 Rachel M. Muti
Rachel M. Muti Craig F. Barrett
Craig F. Barrett Brandon T. Sinn
Brandon T. Sinn- 1Department of Biology and Earth Science, Otterbein University, Westerville, OH, United States
- 2Department of Hematology and Medical Oncology, Emory University, Atlanta, GA, United States
- 3Department of Biology, West Virginia University, Morgantown, WV, United States
- 4Faculty of Biology, University of Latvia, Riga, Latvia
The plastid-targeted transcription factor Whirly1 (WHY1) has been implicated in chloroplast biogenesis, plastid genome stability, and fungal defense response, which together represent characteristics of interest for the study of autotrophic losses across the angiosperms. While gene loss in the plastid and nuclear genomes has been well studied in mycoheterotrophic plants, the evolution of the molecular mechanisms impacting genome stability is completely unknown. Here, we characterize the evolution of WHY1 in four early transitional mycoheterotrophic orchid species in the genus Corallorhiza by synthesizing the results of phylogenetic, transcriptomic, and comparative genomic analyses with WHY1 genomic sequences sampled from 21 orders of angiosperms. We found an increased number of non-canonical WHY1 isoforms assembled from all but the greenest Corallorhiza species, including intron retention in some isoforms. Within Corallorhiza, phylotranscriptomic analyses revealed the presence of tissue-specific differential expression of WHY1 in only the most photosynthetically capable species and a coincident increase in the number of non-canonical WHY1 isoforms assembled from fully mycoheterotrophic species. Gene- and codon-level tests of WHY1 selective regimes did not infer significant signal of either relaxed selection or episodic diversifying selection in Corallorhiza but did so for relaxed selection in the late-stage full mycoheterotrophic orchids Epipogium aphyllum and Gastrodia elata. Additionally, nucleotide substitutions that most likely impact the function of WHY1, such as nonsense mutations, were only observed in late-stage mycoheterotrophs. We propose that our findings suggest that splicing and expression changes may precede the selective shifts we inferred for late-stage mycoheterotrophic species, which therefore does not support a primary role for WHY1 in the transition to mycoheterotrophy in the Orchidaceae. Taken together, this study provides the most comprehensive view of WHY1 evolution across the angiosperms to date.
1 Introduction
The ability to photosynthesize has been lost dozens of times across the angiosperm Tree of Life, and at least 30 independent losses have occurred in the Orchidaceae (Merckx and Freudenstein, 2010; Barrett et al., 2014, Barrett et al, 2019). Mycoheterotrophy, the derivation of carbon nutrition from fungi (Leake, 1994), is common to all orchids during early development and is a nutritional requirement due to the lack of endosperm in their seeds (Rasmussen, 1995, Rasmussen, 2002). In lieu of stored nutrition, orchid seeds have evolved a complex symbiotic relationship with fungi, where orchid seeds germinate only in the presence of an appropriate fungal partner and the nutrition required for embryo development is derived exclusively via the degradation of fungal hyphae which penetrate the orchid cells (Merckx and Merckx, 2013; Zeng et al., 2017; Yuan et al., 2018). The duration of reliance upon their fungal partner for nutrition has been extended in some orchid species, which have evolved to parasitize fungi for the entirety of their lives. Independent shifts to a mycoheterotrophic condition throughout the angiosperms have independently led to plastid genome (plastome) degradation (Barrett et al., 2014; Wicke et al., 2016; Graham et al., 2017; Timilsena et al., 2023), elevated rates of nucleotide substitution (Lemaire et al., 2010; Wicke et al., 2016), and oftentimes the loss of morphological structures such as leaves and roots (Leake, 1994).
Corallorhiza is a North American, temperate genus comprising 12 species of morphologically reduced, mycoheterotrophic orchids for which varying states of plastome degradation and inferred photosynthetic ability have been characterized (Barrett and Freudenstein, 2008; Zimmer et al., 2008; Cameron et al., 2009; Barrett and Davis, 2012; Barrett et al., 2014, Barrett et al, 2018). The presence of relatively intact plastomes containing the expected repertoire of housekeeping genes in Corallorhiza species evidences the clade as a group of early transitional mycoheterotrophs, in contrast with late-stage mycoheterotrophic species which have highly degraded plastomes and lack many or all plastid housekeeping genes, such as Epipogium and Gastrodia species (sensu Barrett and Davis, 2012; see also Barrett et al., 2014). Corallorhiza species parasitize Basidiomycete fungi that are engaged in mycorrhizal relationships with nearby autotrophic plants, predominantly in the families Russulaceae and Thelephoraceae (Taylor and Bruns, 1997; Barrett et al., 2010; Freudenstein and Barrett, 2014; Taylor et al., 2022). In addition to their relationship with fungi, a conspicuous characteristic of Corallorhiza species is the complete loss of both leaf laminae and roots. A recurrent theme of morphological reduction has been documented across parasitic and mycoheterotrophic plant lineages (see Leake, 1994), and work has recently focused on genomic content and gene expression in mycoheterotrophic species in order to improve our understanding of the genomic precursors and consequences of this trophic transition (Barrett et al., 2014; Wicke et al., 2016; Graham et al., 2017; Zhang et al., 2017; Yuan et al., 2018; Cai, 2023; Timilsena et al., 2023).
The integrity of the plastome of parasitic and mycoheterotrophic plants is of particular interest as reduction in gene content, increased number of pseudogenes, structural variation, and a reduction in overall genome length have been found to correlate with the degree of external carbon reliance among parasitic angiosperms. Generally, disruptions to the genome such as double-strand DNA breaks are harmful to the organism, and many mechanisms that help to protect against such occurrences have evolved throughout the Tree of Life (Waterworth et al., 2011). One such genome stabilizing mechanism that is increasingly recognized for its involvement in processes such as plastome double-strand break repair is the activity of the Whirly family of transcription factors (WHY; Desveaux et al., 2005).
Transcription factors are regulatory gene products that function by binding to DNA (Latchman, 1993). The Whirly family comprises WHY1, WHY2, and WHY3, which are three plant-specific, nuclear-encoded genes with DNA-binding domains, that are named for their whirligig-like structural conformation (Desveaux et al., 2002; Cappadocia et al., 2010). Crystal structures of the Whirly transcription factors have been determined as tetramers that have a single-stranded DNA-binding domain that spans two subunits (Cappadocia et al., 2013). Of particular interest is WHY1, the product of which has been implicated to play roles in several processes including mediation of abiotic stressors (Zhuang et al., 2019, Zhuang et al, 2020; Ruan et al., 2022), induction of double-strand DNA break repair (Cappadocia et al., 2010), and plastid biogenesis (Prikryl et al., 2008). Transcription factors (TF) are crucial to many regulatory and developmental processes, which is reflected in the massive expansions of many TF gene families in plants (Lehti-Shiu et al., 2017). Our present understanding of TF evolution has largely been informed through the investigations focused on understanding their roles in morphological or ecological diversification (see de Mendoza et al., 2013; Lai et al., 2020) rather than how they change in systems, which have undergone coincident extreme loss of morphological and genomic features.
WHY1 has been shown to dually localize to both plastids and the nucleus (Krause et al., 2005; Grabowski et al., 2008; Prikryl et al., 2008; Isemer et al., 2012; Ren et al., 2017). In chloroplasts, WHY1 localizes to the boundary between the thylakoid and nucleoid membrane in chloroplasts and has been implicated in retrograde signaling regulating H2O2 homeostasis and as a coordinator of photosynthetic gene expression (Lepage et al., 2013; Foyer et al., 2014; Lin et al., 2019). Species that have undergone the transition to heterotrophy experience elevated levels of oxidative stress compared to autotrophic relatives (Suetsugu et al., 2017; Lallemand et al., 2019). Additionally, WHY1 proteins stabilize plastid genomes by non-specific binding to the genome, which protects against microhomology-mediated DNA rearrangements, including deletions and duplications of sequences (Maréchal et al., 2009; Lepage et al., 2013; Zampini et al., 2015).
WHY1 is involved in complex roles in both plant defense responses and genomic stabilization. For example, mutations which reduce the binding affinity of WHY1 correlate with increased infection by some pathogens (Desveaux et al., 2004). WHY1 has been shown to bind to specific DNA promoter regions that can induce transcription (Zhuang et al., 2020), aid in defense response signaling and accumulation of disease resistance (Desveaux et al., 2004, Desveaux et al, 2005), and also maintain telomere length in the nuclear genome (Desveaux et al., 2005). Recently, WHY1 has even been shown to be capable of negatively regulating the RNA interference response to two geminiviruses (Sun et al., 2023). Taken together, the literature suggests that modulation of WHY1 expression can result in tradeoffs between plant defense and genomic stability.
The roles that WHY1 plays in stabilizing both the nuclear and plastid genomes (Yoo et al., 2007; Zampini et al., 2015), defense response (Desveaux et al., 2005), and chloroplast development (Qiu et al., 2022) are central to our choice to study the evolution of this transcription factor. In particular, experimental work demonstrating plastome destabilization (Lepage et al., 2013; Zampini et al., 2015), albinism, and variegation in WHY1 mutants (Prikryl et al., 2008; Ren et al., 2017; Qiu et al., 2022) makes the gene a compelling target given the reduction in plastome content and length observed across parasitic angiosperms (Barrett et al., 2014; Wicke et al., 2016). To date, work characterizing the sequence and expression diversity of WHY1 has been restricted to model or agricultural systems (Cappadocia et al., 2013; Ruan et al., 2022; Taylor et al., 2022) and no phylo-comparative investigations of sequence evolution and selective regime have been conducted. The implication of WHY1 in processes associated with the mycoheterotrophic condition frames a phylogenetically informed investigation of WHY1 along a trophic gradient as an important step in improving our understanding of the evolution of plastid-targeted TFs in heterotrophic plant lineages.
We focus on four species, C. trifida, C. striata, C. wisteriana, and C. maculata, which together comprise an early transitional trophic gradient to full mycoheterotrophy with sister relationships among partial and full mycoheterotrophs (Figure 1). The well-characterized phylogenetic relationships between these four Corallorhiza species (Barrett et al., 2018) provide a powerful framework upon which to investigate WHY1 evolution during the early stages of transition to full mycoheterotrophy while accounting for phylogenetic non-independence (sensu Felsenstein, 1985). We consider C. trifida and C. wisteriana to be partial mycoheterotrophs, as their tissues contain measurable chlorophyll content and their plastomes are the most genetically intact plastid genomes in the genus, although photosynthesis has only been directly observed in C. trifida (Zimmer et al., 2008; Cameron et al., 2009; Barrett et al., 2014, Barrett et al, 2018). Conversely, we consider C. maculata and C. striata to be fully mycoheterotrophic, evidenced by their highly reduced chlorophyll content and degradation of many photosynthesis-related genes (Barrett et al., 2014, Barrett et al, 2018).
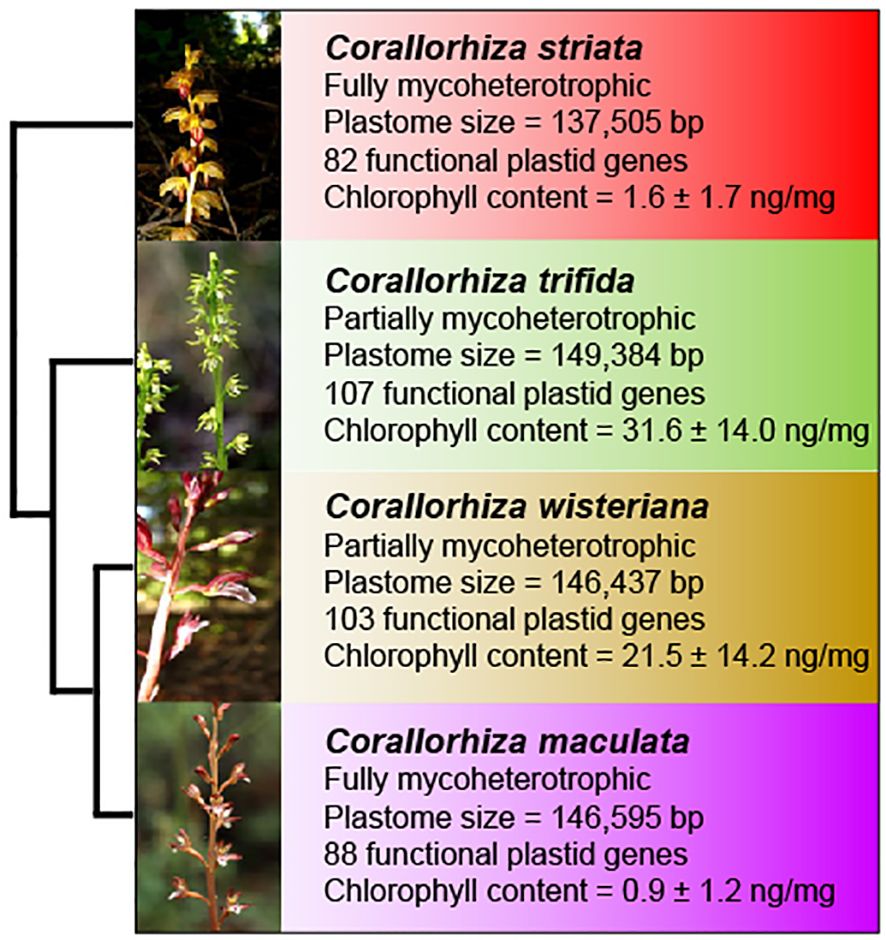
Figure 1 Overview of Corallorhiza species included in this study, showing plastid and nuclear phylogenetic relationships, inflorescence, trophic status (fully vs. partially mycoheterotrophic), plastome size (bp), number of putatively functional plastid genes, and chlorophyll content (mean and standard deviation in nanograms of total chlorophylls per milligram of plant material). Phylogenetic, plastome, and chlorophyll content data are from Barrett et al. (2014).
Here, we characterize the evolution of WHY1 across a mycoheterotrophic gradient, framed by phylogenetic context provided by the broadest taxonomic sampling of the gene to date, including late-stage mycoheterotrophic orchid species from the genera Epipogium and Gastrodia. Our integrative work leverages a combination of novel and publicly available data generated from DNA sequencing, RNA sequencing (RNA-seq), and Oxford Nanopore sequencing from 110 angiosperm species representing 21 orders. Taken together, the results of our analyses of WHY1 sequence, expression, splicing, and selective regime across both a trophic gradient and the angiosperms more broadly suggest that the gene may play a critical role in maintaining plastome stability after the transition to mycoheterotrophy.
2 Materials and methods
2.1 Publicly available sequences
Annotated WHY1 sequences were obtained from the nucleotide and Ref-seq National Center for Biotechnology Information (NCBI) databases and Orchidstra, an orchid-specific database (Chao et al., 2017). Only WHY1 sequences containing canonical WHY1 ORFs (open reading frames) were retained. Additionally, sequences were excluded if they did not contain the ssDNA-binding region (KGKAAL; A. thaliana Q9M9S3) as reported by Cappadocia et al. (2013; PDB 4KOO). Identical sequences were excluded for taxa with multiple database accessions. Stop codons were trimmed from sequences, with the exception of premature stop codons in sequences from known mycoheterotrophs, which were changed to gap characters (-) for compatibility with downstream methods (e.g., HyPhy, see below). In total, 110 species were included in the angiosperm-wide alignment (see Alignment section below; Supplementary Table S1).
2.2 RNA-seq, de-novo assembly of transcripts, and in-silico differential expression
Corallorhiza tissues used for RNA-seq are those referred to in Sinn and Barrett (2020), where complete methodological details can be found. In brief, total RNAs were extracted from pooled tissues using the ZR Plant RNA MiniPrep Kit (Zymo Research, Irvine, California, USA), leveraging a DNA exclusion column and a DNase digestion step. RNA extractions were conducted in an area where both DNA and RNase contamination were actively managed, the later with both RNase Away and RNase Zap (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Pooled tissues were categorized as either aboveground (combined stem, flower, and ovary tissues) or belowground (rhizome tissue, including fungal tissue), and three biological replicates of each tissue type were extracted for all four species. Extracted RNAs were quantified on a 2100 Bioanalyzer (Agilent, Santa Clara, California, USA) and a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Library construction was conducted at the West Virginia University Genomics Core Facility using TruSeq Stranded mRNA kit (Illumina, San Diego, California, USA). Libraries were sequenced on the Illumina HiSeq 1500 platform at the Marshall University Genomics Core Facility, with the exception of aboveground C. maculata (two samples) and belowground C. striata (two samples) for which library preparation of limited material was not successful.
De-novo assembly of transcripts from each Corallorhiza species was conducted using Trinity (version v2.13.2, Grabherr et al., 2011). Reads were trimmed using Trimmomatic (version 0.36, Bolger et al., 2014) with default settings to remove sequencing adapters and low-quality bases from the read ends. Trinity was provided with a samples file with biological replicate relationships, and strand-specific (SS) library type was set to reverse-forward (RF). Trimmed reads were mapped to each assembled transcript using the splice-aware read mapper BBMAP (version 38.96; Bushnell, 2022) with default settings, with mapped reads output to SAM format and converted to sorted BAM-formatted files using SAMtools (version 1.15; Li et al., 2009).
In-silico analysis of differential expression was conducted using scripts provided as components of the Trinity RNA-seq pipeline (Haas et al., 2013). Transcript abundance was estimated using the alignment-free estimation method as implemented in Salmon (version 1.2.0, Patro et al., 2017). The strand-specific library type was set to RF. Salmon output files included transcript abundance estimates at both the transcript and gene level. Matrices were built using the abundance_estimates_to_matrix.pl script for both transcript counts and gene expression. Differential expression analysis was run using the R (R Core Team, 2019) package edgeR (Bioconductor version 3.10, Robinson et al., 2010) via the run_DE_analysis.pl script, which identified differential expression using biological replicates of tissues across the replicate conditions aboveground and belowground. TPM (transcripts per million) values were normalized using the TMM (trimmed mean of M-values; Robinson and Oshlack, 2010) approach in order to normalize expression values while maintaining comparability of expression between samples.
2.3 Identification of WHY1 transcripts
We used the HMMER suite (version 3.3.2; Eddy, 2011) to create a WHY1 hidden Markov model (HMM) profile. The HMM profile was built with hmmbuild, which used the complete angiosperm-wide WHY1 nucleotide alignment. All assembled Corallorhiza transcripts were then searched against the HMM model using nhmmer (Wheeler and Eddy, 2013). Default parameters were used for both programs. All assembled isoforms of a transcript identified with the highest E-value were considered as potential splicing variants of WHY1.
2.4 PCR amplification
Primers for amplification of genomic WHY1 sequence were designed using the Geneious Prime (version 2020.2.4, https://www.geneious.com) plugin for Primer3 (version 2.3.7; Untergasser et al., 2012). Oligos were synthesized by Integrated DNA Technologies (Coralville, Iowa, USA). Genomic sequences presented here were amplified using a forward primer (WHY1_upstreamF: TTC AAA TCG AAG AGT AAA CTA ACC) whose 3’ end binds five nucleotides upstream of exon 1 and a reverse primer (WHY1_exon2R: TTT GGC TCA ACT GAT AGA GC), which binds in the downstream portion of exon 2. PCR amplification of WHY1 for each Corallorhiza species was performed on CTAB extractions (Doyle and Doyle, 1987) of total DNA. PCR was conducted in 25 μl volumes, comprising 12.5 μl of Apex Taq RED Master Mix (Genesee Scientific; Morrisville, North Carolina, USA), 1.25 μl of each forward and reverse primer, 9 μl of water, and 1 μl of template DNA. PCR was conducted in a Bio-Rad T100 Thermocycler (Hercules, California, USA) using the following program: initial template denaturation at 95°C for 3 min, followed by 30 cycles of denaturation at 95°C for 30 s, primer annealing at 52°C for 30 s, and template extension at 72°C for 30 s. The program ended with a final extension at 72°C for 5 min and was held at 4°C until retrieval. PCR cleanup was performed with Agencourt AMPure XP PCR Purification beads (Beckman Coulter Life Sciences; Indianapolis, Indiana, USA). Purified DNA samples were quantified using a NanoDrop OneC spectrophotometer (Thermo Fisher Scientific) and a Qubit fluorometer (Thermo Fisher Scientific) with the dsDNA BR Assay Kit.
2.5 Nanopore sequencing
Purified DNA samples were diluted to equimolar concentrations and libraries for long-read sequencing were prepared according to the Oxford Nanopore Technologies (ONT; Oxford, United Kingdom) End-Prep protocol (SQK-LSK109). The library of each Corallorhiza species received a unique barcode for Nanopore sequencing using the Native Barcoding Expansion 1–12, PCR-free kit (EXP-NBD104). The MinION SpotON flow cell (R9.4.1 FLO-MIN 106; ONT) was used for sequencing. Base calling was performed using the high-accuracy base calling algorithm as implemented in the GPU version of Guppy (version 6.2.1 + 6588110a6; ONT) on an NVIDIA GeForce RTX 2060 graphics card. Nanopore reads were mapped to the Dendrobium catenatum (RefSeq ID: GCF_001605985.2; Zhang et al., 2016) WHY1 genomic sequence in Geneious Prime using Minimap2 (version 2.17, Li, 2018) with a K-Mer length set to 15.
2.6 Alignment
We aligned all Corallorhiza WHY1 isoforms using MAFFT (version 7.3.10; Katoh and Standley, 2013), and the E-INS-i algorithm (Katoh et al., 2002) and the 1PAM scoring matrix, as a Geneious Prime plugin. Visualization and structural annotation against the canonical WHY1 sequence of Arabidopsis thaliana (NCBI Q9M9S3) was also conducted using Geneious Prime. Consensus was determined as majority consensus with a 0% threshold, meaning no minimum frequency was required for a consensus character if the character was shared by most sequences. All Corallorhiza WHY1 isoforms were translated in Geneious Prime to amino acid sequence and manually trimmed to the correct ORF. ORF-trimmed WHY1 translations were aligned with the ORF-trimmed WHY1 sequence of Arabidopsis thaliana (NCBI NM101308) to verify the presence of the expected canonical reading frame.
A translation-aware alignment of the canonical form of WHY1 representing lineages across the angiosperms was generated using a two-step process. Nucleotides were first aligned and translated using the frameshift-aware aligner MASCE2 (version 2.0.6; Ranwez et al., 2011) with default parameters, which inserts gap characters necessary to maintain codon-based statements of homology across the alignment. This approach was necessary due to the presence of frameshift mutations in sequences from Gastrodia elata, Epipogium aphyllum, and C. striata. The MACSE2-processed nucleotide and amino acid alignments were then refined using MAFFT and the E-INS-i algorithm with a BLOSUM 80 substitution matrix.
An alignment including the complete genomic sequences of WHY1 from Phalaenopsis equestris (ASM126359v1) and Dendrobium catenatum (ASM160598v2) was also generated. Phalaenopsis equestris (Cai et al., 2015) and D. catenatum (Zhang et al., 2016) are the closest relatives of Corallorhiza with sequenced genomes (Chen et al., 2022). This DNA alignment was generated to identify the introns of WHY1 and to evaluate the exonic content of assembled transcripts. All Corallorhiza isoforms and Corallorhiza Nanopore consensus sequences were aligned with the sequences of P. equestris and D. catenatum using MAFFT (version 7.3.10; Katoh and Standley, 2013), as a Geneious Prime plugin, and the E-INS-i algorithm (Katoh et al., 2002).
2.7 Visual screening for amino acid substitutions
Nonsynonymous substitutions within the angiosperm WHY1 alignment were surveyed visually in our amino acid alignments via Geneious Prime. Substitutions of interest included those present exclusively in Corallorhiza species, mycoheterotrophs, and the Orchidaceae. The codons of A. thaliana (Q9M9S3) and P. equestris that corresponded to the sites of nonsynonymous substitutions in WHY1 of interest were cross-referenced with the annotated A. thaliana WHY1 sequence for structure and the P. equestris WHY1 genomic sequence as included in the DNA alignment for exon location.
2.8 Phylogenetic inference
IQ-TREE (version 1.6.12; Nguyen et al., 2015) was used to infer phylogenetic relationships among the recovered WHY1 sequences using automated model choice (Kalyaanamoorthy et al., 2017), optimal partitioning assessment (Chernomor et al., 2016), and nearest neighbor interchange search enabled. Node support was estimated using 1,000 ultrafast bootstrap approximation replicates (Hoang et al., 2018). Two partitions were defined, which comprise the signal peptide and the highly variable 5’ portion of the chain (positions 1–516) and the highly conserved chain region (517–1,122), identified using functional annotations on WHY1 sequence per A. thaliana (Q9M9S3). The Amborella trichopoda WHY1 sequence was used for phylogram rooting.
2.9 Selection analyses
The WHY1 nucleotide alignment was tested for statistically significant changes in selection regime using four methods implemented in the command-line, multithreaded version of the Hypothesis Testing using Phylogenies suite (HyPhy; version 2.5.39; Pond and Muse, 2005). We used the Genetic Algorithm for Recombination Detection (GARD; Pond et al., 2006), with default parameters, to test for signal of recombination breakpoints within WHY1. We tested for significant change of selective regime using five test branch sets against null reference branch sets comprising all other species in the phylogram: (1) Corallorhiza species; (2) C. maculata + C. striata; (3) Corallorhiza + Epipogium aphyllum + Gastrodia elata; (4) C. maculata + C. striata + E. aphyllum + G. elata; (5) E. aphyllum + G. elata. The same Amborella trichopoda-rooted maximum likelihood WHY1 topology inferred with IQ-tree was used for all analyses.
RELAX (Wertheim et al., 2015) was used to test for evidence of relaxed selection. RELAX breaks each codon into its three component sites, each with an assigned omega class. Values for omega are calculated as Dn/Ds ratios, using the calculation for the reference branches as the null hypothesis. The value k is the selection intensity parameter and is an exponent value on omega. The alternative model fits a value for k that changes the rate to fit with the test branches. Evidence for intensified selection strength along test branches is indicated by a significant result where the value of k is greater than 1 (k > 1, P < 0.05). Evidence for relaxed selection along test branches is indicated by a significant value of k less than 1 (k < 1, P < 0.05). Strength of selection was assessed simultaneously for all species using Fast Unconstrained Bayesian Approximation (FUBAR, Murrell et al., 2013). FUBAR can detect weak, yet pervasive, purifying or diversifying selection at the codon level (posterior probability >0.90) without the use of test and reference branch sets.
The Branch-Site Unrestricted Statistical Test for Episodic Diversification (BUSTED) was used to test for evidence of gene-wide positive selection (Murrell et al., 2015). BUSTED uses three omega classes defined as ω1 ≤ ω 2 ≤ 1 ≤ ω3. The ω1 class is the proportion of sites with a very low Dn/Ds ratio. The ω2 class is the proportion of sites just below 1, and ω3 are sites above 1. A value of 1 suggests selective neutrality, and therefore defines the null, or constrained, model. BUSTED then calculates the log-likelihood of the data for each of the null and alternative models. These ratios are calculated for each site and are called evidence ratios (ERs). They are used as a threshold (χ2 distribution, P < 0.01) but are not a valid test for site-specific likelihood. The null model is rejected if at least one site on a test branch experienced positive selection. Evidence for positive, or diversifying, selection in a gene of a test branch is indicated by a rejection of the null model.
The adaptive branch-site random effects likelihood (aBSREL) test was used to test for signal of episodic positive selection. aBSREL infers ω values at both the level of sites and branches and can account for rate heterogeneity inherent to complex evolutionary scenarios by partitioning these values into multiple rate classes per branch. aBSREL was conducted in exploratory mode, where all branches were tested and p-values were Holm–Bonferroni corrected, and in the more sensitive a priori mode to test for episodic positive selection in each test branch set.
3 Results
3.1 De-novo assembly and identification of WHY1
The hmmer suite revealed a single, Trinity-identified gene model and its isoforms as representing WHY1 from de-novo assembled transcripts for each Corallorhiza species. Trinity assembled a single isoform representing the expected canonical CDS (coding DNA sequence) of WHY1 for all but C. striata, the latest stage fully mycoheterotrophic species of Corallorhiza sampled. However, mapping reads to each isoform with BBMAP revealed that a five nucleotide indel, the absence of which results in a premature stop codon, was differentially present or absent in the reads of each Corallorhiza species (NCBI BioProject PRJNA984634). Trinity differentially incorporated that indel, hereafter referred to as the GTGAA indel, into the isoform pool of each Corallorhiza species. The absence of the GTGAA indel in Trinity-assembled isoforms of C. striata precluded the recovery of the canonical ORF, but read mapping supports the presence of the five nucleotides necessary to recover the expected ORF in C. striata, at a rate of 67.2% of reads in isoform 2 and 76.1% of reads in isoform 4. Those data support that the canonical variant of WHY1 is also expressed. We analyzed the C. striata isoforms as assembled, rather than manually modifying the C. striata transcripts to conform to a hypothesized canonical sequence. The inclusion of those five nucleotides would not alter any results presented here, aside from whether a canonical WHY1 isoform is transcribed in C. striata.
The canonical WHY1 ORF as assembled in C. trifida, C. wisteriana, and C. maculata is 795 nucleotides in length and comprises 265 amino acids, two fewer amino acids than that of A. thaliana. Non-canonical splicing variants were found in the isoform pools of each Corallorhiza species. A splicing variant containing a 79-nucleotide long sequence of 96.6% mean pairwise identity was recovered from C. wisteriana, C. maculata, and C. striata, at nucleotide position 232. The second major variant is the GTGAA indel discussed earlier, which is variously present at nucleotide position 514 in all four species. A third variant identified from the isoform pool of C. striata represents a modification of the 3’ end of the ORF, immediately downstream of the previously discussed five-nucleotide indel that was variously present in our read pools throughout Corallorhiza.
3.2 Alignment
MAFFT alignment of all raw Trinity-assembled Corallorhiza WHY1 isoforms resulted in a matrix of 1,302 positions. Pairwise percent identity across isoforms was 81.4% and 88.2% and gaps comprised 11.7% and 8.1% of character states for all isoforms and with the exclusion of the early terminating isoform 1 of C. striata, respectively. Alignment of ORF-trimmed transcripts resulted in a matrix of 874 positions (Figure 2). Percent pairwise identity among isoforms within a species was highest for C. trifida (99.5%) and lowest for C. striata (70.6%). A 79-nucleotide-long indel of 96.6% pairwise identity was identified in at least one isoform assembled from all Corallorhiza species except C. trifida. Translation-aware alignment of ORF-trimmed canonical Corallorhiza WHY1 sequences, with the differentially present GTGAA indel manually inserted into the otherwise canonical sequence of C. striata, resulted in an alignment of 795 characters of 98.0% pairwise identity and no gaps. The highest pairwise percent identity was 99.37% between C. wisteriana and C. maculata and the lowest was 96.98% between C. trifida and C. striata.

Figure 2 Wrapped view of open reading frame-trimmed Corallorhiza WHY1 isoform alignment. Disagreements with the consensus sequence are highlighted. An * denotes a canonical isoform, while ψ denotes an isoform with a retained intron.
The WHY1 genomic DNA alignment of Corallorhiza isoforms, Corallorhiza Nanopore sequences, and P. equestris and D. catenatum genomic sequences had a total length of 12,000 nucleotides (Figure 3), in which the assembled canonical isoforms from each Corallorhiza species contained the expected exons of WHY1. One non-canonical isoform of C. wisteriana, C. maculata, and C. striata each contained intron 1, for which pairwise percent identity was 96.6% across those species. Percent pairwise identity of intron 1 between that of D. catenatum and P. equestris was 82.3%, and similarity between retained introns and that of P. equestris ranged from 78.5% to 82.3% for C. maculata and C. striata, respectively. The Nanopore-sequenced and Trinity-assembled WHY1 intronic sequences of C. striata were identical, while those of C. maculata were 94.9% similar, due to the relative lack of two thiamine nucleotides in the Nanopore sequence.
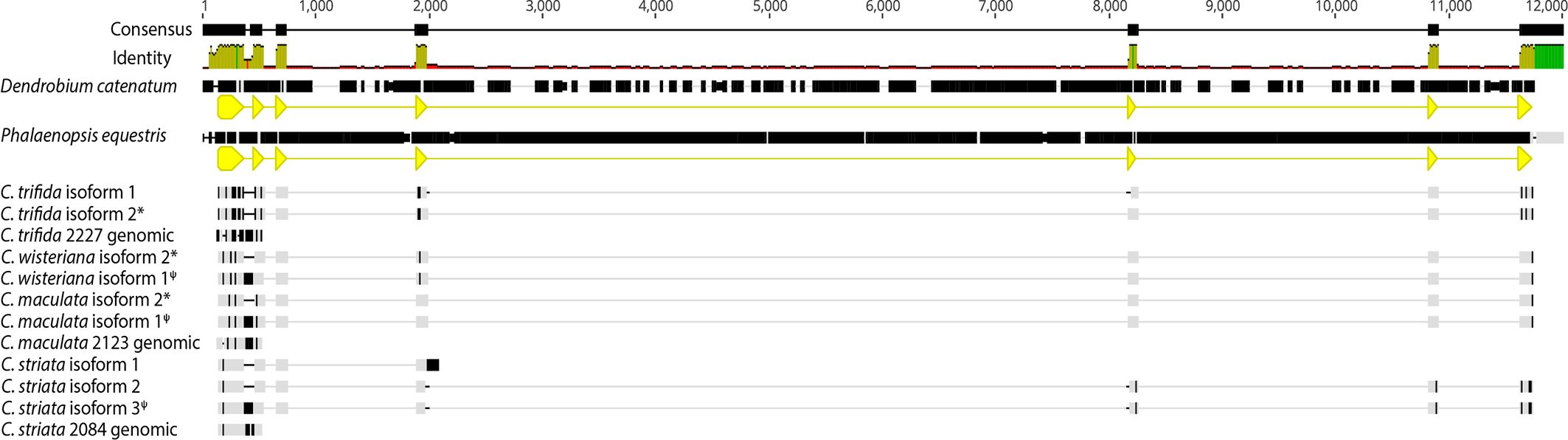
Figure 3 DNA alignment of canonical isoforms and Nanopore-sequenced genomic sequence from each Corallorhiza species with annotated WHY1 sequence from the Phalaenopsis equestris and Dendrobium catenatum reference genomes. Coding sequences for each reference genome are depicted by yellow annotations. Disagreements with the consensus sequence are shown in black. Percent identity of aligned sites is depicted as a histogram. An * denotes a canonical isoform, while ψ denotes an isoform with a retained intron.
The angiosperm-scale WHY1 nucleotide alignment contained WHY1 sequences from 110 species, including 22 orchid species. The total length of the alignment was 1,122 positions, of which 150 were of identical states across all species. Two partitions roughly corresponding to the transit peptide and chain regions as annotated in A. thaliana (Q9M9S3) were conspicuously visible in the consensus sequence of the alignment. The first was a highly variable, lineage-specific portion of sequence ranging from positions 1–516 in the alignment. The second was a highly conserved portion spanning positions 517–1,122. The mean pairwise percent identity of the transit region of Orchidaceae was 51.3%, while that of Apostasia shenzhenica compared to either G. elata or E. aphyllum was 21.5% and 31.1%, respectively. The mean pairwise percent identity of the transit region of Corallorhiza was 94.4%. The mean pairwise percent identity of the four Corallorhiza species in the alignment was 97.7% and 81.0% across the Orchidaceae. The grasses had the lowest mean percent pairwise similarity of any clade, which was 49.36% when each sequence was compared to each non-grass species in the alignment.
The angiosperm-wide WHY1 amino acid alignment comprised 376 positions, of which 48 were of identical states across all species. The two partitions in the nucleotide alignment corresponding to functional annotation in A. thaliana (Q9M9S3) were more conspicuous in the amino acid alignment. Positions 1–115 and 116–376 corresponded to the transit and chain regions of A. thaliana (Q9M9S3), respectively. The pairwise percent identity of the four Corallorhiza species in the alignment was 97.4% for WHY1, and across the Orchidaceae it was 77.8%.
3.3 Nanopore sequencing
Nanopore sequencing of genomic WHY1 sequence from three of the four Corallorhiza species confirmed that the unique sequence in the non-canonical transcripts of C. wisteriana, C. striata, and C. maculata represented retention of WHY1 intron 1. Sequencing of WHY1 amplicons generated read pools ranging from 25,546 to 74,261 reads in C. trifida and C. maculata, respectively. Mapping of Nanopore reads against the D. catenatum genomic WHY1 sequence resulted in a mean coverage depth for the exon 1–2 region ranging from 4,034.9 to 10,617.6 in C. trifida and C. striata, respectively (Supplementary Table S2). Library preparation for Nanopore sequencing of C. wisteriana WHY1 amplicons was deemed unsuccessful since the resulting sequence pool contained presumably off target reads that precluded confident assembly of a WHY1 consensus sequence from that sample. Alignment of the Nanopore-sequenced WHY1 amplicon consensus reads with the Corallorhiza transcript isoforms and the full WHY1 genomic sequences of P. equestris and D. catenatum provided evidence that the non-canonical isoforms of WHY1 in Corallorhiza were a result of alternative splicing (Figure 4). At least one non-canonical isoform from all but C. trifida contains intron 1 of WHY1.
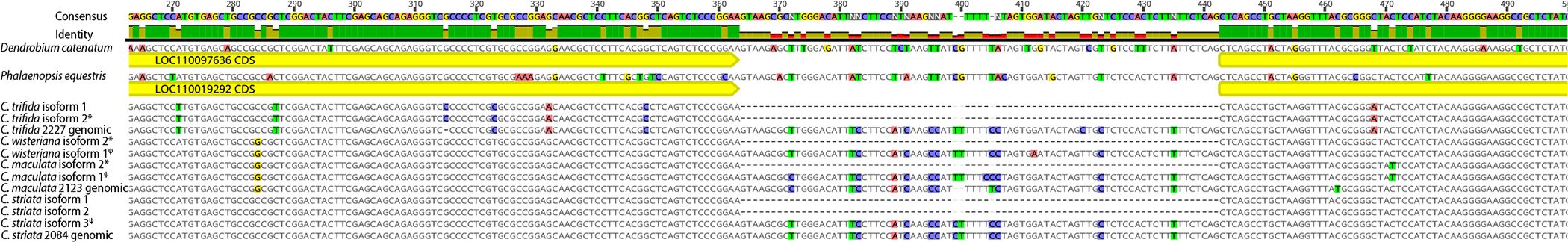
Figure 4 DNA alignment of Corallorhiza isoforms assembled from each Corallorhiza species and Nanopore sequences for C. trifida, C. maculata, and C. striata with annotated WHY1 sequence from the Phalaenopsis equestris and Dendrobium catenatum reference genomes. WHY1 exons 1 and 2 are annotated in yellow and the locus ID tags for each reference genome are provided in the coding sequence (yellow) annotations. Disagreements with the consensus sequence are highlighted. Percent identity of aligned sites is depicted as a histogram. An * denotes a canonical isoform, while ψ denotes an isoform with a retained intron.
The sequences obtained from Corallorhiza species via Nanopore and RNA-seq shared a high degree of similarity with each other and the WHY1 sequence in the previously published P. equestris and D. catenatum genomes. Intron 1 of WHY1 was found to have a pairwise percent similarity of 82.3% between P. equestris and D. catenatum. Corallorhiza WHY1 intron 1 sequences obtained via Nanopore sequencing and RNA-seq had a mean pairwise percent similarity of 96.9%, while that of C. striata obtained via both RNA-seq and Nanopore sequencing had a pairwise percent similarity of 82.3% with P. equestris.
3.4 Select amino acid substitutions of interest
Substitutions exclusive to mycoheterotrophic species were evident and, in some cases, were exclusive to fully mycoheterotrophic species. The transit regions of both E. aphyllum and G. elata contained some substitutions that were unique within the Orchidaceae. A phenylalanine-glycine residue at positions 76–77 was exclusive to C. trifida, while a leucine-arginine residue was present for the remainder of Orchidaceae at those positions. While pairwise percent identity of positions 76–77 was only 13.3% throughout angiosperms, the leucine-arginine residue in the Orchidaceae had a pairwise percent identity of 90.9% when including the residue from C. trifida and was identical when that taxon was excluded. Amino acid substitutions found in the transit regions of both C. wisteriana (positions 46 and 95) and C. trifida (position 113) were not identified in other orchids but were found in some non-orchid autotrophs.
At least one substitution that could impact WHY1 conformation was identified in Corallorhiza. The terminal codon of an alpha helix annotated in the structure of A. thaliana (9M9S3) WHY1 (position 336) has been substituted to an isoleucine in C. trifida and to a phenylalanine in C. wisteriana, C. maculata, and C. striata (Figure 5). We infer that the ancestral state of position 336 is leucine, on the basis of that codon state for 89.1% of sampled taxa, including Amborella trichopoda. The only other substitutions at that position were to isoleucine, which was identified in eight autotrophic angiosperms outside the Orchidaceae, and in C. trifida. Additionally, a substitution from alanine to valine at position 359 was exclusive to C. striata, which represents the only occurrence of that state for this position for 109 other angiosperm species, and the only subgeneric polymorphism observed in that position.
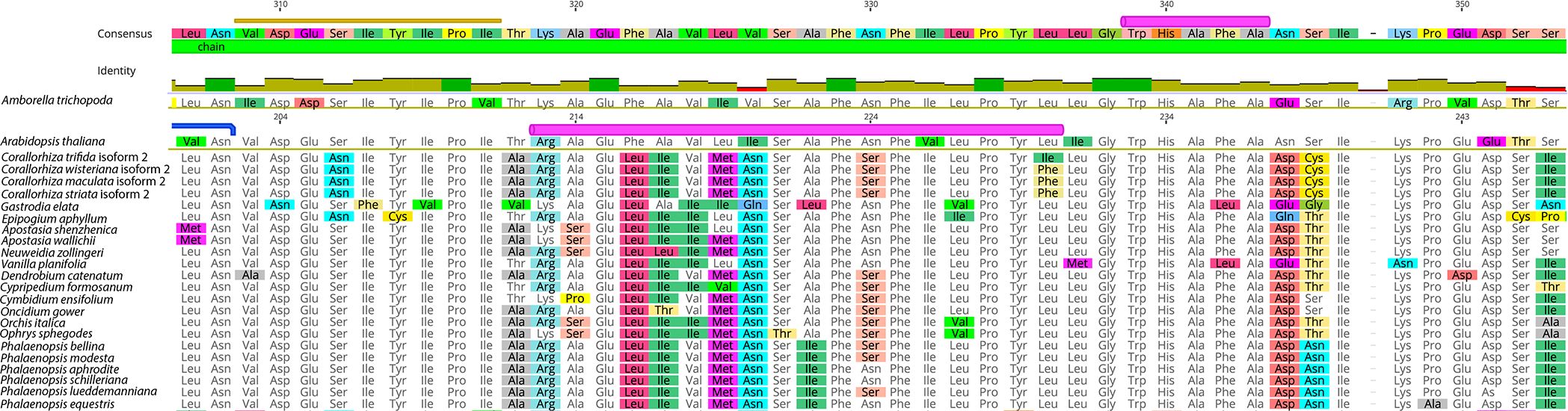
Figure 5 Detailed view of amino acid alignment of WHY1 sequences of orchid species. Note the phenylalanine substitutions unique to Corallorhiza wisteriana, C. maculata, and C. striata, relative to the other species sampled. The amino acid substitutions of interest are located at the 3’ portion of a region inferred to conform into an alpha helix structure (pink annotation) in Arabidopsis thaliana WHY1 (alignment row two).
A five-nucleotide indel resulting in a frameshift (amino acid alignment positions 269 and 270) was found in C. striata, but the pairwise percent identity of downstream sequence with that of C. maculata was high (97.8%) and reads containing the corresponding five nucleotides were identified in the RNA-seq read pool. In fact, RNA-seq reads containing the indel were found in the read pools of all Corallorhiza species, suggesting that isoform diversity was conservatively interpreted by our methods.
We found that the chain region of E. aphyllum contained a glycine to arginine substitution in the characteristic WHY1 ssDNA-binding motif (positions 188–193), a site that was otherwise conserved throughout the remainder of samples. Additionally, we found that the E. aphyllum sequence contained a residue comprising seven amino acids (positions 294–300), the last of which was a premature stop codon. MAFFT resolved those seven amino acids as an insertion with no homology to other angiosperm sequences. Six substitutions downstream of that premature stop codon are exclusive to E. aphyllum.
3.5 Phylogenetic inference
Both the Eudicots and Monocots were recovered as monophyletic (Figure 6; BS = 100%). Orchidaceae was recovered as a monophyletic group (BS = 94%), with D. catenatum, G. elata and E. aphyllum as an early diverging paraphyletic grade within the Epidendroids (BS = 98%). However, the positions of D. catenatum (BS = 79%) and G. elata (BS = 76%) were not robustly supported. Corallorhiza species were recovered as a monophyletic group (BS = 100%) with C. trifida sister to C. striata (BS = 100%) + (C. wisteriana, C. maculata; BS = 100%), rather than those inferred in previous, genomic-scale work depicted in Figure 1.
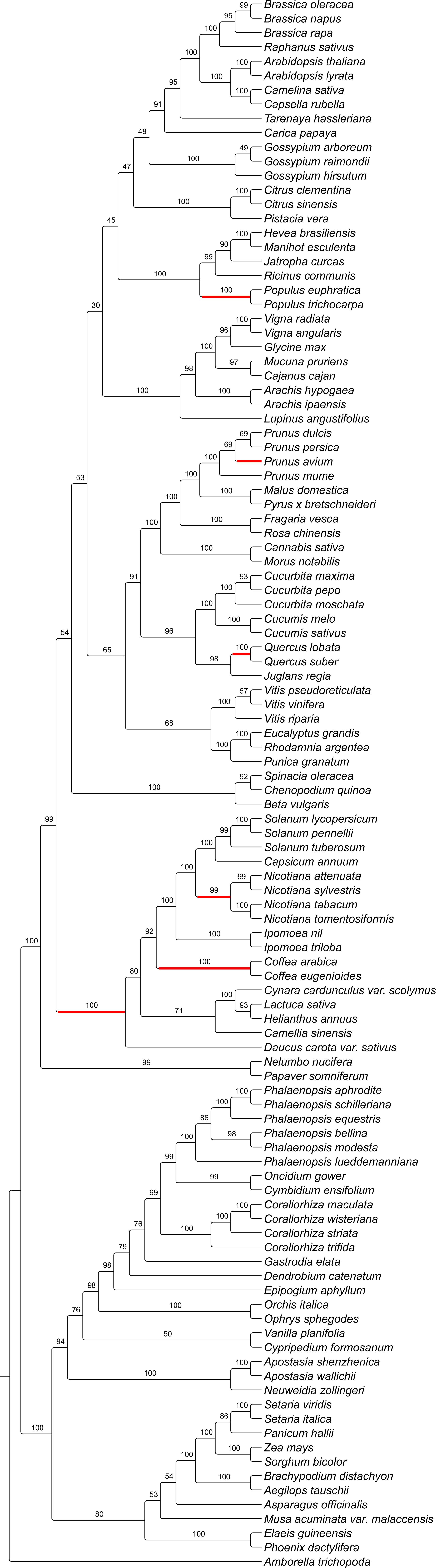
Figure 6 Maximum likelihood inferred topology of WHY1 evolution with support values derived from 1,000 non-parametric bootstrap replicates. Lineages highlighted in red represent those for which statistically significant signal of episodic diversifying selection of WHY1 was detected by the adaptive branch-site random effects likelihood test.
3.6 Differential expression
Canonical isoforms of WHY1 were most highly expressed in all four species (see Table 1), but statistically significant elevated expression in aboveground tissues relative to those belowground was only detected in C. trifida. Gene-level expression of WHY1 across both tissues was highest for C. wisteriana (244.5) and C. trifida (175.4) and lowest for C. maculata (131.9) and C. striata (95.3). Expression of the canonical isoform of WHY1 in C. trifida ranged from 4.15 to 9.64 TMM in belowground tissues (median = 7.17 TMM) and from 31.353 to 56.389 TMM in the aboveground tissues (median = 43.273 TMM). The log fold-change value was 2.56 between belowground and aboveground tissues for the canonical isoform of C. trifida, with a P-value of 0.00018 and a false discovery rate (FDR) of 0.00374. Expression of the WHY1 nearly canonical isoform in C. striata ranged from 5.547 to 16.859 TMM in belowground tissues (median = 12.31 TMM) and 17.28 and 30.538 TMM in aboveground tissues (median = 23.909 TMM). The C. striata canonical WHY1 isoform had a log fold-change value of 0.396 between belowground and aboveground tissues, with a P-value of 0.69 and FDR of 0.91. Expression of the canonical isoform of WHY1 in C. wisteriana ranged from 9.256 to 23.023 TMM in belowground tissues (median = 15.663 TMM) and 34.792 to 74.955 TMM in aboveground tissues (median = 60.80 TMM). The C. wisteriana canonical isoform had a log fold-change value of 1.96 between belowground and aboveground tissues, with P-value of 0.02 and FDR of 0.28. The expression of the canonical isoform in C. maculata had values of 11.064 and 13.947 TMM in belowground tissues (median = 12.51 TMM) and ranged from 26.179 to 45.999 TMM in aboveground tissues (median = 33.01 TMM). The C. maculata canonical isoform had a log fold-change value of 1.17 between belowground and aboveground tissues, with P-value of 0.16 and FDR of 0.62.
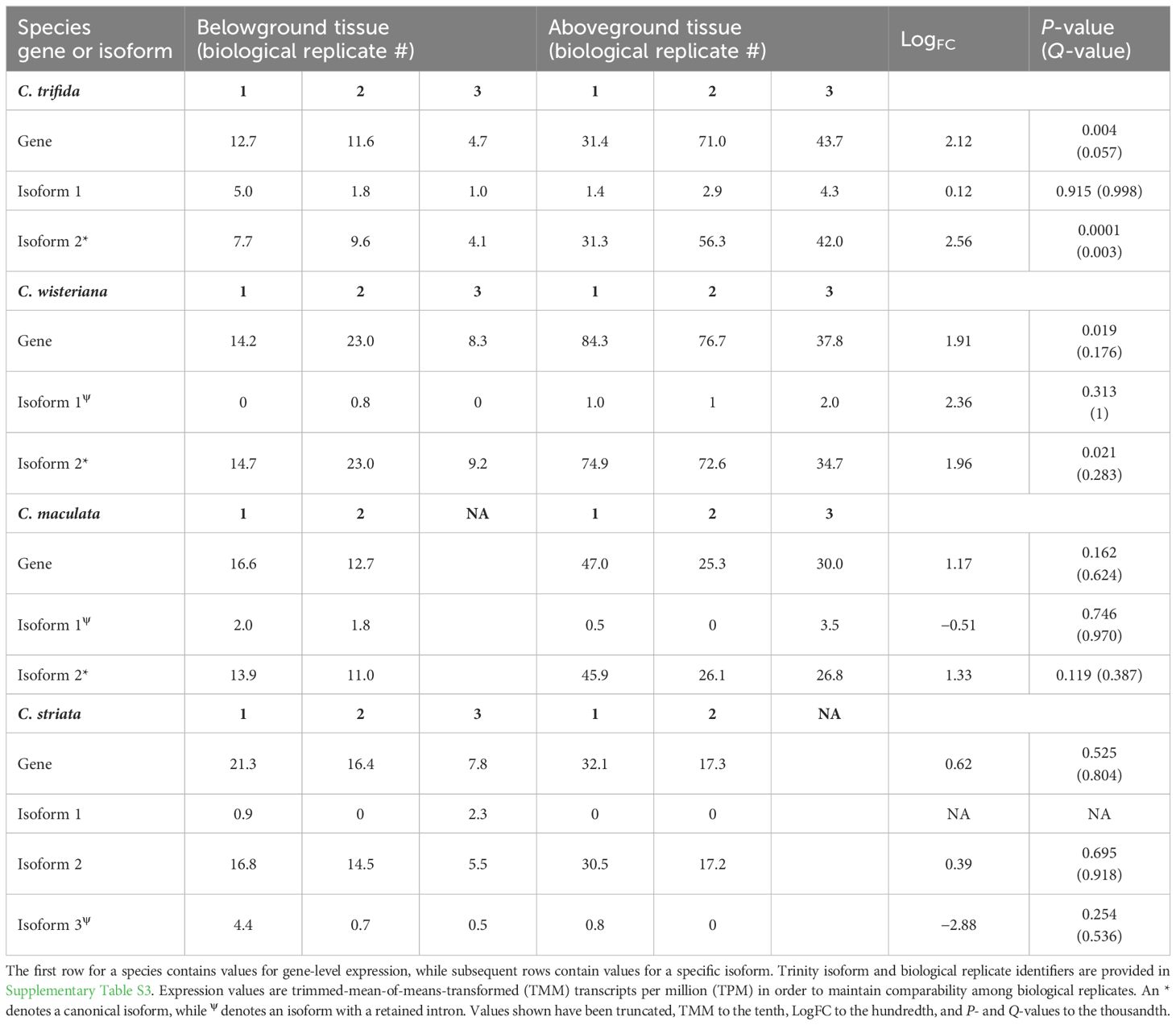
Table 1 In silico expression of WHY1 across species, tissues, and biological replicates.
The expression of non-canonical isoforms assembled from the read pools of all four species varied across tissues and samples, with some tissues or individuals not expressing splicing variants, and expression was not statistically different between the two tissue types in any of the four species. The C. trifida non-canonical isoform was expressed at relatively low levels across all aboveground and belowground tissues, the TMM of which ranged from 1.02 belowground to 4.374 aboveground. Contrastingly, the TMM of the C. wisteriana non-canonical isoform ranged from 0 to 2.017 and was not detected in two of the belowground replicates for this species. The noncanonical C. maculata and C. striata isoforms were sporadically expressed across tissue types, with the TMM values of one isoform of C. maculata ranging from 0 to 3.564 and those of C. striata ranging from 0 to 4.471. Even in the case of the C. striata isoform for which the TMM value was 4.471 in one belowground sample, the value was either 0 or less than 1 in the remainder of samples.
3.7 Selection analyses
GARD evaluated 2,393 models and inferred a single potential recombination breakpoint, separating the signal peptide and chain portions of WHY1, but AICc was not significantly improved for the partitioned analysis (75,130.4) versus unpartitioned (75,111.5). The RELAX test inferred statistically significant signal for relaxation of selection pressure for the following test branch sets: Corallorhiza + E. aphyllum + G. elata (p = 0.001); C. maculata + C. striata + E. aphyllum + G. elata (p = 0.002); and E. aphyllum + G. elata (p = 0.001). However, runs of RELAX analyzing test branch sets comprising only Corallorhiza (p = 0.30) or C. maculata + C. striata (p = 0.62) did not infer significant signal for selection relaxation.
The FUBAR analysis inferred that 254 of 373 codons were under pervasive purifying selection (posterior probability threshold ≥0.9) and that no codons were under pervasive diversifying selection. Likewise, the BUSTED analyses did not infer statistically significant signal of gene-wide episodic diversifying selection for any test branch set, where p-values ranged from p = 0.15 for E. aphyllum + G. elata to p = 0.50 for Corallorhiza.
The aBSREL analysis recovered signal of episodic diversifying selection in seven of 217 branches in the WHY1 tree (Figure 6) but did not infer statistically significant signal in any test branch set during analyses conducted in a priori mode. The terminal node leading to Sorghum bicolor (p = 2.76 × 10−6) was the only branch in the monocots that showed significant signal of episodic diversifying selection. Within the eudicots, the branches leading to Prunus avium (p = 2.5 × 10−5), the Populus clade (p = 1.17 × 10−7), the Quercus clade (2.35 × 10−3), the Nicotiana clade (p = 5.42 × 10−3), the Coffea clade (6.96 × 10−5), and the asterid clade (p = 3.7 × 10−4) were inferred to contain signal of episodic diversifying selection.
4 Discussion
Our work represents the largest scale investigation of WHY1 evolution to date and reveals strong phylogenetic signal for this gene across multiple taxonomic levels in the angiosperms. Phylogenetic methods have emerged as the gold-standard for the inference of gene orthology (Münster et al., 1997; Emms and Kelly, 2015, Emms and Kelly, 2019) by which hundreds of genes have been determined to not only be highly conserved across plant lineages but to exist as low- or single-copy (Wu et al., 2006; Duarte et al., 2010; De Smet et al., 2013). The inferred evolutionary relationships of WHY1 (Figure 6) were largely congruent with our contemporary understanding of angiosperm phylogenetic relationships and recent inferences based on genomic-scale datasets (The Angiosperm Phylogeny Group, 2016; Guo et al., 2021; Zhao et al., 2021). Likewise, the inferred relationships within the Orchidaceae were largely consistent with those inferred in other studies (e.g., Givnish et al., 2016; Li et al. 2019; Pérez-Escobar et al., 2021; Serna-Sánchez et al., 2021; Zhang et al., 2023; Barrett et al., 2024; Pérez-Escobar et al., 2024), apart from D. catenatum, G. elata, and E. aphyllum forming an early diverging, paraphyletic grade within the Epidendroids. However, our recovery of C. trifida as sister to the remainder of the species sampled from Corallorhiza, rather than C. striata, was not congruent with relationships inferred previously on the basis of other loci and even genomic-scale datasets (Barrett and Freudenstein, 2008; Barrett et al., 2014, Barrett et al, 2018). We are not surprised to infer discordance between the evolutionary history of WHY1 and that of the phylogenetic history of Corallorhiza, since it is accepted that the evolutionary history of any gene can differ from that of the genome within which it is found (Pamilo and Nei, 1988; Maddison, 1997). That said, the recovery of the Corallorhiza species with the most intact plastome as sister to those with more degraded plastomes may indicate that the gene-species tree discordance we infer is due to functional convergence in WHY1 sequence. For example, the phenylalanine substitution at position 336 shared by all Corallorhiza species aside from C. trifida may affect protein conformation and therefore WHY1 function. Our findings of general congruence between the evolutionary history of WHY1 across angiosperms supports the gene as single- or low-copy across the 110 angiosperm taxa sampled.
Differences in the degree of nucleotide conservation were evident among WHY1 exons, with the transit peptide region consistently the most divergent throughout the lineages sampled. The diversity and evolution of transit peptides, and the apparent discrepancy between our perception of their functional importance and high-sequence divergence, have long been of interest (Bruce, 2001; Patron and Waller, 2007; Christian et al., 2020). While the functional nature of a transit peptide might lead to an expectation of conservatism, low-sequence similarity and patterns of mutation that we describe in WHY1 are emerging as generalizable properties of plant transit peptides. For example, Christian et al. (2020) found that the transit peptides in the genomes of 15 genera sampled throughout the angiosperms had a mean pairwise percent identity of just 37.9% and that random indels drive transit peptide evolution. Our results provide evidence that the transit peptide of WHY1 evolves similarly, with a pairwise percent identity of 32.5% across the angiosperms, and with evident substitutions and indels downstream of a homologous start codon being the most likely drivers of divergence in the gene region. In contrast, the portion of the chain encoding the ssDNA binding motif was the most conserved with a mean percent pairwise similarity of 72.1%. The only sizable indel observed in the gene region was a seven-codon insertion in E. aphyllum, a late-stage mycoheterotrophic orchid. It is likely that E. aphyllum WHY1 results in a non-functional product, given that it encodes a premature stop codon that would result in a protein 70 amino acids shorter than that of any other sampled species. Interestingly, WHY1 was more conserved overall at the nucleotide level among Corallorhiza species than were either expression patterns across tissues or exon inclusion in sequenced mRNAs across the trophic gradient. Among autotrophic species, nucleotide divergence of WHY1 is particularly pronounced in the grasses, with the mean pairwise percent identity among all members of that clade versus the remainder of samples being more than 5% lower than that of the same comparison made for either of the late-stage mycoheterotrophic species sampled. The high substitution rate we inferred for WHY1 in the grasses is interesting because the plastomes of many species in the clade are known to contain inversions and structural heteroplasmy within individual plants, of which the latter has only recently been described from their relatives the Cyperaceae (Doyle et al., 1992; Lee et al., 2020). The contrasting levels of nucleotide conservation in the transit and chain portions of WHY1 across lineages suggest that lineage-specific functions of the transit peptide sequence could be a fruitful line of investigation, especially given that Christian et al. (2020) identified bias in amino acid usage between the plastid transit peptide sequences of monocot and eudicot lineages.
Alignment of WHY1 revealed lineage-specific substitutions at sites inferred to be involved in structural conformation in both poorly and highly conserved gene regions. For example, the transit regions of both late-stage mycoheterotrophic species contained indels not found in other samples, and the sequence of E. aphyllum contained a premature stop codon in the typically highly conserved chain region followed downstream by four autapomorphic amino substitutions in an eight amino acid span. We identified substitutions that could underlie functional change in each Corallorhiza species, the most interesting of which was a substitution involving an alpha helix (position 336) inferred in previous work conducted in A. thaliana (Cappadocia et al., 2013). Our phylogenetic framework supports the plesiomorphic codon state of position 336 as a leucine, which we infer was substituted to a phenylalanine in Corallorhiza and then to an isoleucine in C. trifida, on the basis of the relationships depicted in Figure 1. We additionally infer that the phenylalanine substitution present in Corallorhiza species aside from C. trifida would result in the introduction of a benzene ring in which there were previously only aliphatic hydrocarbons. However, comprehensive taxonomic sampling of WHY1 across Corallorhiza is needed to determine sequence diversity and substitution patterns. While our work revealed many nonsynonymous substitutions throughout Corallorhiza sequences, we did not identify nucleotide substitutions that obviously result in loss of function in these transitional mycoheterotrophic species.
Statistically significant shifts of selective regime were detected for late-stage mycoheterotrophic and some non-orchid autotrophic lineages. Our inference that 68.1% of WHY1 codons are under pervasive purifying selection and none are under pervasive positive selection, despite the relatively low levels of nucleotide conservatism we documented, are congruent with the critical function of the gene across the angiosperms as has been documented in model and agricultural species in previous work (Cappadocia et al., 2013; Lepage et al., 2013; Zampini et al., 2015; Ren et al., 2017; Qiu et al., 2022; Ruan et al., 2022; Taylor et al., 2022; Sun et al., 2023). While we inferred disproportionately high substitution rates for WHY1 in the grasses, we did not find evidence that divergence of those sequences was associated with diversifying selection. Despite the lack of pervasive positive selection in WHY1 across the angiosperms, our identification of significant episodic diversifying selection in seven of 100 autotrophic lineages sampled suggests that a lineage-specific adaptive role of WHY1 may be relatively common, though similar signal was not identified in sampled mycoheterotrophs. The lack of diversifying signal in any of our mycoheterotrophic species does not support neofunctionalization of the gene, which we find interesting given the multiple functions of WHY1 and the diversifying signal identified in lineages across the angiosperms. Our findings together suggest that relaxation of WHY1 selective constraint occurs after the transition to full mycoheterotrophy, as significant signal of relaxed selection was only detected in branch sets containing E. aphyllum and G. elata, two late-stage fully mycoheterotrophic orchids.
Our analyses suggest that the expression of WHY1 in Corallorhiza may differ by both tissue type and across the mycoheterotrophic gradient. Multiple studies have induced and characterized the effects of differential expression of WHY1 by exposure to biotic and abiotic stimuli, implicating roles for the gene ranging from mediating drought stress (Zhao et al., 2018; Ruan et al., 2022) to pathogen response (Desveaux et al., 2000; Sun et al., 2023). A minimum level of WHY1 expression could be expected due to roles of WHY1 that are not involved in photosynthesis, such as the maintenance of telomere length of nuclear chromosomes (Yoo et al., 2007). Our analyses are the first to characterize WHY1 expression in non-model or non-cultivated plant species, and therefore baseline expectations for tissue-specific expression levels for wild species have not yet been established. However, the estimations of gene expression we inferred for Corallorhiza species are within the expression ranges reported in studies of A. thaliana (4–76 TPM; Liu et al., 2012; Mergner et al., 2020) and Solanum tuberosum (7–50 TPM; The Potato Genome Sequencing Consortium, 2011). Additionally, the Klepikova Arabidopsis Atlas (Klepikova et al., 2016) and 1,122 tissue-specific samples available via the Arabidopsis RNA-seq Database (http://ipf.sustech.edu.cn/pub/athrdb/ accessed 8 March 2023) evidence that WHY1 expression should be expected to be lower in roots or rhizomes than in leaves, which is congruent with expression patterns in C. trifida. Previous studies reporting WHY1 expression have been conducted in species with larger individuals with typical, non-reduced morphologies allowing for finer scale investigations of tissue-specific expression than can be conducted in Corallorhiza, due to a lack of leaf laminae and roots in the latter. However, the patterns of expression we inferred across Corallorhiza tissues are congruent with those known for WHY1. While gene-level expression inferred across tissues was highest for C. wisteriana and C. trifida, tissue-specific expression was only statistically significant in C. trifida, after correcting for repeated testing. Our inferences of WHY1 expression among the belowground tissues of Corallorhiza provide for a hypothesized minimum expression level of canonical WHY1, which is similar to aboveground levels of expression in Corallorhiza species aside from C. trifida. Taken together, our results provide evidence for a trajectory beginning with differential expression of WHY1 between aboveground and belowground tissues of the most photosynthetically capable Corallorhiza species to similar expression levels between above and belowground tissues of the latest stage mycoheterotrophic members of the genus. However, future work leveraging qPCR-derived estimates of WHY1 expression across species and tissues is needed to corroborate the trends in expression patterns we characterize here. Taken together, our results suggest that alteration in expression or splicing of WHY1 is unlikely to underlie the transition to mycoheterotrophy since the tissue-level expression patterns of the gene in C. trifida are similar to those described from autotrophic plants.
Our work is the first to provide evidence for alternative splicing of, and intron retention in, WHY1. Approximately 70% of plant genes with multiple exons can be expected to be alternatively spliced (Reddy et al., 2013; Chamala et al., 2015), and intron retention is a common form of alternative splicing in plants (Ner-Gaon et al., 2004). Our finding of intron retention in one isoform from each Corallorhiza species aside from C. trifida is the first such event described for the gene. It has long been recognized that intron retention is most common in transcripts of genes like WHY1 which serve roles related to photosynthesis and stress response (Ner-Gaon et al., 2004), a finding also supported by work investigating the effects of plant stressors on levels of alternative splicing (Filichkin et al., 2018; Jabre et al., 2019). In fact, tissue-specific differential intron retention has been shown to be an inducible stress response in Populus trichocarpa (Filichkin et al., 2018). Modification of WHY1, including inserted sequence, has long been used to study the effects of mutations on the function of the gene and to induce knockouts (Desveaux et al., 2004; Yoo et al., 2007; Maréchal et al., 2009), work which helped identify the many pathways that WHY1 is involved in. For example, Yoo et al. (2007) and Maréchal et al. (2009) leveraged knockouts caused by T-DNA insertions into WHY1 to reveal the critical role the gene plays in maintaining telomere length and plastome stability in Arabidopsis thaliana, respectively. Similarly, Prikryl et al. (2008) and Qiu et al. (2022) found that double knockout WHY1 mutants were characterized by a lethal albino phenotype after the development of a few leaves, with Qiu et al. (2022) also characterizing divergent splicing and mRNA editing of plastid genes in WHY1 mutants. The assembly of intron-retaining transcripts from all Corallorhiza species aside from C. trifida, and more transcriptional isoforms in Corallorhiza species in later stages of mycoheterotrophy, is suggestive of a negative correlation between increased splicing variation and both plastome stability and chlorophyll concentration in tissues (Barrett et al., 2014). However, future long-read sequencing of isoforms is needed to definitively verify assembled isoform variants and the presence of retained introns. We propose that it is unlikely that the intron-containing isoforms result in functional products, since a premature stop codon results from intron retention. The recovery of non-canonical and intron-retaining WHY1 isoforms across individuals and tissues of Corallorhiza could be signal of idiosyncratic spliceosome regulation in mycoheterotrophic species, epitranscriptomic differences (Jabre et al., 2019), differential responses to stress in sampled tissues (Filichkin et al., 2018), or the expression of multiple, divergent copies of the gene. However, we hypothesize that reduced fidelity in the spliceosome of a mycoheterotrophic plant is the most likely cause of the observed splicing variation, given the phylogenetic and sequencing data at hand. We predict that changes in the expression and splicing of WHY1 across Corallorhiza would likely be due to the alteration of one or more pathways involved in gene regulation, since we did find nucleotide-level changes that could be responsible.
Our work is the first to characterize the evolution of a transcription factor that could impact the genetic and phenotypic changes that occur along the path to full mycoheterotrophy. The previously characterized roles that WHY1 plays in plastome stability (Parent et al., 2011; Lepage et al, 2013), defense responses (Lin et al., 2020), and leaf senescence (Lin et al., 2019), together position the gene as a worthwhile target for the study of the molecular underpinnings of the transition to mycoheterotrophy. Heterotrophy in plants is associated with genomic restructuring, where a trend of plastome contraction and nuclear genome expansion via rampant repetitive element accumulation has commonly been observed (Barrett et al., 2014; Lyko and Wicke, 2021). Dramatic reductions of plastome length and gene content of mycoheterotrophic plants have been documented, ranging from minimal degradation in early transitional orchids such as Corallorhiza (Barrett and Davis, 2012) to pronounced degradation in late-transitional orchids such as Epipogium and Pogoniopsis (Schelkunov et al., 2015; Klimpert et al., 2022). Our finding of putatively functional WHY1, but the putative presence of increasingly alternatively spliced WHY1 isoforms across the Corallorhiza trophic gradient is not necessarily surprising, given the minimally destabilized plastomes of the group. For example, Barrett and Davis (2012) found that the plastome of C. striata, the most destabilized of the Corallorhiza species included here, is only about 6% reduced relative to that of a leafy, autotrophic relative. Despite their relatively intact states, Corallorhiza plastomes are in various stages of degradation (Barrett et al., 2014), and our work here together suggests a negative correlation between both increased putatively aberrant splicing and nucleotide-level divergence of WHY1 with plastome stability across the sampled species. Likewise, the plastomes of Gastrodia elata and Epipogium aphyllum, both late-stage mycoheterotrophs for which our analyses show that WHY1 contains premature stop codons and significant signal of relaxed selection, are both extremely reduced and syntenically disrupted (Yuan et al., 2018; Chen et al., 2020; Xu et al., 2021). Taken together, our findings provide the first evidence of a potential negative correlation between increased divergence in sequence, splicing of WHY1, and plastome stability in early to late-stage mycoheterotrophic orchids.
5 Conclusions
Our work showcases the rich opportunities afforded by mycoheterotrophic plants not just for the study of the evolution of WHY1 but for any gene of which homozygous knockouts can result in a fatal phenotype in autotrophic plants. Continued investigation of non-autotrophic plant lineages promises to fill gaps in our understanding of the precursors and consequences of genomic instability, and even the minimum gene space of land plants. Here we presented findings of non-synonymous nucleotide substitutions in functionally annotated regions in Corallorhiza WHY1 sequence, and a high degree of divergence in WHY1 in late-stage fully mycoheterotrophic orchids. However, our results together suggest that changes to the expression and splicing of WHY1 may occur prior to the establishment of obviously deleterious genomic substitutions that would render the TF non-functional in late stage mycoheterotrophic orchids. In sum, our work characterizes WHY1 variation and evolution throughout the angiosperms and serves as the first evidence of a potential correlation between decreased expression and increased alternative splicing of WHY1 concomitant with plastome degradation in a group of early transitional mycoheterotrophic orchids. However, our results do not implicate divergent WHY1 function as a primary factor in the transition from partial to full mycoheterotrophy. Future work documenting differential non-canonical splicing and tissue-level expression of WHY1 in vivo are necessary to fully confirm the results of our transcriptomic analyses.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Bioproject accession number: PRJNA984634.
Author contributions
BS and CB contributed to the conception and design of the study. CB collected Corallorhiza samples for RNA-seq. RM and BS collected all publicly available samples, conducted wet-lab work, Nanopore sequencing, and bioinformatic analyses. RM wrote the first manuscript draft. All authors contributed to the article and approved the submitted version.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding was provided by the Undergraduate Student Research Award and the Bert and Jane Horn Endowed Student Research Award from Otterbein University to RM; US National Science Foundation (DEB#0830020), the California State University Program for Research and Education in Biotechnology, and the West Virginia University Program to Stimulate Competitive Research to CB, and a Faculty Scholarship and Development Fund Award from Otterbein University to BS.
Acknowledgments
We would like to acknowledge John V. Freudenstein for supplying DNAs used for Nanopore sequencing, the West Virginia University Genomics Core Facility for their services and technical expertise. We also thank the USDA Forest Service and California State Parks for permission to collect material.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1241515/full#supplementary-material
References
Barrett, C. F., Davis, J. I. (2012). The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. Am. J. Bot. 99, 1513–1523. doi: 10.3732/ajb.1200256
Barrett, C. F., Freudenstein, J. V. (2008). Molecular evolution of rbcL in the mycoheterotrophic coralroot orchids (Corallorhiza Gagnebin, Orchidaceae). Mol. Phylogenet. Evol. 47, 665–679. doi: 10.1016/j.ympev.2008.02.014
Barrett, C. F., Freudenstein, J. V., Lee Taylor, D., Kõljalg, U. (2010). Rangewide analysis of fungal associations in the fully mycoheterotrophic Corallorhiza striata complex (Orchidaceae) reveals extreme specificity on ectomycorrhizal Tomentella (Thelephoraceae) across North America. Am. J. Bot. 97, 628–643. doi: 10.3732/ajb.0900230
Barrett, C. F., Freudenstein, J. V., Li, J., Mayfield-Jones, D. R., Perez, L., Pires, J. C., et al. (2014). Investigating the path of plastid genome degradation in an early transitional clade of heterotrophic orchids, and implications for heterotrophic angiosperms. Mol. Biol. Evol. 31, 3095–3112. doi: 10.1093/molbev/msu252
Barrett, C. F., Pace, M. C., Corbett, C. W., Kennedy, A. H., Thixton-Nolan, H. L., Freudenstein, J. V. (2024). Organellar phylogenomics at the epidendroid orchid base, with a focus on the mycoheterotrophic Wullschlaegelia. Ann. Bot. doi: 10.1093/aob/mcae084
Barrett, C. F., Sinn, B. T., Kennedy, A. H. (2019). Unprecedented parallel photosynthetic losses in a heterotrophic orchid genus. Mol. Biol. Evol. 36, 1884–1902. doi: 10.1093/molbev/msz111
Barrett, C. F., Wicke, S., Sass, C. (2018). Dense infraspecific sampling reveals rapid and independent trajectories of plastome degradation in a heterotrophic orchid complex. New Phytol. 218, 1192–1204. doi: 10.1111/nph.15072
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illuminasequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bruce, B. D. (2001). The paradox of plastid transit peptides: conservation of function despitedivergence in primary structure. Biochim. Biophys. Acta 1541, 2–21. doi: 10.1016/S0167-4889(01)00149-5
Bushnell, B. B. (2022) bbtools. Available online at: https://sourceforge.net/projects/bbmap/files.
Cai, L. (2023). Rethinking convergence in plant parasitism through the lens of molecular and population genetic processes. Am. J. Bot. 110 (5), e16174. doi: 10.1002/ajb2.16174
Cai, J., Liu, X., Vanneste, K., Proost, S., Tsai, W.-C., Liu, K.-W., et al. (2015). The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 47, 65–72. doi: 10.1038/ng.3149
Cameron, D. D., Preiss, K., Gebauer, G., Read, D. J. (2009). The chlorophyll-containingorchid Corallorhiza trifida derives little carbon through photosynthesis. New Phytol. 183, 358–364. doi: 10.1111/j.1469-8137.2009.02853.x
Cappadocia, L., Maréchal, A., Parent, J.-S., Lepage, É. (2010). Crystal structures of DNA Whirly complexes and their role in Arabidopsis organelle genome repair. Plant Cell 22, 1849–1867. doi: 10.1105/tpc.109.071399
Cappadocia, L., Parent, J.-S., Sygusch, J., Brisson, N. (2013). A family portrait: structural comparison of the Whirly proteins from Arabidopsis thaliana and Solanum tuberosum. Acta Crystallographica Section F69, 1207–1211. doi: 10.1107%2FS1744309113028698
Chamala, S., Feng, G., Chavarro, C., Barbazuk, W. B. (2015). Genome-wide identification of evolutionarily conserved alternative splicing events in flowering plants. Front. Bioengineering Biotechnol. 3, 33. doi: 10.3389/fbioe.2015.00033
Chao, Y.-T., Yen, S.-H., Yeh, J.-H., Chen, W.-C., Shih, M.-C. (2017). Orchidstra 2.0 – a transcriptomics resource for the orchid family. Plant Cell Physiol. 58, e9. doi: 10.1093/pcp/pcw220
Chen, Y.-Y., Li, C.-I., Hsiao, Y.-Y., Ho, S.-Y., Zhang, Z.-B., Liao, C.-C., et al. (2022). OrchidBase 5.0: updates of the orchid genome knowledgebase. BMC Plant Biol. 22, 557. doi: 10.1186/s12870-022-03955-5
Chen, S., Wang, X., Wang, Y., Zhang, G., Song, W., Dong, X., et al. (2020). Improved de novo assembly of the achlorophyllous orchid Gastrodia elata. Front. Genet. 11, 580568. doi: 10.3389/fgene.2020.580568
Chernomor, O., von Haeseler, A., Minh, B. Q. (2016). Terrace aware data structure for phylogenomic inference from supermatrices. Systematic Biol. 65, 997–1008. doi: 10.1093/sysbio/syw037
Christian, R. W., Hewitt, S. L., Nelson, G., Roalson, E. H., Dhingra, A. (2020). Plastid transit peptides—where do they come from and where do they all belong? Multi-genome and pan-genomic assessment of chloroplast transit peptide evolution. PeerJ 8, e9772. doi: 10.7717/peerj.9772
de Mendoza, A., Sebé-Pedrós, A., Ŝestak, M. S., Matejčić, M., Torruella, G., Domazet-Lošo, T., et al. (2013). Transcription factor evolution in eukaryotes and the assembly of the regulatory toolkit in multicellular lineages. Proc. Natl. Acad. Sci. United States America 110, E4858–E4866. doi: 10.1073/pnas.1311818110
De Smet, R., Adams, K. L., Vandepoele, K., Van Montagu, M. C. E., Maere, S., Van de Peer, Y. (2013). Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc. Acad. Sci. United States America 110, 2898–2903. doi: 10.1073/pnas.1300127110
Desveaux, D., Allard, J., Brisson, N., Sygusch, J. (2002). A new family of plant transcriptionfactors displays a novel ssDNA-binding surface. Nat. Struct. Mol. Biol. 9, 512–517. doi: 10.1038/nsb814
Desveaux, D., Després, C., Joyeux, A., Subramaniam, R., Brisson, N. (2000). PBF-2 is a novel single-stranded DNA binding factor implicated in PR-10a gene activation in potato. Plant Cell 12, 1477–1489. doi: 10.1105/tpc.12.8.1477
Desveaux, D., Maréchal, A., Brisson, N. (2005). Whirly transcription favors: defense gene regulation and beyond. Trends Plant Sci. 10, 1360–1385. doi: 10.1016/j.tplants.2004.12.008
Desveaux, D., Subramaniam, R., Després, C., Mess, J.-N., Lévesque, C., Fobert, P. R., et al. (2004). A “whirly” transcription factor is required for salicylic acid dependent disease resistance in Arabidopsis. Dev. Cell 6, 229–240. doi: 10.1016/S1534-5807(04)00028-0
Doyle, J. J., Davis, J. I., Soreng, R. J., Garvin, D., Anderson, M. J. (1992). Chloroplast DNA inversions and the origin of the grass family. Proc. Natl. Acad. Sci. United States America 89, 7722–7726. doi: 10.1073%2Fpnas.89.16.7722
Doyle, J. J., Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bull. 19, 11–15.
Duarte, J. M., Wall, P. K., Edger, P. P., Landherr, L. L., Ma, H., Pires, J. C., et al. (2010). Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evolutionary Biol. 10, 61. doi: 10.1186/1471-2148-10-61
Eddy, S. R. (2011). Accelerated profile HMM searches. PloS Comput. Biol. 7, e1002195. doi: 10.1371/journal.pcbi.1002195
Emms, D. M., Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. doi: 10.1186/s13059-015-0721-2
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Felsenstein, J. (1985). Phylogenies and the comparative method. Am. Nat. 125, 1–15. doi: 10.1086/284325
Filichkin, S. A., Hamilton, M., Dharmawardhana, P. D., Singh, S. K., Sullivan, C., Ben-Hur, A., et al. (2018). Abiotic stresses modulate landscape of Popular transcriptome via alternative splicing, differential intron retention, and isoform ratio switching. Front. Plant Sci. 9, 5. doi: 10.3389/fpls.2018.00005
Foyer, C. H., Karpinska, B., Krupinska, K. (2014). The functions of WHIRLY1 and REDOXRESPONSIVE TRANSCRIPTION FACTOR 1 in cross tolerance responses in plants: a hypothesis. Philos. Trans. R. Soc. B 369, 20130226. doi: 10.1098%2Frstb.2013.0226
Freudenstein, J. V., Barrett, C. F. (2014). Fungal host utilization helps circumscribe leafless Coralroot orchid species: an integrative analysis of Corallorhiza odontorhiza and C. wisteriana. Taxon 63 (4), 759–772. doi: 10.12705/634.3
Givnish, T. J., Spali nk, D., Ames, M., Lyon, S. P., Hunter, S. J., Zuluaga, A., et al. (2016). Orchid historical biogeography, diversification, Antarctica and the paradox of orchid dispersal. J. Biogeography 43, 1905–1916. doi: 10.1111/jbi.12854
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Grabowski, E., Miao, Y., Mulisch, M., Krupinska, K. (2008). Single-stranded DNA-binding protein Whirly1 in barley leaves is located in plastids and the nucleus of the same cell. Plant Physiol. 147, 1800–1804. doi: 10.1104/pp.108.122796
Graham, S. W., Lam, V. K. Y., Merckx, V. S. F. T. (2017). Plastomes on the edge: the evolutionary breakdown of mycoheterotroph plastid genomes. New Phytol. 214, 48–55. doi: 10.1111/nph.14398
Guo, X., Fang, D., Sahu, S. K., Yang, S., Guang, X., Folk, R., et al. (2021). Chloranthus genome provides insights into the early diversification of angiosperms. Nat. Commun. 12, 6930. doi: 10.1038/s41467-021-26922-4
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., Vinh, L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Isemer, R., Mulisch, M., Schäfer, A., Kirchner, S., Koop, H.-U., Krupinksa, K. (2012). Recombinant Whirly1 translocates from transplastomic chloroplasts to the nucleus. Fed. Eur. Biochem. Societies Lett. 586, 85–88. doi: 10.1016/j.febslet.2011.11.029
Jabre, I., Reddy, A. S. N., Kalyna, M., Chaudhary, S., Khokhar, W., Byrne, L. J., et al. (2019). Does co-transcriptional regulation of alternative splicing mediate plant stress responses? Nucleic Acids Res. 47, 2716–2726. doi: 10.1093/nar/gkz121
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., Misawa, K., Kuma, K., Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Klepikova, A. V., Kasianov, A. S., Gerasimov, E. S., Logacheva, M. D., Penin, A. A. (2016). A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 88, 1058–1070. doi: 10.1111/tpj.13312
Klimpert, N. J., Mayer, J. L. S., Sarzi, D. S., Prosdocimi, F., Pinheiro, F., Graham, S. W. (2022). Phylogenomics and plastome evolution of a Brazilian mycoheterotrophic orchid, Pogoniopsis schenckii. Am. J. Bot. 109, 2030–2050. doi: 10.1002/ajb2.16084
Krause, K., Kilbienski, I., Mulisch, M., Rödiger, A., Schäfer, A., Krupinksa, K. (2005). DNA-binding proteins of the Whirly family in Arabidiopsis thaliana are targeted to the organelles. Fed. Eur. Biochem. Societies Lett. 579, 3707–3712. doi: 10.1016/j.febslet.2005.05.059
Lai, X., Chahtane, H., Martin-Arevalillo, R., Zubita, C., Parcy, F. (2020). Contrasted evolutionary trajectories of plant transcription factors. Curr. Opin. Plant Biol. 54, 101–107. doi: 10.1016/j.pbi.2020.03.002
Lallemand, F., Martin-Magniette, M.-L., Gilard, F., Gakière, B., Launay-Avon, A., Delannoy, É., et al. (2019). In situ transcriptomic and metabolomic study of the loss of photosynthesis in the leaves of mixotrophic plants exploiting fungi. Plant J. 98, 826–841. doi: 10.1111/tpj.14276
Leake, J. R. (1994). The biology of myco-heterotrophic (“saprophytic”) plants. New Phytol. 127, 171–216. doi: 10.1111/j.1469-8137.1994.tb04272.x
Lee, C., Ruhlman, T., Jansen, R. K. (2020). Unprecedented intraindividual structural heteroplasmy in Eleocharis (Cyperaceae, Poales) Plastomes. Genome Biol. Evol. 12, 641–655. doi: 10.1093/gbe/evaa076
Lehti-Shiu, M. D., Panchy, N., Wang, P., Uygun, S., Shiu, S.-H. (2017). Diversity, expansion, and evolutionary novelty of plant DNA-binding transcription factor families. Biochim. Biophys. Acta 1860, 3–20. doi: 10.1016/j.bbagrm.2016.08.005
Lemaire, B., Huysmans, S., Smets, E., Merckx, V. S. F. T. (2010). Rate accelerations in nuclear 18S rDNA of mycoheterotrophic and parasitic angiosperms. J. Plant Res. 124, 561–576. doi: 10.1007/s10265-010-0395-5
Lepage, É., Zampini, É., Brisson, N. (2013). Plastid genome instability leads to reactive oxygen species production and plastid-to-nucleus retrograde signaling in Arabidopsis. Plant Physiol. 163, 867–881. doi: 10.1104/pp.113.223560
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, Y. X., Li, Z. H., Schuiteman, A., Chase, M. W., Li, J.-W., Huang, W. C., et al. (2019). Phylogenomics of Orchidaceae based on plastid and mitochondrial genomes. Mol. Phylogenet. Evol. 139, 106540. doi: 10.1016/j.ympev.2019.106540
Lin, W., Huang, D., Shi, X., Deng, B., Ren, Y., Lin, W., et al. (2019). H2O2 as a feedback signal on dual-located WHIRLY1 associates with leaf senescence in Arabidopsis. Cells 8 (12), 1585. doi: 10.3390/cells8121585
Lin, W., Zhang, H., Huang, D., Schenke, D., Cai, D., Wu, B., et al. (2020). Dual-localized WHIRLY1 affects alicyclic acid biosynthesis via coordination of ISOCHORISMATE SYNTHASE1, PHENYLALANINE AMMONIA LYASE1, and S-ADENOSYL-L-METHIONINE-DEPENDENT METHYLTRANSFERASE1. Plant Physiol. 1840, 1884–1899. doi: 10.1104/pp.20.00964
Liu, Jung, J. C., Xu, J., Wang, H., Deng, S., Bernad, L., et al. (2012). Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 24, 4333–4345. doi: 10.1105/tpc.112.102855
Lyko, P., Wicke, S. (2021). Genomic reconfiguration in parasitic plants involves considerable gene losses alongside global genome size inflation and gene births. Plant Physiol. 186, 1412–1423. doi: 10.1093/plphys/kiab192
Maddison, W. P. (1997). Gene trees in species trees. Systematic Biol. 46, 523–536. doi: 10.1093/sysbio/46.3.523
Maréchal, A., Parent, J. S., Véronneau-Lafortune, F., Joyeux, A., Lang, B. F., Brisson, N. (2009). Whirly proteins maintain plastid genome stability in Arabidopsis. Proc. Natl. Acad. Sci. United States America 106, 14693–14698. doi: 10.1073/pnas.0901710106
Merckx, V. S. F. T., Freudenstein, J. V. (2010). Evolution of mycoheterotrophy in plants: a phylogenetic perspective. New Phytol. 185, 605–609. doi: 10.1111/j.1469-8137.2009.03155.x
Merckx, V. S. F. T., Merckx, V. S. F. T. (2013). “Mycoheterotrophy: an introduction,” in Mycoheterotrophy: the biology of plants living on fungi, 1st ed (Springer, NY). doi: 10.1007/978-1-4614-5209-6
Mergner, J., Fresno, M., List, M., Papacek, M., Chen, X., Chaudhary, A., et al. (2020). Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 579, 409–414. doi: 10.1038/s41586-020-2094-2
Münster, T., Pahnke, J., Di Rosa, A., Kim, J. T., Martin, W., Saedler, H., et al. (1997). Floral homeotic genes were recruited from homologous MADS-box genes preexisting in the common ancestor of ferns and seed plants. Proc. Natl. Acad. United States America 94, 3415–2420. doi: 10.1073%2Fpnas.94.6.2415
Murrell, B., Moola, S., Mabona, A., Weighill, T., Sheward, D., Pond, S. L. K., et al. (2013). FUBAR: A Fast, Unconstrained Bayesian AppRoximation for inferring selection. Mol. Biol. Evol. 30), 1196–1205. doi: 10.1093/molbev/mst030
Murrell, B., Weaver, S., Smith, M. D., Wertheim, J. O., Murrell, S., Aylward, A., et al. (2015). Gene-wide identification of episodic selection. Mol. Biol. Evol. 32, 1365–1371. doi: 10.1093/molbev/msv035
Ner-Gaon, H., Halachmi, R., Savaldi-Goldstein, S., Rubin, E., Ophir, R., Fluhr, R. (2004). Intronretention is a major phenomenon in alternative splicing in Arabidopsis. Plant J. 39, 877–885. doi: 10.1111/j.1365-313x.2004.02172.x
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Pamilo, P., Nei, M. (1988). Relationships between gene trees and species trees. Mol. Biol. Evol. 5, 568–583. doi: 10.1093/oxfordjournals.molbev.a040517
Parent, J.-S., Lepage, E., Brisson, N. (2011). Divergent roles for the two polI-like organelle DNA polymerases of Arabidopsis. Plant Physiol. 156, 254–262. doi: 10.1104/pp.111.173849
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Patron, N. J., Waller, R. F. (2007). Transit peptide diversity and divergence: a global analysis of plastid targeting signals. BioEssays 29, 1048–1058. doi: 10.1002/bies.20638
Pérez-Escobar, O. A., Bogarín, D., Przelomska, N. A. S., Ackerman, J. D., Balbuena, J. A., Bellot, S., et al. (2024). The origin and speciation of orchids. New Phytol. 242, 700–716. doi: 10.1111/nph.19580
Pérez-Escobar, O. A., Dodsworth, S., Bogarín, D., Bellot, S., Balbuena, J. A., Schely, R. J., et al. (2021). Hundreds of nuclear and plastid loci yield novel insights into orchid relationships. Am. J. Bot. 108, 1166–1180. doi: 10.1002/ajb2.1702
Pond, S. L. K., Muse, S. V. (2005). “HyPhy: hypothesis testing using phylogenies,” in Statistical methods in molecular evolution, 1st ed. Ed. Nielsen, R. (Springer, NY).
Pond, S. L. K., Posada, D., Gravenor, M. B., Woelk, C. H., Frost, S. D. W. (2006). GARD: a genetic algorithm for recombination detection. Bioinformatics 22, 3096–3098. doi: 10.1093/bioinformatics/btl474
Prikryl, J., Watkins, K. P., Friso, G., van Wijk, K. J., Barkan, A. (2008). A member of the Whirly family is a multifunctional RNA- and DNA-binding protein that is essential for chloroplast biogenesis. Nucleic Acids Res. 36, 5152–5165. doi: 10.1093/nar/gkn492
Qiu, Z., Chen, D., Teng, L., Guan., P., Yu, G., Zhang, P., et al. (2022). OsWHY1 interacts with OsTRXz and is essential for early chloroplast development in rice. Rice 15, 50. doi: 10.1186/s12284-022-00596-y
Ranwez, V., Harispe, S., Delsuc, F., Douzery, E. J. P. (2011). MACSE: multiple alignment ofcoding sequences accounting for frameshifts and stop codons. PloS One 6, e22594. doi: 10.1371/journal.pone.0022594
Rasmussen, H. N. (1995). Terrestrial orchids: from seed to mycotrophic plant (Cambridge: Cambridge University Press). doi: 10.1017/CBO9780511525452
Rasmussen, H. N. (2002). Recent developments in the study of orchid mycorrhiza. Plant Soil 244, 149–163. doi: 10.1023/A:1020246715436
R Core Team (2019). R: A language and environment for statistical computing (Vienna, Austria: The R Foundation). Available at: https://www.R-project.org/.
Reddy, A. S., Marquez, Y., Kalyna, M., Barta, A. (2013). Complexity of the alternative splicing landscape in plants. Plant Cell 25, 3657–3683. doi: 10.1105/tpc.113.117523
Ren, Y., Li, Y., Jiang, Y., Wu, B., Miao, Y. (2017). Phosphorylation of WHIRLY1 by CIPK14 shifts its localization and dual functions in Arabidopsis. Mol. Plant 10, 749–763. doi: 10.1016/j.molp.2017.03.011
Robinson, M. D., McCarthy, D. J., Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Robinson, M. D., Oshlack, A. (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. doi: 10.1186/gb-2010-11-3-r25
Ruan, Q., Wang, Y., Xu, H., Wang, B., Zhu, X., Wei, B., et al. (2022). Genome-wide identification, phylogenetic, and expression analysis under abiotic stress conditions of Whirly (WHY) gene family in Medicago sativa L. Sci. Rep. 12, 18676. doi: 10.1038/s41598-022-22658-3
Schelkunov, M. I., Shtratnikova, V. Y., Nuraliev, M. S., Selosse, M.-A., Penin, A. A., D.Logacheva, M. (2015). Exploring the limits for reduction of plastid genomes: a case study of the mycoheterotrophic orchids Epipogium aphyllum and Epipogium roseum. Genome Biol. Evol. 7, 1179–1191. doi: 10.1093/gbe/evv019
Serna-Sánchez, M. A., Pérez-Escobar, O. A., Bogarın, D., Torres-Jimenez, F. F., Alvarez-Yela, A. C., Arcila-Galvis, J. E., et al. (2021). Plastid phylogenomics resolves ambiguous relationships within the orchid family and provides a solid timeframe for biogeography and macroevolution. Sci. Rep. 11, 6858. doi: 10.1038/s41598-021-83664-5
Sinn, B. T., Barrett, C. F. (2020). Ancient mitochondrial gene transfer between fungi and theorchids. Mol. Biol. Evol. 37, 44–57. doi: 10.1093/molbev/msz198
Suetsugu, K., Yamato, M., Miura, C., Yamaguchi, K., Takahashi, K., Ida, Y., et al. (2017). Comparison of green and albino individuals of the partially mycoheterotrophic orchid Epipactis helleborine on molecular identities of mycorrhizal fungi, nutritional modes and gene expression in mycorrhizal roots. Mol. Ecol. 26, 1652–1669. doi: 10.1111/mec.14021
Sun, S., Li, S., Zhou, X., Yang, X. (2023). WRKY1 represses the WHIRLY1 transcription factor to positively regulate plant defense against geminivirus infection. PloS Pathog. 19, e1011319. doi: 10.1371/journal.ppat.1011319
Taylor, D. L., Bruns, T. D. (1997). Independent, specialized invasions of ectomycorrhizal mutualism by two nonphotosynthetic orchids. Proc. Natl. Acad. United States America. 94, 4510–4515. doi: 10.1073/pnas.94.9.4510
Taylor, R. E., West, C. E., Foyer, C. H. (2022). WHIRLY protein functions in plants. Food Energy Secur. 00, e379. doi: 10.1002/fes3.379
The Angiosperm Phylogeny Group, Chase, M. W., Christenhusz, M. J. M., Fay, M. F., Byng, J. W., Judd, W. S., et al. (2016). An update of the Angiosperm Phylogeny Classification for the orders and families of flowering plants: APG IV. Botanical J. Linn. Society. 181, 1–20. doi: 10.1111/boj.12385
The Potato Genome Sequencing Consortium (2011). Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195. doi: 10.1038/nature10158
Timilsena, P. R., Barrett, C. F., Piñeyro-Nelson, A., Wafula, E. K., Ayyampalayam, S., McNeal, J. R., et al. (2023). Phylotranscriptomic analyses of mycoheterotrophic monocots show a continuum of convergent evolutionary changes in expressed nuclear genes from three independent nonphotosynthetic lineages. Genome Biol. Evol. 15, evac183. doi: 10.1093/gbe/evac183
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3-new capabilities and interfaces. Nucleic Acids Res. 40, e115. doi: 10.1093/nar/gks596
Waterworth, W. M., Drury, G. E., Bray, C. M., West, C. E. (2011). Repairing breaks in the plant genome: the importance of keeping it together. New Phytol. 192, 805–822. doi: 10.1111/j.1469-8137.2011.03926.x
Wertheim, J. O., Murrell, B., Smith, M. D., Pond, S. L. K., Scheffler, K. (2015). RELAX:detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 32), 820–832. doi: 10.1093/molbev/msu400
Wheeler, T. J., Eddy, S. R. (2013). nhmmer: DNA homology search with profile HMMs. Bioinformatics 29 (19), 2487–2489. doi: 10.1093/bioinformatics/btt403
Wicke, S., Müller, K. F., dePamphilis, C. W., Quandt, D., Bellot, S., Schneeweiss, G. M. (2016). Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proc. Natl. Acad. Sci. United States America 113, 9045–9050. doi: 10.1073/pnas.1607576113
Wu, Mueller, F. L. A., Crouzillat, D., Pétiard, V., Tanksley, S. D. (2006). Combining bioinformatics and phylogenetics to identify large sets of single-copy orthologous genes (COSII) for comparative, evolutionary, and systematic studies: a test case in the euasterid plant clade. Genetics 174, 1407–1420. doi: 10.1534/genetics.106.062455
Xu, Y., Lei, Y., Su, Z., Zhao, M., Zhang, J., Shen, G., et al. (2021). A chromosome-scale Gastrodia elata genome and large-scale comparative genomic analysis indicate convergent evolution by gene loss in mycoheterotrophic and parasitic plants. Plant J. 108, 1609–1623. doi: 10.1111/tpj.15528
Yoo, H. H., Kwon, C., Lee, M. M., Chung, I. K. (2007). Single-stranded DNA binding factor AtWHY1 modulates telomere length homeostasis in Arabidopsis. Plant J. 49, 442–451. doi: 10.1111/j.1365-313X.2006.02974.x
Yuan, Y., Jin, X., Liu, J., Zhao, X., Zhou, J., Wang, X., et al. (2018). The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 9, 1615. doi: 10.1038/s41467-018-03423-5
Zampini, É., Lepage, É., Tremblay-Belzile, S., Truche, S., Brisson, N. (2015). Organelle DNA rearrangement mapping reveals U-turn-like inversions as a major source of genomic instability in Arabidopsis and humans. Genome Res. 25, 645–654. doi: 10.1101/gr.188573.114
Zeng, X., Li, Y., Ling, H., Liu, S., Liu, M., Chen, J., et al. (2017). Transcriptomic analyses reveal clathrin-mediated endocytosis involved in symbiotic seed germination of Gastrodia elata. Botanical Stud. 58, 31. doi: 10.1186/s40529-017-0185-7
Zhang, G., Hu, Y., Huang, M.-Z., Huang, W.-C., Liu, D.-K., Zhang, D., et al. (2023). Comprehensive phylogenetic analyses of Orchidaceae using nuclear genes and evolutionary insights into epiphytism. J. Integr. Plant Biol. 65, 1204–1225. doi: 10.1111/jipb.13462
Zhang, G.-Q., Liu, K.-W., Lohaus, R., Hsiao, Y.-Y., Niu, S.-C., Wang, J.-Y., et al. (2017). The Apostasia genome and the evolution of orchids. Nature 549 (7672), 379–383. doi: 10.1038/nature23897
Zhang, G.-Q., Xu, Q., Bian, C., Tsai, W.-C., Yeh, C.-M., Liu, K.-W., et al. (2016). The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 6, 19029. doi: 10.1038/srep19029
Zhao, S. Y., Wang, G. D., Zhao, W. Y., Zhang, S., Kong, F. Y., Dong, X. C., et al. (2018). Overexpression of tomato WHIRLY protein enhances tolerance to drought stress and resistance to Pseudomonas solanacearum in transgenic tobacco. Biol. Plantarum 62, 55–68. doi: 10.1007/s10535-017-0714-y
Zhao, T., Zwaenepoel, A., Xue, J.-Y., Kao, S.-M., Li, Z., Schranz, M. E., et al. (2021). Whole-genome microsynteny-based phylogeny of angiosperms. Nat. Commun. 12, 3498. doi: 10.1038/s41467-021-23665-0
Zhuang, K., Kong, F., Zhang, S., Meng, C., Yang, M., Liu, Z., et al. (2019). Whirly1 enhances tolerance to chilling stress in tomato via protection of photosystem II and regulation of starch degradation. New Phytol. 221, 1998–2012. doi: 10.1111/nph.15532
Zhuang, K., Wang, J., Jiao, B., Chen, C., Zhang, J., Ma, N., et al. (2020). WHIRLY1 maintains leaf photosynthetic capacity in tomato by regulating the expression of RbcS1 under chilling stress. J. Exp. Bot. 12, 3653–3663. doi: 10.1093/jxb/eraa145
Keywords: mycoheterotrophy, Corallorhiza, orchid, genomic stability, intron retention, transcription factor, plastome evolution, Whirly1
Citation: Muti RM, Barrett CF and Sinn BT (2024) Evolution of Whirly1 in the angiosperms: sequence, splicing, and expression in a clade of early transitional mycoheterotrophic orchids. Front. Plant Sci. 15:1241515. doi: 10.3389/fpls.2024.1241515
Received: 16 June 2023; Accepted: 07 June 2024;
Published: 28 June 2024.
Edited by:
Oscar Alejandro Pérez-Escobar, Royal Botanic Gardens, Kew, United KingdomReviewed by:
Dewi Pramanik, National Research and Innovation Agency (BRIN), IndonesiaKatharina Nargar, Commonwealth Scientific and Industrial Research Organisation (CSIRO), Australia
Copyright © 2024 Muti, Barrett and Sinn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brandon T. Sinn, c2lubjFAb3R0ZXJiZWluLmVkdQ==