 Guglielmo Puccio1,2*
Guglielmo Puccio1,2* Rosolino Ingraffia1
Rosolino Ingraffia1 Dario Giambalvo1
Dario Giambalvo1 Alfonso S. Frenda1
Alfonso S. Frenda1 Alex Harkess3
Alex Harkess3 Francesco Sunseri2,4*
Francesco Sunseri2,4* Francesco Mercati2*
Francesco Mercati2*- 1Department of Agricultural, Food and Forestry Sciences, University of Palermo, Palermo, Italy
- 2Institute of Biosciences and BioResources (IBBR), National Research Council, Palermo, Italy
- 3HudsonAlpha Institute for Biotechnology, Huntsville, AL, United States
- 4Department Agraria , University Mediterranea of Reggio Calabria, Reggio Calabria, Italy
Nitrate uptake by plants primarily relies on two gene families: Nitrate transporter 1/peptide transporter (NPF) and Nitrate transporter 2 (NRT2). Here, we extensively characterized the NPF and NRT2 families in the durum wheat genome, revealing 211 NPF and 20 NRT2 genes. The two families share many Cis Regulatory Elements (CREs) and Transcription Factor binding sites, highlighting a partially overlapping regulatory system and suggesting a coordinated response for nitrate transport and utilization. Analyzing RNA-seq data from 9 tissues and 20 cultivars, we explored expression profiles and co-expression relationships of both gene families. We observed a strong correlation between nucleotide variation and gene expression within the NRT2 gene family, implicating a shared selection mechanism operating on both coding and regulatory regions. Furthermore, NPF genes showed highly tissue-specific expression profiles, while NRT2s were mainly divided in two co-expression modules, one expressed in roots (NAR2/NRT3 dependent) and the other induced in anthers and/ovaries during maturation. Our evidences confirmed that the majority of these genes were retained after small-scale duplication events, suggesting a neo- or sub-functionalization of many NPFs and NRT2s. Altogether, these findings indicate that the expansion of these gene families in durum wheat could provide valuable genetic variability useful to identify NUE-related and candidate genes for future breeding programs in the context of low-impact and sustainable agriculture.
1 Introduction
Nitrogen (N) is a crucial nutrient for plant growth and development. Suboptimal nitrogen utilization can lead to diminished yields and significant environmental repercussions. Excessive or misapplied nitrogen fertilizers often lead to an increased risk of nitrogen escaping into the environment through processes like denitrification, leaching, or volatilization. This contributes to higher levels of nitrate in both surface and groundwater, as well as the release of N2O and NH3 into the atmosphere. Therefore, improving the efficiency of nitrogen utilization is crucial to address issues such as environmental degradation, climate change, and food security (Javed et al., 2022). Despite valuable research efforts in this field and the development of various technologies (i.e., slow-release fertilizers, inhibitors for nitrification and urease, fertigation, and advanced precision agriculture techniques) nitrogen efficiency remains relatively low for many crops, particularly for cereals where it typically ranges between 25% and 50% of the applied nitrogen (Giambalvo et al., 2018; Javed et al., 2022). This can be attributed to the complexity of Nitrogen Use Efficiency (NUE), which involves a multitude of factors related to agronomy, physiology, and molecular biology. Nitrate (NO3-) is one of the major N-forms taken up by plants from the soil. NO3- availability in the soil is highly variable and its uptake is governed by at least two transport systems, depending on soil NO3- concentrations: the low affinity NO3- transport (LATS) and the high affinity NO3- transport (HATS) systems. LATS is mediated by the NO3- transporter 1/peptide transporter (NRT1/NPF) family, which comprises a diverse array of membrane transport proteins found within multiple cell types and tissues, whereas HATS is facilitated by the NRT2 family, and is specific for NO3-. These two transport systems are responsible for the uptake of NO3- at different range of concentrations from millimolar to micromolar. N uptake is an important component of NUE, defined as the total biomass (or yield) per unit of N supplied (Moll et al., 1982), it is a complex trait influenced by interacting environmental factors and controlled by gene networks involved in N uptake, assimilation, and remobilization. NUE is divided in two main components, the Nitrogen Uptake Efficiency (NUpE), referred to the ability of the plant to take up N from the soil, and the Nitrogen Utilization Efficiency (NUtE), which encompasses the ability of the plant to assimilate, transfer, and utilize N to the harvestable part of the crop (Good et al., 2004; Xu et al., 2012).
The NPF and NRT2 families differ in both their structure and copy number across angiosperms. The NPF family harbors a conserved structural arrangement consisting of twelve transmembrane domains (TM) connected by short peptides and a central hydrophilic loop of about 90 amino acids between the sixth and the seventh TM domains (Wang et al., 2018b). They were previously known as NRT1s (NO3- transporters) and/or PTRs (peptide transporters) depending on their first discovered substrates. Based on a wide multi-species phylogenetic analysis, Léran et al. (2014) proposed a unified nomenclature for the NO3- transporter/Peptide transporter family (NPF), defining eight subfamilies (NPF1-8). The first NPF gene member isolated in plants and one of the most studied is the Arabidopsis thaliana NPF6.3 (AtNPF6.3), previously known as CHL1/AtNRT1.1. It is considered a dual-affinity NO3- transporter contributing to root NO3- uptake at both low (LATS) and high (HATS) NO3- availability, acting also as an NO3- sensor or ‘transceptor’ (Liu et al., 1999; Gojon et al., 2011; Xuan et al., 2017). AtNPF6.3 can also act as a chlorate transporter (per the old name CHL1 was awarded) when NO3- is less available and as an auxin transporter, a process negatively regulated by NO3- (Mounier et al., 2014; Maghiaoui et al., 2020; Meier et al., 2020). The interaction between auxin and NO3- is associated to NO3- sensing and it is involved in the regulation of N-dependent root development (Bouguyon et al., 2015). NPF proteins can transport a high number of different substrates other than NO3-, including phytohormones such as ABA and auxin, but also peptides, potassium, and secondary metabolites (Chiba et al., 2015; Tal et al., 2016; Kanstrup and Nour-Eldin, 2022). Although the NPF family is often involved in the LATS, many members also show high affinity transport in many species such as ZmNPF6.6 and MtNPF6.8 in maize and Medicago truncatula, respectively (Bagchi et al., 2012; Wen et al., 2017).
Land plant genomes typically contain a higher number of NPF/PTR genes compared to bacteria, animals, and algae, with 20 members in the moss Physcomitrella patens, 52 members in Arabidopsis thaliana and even more members in polyploid species such as Brassica napus (199) and Triticum aestivum (331) (Bajgain et al., 2018; Longo et al., 2018). In Brassica napus, allopolyploidy greatly contributed to the gene family expansion of the NPF family (Wen et al., 2020). A recent characterization of the NPF and NRT2 families in bread wheat also showed an expansion of these families (331 and 46, respectively) mainly due to tandem and segmental duplication (Bajgain et al., 2018; Li et al., 2021). The retention of multiple gene copies, after duplication, can be associated with the acquisition of new beneficial functions or the reduction of their full capacity, compared to that of the single-copy ancestral gene (Lynch and Conery, 2000). The high number of NPF genes in allopolyploid species suggested that the transporters encoded by these genes may have evolved for new unknown roles in plants (Corratg’e-Faillie and Lacombe, 2017; Longo et al., 2018). Thus, exploration for novel functions within these large gene families in polyploid crops is necessary. The NRT2 genes are primarily involved in HATS, and mainly active in roots, although some members are expressed in other tissues such as seeds or leaves to allow NO3- remobilization and storage (Chopin et al., 2007; Miller et al., 2007). Seven members were characterized in Arabidopsis thaliana, while five were detected in Oryza sativa. Similarly to the NPF family, a higher number of NRT2 members were discovered in allopolyploid species such as Triticum aestivum and Brassica napus with 47 and 17 genes, respectively (Tong et al., 2020; Li et al., 2021). This family was deeply studied in Arabidopsis thaliana, AtNRT2.1 resulted the most studied member due to its main role in high affinity NO3- uptake in roots (Li et al., 2007). The NRT2 genes are usually identified based on the sequence homology to known NO3- transporters, then their functions are predicted through gene expression analysis and heterologous expression in Xenopus oocytes. Nonetheless, several studies on monocot species such as wheat and rice have highlighted high sequence divergence with dicot species, making it hard to directly infer gene functions relying only on sequence identity (Plett et al., 2010; Pellizzaro et al., 2015; Wang et al., 2019a). Therefore, the utilization of multi-tissue and -condition expression data become mandatory to fully characterize these genes in monocot crops, mainly in the allopolyploids.
The hexaploid bread wheat (Triticum aestivum L.; genome AABBDD) is among the most important global crop species, shaped heavily by polyploidy and hybridization between the tetraploid durum wheat (Triticum turgidum L.; genome AABB) and Aegilops tauschii (genome DD). The NPF and NRT2 gene families have been investigated mainly in bread wheat, exploring their expression levels under different abiotic stresses, such as drought, salt and N deficiency, in response to Arbuscular Mycorrhizal Fungi (AMF), and in several tissues and development stages (Buchner and Hawkesford, 2014; Duan et al., 2016; Tian et al., 2017; Bajgain et al., 2018). Recently, the increase of grain NO3- uptake through the TaNRT2.5 overexpression, localized in the grain cell tonoplast, was reported (Li et al., 2020). Many other studies highlighted improved crop yield, shoot biomass, and N uptake when NPF or NRT2 genes were overexpressed (Hu et al., 2015; Fan et al., 2016; Sol et al., 2019; Wang et al., 2021). Furthermore, the nucleotide variability in protein-coding regions of the NPF genes seems to affect NUE related traits such as yield and shoot N content (Li et al., 2021). These findings suggested that further efforts in the detection and functional characterization of both gene families may greatly aid the selection of N-use efficient wheat cultivars. The high wheat genetic variability, the high number of duplicated genes, and its economic relevance make this plant a key species for the screening of potentially beneficial genes.
Compared to bread wheat, much less is known about the phylogenetic diversity, evolution, and expression of the NPF and NRT2 gene families in tetraploid durum wheat, which is an important crop in the Mediterranean basin (Hawkesford, 2017; Hawkesford and Griffiths, 2019; Lupini et al., 2021). The detection of key genes involved in NO3- transport is a primary goal for NUE improvement, and a gene family comparison between durum and bread wheat can elucidate the impact of polyploidy on NUE components. Hence, it is crucial to undertake a thorough characterization and annotation of nitrate transporters in the durum wheat genome. In this study, we have identified and analyzed both NPF and NRT2 gene families, exploring their phylogenetic relationships, gene and protein structures, regulatory elements, and expression profiles within the durum wheat genome.
2 Materials and methods
2.1 NPF and NRT2 identification in durum wheat genome
To identify NPF and NRT2 genes in the durum wheat genome, the protein sequences of NPF and NRT2 genes of Arabidopsis thaliana, barley (Hordeum vulgare), maize (Zea mays), rice (Oryza sativa), and bread wheat (Triticum aestivum) were downloaded from Ensembl plants (http://plants.ensembl.org/). These sequences were used for a BLASTP search against the entire durum wheat proteome, also downloaded from Ensembl plants, using an e-value threshold of 1e-10 and a minimum sequence identity of 50%. The durum wheat BLASTP best hits were then used as input for HMMER3 (Mistry et al., 2013), using the hmmscan command and the ‘Proton-dependent oligopeptide transporter family’ (IPR000109) HMM profile with an e-value cut-off of 1e-05 for the NPF genes. Furthermore, Pfam (Bateman et al., 2004) and NCBI protein sequence analysis tools were used to check that all the NPF protein sequences belonged to the PTR2 family (PF00854) and that all the NRT2 protein sequences contained the NCBI conserved domain PLN00028. The final set of genes was then used to identify homologous groups. These were defined through a reciprocal BLASTN using nucleotide sequence identity >95%.
2.2 Motif discovery, TF binding site, CREs prediction, and gene structure analysis
Gene structure of both TdNPF and TdNRT2 family members using Webscipio2 was analyzed (Hatje et al., 2011). The Multiple Em for Motif Elicitation (MEME) suite (Bailey et al., 2015) was used to identify conserved motifs with the following parameters: classic mode algorithm, 6 and 100 for minimum and maximum motif width, and a maximum number of 30 motifs per sequence. Conserved motifs were further analyzed through the NCBI protein domain search tool (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and the Conserved Domain Database (CDD), using an e-value threshold of 0.01. Transmembrane helices and protein localization prediction was performed using the TMHMM2.0 tool (Krogh et al., 2001) and both WoLF PSORT (Horton et al., 2007) and PProwler1.2 (Hawkins and Bodén, 2006), respectively. Chromosome location was extracted from the durum wheat genome annotation v1.0 and then displayed using the MG2C online tool (Chao et al., 2021). Significantly enriched chromosomal locations for both NPF and NRT2 were detected with ShinyGO (Ge et al., 2020) using a sliding window size of 6Mb and an FDR cutoff of 1e-05. The same tool was used to perform a GO enrichment analysis of both TdNPF and TdNRT2 genes.
Transcription Factor (TF) binding site prediction was performed on the promoter region using the binding site prediction tool of the Plant Transcription Factor Database (http://plantregmap.gao-lab.org/binding_site_prediction.php) with a p-value threshold of 1e-06 and the Triticum aestivum orthologs. The UniProtKB database (www.uniprot.org) was then used to extract protein domain information and annotation of the predicted TFs. Cis-regulatory Elements (CREs) in upstream promoter regions (− 2000 bp) of TdNPFs and TdNRT2s were predicted using PlantCARE (Lescot et al., 2002).
2.3 Collinearity and gene duplications analysis
The intraspecific collinearity was analyzed using both TdNPF and TdNRT2 gene sets. A reciprocal BLASTP was performed using an e-value threshold of 1e-10. MCScanX was used to evaluate collinearity and duplication events using an e-value threshold of 1e-05 and a match score of 50. MCScanX was also used to display the collinear blocks among five Poaceae species selected on the knowledge about the genesis of both durum and bread wheat (Kimber and Feldman, 1987; Matsuoka, 2011) (Aegilops speltoides Tausch: closer to B genome, Triticum urartu: A genome, Triticum durum: A and B genomes, Triticum aestivum: A, B and D genomes, and Aegilops tauschii: D genome). Collinear blocks between species were used for the evaluation of non-synonymous (Ka) and synonymous (Ks) values using TBtools (Chen et al., 2020). Tandem and collinear gene pairs inside the durum wheat genome were further used to evaluate both Ka and Ks using TBtools.
2.4 Phylogenetic analyses
Phylogenetic trees including Arabidopsis thaliana and Oryza sativa NPF and NRT2 genes and those here identified on durum wheat were constructed. The final dataset included 357 and 31 protein sequences for NPF and NRT2 families, respectively. Alignment was performed with the online tool CLUSTALW (Sievers et al., 2011) with default parameters. The unrooted phylogenetic tree was generated through the IQ-TREE software v. 2.2 (Nguyen et al., 2015) with the maximum likelihood method, 1000 bootstrap replicates, and the JTT + G4 model for both NPF and NRT2 trees, selected by the IQ-TREE best-fit model selection. Gene trees were visualized and analyzed through FigTree v. 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
2.5 Expression profiles of TdNPF and TdNRT2 genes and co-expression analysis
A total of 195 wheat RNA-Seq datasets were downloaded from the Sequence Read Archive (SRA). These included 13 durum cultivars, 9 tissues, and 25 phenological stages (Zadoks scale: from Z12 to Z90) (Table S1). Raw reads were trimmed with the Trimmomatic tool (Bolger et al., 2014) using the options: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:50. Clean reads were then quantified using Salmon (Patro et al., 2017) with default parameters and normalized through DESeq2 (Love et al., 2014). Reads were further filtered using the SVA package (Leek et al., 2012) to remove any batch effect or unwanted sources of variation using 10 surrogate variables. A co-expression network analysis was carried out by using the Weighted Gene Co-Expression Analysis (WGCNA) method (Langfelder and Horvath, 2008) with the following parameters: soft threshold=12, minimum module size=100, mergeCutHeight=0.3. Co-expression networks for each module were analyzed using Cytoscape (Shannon, 2003) and the hub genes for each network were selected using the CytoHubba pluging (Chin et al., 2014). Furthermore, module-trait (conditions) relationship was evaluated as correlation between the eigengenes of each module and a binary matrix representing each condition. Heatmaps were generated using the Pheatmap R package (Kolde and Kolde, 2018) using log-transformed normalized counts.
2.6 Data retrieval
The sequences and annotation files of all genomes were downloaded from the Ensembl plants database (http://plants.ensembl.org) (Bolser et al., 2016). The Aegilops speltoides Tausch. genome was obtained from the e!DAL - Plant Genomics & Phenomics Research Data Repository (https://doi.org/10.5447/ipk/2022/0) (Avni et al., 2022). The RNA-Seq datasets used for the expression profile and the co-expression analyses were obtained from the SRA archive (Leinonen et al., 2010) (Table S1).
3 Results
3.1 Durum wheat NPF and NRT2 genes identification
To identify NRT2 and NPF genes in durum wheat, a BLASTP search against all predicted protein sequences of the genome using the full-length amino acid sequences from five different plant species was carried out. The output of the BLAST search was further scanned using the HMMER3 tool with the ‘Proton-dependent oligopeptide transporter family’ profile (IPR000109) and the PLN00028 NCBI domain, and finally 211 and 20 NPF and NRT2 genes, respectively in the durum wheat genome were identified. NPFs and NRT2s showed 103 and 6 homologous groups, respectively, between A and B genomes.
The TdNPFs showed high variability in both gene length and amino acids content. The nucleotide sequences of the 211 genes showed a 3400 bp average gene length and encoded proteins ranging from 71 to 943 amino acids, with an average length of 583 amino acids, and molecular weights ranging from 7 to 105 kDa.Eighty percent of the TdNPF proteins showed 12 predicted transmembrane domains, while almost 95% of these proteins were localized in the plasma membrane (Figure S1). Like in durum wheat, NRT2 is a smaller gene family with a lower variability compared to the NPF family. The twenty TdNRT2 members showed a 1600 bp average gene length and encoded proteins ranging from 113 to 573 amino acids, with a mean length of 509 amino acids. Their molecular weights ranged from 12 to 62 kDa. Seventy-five percent of the TdNRT2 proteins showed 12 predicted transmembrane domains, while 90% were predicted to be localized in the plasma membrane (Table S2).
3.2 TdNPFs and TdNRT2s phylogenetic analysis
To explore the molecular evolution and the TdNPF gene family organization, we performed a phylogenetic analysis including protein sequences from Arabidopsis thaliana (53 AtNPFs), Oryza sativa (93 OsNPFs), and the 211 TdNPFs here identified in Triticum durum for a total of 357 NPF sequences. The Multiple Sequence Alignment (MSA) performed by CLUSTALW was used as input to IQ-TREE for both the model selection and the maximum-likelihood tree estimation. The phylogenetic tree showed a distinct clustering among the eight known NPF sub-families (Figure 1A). All the key nodes between sub-families are well supported with bootstrap values > 98 and all the genes from Arabidopsis and rice belonging to the same sub-family clustered together (Figure 1B). These results ensured the accuracy and reliability of the tree construction, suggesting a higher sequence variability between sub-families compared to the interspecific variability of each sub-family. The TdNPFs were assigned to the eight sub-families, namely from TdNPF1 to TdNPF8, following the tree topology and the previous classifications from other species. The sub-families TdNPF5 and TdNPF8 included the highest numbers of members (63 and 52, respectively), while TdNPF1 and TdNPF3 were the smaller sub-families with four and 8 genes, respectively (Table S2). TdNPF4, TdNPF5 and TdNPF6 were the only monophyletic groups, while the sub-families TdNPF1, TdNPF2, TdNPF3 and TdNPF7, TdNPF8 formed two distinct clusters with TdNPF1 clustering inside the TdNPF2 branch.
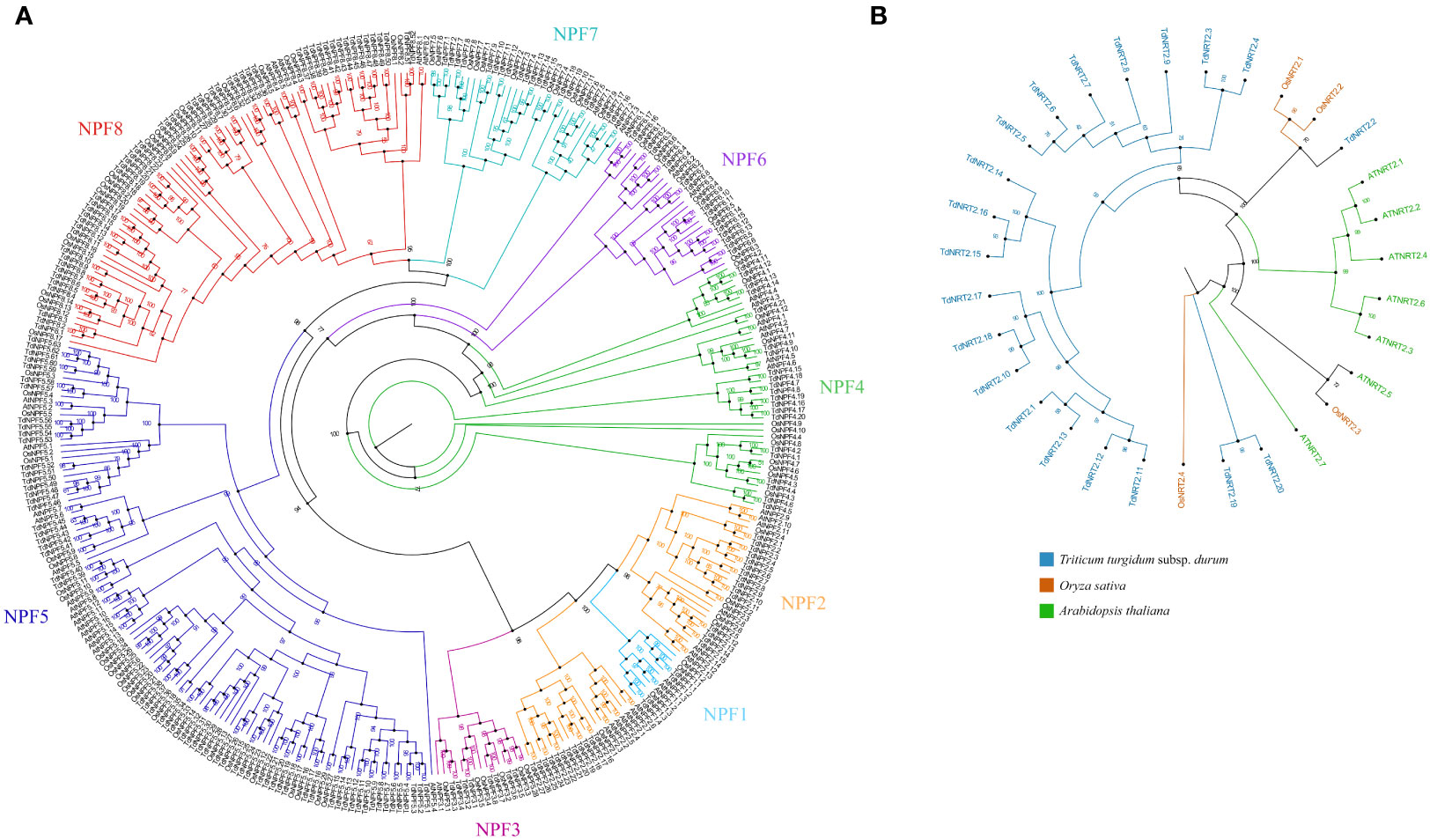
Figure 1 Phylogenetic analysis of the two gene families from Arabidopsis thaliana, Oryza sativa and Triticum durum. (A) Maximum likelihood tree of the NPFs full length protein sequences. The eight NPF sub-families are highlighted using colors: 1:Turquoise – 2:Orange – 3:Pink – 4:Green – 5:Blue – 6:Violet – 7:Cyan – 8:Red. (B) Maximum likelihood tree of the NRT2s full length protein sequences. The three species were highlighted using colors as indicated in the figure legend. Species specific branches were further highlighted using the same colors. Both trees were constructed using iqtree2.2, visualized, and modified using Figtree. Bootstrap values (1000 replicates) are shown for each node.
The NRT2 gene family was analyzed by using a similar approach; the maximum-likelihood phylogenetic tree was constructed based on the 31 NRT2 protein sequences, of which 7, 4 and 20 from Arabidopsis, rice and durum wheat, respectively. The phylogenetic tree revealed distinct evolutionary relationships among the NRT2 proteins of durum wheat and the other two species. Specifically, almost all the durum wheat NRT2 proteins formed a separate cluster, with only one protein (TdNRT2.2) closely grouped with the NRT2 proteins of Oryza sativa (OsNRT2.1 and 2.2) (Figure 1B). More interestingly, two other proteins (TdNRT2.19 and TdNRT2.20) showed a more ancient evolutionary divergence compared to all other TdNRT2 proteins, forming adistinct basal cluster, while all the AtNRT2 proteins clustered in three sub-clusters.
3.3 Chromosome location
The TdNPFs were evenly distributed along chromosomes in the A and B genomes (Figure S2A). The 2B chromosomal region (R2B) showed the highest gene density while the central chromosomal regions showed a lower gene density on average. Interestingly, four NPFs-enriched regions in chromosomes 2B, 3A, 3B, and 4B, also located in the R2B, were found, ranging from 5 to 7 genes per window (6Mb).
By contrast, the TdNRT2s were unevenly distributed along the genome, with chromosome 6 in both genomes (A and B) significantly enriched with 9 and 8 genes in 6B and 6A chromosomes, respectively (Figure S2B). Interestingly, all the TdNRT2s on chromosome 6 were located in the R1 in a significantly enriched window in both genomes (A and B) while only two gene members were found in the R2B (Figure S2A).
3.4 Gene structure and conserved motifs prediction
The gene structure of both gene families showed a significant difference in the number of exons and transcript isoforms (Figure S3). Most of the 211 TdNPFs exhibited more than two exons, with 85% of genes ranging from 3 to 6. The distribution of the transcript isoforms number was significantly different compared to the genome, with an average number of 3 transcript isoforms per gene. The TdNRT2 gene family showed a lower number of exons (9 out of 20 genes with one exon) and a lower number of transcripts per gene with more than 50% of genes showing only one transcript isoform.
To highlight conserved motifs and analyze their distribution among sub-families, protein motif analysis was carried out using the MEME tool. All the TdNPF proteins showed highly conserved motifs patterns (Figure S4). Despite that, the spatial organization and distance between conserved motifs were highly variable. The intra-motif variability was very high, with only few positions conserved in almost all the protein sequences (Figure S5).
Functional characterization of these motifs was performed using the NCBI protein domain search using the most represented sequence for each of the 25 conserved motifs. 14 motifs were assigned to the Major Facilitator Superfamily (MFS) while the remaining (11) were not assigned to any known protein domain. The motif#1 was identified in all the 211 NPF proteins, while the less conserved motif#19 and motif#21 were found only in 98 and 99 proteins, respectively. Furthermore, motif#18 (FILGNEFFERLAYYG), shared by 147 TdNPF proteins, contains the highly conserved ExxER/K peptide, suggesting its involvement in proton binding and transport. Among these sequences, both glutamic acids (E) were conserved in 80% of sequences, while the arginine residue (R) is less conserved. Rare motif variants such as ExxDR and ExxEE were also detected.
15 conserved motifs were identified in the TdNRT2 gene family (Figure S6). Nine out of 15 were assigned to the NO3- transmembrane transporter superfamily (PLN00028). Moreover, the distribution and position of the motifs created regular patterns and showed lower sequence variability compared to TdNPF family. Almost all the NRT2 genes shared many of the conserved motifs, except four highly variable genes (TRITD7Av1G231010, TRITD7Bv1G180680, TRITD2Av1G017380, TRITD6Bv1G008700).
3.5 Transcription factor binding sites, and CREs prediction
Transcription Factors (TFs) are essential for modulating gene transcription levels and many TFs directly regulate the expression of NPF and NRT2 genes (Marchive et al., 2013; Liu et al., 2017). We predicted the TF binding sites in promoter regions (3,000 bp upstream of transcription start site) of TdNPFs and TdNRT2 using the Binding Site Prediction tool of the PlantTFDB, and more than four thousand (4,072) binding sites for 163 TFs were identified in the promoter regions of 197 TdNPFs. The most abundant families of TFs were MYB, AP2, and NAC (Figure S7). One hundred twenty (120) binding sites for 53 TFs were detected in the promoter region of 19 TdNRT2s, of which the AP2 family resulted the most abundant. Interestingly, almost 96% (51 out of 53) of the TFs families were shared between NPF and NRT2 genes promoter region.
Cis-regulatory elements (CRE) are non-coding DNA regions also involved in the transcription regulation of neighboring genes (Bai et al., 2013). Here, we predicted CREs in the promoter regions of both TdNPFs and TdNRT2s using PlantCARE. Five thousand one hundred and twenty-one (5,121) CREs of 27 different types in the 211 promoter regions of TdNPF were found (Figure S7). The most abundant sites were the ABA responsive element (ABRE), DRE and MYB binding sites, activation sequence-1 (as-1), and the stress response element STRE, accounting for 70% of all the CREs. Other less abundant CREs were involved in light-response (G-box), biotic and abiotic stress response (MYC), and the common TATA-box and CAAT-box. One thousand five hundred and eighteen (1,518) CREs were predicted in the promoter regions of TdNRT2s. They were highly enriched in MYB and MYC binding sites, with many genes showing more than 5 sites in their upstream sequence accounting for almost 40% of CREs, in agreement with the previously described TF binding site prediction.
3.6 Expression profiles and co-expression analysis
The expression profiles of 211 TdNPF and 20 TdNRT2 genes were detected using publicly available RNA-seq datasets from the Sequence Reads Archive (SRA) covering 9 tissues at different growth stages from 13 different cultivars (Figure 2A). The hierarchical clustering based on TdNPF genes showed a clear tissue-specific signal, with almost all the samples from the same tissue clustering together (Figure 2B). The 211 NPF genes were further divided roughly into 9 clusters based on their expression patterns. These clusters ranged from 7 to 79 genes, with an average of 18 genes. Almost all the clusters showed the highest expression in roots, stem, and leaf, with five clusters being the most expressed (Cluster1-5). Interestingly, the cluster with the higher number of genes (Cluster 6) showed very constant low gene expression levels in almost all samples, except two small groups of genes induced in anthers, endosperm and roots.
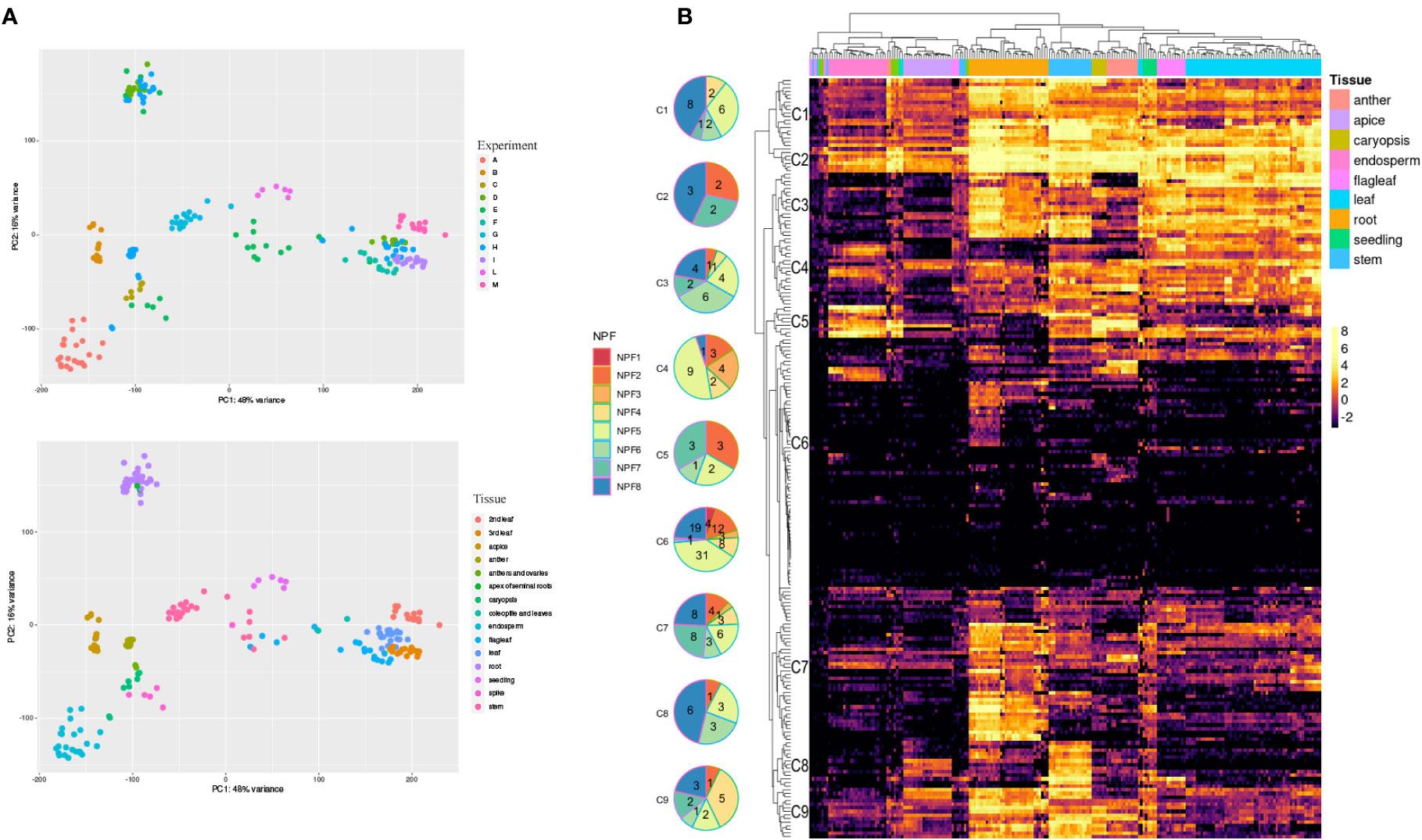
Figure 2 RNA-Seq of 195 samples obtained from the Short Reads Archive (SRA). (A) Principal Component Analysis performed with DESeq2 after the SVA correction. We highlighted SRA experiments (Top) and tissues (bottom). (B) Expression profiles of TdNPF genes in 9 tissues. Hierarchical clustering was performed both on rows and columns. Rows were roughly divided into 9 groups according to the similar expression levels. Pie charts were used to highlight the abundance of each NPF sub-family in each cluster. Tissues were highlighted as colored bars at the top of the heatmap.
TdNRT2 showed an opposite trend compared to TdNPF, with limited correlation between gene expression and tissue, except in the roots and seedlings (Figure S8). As might be expected, the higher gene expression was detected in roots, with 7 NRT2 members, highly similar to AtNRT2.1 and AtNRT2.4, that showed higher expression (TdNRT2.3,.4,.5,.6,.7,.8,.9) (Figure S8).
The same dataset was used to evaluate the co-expression of both NRT2 and NPF gene families adopting the Weighted Gene Co-expression Network Analysis (WGCNA) method. We detected fourteen co-expression modules which showed highly variable expression profiles. 87.2% of durum wheat genes were assigned to co-expression modules, with four modules, colored salmon, red, blue and brown, showing a significantly higher number of genes ranging from roughly thirteen thousand to eight thousand. Module-tissue relationships were evaluated to highlight each module expression profile (Figure 3A). TdNRT2s were assigned to modules brown (8), green (8), red (2) and turquoise (2) which were highly induced in roots, anthers, endosperm-apex and leaves-flag leaves, respectively (Table S7). These expression profiles closely correlated to the phylogenetic tree distribution of the NRT2 genes, with almost all TdNRT2 in the same co-expression module clustering together (Figure 3B). Furthermore, all six NAR2/NRT3 genes in the durum wheat genome were assigned to the brown module, highlighting their combined action mainly in roots.
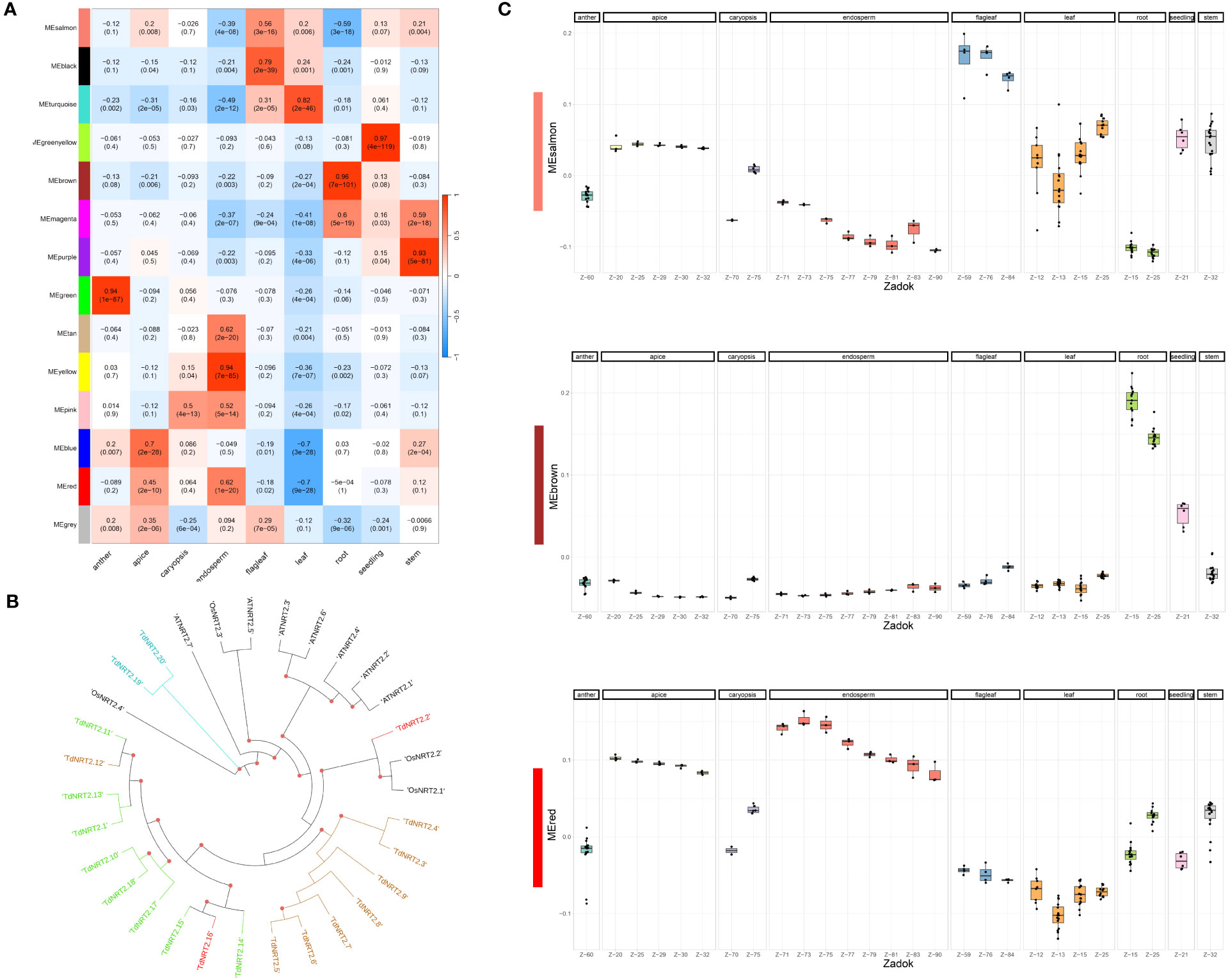
Figure 3 Weighted Gene Co-expression Network Analysis (WGCNA). (A) Heatmap of the Correlation coefficients between Module Eigengenes (WGCNA) and tissues. Pearson Correlation coefficient was evaluated using a binary matrix representing each tissue (1) against all the others (0). (B) Phylogenetic tree of TdNRT2 genes. Genes within each co-expression module form distinct clusters on the tree, visually distinguished by the use of module-specific colors (turquoise, red, brown and green). (C) Expression profile of the three most abundant co-expression modules visualized using the Boxplots of Module Eigengenes. Each tissue was highlighted in facets, while the developmental stage (Zadok) was highlighted on the X-axis.
TdNPFs showed a wider range of expression patterns, in agreement with the hierarchical clustering. They were assigned to ten of the fourteen modules detected. The majority of NPF genes belonged to the brown (52) and salmon (48) modules which were induced in root and flag leaf, respectively (Figure 3C). Interestingly 35 TdNPFs were assigned to three modules, red (23), yellow (9) and pink (3), highly upregulated in the caryopsis, especially in the endosperm, with three slightly different expression profiles (Figure S9). Furthermore, the network analysis allowed us to detect the hub-genes in each module. Among these we detected three NPF genes, TdNPF6.12, TdNPF6.8 and TdNPF5.61, belonging to brown, red and salmon modules, respectively. We further used co-expression modules to detect expression patterns in homologous genes among the two genomes (A and B). The half (50.4%) of the TdNPFs homologous belonged to different co-expression modules while only two NRT2 genes did not cluster in the same module.
3.7 NPF gene sequence divergence and collinearity in five species of the Triticeae tribe
Evolutionary constraints of durum wheat NPF and NRT2 genes was evaluated through pairwise comparisons of Ka/Ks values from five species belonging to the Triticeae tribe (Figure 4). In detail, the durum wheat TdNPFs were compared to their orthologs in T. urartu, Ae. speltoides, Ae. tauschii and T. aestivum genomes. Furthermore, the Ka/Ks values of each duplicated TdNPF gene pairs were also evaluated. Ka/Ks was evaluated for 170, 91, 83, 74 orthologs in the durum/aestivum, durum/speltoides, durum/urartu and aestivum/tauschii comparisons, respectively. Interestingly, both the durum/speltoides and the aestivum/tauschii comparison showed very low Ka/Ks values with an average of 0.19 and 0.23, respectively, by contrast, the highest values were detected in the durum/aestivum comparison with an average of 0.49. Five genes (TdNPF3.5, TdNPF4.7, TdNPF5.42, TdNPF7.12, TdNPF8.38) exhibited Ka/Ks values greater than 1.5, indicating a substantial positive selection acting on these genes. On the other hand, ten genes displayed a Ka/Ks value close to 1, suggesting a relatively neutral selection. Among the five genes with Ka/Ks > 1.5, three were associated with the brown co-expression module, indicating their upregulation specifically in roots. Based on these results, the significant difference between durum/aestivum and the other three comparisons was confirmed by Tukey’s test (Figure S10).
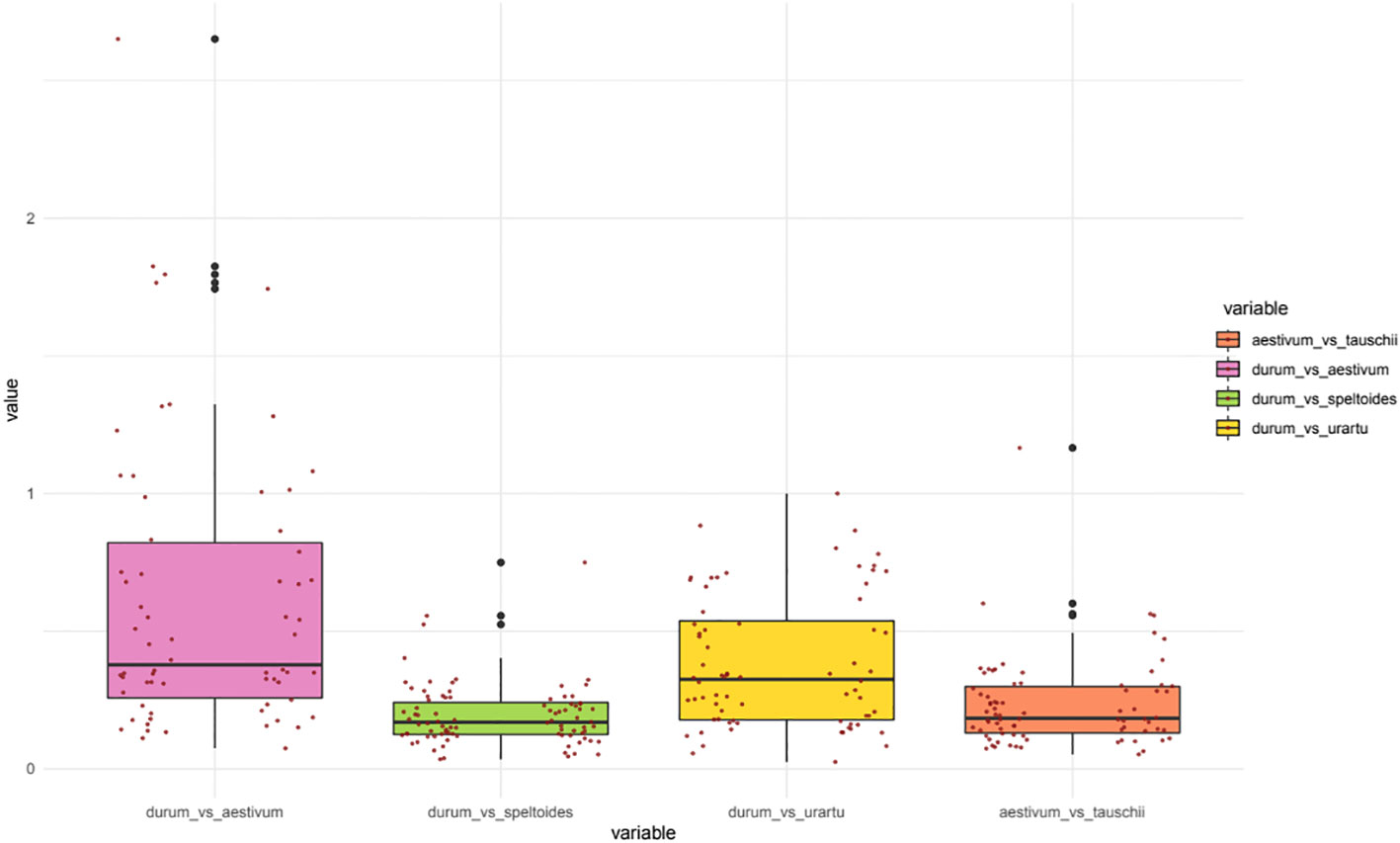
Figure 4 Boxplots of the Ka/Ks values for the orthologous NPF genes between five poaceae species: Durum wheat (Triticum turgidum subsp. durum), Bread wheat (Triticum aestivum), Aegilops speltoides Tausch, Triticum urartu and Aegilops tauschii. Two scatter plots (left: TdNPF on the A sub-genome; right: TdNPF on the B sub-genome) highlights the single Ka/Ks for each gene pairs.
Ka/Ks was evaluated also on durum wheat NPF and NRT2 tandem duplicated genes and NPF collinear genes between the two sub-genomes. All the gene pairs comparisons showed Ka/Ks values lower than 1, rarely higher than 0.5, suggesting strong purifying selection acting on duplicated genes, regardless the duplication event type. In particular, tandem duplications showed a slightly higher Ka/Ks among the NPF genes with an average of 0.31 compared to collinear duplicated NPF genes between sub-genome A and B (average 0.26). Finally, NRT2 showed a drastically lower Ka/Ks value ranging from 0.1 to 0.01.
Furthermore, using collinearity analysis through MCScanX we were able to characterize the relationships and the duplication events of both gene families inside the durum wheat genome and between these five species (Figure 5). In the durum wheat genome, almost 45% of TdNPFs were included in collinear pairs detected between A and B genomes. In detail, 77 segmental and 94 tandem duplications, as well as fewer dispersed (30) and proximal duplication (10) were detected; 42% of TdNPFs formed tandem blocks, with 11 blocks including three or more genes.
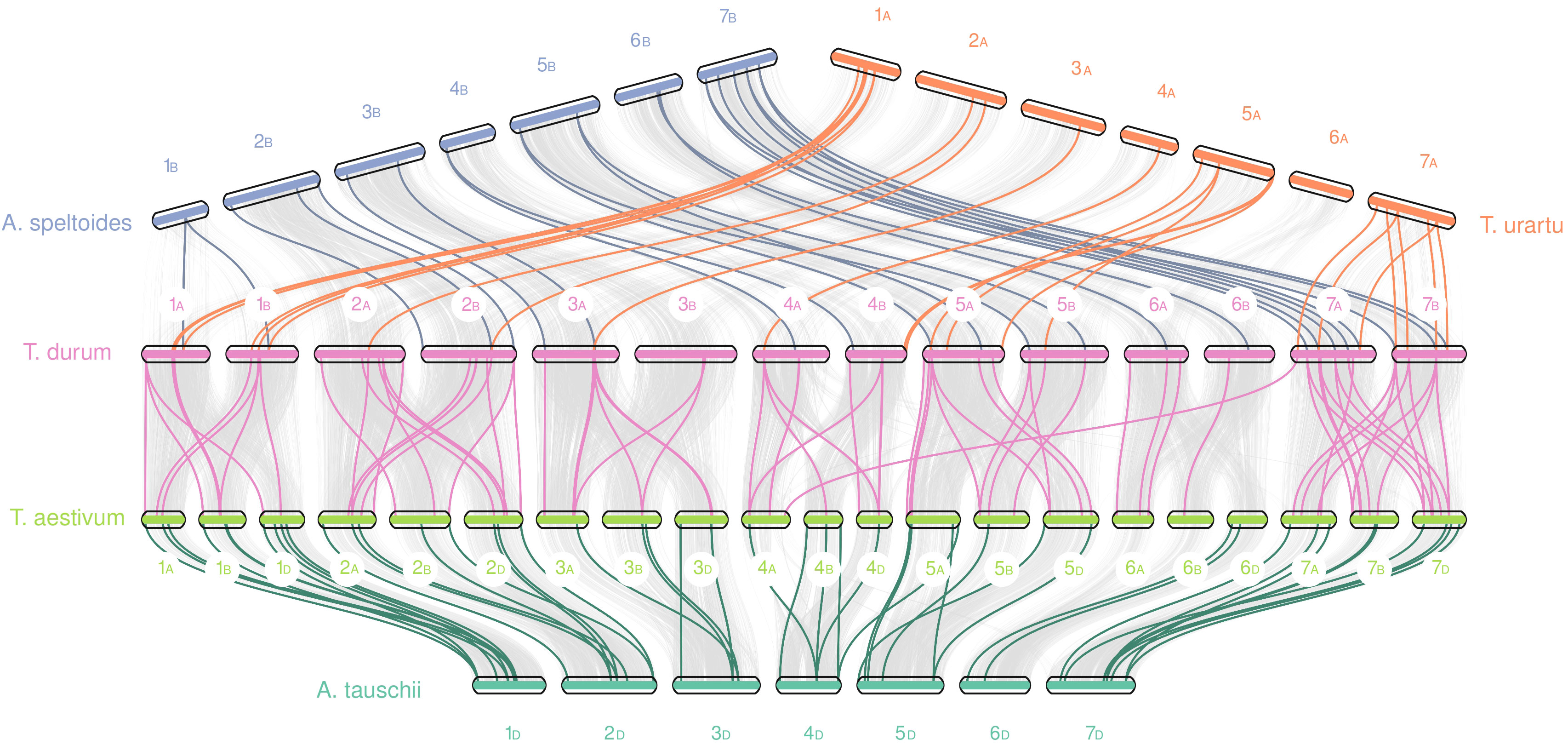
Figure 5 Collinearity analysis of NPF genes between five species from the Trititceae tribe. Grey lines indicate collinear blocks between genomes, while the colored lines indicate NPF genes detected inside collinear blocks. Sub-genomes were highlighted using the A, B or D letters.
TdNRT2s are mainly located in two enriched regions on chromosomes 6A and 6B, as previously highlighted. These formed 5 tandem blocks, 3 and 2 located on 6B and 6A chromosomes, respectively. Furthermore, 14 tandem and no segmental duplications were detected.
Interspecific analysis of NPF genes revealed 23 and 25 collinear blocks in durum-speltoides and durum-urartu comparisons, respectively, 48 and 75 pairs in aestivum-aegilops and durum-aestivum comparisons, respectively. Almost all the blocks were detected between homologous chromosomes among the five genomes, significant differences in the number of blocks between A, B or D sub-genomes were not detected.
4 Discussion
In plants, both the NPF and the NRT2 gene families are involved in nitrate/nitrite uptake, translocation and remobilization. NPFs are also involved in the transport of many other substrates such as hormones, secondary metabolites, peptides, chloride and potassium. A deeper characterization of these gene families is crucial to understand plant nitrate and metabolite transport.
In the present study, 211 TdNPFs and 20 TdNRT2s were identified in the Triticum turgidum L. subsp. durum (Desf.) Husn. genome. These numbers were comparable to other allopolyploid species such as Brassica napus (199 NPFs, 17 NRT2s), Saccharum spontaneum (178, 20), and Triticum aestivum (331, 46) and significantly higher than many diploid monocots and dicots such as Arabidopsis thaliana (53 and 7), Oryza sativa (93, 4) and Zea mays (79, 1). The NPF gene family expansion in plants seems to have arisen from neo- and sub-functionalization, as suggested by many reports (Lynch and Conery, 2000; O’Brien et al., 2016; Jørgensen et al., 2017; Wang et al., 2019a). In wheat, the large number of members in both gene families could be involved in highly differentiated responses to the availability of various substrates. Indeed, the high number of TdNPF and TdNRT2 genes, deriving from recent polyploidization and duplication events, may provide a higher modularity in terms of substrate affinity, condition or tissue specific gene expression induction and new protein-protein interactions. Similar effects were reported in many allopolyploid species such as rice, soybean, cotton and, in the MIKC-type MADS-box gene group, in bread wheat (Flagel et al., 2008; Schilling et al., 2020; Lee and Szymanski, 2021). In Triticum aestivum, the diversification of both NPF and NRT2 gene families was likely due to drift, leading to significant differences in the N-use efficiency of subpopulations clustered based on Single Nucleotide Polymorphisms (SNPs) within the NPF and NRT2 genes (Li et al., 2021).
Our phylogenetic analysis highlighted a high divergence among NRT2 genes from durum wheat, rice and Arabidopsis with genes from each species clustering together in distinguished groups. The best hit (BLASTP) of many TdNRT2s against the Arabidopsis NRT2 protein sequences is often AtNRT2.1, making it hard to functionally characterize these transporters solely based on their sequence homology, thus the association of previous functional annotations from model species to the newly identified genes in durum wheat become challenging. Here, we focused on sequence, gene expression and protein domains characterization, but further studies will be needed to fully describe TdNRT2s at a functional level.
The phylogenetic analysis on the NPF genes yielded more informative results, with all the orthologous genes belonging to the same sub-family clustering together, allowing us a much more reliable annotation of the novel TdNPFs. These results support the hypothesis of NPF family divergence before the separation of monocots and dicots, as suggested by Wang et al. (2019b). Furthermore, the NPF1 branch clustered inside the NPF2 branch, breaking it in two sub-classes. This feature was observed in other species such as Brassica napus and often led to the definition of more than eight sub-families, with NPF2 split into NPF2-1 and NPF2-2(Wen et al., 2020). Interestingly, this split was not detected in Triticum aestivum (Li et al., 2021; Kumar et al., 2022) potentially due to slightly different plant species utilized or to slightly different clustering method.
The chromosome location of both transporter families showed a non-random distribution inside the genome. Interestingly, TdNRT2s are highly concentrated on chromosomes 6 from both genomes (A and B), probably due to the multiple tandem duplication events, as shown by the results of collinearity analysis. In Arabidopsis, the AtNRT2.1 and AtNRT2.2 genes are adjacent, and end to end on chromosome 1, and this apparent duplication has been seen for orthologues in other species. AtNRT2.5 is also located on chromosome 1. Three other NRT2s are located on chromosome 5, with only AtNR2.6 located on chromosome 3. A similar enrichment on chromosome 6 of all three genomes (A, B and D) was detected in bread wheat, also deriving from tandem duplication that was suggested to have arisen from unequal crossing-over events (Li et al., 2021). Although similar, the number of NRT2 genes in these genomic regions is higher in bread wheat, suggesting that some of these duplication events should have occurred after or during the hybridization of durum wheat with Aegilops tauschii (genome D) as supposed by the International Wheat Genome Sequencing Consortium (IWGSC) (2014). Further studies on the intraspecific variability of these gene families among the main wheat species could help to deeply understand how their expansions occurred and what type of mechanisms underlie their preservation after duplication.
In Arabidopsis, several putative NO3− response cis-regulatory elements (CREs) have been detected in many promoters of N-related genes, while limited information is available for other plant species (Konishi and Yanagisawa, 2010; Wang et al., 2010; Rolly and Yun, 2021). Here, a high number of CREs related to ABA signaling and binding of Drought Responsive Element (DRE) and MYB TFs were detected in the TdNPFs promoter regions. Interestingly, the TdNRT2s upstream region was also highly enriched with MYB binding sites, as shown by both CREs and TF binding site prediction. MYB TFs are often involved in abiotic and biotic stress responses as well as in plant development, root and flower development (Kaur et al., 2017), although their role in NO3–related regulation has also been reported (Todd et al., 2004; Wang et al., 2018a; Zhang et al., 2021; Puccio et al., 2022). Interestingly, both gene family promoters showed multiple putative MYB binding sites for many genes. The presence of multiple binding sites for the same TF on the promoter of one or more genes has been often associated with a higher sensitivity to specific TFs (Howard and Davidson, 2004; Yáñez-Cuna et al., 2013; Brendolise et al., 2017).
The presence of many CREs and the partial overlap of their functions between the two gene families suggested that a complex regulatory network may be involved in modulating and fine-tuning their expression, with some TFs putatively involved in the regulation of members of both families. These could be involved in the spatiotemporal- or tissue-specific activation of transporter genes or may take part in the signaling cascade in response to the fluctuations of specific substrates concentration into the soil. Interestingly, the same analysis performed on the NPFs from Brassica napus yielded similar results on NPF genes (Wen et al., 2020), suggesting that the regulation of this gene family may involve the same TFs classes and could be evolutionarily conserved.
Fourteen motifs were assigned to the Major Facilitator Superfamily (MFS), the remaining 11 were not assigned to any known protein domain, suggesting a highly specific function for these peptides (putatively species-specific). Interestingly, 11 TdNPF7 proteins showed the monocot-specific variant ExxES, which is associated to non-proton dependent nitrate uptake and specific to the NPF7 sub-family (Longo et al., 2018). These genes were defined as NPF7a in rice, involved in the low-affinity nitrate transport system with some being tonoplast located (Hu et al., 2016).
Overall, NPFs showed a more variable gene structure and sequence variability in their conserved motifs compared to NRT2s. Interestingly, the NRT2s showed a simpler gene structure, with only one or two exons and highly clustered genomic locations. Both the gene structure and the chromosome locations of NRT2s seemed highly conserved among many monocots such as Brachypodium distachyon, Saccharum spontaneum, and bread wheat, mainly distributed on two chromosomes and harboring mainly one or two exons (Wang et al., 2019a; Li et al., 2021).
The high percentage of NPFs deriving from segmental and tandem duplication (37% and 44%, respectively) found in durum wheat supported the role of these genomic events in the expansion of NPF genes already reported in bread wheat (Li et al., 2021). Furthermore, the collinear analysis between durum and bread wheat and the putative A, B and D genomes was not able to detect a significantly higher number of collinear blocks between each putative sub-genome donor and the respective sub-genomes in durum or bread wheat. This observation does not directly support the idea that the expansion of these families derives from ancient duplication events in diploid wheat species, which should have occurred before hybridization into allopolyploid species (Salse et al., 2008). Instead, our results indirectly support the idea that the substantial increase in gene members in both these families is mainly due to tandem and segmental duplications in the tetraploid or hexaploid ancestral genomes, and not in the diploid ancestral genomes. These duplications seemed favored by polyploidization events, with bread wheat showing a higher number of duplication events (Buchner and Hawkesford, 2014; Kumar et al., 2022). Furthermore, tandem duplicated NPF genes in durum wheat genomes showed strong purifying selection, suggesting preserved function after duplication, in agreement with many studies on other gene families (Hu et al., 2018; Hajiahmadi et al., 2020; Zhu et al., 2020).
In allopolyploid species, gene expression patterns can be significantly altered and this is one of the main sources of phenotypic variation (Jackson and Chen, 2010). Here, by using 195 RNA-seq durum wheat datasets the expression profiles highlighted different trends in both gene families. The TdNPFs expression patterns resulted highly tissue-specific, with most samples from specific tissue forming distinct clusters. By contrast, NRT2 genes were predominantly expressed in roots and anthers, being assigned to brown and green modules. The distinctiveness of these two groups of TdNRT2s becomes even more evident, as we observed that all the NAR2/NRT3 genes are present in the brown module. This finding implies that most of the NRT2 genes in wheat are either engaged in root uptake, facilitated by NAR2/NRT3, or have undergone evolutionary adaptations for translocation or accumulation in anthers/seeds. Additionally, four members of TdNRT2 showed a more complex expression profile, with TdNRT2.2 and TdNRT2.16 being highly induced in apex, grain and endosperm during maturation, while TdNRT2.19 and TdNRT2.20 being expressed in leaves and flag-leaves. The detection of NRT2 genes responsible for seed N-accumulation, such as TdNRT2.2 and TdNRT2.16, could be crucial to increase yield and higher N content, as already demonstrated by their overexpression in bread wheat (Li et al., 2020). TdNRT2.19 and TdNRT2.20 were the most basal genes in our phylogenetic analysis together with OsNRT2.4 and AtNRT2.7 in agreement with recent reports (Li et al., 2021; Deng et al., 2023; Kumar et al., 2023). Interestingly, both OsNRT2.4 and AtNRT2.7 are mainly expressed in the tonoplast of maturing seeds and roots, which seems to suggest differentiated functions of these basal genes in the vacuole (Chopin et al., 2007; Wei et al., 2018) and contrasting with most other family members located in the plasma membrane.
TdNRT2.2 was closely related to both OsNRT2.1 and OsNRT2.2, which are usually expressed in root and germinating seeds (Feng et al., 2011). Furthermore, both phylogenetic and co-expression clustering yielded mostly the same results, with almost all the TdNRT2s from the same phylogenetic branch belonging to the same co-expression module. These results highlighted a close relationship between nucleotide variation and gene expression in this family, suggesting a shared selection mechanism between coding and regulatory regions. Similar coordinated evolution has been already observed in many gene families in mammals and plants (Necsulea and Kaessmann, 2014; Wang et al., 2020; Winkelmüller et al., 2021). Furthermore, duplication events may induce expression shifts favored by gene neo-functionalization as suggested by Fukushima and Pollock (2020), and this hypothesis could enhance the co-evolution of genome and transcriptome in the NRT2 gene family in durum wheat.
The expression profiles of homologous genes showed significant variation, mainly within the NPF family. Indeed, about half of the NPF homologues exhibited dissimilar expression patterns. Although these differences may have already been present in ancestral genomes, the maintenance or development of highly similar genes with different expression patterns may provide a greater degree of modularity for regulation.
5 Conclusions
Our approach led to a comprehensive characterization of the NPF and NRT2 gene families in the durum wheat genome. Manual annotation of these transporters is crucial for understanding NO3- and N dynamics and their impact on NUE in durum wheat. This study identified 211 TdNPF and 20 TdNRT2 genes for the first time, providing detailed insights into their protein sequences and conserved domains and on their regulatory elements. By extensively analyzing nearly all publicly available RNA-seq datasets, we achieved the most comprehensive characterization of both gene expression profiles and co-expression relationships. This investigation confirmed that a considerable number of these genes underwent neo- or sub-functionalization following small-scale duplication events. These findings indicate that the expansion of these gene families in wheat holds promising potential as a valuable resource for identifying NUE-related genes and as potential candidates for molecular markers and the development of transgenic plants. By understanding the key players involved in durum wheat production and incorporating these findings into future research, we can take significant steps towards more eco-friendly and sustainable durum wheat fertilization management, addressing a critical challenge in modern agriculture.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
GP: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. RI: Conceptualization, Supervision, Validation, Visualization, Writing – review & editing. DG: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. AF: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. AH: Conceptualization, Formal Analysis, Investigation, Methodology, Supervision, Writing – review & editing. FS: Conceptualization, Formal Analysis, Investigation, Methodology, Resources, Supervision, Validation, Writing – review & editing. FM: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Agritech National Research Center funded within the European Union Next-Generation EU program (the National Recovery and Resilience Plan, mission 4, component 2, investment 1.4 – D.D. 1032 del 17/06/2022, project CN00000022). This manuscript reflects only the authors’ views and opinions, neither the European Union nor European Commission can be considered responsible for them. This work was also partially supported by the ANCIENT project (n. 942500454), granted by the Assessorato Regionale dell’agricoltura, dello sviluppo rurale e della pesca mediterranea, Regione Sicilia (P.S.R. Sicilia. 2014/2020, Sottomisura 16.1, DRS n. 3390/2022 del 11.08.2022). This research was also partially funded by BIAS - Innovative biofertilizers for sustainable agriculture to protect human health and the environment (project number 082015000275), funded by Sicily Region through the European Regional Development Fund (PO-FESR Sicilia 2014-2020).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1302337/full#supplementary-material
References
Avni, R., Lux, T., Minz-Dub, A., Millet, E., Sela, H., Distelfeld, A., et al. (2022). Genome sequences of three Aegilops species of the section Sitopsis reveal phylogenetic relationships and provide resources for wheat improvement. Plant J. 110, 179–192. doi: 10.1111/tpj.15664
Bagchi, R., Salehin, M., Adeyemo, O. S., Salazar, C., Shulaev, V., Sherrier, D. J., et al. (2012). Functional assessment of the Medicago truncatula NIP/LATD protein demonstrates that it is a high-affinity nitrate transporter. Plant Physiol. 160, 906–916. doi: 10.1104/pp.112.196444
Bai, H., Euring, D., Volmer, K., Janz, D., Polle, A. (2013). The nitrate transporter (NRT) gene family in poplar. PloS One 8, e72126. doi: 10.1371/journal.pone.0072126
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Bajgain, P., Russell, B., Mohammadi, M. (2018). Phylogenetic analyses and in-seedling expression of ammonium and nitrate transporters in wheat. Sci. Rep. 8, 1–13. doi: 10.1038/s41598-018-25430-8
Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., et al. (2004). The Pfam protein families database. Nucleic Acids Res. 32, D138–D141. doi: 10.1093/nar/gkh121
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolser, D., Staines, D. M., Pritchard, E., Kersey, P. (2016). Ensembl plants: integrating tools for visualizing, mining, and analyzing plant genomics data. Plant Bioinformatics: Methods Protoc. 1374, 115–140. doi: 10.1007/978-1-4939-3167-5_6
Bouguyon, E., Brun, F., Meynard, D., Kubeš, M., Pervent, M., Leran, S., et al. (2015). Multiple mechanisms of nitrate sensing by Arabidopsis nitrate transceptor NRT1.1. Nat. Plants 1, 15015. doi: 10.1038/nplants.2015.15
Brendolise, C., Montes, E., Hummel, I., Gouesnard, B., Glöckler, J., Martin, O., et al. (2017). Multiple copies in maize of Male Transmission Ratio Distortion (MTRD) region corresponding to an insertion in the reference genome. J. Exp. Bot. 68, 2075–2086. doi: 10.1093/jxb/erx090
Buchner, P., Hawkesford, M. J. (2014). Complex phylogeny and gene expression patterns of members of the NITRATE TRANSPORTER 1/PEPTIDE TRANSPORTER family (NPF) in wheat. J. Exp. Bot. 65, 5697–5710. doi: 10.1093/jxb/eru231
Chao, H., He, J., Cai, Q., Zhao, W., Fu, H., Hua, Y., et al. (2021). The expression characteristics of NPF genes and their response to vernalization and nitrogen deficiency in rapeseed. Int. J. Mol. Sci. 22, 4944. doi: 10.3390/ijms22094944
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chiba, Y., Shimizu, T., Miyakawa, S., Kanno, Y., Koshiba, T., Kamiya, Y., et al. (2015). Identification of Arabidopsis thaliana NRT1/PTR FAMILY (NPF) proteins capable of transporting plant hormones. J. Plant Res. 128, 679–686. doi: 10.1007/s10265-015-0710-2
Chin, C. H., Chen, S. H., Wu, H. H., Ho, C. W., Ko, M. T., Lin, C. Y. (2014). cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, 1–7. doi: 10.1186/1752-0509-8-S4-S11
Chopin, F., Orsel, M., Dorbe, M. F., Chardon, F., Truong, H. N., Miller, A. J., et al. (2007). The Arabidopsis ATNRT2. 7 nitrate transporter controls nitrate content in seeds. Plant Cell 19, 1590–1602. doi: 10.1105/tpc.107.050542
Corratg’e-Faillie, C., Lacombe, B. (2017). Substrate (un)specificity of arabidopsis NRT1/PTR FAMILY (NPF) proteins. J. Exp. Bot. 68, 3107–3113. doi: 10.1093/jxb/erw499
Deng, Q.-Y., Luo, J.-T., Zheng, J.-M., Tan, W.-F., Pu, Z.-J., Wang, F. (2023). Genome-wide systematic characterization of the NRT2 gene family and its expression profile in wheat (Triticum aestivum L.) during plant growth and in response to nitrate deficiency. BMC Plant Biol. 23, 353. doi: 10.1186/s12870-023-04333-5
Duan, J., Tian, H., Gao, Y. (2016). Expression of nitrogen transporter genes in roots of winter wheat (Triticum aestivum L.) in response to soil drought with contrasting nitrogen supplies. Crop Pasture Sci. 67, 128–136. doi: 10.1071/CP15152
Fan, X., Tang, Z., Tan, Y., Zhang, Y., Luo, B., Yang, M., et al. (2016). Overexpression of a pH-sensitive nitrate transporter in rice increases crop yields. Proc. Natl. Acad. Sci. 113, 7118–7123. doi: 10.1073/pnas.1525184113
Feng, H., Yan, M., Fan, X.-L., Li, B.-J., Shen, Q., Miller, A. J., et al. (2011). Spatial expression and regulation of rice high-affinity nitrate transporters by nitrogen and carbon status. J. Exp. Bot. 62, 2319–2332. doi: 10.1093/jxb/erq403
Flagel, L. E., Udall, J. A., Nettleton, D., Wendel, J. F. (2008). Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC Biol. 6, 16. doi: 10.1186/1741-7007-6-16
Fukushima, K., Pollock, D. D. (2020). Amalgamated cross-species transcriptomes reveal organ-specific propensity in gene expression evolution. Nat. Commun. 11, 4459. doi: 10.1038/s41467-020-18090-8
Ge, S.-X., Jung, D., Yao, R. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. doi: 10.1093/bioinformatics/btz931
Giambalvo, D., Amato, G., Badagliacca, G., Ingraffia, R., Di Miceli, G., Frenda, A. S., et al. (2018). Switching from conventional tillage to no-tillage: Soil N availability, N uptake, 15N fertilizer recovery, and grain yield of durum wheat. Field Crops Res. 218, 171–181. doi: 10.1016/j.fcr.2018.01.018
Gojon, A., Krouk, G., Perrine-Walker, F., Laugier, E. (2011). Nitrate transceptor(s) in plants. J. Exp. Bot. 62, 2299–2308. doi: 10.1093/jxb/erq419
Good, A. G., Shrawat, A. K., Muench, D. G. (2004). Can less yield more? Is reducing nutrient input into the environment compatible with maintaining crop production? Trends Plant Sci. 9, 597–605. doi: 10.1016/j.tplants.2004.10.008
Hajiahmadi, Z., Abedi, A., Wei, H., Sun, W., Ruan, H., Zhuge, Q., et al. (2020). Identification, evolution, expression, and docking studies of fatty acid desaturase genes in wheat (Triticum aestivum L.). BMC Genomics 21, 778. doi: 10.1186/s12864-020-07199-1
Hatje, K., Keller, O., Hammesfahr, B., Pillmann, H., Waack, S., Kollmar, M. (2011). Cross-species protein sequence and gene structure prediction with fine-tuned Webscipio 2.0 and Scipio. BMC Res. Notes 4, 1–20. doi: 10.1186/1756-0500-4-265
Hawkesford, M. J. (2017). Genetic variation in traits for nitrogen use efficiency in wheat. J. Exp. Bot. 68, 2627–2632. doi: 10.1093/jxb/erx079
Hawkesford, M. J., Griffiths, S. (2019). Exploiting genetic variation in nitrogen use efficiency for cereal crop improvement. Curr. Opin. Plant Biol. 49, 35–42. doi: 10.1016/j.pbi.2019.05.003
Hawkins, J., Bodén, M. (2006). Detecting and sorting targeting peptides with neural networks and support vector machines. J. Bioinf. Comput. Biol. 4, 1–18. doi: 10.1142/S0219720006001771
Horton, P., Park, K.-J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Howard, M. L., Davidson, E. H. (2004). cis-Regulatory control circuits in development. Dev. Biol. 271, 109–118. doi: 10.1016/j.ydbio.2004.03.031
Hu, R., Qiu, D., Chen, Y., Miller, A. J., Fan, X., Pan, X., et al. (2016). Knock-down of a tonoplast localized low-affinity nitrate transporter osNPF7.2 affects rice growth under high nitrate supply. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01529
Hu, B., Wang, W., Ou, S., Tang, J., Li, H., Che, R., et al. (2015). Variation in NRT1. 1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 47, 834–838. doi: 10.1038/ng.3337
Hu, R., Xiao, J., Gu, T., Yu, X., Zhang, Y., Chang, J., et al. (2018). Genome-wide identification and analysis of WD40 proteins in wheat (Triticum aestivum L.). BMC Genomics 19, 803. doi: 10.1186/s12864-018-5157-0
International Wheat Genome Sequencing Consortium (IWGSC), Mayer, K. F., Rogers, J., Doležel, J., Pozniak, C., Eversole, K., et al. (2014). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 1251788. doi: 10.1126/science.1251788
Jackson, S., Chen, Z. J. (2010). Genomic and expression plasticity of polyploidy. Curr. Opin. Plant Biol. 13, 153–159. doi: 10.1016/j.pbi.2009.11.004
Javed, T., Singhal, R., Shabbir, R., Kumar, P., Shah, A. N., Jinger, D., et al. (2022). Recent advances in agronomic and physio-molecular approaches for improving nitrogen use efficiency in crop plants. Front. Plant Sci. 13, 877544. doi: 10.3389/fpls.2022.877544
Jørgensen, M. E., Xu, D., Crocoll, C., Ernst, H. A., Ramírez, D., Motawia, M. S., et al. (2017). Origin and evolution of transporter substrate specificity within the NPF family. eLife 6, e19466. doi: 10.7554/eLife.19466
Kanstrup, C., Nour-Eldin, H. H. (2022). The emerging role of the nitrate and peptide transporter family: NPF in plant specialized metabolism. Curr. Opin. Plant Biol. 68, 102243. doi: 10.1016/j.pbi.2022.102243
Kaur, A., Pati, P. K., Pati, A. M., Nagpal, A. K. (2017). In-silico analysis of cis-acting regulatory elements of pathogenesis-related proteins of Arabidopsis thaliana and Oryza sativa. PloS One 12, e0184523. doi: 10.1371/journal.pone.0184523
Kimber, G., Feldman, M. (1987). Wild wheat. An introduction (Columbia, Mo, U.S.A.: College of Agriculture University of Missouri). 353, Wild wheat. An introduction.
Konishi, M., Yanagisawa, S. (2010). Identification of a nitrate-responsive cis-element in the Arabidopsis NIR1 promoter defines the presence of multiple cis-regulatory elements for nitrogen response. Plant J. 63, 269–282. doi: 10.1111/j.1365-313X.2010.04239.x
Krogh, A., Larsson, B., Von Heijne, G., Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kumar, A., Jayaswal, P. K., Mahato, A. K., Arya, A., Mandal, P. K., Singh, N. K., et al. (2023). Growth stage and nitrate limiting response of NRT2 and NAR2 gene families of bread wheat, and complementation and retrieval of nitrate uptake of atnrt2. 1 mutant by a wheat NRT2 gene. Environ. Exp. Bot. 207, 105205. doi: 10.1016/j.envexpbot.2022.105205
Kumar, A., Sandhu, N., Kumar, P., Pruthi, G., Singh, J., Kaur, S., et al. (2022). Genome-wide identification and in silico analysis of NPF, NRT2, CLC and SLAC1/SLAH nitrate transporters in hexaploid wheat (Triticum aestivum). Sci. Rep. 12, 11227. doi: 10.1038/s41598-022-15202-w
Langfelder, P., Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 9, 559. doi: 10.1186/1471-2105-9-559
Lee, Y., Szymanski, D. B. (2021). Multimerization variants as potential drivers of neofunctionalization. Sci. Adv. 7, eabf0984. doi: 10.1126/sciadv.abf0984
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E., Storey, J. D. (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. doi: 10.1093/bioinformatics/bts034
Leinonen, R., Sugawara, H., Shumway, M., Collaboration, I. S. D. (2010). The sequence read archive. Nucleic Acids Res. 39, D19–D21. doi: 10.1093/nar/gkq1019
Léran, S., Varala, K., Boyer, J.-C., Chiurazzi, M., Crawford, N., Daniel-Vedele, F., et al. (2014). A unified nomenclature of NITRATE TRANSPORTER 1/PEPTIDE TRANSPORTER family members in plants. Trends Plant Sci. 19, 5–9. doi: 10.1016/j.tplants.2013.08.008
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Li, W., He, X., Chen, Y., Jing, Y., Shen, C., Yang, J., et al. (2020). A wheat transcription factor positively sets seed vigour by regulating the grain nitrate signal. New Phytol. 225, 1667–1680. doi: 10.1111/nph.16234
Li, M., Tian, H., Gao, Y. (2021). A genome-wide analysis of NPF and NRT2 transporter gene families in bread wheat provides new insights into the distribution, function, regulation and evolution of nitrate transporters. Plant Soil 465, 47–63. doi: 10.1007/s11104-021-04927-8
Li, W., Wang, Y., Okamoto, M., Crawford, N. M., Siddiqi, M. Y., Glass, A. D. M. (2007). Dissection of the AtNRT2.1–AtNRT2.2 inducible high-affinity nitrate transporter gene cluster. Plant Physiol. 143, 425–433. doi: 10.1104/pp.106.091223
Liu, K.-H., Huang, C.-Y., Tsay, Y.-F. (1999). CHL1 is a dual-affinity nitrate transporter of arabidopsis involved in multiple phases of nitrate uptake. Plant Cell 11, 865–874. doi: 10.1105/tpc.11.5.865
Liu, K. H., Niu, Y., Konishi, M., Wu, Y., Du, H., Chung, H. S. (2017). Discovery of nitrate–CPK–NLP signalling in central nutrient–growth networks. Nature 545, 311–316. doi: 10.1038/nature22077
Longo, A., Miles, N. W., Dickstein, R. (2018). Genome mining of plant NPFs reveals varying conservation of signature motifs associated with the mechanism of transport. Front. Plant Sci. 9, 1668. doi: 10.3389/fpls.2018.01668
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Lupini, A., Preiti, G., Badagliacca, G., Abenavoli, M. R., Sunseri, F., Monti, M., et al. (2021). Nitrogen use efficiency in durum wheat under different nitrogen and water regimes in the mediterranean basin. Front. Plant Sci. 11, 607226. doi: 10.3389/fpls.2020.607226
Lynch, M., Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Maghiaoui, A., Bouguyon, E., Cuesta, C., Perrine-Walker, F., Alcon, C., Krouk, G., et al. (2020). The Arabidopsis NRT1.1 transceptor coordinately controls auxin biosynthesis and transport to regulate root branching in response to nitrate. (A Murphy, Ed.). J. Exp. Bot. 71, 4480–4494. doi: 10.1093/jxb/eraa242
Marchive, C., Roudier, F., Castaings, L., Brehaut, L., Blondet, E., Colot, V. (2013). Nuclear retention of the transcription factor NLP7 orchestrates the early response to nitrate in plants. Nat. Commun. 4, 1713. doi: 10.1038/ncomms2650
Matsuoka, Y. (2011). Evolution of polyploid Triticum wheats under cultivation: the role of domestication, natural hybridization and allopolyploid speciation in their diversification. Plant Cell Physiol. 52, 750–764. doi: 10.1093/pcp/pcr018
Meier, M., Liu, Y., Lay-Pruitt, K. S., Takahashi, H., Von Wirén, N. (2020). Auxin-mediated root branching is determined by the form of available nitrogen. Nat. Plants 6, 1136–1145. doi: 10.1038/s41477-020-00756-2
Miller, A. J., Fan, X., Shen, Q., Smith, S. J. (2007). Expression and functional analysis of rice NRT2 nitrate transporters. Comp. Biochem. Physiol. Part A: Mol. Integr. Physiol. 146, S241. doi: 10.1016/j.cbpa.2007.01.618
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121–e121. doi: 10.1093/nar/gkt263
Moll, R. H., Kamprath, E. J., Jackson, W. A. (1982). Analysis and interpretation of factors which contribute to efficiency of nitrogen utilization1. Agron. J. 74, 562–564. doi: 10.2134/agronj1982.00021962007400030037x
Mounier, E., Pervent, M., Ljung, K., Gojon, A., Nacry, P. (2014). Auxin-mediated nitrate signalling by NRT1.1 participates in the adaptive response of Arabidopsis root architecture to the spatial heterogeneity of nitrate availability: Nitrate signalling by NRT1.1. Plant Cell Environ. 37, 162–174. doi: 10.1111/pce.12143
Necsulea, A., Kaessmann, H. (2014). Evolutionary dynamics of coding and non-coding transcriptomes. Nat. Rev. Genet. 15, 734–748. doi: 10.1038/nrg3802
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
O’Brien, J. A., Vega, A., Bouguyon, E., Krouk, G., Gojon, A., Coruzzi, G., et al. (2016). Nitrate transport, sensing, and responses in plants. Mol. Plant 9, 837–856. doi: 10.1016/j.molp.2016.05.004
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Pellizzaro, A., Clochard, T., Planchet, E., Limami, A. M., Mor`ere-Le Paven, M. (2015). Identification and molecular characterization of Medicago truncatula NRT2 and NAR2 families. Physiologia Plantarum 154, 256–269. doi: 10.1111/ppl.12314
Plett, D., Toubia, J., Garnett, T., Tester, M., Kaiser, B. N., Baumann, U. (2010). Dichotomy in the NRT gene families of dicots and grass species. PloS One 5, e15289. doi: 10.1371/journal.pone.0015289
Puccio, G., Crucitti, A., Tiberini, A., Mauceri, A., Taglienti, A., Palumbo Piccionello, A., et al. (2022). WRKY gene family drives dormancy release in onion bulbs. Cells 11, 1100. doi: 10.3390/cells11071100
Rolly, N. K., Yun, B. W. (2021). Regulation of nitrate (NO3) transporters and glutamate synthase-Encoding genes under drought stress in arabidopsis: the regulatory role of atbZIP62 transcription factor. Plants 10, 2149. doi: 10.3390/plants10102149
Salse, J., Bolot, S., Throude, M., Jouffe, V., Piegu, B., Quraish, U. M., et al (2008). Identifica-tion and characterization of shared duplications between rice and wheat provide new in-sight into grass genome evolution. Plant Cell 20, 11–24. doi: 10.1105/tpc.107.056309
Schilling, S., Kennedy, A., Pan, S., Jermiin, L. S., Melzer, R. (2020). Genome-wide analysis of MIKC-type MADS-box genes in wheat: pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 225, 511–529. doi: 10.1111/nph.16122
Shannon, P. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi: 10.1038/msb.2011.75
Sol, S., Valkov, V. T., Rogato, A., Noguero, M., Gargiulo, L., Mele, G., et al. (2019). Disruption of the Lotus japonicus transporter LjNPF2.9 increases shoot biomass and nitrate content without affecting symbiotic performances. BMC Plant Biol. 19, 1–14. doi: 10.1186/s12870-019-1978-5
Tal, I., Zhang, Y., Jørgensen, M. E., Pisanty, O., Barbosa, I. C., Zourelidou, M., et al. (2016). The Arabidopsis NPF3 protein is a GA transporter. Nat. Commun. 7, 1–11. doi: 10.1038/ncomms11486
Tian, H., Yuan, X., Duan, J., Li, W., Zhai, B., Gao, Y., et al. (2017). Influence of nutrient signals and carbon allocation on the expression of phosphate and nitrogen transporter genes in winter wheat (Triticum aestivum L.) roots colonized by arbuscular mycorrhizal fungi. PloS One 12, e0172154. doi: 10.1371/journal.pone.0172154
Todd, C. D., Zeng, P., Huete, A. M. R., Hoyos, M. E., Polacco, J. C. (2004). Transcripts of MYB-like genes respond to phosphorous and nitrogen deprivation in Arabidopsis. Planta 219, 1003–1009. doi: 10.1007/s00425-004-1305-7
Tong, J., Walk, T. C., Han, P., Chen, L., Shen, X., Li, Y., et al. (2020). Genome-wide identification and analysis of high-affinity nitrate transporter 2 (NRT2) family genes in rapeseed (Brassica napus L.) and their responses to various stresses. BMC Plant Biol. 20, 464. doi: 10.1186/s12870-020-02648-1
Wang, X. F., An, J. P., Liu, X., Su, L., You, C. X., Hao, Y. J. (2018a). The nitrate-responsive protein MdBT2 regulates anthocyanin biosynthesis by interacting with the MdMYB1 transcription factor. Plant Physiol. 178, 890–906. doi: 10.1104/pp.18.00244
Wang, X., Cai, X., Xu, C., Wang, Q. (2021). Identification and characterization of the NPF, NRT2 and NRT3 in spinach. Plant Physiol. Biochem. 158, 297–307. doi: 10.1016/j.plaphy.2020.11.017
Wang, R., Guan, P., Chen, M., Xing, X., Zhang, Y., Crawford, N. M. (2010). Multiple regulatory elements in the Arabidopsis NIA1 promoter act synergistically to form a nitrate enhancer. Plant Physiol. 154, 423–432. doi: 10.1104/pp.110.162586
Wang, J., H"uner, N., Tian, L. (2019a). Identification and molecular characterization of the Brachypodium distachyon NRT2 family, with a major role of BdNRT2.1. Physiologia plantarum 165, 498–510. doi: 10.1111/ppl.12716
Wang, W., Hu, B., Yuan, D., Liu, Y., Che, R., Hu, Y., et al. (2018b). Expression of the nitrate transporter gene OsNRT1.1A/OsNPF6.3 confers high yield and early maturation in rice. Plant Cell 30, 638–651. doi: 10.1105/tpc.17.00809
Wang, Z., Leushkin, E., Liechti, A., Ovchinnikova, S., Mößinger, K., Brüning, T., et al. (2020). Transcriptome and translatome co-evolution in mammals. Nature 588, 642–647. doi: 10.1038/s41586-020-2899-z
Wang, J., Li, Y., Zhu, F., Ming, R., Chen, L.-Q. (2019b). Genome-wide analysis of nitrate transporter (NRT/NPF) family in sugarcane saccharum spontaneum L. Trop. Plant Biol. 12, 133–149. doi: 10.1007/s12042-019-09220-8
Wei, J., Zheng, Y., Feng, H., Qu, H., Fan, X., Yamaji, N., et al. (2018). OsNRT2. 4 encodes a dual-affinity nitrate transporter and functions in nitrate-regulated root growth and nitrate distribution in rice. J. Exp. Bot. 69, 1095–1107. doi: 10.1093/jxb/erx486
Wen, J., Li, P., Ran, F., Guo, P., Zhu, J., Yang, J., et al. (2020). Genome-wide characterization, expression analyses, and functional prediction of the NPF family in Brassica napus. BMC Genomics 21, 1–17. doi: 10.1186/s12864-020-07274-7
Wen, Z.-Y., Tyerman, S. D., Dechorgnat, J., Ovchinnikova, E., Dhugga, K. S., Kaiser, B. N. (2017). Maize NPF6 proteins are homologs of Arabidopsis CHL1 that are selective for both nitrate and chloride. Plant Cell 29, 2581–2596. doi: 10.1105/tpc.16.00724
Winkelmüller, T. M., Entila, F., Anver, S., Piasecka, A., Song, B., Dahms, E., et al. (2021). Gene expression evolution in pattern-triggered immunity within Arabidopsis thaliana and across Brassicaceae species. Plant Cell 33, 1863–1887. doi: 10.1093/plcell/koab073
Xu, G., Fan, X., Miller, A. J. (2012). Plant nitrogen assimilation and use efficiency. Annu. Rev. Plant Biol. 63, 153–182. doi: 10.1146/annurev-arplant-042811-105532
Xuan, W., Beeckman, T., Xu, G. (2017). Plant nitrogen nutrition: sensing and signaling. Curr. Opin. Plant Biol. 39, 57–65. doi: 10.1016/j.pbi.2017.05.010
Yáñez-Cuna, J. O., Kvon, E. Z., Stark, A. (2013). Deciphering the transcriptional cis-regulatory code. Trends Genet. 29, 11–22. doi: 10.1016/j.tig.2012.09.007
Zhang, Z., Li, Z., Wang, W., Jiang, Z., Guo, L., Wang, X., et al. (2021). Modulation of nitrate-induced phosphate response by the MYB transcription factor RLI1/HINGE1 in the nucleus. Mol. Plant 14, 517–529. doi: 10.1016/j.molp.2020.12.005
Keywords: durum wheat, nitrogen, N uptake, nitrate transporters, NPF and NRT2 family, Nitrogen Use Efficiency (NUE), N uptake, Weighted Gene Co-expression Network Analysis (WGCNA)
Citation: Puccio G, Ingraffia R, Giambalvo D, Frenda AS, Harkess A, Sunseri F and Mercati F (2023) Exploring the genetic landscape of nitrogen uptake in durum wheat: genome-wide characterization and expression profiling of NPF and NRT2 gene families. Front. Plant Sci. 14:1302337. doi: 10.3389/fpls.2023.1302337
Received: 26 September 2023; Accepted: 25 October 2023;
Published: 09 November 2023.
Edited by:
Manohar Chakrabarti, The University of Texas Rio Grande Valley, United StatesReviewed by:
Sanjay Joshi, University of Kentucky, United StatesSareena Sahab, Department of Economic Development Jobs Transport and Resources, Australia
Copyright © 2023 Puccio, Ingraffia, Giambalvo, Frenda, Harkess, Sunseri and Mercati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guglielmo Puccio, Z3VncHVjY2lvQGdtYWlsLmNvbQ==; Francesco Sunseri, ZnJhbmNlc2NvLnN1bnNlcmlAdW5pcmMuaXQ=; Francesco Mercati, ZnJhbmNlc2NvLm1lcmNhdGlAaWJici5jbnIuaXQ=