 Lucio D’Andrea1
Lucio D’Andrea1 Nicolas Sierro2
Nicolas Sierro2 Sonia Ouadi2
Sonia Ouadi2 Tomas Hasing3
Tomas Hasing3 Elijah Rinaldi4
Elijah Rinaldi4 Nikolai V. Ivanov2
Nikolai V. Ivanov2 Aureliano Bombarely1,5*
Aureliano Bombarely1,5*- 1Department of Bioscience, Universita degli Studi di Milano, Milan, Italy
- 2PMI R&D, Philip Morris Products S.A., Quai Jeanrenaud 5, Neuchâtel, Switzerland
- 3Elo Life Systems, Durham, NC, United States
- 4School of Plant and Environmental Sciences, Virginia Tech, Blacksburg, VA, United States
- 5Instituto de Biologia Molecular y Celular de Plantas (IBMCP) (CSIC-UPV), Valencia, Spain
Introduction: Nicotiana section Suaveolentes is an almost all-Australian clade of allopolyploid tobacco species that emerged through hybridization between diploid relatives of the genus. In this study, we aimed to assess the phylogenetic relationship of the Suaveolentes section with several Nicotiana diploid species based on both plastidial and nuclear genes.
Methods: The Nicotiana plastome-based phylogenetic analysis representing 47 newly re-built plastid genomes suggested that an ancestor of N. section Noctiflorae is the most likely maternal donor of the Suaveolentes clade. Nevertheless, we found clear evidence of plastid recombination with an ancestor from the Sylvestres clade. We analyzed 411 maximum likelihood-based phylogenetic trees from a set of conserved nuclear diploid single copy gene families following an approach that assessed the genomic origin of each homeolog.
Results: We found that Nicotiana section Suaveolentes is monophyletic with contributions from the sections Alatae, Sylvestres, Petunioides and Noctiflorae. The dating of the divergence between these sections indicates that the Suaveolentes hybridization predates the split between Alatae/Sylvestres, and Noctiflorae/Petunioides.
Discussion: We propose that Nicotiana section Suaveolentes arose from the hybridization of two ancestral species from which the Noctiflorae/Petunioides and Alatae/Sylvestres sections are derived, with Noctiflorae the maternal parent. This study is a good example in which the use of genome wide data provided additional evidence about the origin of a complex polyploid clade.
1 Introduction
Nicotiana, an herbaceous plant genus, belongs to the Solanaceae family. The genus includes 90 naturally occurring species divided into 13 sections, of which five contain allopolyploids formed by interspecific hybridization between diploids (Knapp et al., 2004; Bally et al., 2021; Chase et al., 2021; Augsten et al., 2022; Santilli et al., 2022). The species of the Nicotiana genus vary greatly in genome size and chromosome number based on their sections (Leitch et al., 2008). For example, although the haploid genome size (1C) of diploid N. obtusifolia (2n = 2x = 22) is 1.51 Gb (Leitch et al., 2008), that of the allopolyploid N. tabacum (2n = 4x = 48) is approximately 4.5 Gb. Suaveolentes is one of the oldest polyploid sections of the Nicotiana genus and includes 38 species (Chase et al., 2021), such as the well-studied Nicotiana benthamiana (Cauz-Santos et al., 2022). Molecular dating and phylogenetic analysis indicate that the Suaveolentes clade arose from a single hybridization event (circa 6 Mya) (Chase et al., 2003; Chase et al., 2018). Biogeographic surveys have revealed that the majority of Suaveolentes species exist in Australia, with a few present in other areas such as Namibia, New Caledonia, and a group of isolated islands in the Pacific Ocean (Ladiges et al., 2011). Australian Nicotiana species are widely distributed across the country and in several bioregions. This extensive distribution of this section has been attributed to a single allopolyploidization event followed by a rapid adaptation and a high speciation rate rather than features coming from its ancestors (Leitch et al., 2008; Ladiges et al., 2011; Clarkson et al., 2017; Chase et al., 2018; Dodsworth et al., 2020)
Several efforts have been made to elucidate the origin of the section Suaveolentes (Goodspeed, 1954; Clarkson et al., 2010; Kelly et al., 2013; Schiavinato et al., 2020; Schiavinato et al., 2021). The first attempts, based on morphological and/or genetic characterization, revealed that this section was derived from ancestors of the present-day sections Alatae and Sylvestres and that of the sections Petunioides and Noctiflorae (Goodspeed, 1954; Clarkson et al., 2010; Kelly et al., 2013). Recently, two articles describing phylogenetic analyses focused on the origin of N. benthamiana identified the sections Noctiflorae and Sylvestres as genome donors (Schiavinato et al., 2020; Schiavinato et al., 2021). Plastome phylogenetic analysis was conducted to verify the maternal contribution of the Suaveolentes section (Chase et al., 2003; Clarkson et al., 2004; Kelly et al., 2013; Schiavinato et al., 2020), although independent studies have provided conflicting evidence. Most of the published studies proposed section Noctiflorae/Petunioides as the most likely maternal donor (Chase et al., 2003; Clarkson et al., 2004; Kelly et al., 2013; Schiavinato et al., 2020), with one exception in which section Sylvestres is suggested as the most likely maternal ancestor using a different set of genetic markers (Clarkson et al., 2010). This inconsistency could have been due to different processes, such as incomplete lineage sorting (ILS) and/or hybridization (Kelly et al., 2013). The same study, involving maximum parsimony analysis and phylogenetic supernetworks, proposed that the recurrent gene tree discordances are more likely due to hybridization (and/or introgression) between the progenitors. Thus, Kelly et al. (Kelly et al., 2013) proposed that section Suaveolentes arose from a single allopolyploidization event involving an ancestral member of section Sylvestres and a hypothetical diploid species that contains alleles from sections Petunioides and Noctiflorae.
The complex genomic evolution of the clade, the limited number of evaluated loci per taxa, and the total number of species analyzed are likely responsible for these contradictions. Hence, to elucidate the origins of the Suaveolentes section, we performed the following analyses: (1) phylogenetic analysis based on 54 whole-plastome sequences representing all Nicotiana sections; (2) high-throughput phylogenetic analysis based on nuclear gene families that distinguish the origins of each homoelog gene in four polyploids species of the Suaveolentes section; and (3) dating of the ancestral speciation event for the sections Alatae, Noctiflorae, Petunioides, Sylvestres and Suaveolentes by two independent approaches.
Whole-plastome-based maximum-likelihood (ML) and Bayesian inference analyses (BI) assigned sections Noctiflorae and Sylvestres as respective maternal donors. This contradictory result is due to a recombination event in which the inverted repeats were derived from Sylvestres and the rest of the plastid genome of Noctiflorae. Nuclear gene-based high-throughput analysis revealed the genetic contribution of each present-day diploid species in the Suaveolentes section. Section Suaveolentes possesses nuclear gene families from sections Alatae, Noctiflorae, Petunioides, and, to a lesser extent, Sylvestres. The dating of the different diversification events suggests that the Suaveolentes section arose from the hybridization of common ancestors of section Alatae and Sylvestres, as one parent and section Noctiflorae and Petunioides as the other parent.
2 Material and methods
2.1 Plant material
Seeds for the whole-genome sequencing of 18 Nicotiana species as well as other diploid species (Supplementary Table S1) were obtained from the Agricultural Research System (ARS) Germplasm Resources Information Network (GRIN, Beltsville, MD, USA). After sowing the seeds in a standard soil, the plants were grown in a greenhouse at a 16-h light:8-h dark cycle. Leaf samples were collected from adult plants, ground in liquid nitrogen, lyophilized, and stored at room temperature until DNA extraction.
2.2 DNA extraction, library preparation, and sequencing
DNA for whole-genome sequencing of the 18 Nicotiana species (Supplementary Table S2) was extracted from aerial parts of one single plant of each species using a Qiagen DNeasy Plant Maxi Kit (Qiagen, Hilden, Germany), and the quality and quantity of the extracted DNA were verified using a Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Following fragmentation using a Covaris E220 Focused-ultrasonicator (LGC Genomics, Berlin, Germany), paired-end sequencing libraries were prepared using an Illumina TruSeq DNA Sample Preparation Kit v2 (Illumina, San Diego, CA, USA). All libraries were sequenced on an Illumina HiSeq-2500 sequencer (Illumina) using v3 chemistry and flow cells with runs of 2 × 100 bases. Each accession was sequenced with a minimum genome coverage of 40X according to the genome size previously estimated by flow cytometry (Supplementary Table S1). Except for N. hesperis, all the samples reached the sequencing goal (Supplementary Table S2).
2.3 NCBI Data mining
Illumina pair end reads were downloaded from the NCBI SRA database as follows: 1) navigating to “Nicotiana” in the Taxonomy NCBI database; 2) selecting the “SRA Experiments” box and clicking on the display button; 3) selecting the number of SRA experiments and clicking on “Send results to Run selector”; 4) filtering SRA runs by Platform = “Illumina”, Assay Type = “wgs” and “rna-seq” and Library Layout = “paired”; and 5) selecting the datasets and clicking on “Run Info Table.” Once the table was downloaded, the lines containing the species that were sequenced at the time of data mining were filtered out (“N. tabacum,” “N. benthamiana,” “N. attenuata,” “N. sylvestris,” and “N. tomentosiformis”). Then, three columns were selected: Run, Assay Type, and Organism. For the plastid genome assembly, the reads were downloaded through a bash script using the OrganelleSRABuild tool (see below). For the BUSCO nuclear gene reconstruction, the reads were downloaded with Fastq-dump v2.8.1. The mined data are summarized in the table describing the results of the plastome genome assemblies (Supplementary Table S3).
2.4 Plastid genome reference-based assembly and phylogenetic analysis
Two different methodologies were evaluated for the plastid genome assembly (see section 3.1. for the comparison of both methodologies). The de-novo plastid genome assembly was performed with the tool GetOrganelle v1.7.6.1 (Jin et al., 2020). See the reference-based approach (OrganelleSRABuild pipeline) in the next paragraph for more details. Both methodologies were compared by an alignment derived from a Smith-Waterman algorithm with the reference sequence using BlastN v2.12.0 (Camacho et al., 2009). They were also compared mapping the reads used in the assembly using BWA v0.7.17-r1188, and calling variants using FreeBayes v0.9.20-17-g5f1bc44-dirty (Garrison and Marth, 2012). Homozygous and heterozygous variants were counted with the following Linux command line: “grep -v “#” <my_variants.vcf> | cut -f8,10 | sed -r ‘s/.+=//’ | sed -r ‘s/:.+//’ | sort | uniq -c”.
The plastid genome assembly was performed using a reference-based approach. Currently, the following Nicotiana species plastome sequences are available at NCBI: N. attenuata (PRJNA412786), N. otophora (PRJNA359948), N. sylvestris (PRJNA257217), N. tabacum (PRJNA319578), N. tomentosiformis (PRJNA257218), and N. undulata (PRJNA74317). Additionally, the N. benthamiana plastome was available in-house. Briefly, we mapped the processed reads from publicly available Nicotiana sequencing experiments (SRA) against the available and phylogenetically closer Nicotiana plastome using Bowtie2 v2.1.0 (Langmead and Salzberg, 2012) and the default parameters. Mapped reads were filtered from the SAM output file using Samtools v1.5-1-g27b628e (Li et al., 2009). Then, the variants were calculated with Freebayes v0.9.20-17-g5f1bc44-dirty (Garrison and Marth, 2012). The VCF file was compressed and indexed with Bgzip v1.3.1 and Tabix v1.3.1. Before the consensus call, the regions of zero coverage were calculated using Bedtools v2.27.1 (Quinlan and Hall, 2010) and the output was filtered with the command “grep -w 0$ my_cov.bed | cut -f1,2,3 > my_cov0.bed.” The consensus sequences were calculated with Bcftools v1.3.1 (Narasimhan et al., 2016) using the command “cat N_benthamiana_Chl_reference.fasta | bcftools consensus -m my_cov0.bed my_variants.vcf.gz | sed -r ‘s/>Niben/>MySpecies/’ > MySpecies_CHLMAP.fasta.” Finally, the consensus sequence was polished using Pilon, for which the following steps were followed: the reads were remapped to the sequence consensus (MySpecies_CHLMAP.fasta) using Bowtie2 v2.1.0 and run on Pilon v1.22 (Walker et al., 2014) using the remapped reads. All these commands were wrapped in a Perl script, OrganelleSRABuild, publicly available from a Github repository (https://github.com/aubombarely/OrganelleSRABuild.)
Plastid genome sequences were aligned with Kalign v2.04 (Lassmann and Sonnhammer, 2005) using default parameters. Two phylogenetic approaches and three different programs were used to build the phylogenetic tree: 1.1) maximum likelihood (ML) using iQTree v2.1.4 (Nguyen et al., 2015) and the substitution model TVM+F+R6 with 1,000 bootstraps. The substitution model was previously generated using jModelTest2 v2.1.7 (Darriba et al., 2012). 1.2) Maximum Likelihood using iQTree v2.1.4. The number of cores was set to AUTO (-nt AUTO), allowing the program to optimize the number of cores for a long alignment. 2) Bayesian inference (BI) using BEAST v2.5.2 (Bouckaert et al., 2019) with the same substitution model as used for 1.1. Phylogenetic trees were generated with FigTree v1.4.0 (Rambaut, 2012).
2.5 De-novo transcriptome assembly
Reads were downloaded from the NCBI SRA database using Fastq-dump v2.9.6 and then processed with Fastq-mcf v1.04.676 from the Ea-utils package (Aronesty, 2011) (minimum qscore of 30 and minimum length of 50 bp). The processed reads were assembled using Trinity v2.8.5 (Haas et al., 2013) with the default parameters. The transcripts were combined in “supertranscripts” following the recommendations of the Trinity pipeline. The CDS and protein were predicted using Transdecoder v5.5.0 from the Trinity pipeline with the default values.
2.6 Reference-based gene model reconstruction
Reads from each of the Suaveolentes species were assembled using SOAPdenovo v2.04 (Luo et al., 2012) with kmer sizes of 31, 39, 47, 55, 63, 71, 79, 87, and 95. The assembly was selected based on the longest scaffold, the longest N50, and the largest assembly size. One round of gap filling was performed with GapCloser v2.04 (Luo et al., 2012) (defined as assembly v0.0.5). Scaffolds were broken into contigs again with an in-house Perl script (BreakScaffolds) and re-scaffolded using the N. benthamiana reference genome v2.6.1 and RaGOO v1.1. (Alonge et al., 2019) (defined as chimeric assembly v0.1.1). Then another round of gap filling and scaffold break was performed. The final contigs were re-scaffolded with SOAPdenovo v2.04 and a final step of gap filling was run (assembly v0.1.5). The quality of the assemblies was evaluated with BUSCO v4.1.4 (Simão et al., 2015), QUAST v5.0.2 (Gurevich et al., 2013), and Merqury v2020-01-29 (Rhie et al., 2020). Although a basic assembly was performed for all the Suaveolentes species (v0.0.5), RaGOO scaffolding was performed only for N. africana, N. amplexicaulis, N. forsteri (formerly N. debneyi, (Marks, 2010)), N. cavicola, and N. suaveolens (PI230960) due to the long computing time.
2.7 Whole-genome assembly annotation
Whole-genome assemblies were downloaded from the NCBI genome database. For the genomes without available annotations (N. glauca, N. knightiana, N. obtusifolia, N. paniculata and N. undulata), an RNA-Seq dataset was also downloaded from NCBI and processed as described in the previous section. The processed RNA-Seq reads were mapped to their corresponding references using Hisat2 v2.1.0 (Kim et al., 2015), transcript models were produced with Stringtie v1.3.3 (Pertea et al., 2015), and repeats were annotated with RepeatModeler v2.0.3 (Flynn et al., 2020) (http://www.repeatmasker.org/RepeatModeler/). A set of proteins for the species S. lycopersicum, N. sylvestris, N. tomentosiformis, and N. attenuata were downloaded from NCBI and used as closed related protein sets for gene model annotation. From the Suaveolentes species, N. africana, N. forsteri, and N. cavicola were annotated with Maker-P v2.31.10 on an Ubuntu server 3.19.0-84 with 256 Gb of RAM, 4 Tb of hard drive, and 64 threads.
2.8 Phylogenetic analysis by gene families
All the CDS sequence files were merged with a simple cat command and then gene families were built using program WGD v2018 (Zwaenepoel and Van De Peer, 2019). Specifically, the command “wgd mcl –cds –mcl -s All.CDS.fasta -o All.CDS” was run to create the gene families and then command “wgd ksd –preserve All.CDS.mcl All.CDS.fasta” was run to calculate the Ks distributions. This command kept the intermediate files, such as the alignments produced by MAFFT and the trees produced by Codeml, as part of the WGD script. Different inflation values were assessed (1.5, 2.0, 2.5, 3.0, 3.5, and 4.0) to maximize the number of clusters in which each of the diploid species have one gene and the tetraploid species have two. Alignments with at least one species of the different clades, two N. benthamiana sequences and one S. lycopersicum sequence, were selected and realigned using Guidance v2.02 (Sela et al., 2015). Then, a first run of iQTree v1.6.12 (Nguyen et al., 2015) was performed on each of the alignments with the default parameters to optimize the substitution model, after which it was run again with the optimal substitution model and 1,000 bootstraps (-bb 1000), a likelihood ratio test (-alrt 1000), and optimization of the UFBoot trees (–bnni). Guidance and iQTree were run for all the alignments using an in-house script called FromAlign2Trees.
2.9 Origin and divergence dating of Suaveolentes
Identification of the origin and polyploid event dating were performed using the phylogenetic alignments and trees derived from the gene family analysis. The code can be found at the repository: https://github.com/aubombarely/GenoToolBox/tree/master/GeneFamilies In brief, phylogenetic trees were selected based on the following requirements: 1) they should contain at least one sequence of each of the diploid species (Nicotiana as well as Solanum); 2) they should contain two sequences from N. benthamiana and at least another one of each of the other Suaveolentes species used to produce the gene families (N. africana, N. forsteri, and N. cavicola); 3) all the nodes of the phylogenetic tree produced by iQTree should have a bootstrap percentage >70. Each of the trees were analyzed using an in-house script called MultiTreeAnalyzer. This script has several functions. First, it tags each of the leaves for polyploid species (N. africana, N. benthamiana, N. cavicola, N. forsteri, and N. tabacum) with the closest diploid section for which it 1) checks that the different diploid sections are monophyletic, 2) retrieves the common ancestor node for the polyploid leaves and the diploid section, and 3) assigns a tag based on the closest diploid section. If more than one diploid section is under the same node, it will assign multiple origins (e.g., if Sylvestres and the Alatae clades have a common ancestor and this node is connected with the common recent ancestor of polyploid leaves, it will be assigned as Sylvestres, Alatae). Second, the script produces a list of ancestral nodes and the clades under these nodes. Then it counts the number of nodes normalized according to the number of leaves under each node, which represents the frequency for which specific clades are related under a multi-tree analysis.
Once each of the possible polyploid ancestors were identified, each of the gene clusters was classified according to their diploid origin into different types (e.g., Type001 were trees where Suaveolentes genes had either Alatae or Noctiflorae origins) using the ClusterClassification.txt file. Each of the gene family trees were concatenated into a multi-tree file according to their type. An Astral species mapping file was created for each of the types with clear diploid assignment using the PrepareASTRALTaxamap script and Astral v.5.7.8. (Zhang et al., 2018) was run for each of the cluster types with the default parameters. Each of the consensus trees was visualized with Figtree v1.4.4.
Plastid phylogeny dating was performed with BEAST v2.5.2 (Bouckaert et al., 2019), using the split between Solanum and Nicotiana to calibrate the tree. The gamma site model with a gamma category of 4 and HKY substitution model was selected. The gene tree model was a calibrated Yule Model with previously based on the divergence between the Nicotiana and Solanum genera. The Solanum-Nicotiana split date was estimated at 29.5 Mya based on 21 studies (www.timetree.org). Additionally, gamma distributions with Alpha = 0.001 and Beta = 1,000 were selected for the birth and clock rates. The simulation was run for a chain length of 10 million.
Polyploid event dating using nuclear genes was performed using each of the alignments of the trees classified according to their type. Alignments were converted into FASTA format to change the ID of the sequence using their corresponding species assignment (e.g., Nafr000004 was Nafr_S, Ntom000007 was Ntom) and a simple Perl command. Each of the alignments were changed back to the Phylip format and a BEAST tree phylogeny was estimated for each of the alignments in which the Suaveolentes polyploid genes had a clear diploid origin using the same parameters as for the plastid tree. Dating of the different nodes was assigned with Figtree v1.4.4, in which the root node (divergence between Solanum and Nicotiana) was annotated as 29.5 Mya. Any BA trees that disagreed with the ML trees calculated with iQTree were discarded from the analysis. The Suaveolentes ancestor and diploid relative divergence date was calculated as an average of the dating for the different nodes.
An alternative dating was performed based on the Ks distribution obtained from the WGD program (see previous section). The Ks tables were uploaded into R v4.1.2 and the distribution was modeled using function kde() from the package Ks v1.13.5 (Chacón and Duong, 2018). Peaks were found with the function which.max() for the standard R package. The phylogenetic tree produced from the Ks peaks between the different species was produced with the function upgma() from the package Phangorn v2.8.1 (Schliep, 2011) after the KS peak matrix was transformed into a distance matrix using the function as.dist() from the standard R toolbox.
3 Results
3.1 Plastid genome reconstruction pipeline.
At the time of the publication of this article there were several pipelines to reconstruct plastid genome sequences using short read data, of which GetOrganelle is the most popular one (Jin et al., 2020). We assessed this tool on our whole genome sequencing (WGS) and RNA-Seq data (Supplementary Table S3). Although, in general we obtained good results for the WGS data for the datasets with enough coverage (> 20X for the plastid genome), this pipeline failed for all the RNA-Seq data. Due to these results, we decided to develop a reference-guided plastid genome assembly approach called OrganelleSRABuild, in which the reads are aligned with a plastid reference genome and then, variants are called and used to produce a new consensus (see material and methods for more details about the pipeline implementation). The pipeline is summarized in the Figure 1.
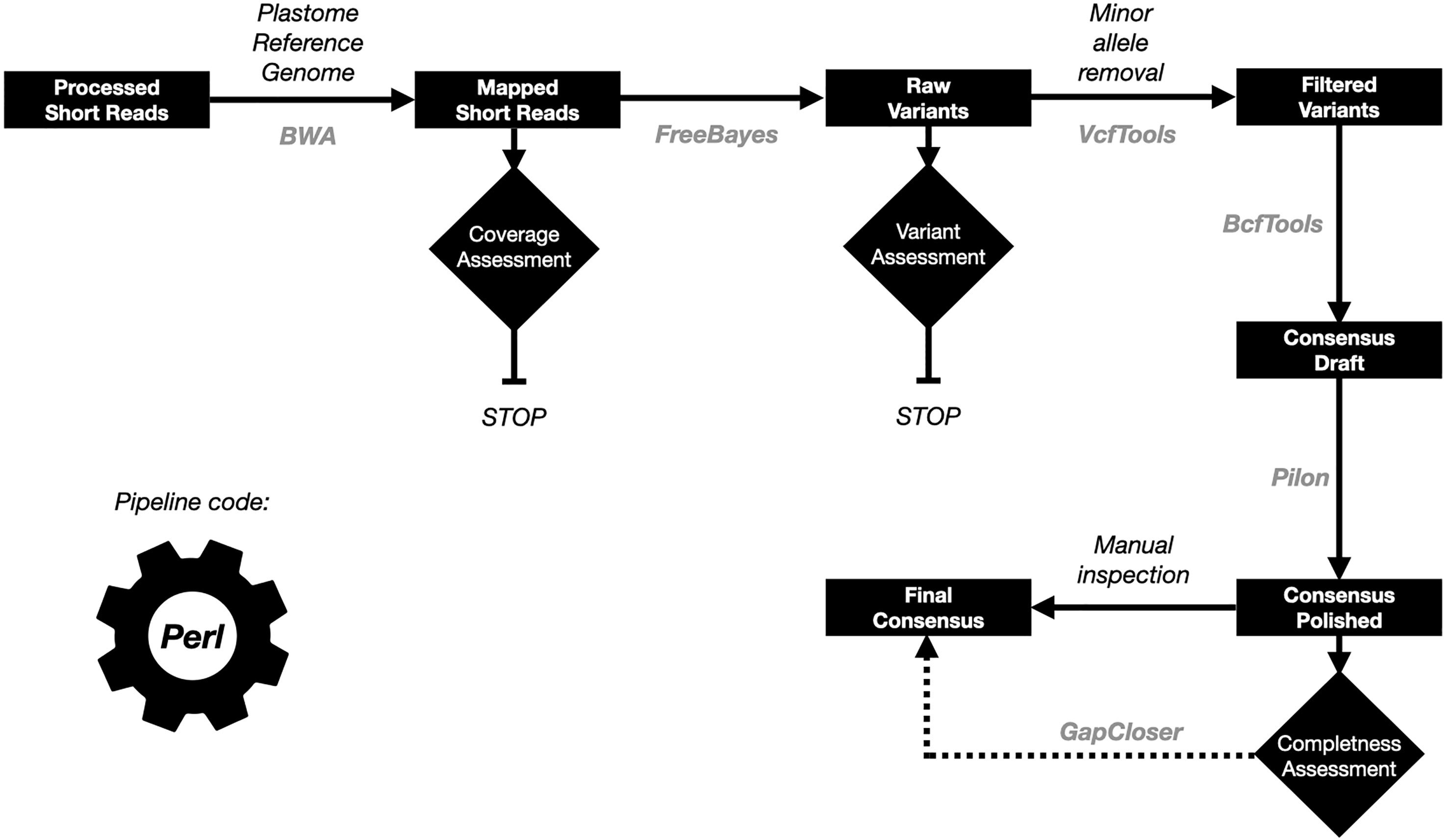
Figure 1 Summary of the plastid genome assembly pipeline. The data is represented in black boxes. The control processes as diamonds. The tools names used in each of the steps are in gray color.
The OrganelleSRABuild (OSB) pipeline was compared with the GetOrganelle (GO) to assess the accuracy in the plastome genome reconstruction. We assembled the Arabidopsis thaliana and the Nicotiana benthamiana and N. africana plastome genomes. The accuracy was evaluated through the number of homozygous variants identified after the remapping of the assembled reads. More accurate assemblies will have less homozygous variants. We considered that heterozygous variants were produced by NUPTs, especially if the alternative alleles were supported by less than 1/10 of the reads supporting the reference allele. The results are summarized in the Supplementary Table S4. Both pipelines delivered the same assemblies for the A. thaliana plastome genomes. For the N. benthamiana plastome genome assemblies, the GO pipeline produced better results with only one variant (one SNP) compared with the 15 variants (of which five were SNPs) obtained by the OSB pipeline. The use of a reference plastome genome of a species close to our target species was evaluated through the N. africana plastome genome assembly using the N. benthamiana plastome as reference. Both tools delivered similar results (eight homozygous variants for GO and 10 for OSB), although the number of homozygous SNPs were lower in the OSB assembly (seven compared with the eight of the GO assembly). Considering that insertion/deletion variants produce gaps for the multisequence alignment and that those are ignored for the phylogenetic methodologies used in this work, we decided that the OSB tool produce assemblies better tuned for a phylogenetic analysis. Additionally, GetOrganelle failed to reconstruct any of the plastome genomes derived from RNA-Seq data.
3.2 Plastid genome phylogenetic tree present different topologies for the different structural regions
We assessed maternal inheritance of the Suaveolentes section through a comparison of the whole-plastome sequences of diploid Nicotiana species from all sections. Whole-genome sequencing was performed for 18 Suaveolentes species (Supplementary Table S2). Plastome genome sequences were obtained and/or reconstructed for 62 taxa (including one outgroup, Solanum lycopersicum, and several accessions for three species: N. amplexicaulis, two N. suaveolens, three; N. forsteri, two; N. glauca, two; N. alata, two; and N. stocktonii, two), representing members of all Nicotiana clades. Plastome re-assembly was performed for 47 species using the in-house tool OrganelleSRABuild. All assembled plastomes were approximately 0.15 Mb in size. The plastome sequences were assembled without any gap except for N. petunioides (1 nt, position 155,690) and N. raimondii (1 nt, position 436), (Supplementary Table S3). None of the small gaps were in the junction between the IR and the LSC/SSC regions, so they may represent a structural variation in which the polishing methodology may have failed.
The whole-plastome alignment was comprised of 62 taxa with 161,044 columns, 4,311 distinct patterns, 3,813 parsimony-informative-, 5,104 singleton-, and 151,927 constant sites. Topology of the phylogenetic tree of the Nicotiana genus was independently assessed by ML and BI (Figure 2). Both approaches confirmed the previously reported monophyletic nature of the Suaveolentes section, with N. africana as the oldest ancestor. The ML analysis revealed that most branches were well-supported (B > 90), with only three nodes with B < 90: the N. megalosiphon and N. rotundifolia ancestral node in the Suaveolentes clade (77); the N. undulata and N. setchellii ancestral node (84); and the ancestral node for the Repandae clade closely nested with the Alatae section (84) (Figure 2A). BEAST-based BA calculated high posterior probabilities (PP > 0.9) for all nodes (Figure 2B). Both topologies revealed Noctiflorae as the closest diploid section sister to the Suaveolentes section.
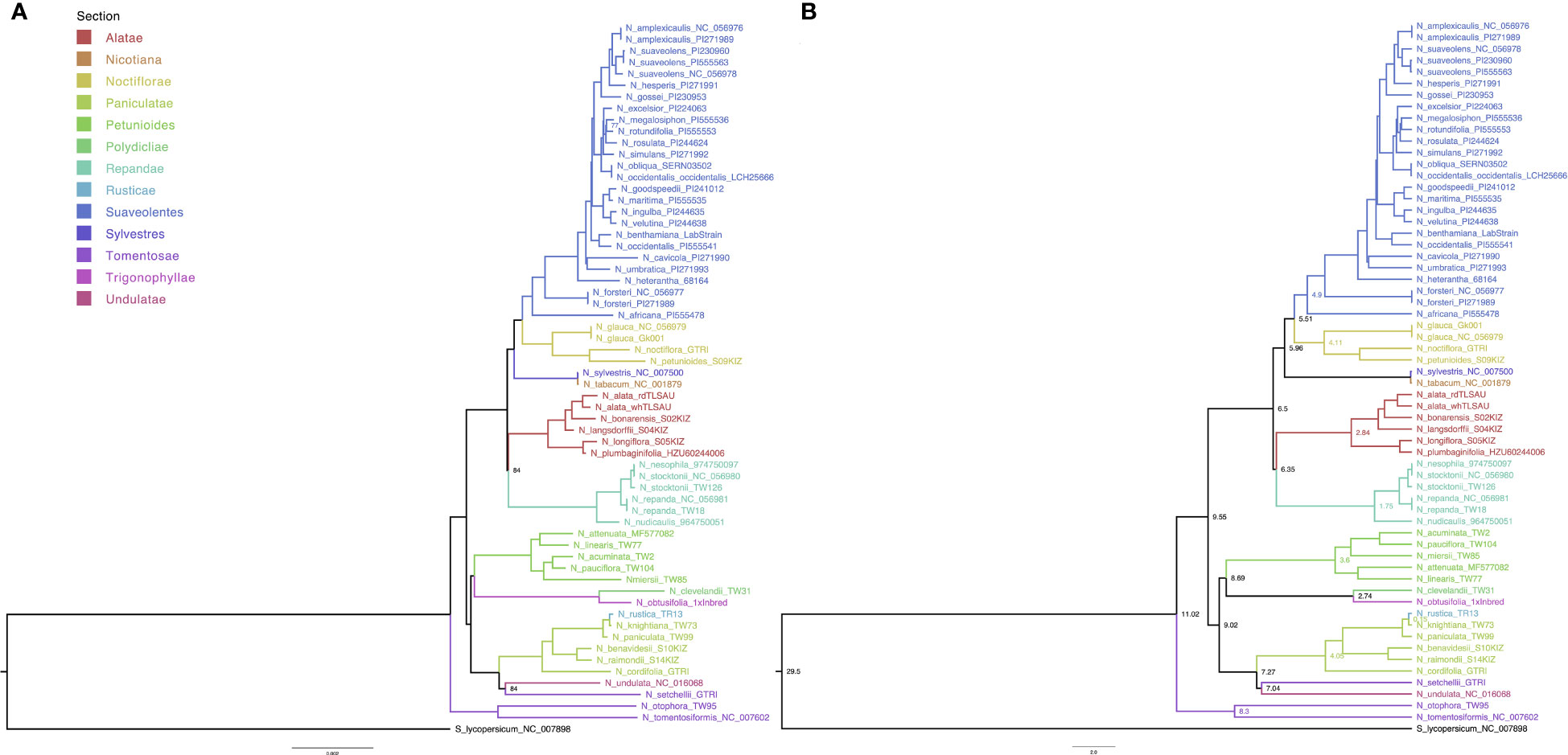
Figure 2 Maternal inheritance analysis of Nicotiana section Suaveolentes based on whole plastome genome sequence phylogenetic analysis. (A) Consensus tree obtained from the maximum likelihood (ML) analysis; B < 70 is indicated in the branch. (B) Consensus tree obtained from Bayesian inference (BI) analysis; PP > 0.9; scale bar depicts the years in Million years ago (Mya). Divergence time of the Suaveolentes section from the sister clade Sylvestres is depicted in a light-blue circle (Mya). Splitting times in Mya between selected sister clades (Petunioides/Trygonophyllae, Sylvestres/Nicotiana, and Alatae/Noctiflorae) are indicated at the bottom in black numbers (10, 5.5, and 0.15). Species are color-coded based on taxonomy (Section), as indicated in the legend. Solanum lycopersicum (black) was used as an outgroup. Different species accessions are tagged with accession numbers (Supplementary Table S3). Where relevant, the subspecies (subsp.) is indicated.
Both analyses produced the same topology. To compare our results with a previously published phylogeny, we selected the markers trnL-F, matK, trnS-G, and ndhF for a new ML-based phylogenetic analysis (4,260 nucleotides and 124 parsimony informative sites). Our results were like those of previous studies (Supplementary Figure S1). In previous analysis in which we used 54 taxa, we obtained topologies in which the Sylvestres clade was sister to the Suaveolentes section (data no shown). Intrigued by a possible Sylvestres phylogenetic signal, we divided the chloroplast genome alignment into three major structural regions: large single copy (LSC), small single copy (SSC), and inverted repeats (IR). Each alignment had 86,293, 25,643, and 21,877 nucleotides with 2,232, 724, and 207 parsimony informative sites, respectively. The LSC and SSC regions produced a ML and BA phylogenetic tree topology in which the Suaveolentes and Noctiflorae sections were sister groups, whereas the IR region showed similar results to our initial topology, in which Suaveolentes and Sylvestres were sister groups (Supplementary Figure S2). We tested a possible recombination between the Sylvestres and the Noctiflorae sections on the Suaveolentes plastid genomes with the Phi test using two different tools SplitTree (Supplementary Figure S3) and PhiPack-Phi. In both cases the phi test did find statistically significant evidence for recombination (p = 1.88E-8 and p = 2.76E-20 respectively) (Supplementary Table S5). The significant recombinant regions identified by PhiPack-Phi (p-value < 0.01, length > 5 Kb) were from 89950 to 97700, from 100150 to 111625, from 134075 to 139875, from 140125 to 145525 and from 148000 to 155750 associated with the IR regions.
We estimated the divergence date in the BI dataset (Figure 2B) using the Nicotiana-Solanum divergence time as calibration in a relaxed-clock model and validated the analysis based on the N. tabacum divergence time. Similar to previous results, we dated N. tabacum divergence as circa 0.06 Mya (Sierro et al., 2014; Edwards et al., 2017). Consistent with previous data (Clarkson et al., 2017; Dodsworth et al., 2020), we estimated the Suaveolentes section divergence from the Sylvestres/Nicotiana sections to be circa 6.34 Mya. We also analyzed the divergence time of the other clades associated with the origins of Suaveolentes (Figure 2B). While the BI sister clades Alatae and Noctiflorae split after the divergence of Suaveolentes circa 6.50 Mya, the Petunioides section branched off with the Trigonophyllae section around 9.99 Mya. Our analysis supports N. knightiana as the maternal ancestor of N. rustica as was reported before (Sierro et al., 2018). Nevertheless our analysis date the polyploidization event as an oldest event (1.48 Mya) compared with the previously published estimates (0.6 Mya (Clarkson et al., 2017)) probably due the use of the whole chloroplast genome, including more variable regions.
3.3 Suaveolentes nuclear genes have clear contributions from Alatae, Noctiflorae, Sylvestres and Petunioides sections
To elucidate the complex origin of genome of the members of the Suaveolentes section, we performed a phylogenetic analysis of nuclear genes. We selected sequencing data from 19 species representing all the diploid sections of the Nicotiana genus, two polyploid sections (Suaveolentes and Nicotiana), and an outgroup (S. lycopersicum) (Supplementary Table S6). Our dataset included 4 transcriptome-, 12 genome-, and 3 in-house genome assemblies, including those of N. africana, N. cavicola, and N. forsteri; 9 out 11 DNA whole genome wequence (WGS) datasets were annotated in-house. The genome size of each of the Suaveolentes species were estimated by flow cytometry (Supplementary Table S1) to estimate the sequencing depth of each of the species. The completeness of the annotations was assessed using the BUSCO set (Supplementary Table S6). DNA-based sequencing annotations showed a high degree of completeness. Two species displayed low completeness values (N. forsteri, C: 78.1%; N. otophora, C: 76%), whereas the rest of the annotations had scores greater than 82.5%. Although some BUSCO values were below the recommended value, we assumed that they were representative of the genome. However, transcriptome-based annotations showed greater variability in completeness (N. plumbaginifolia, C: 56.4%; N. pauciflora, C: 87.6%), possibly due to the lack of tissue diversity in the datasets. Hence, all datasets were used for further analysis.
Our first analytical approach was based on a gene family analysis (Figure 3). First, genes from all previous datasets were clustered in gene families using the Markov cluster algorithm (MCL) (Schaeffer, 2007). In the MCL analysis, the inflation value (I) controls the granularity of the clustering. Thus, variables such as the number of output clusters, number of genes in each cluster, and number of clusters with species-specific genes are controlled by I. We optimized MCL clustering by selecting a I of 2, which displayed the best balance between clusters with single copy genes in diploids and clusters with all the species. We obtained 46,429 clusters, including 8,098 clusters containing all the taxa. The biggest gene family contained 2,346 protein sequences with more than 100 sequences for the Suaveolentes species: N. africana, N. benthamiana, N. cavicola, and N. forsteri. Other Nicotiana genomes showed few members (e.g., N. attenuata, 8; N. knightiana, 1; N. obtusifolia, 9; N. paniculata, 2; N. pauciflora, 1; N. tabacum, 59; and N. undulata. Second, further analysis revealed that the gene family cluster was a retrotransposon, which was wrongly annotated; the protein encoded by this cluster had the retrotrans_gag_dom (IPR005162) protein domain. Although we did not re-annotate the genomes, the gene family analysis aided in the identification of misannotated repetitive elements. We selected 423 clusters in which diploid species presented one single copy and polyploid species presented at least two copies for N. benthamiana and N. tabacum and one or two copies for the other Suaveolentes species. We used an in-house script to perform the gene family analysis (See Material and Methods). The optimal model in most cases was MGK+F1X4+G4, with or without I. We obtained a total of 411 trees after the filtering (Supplementary Table S7).
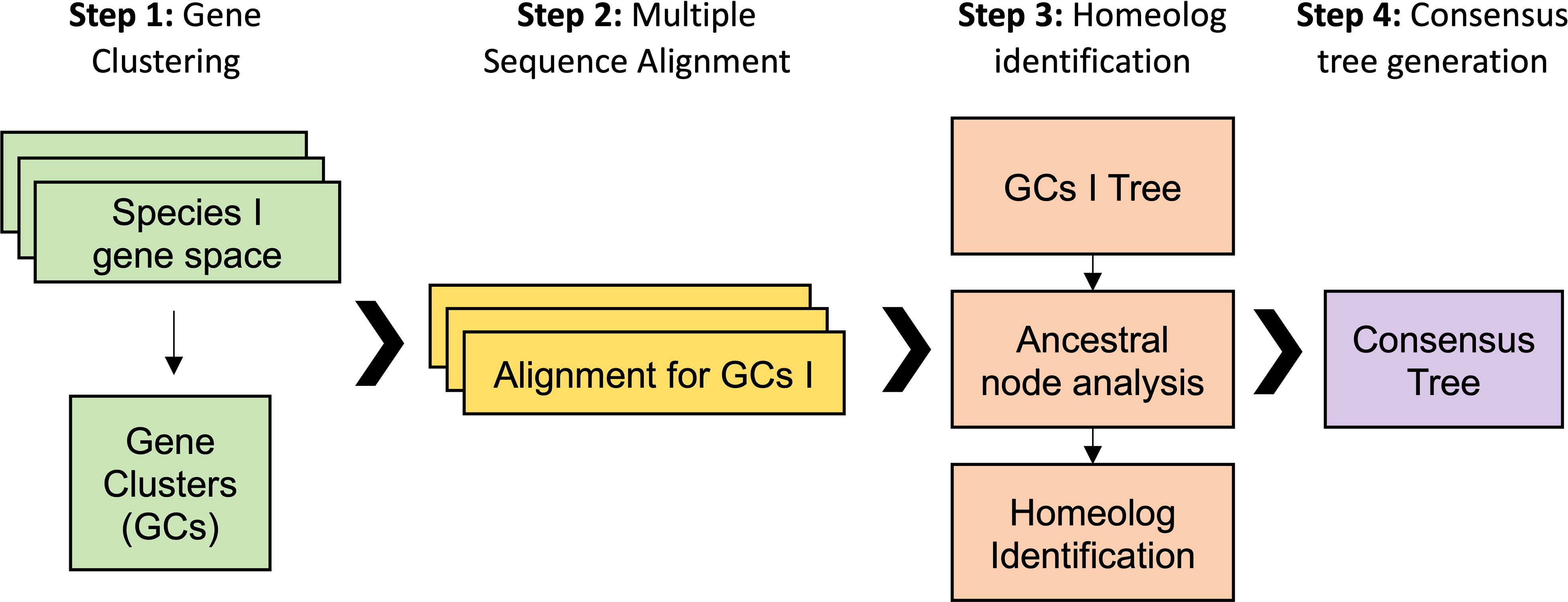
Figure 3 Workflow for the analysis of the origin of homeolog genes in polyploid Nicotiana species. The gene family analysis pipeline is divided into four steps. First, gene clustering using protein sequenced. The inflation value (I) was optimized to maximize the number of clusters in which the diploid species have one single copy gene. Second, the mRNA sequence of each gene was aligned for each of the gene clusters. Third, a ML tree was performed for each alignment from which was derived an ancestral node analysis and the homolog identification. Fourth, each of the clusters was classified according its topology and the polyploid homoelog, then a consensus was built for each topology.
We retrieved each of the nodes to analyze the frequency of the associated sections. As expected, the nodes with a significant weight for the N. section (N. tabacum) were associated with N. sylvestris (719) and N. tomentosiformis (1,532) (Figures 4A, B; Supplementary Table S8, Supplementary Figure S3). We also found strong support for Alatae-Sylvestres (270) and Noctiflorae-Petunioides (815) as diploid sister clades. The number of nodes supporting the structure in which the diploid sections Alatae-Sylvestres-Noctiflorae had a common ancestor and the Petunioides clade was an outgroup (60) was as probable as the sections Alatae-Sylvestres-Petunioides having the same ancestor and the Noctiflorae section being the outgroup (77). Similarly, Undulatae-Paniculatae had high support as sister diploid clades (426), in agreement with the chloroplast phylogeny. Nevertheless, Trigonophyllae-Tomentosae appeared as sister clades (276), instead of Trigonophyllae-Petunioides (4). The Suaveolentes section showed significant relatedness with Alatae (1,059), Noctiflorae (226), Petunioides (193), and Sylvestres (189). The number of nodes in which Suaveolentes was a sister group to the ancestor of Noctiflorae-Petunioides was also high compared with other nodes (281), which was likely derived from the ancestor of these species instead of a hybridization event between the ancestors of both sections. Nevertheless, the same case was less frequent for the Alatae-Sylvestres ancestor (97), compared with the hypothesis in which both sections had independent hybridization events.
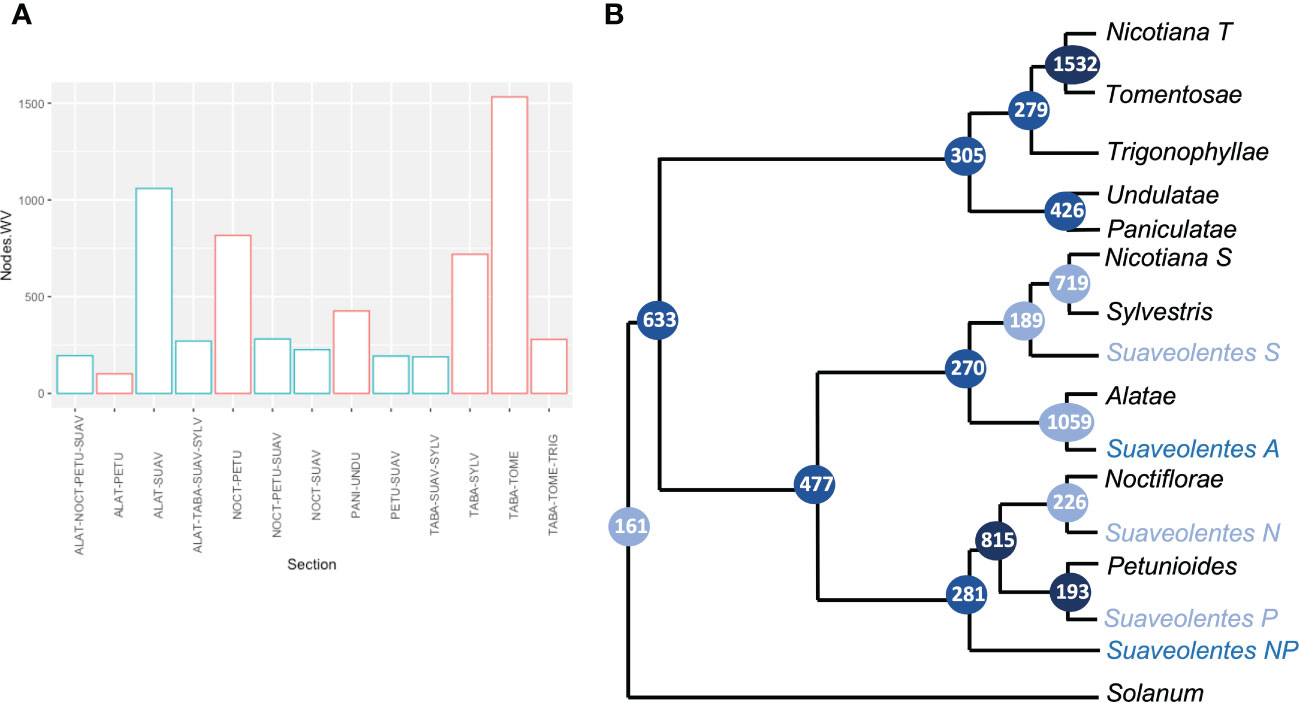
Figure 4 Quantitative analysis of the origin of Nicotiana section Suaveolentes. (A) The number of gene clusters that grouped two, three, or four Nicotiana sections together (node weight values). Nodes involving Suaveolentes species are colored in light blue. Sections are named as follows: Alatae (ALAT), Petunioides (PETU), Suaveolentes (SUAV), Noctiflorae (NOCT), Nicotiana (TABA), Sylvestres (SYLV), Tomentosae (TOME), and Trigonophyllae (TRIG). (B) Phylogenetic tree diagram based on the gene-family quantification approach. Branch labels represent the number of gene families that cluster in each set of Nicotiana sections. Color intensity represents the weight values. Polyploid species homeologs are tagged based on the phylogenetically closer diploid section. P (Petunioides), N (Noctiflorae), A (Alatae), S (Sylvestres), and T (Tomentosae).
Once the tags were assigned to each of the polyploid leaves, we classified 130 types of trees according to the possible ancestors of each polyploid leaf (Supplementary Table S9). We discarded 45 trees in which the N. tabacum leaves did not have the expected ancestor tags. Of the 130 types, only 31 had 3 or more trees, and these were used for further analysis. We used ASTRAL to calculate the consensus trees for each of the types. The types for which the Suaveolentes leaves did not have a clear assignment failed to produce a consensus tree (e.g., Type029). The ASTRAL consensus tree with more phylogenetic trees (Type001, 23 trees), presented a phylogenetic structure in which Alatae and Sylvestres were sister clades (Figure 5A), consistent with the node weight analysis and previously published Nicotiana phylogenies, though there were some trees wherein Noctiflorae was not a sister clade to the ancestor Alatae-Sylvestres (bootstrap 0.58). The contribution of Alatae and Noctiflorae to the Suaveolentes section was well supported (bootstrap > 70); although for Alatae derived leaves, N. forsteri was the oldest divergent Suaveolentes species, and that for Noctiflorae was N. africana. The second-most frequent topology (Type002, 19 trees) presented only one homeolog for the Suaveolentes clade, derived from Alatae (Figure 5B). Noctiflorae and Petunioides were well supported sister groups (bootstrap = 0.99). Alatae and Suaveolentes were also identified as sister groups, though the bootstrap value was low (0.21) compared to that of other topologies with Alatae and Sylvestres as sister clades. Like Type001, N. forsteri was the oldest divergent Suaveolentes species in the Type002 topology. The Type003 topology was calculated as a consensus of 16 trees (Figure 5C). Suaveolentes were represented by one homeolog closely related with the Noctiflorae clade. In this topology, Noctiflorae and Sylvestres were sister clades, and shared the closest ancestor first with Petunioides and then with Alatae (bootstrap = 0.62 and 0.77, respectively). Like Type001, N. africana was the oldest divergent Suaveolentes. This was also consistent with the trend in the Type005 consensus tree (Figure 5E). However, for the Alatae derived Suaveolentes, N. forsteri usually appeared as an outgroup (Figures 5A, B, D, F). Few trees presented Alatae/Sylvestres and the Noctiflorae/Petunioides ancestral nodes as the most close related to the Suaveolentes clade (Figures 5G). The ASTRAL consensus tree for Sylvestres-derived Suaveolentes homeologs had low bootstrap support (Figures 5D, E, H). The consensuses trees had good bootstrapping values (>90) (Supplementary Tables S7, S9), although the topology varied between trees. For example, in tree CL13177, one of the N. forsteri homeologs was a sister taxon to N. africana under the Sylvestres relationship, whereas the other homeolog was associated with the Alatae clade. The homeologs of the other Suaveolentes taxa were sister groups to the Sylvestres clade without Alatae derived homeologs. Similar results were observed for CL13194, CL13356, and CL14077, although some bootstrap values were low. Nevertheless, in CL13605 with Alatae-derived Suaveolentes, N. africana was the oldest Suaveolentes taxon, whereas that for the Sylvestres-derived taxa was N. forsteri.
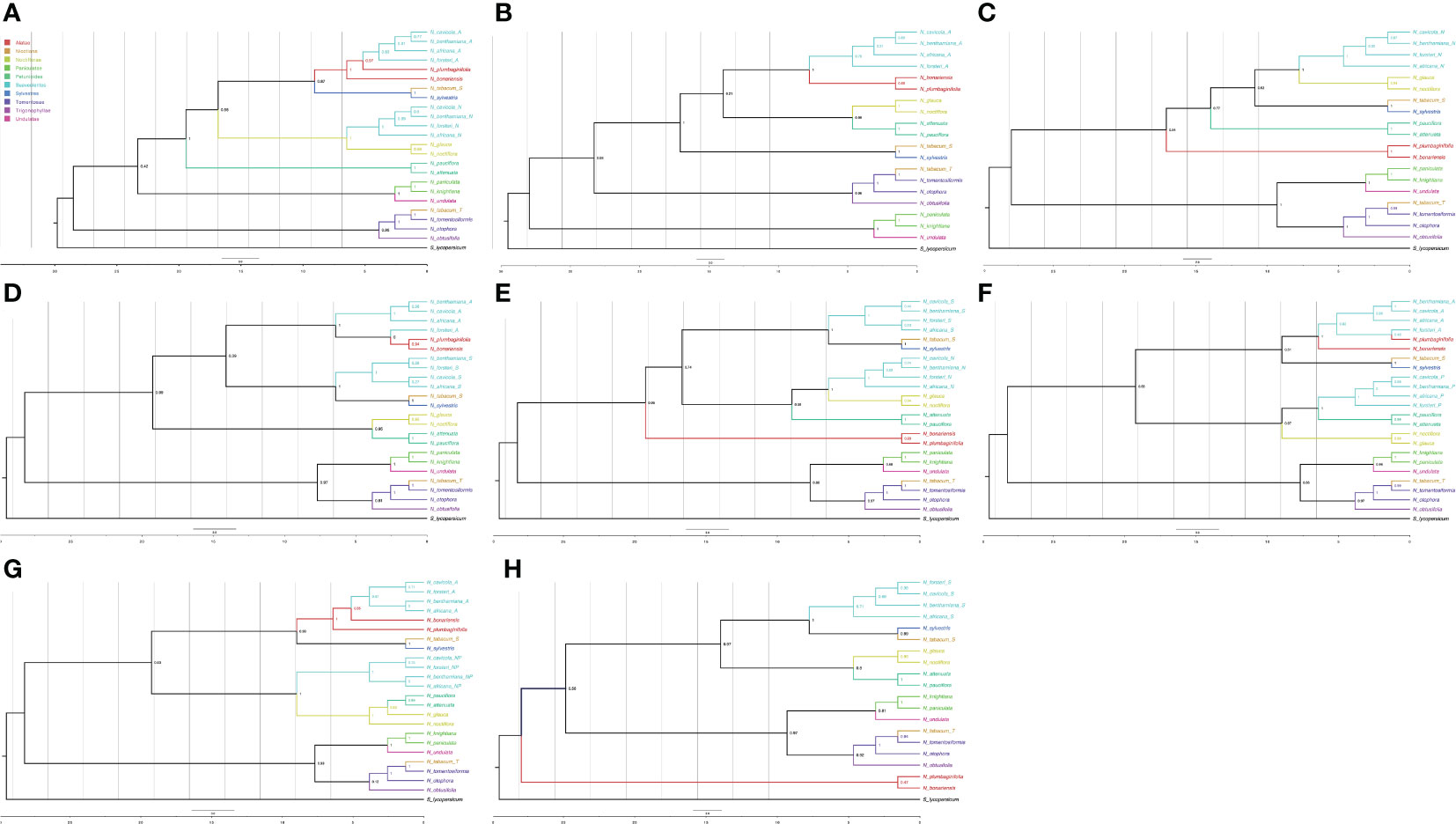
Figure 5 ASTRAL consensus trees for the different ancestral assignments of polyploid leaves. Only the consensus trees produced with more than 10 trees are represented. (A) Consensus tree Type001, (B) Type002, (C) Type003, (D) Type004, (E) Type005, (F) Type006, (G) Type007, and (H) Type010; suffixes for the Suaveolentes leaves are A, Alatae; N, Noctiflorae; P, Petunioides; S, Sylvestres; and NP, Noctiflorae-Petunioides ancestor. The color for each leaf is indicated in the legend.
To elucidate the relationship between the different homeologs in the Suaveolentes genes, we painted the chromosomes of the N. benthamiana reference genome (Niben2.6.0) according to the origin of its homeolog (Supplementary Figure S5; Supplementary Table S10). Although the chromosomes were a mosaic of different origins, some trends could be observed. Chromosomes were generally enriched in Alatae (e.g., 6 and 16; total genome 26.34%), Sylvestres (e.g., 7, 9, and 10; total genome 24.36%), and Noctiflorae (e.g., 8 and 11; total genome 23.76%) homeologs, with lower contributions from the Petunioides homeolog (e.g., 12, total genome 11.48%). Considering enrichments >20% of the homeologs, the most frequent combinations were Alatae-Sylvestres (chromosomes 4, 5, 9, 15, and 19), Alatae-Noctiflorae (chromosomes 1, 2, 3, and 14), and Sylvestres-Noctiflorae (chromosomes 10 and 17). The triple combination Alatae-Noctiflorae-Sylvestres was present in two chromosomes (15 and 18) and Petunioides-Noctiflorae was present in >20% of the homeologs for chromosome 12 (Figure 6; Supplementary Table S11).
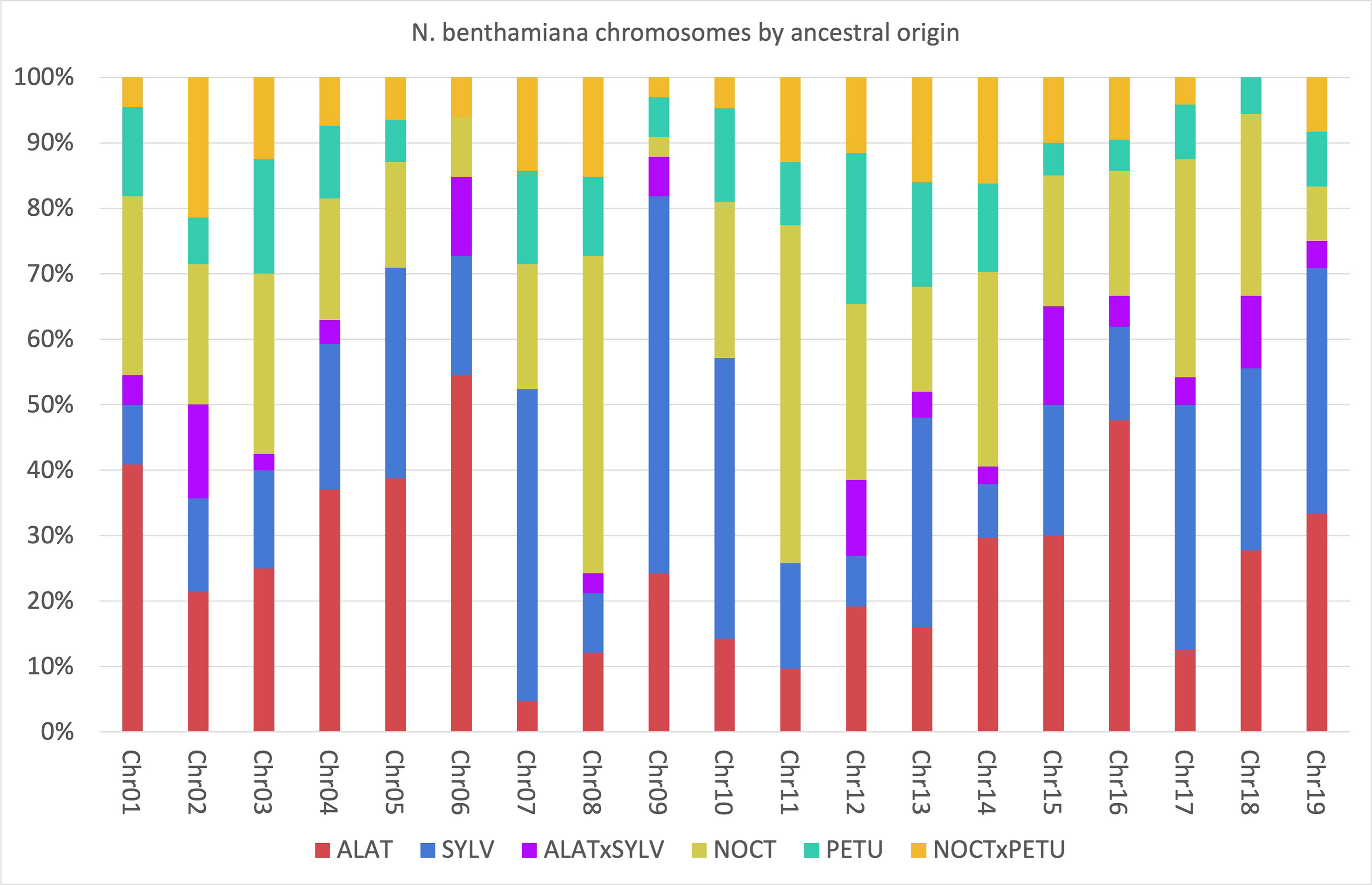
Figure 6 Percentage of the Nicotiana benthamiana chromosomes associated to each of the possible origins. Analysis based on the 505 genes for which the origin was identified. Blue, Sylvestres; Green, Petunioides; Red, Alatae; Yellow, Noctiflorae; Orange, Noctiflorae-Petunioides ancestor; Purple, Alatae-Sylvestres ancestor.
3.4 Molecular dating of the origin of the Suaveolentes clade
Our results show different hybridization/polyploidization events that could have generated the Suaveolentes section. The split between the Suaveolentes homeologs and the different diploid ancestors, Alatae, Noctiflorae, Petunioides, and Sylvestres, was dated to 5.37, 6.16, 5.45, and 5,56 Mya, respectively. The diversification event for the Suaveolentes section, based on the split of N. africana, started around 3.43 Mya (Figure 7; Supplementary Table S12), which was consistent with chloroplast dating results (5.43 Mya for the split of Sylvestres-Suaveolentes and 4.48 Mya for the split of N. africana with the rest of the species).
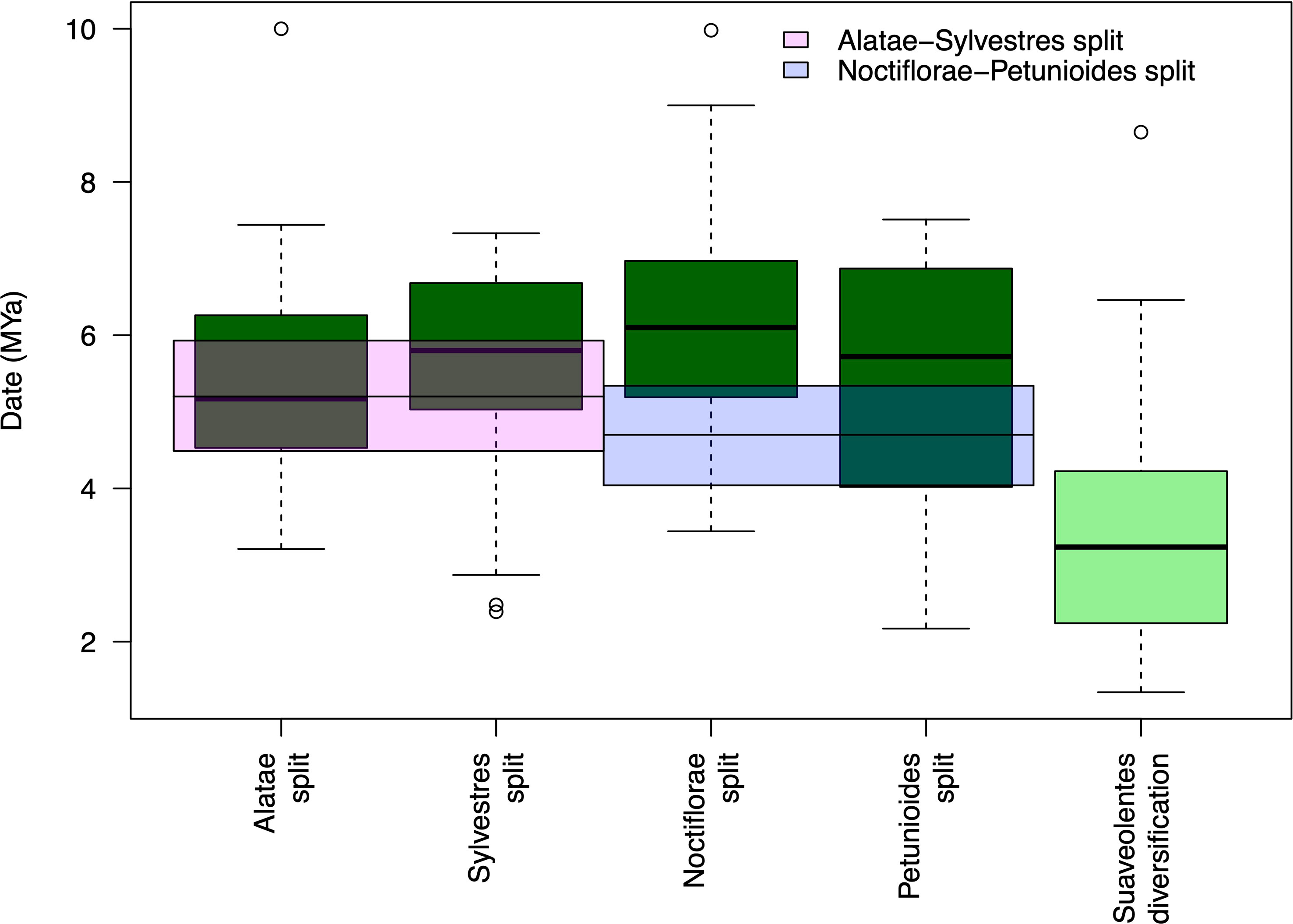
Figure 7 Suaveolentes polyploid event dating. The date distribution for the diploid ancestor split with the Suaveolentes homeologs as well as the diversification date of the Suaveolentes clade calculated from 59 BI trees. The divergency estimate for the diploid species are represented as pink (Alatae-Sylvestres) and blue (Noctiflorae-Petunioides) boxes.
However, it is unclear when the Alatae, Noctiflorae, Petunioides, or Sylvestres sections diverged and whether this occurred before the Suaveolentes polyploidization event. We estimated the phylogenetic relationship between the diploid accessions and divergence time using a BI tree composed of the 411 gene clusters previously used. This nuclear tree differed in several ways from the whole-chloroplast genome sequence tree. Specifically, Noctiflorae was a sister clade to Petunioides, whereas Alatae was sister to Sylvestres (Supplementary Figure S6). The divergence date for Noctiflorae-Petunioides was 4.70 Mya (height_95%_HPD: 4.04, 5.34) and that for Alatae-Sylvestres was 5.20 Mya (height_95%_HPD: 4.49, 5.93), predating the estimated polyploidization event for Suaveolentes.
We used a synonymous ratio (Ks) distribution analysis of orthologous genes to estimate the date of the speciation events for the diploid clades and Suaveolentes homeologs, as an alternative to the phylogenetic method (Blanc and Wolfe, 2004; Barker et al., 2008; Rabier et al., 2014) (Supplementary Figure S7). The split date between the Alatae and Sylvestres sections (Ks = 0.068) was close to that for Noctiflorae and Petunioides (Ks = 0.052). Both events were more recent than the divergence between the N. benthamiana (Ks = 0.095), N. africana (Ks = 0.096), N. forsteri (Ks = 0.101), and N. cavicola homeologs (Ks = 0.107) as well as the Solanum-Nicotiana split (Ks = 0.279) (Supplementary Figure S8). The estimated dates are earlier than those estimated with the phylogenetic approach: 7.20, 5.51, 10.02, 10.13, 10.71, 11.39, and 29.5 Mya, respectively (Supplementary Table S13). Finally, we constructed a phylogenetic tree with the Ks peak values for the homolog pairs of two representatives of each section (Supplementary Figure S9) and dated the nodes according to the Solanum-Nicotiana split. Based on the tree, the divergence dates were more recent than in the previous methods; however, the divergence between Suaveolentes (5.31 and 5.55 Mya) and its progenitor predated the split between Alatae and Sylvestres (4.5 Mya) and Noctiflorae and Petunioides (4.63 Mya).
4 Discussion
Our plastid phylogenetic tree results allow us to hypothesize that although Noctiflorae is the maternal ancestor, paternal plastid populations from the Sylvestres ancestor could have leaked into the newly formed polyploid cells. Then, the recombination of both genomes drove to the evolution of Suaveolentes plastid, where the LSC and the SSC are derived from that of the Noctiflorae section and the IR from that of the Sylvestres section. Although this scenario may appear improbable, the two processes needed for this to occur, i.e., biparental inherence and plastid genome recombination, have previously been described.
Many studies have described heteroplasmic events in angiosperms (McCauley et al., 2005; Ellis et al., 2008; Lambertini, 2016; Ramsey et al., 2019). Potential biparental plastid inheritance (PBPI) has been described in up to 20% of angiosperm genera (Zhang and Sodmergen, 2010). Furthermore, several studies show that Nicotiana species present PBPI (Horlow et al., 1990; Svab and Maliga, 2007), including N. sylvestris (Thyssen et al., 2012). The hybridization between N. sylvestris and N. undulata plants showed paternal inheritance for 0.002% of the seedlings, proving that this type of event occurs in modern Nicotiana species. Under this scenario, it is reasonable to think that ancestral Noctiflorae and Sylvestres plastid genomes shared the same polyploid cell in at least a few individuals.
Biparental plastid inheritance is not the only event needed to produce chimeric plastid genomes, recombination is also necessary. Our sequence recombination analysis on the plastid genomes indicates that recombination may occurred during the polyploidization event in the Suaveolentes clade. The application of new generation sequencing techniques, such as long read sequencing, has showed that most plant species have two different chloroplast structures with different orientations of the single copy regions (Wang et al., 2019). The two structures have been interpreted as consequences of a “BIR-like, recombination-dependent replication mechanism between different linear copies of the plastome” (Maréchal and Brisson, 2010), which could explain the recombination between two different chloroplast genomes. Transplastomic N. tabacum plants proved that these types of mechanisms are active in Nicotiana chloroplasts. After three generations, the deleterious mutations introduced in the chloroplast genome were removed by gene conversions (Khakhlova and Bock, 2006). This is not the only experiment proving plastid genome recombination in Nicotiana species. It was already described in Nicotiana hybrids between N. tabacum and N. plumbaginifolia more than 35 years ago (Medgyesy et al., 1985). Although we do not know if the recombined Noctiflorae-Sylvestres plastid genomes were favored by natural selection in the recently formed Suaveolentes polyploids, previous studies elucidated the evidence of the occurrence of these two events in Nicotiana species.
Although we have some evidence about the Noctiflorae-Sylvestres plastid genome recombination, these results need to be considered carefully due the technical limitations of our approach. NUclear PlasTid insertions (NUPTs) are difficult to identify with short reads (e.g., 66% of the Arabidopsis thaliana NUPTs are longer than 150 bp, (Michalovova et al., 2013)). Our assembly approach assumed that reads derived from the plastid genome will be found in a much higher proportion than reads derived from the nuclear genome (~20% of sequenced reads from leaf tissues are derived from the plastid DNA) (Soorni et al., 2017). As such, the variants derived from NUPTs will be filtered out as minor alleles before a consensus sequence is called on a guided assembly. Nevertheless, a lower proportion of reads derived from the plastid genome may drive to the introduction of variants derived from the nuclear genome producing chimeric assemblies. Long reads are a better approach to detect NUPTs and filter out of the plastid genome assembly but in our case, we only have them for the N. benthamiana genome, so we can’t fully discard the introduction of NUPTs variants in the plastid consensus genomes.
Our phylogenomic approach on the Nicotiana nuclear genes identified the following four sections as possible ancestors: Noctiflorae, Petunioides, Alatae, and Sylvestres. Noctiflorae and Sylvestres have been identified as possible ancestors in many previous studies (Aoki and Ito, 2000; Kelly et al., 2013; Clarkson et al., 2017; Schiavinato et al., 2020), sometimes with signals for Alatae (e.g., ITS marker (Chase et al., 2003; Kelly et al., 2013)) and Petunioides (e.g., GS3’ marker (Kelly et al., 2013)) (Summary on Supplementary Table S14). Some of our results agree with those previously published, but some also suggest that Alatae and Petunioides are involved in the formation of Suaveolentes genomes, which is in disagreement with the results of most recent studies (Clarkson et al., 2017; Schiavinato et al., 2020). The elucidation of complex phylogenetic histories requires not only the use of hundreds of genes, but also extensive sampling. Although analysis of the N. benthamiana genome revealed the origin of the Suaveolentes clade (Schiavinato et al., 2020), the same analysis lacked a representative sample size (some diploid sections were not represented). The absence of any Alatae representative drives the unequivocal conclusion of both Sylvestres and Noctiflorae being the ancestor. In our study, Alatae had the same weight as Sylvestres, and although not many gene families supported Petunioides as an ancestor, there was enough evidence to consider that this section contributed to the origin of the Suaveolentes clade, as previously proposed (Kelly et al., 2013; Clarkson et al., 2017). Under this scenario, two hypotheses exist. One, Suaveolentes was derived from the hybridization of two hybrids: Noctiflorae-Petunioides (with a higher proportion of the maternal genome from Noctiflorae) and Alatae-Sylvestres (with equal proportions of both). Two, hybridization occurred between the ancestors of each of the two clades (Noctiflorae-Petunioides and Alatae-Sylvestres) before they split into the modern sections. There is also a possibility in which one hybrid (Alatae-Sylvestres) hybridized with the ancestor of the two other sections (Noctiflorae-Petunioides).
We used two different molecular dating approaches. We dated each of the speciation events using a multi-coalescent species model (MSCM) with BEAST (Bouckaert et al., 2019) after identifying the possible homeologs in the tree topology. Alternatively, we analyzed the Ks distribution between different gene pairs for each pair of genomes (Blanc and Wolfe, 2004; Zwaenepoel and Van De Peer, 2019). Both approaches have their strengths and weaknesses, though their inherent differences could provide complementary perspectives. With both approaches, the date of the split between Suaveolentes homeologs predates the Noctiflorae-Petunioides split (Figure 7, Supplementary Figures S8, S9; Supplementary Tables S12, S13). The split of the Suaveolentes homeologs overlapped with the Alatae-Sylvestres split determined in the phylogenetic approach, though it also predates this split according to the Ks approach. Considering that two rounds of hybridizations in less than 1 Mya is more unlikely than just between two ancestors before they split into the major sections, we propose that the Suaveolentes section originated from the hybridization of the Noctiflorae-Petunioides and Alatae-Sylvestres ancestors before their split into these sections.
The divergence date between the Suaveolentes homeologs and diploid species derived from the Suaveolentes ancestors (5.37 to 6.36 Mya) estimated by a calibrated MSCM tree agreed with previously published results (Clarkson et al., 2017; Schiavinato et al., 2021). Unfortunately, the absence of fossil records limits the possibility of validating our results using a more accurate type of evidence for the dating for which we used an alternative molecular dating methodology. Our estimations based on Ks distributions between homologs generated earlier dates than those obtained by the calibrated MSCM phylogenetic tree (Supplementary Figures S6, S8, S9, Supplementary Table S13). The dating differences in both methodologies are greater between the Suaveolentes homeologs (~10 Mya compared with 5.37 to 6.36 Mya) than the diploid divergence dates (5.51 and 7.20 Mya for the split in Noctiflorae-Petunioides and Alatae-Sylvestres, respectively). Although we could attribute some of these differences to an incomplete assembly in the N. africana, N. forsteri, and N. cavicola genomes (Supplementary Table S6), our N. benthamiana genome delivered similar results with a comparatively complete assembly (C: 97.7% by BUSCO, C: 98.13% by Merqury). Dating whole-genome duplication events by homolog Ks distribution has some important limitations, though older events are generally less accurate than those considered in this work (Vanneste et al., 2013). Nevertheless, it has been proposed that the ratio of synonymous substitutions is higher between homeologs in a polyploid than two diploid species in the overestimation of WGD event dating (Vanneste et al., 2013). We have also to consider alternative scenarios in which the WGD event dating has been underestimated by the molecular dating methodologies. Our dates assume that the Suaveolentes dispersal from South America to Australia was produced through long distances by wind via the Atlantic and Indian oceans (Clarkson et al., 2004), and the Pacific ocean (Clarkson et al., 2010). Nevertheless, it also has been proposed that the Suaveolentes species arrived to Australia through an Antarctic land bridge that connected South America and Australia (Goodspeed, 1954). Based on the disjunct distribution of many related Australian and South American taxa, it has been proposed that this Antarctic land bridge existed at least until 30 Mya (van den Ende et al., 2017) which could push the origin of the Suaveolentes clade to the Oligocene.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA853913.
Author contributions
LD’A performed the chloroplast phylogenetic analysis and article writing. SO performed the DNA extraction and sequencing. NS participated to the DNA extraction and sequencing and on the article writing and project design. TH and ER performed initial experiments associated to this study and participated in the experimental design. NI participated on the article writing and project design. AB performed the genome assemblies and annotations, nuclear phylogenies, script writing, article writing and project design. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Philip Morris International (PMI R&D., CH-2000 Neuchâtel, Switzerland) when AB was affiliated with Virginia Polytechnic Institute and State University (Virginia Tech, Blacksburg, VA, US).
Acknowledgments
The authors would like to acknowledge the BTI Nicotiana benthamiana genome consortium (https://btiscience.org/our-research/research-facilities/research-resources/nicotiana-benthamiana/) for giving access to the last N. benthamiana genome version. This version is an improved version of the published version 1.0.1. thanks to the data contribution provided by Silin Zhong and Brian Kivtko. The authors would like to thank also to the Virginia Tech Advanced Resource Computing (ARC) center for the use of the Blueridge system for some assemblies performed in this work. Finally the authors would like to thank the reviewers for their useful comments and the editor for his support accommodating the editorial process to our timing.
Conflict of interest
NI, NS, and SO are employees of Philip Morris International.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.999887/full#supplementary-material
References
Alonge, M., Soyk, S., Ramakrishnan, S., Wang, X., Goodwin, S., Sedlazeck, F. J., et al. (2019). RaGOO: Fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20, 224. doi: 10.1186/s13059-019-1829-6
Aoki, S., Ito, M. (2000). Molecular phylogeny of nicotiana (Solanaceae) based on the nucleotide sequence of the matK gene. Plant Biol. 2, 316–324. doi: 10.1055/s-2000-3710
Aronesty, E. (2011). Ea-utils: Command-line tools for processing biological sequencing data. Available at: http://expressionanalysis.github.io/ea-utils/.
Augsten, M., Meyer, P. B., Freitas, L. B., Batista, J. A. N., Stehmann, J. R. (2022). Nicotiana gandarela (Solanaceae), a new species of ‘tobacco’ highly endangered from the quadrilátero ferrífero in Brazil. PhytoKeys 190, 113–129. doi: 10.3897/phytokeys.190.76111
Bally, J., Marks, C. E., Jung, H., Jia, F., Roden, S., Cooper, T., et al. (2021). Nicotiana paulineana, a new Australian species in nicotiana section suaveolentes. Aust. Syst. Bot. 34, 477–484. doi: 10.1071/SB20025
Barker, M. S., Kane, N. C., Matvienko, M., Kozik, A., Michelmore, R. W., Knapp, S. J., et al. (2008). Multiple paleopolyploidizations during the evolution of the compositae reveal parallel patterns of duplicate gene retention after millions of years. Mol. Biol. Evol. 25 (11), 2445–2455. doi: 10.1093/molbev/msn187
Blanc, G., Wolfe, K. H. (2004). Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16, 1667–1678. doi: 10.1105/tpc.021345
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchêne, S., Fourment, M., Gavryushkina, A., et al. (2019). BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PloS Comput. Biol. 15 (4), e1006650. doi: 10.1371/journal.pcbi.1006650
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: Architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Cauz-Santos, L. A., Dodsworth, S., Samuel, R., Christenhusz, M. J. M., Patel, D., Shittu, T., et al. (2022). Genomic insights into recent species divergence in nicotiana benthamiana and natural variation in Rdr1 gene controlling viral susceptibility. Plant J. 111, 7–18. doi: 10.1111/tpj.15801
Chacón, J. E., Duong, T. (2018). Multivariate kernel smoothing and its applications (1st ed.). Chapman and Hall/CRC. doi: 10.1201/9780429485572
Chase, M. W., Christenhusz, M. J. M., Conran, J. G., Dodsworth, S., Medeiros de Assis, F. N., Felix, L. P., et al. (2018). Unexpected diversity of Australian tobacco species (Nicotiana section Suaveolentes, Solanaceae). Curtis’s Bot. Mag. 35 (3), 212-227. doi: 10.1111/curt.12241
Chase, M. W., Christenhusz, M. J. M., Palsson, R. L., Fay, M. F., Dodsworth, S., Conran, J. G., et al. (2021). SSpecies delimitation in Nicotiana sect. Suaveolentes (Solanaceae): reciprocal illumination leads to recognition of many new species. Curtis’s Bot. Mag. 38 (3), 266-228. doi: 10.1111/curt.12410
Chase, M. W., Knapp, S., Cox, A. V., Clarkson, J. J., Butsko, Y., Joseph, J., et al. (2003). Molecular systematics, GISH and the origin of hybrid taxa in nicotiana (Solanaceae). Ann. Bot. 92, 107–127. doi: 10.1093/aob/mcg087
Clarkson, J. J., Dodsworth, S., Chase, M. W. (2017). Time-calibrated phylogenetic trees establish a lag between polyploidisation and diversification in nicotiana (Solanaceae). Plant Syst. Evol 303, 1001-1012. doi: 10.1007/s00606-017-1416-9
Clarkson, J. J., Kelly, L. J., Leitch, A. R., Knapp, S., Chase, M. W. (2010). Nuclear glutamine synthetase evolution in nicotiana: phylogenetics and the origins of allotetraploid and homoploid (diploid) hybrids. Mol. Phylogenet. Evol. 55, 99–112. doi: 10.1016/j.ympev.2009.10.003
Clarkson, J. J., Knapp, S., Garcia, V. F., Olmstead, R. G., Leitch, A. R., Chase, M. W. (2004). Phylogenetic relationships in nicotiana (Solanaceae) inferred from multiple plastid DNA regions. Mol. Phylogenet. Evol. 33, 75–90. doi: 10.1016/j.ympev.2004.05.002
Darriba, D., Taboada, G. L., Doallo, R., Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
Dodsworth, S., Christenhusz, M. J. M., Conran, J. G., Guignard, M. S., Knapp, S., Struebig, M., et al. (2020). Extensive plastid-nuclear discordance in a recent radiation of nicotiana section suaveolentes (Solanaceae). Bot. J. Linn. Soc. 193 (4), 546-559. doi: 10.1093/botlinnean/boaa024
Edwards, K. D., Fernandez-Pozo, N., Drake-Stowe, K., Humphry, M., Evans, A. D., Bombarely, A., et al. (2017). A reference genome for nicotiana tabacum enables map-based cloning of homeologous loci implicated in nitrogen utilization efficiency. BMC Genomics 18, 448. doi: 10.1186/s12864-017-3791-6
Ellis, J. R., Bentley, K. E., McCauley, D. E. (2008). Detection of rare paternal chloroplast inheritance in controlled crosses of the endangered sunflower helianthus verticillatus. Heredity (Edinb). 100 (6), 574-580. doi: 10.1038/hdy.2008.11
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117 (17), 9451-9457. doi: 10.1073/pnas.1921046117
Garrison, E., Marth, G. (2012). Haplotype-based variant detection from short-read sequencing – free bayes – variant calling – longranger. arXiv Prepr. arXiv1207.3907 9. doi: 10.48550/arXiv.1207.3907
Goodspeed, T. H. (1954). The genus nicotiana: Origins, relationships, and evolution of its species in the light of their distribution, morphology, and cytogenetics (Waltham, USA: Chronica Botanica Company), 1–536.
Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29 (8), 1072-1075. doi: 10.1093/bioinformatics/btt086
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 8 (8), 1494-1512. doi: 10.1038/nprot.2013.084
Horlow, C., Goujaud, J., Lépingle, A., Missonier, C., Bourgin, J. P. (1990). Transmission of paternal chloroplasts in tobacco (Nicotiana tabacum). Plant Cell Rep. 9, 249-252. doi: 10.1007/BF00232294
Jin, J. J., Yu, W., Yang, J. B., Song, Y., Depamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Kelly, L. J., Leitch, A. R., Clarkson, J. J., Knapp, S., Chase, M. W. (2013). Reconstructing the complex evolutionary origin of wild allopolyploid tobaccos (Nicotiana section suaveolentes). Evol. (N. Y) 67 (1), 80-94. doi: 10.1111/j.1558-5646.2012.01748.x
Khakhlova, O., Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Knapp, S., Chase, M. W., Clarkson, J. J. (2004). Nomenclatural changes and a new sectional classification in nicotiana (Solanaceae). Taxon 53, 73–82. doi: 10.2307/4135490
Ladiges, P. Y., Marks, C. E., Nelson, G. (2011). Biogeography of nicotiana section suaveolentes (Solanaceae) reveals geographical tracks in arid Australia. J. Biogeogr. 38, 2066–2077. doi: 10.1111/j.1365-2699.2011.02554.x
Lambertini, C. (2016). Heteroplasmy due to chloroplast paternal leakage: another insight into phragmites haplotypic diversity in north America. Biol. Invasions 18, 2443-2455. doi: 10.1007/s10530-016-1193-3
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9 (4), 357-359. doi: 10.1038/nmeth.1923
Lassmann, T., Sonnhammer, E. L. L. (2005). Kalign - an accurate and fast multiple sequence alignment algorithm. BMC Bioinf. 6, 298. doi: 10.1186/1471-2105-6-298
Leitch, I. J., Hanson, L., Lim, K. Y., Kovarik, A., Chase, M. W., Clarkson, J. J., et al. (2008). The ups and downs of genome size evolution in polyploid species of nicotiana (Solanaceae). Ann. Bot. 101, 805–814. doi: 10.1093/aob/mcm326
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. doi: 10.1186/2047-217X-1-18
Maréchal, A., Brisson, N. (2010). Recombination and the maintenance of plant organelle genome stability. New Phytol. 186 (2), 299-317. doi: 10.1111/j.1469-8137.2010.03195.x
Marks, C. E. (2010). Definition of south pacific taxa of nicotiana section suaveolentes (Solanaceae). Muelleria 28 (1), 74-84. doi: 10.5962/p.337574
McCauley, D. E., Bailey, M. F., Sherman, N. A., Darnell, M. Z. (2005). Evidence for paternal transmission and heteroplasmy in the mitochondrial genome of silene vulgaris, a gynodioecious plant. Heredity (Edinb). 95 (1), 50-58. doi: 10.1038/sj.hdy.6800676
Medgyesy, P., Fejes, E., Maliga, P. (1985). Interspecific chloroplast recombination in a nicotiana somatic hybrid. Proc. Natl. Acad. Sci. 82 (20), 6960-6964. doi: 10.1073/pnas.82.20.6960
Michalovova, M., Vyskot, B., Kejnovsky, E. (2013). Analysis of plastid and mitochondrial DNA insertions in the nucleus (NUPTs and NUMTs) of six plant species: size, relative age and chromosomal localization. Heredity (Edinb). 111, 314–320. doi: 10.1038/hdy.2013.51
Narasimhan, V., Danecek, P., Scally, A., Xue, Y., Tyler-Smith, C., Durbin, R. (2016). BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749–1751. doi: 10.1093/bioinformatics/btw044
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268-274. doi: 10.1093/molbev/msu300
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Quinlan, A. R., Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rabier, C. E., Ta, T., Ané, C. (2014). Detecting and locating whole genome duplications on a phylogeny: A probabilistic approach. Mol. Biol. Evol. 31, 750–762. doi: 10.1093/molbev/mst263
Rambaut, A. (2012). FigTree version 1.4.0. Available at: https://github.com/rambaut/figtree/.
Ramsey, A. J., McCauley, D. E., Mandel, J. R. (2019). Heteroplasmy and patterns of cytonuclear linkage disequilibrium in wild carrot. in. Integr. Comp. Biol. 59 (4), 1005-1015. doi: 10.1093/icb/icz102
Rhie, A., Walenz, B. P., Koren, S., Phillippy, A. M. (2020). Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245. doi: 10.1186/s13059-020-02134-9
Santilli, L., Pérez, F., de Schrevel, C., Dandois, P., Mondaca, H., Lavandero, N. (2022). Nicotiana rupicola sp. nov. and nicotiana knightiana (sect. paniculatae, solanaceae), a new endemic and a new record for the flora of Chile. PhytoKeys 188, 83–103. doi: 10.3897/phytokeys.188.73370
Schaeffer, S. E. (2007). Graph clustering by flow simulation. Comput. Sci. Rev. 1, 27–64. doi: 10.1016/j.cosrev.2007.05.001
Schiavinato, M., Bodrug-Schepers, A., Dohm, J. C., Himmelbauer, H. (2021). Subgenome evolution in allotetraploid plants. Plant J. 106, 672–688. doi: 10.1111/TPJ.15190
Schiavinato, M., Marcet-Houben, M., Dohm, J. C., Gabaldón, T., Himmelbauer, H. (2020). Parental origin of the allotetraploid tobacco nicotiana benthamiana. Plant J. 102 (3), 541-554. doi: 10.1111/tpj.14648
Schliep, K. P. (2011). Phangorn: Phylogenetic analysis in r. Bioinformatics 27 (4), 592-593. doi: 10.1093/bioinformatics/btq706
Sela, I., Ashkenazy, H., Katoh, K., Pupko, T. (2015). GUIDANCE2: Accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 43 (W1), W7-W14. doi: 10.1093/nar/gkv318
Sierro, N., Battey, J. N. D., Bovet, L., Liedschulte, V., Ouadi, S., Thomas, J., et al. (2018). The impact of genome evolution on the allotetraploid nicotiana rustica - an intriguing story of enhanced alkaloid production. BMC Genomics 19, 855. doi: 10.1186/s12864-018-5241-5
Sierro, N., Battey, J. N. D., Ouadi, S., Bakaher, N., Bovet, L., Willig, A., et al. (2014). The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 5, 3833. doi: 10.1038/ncomms4833
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Soorni, A., Haak, D., Zaitlin, D., Bombarely, A. (2017). Organelle_PBA, a pipeline for assembling chloroplast and mitochondrial genomes from PacBio DNA sequencing data. BMC Genomics 18, 49. doi: 10.1186/s12864-016-3412-9
Svab, Z., Maliga, P. (2007). Exceptional transmission of plastids and mitochondria from the transplastomic pollen parent and its impact on transgene containment. Proc. Natl. Acad. Sci. USA 104 (17), 7003-7008. doi: 10.1073/pnas.0700063104
Thyssen, G., Svab, Z., Maliga, P. (2012). Exceptional inheritance of plastids via pollen in nicotiana sylvestris with no detectable paternal mitochondrial DNA in the progeny. Plant J. 72, 84–88. doi: 10.1111/j.1365-313X.2012.05057.x
van den Ende, C., White, L. T., van Welzen, P. C. (2017). The existence and break-up of the Antarctic land bridge as indicated by both amphi-pacific distributions and tectonics. Gondwana Res. 44, 219-227. doi: 10.1016/j.gr.2016.12.006
Vanneste, K., de Peer, Y., Maere, S. (2013). Inference of genome duplications from age distributions revisited. Mole. Biol. Evol.30 (1), 177–190. doi: 10.1093/molbev/mss214vvvvv
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9 (11), e112963. doi: 10.1371/journal.pone.0112963
Wang, W., Lanfear, R., Gaut, B. (2019). Long-reads reveal that the chloroplast genome exists in two distinct versions in most plants. Genome Biol. Evol. 11 (12), 3372-3381. doi: 10.1093/gbe/evz256
Zhang, C., Rabiee, M., Sayyari, E., Mirarab, S. (2018). ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinf. 19 (6), 15-30. doi: 10.1186/s12859-018-2129-y
Zhang, Q., Sodmergen (2010). Why does biparental plastid inheritance revive in angiosperms? J. Plant Res. 123, 201–206. doi: 10.1007/s10265-009-0291-z
Keywords: Nicotiana, polyploidy, phylogenomic, phylogenetic dating, plastidgenome recombination
Citation: D’Andrea L, Sierro N, Ouadi S, Hasing T, Rinaldi E, Ivanov NV and Bombarely A (2023) Polyploid Nicotiana section Suaveolentes originated by hybridization of two ancestral Nicotiana clades. Front. Plant Sci. 14:999887. doi: 10.3389/fpls.2023.999887
Received: 21 July 2022; Accepted: 30 March 2023;
Published: 08 May 2023.
Edited by:
Steven Dodsworth, University of Portsmouth, United KingdomReviewed by:
Luiz Augusto Cauz Dos Santos, University of Vienna, AustriaAles Kovarik, Academy of Sciences of the Czech Republic (ASCR), Czechia
Copyright © 2023 D’Andrea, Sierro, Ouadi, Hasing, Rinaldi, Ivanov and Bombarely. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aureliano Bombarely, YWJvbWJhcmVseUBpYm1jcC51cHYuZXM=