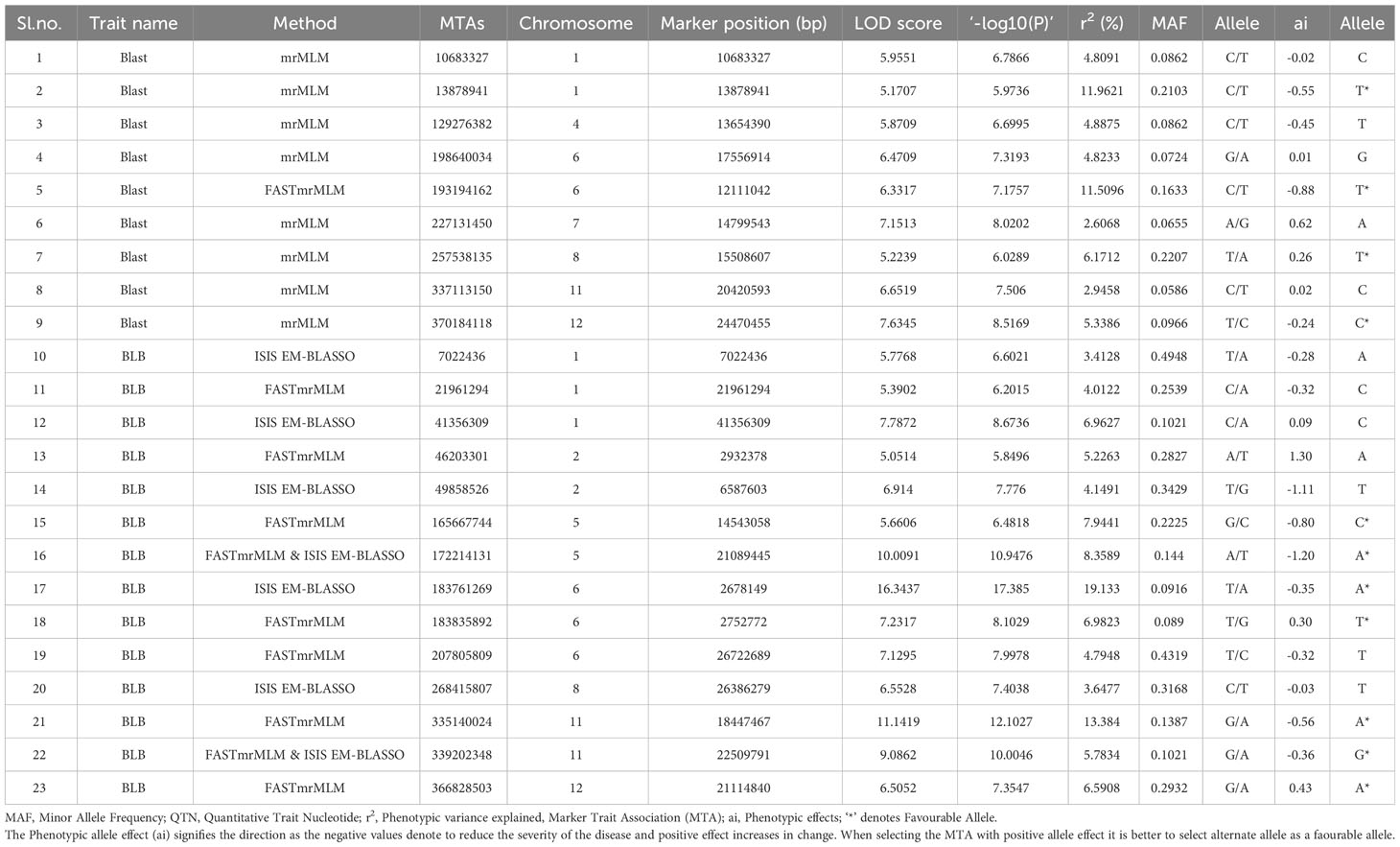
Table 1 MTAs significantly associated with resistance to Blast and BLB, PVE-Phenotypic variance explained, MTA) in red colour font are identified by more than one model.
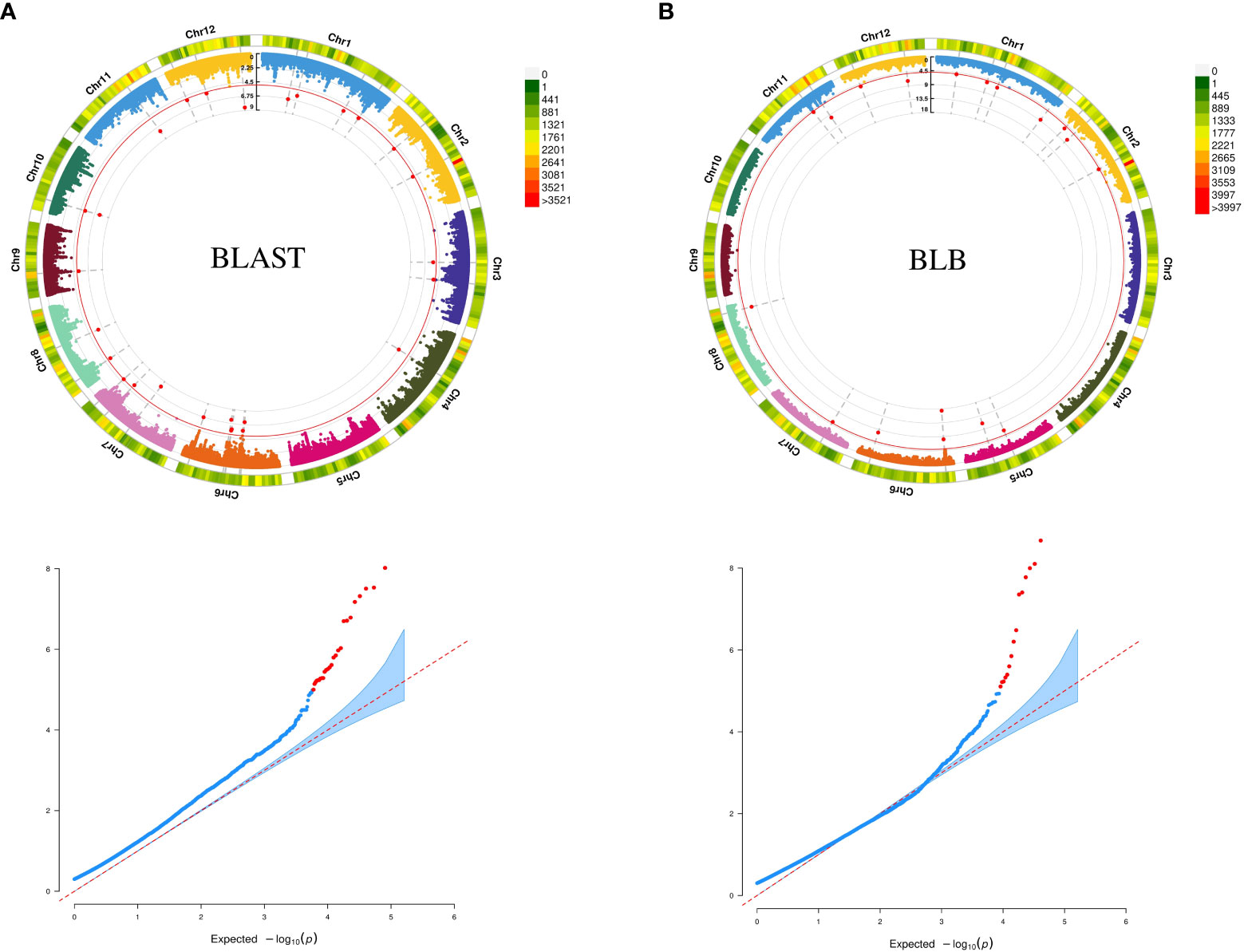
Figure 1 (A) Circular Manhattan and QQ-plot for MTAs associated with resistance to BLAST (B) Circular Manhattan and QQ-plot for MTAs associated with resistance to BLB.
3.4 Prediction and annotation of potential candidate genes
A comprehensive search yielded 107 candidate genes from the nine significant MTAs associated with blast resistance. Similarly, for BLB, 210 candidate genes emerged from the 14 significant MTAs. To enhance our understanding, all identified candidate genes for both blast and BLB were annotated using the RAP-DB database (Supplementary Table 2)
3.5 GWAS findings aligned with known resistant genes
We compared the SNP positions of the prime MTAs pertaining to the target trait with those of established blast and BLB resistance genes or QTLs. By assessing candidate genes positioned within a range of −/+ 50kbp sequences from the peak SNP marker, we sought connections with known R-genes/QTLs. It’s noteworthy that all the significant MTAs for both blast and BLB were distinct, as there were no prior reports of R-genes or QTLs from these regions, except for the presence of the blast resistance gene Pi40 (16274830–17531111) in close proximity to MTA-17556914 on chromosome 6. Nonetheless, several other genes and transcription factors associated with defense response were successfully identified. For instance, among the 107 candidate genes related to blast resistance, 18 corresponded with already documented genes associated with defense response mechanisms. In contrast, for BLB, a substantial number of 54 candidate genes aligned with previously reported defense response genes were identified. However, when we expanded the search to include sequences within −/+ 100 kb from the significant MTAs, we found four overlapping resistant genes on chromosome 11 associated with bacterial blight: Xa23 (LOC_Os11g37620), Xa39 (LOC_Os11g37759), Xa10 (LOC_Os11g37570), and Xa46 (LOC_Os11g37540). These findings contribute to a deeper understanding of the genetic basis of blast and bacterial blight resistance, offering valuable insights for the development of improved disease-resistant varieties through targeted breeding programs.
3.6 Identification of favourable SNP alleles linked to blast and BLB resistance
All MTAs associated with blast and BLB resistance underwent a detailed exploration to uncover favourable SNP alleles. This investigation categorized SNPs demonstrating a negative SNP effect (ai) leading to increased resistance against blast and BLB diseases as “favourable alleles.” Conversely, alleles resulting in positive SNP effect (ai) to blast and BLB diseases were classified as “unfavorable alleles. Allele with more than 5% PVE with high phenotypic allele effect (ai) were selected as favourable allele. For blast and BLB traits, MTAs showcasing a reduction in phenotypic allele effect were recognized as harboring favorable alleles. For instance, for blast resistance, (MTA-193194162 and MTA-13878941 exhibiting high PVE with 11.5% and 11.96%, carrying ‘T’ and ‘T’ alleles denoting a decrease in phenotypic allele effect by -0.88 and -0.56 respectively. Similarly, for BLB, (MTA-335140024 and MTA-183761269), possessing the ‘A’ and ‘A’ alleles respectively, demonstrated a reduction in phenotype by -0.56 and -0.35 with high PVE of 13.38% and 19.13%. However, in case of blast and BLB, a total of four and nine MTAs were identified respectively as favourable alleles with high PVE. This study effectively illustrated that genotype possessing favourable SNPs for the target trait exhibited a substantial decrease in the phenotypic effect of the trait (Table 1).
3.7 Haplo−pheno analysis for the identification of superior haplotypes
To determine the haplotype diversity, haplotype analysis was conducted on the 3K RG panel sub-set, correlating phenotypic outcomes against SNP data for all 317 identified candidate genes (107 for blast and 210 for BLB). This comprehensive analysis aimed to delineate the diversity of haplotypes within the 3K-RG panel. Significant haplotype diversity across 107 genes for blast and 210 genes for BLB were observed (Supplementary Figure 5).
For a better comprehension of the phenotypic performance associated with specific haplotypes, the trait mean was calculated for each individual haplotype. The objective was to identify superior haplotypes, defined as those leading to significantly lower blast scores or lesion lengths for BLB compared to other haplotypes. Statistically validated by Duncan’s test, all candidate loci possessing more than two haplotypes were scrutinized to pinpoint superior haplotypes (Table 2). A number of superior haplotypes linked to resistance traits were successfully detected. In the case of blast, eight superior haplotypes were defined by blast scores ≤2. Among the identified superior haplotypes, eight candidate genes exhibited blast scores ranging from 0.00 to 1.33. LOC_OS12G39700 and LOC_Os06g30440 stood out with superior haplotypes H4 and H33, showing the lowest average blast scores of 0.00-0.67 (Figure 2; Supplementary Figure 3). Furthermore, the superior haplotype H29, associated with candidate gene LOC_Os01g18910, displayed an average blast score of 0.33-1.00. Similarly, H18 emerged as the superior haplotype for LOC_Os01g24540, LOC_Os01g24640, and LOC_Os01g24750, manifesting an average blast score within the range of 0.33-1.33. Additionally, candidate gene LOC_Os04g23960 featured the superior haplotype H17, while LOC_Os06g21040 harbored H177 and H60 as superior haplotypes with scores of 0.67-1.33.
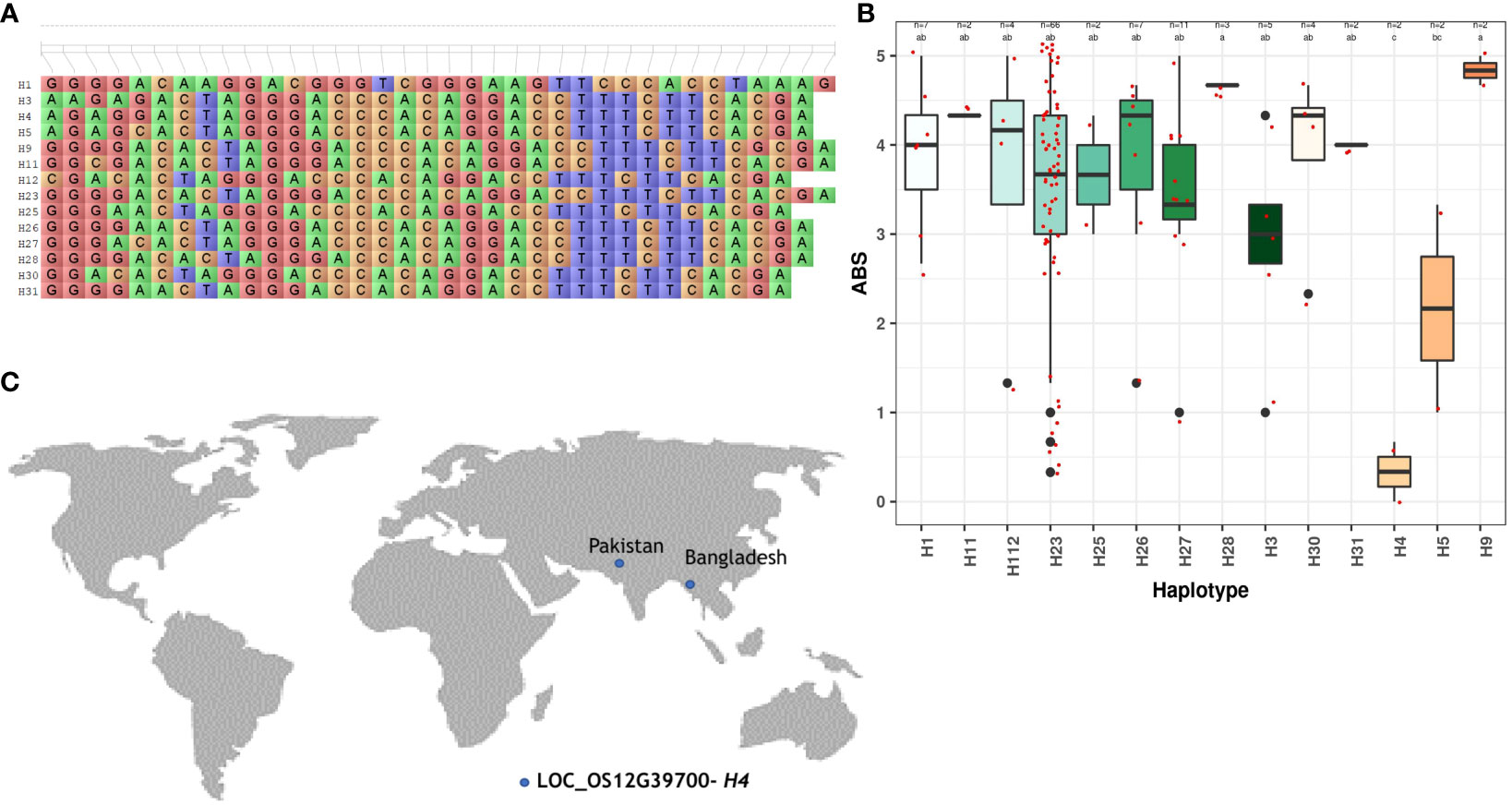
Figure 2 Haplotype analysis of LOC_OS12G39700 across the sub-set panel. (A) Haplotypic variation of LOC_OS12G39700, a gene associated with blast resistance. (B) Boxplot showing variation in blast resistance among 147 accessions of 3K RGP (C) The geographical distribution of superior haplotype.
For BLB, superior haplotypes were characterized by average lesion lengths ≤3. Out of the 210 candidate genes analyzed, four genes located on chromosomes 2, 5, and 6 yielded superior haplotypes (Table 2). Among these genes, a total of five superior haplotypes with BLB scores spanning from 1.52cm to 4.86cm were identified. Notably, LOC_Os02g12660 contained the superior haplotype H39, showcasing the lowest average lesion length of 1.88 – 2.06cm (Figure 3; Supplementary Figure 4). Identifying these superior haplotypes in the context of the studied traits holds promising implications for developing next-generation disease-resistant cultivars.
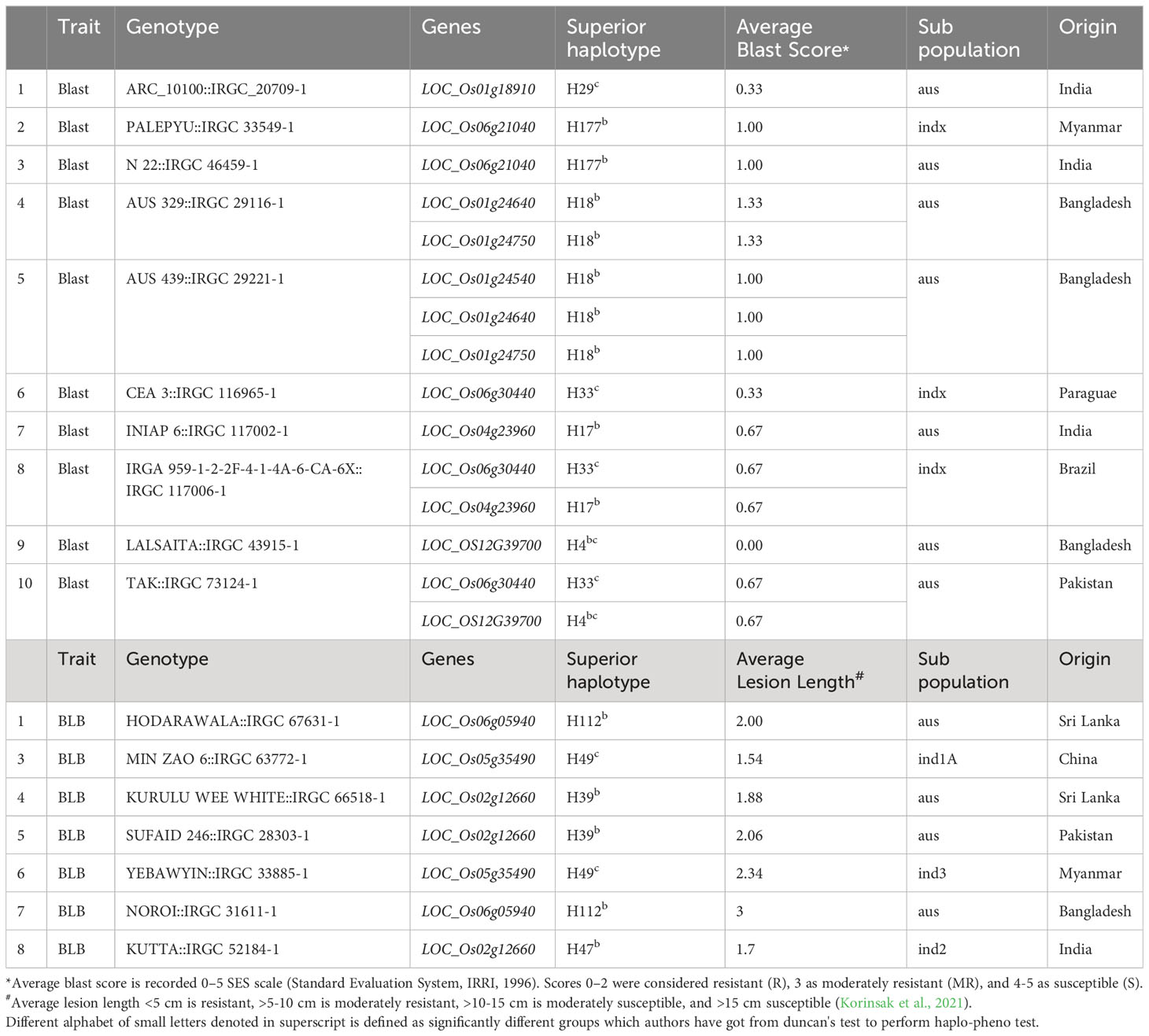
Table 2 Accessions having superior haplotypes for resistance to blast and BLB.
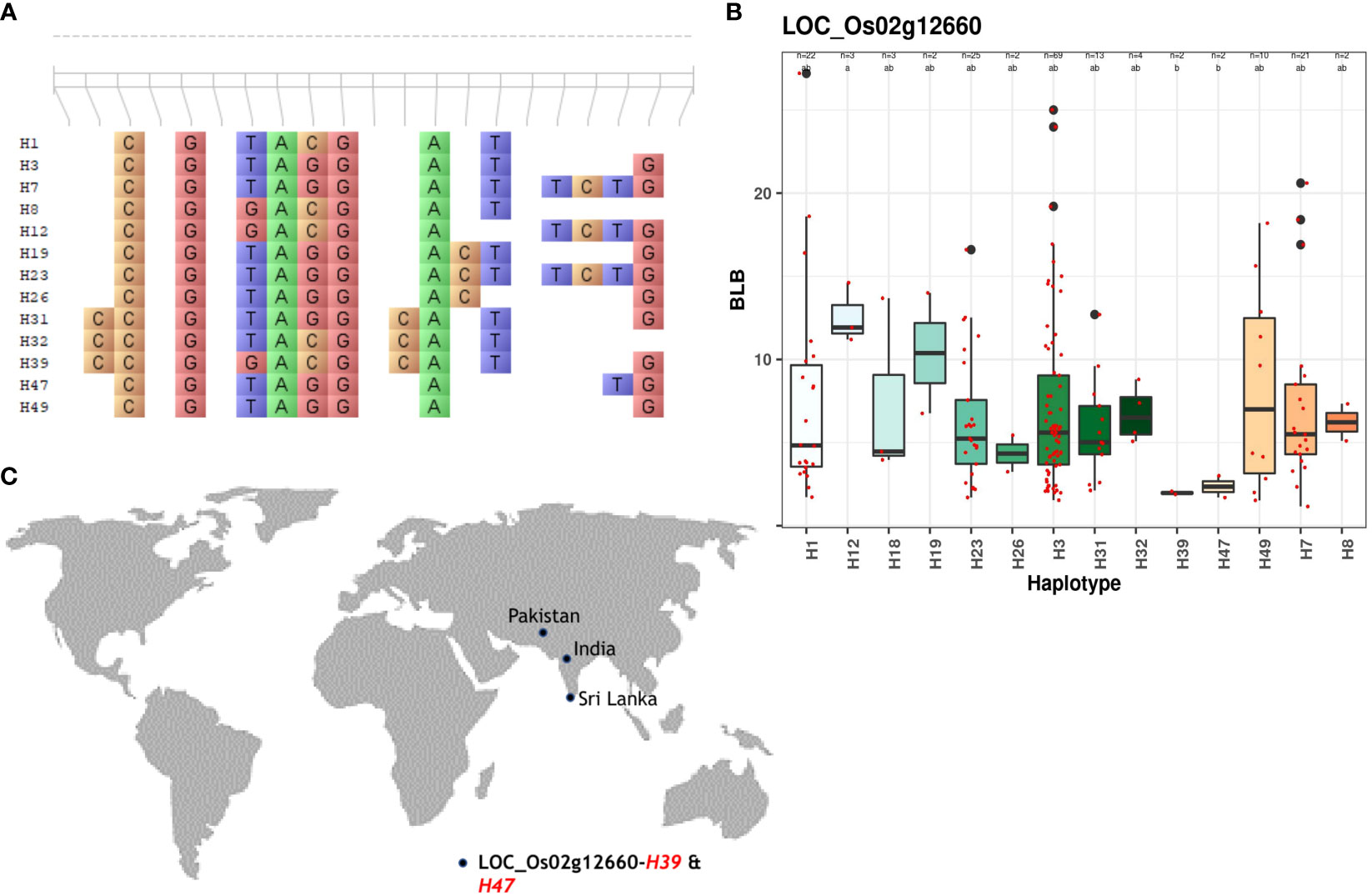
Figure 3 Haplotype analysis of LOC_Os02g12660 across the sub-set panel. (A) Haplotypic variation of LOC_Os02g12660, a gene associated with BLB resistance. (B) Boxplot showing variation in BLB resistance among 147 accessions of 3K RGP (C) The geographical distribution of superior haplotype.
3.8 Identification of accessions carrying superior haplotype
A selection of 10 accessions was successfully identified carrying superior haplotypes corresponding to eight candidate genes associated with blast resistance. These accessions fall into the aus and indx subpopulations across five distinct countries: Bangladesh, Brazil, India, Myanmar, and Pakistan. For instance, accession AUS 439:IRGC 29221-1 (LOC_Os01g24540-H18, LOC_Os01g24640-H18, and LOC_Os01g24750-H18) was recognized as bearing the superior haplotype for three candidate genes:. Similarly, two accessions, namely AUS 329:IRGC 29116-1 (LOC_Os01g24640-H18, LOC_Os01g24540-H18) and TAK::IRGC 73124-1 (LOC_Os06g30440-H33, and LOC_OS12G39700-H4) carried the superior haplotype of two candidate genes. The remaining accessions were carriers of superior haplotypes from a single gene (Figure 4, Table 3).
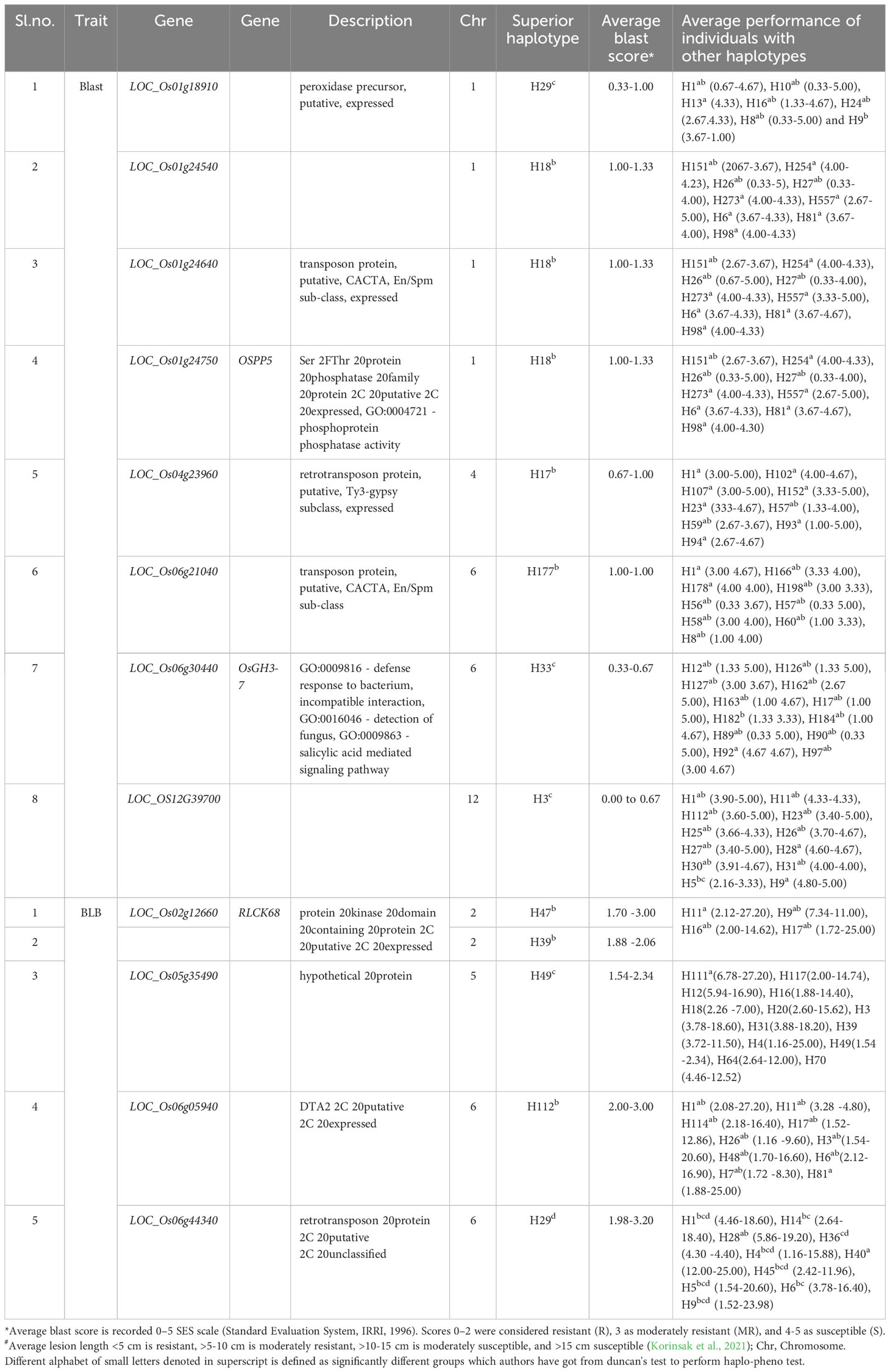
Table 3 Haplo-Pheno analysis of candidate gene carrying superior haplotype and functional annotation of the candidate genes.
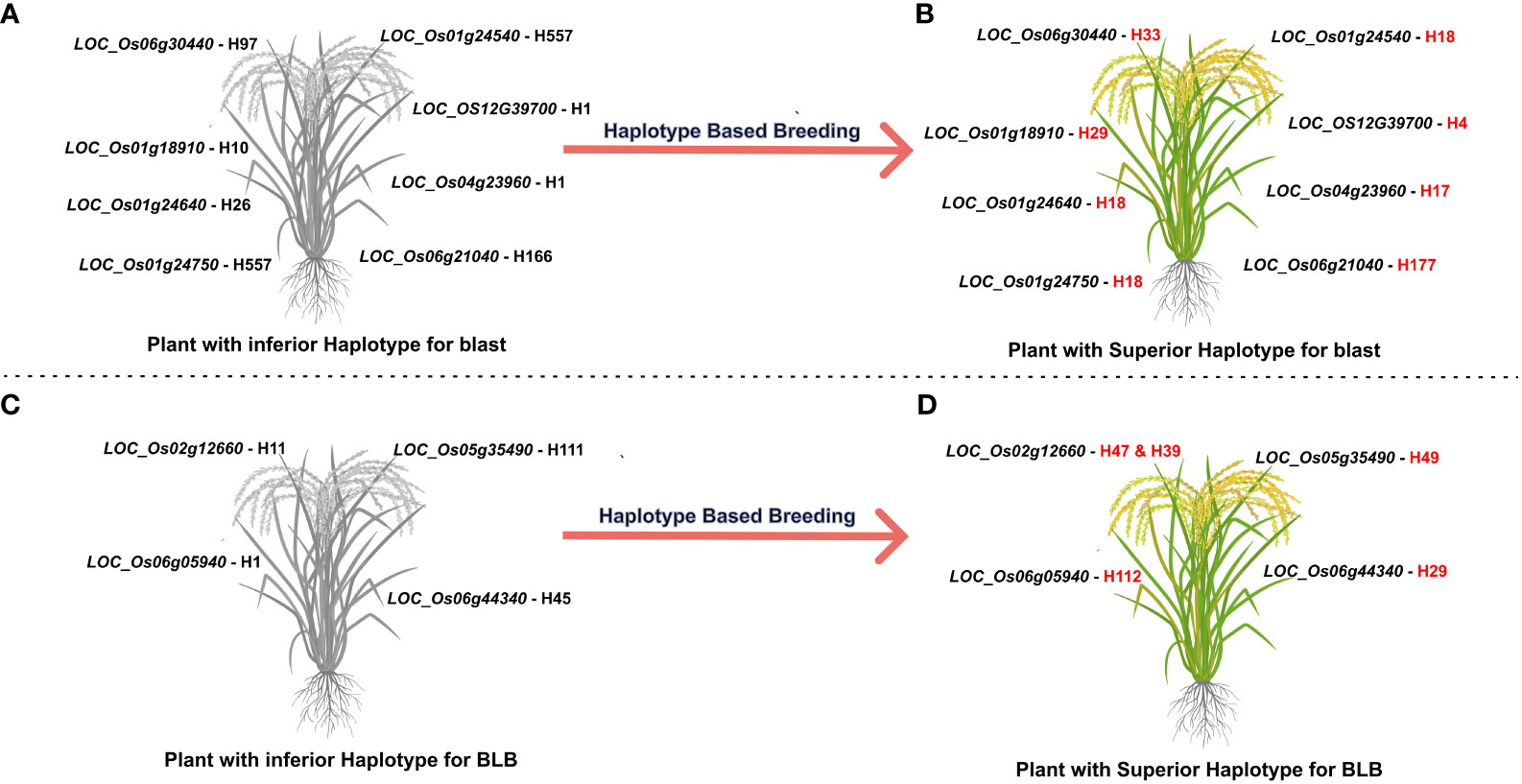
Figure 4 Towards developing tailored made rice with superior haplotypes for blast and BLB resistance. (A, C) The most inferior haplotype combination for blast and BLB resistance (B, D) the most superior haplotype combination for enhanced blast resistance. Through haplotype-based breeding, new breeding lines can be developed with the most superior haplotype combination.
Eight accessions coming from six diverse countries (Bangladesh, China, India, Myanmar, Pakistan, and Sri Lanka) and spanning four subpopulations (aus, ind1A, ind2, and ind3) were identified as carrying superior haplotypes for BLB resistance. The average lesion length of these accessions ranged from 1.54 to 3.00 cm. Notably, accession MIN ZAO 6::IRGC 63772-1, possessing the superior haplotype of the candidate gene LOC_Os05g35490-H49, exhibited the lowest average lesion length of 1.54 cm. Three of the eight accessions—MIN ZAO 6::IRGC 63772-1 (1.54 cm), KUTTA::IRGC 52184-1 (1.70 cm), and KURULU WEE WHITE::IRGC 66518-1 (1.88 cm)—displayed the lowest average lesion lengths for BLB (Figure 4, Table 3).
The superior haplotypes identified through this study hold promising potential for the development of next-generation disease-resistant varieties via haplotype-based breeding approaches.
4 Discussion
Despite the utilization of numerous resistant genes, rice blast, and bacterial blight are continue to be the most devastating and widespread diseases globally. Although more than 100 blast and 42 BLB resistance genes have been identified and employed in resistance breeding programs (Sharma et al., 2012; Alam et al., 2017). Emergence of new pathotypes or races have led to the breakdown of resistance. Consequently, there is a pressing need to discover novel resistance genes or sources to develop durable and sustainable resistant rice lines. The wealth of genetic diversity found in diverse germplasm collections, such as the 3K-RG panel, along with publicly available resequencing data, serves as a valuable resource for identifying novel alleles and donors associated with the trait of interest. GWAS for blast and bacterial blight, using high-density marker information from highly diverse germplasm like the 3K-RG panel, holds tremendous importance in pinpointing the genomic regions associated with these major diseases in rice.
Conventional exploration of novel resistance genes/QTLs has primarily relied on a linkage mapping approach. However, this method has its limitations, including prolonged population development time and the detection of only a few segregating alleles. In contrast, GWAS have emerged as a powerful alternative for mapping genomic regions associated with biotic stress, surpassing biparental or linkage mapping approaches. GWAS involves identifying genomic regions related to biotic stress in a diverse set of germplasm. Over the past few decades, substantial efforts have been dedicated to identifying genes or QTLs that confer resistance to blast and BLB in rice, resulting in numerous noteworthy findings. The genotyping data from the 3K RG project has proven to be a valuable resource for several researchers in pinpointing genomic regions associated with both abiotic and biotic stress in rice (Shi et al., 2017; Phan et al., 2023; Zhang et al., 2017; Zhang et al., 2019). By leveraging the information obtained through GWAS and the comprehensive genotyping data available in the 3K-RG, have made significant strides in understanding and enhancing resistance to biotic stresses, paving the way for the development of more resilient and disease-resistant varieties.
In this study, we focused on a sub-set of rice accessions from the 3K-RG to investigate blast and bacterial blight diseases. Our aim was to identify significant MTAs and unravel the haplotype diversity of candidate genes associated with these diseases. Through an in-house multi-locus GWAS analysis, we discovered a total of 23 MTAs, with 8 linked to blast resistance and 14 to bacterial blight resistance.
What makes our findings particularly intriguing is that all the identified MTAs were novel, with no previously reported genes for blast and bacterial blight within these loci, except for one blast resistance gene, Pi40, which was located near MTA-17556914 on chromosome 6. However, when we expanded the search to include sequences within −/+ 100 kb from the significant MTAs, we found four overlapping resistant genes on chromosome 11 associated with bacterial blight: Xa23 (LOC_Os11g37620), Xa39 (LOC_Os11g37759), Xa10 (LOC_Os11g37570), and Xa46 (LOC_Os11g37540). Furthermore, our comprehensive analysis led to the detection of 107 candidate genes on chromosomes 1, 4, 6, 7, 8, 11, and 12 associated with blast resistance and 324 candidate genes on chromosomes 1, 3, 4, 5, 7, 8, 10, 11, and 12 associated with bacterial blight resistance. These findings contribute to a deeper understanding of the genetic basis of blast and bacterial blight resistance, offering valuable insights for the development of improved disease-resistant varieties through targeted breeding programs.
In pursuit of developing durable resistant cultivars, we employed the identification and deployment of superior haplotypes for pest and disease resistance, which is a promising approach. Through haplotype analysis of candidate genes, we successfully identified several superior haplotypes for the target traits, with 8 superior haplotypes for blast resistance and 4 for bacterial blight resistance. Interestingly, four candidate genes (LOC_Os06g21040, LOC_Os04g23960, LOC_Os12g39700, and LOC_Os01g24640) encoding transposon and retrotransposon proteins (CACTA, En/Spm sub-class, putative, Ty3-gypsy subclass) were among those with superior haplotypes. These genes are known to play a crucial role in plant defense responses (Todorovska, 2007). Furthermore, one of the candidate genes, LOC_Os01g24640, encodes the peroxidase gene, which has essential functions in the oxidation of various components and is involved in the biosynthesis and degradation of lignin in cell walls. The peroxidase gene is critical in the host plant’s resistance during the basal (PTI) defense response (Yoshida et al., 2003). These findings shed light on the genetic mechanisms underlying the resistance traits and provide valuable insights for developing disease-resistant cultivars by strategically deploying superior haplotypes.
We discovered a superior haplotype of the candidate gene LOC_Os06g30440 on chromosome 6, which encodes for OsGH3-7. This gene plays a crucial role in the salicylic acid (SA)-mediated signaling pathway. SA is a key plant hormone responsible for triggering host responses against microbial pathogens. Specifically, it induces a defense response against biotrophic pathogens like blast pathogens, which rely on living host tissues for their growth and reproduction. On the other hand, the jasmonic acid (JA)-activated defense response is targeted toward wounding and necrotrophic pathogens. Our identification of LOC_Os06g30440’s involvement in the salicylic acid-mediated signaling pathway highlights its significance in combating the blast pathogen, which is a biotrophic pathogen. Additionally, on chromosome 1, we identified the gene LOC_Os01g24750, which codes for OSPP5, a serine-threonine family protein. This protein has been previously reported to play a role in plant defense responses. These findings contribute to a better understanding of the genetic mechanisms involved in the plant’s defense against pathogens and offer potential targets for enhancing disease resistance through genetic manipulation or breeding programs.
Superior haplotypes were identified in four candidate genes associated with bacterial blight (BLB). Upon functional annotation of these genes obtained from RAP-DB, one candidate gene, LOC_Os02g12660 on chromosome 2, was found to encode a receptor-like cytoplasmic kinase (RLCK). RLCKs belong to a significant subfamily of proteins that play a crucial role in regulating plant immunity against bacterial and fungal pathogens in various plant species, including rice, tomato, and Arabidopsis (AbuQamar et al., 2008; Vij et al., 2008; Ao et al., 2014). In the case of bacterial blight resistance in rice, the function of Receptor-like cytoplasmic kinase has already been reported. RLCKs are involved in PAMP-triggered immunity, a key defense mechanism in plants. Specifically, in rice, the RLCK gene OsRLCK185 encodes an RLCK that is directly phosphorylated by the lysine motif-containing PAMP-receptor OsCERK1. Suppression of OsRLCK185 expression has been observed to result in reduced MAP kinase activation and reduced expression of chitin-induced genes PBZ1 and PAL1, which are important components of the plant’s immune response (Yamaguchi et al., 2013; Wang et al., 2017).
In this study, it was found that a Xanthomonas oryzae effector called Xoo1488 can suppress the interaction between OsRLCK185 and OsCERK1, indicating the essential role of the OsRLCK185/OsCERK1 complex in plant immune responses, particularly PTI (PAMP-triggered immunity). However, the other three candidate genes identified in the study were not directly linked to disease resistance.
Overall, these findings underscore the significance of the identified candidate genes in the context of blast and BLB resistance and offer potential targets for enhancing the plant’s ability to combat these devastating diseases. The study revealed several novel genomic regions (MTAs) associated with blast and BLB diseases, along with their corresponding superior haplotypes. These superior haplotypes could be effectively integrated into a single genetic background using a haplotype-based breeding approach, ultimately leading to the development of varieties with a broader and more effective resistance spectrum against both blast and bacterial blight diseases. This approach holds promise for enhancing rice cultivars’ durability and resilience to these devastating diseases.
5 Conclusions
In conclusion, rice blast and bacterial blight poses a global threat to rice yield, making the identification of resistance genes and haplotypes a crucial strategy for disease management. Our GWAS on a diverse set of 147 rice accessions led to the discovery of 23 significant marker-trait associations (9 for blast and 14 for BLB), corresponding to 107 and 210 candidate genes for blast and BLB, respectively. Haplo-pheno analysis revealed eight superior haplotypes for blast and five for BLB, with remarkable SES and lesion length scores. Notably, the candidate genes possessing the superior haplotypes are known to play vital roles in plant defense responses. The identified superior haplotypes, sourced from diverse subpopulations and countries, hold promise to be incorporated into a single genetic background through haplotype-based breeding, providing a broader resistance spectrum against blast and bacterial blight diseases.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
SA: Conceptualization, Project administration, Data curation, Investigation, Methodology, Validation, Writing – original draft. KS: Data curation, Formal analysis, Software, Writing – review & editing. US: Data curation, Formal analysis, Writing – review & editing, Methodology. MS: Writing – review & editing, Resources. GL: Resources, Writing – review & editing. PS: Formal analysis, Resources, Writing – review & editing, Conceptualization, Data curation, Methodology, Project administration. VS: Conceptualization, Project administration, Writing – review & editing, Funding acquisition, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was supported by grants from the Department of Biotechnology (DBT), Government of India, New Delhi and the CGIAR Research Program on Rice.
Acknowledgments
Authors are highly thankful to the Department of Biotechnology (DBT), Government of India, New Delhi and the CGIAR Research Program on Rice for providing financial support. This research has been conducted as part of the collaborative research project between ICAR and IRRI, with IRRI being a valued member of the CGIAR Consortium. The authors are grateful to Mr. G Anil for their invaluable assistance in supporting in screening 3K-RGP and pathological work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1272326/full#supplementary-material
References
3,000 rice genomes project (2014). The 3,000 rice genomes project. GigaScience 3, 7. doi: 10.1186/2047-217X-3-7
AbuQamar, S., Chai, M. F., Luo, H., Song, F., Mengiste, T. (2008). Tomato protein kinase 1b mediates signaling of plant responses to necrotrophic fungi and insect herbivory. Plant Cell 20, 1964–1983. doi: 10.1105/tpc.108.059477
Alam, S., Imam, J., Nitin, M., Prasad, C., Variar, M. (2017). Molecular screening of Blast Resistance Gene Pi2 in Indian rice landraces (Oryza sativa L.) and its verification by virulence analysis. Proc. Natl. Acad. Sciences India Section B 87, 67–72. doi: 10.1007/s40011-015-0548-3
Ao, Y., Li, Z., Feng, D., Xiong, F., Liu, J., Li, J. F., et al. (2014). OsCERK1 and OsRLCK176 play important roles in peptidoglycan and chitin signaling in rice innate immunity. Plant Journal: Cell Mol. Biol. 80, 1072–1084. doi: 10.1111/tpj.12710
Ashkani, S., Rafii, M. Y., Shabanimofrad, M., Ghasemzadeh, A., Ravanfar, S. A., Latif, M. A. (2016). Molecular progress on the mapping and cloning of functional genes for blast disease in rice (Oryza sativa L.): Current status and future considerations. Crit. Rev. Biotechnol. 36, 353–367. doi: 10.3109/07388551.2014.961403
Chen, L. C., Huang, S. H., Cheng, C. H. (2009). “Review of the screening tests for rice varietal resistance to major diseases and insect pests in Taiwan,” in Ni HF. Proceedings of the symposium on achievements and perspectives of rice protection in Taiwan (1st ed), 2009 Chiayi agricultural experiment branch. Ed. Yang, H. R. (Taiwan: Taiwan Agricultural Research Institute), 83–103.
Chukwu, S. C., Rafii, M. Y., Ramlee, S. I., Ismail, S. I., Oladosu, Y., Kolapo, K., et al. (2019). Marker-assisted introgression of multiple resistance genes confers broad spectrum resistance against bacterial leaf blight and blast diseases in PUTRA-1 rice variety. Agronomy. 10, 42. doi: 10.3390/agronomy10010042
de Mendiburu, M. F. (2018). Package ‘Agricolae.’ Statistical Procedures for Agricultural Research, Version 1, 3-0. http://CRAN.R-project.org/package=agricolae
Devanna, B. N., Jain, P., Solanke, A. U., Das, A., Thakur, S., Singh, P. K., et al. (2022). Understanding the dynamics of blast resistance in Rice-Magnaporthe oryzae interactions. J. Fungi. 8, 584. doi: 10.3390/jof8060584
Dilla-Ermita, C. J., Tandayu, E., Juanillas, V. M., Detras, J., Lozada, D. N., Dwiyanti, M. S., et al. (2017). Genome-wide association analysis tracks bacterial blight resistance loci in rice diverse germplasm. Rice 10, 8. doi: 10.1186/s12284-017-0147-4
Dixit, S., Singh, U. M., Singh, A. K., Alam, S., Venkateshwarlu, C., Nachimuthu, V. V., et al. (2020). Marker assisted forward breeding to combine multiple biotic-abiotic stress resistance/tolerance in rice. Rice 13, 29. doi: 10.1186/s12284-020-00391-7
FAO. (2019). New standards to curb the global spread of plant pests and diseases. Available online at: https://www.fao.org/news/story/en/item/1187738/icode/.
Frontini, M., Boisnard, A., Frouin, J., Ouikene, M., Morel, J. B., Ballini, E. (2021). Genome-wide association of rice response to blast fungus identifies loci for robust resistance under high nitrogen. BMC Plant Biol. 21, 99. doi: 10.1186/s12870-021-02864-3
IRRI (International Rice Research Institute) (1996). Standard evaluation system for rice (SES). 5th (Philippines: International Rice Research Institute).
Jamaloddin, M., Durga Rani, C. V., Swathi, G., Anuradha, C., Vanisri, S., Rajan, C. P. D., et al. (2020). Marker assisted gene pyramiding (MAGP) for bacterial blight and blast resistance into mega rice variety “Tellahamsa”. PloS One 15, e0234088. doi: 10.1371/journal.pone.0234088
Janaki Ramayya, P., Vinukonda, V. P., Singh, U. M., Alam, S., Venkateshwarlu, C., Vipparla, A. K., et al. (2021). Marker-assisted forward and backcross breeding for improvement of elite Indian rice variety Naveen for multiple biotic and abiotic stress tolerance. PloS One 16, e0256721. doi: 10.1371/journal.pone.0256721
Jiang, N., Fu, J., Zeng, Q., Liang, Y., Shi, Y., Li, Z., et al. (2021). Genome-wide association mapping for resistance to bacterial blight and bacterial leaf streak in rice. Planta 253, 94. doi: 10.1007/s00425-021-03612-5
Jiang, N., Yan, J., Liang, Y., Shi, Y., He, Z., Wu, Y., et al. (2020). Resistance Genes and their Interactions with Bacterial Blight/Leaf Streak Pathogens (Xanthomonas oryzae) in Rice (Oryza sativa L.)—an Updated Review. Rice 13 (1), 3. doi: 10.1186/s12284-019-0358-y
Kang, H., Wang, Y., Peng, S., Zhang, Y., Xiao, Y., Wang, D., et al. (2016). Dissection of the genetic architecture of rice resistance to the blast fungus Magnaporthe oryzae. Mol. Plant Pathol. 17, 959–972. doi: 10.1111/mpp.12340
Kauffman, H. E., Reddy, A. P. K., Hsieh, S. P. Y., Merca, S. D. (1973). A improved technique for evaluation of resistance of rice varieties to Xanthomonas oryzae. Plant Dis. Rep. 57, 537–541.
Khanna, A., Sharma, V., Ellur, R. K., Shikari, A. B., Krishnan, S. G., Singh, U. D., et al. (2015). Marker assisted pyramiding of major blast resistance genes Pi9 and Pita in the genetic background of an elite Basmati rice variety, Pusa Basmati 1. Indian J. Genet. Plant Breed. 75, 417–425. doi: 10.5958/0975-6906.2015.00068.1
Khush, G. S. (2005). What it will take to Feed 5.0 Billion Rice consumers in 2030. Plant Mol. Biol. 59, 1–6. doi: 10.1007/s11103-005-2159-5
Korinsak, S., Darwell, C. T., Wanchana, S., Praphaisal, L., Korinsak, S., Thunnom, B., et al. (2021). Identification of bacterial blight resistance loci in rice (Oryza sativa L.) against diverse Xoo Thai Strains by genome-wide association study. Plants 10, 518. doi: 10.3390/plants10030518
Li, C., Wang, D., Peng, S., Chen, Y., Su, P., Chen, J., et al. (2019). Genome-wide association mapping of resistance against rice blast strains in South China and identification of a new Pik allele. Rice 12, 47. doi: 10.1186/s12284-019-0309-7
Lin, H. A., Chen, S. Y., Chang, F. Y., Tung, C. W., Chen, Y. C., Shen, W. C., et al. (2018). Genome-wide association study of rice genes and loci conferring resistance to Magnaporthe oryzae isolates from Taiwan. Botanical Stud. 59, 32. doi: 10.1186/s40529-018-0248-4
Lu, J., Wang, C., Zeng, D., Li, J., Shi, X., Shi, Y., et al. (2021). Genome-wide association study dissects resistance loci against bacterial blight in a diverse rice panel from the 3000 rice genomes project. Rice 14, 22. doi: 10.1186/s12284-021-00462-3
Ma, J., Lei, C., Xu, X., Hao, K., Wang, J., Cheng, Z., et al. (2015). Pi64, encoding a novel CC-NBS-LRR protein, confers resistance to leaf and neck blast in rice. Mol. Plant-Microbe Interactions: MPMI 28, 558–568. doi: 10.1094/MPMI-11-14-0367-R
Madhan Mohan, K. (2011). Molecular characterization of pathogenic variability of Pyricularia grisea (Rice Blast fungus) (Doctoral dissertation, Ph. D. thesis Faculty of Biotechnology (Hyderabad, Telangana, India: Jawaharlal Nehru Technological University).
McCouch, S. R., Wright, M. H., Tung, C. W., Maron, L. G., McNally, K. L., Fitzgerald, M., et al. (2016). Open access resources for genome-wide association mapping in rice. Nat. Commun. 7, 10532. doi: 10.1038/ncomms10532
Mgonja, E. M., Balimponya, E. G., Kang, H., Bellizzi, M., Park, C. H., Li, Y., et al. (2016). Genome-wide association mapping of rice resistance genes against Magnaporthe oryzae isolates from four African countries. Phytopathology 106, 1359–1365. doi: 10.1094/PHYTO-01-16-0028-R
Neelam, K., Mahajan, R., Gupta, V., Bhatia, D., Gill, B. K., Komal, R., et al. (2020). High-resolution genetic mapping of a novel bacterial blight resistance gene xa-45(t) identified from Oryza glaberrima and transferred to Oryza sativa. TAG. Theor. Appl. Genet. Theoretische und Angewandte Genetik 133, 689–705. doi: 10.1007/s00122-019-03501-2
Phan, N. T. H., Draye, X., Pham, C. V., Bertin, P. (2023). Identification of quantitative trait loci controlling nitrogen use efficiency related traits in rice at the seedling stage under salt condition by genome-wide association study. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1197271
Pradhan, S. K., Nayak, D. K., Mohanty, S., Behera, L., Barik, S. R., Pandit, E., et al. (2015). Pyramiding of three bacterial blight resistance genes for broad-spectrum resistance in deepwater rice variety, Jalmagna. Rice 8, 51. doi: 10.1186/s12284-015-0051-8
Raboin, L. M., Ballini, E., Tharreau, D., Ramanantsoanirina, A., Frouin, J., Courtois, B., et al. (2016). Association mapping of resistance to rice blast in upland field conditions. Rice 9, 59. doi: 10.1186/s12284-016-0131-4
Savary, S., Willocquet, L., Elazegui, F. A., Castilla, N. P., Teng, P. S. (2000). Rice pest constraints in tropical Asia: Quantification of yield losses due to rice pests in a range of production situations. Plant Dis. 84, 357–369. doi: 10.1094/PDIS.2000.84.3.357
Sharma, T. R., Rai, A. K., Gupta, S. K., Vijayan, J., Devanna, B. N., Ray, S. (2012). Rice blast management through host-plant resistance: Retrospect and prospects. Agric. Res. 1, 37–52. doi: 10.1007/s40003-011-0003-5
Shi, Y., Gao, L., Wu, Z., Zhang, X., Wang, M., Zhang, C., et al. (2017). Genome-wide association study of salt tolerance at the seed germination stage in rice. BMC Plant Biol. 17, 92. doi: 10.1186/s12870-017-1044-0
Singh, U. M., Dixit, S., Alam, S., Yadav, S., Prasanth, V. V., Singh, A. K., et al. (2022). Marker-assisted forward breeding to develop a drought-, bacterial-leaf-blight, and blast-resistant rice cultivar. Plant Genome 15, e20170. doi: 10.1002/tpg2.20170
Singh, V. K., Singh, A., Singh, S. P., Ellur, R. K., Choudhary, V., Sarkel, S., et al. (2013). Incorporation of blast resistance into “PRR78”, an elite Basmati rice restorer line, through marker assisted backcross breeding. Field Crops Res. 128, 8–16. doi: 10.1016/j.fcr.2011.12.003
Su, J., Wang, W., Han, J., Chen, S., Wang, C., Zeng, L., et al. (2015). Functional divergence of duplicated genes results in a novel blast resistance gene Pi50 at the Pi2/9 locus. TAG. Theor. Appl. Genet. Theoretische und Angewandte Genetik 128, 2213–2225. doi: 10.1007/s00122-015-2579-9
Suh, J. P., Cho, Y. C., Won, Y. J., Ahn, E. K., Baek, M. K., Kim, M. K., et al. (2015). Development of resistant gene-pyramided Japonica rice for multiple biotic stresses using molecular marker-assisted selection. Plant Breed. Biotechnol. Biotech. Publishing 3, 333–345. doi: 10.9787/PBB.2015.3.4.333
Swathi, G., Durga Rani, C. V., Md, J., Madhav, M. S., Vanisree, S., Anuradha, C., et al. (2019). Marker-assisted introgression of the major bacterial blight resistance genes, Xa21 and xa13, and blast resistance gene, Pi54, into the popular rice variety, JGL1798. Mol. Breed. 39, 58. doi: 10.1007/s11032-019-0950-2
Tamba, C. L., Zhang, Y. M. (2018). A fast mrMLM algorithm for multi-locus genome-wide association studies (biorxiv.org). doi: 10.1101/341784
Todorovska, E. (2007). Retrotransposons and their role in plant—Genome evolution. Biotechnol. Biotechnol. Equip. 21, 294–305. doi: 10.1080/13102818.2007.10817464
Varshney, R. K., Graner, A., Sorrells, M. E. (2005). Genomics-assisted breeding for crop improvement. Trends Plant Sci. 10, 621–630. doi: 10.1016/j.tplants.2005.10.004
Varshney, R. K., Nayak, S. N., May, G. D., Jackson, S. A. (2009). Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 27, 522–530. doi: 10.1016/j.tibtech.2009.05.006
Vera Cruz, C. M., Bai, J., Ona, I., Leung, H., Nelson, R. J., Mew, T. W., et al. (2000). Predicting durability of a disease resistance gene based on an assessment of the fitness loss and epidemiological consequences of avirulence gene mutation. Proc. Natl. Acad. Sci. United States America 97, 13500–13505. doi: 10.1073/pnas.250271997
Vij, S., Giri, J., Dansana, P. K., Kapoor, S., Tyagi, A. K. (2008). The receptor-like cytoplasmic kinase (OsRLCK) gene family in rice: Organization, phylogenetic relationship, and expression during development and stress. Mol. Plant 1, 732–750. doi: 10.1093/mp/ssn047
Volante, A., Tondelli, A., Desiderio, F., Abbruscato, P., Menin, B., Biselli, C., et al. (2020). Genome wide association studies for japonica rice resistance to blast in field and controlled conditions. Rice 13, 71. doi: 10.1186/s12284-020-00431-2
Wang, S. B., Feng, J. Y., Ren, W. L., Huang, B., Zhou, L., Wen, Y. J., et al. (2016). Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 6, 19444. doi: 10.1038/srep19444
Wang, X., Pang, Y., Zhang, J., Wu, Z., Chen, K., Ali, J., et al. (2017). Genome-wide and gene-based association mapping for rice eating and cooking characteristics and protein content. Sci. Rep. 7, 17203. doi: 10.1038/s41598-017-17347-5
Webb, K. M., Garcia, E., Vera Cruz, C. M., Leach, J. E. (2010). Influence of rice development on the function of bacterial blight resistance genes. Eur. J. Plant Pathol. 128, 399–407. doi: 10.1007/s10658-010-9668-z
Xiao, W., Luo, L., Wang, H., Guo, T., Liu, Y., Zhou, J., et al. (2016). Pyramiding of Pi46 and Pita to improve blast resistance and to evaluate the resistance effect of the two R genes. J. Integr. Agric. 15, 2290–2298. doi: 10.1016/S2095-3119(16)61415-6
Xu, X., Hayashi, N., Wang, C. T., Fukuoka, S., Kawasaki, S., Takatsuji, H., et al. (2014). Rice blast resistance gene Pikahei-1(t), a member of a resistance gene cluster on chromosome 4, encodes a nucleotide-binding site and leucine-rich repeat protein. Mol. Breed. 34, 691–700. doi: 10.1007/s11032-014-0067-6
Yamaguchi, K., Yamada, K., Kawasaki, T. (2013). Receptor-like cytoplasmic kinases are pivotal components in pattern recognition receptor-mediated signaling in plant immunity. Plant signaling & behavior 8 (10), e25662.
Yang, F., Zhang, J., Zhang, H., Ji, G., Zeng, L., Li, Y., et al. (2020). Bacterial blight induced shifts in endophytic microbiome of rice leaves and the enrichment of specific bacterial strains with pathogen antagonism. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.00963
Yang, Y., Zhou, Y., Sun, J., Liang, W., Chen, X., Wang, X., et al. (2022). Research progress on cloning and function of xa genes against rice bacterial blight. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.847199
Yoshida, K., Kaothien, P., Matsui, T., Kawaoka, A., Shinmyo, A. (2003). Molecular biology and application of plant peroxidase genes. Appl. Microbiol. Biotechnol. 60, 665–670. doi: 10.1007/s00253-002-1157-7
Yugander, A., Sundaram, R. M., Ladhalakshmi, D., Hajira, S. K., Prakasam, V., Prasad, M. S., et al. (2017). Virulence profiling of Xanthomonas oryzae pv. oryzae isolates, causing bacterial blight of rice in India. Eur. J. Plant Pathol. 149, 171–191. doi: 10.1007/s10658-017-1176-y
Zhang, T., Qian, N., Zhu, X., Chen, H., Wang, S., Mei, H., et al. (2013). Variations and transmission of QTL alleles for yield and fiber qualities in upland cotton cultivars developed in China. PloS One 8 (2), e57220. doi: 10.1371/journal.pone.0057220
Zhang, J., Feng, J. Y., Ni, Y. L., Wen, Y. J., Niu, Y., Tamba, C. L., et al. (2017a). pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 118, 517–524. doi: 10.1038/hdy.2017.8
Zhang, F., Wu, Z. C., Wang, M. M., Zhang, F., Dingkuhn, M., Xu, J. L., et al. (2017b). Genome-wide association analysis identifies resistance loci for bacterial blight in a diverse collection of indica rice germplasm. PloS One 12, e0174598. doi: 10.1371/journal.pone.0174598
Zhang, F., Zeng, D., Zhang, C. S., Lu, J. L., Chen, T. J., Xie, J. P., et al. (2019). Genome-wide association analysis of the genetic basis for sheath blight resistance in rice. Rice 12, 93. doi: 10.1186/s12284-019-0351-5
Zhao, K., Tung, C. W., Eizenga, G. C., Wright, M. H., Ali, M. L., Price, A. H., et al. (2011). Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2, 467. doi: 10.1038/ncomms1467
Keywords: rice, blast, BLB, GWAS, haplo-pheno, superior haplotype
Citation: Alam S, Sundaram KT, Singh UM, Srinivas Prasad M, Laha GS, Sinha P and Singh VK (2024) Superior haplotypes towards the development of blast and bacterial blight-resistant rice. Front. Plant Sci. 15:1272326. doi: 10.3389/fpls.2024.1272326
Received: 03 August 2023; Accepted: 31 January 2024;
Published: 28 February 2024.
Edited by:
Ying Sun, Cornell University, United StatesReviewed by:
Shimin Zuo, Yangzhou University, ChinaApichart Vanavichit, Kasetsart University, Thailand
Copyright © 2024 Alam, Sundaram, Singh, Srinivas Prasad, Laha, Sinha and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vikas Kumar Singh, di5rLnNpbmdoQGlycmkub3Jn; Pallavi Sinha, cC5zaW5oYUBpcnJpLm9yZw==