 Guilong Lu1,2
Guilong Lu1,2 Qing Li1*
Qing Li1*- 1Institute of Vegetables, Tibet Academy of Agricultural and Animal Husbandry Sciences, Lhasa, China
- 2College of Horticulture and Landscape Architecture, Henan Institute of Science and Technology, Xinxiang, China
Wax apple (Syzygium samarangense) is a commercial fruit that belongs to one of the most species-rich tree genera in the world. We report here the first complete S. samarangense mitogenome obtained using a hybrid assembly strategy. The mitogenome was a 530,242 bp circular molecule encoding 61 unique genes accounting for 7.99% of the full-length genome. Additionally, 167 simple sequence repeats, 19 tandem repeats, and 529 pairs of interspersed repeats were identified. Long read mapping and Sanger sequencing revealed the involvement of two forward repeats (35,843 bp and 22,925 bp) in mediating recombination. Thirteen homologous fragments in the chloroplast genome were identified, accounting for 1.53% of the mitogenome, and the longest fragment was 2,432 bp. An evolutionary analysis showed that S. samarangense underwent multiple genomic reorganization events and lost at least four protein-coding genes (PCGs) (rps2, rps7, rps11, and rps19). A total of 591 RNA editing sites were predicted in 37 PCGs, of which nad1-2, nad4L-2, and rps10-2 led to the gain of new start codons, while atp6-1156, ccmFC-1315 and rps10-331 created new stop codons. This study reveals the genetic features of the S. samarangense mitogenome and provides a scientific basis for further studies of traits with an epistatic basis and for germplasm identification.
Introduction
Wax apple (Syzygium samarangense, also known as java apple, rose apple, wax jambu, and bell fruit) is a small evergreen tree in the family Myrtaceae that is native to the Andaman and Nicobar islands and to the Malaysian Archipelago (Khandaker and Boyce, 2016; Banadka et al., 2022). The genus is extremely rich in germplasm resources, with 1,193 recognized species worldwide. It is mainly grown in tropical and subtropical regions and is distributed from Africa to India, across Southeast Asia, and extending to Hawaii in the Pacific (Govaerts et al., 2008). Wax apple fruit is rich in proteins, dietary fiber, sugar, vitamins, flavonoids, phenolic acids, and other nutrients. Phenolic acids antioxidant, anti-cancer, anti-bacterial, anti-diabetic, and other biological activities (Banadka et al., 2022). In addition, the extracts of leaves, flower buds, bark, and roots of wax apple are effective in removing fire and toxins, drying dampness, and relieving itching (Chinese Herbalism Editorial Board, 1999; Gurib-Fakim, 2006). The species also has high ornamental value, mostly owing to its beautiful shape, fragrant and beautiful flowers, bright fruit color, beautiful fruit shape, and long hanging period. In recent years, the rapid development of the wax apple industry has increased the demand for high quality, high yield, and high resistance varieties. Genome and transcriptome research on wax apple has laid a scientific foundation for the utilization of excellent germplasm resources and improvement of varietal traits (Chen et al., 2017; Liu et al., 2018; Low et al., 2022).
Mitochondria, which provide energy for various physiological activities, are important organelles in eukaryotic cells (Møller et al., 2021). Originally as an endosymbiotic form of alphaproteobacteria (Lang et al., 1999), mitochondria are semi-autonomous organelles exhibiting predominantly matrilineal inheritance (Greiner et al., 2015). Previous studies have shown that mitochondria play an important role in plant growth and development and are closely associated with growth vigor, chloroplast function (Chevigny et al., 2020), stress tolerance (Liberatore et al., 2016). Compared to animal mitogenomes, plant mitogenomes exhibit complex and unique genetic features. Their structures are remarkably diverse, with circular, linear, highly branched, sigma-like, and networked types; and the genome sizes vary widely among species, ranging from 66 Kb (Viscum scurruloideum) (Skippington et al., 2015) to 11.7 Mb (Larix sibirica Ledeb.) (Putintseva et al., 2020). During long-term evolution, the number of mitochondrial genes in extant plants is very low, and most of them have been transferred into the nucleus. There are generally 32-67 genes that are sparsely distributed and have a highly conserved coding sequence, with a large number of repetitive sequences distributed among them (Mower et al., 2012; Richardson et al., 2013). Accumulation of repetitive sequences may lead to frequent recombination events and structural variation in the mitogenome (Sloan, 2013; Wu et al., 2022), whereas active genomic rearrangements can even lead to cytoplasmic male sterility (Kubo et al., 2011; Liao et al., 2020; Wang et al., 2022). The plant mitogenome can integrate plastome and nuclear DNA fragments and even exogenous mitogenomes, which is a major driver of the enlargement and rapid evolution of plant mitogenomes (Rice et al., 2013; Prasad et al., 2022). In addition, RNA editing events are widespread in plant mitochondrial transcripts, where they are critical for the production of functional proteins and adaptive evolution (Gommans et al., 2009; Ichinose and Sugita, 2017) and are closely associated with cytoplasmic male sterility (Gallagher et al., 2002; Wei et al., 2010). In summary, plant mitogenomes have become an important tool for species classification, evolutionary analyses, and parental traceability (Sibbald et al., 2021; Wang et al., 2022; Zhang et al., 2023). However, related research lags far behind studies on chloroplast and plastid genomes owing to the complexity of the mitogenome. The complete mitochondrial genome of S. samarangense has not been reported in public databases (https://www.ncbi.nlm.nih.gov/genome/browse/#!/organelles/).
In this study, the main S. samarangense cultivar in China, Black Diamond (Zheng, 2011), was selected for the following investigations: 1) the mitogenome was assembled, annotated, and comparatively analyzed; 2) the repetitive sequences present in this genome were identified, and the possible structure of the repeat-mediated genomic recombination was verified using Sanger sequencing and polymerase chain reaction (PCR) validation; 3) the chloroplast genome of the cultivar was assembled, and the transfer of homologous fragments between this genome and the mitogenome was identified; 4) phylogeny and collinearity analyses were conducted to identify the relationships between this species and closely related species; and 5) RNA editing events were predicted to be present in the protein-coding gene (PCG) transcripts, and editing sites that could create new start and stop codons were validated. This study aims to provide a scientific and theoretical basis for the in-depth understanding of the genetic characteristics of S. samarangense and further germplasm identification.
Materials and methods
Plant material, DNA and RNA preparation, and sequencing
The young leaves of wax apple (Black Diamond) were harvested from the National Agricultural Science and Technology Park, located in Lhasa, Tibet Autonomous Region, China (Coordinates: 91°2’8’’E, 29°38’15’’N; Altitude: 3650 m). The gDNA and RNA were separately extracted using the TianGen Plant Genomic DNA Kit and the RNAprep Pure Plant Kit, respectively (Beijing, China). The NanoDrop One Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific, Massachusetts, USA) was used to ensure the integrity of the samples. Samples meeting quality thresholds were then sent to Wuhan Benagen Tech Solutions Company Limited (Wuhan, China) for sequencing, after packing in dry ice. Using DNBSEQ-T7 (developed by Shenzhen Huada Intelligent Technology Co., Ltd., Shenzhen, China), short reads were sequenced, followed by a quality control filtering process on the raw reads using fastp v0.21.0 (Chen, 2023). The Nanopore PromethION sequencer from Oxford Nanopore Technologies (Oxford, UK) was employed to sequence long reads, and NanoFilt v2.8.0 (De Coster et al., 2018) was used for quality control. Long non-coding RNA (lncRNA) sequencing was performed via MGISEQ-2000 (by Shenzhen Huada Intelligent Technology Co., Ltd., Shenzhen, China), and the raw reads were subjected to quality control filtering using SOAPnuke v2.0 (Chen et al., 2018).
Mitogenome assembly
Initially, Flye v2.9.2 software (University of California, San Diego, USA) (Kolmogorov et al., 2019) was utilized to assemble the long-read sequencing data. Following this, the Arabidopsis thaliana mitogenome was used as a reference sequence to identify contig fragments containing mitochondrial genomes via the BLASTn program. Both long-read and short-read data were then aligned to mitogenome contigs using BWA v0.7.17 (Li and Durbin, 2010), after which matched reads were filtered, extracted, and stored separately for subsequent assembly. In the final step, short-read and long-read sequencing data were merged using Unicycler v0.4.7 (The University of Melbourne, Victoria, Australia) (Wick et al., 2017), facilitating hybrid assembly with parameters “- -kmers 57,67,77”, and visualized graphics were obtained using Bandage v0.8.1 (Wick et al., 2015).
Mitogenome annotation and analysis
GeSeq v2.03 (https://chlorobox.mpimp-golm.mpg.de/geseq.html) (Tillich et al., 2017) was utilized to annotate PCGs of the S. samarangense mitogenome, with the mitogenomes of A. thaliana (NC_037304), Liriodendron tulipifera (NC_021152.1), and Rhodomyrtus tomentosa (NC_071968.1) used as reference genomes. tRNA and rRNA genes were annotated using tRNAscan-SE v.2.0.11 (Chan et al., 2021) and BLASTN v2.13.0 (Chen et al., 2015), respectively, and errors in annotation were manually fixed using Apollo v1.11.8 (Dunn et al., 2019). For the extraction of protein-coding gene sequences of the S. samarangense mitogenome, PhyloSuite v1.2.2 (Xiang et al., 2023) was employed. Mega software (Tamura et al., 2021) was utilized for the codon preference analysis of PCGs and the calculation of the relative synonymous codon usage values (RSCU). RSCU > 1 signifies that the codon is preferentially used by amino acids, whereas RSCU < 1 denotes the opposite.
Mitogenome repeat elements
The simple sequence repeats (SSRs) were identified using MISA v2.1 (https://webblast.ipk-gatersleben.de/misa/) with the parameter “1-10 2-5 3-4 4-3 5-3 6-3” (Beier et al., 2017). Tandem repeats were recognized using TRF v4.09 (https://tandem.bu.edu/trf/trf.unix.help.html) with the parameter “2 7 7 80 10 50 500 -f -d -m” (Behboudi et al., 2023). Interspersed repeats were detected using the REPuter web server (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) (Kurtz and Schleiermacher, 1999) with the minimum repeat size set at 30 bp.
Validation of repeat-mediated recombination
To confirm the proposed genomic structure of the S. samarangense mitogenome, each set of repeats and their sequences extending 500 bp upstream and downstream were extracted as reference points. Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast) was used to design primers for the four pathways of the dual bifurcation structure, and PCR amplification along with Sanger sequencing (You et al., 2023) was used to verify the validity of the junction sequences. The PCR amplification procedure was carried out in a 50 μL total volume, containing 2 μL of gDNA template, 2 μL each of upstream and downstream primer (10 μmol/L), 25 μL of 2× Rapid Taq Master Mix (Vazyme Biotech Co., Ltd., Nanjing, China), and 19 μL of ddH2O. The PCR steps involved a 95°C pre-denaturation step for 3 min, 35 cycles of 95°C denaturation for 15 s, 55°C annealing for 15 s, and 72°C extension for 30 s, and a final 72°C extension for 15 min.
Chloroplast genome assembly and gene transfer analysis
GetOrganelle v1.7.7.0 (Freudenthal et al., 2020) was used to assemble the S. samarangense chloroplast genome with parameters set to “-R 15 -k 21,45,65,85,105 -F embplant_pt”. The chloroplast genome was then annotated with CPGAVAS2 (Liu et al., 2012) and the annotation results were corrected using CPGView software (Liu et al., 2023). Homologous fragments in the mitochondrial and chloroplast genomes were examined using BLASTN v2.13.0 (Chen et al., 2015) with an e-value threshold of 1e-5. The results were visualized via Excel v2021 and Circos v0.69-9 (Zhang et al., 2013).
Phylogenetic and collinearity analysis
The mitogenomes of 30 species closely related to S. samarangense were downloaded from NCBI for a phylogenetic analysis (Supplementary Table 1), with Tribulus terrestris (MK431825.1) and Zygophyllum fabago (MK431827.1) of Zygophyllales assigned as outgroups. PhyloSuite software was used to extract shared genes in the mitogenomes. A multiple sequence alignment was generated using MAFFT v7.505 (Katoh et al., 2019). The “GTR +F+I+I+R2” model was selected for the phylogenetic analysis using IQ-TREE v2.1.3 (Minh et al., 2020), and the maximum likelihood tree was visualized using iTOL v6 (https://itol.embl.de/) (Letunic and Bork, 2007). The mitogenome sequence information for S. samarangense and seven closely related species within the same order were analyzed via BLASTn. Homologous sequences exceeding 500 bp were maintained as conserved co-linear blocks, and a multiple synteny plot was drafted using MCscanX (Tang et al., 2008).
Identification and validation of RNA-editing sites
By using TopHat2 (Mehmood et al., 2020), mitochondrial DNA sequences from the S. samarangense mitogenome were mapped to transcriptomic data to obtain transcripts. The potential RNA editing sites were identified by REDItools v2.0 (Flati et al., 2020) by a comparison of DNA and RNA sequences and the selection of positions with a coverage depth ≥ 100× and editing frequency ≥ 0.1. Primer-BLAST was used to design primers for the RNA editing sites that generate start and stop codons (Supplementary Table 2). A HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, Nanjing, China) was employed to convert the extracted wax apple RNA into cDNA. Both gDNA and cDNA were used as templates for PCR amplification, employing the same system and procedure described in the validation of repeat-mediated recombination. To confirm the RNA editing sites, the amplified products were sequenced and compared.
Results
Mitogenome assembly, gene annotation, and analysis
The S. samarangense mitogenome was assembled using 10.39 Gb short reads and 11.23 Gb long reads by a hybrid assembly strategy. It contained six contigs, including two double bifurcating structures (Figure 1A). The longest contig (221,760 bp) and the shortest contig (12,514 bp) were single-copy regions. The repetitive sequences were determined based on long-read data and one master circular structure was obtained (Figure 1B) with a total length of 530,242 bp (27.43% Adenine, A; 27.53% Thymine, T; 22.63% Cytosine, C; and 22.41% Guanine, G). The size of this genome differs slightly from those of other species of the same family: Rhodomyrtus tomentosa (400,547 bp, NC_071968.1), Eucalyptus grandis (478,813 bp, NC_040010.1), and E. polybractea (580,440 bp, CM048836.1).
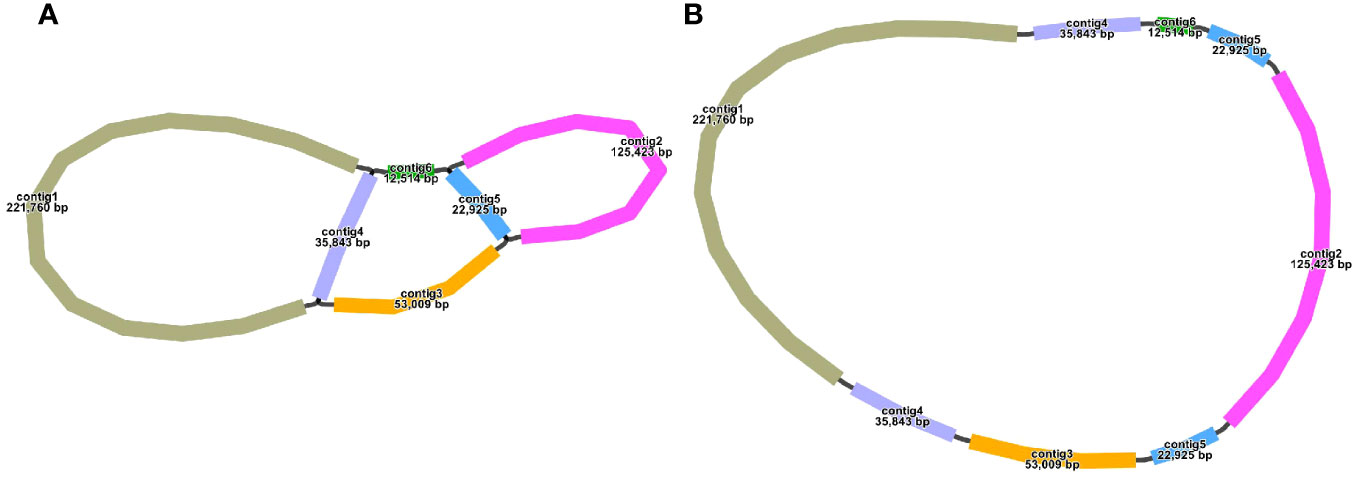
Figure 1 Mitogenome sketch (A) and master circular structure (B) in S. samarangense.
The S. samarangense mitogenome was annotated with 37 unique PCGs, including 24 core genes and 13 non-core genes, 21 tRNA genes (four tRNAs were multiple copies), and three rRNA genes (Figure 2, Table 1). S. samarangense lost four genes (rps2, rps7, rps11, and rps19) compared to the PCGs of the “fossilized” L. tulipifera mitogenome (Richardson et al., 2013). The total length of coding sequences in the S. samarangense mitochondrial genome (PCGs, tRNA, and rRNA genes) was 42,391 bp. This accounted for 7.99% of the entire genome, while over 90% belonged to intergenic regions.
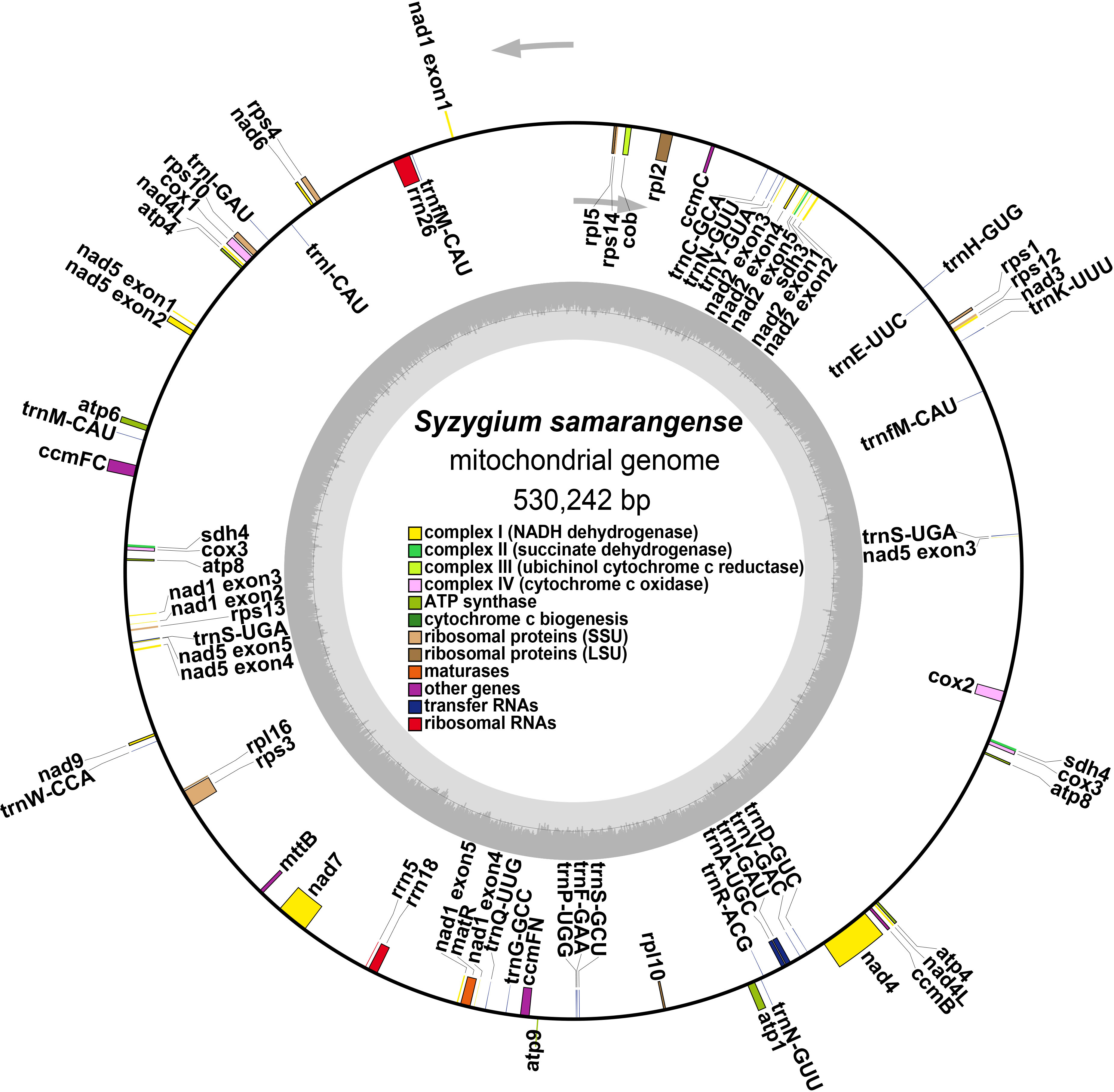
Figure 2 Circular maps of the S. samarangense mitogenome.
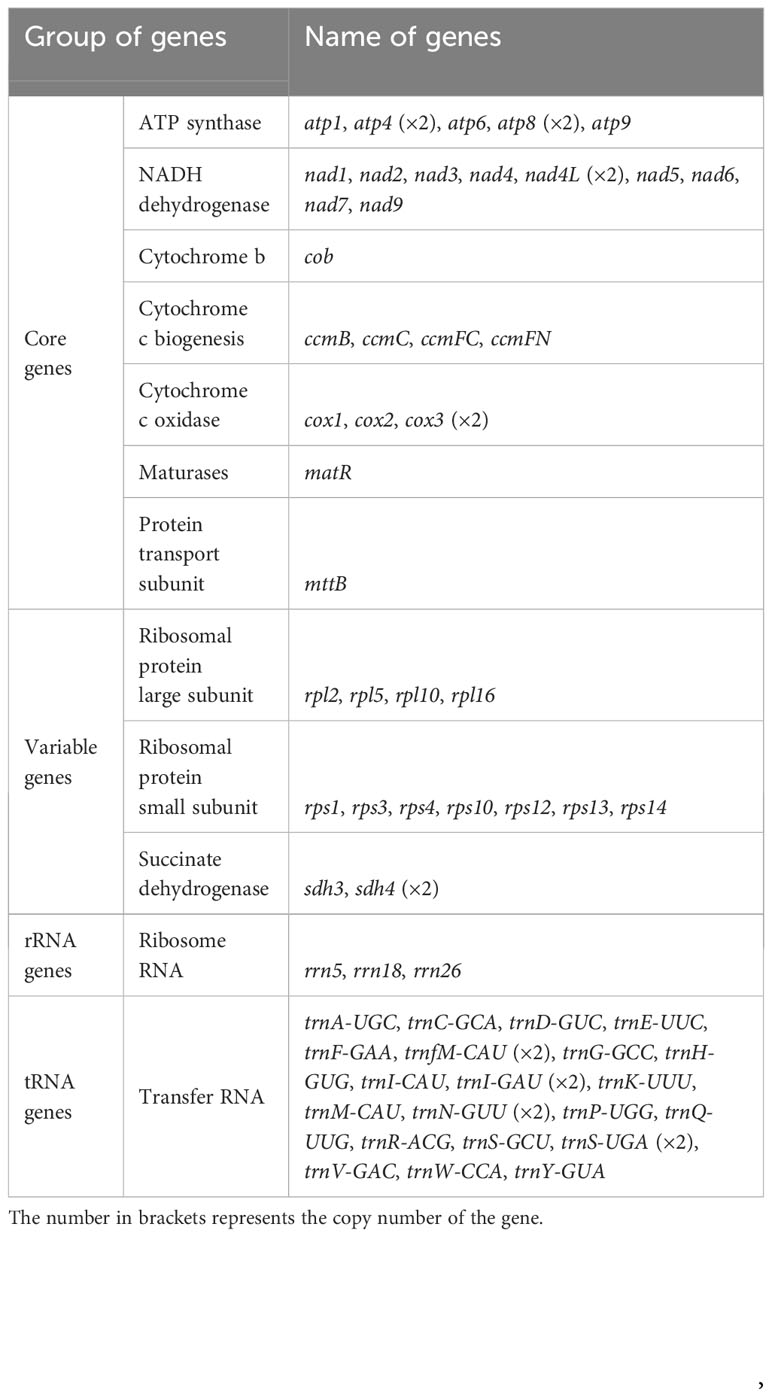
Table 1 Genes in the S. samarangense mitogenome.
RSCU, which reflects the optimized results of the evolution of plant ecological adaptations, is the “dialect” of a gene or genome (Tyagi et al., 2023). The amino acids encoded by 37 PCGs from the S. samarangense mitogenome generally showed codon usage bias, except for the RSCU of 1.00 for the initiation codon (AUG) and tryptophan (UGG) (Supplementary Figure 1, Supplementary Table 3). For example, alanine (Ala) had a high codon usage preference for GCU with the highest RSCU value of 1.56. Histidine (His) had a usage preference for CAU, while tyrosine (Tyr) had a usage preference for UAU, both of which had an RSCU value of 1.52. In addition, cysteine (Cys), lysine (Lys), phenylalanine (Phe), and valine (Val) had maximum RSCU values below 1.20 and did not have strong codon usage preference. However, the third codon of these amino acids has a greater preference for the use of U or A than other nucleotides.
Repeat sequence analysis and repeat-mediated recombination
There were many repetitive sequences in the S. samarangense mitogenome (Supplementary Figure 2). A total of 167 simple sequence repeats (SSRs) were identified throughout the genome (Figure 3A, Supplementary Table 4), and monomeric and dimeric forms of SSRs accounted for 49.10% of the total SSRs. T monomeric repeat sequences (No.35) accounted for 57.38% of the 61 monomeric SSRs, and the TA repeat was the most common type of dimeric SSR, accounting for 38.10% of dimeric SSRs; no hexamer SSRs were detected. Nineteen tandem repeats with lengths between 15 bp and 33 bp and ≥76% match were identified (Supplementary Table 5). In addition, 529 pairs of dispersed repeats with ≥30 bp were detected (Figure 3B, Supplementary Table 6), including 249 pairs of palindromic repeats, 280 pairs of forward repeats, and no reverse and complementary repeats. The total length of the above three types of SSRs, tandem repeats, and interspersed repeats were 1,951 bp, 799 bp and 177,672 bp, accounting for 0.37%, 0.15%, and 33.51% of the mitogenome, respectively.
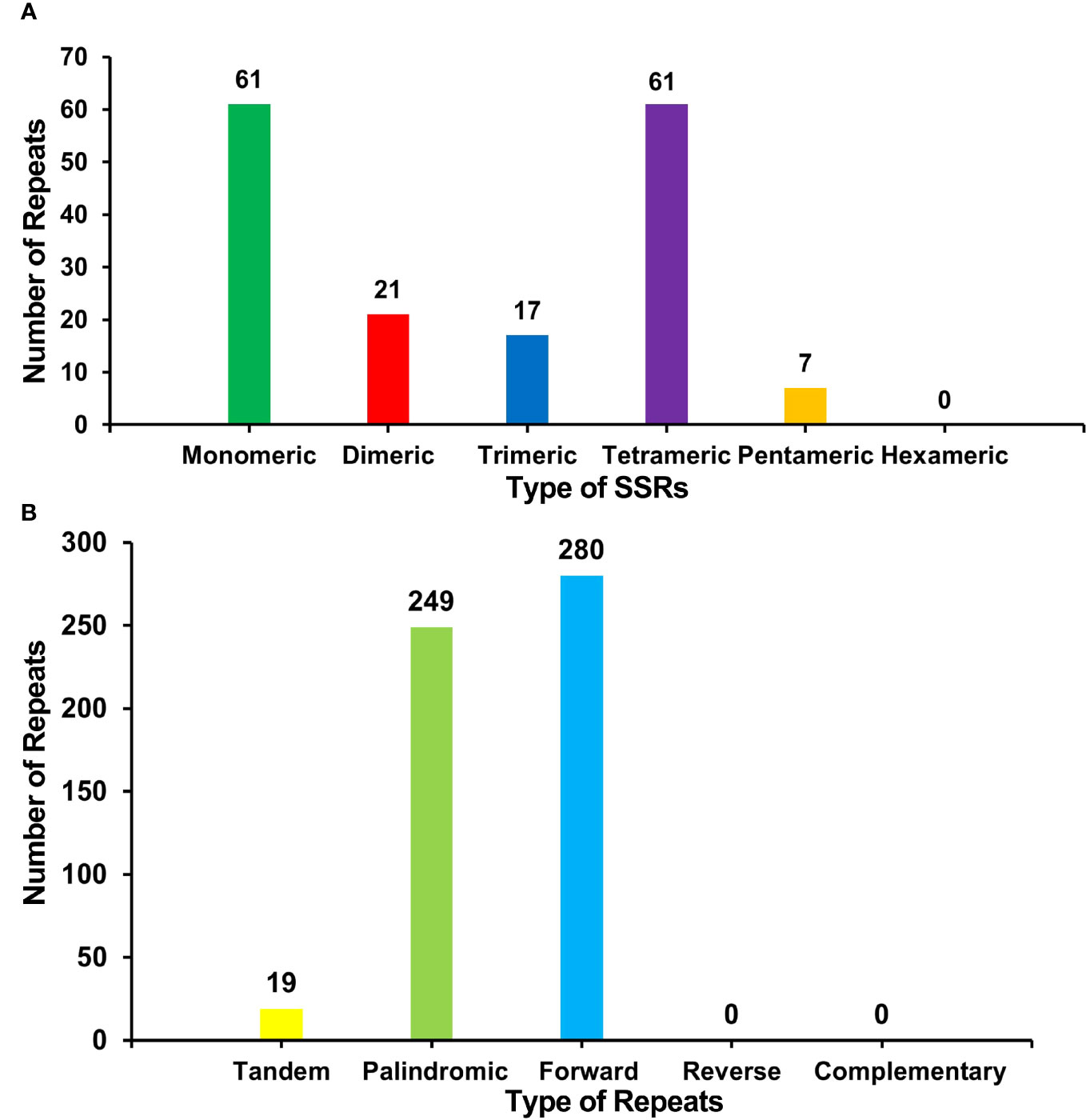
Figure 3 (A) Type and number of SSRs in the S. samarangense mitogenome. Green, red, blue, purple, and yellow indicate monomeric, dimeric, trimeric, tetrameric, and pentameric, respectively. (B) Type and number of repeat sequences in the S. samarangense mitogenome. Yellow, light green, and blue indicate tandem repeats, palindromic repeats, and forward repeats, respectively.
The structure of the plant mitogenome is dynamic owing to long repetitive sequences that can mediate genome recombination causing conformational changes (Gualberto and Newton, 2017; Kozik et al., 2019). It can be reasonably deduced that a single circular molecule is insufficient for the comprehensive display of the S. samarangense mitogenome structure. There may be multiple connections at branch nodes, and potential secondary conformations mediated by R1 and/or R2 (Table 2), as shown in Figures 4A–C. R1 and R2-mediated recombination was identified using a junction approach with validation using newly designed primers (Supplementary Table 7) and electrophoresis (Figures 4D, E). Detailed sequencing comparisons are shown in Supplementary Figure 3. In summary, the S. samarangense mitogenome had a variety of potential recombination conformations.
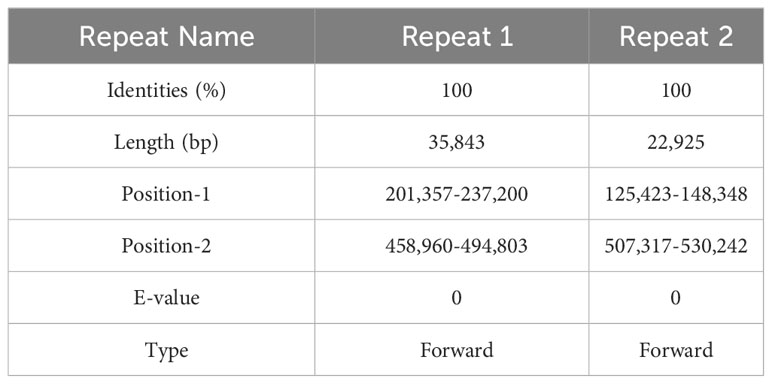
Table 2 List of two repeat sequences mediated recombination in the S. samarangense mitogenome.
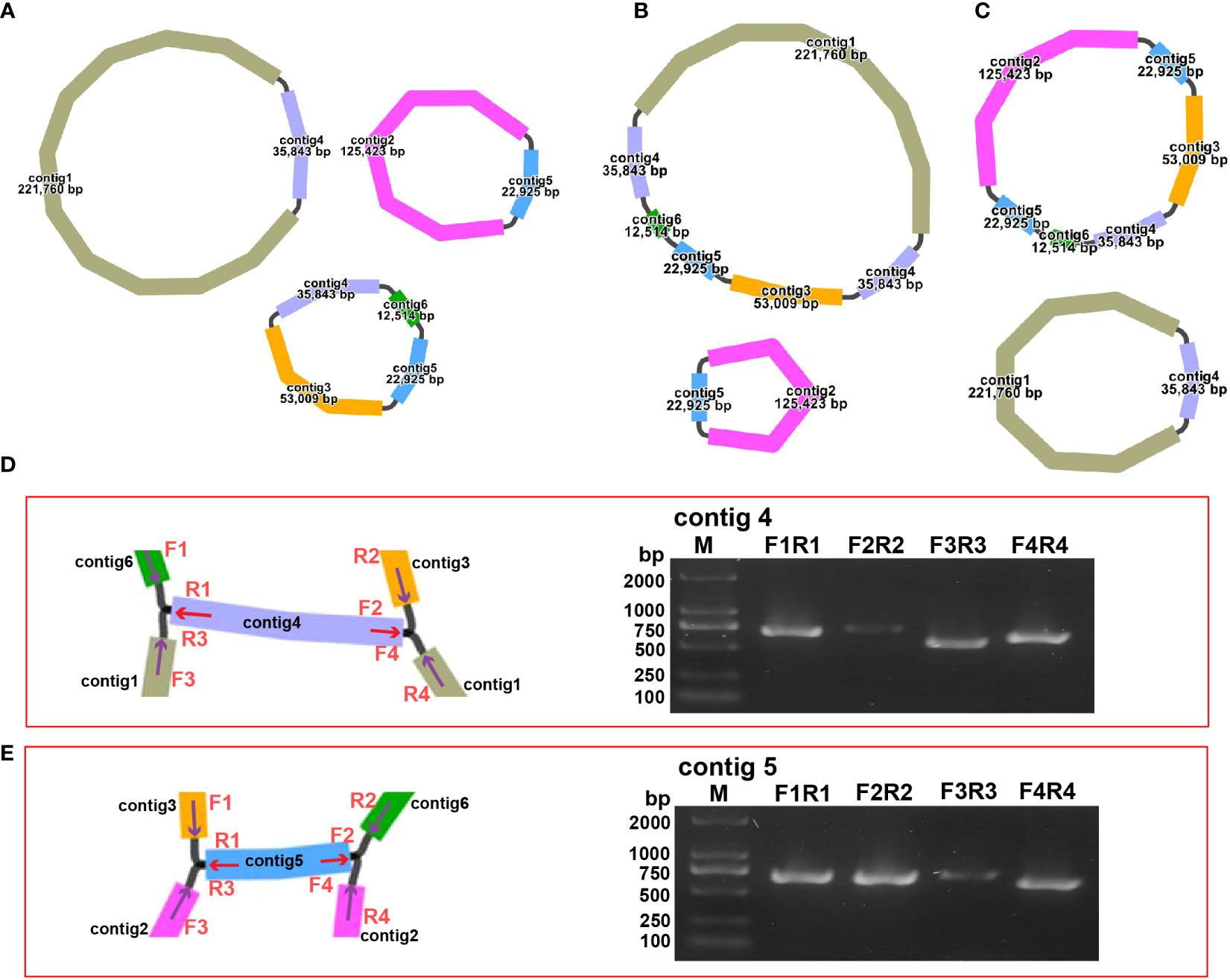
Figure 4 Recombination structure prediction and verification in S. samarangense. (A) Secondary conformation 1 (including 3 small circles) mediated by R1 and R2; (B) Secondary conformation 2 (including 2 small circles) mediated by R2; (C) Secondary conformation 3 (including 2 small circles) mediated by R1. (D) Verification of junction sequences in R1. (E) Verification of junction sequences in R2.
Chloroplast genome and sequence migration analysis
Sequence migration between the chloroplast and plant mitochondrial genomes has been prevalent throughout their long-term evolution (Prasad et al., 2022). The chloroplast genome of S. samarangense was assembled using the same sequencing data, with a size of 159,109 bp (Figure 5A). Thirteen homologous fragments of the chloroplast genome and mitochondrial genome were identified based on sequence similarity (Figure 5B, Supplementary Table 8), with a total length of 8,100 bp accounting for 1.53% of the total mitogenome length. Mitochondrial plastid sequence seven (MTPT7) was the longest (2,432 bp). Eighteen unique genes were identified after annotating these homologous sequences, of which ten were complete, including six PCGs (psbJ, psbL, psbF, psbE, rps19, and rpl2) and four tRNA genes (trnD-GUC, trnH-GUG, trnM-CAU, and trnN-GUU). The eight incomplete genes included ndhC, psaD, rpl22, rpl23, rpoB, ycf2, trnI-GAU, and trnV-UAC. However, these chloroplast genes transferred to the mitochondrial genome have undergone the varying degrees of sequence duplication, substitution, mutation, or loss during evolution, which have hindered their ability to perform their normal functions and caused them to evolve into pseudogenes (Mower et al., 2010; Mower et al., 2012).
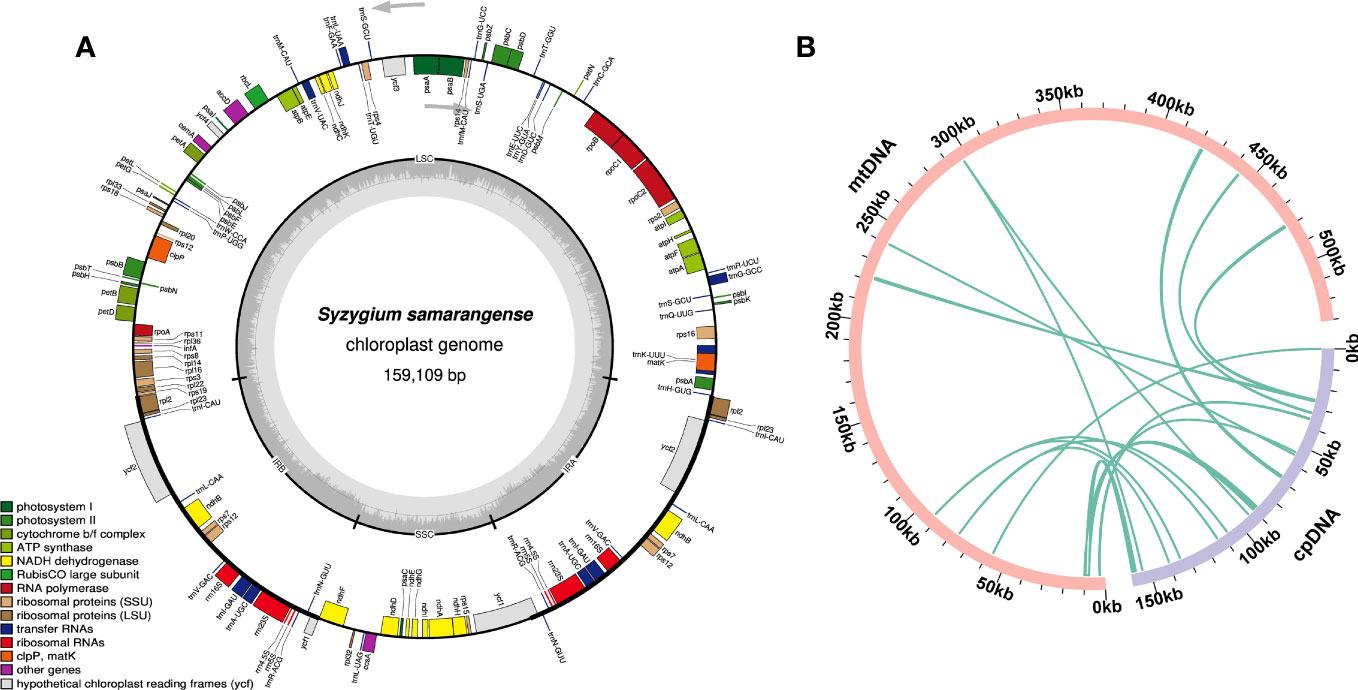
Figure 5 Chloroplast genome of S. samarangense (A) and sequence migration analysis between the chloroplast genome and mitochondrial genome (B). Pink and light purple arcs represent the mitogenome and the chloroplast genome, respectively, and the dark green lines between the arcs correspond to the homologous fragments.
Phylogeny and collinearity analysis
The loss or gain of PCGs commonly occurs during plant mitogenome evolution (Skippington et al., 2015; Wu et al., 2022). Only 23 out of 37 PCGs from the genomes of closely related species included in the phylogenetic analysis were shared: atp1, atp4, atp6, atp8, ccmB, ccmC, ccmFC, ccmFN, cob, cox1, cox2, cox3, nad1, nad2, nad4, nad5, nad6, nad7, nad9, rpl5, rpl16, rps3, and sdh4. The phylogenetic tree showed that S. samarangense was most closely related to Eucalyptus grandis and Rhodomyrtus tomentosa in the Myrtle family (Figure 6A). Moreover, the topology of the phylogeny of the above species (based on mitochondrial DNA) coincided with the latest classification of the Angiosperm Phylogeny Group.
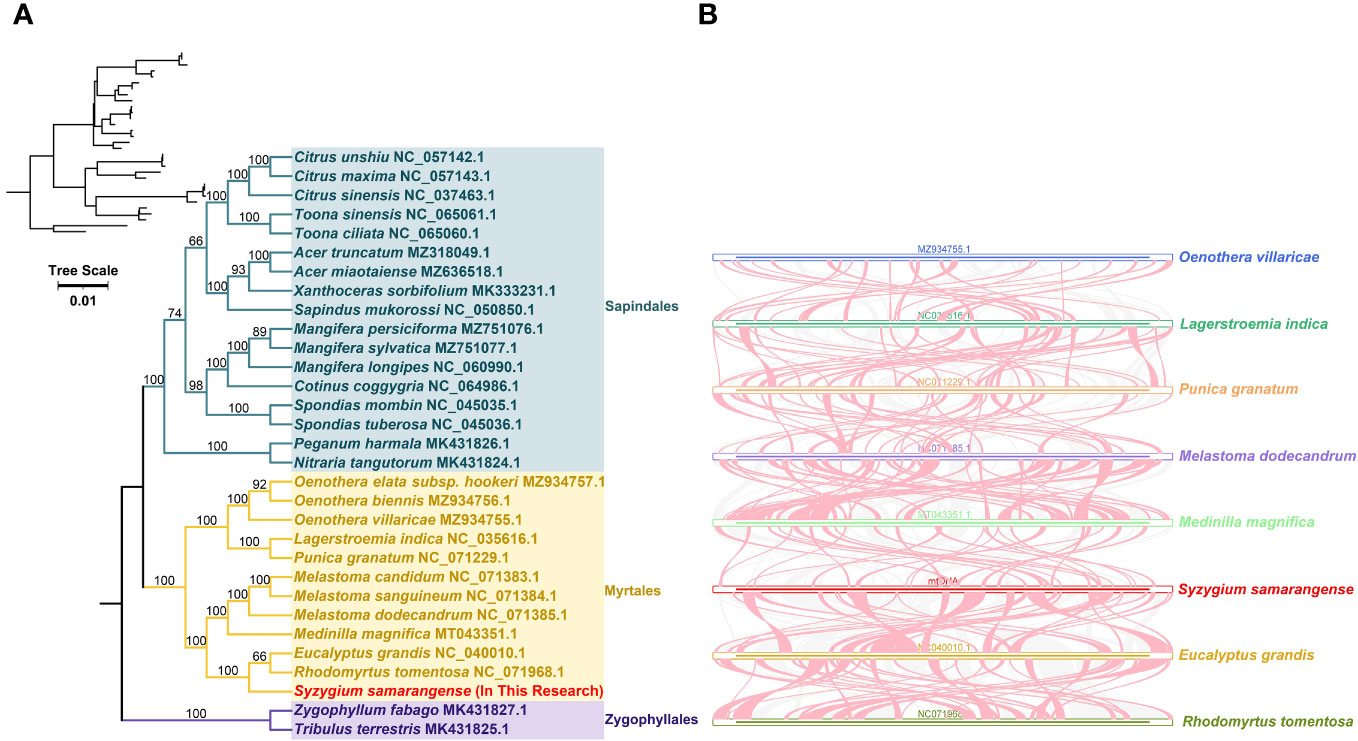
Figure 6 (A) Phylogenetic analysis of S. samarangense and related genera. Numbers at nodes show the bootstrap support values, and different colors indicate different Orders. (B) Collinearity analysis between S. samarangense and related genera. Blue, dark green, orange, purple, light green, red, yellow, and yellow-green lines represent Oenothera villaricae, Lagerstroemia indica, Punica granatum, Melastoma dodecandrum, Medilla magnifica, Syzygium samarangense, Eucalyptus grandis, and Rhodomyrtus tomentosa, respectively. Red curved areas indicate regions where inversions occur, and the gray areas indicate regions with high homology.
A collinearity analysis of S. samarangense and seven Myrtales species (Figure 6B, Supplementary Table 9) revealed many homologous co-linear blocks, although the sizes of these co-linear blocks were generally short. Moreover, the co-linear block arrangement between the individual mitogenomes was inconsistent. It was hypothesized that extensive genomic reorganization occurred during the evolution of S. samarangense and closely related species. In addition, the identification of unique regions in species that lack homology with other species indicated species specificity in evolution.
RNA editing events
RNA editing, one of the steps required for gene expression, is prevalent in higher plant mitochondria (Takenaka et al., 2013). RNA editing events were predicted for 37 PCGs of the S. samarangense mitogenome based on transcriptome data. They were observed in all of those genes, with a total of 591 RNA editing sites (Figure 7A), of which 473 had an editing frequency ≥ 0.80 (Supplementary Table 10). All were base C to U edits, and their RNA-seq mapping results were uploaded to the figshare platform (doi: 10.6084/m9.figshare.22650238). Among them, 47 RNA editing sites were identified in the ccmB gene, which had the highest number of edits among all mitochondrial genes. This was followed by the nad4 gene, with 44 RNA editing sites. Furthermore, these editing sites were primarily observed in the second and first bases of the coding amino acids, and the number was 336 (56.85% of the total) and 187 (31.64%), respectively.
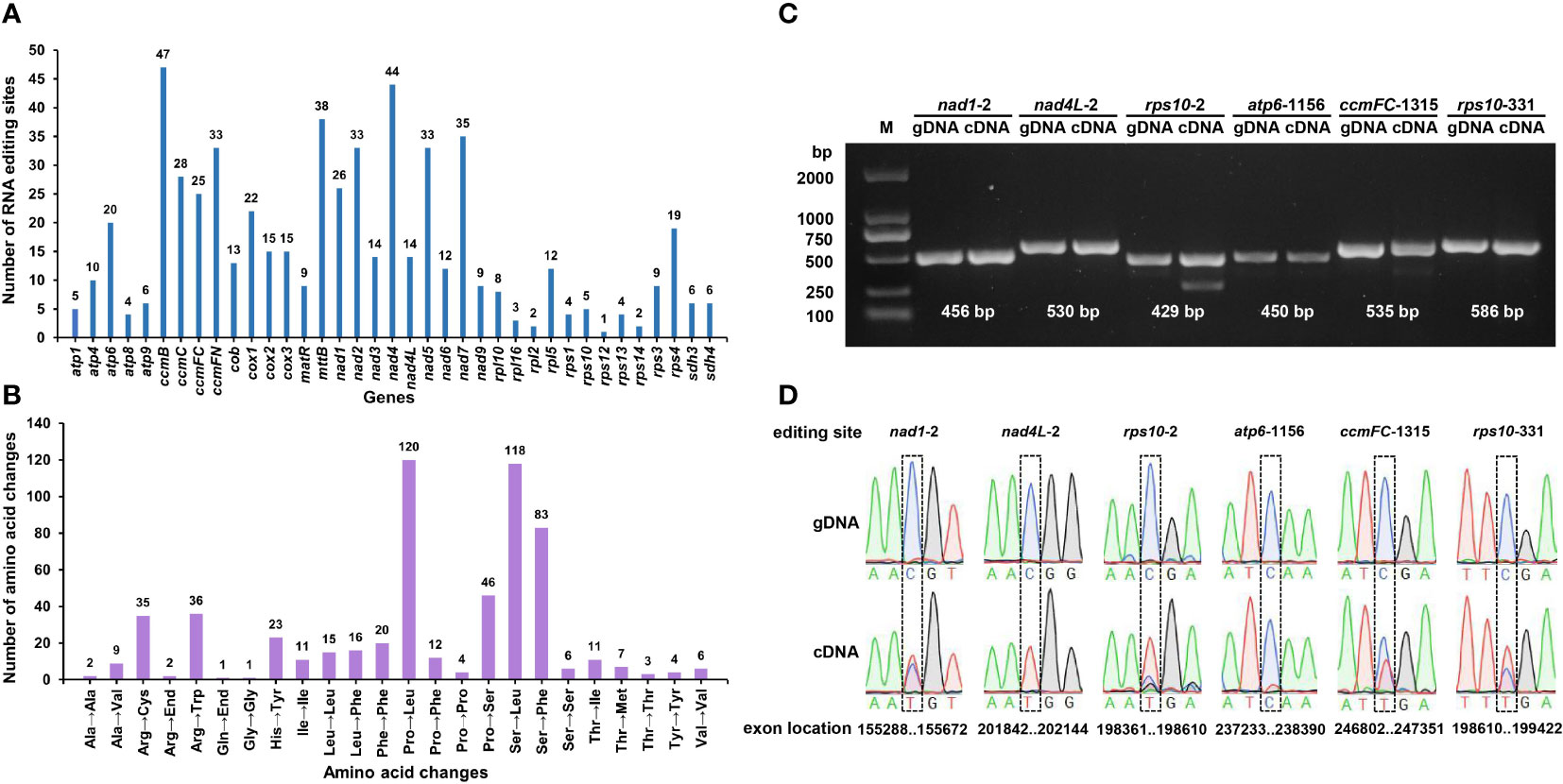
Figure 7 Predicted RNA editing sites (A) and corresponding amino acid changes (B) in S. samarangense mitogenome PCGs. PCR products (C) and sequence comparisons (D) for six specific editing sites.
Most of the editing events resulted in non-synonymous codon changes, primarily involving the following three amino acid changes: Pro to Leu (No.120), Ser to Leu (No.118), and Ser to Phe (No.83) (Figure 7B). We predicted one ACG (Thr) to AUG (Met) editing event in nad1, nad4L, and rps10; one CAA (Gln) to UAA (End) in atp6; and one CGA (Arg) to UGA (End) in ccmFC and rps10. Further, we validated these six editing sites by PCR (Figure 7C) and Sanger sequencing (Figure 7D, Supplementary file 1). The editing frequency of the atp6-1156 editing site was low (the red line indicates base T in transcription). However, the specific effects of changes in the start and stop codons caused by RNA editing events in the S. samarangense mitogenome require in-depth studies.
Discussion
A high-quality S. samarangense mitogenome with a length of 530,242 bp was assembled. The size of this genome varies slightly from that of other Myrtaceae species and differs significantly from those of Fragaria (275.2–351.2 Kb) (Fan et al., 2022), Mangifera indica (714.4–750.9 Kb) (Niu et al., 2022), and Rhynchospora (1.7–2.2 Mb) (Hofstatter et al., 2022). The 45.03% GC content in the S. samarangense mitogenome was not significantly different from those of the above species. This indicated that the GC content of the plant mitogenome was relatively conserved during evolution. L. tulipifera is an ancient legacy plant whose mitogenome evolved very slowly, retaining genes frequently lost in angiosperms and containing 41 PCGs (Richardson et al., 2013). At least four PCGs (rps2, rps7, rps11, and rps19) were lost in the S. samarangense genome. This indicated that a gene loss or transfer event occurred in this species, and ribosomal protein genes are more likely to be lost (Adams and Palmer, 2003). The coding region of the S. samarangense mitogenome only accounted for 7.99% of its full length with >90% non-coding regions; the gene distribution was very sparse, with highly conserved coding sequences. This proportion of the genome that is coding was similar to those in L. tulipifera (7.7%, excluding cis-spliced introns) (Richardson et al., 2013) and Populus simonii (8.25%) (Bi et al., 2022).
Repeats are identical or symmetrical DNA sequence fragments that frequently occur in plant genomes, occupy a large proportion of plant mitogenomes, and have important roles in genome size evolution, gene expression regulation, and responses to stress (Wendel et al., 2016). They are classified into tandem repeats and interspersed repeats according to whether they are adjacent or not. SSRs are a special kind of tandem repeat (generally not more than 6 bp) that are often used as genetic markers in plants (Khera et al., 2015). This study identified a total of 167 SSRs in the S. samarangense mitogenome, providing a large number of referenceable markers for germplasm diversity evaluation and species identification. Interspersed repeat sequences (also known as transposable elements) are a class of DNA sequences that can move their position on the genome, leading to gradual changes in functional genes and contributing to evolution (Bennetzen and Wang, 2014). They can regulate gene expression and affect plant phenotypic traits (Lisch, 2013), such as plant height and ear height in maize (Li et al., 2020), pigment formation in blood orange (Butelli et al., 2012), fruit shape in tomato (Xiao et al., 2008), and aluminum tolerance in wheat (Tovkach et al., 2013). In this study, 529 pairs of interspersed repeats were detected in the S. samarangense mitogenome; however, their effects on plant phenotypic traits require further in-depth study.
Repeat-mediated genome rearrangement is one of the main drivers of changes in mitogenome structure in plants, affecting gene organization and creating gene chimeras. This process affects plant phenotypes and evolution (Sanchez-Puerta et al., 2015; Gualberto and Newton, 2017). Large repetitive sequences over 1000 bp are more prone to recombination in the genome (Kozik et al., 2019). This study identified multiple potential conformations due to R1 (35,843 bp) and/or R2 (22,925 bp) repeat-mediated recombination in the S. samarangense mitogenome, although a single circle conformation containing information about the entire genome predominates (Figures 1B, 4A–C). Therefore, rearrangement events in S. samarangense resulted in differentiation within the Syzygium genome.
Horizontal gene transfer is the transmission of genes or DNA fragments within cells or across species boundaries by asexual means; it is one of the main drivers of land plant evolution (Prasad et al., 2022). However, the introduced genes usually degenerate into pseudogenes (Mower et al., 2010). This study identified a total of 13 homologous fragments between the chloroplast genome and the S. samarangense mitogenome, accounting for 1.53% of the total mitogenome length. Among these homologous sequences, ten complete mitochondrial genes were identified, including six PCGs and four tRNA genes. The migration of gene sequences led to mitogenome expansion, which contributed to plant mitochondrial genetic diversity, although most of these migrating sequences lost their integrity during evolution (Alverson et al., 2011). Furthermore, only 23 PCGs were identical among the 31 closely related species in this study. This further suggested that gene gain/loss events occurred in closely related plants during evolution (Allen et al., 2007; Kubo and Newton, 2008).
RNA editing is a kind of nucleotide modification at the RNA level in higher plants and plays an essential role in plant adaptation and development. Changes in nonsynonymous codons caused by RNA editing may result in significant changes in gene function, especially with the creation of new start and stop codons (Ichinose and Sugita, 2017; Hao et al., 2021). RNA editing produces an ACG (Thr) to AUG (Met) editing events that can serve as the starting point for gene transcription (Quiñones et al., 1995; Zandueta-Criado and Bock, 2004). Similarly, RNA editing may produce stop codons that cause the premature termination of protein synthesis. Mitochondrial RNA editing truncated the orf77 chimeric open reading frame associated with S cytoplasmic male sterility in maize, resulting in pollen abortion (Gallagher et al., 2002). Meanwhile, RNA editing changes from CAA (Gln) to UAA (End) at the atp6-1003 site ensured the normal synthesis of the polypeptide encoded by atp6 in Yunnan purple rice maintenance lines, while sterile lines had no RNA editing at this site (Wei et al., 2010). Interestingly, this study identified six editing sites that could create start or stop codons: nad1-2 (Thr to Met), nad4L-2 (Thr to Met), rps10-2 (Thr to Met), atp6-1156 (Gln to End), ccmFC-1315 (Arg to End), and rps10-331 (Arg to End). Their specific roles in plant growth and development require in-depth exploration.
Conclusions
In this study, we assembled the first complete mitogenome of S. samarangense. It was 530,242 bp in length, with a GC content of 45.03%, and encoded 61 unique genes. The many repetitive sequences and homologous segments of the chloroplast genome in the mitogenome of S. samarangense suggests that it may underwent multiple genomic recombination events during evolution. Moreover, two forward repeats were involved in mediating recombination. Therefore, multiple secondary conformations may exist in addition to the master circular structure. A total of 591 RNA editing sites were identified in the PCGs, among which six sites create start or stop codons and should be the focus of future studies. The newly obtained mitogenome can be used as a reference genome for analyses of other Syzygium species and provides important information for the molecular breeding of S. samarangense.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. The mitogenome sequence supporting the conclusions of this article is available in GenBank (https://www.ncbi.nlm.nih.gov/) under accession number: OQ701348. The chloroplast genome sequence information has been uploaded to NCBI (accession number: OR766332). The Illumina, Nanopore, and transcriptome data of Syzygium samarangense have been deposited to the Sequence Read Archive repository under SRR26670650, SRR26670651, and SRR24058007, respectively.
Author contributions
GL: Conceptualization, Data curation, Formal analysis, Methodology, Software, Validation, Writing – original draft. QL: Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Central Guided Local Development of Science and Technology Project (YDZX20195400004007), the Science and Technology Program of Tibet Autonomous Region, China (XZ2019NKNS009) and the Scientific Research Start-up Fund for High-level Introduced Talents of Henan Institute of Science and Technology.
Acknowledgments
The authors sincerely thank the experimental personnel and bioinformatics analysis at Wuhan Benagen Technology Co., Ltd (www.benagen.com) and MitoRun research group participated in this project. The authors are thankful to Youxiong Que for reviewing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1301164/full#supplementary-material
References
Adams, K. L., Palmer, J. D. (2003). Evolution of mitochondrial gene content: Gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. doi: 10.1016/S1055-7903(03)00194-5
Allen, J. O., Fauron, C. M., Minx, P., Roark, L., Oddiraju, S., Lin, G. N., et al. (2007). Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics 177, 1173–1192. doi: 10.1534/genetics.107.073312
Alverson, A. J., Rice, D. W., Dickinson, S., Barry, K., Palmer, J. D. (2011). Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 23, 2499–2513. doi: 10.1105/tpc.111.087189
Banadka, A., Wudali, N. S., Al-Khayri, J. M., Nagella, P. (2022). The role of Syzygium samarangense in nutrition and economy: An overview. S. Afr. J. Bot. 145, 481–492. doi: 10.1016/j.sajb.2022.03.014
Behboudi, R., Nouri-Baygi, M., Naghibzadeh, M. (2023). RPTRF: A rapid perfect tandem repeat finder tool for DNA sequences. Biosystems 226, 104869. doi: 10.1016/j.biosystems.2023.104869
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bennetzen, J. L., Wang, H. (2014). The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 65, 505–530. doi: 10.1146/annurev-arplant-050213-035811
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.914635
Butelli, E., Licciardello, C., Zhang, Y., Liu, J., Mackay, S., Bailey, P., et al. (2012). Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 24, 1242–1255. doi: 10.1105/tpc.111.095232
Chan, P. P., Lin, B. Y., Mak, A. J., Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Chen, S. (2023). Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107. doi: 10.1002/imt2.107
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, gix120. doi: 10.1093/gigascience/gix120
Chen, F., Hao, Y., Yin, Z., Wu, G., Jiang, X. (2017). Transcriptome of wax apple (Syzygium samarangense) provides insights into nitric oxide-induced delays of postharvest cottony softening. Acta Physiol. Plant 39, 273. doi: 10.1007/s11738-017-2569-4
Chen, Y., Ye, W., Zhang, Y., Xu, Y. (2015). High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 43, 7762–7768. doi: 10.1093/nar/gkv784
Chevigny, N., Schatz-Daas, D., Lotfi, F., Gualberto, J. M. (2020). DNA repair and the stability of the plant mitochondrial genome. Int. J. Mol. Sci. 21, 328. doi: 10.3390/ijms21010328
Chinese Herbalism Editorial Board (1999). Chinese Herbalism Vol. 5 (Shanghai: Shanghai Scientific and Technical Publishers), 658.
De Coster, W., D’hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Dunn, N. A., Unni, D. R., Diesh, C., Munoz-Torres, M., Harris, N. L., Yao, E., et al. (2019). Apollo: Democratizing genome annotation. PloS Comput. Biol. 15, e1006790. doi: 10.1371/journal.pcbi.1006790
Fan, W., Liu, F., Jia, Q., Du, H., Chen, W., Ruan, J., et al. (2022). Fragaria mitogenomes evolve rapidly in structure but slowly in sequence and incur frequent multinucleotide mutations mediated by microinversions. New Phytol. 236, 745–759. doi: 10.1111/nph.18334
Flati, T., Gioiosa, S., Spallanzani, N., Tagliaferri, I., Diroma, M. A., Pesole, G., et al. (2020). HPC-REDItools: a novel HPC-aware tool for improved large scale RNA-editing analysis. BMC Bioinf. 21, 353. doi: 10.1186/s12859-020-03562-x
Freudenthal, J. A., Pfaff, S., Terhoeven, N., Korte, A., Ankenbrand, M. J., Förster, F. (2020). A systematic comparison of chloroplast genome assembly tools. Genome Biol. 21, 254. doi: 10.1186/s13059-020-02153-6
Gallagher, L. J., Betz, S. K., Chase, C. D. (2002). Mitochondrial RNA editing truncates a chimeric open reading frame associated with S male-sterility in maize. Curr. Genet. 42, 179–184. doi: 10.1007/s00294-002-0344-5
Gommans, W. M., Mullen, S. P., Maas, S. (2009). RNA editing: a driving force for adaptive evolution? Bioessays 31, 1137–1145. doi: 10.1002/bies.200900045
Govaerts, R. H. A., Sobral, M., Ashton, P., Barrie, F. R., Holst, B. K., Landrum, L., et al. (2008). World Checklist of Myrtaceae (London: Royal Botanic Gardens).
Greiner, S., Sobanski, J., Bock, R. (2015). Why are most organelle genomes transmitted maternally? Bioessays 37, 80–94. doi: 10.1002/bies.201400110
Gualberto, J. M., Newton, K. J. (2017). Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 68, 225–252. doi: 10.1146/annurev-arplant-043015-112232
Gurib-Fakim, A. (2006). Medicinal plants: traditions of yesterday and drugs of tomorrow. Mol. Aspects Med. 27, 1–93. doi: 10.1016/j.mam.2005.07.008
Hao, W., Liu, G., Wang, W., Shen, W., Zhao, Y., Sun, J., et al. (2021). RNA editing and its roles in plant organelles. Front. Genet. 12. doi: 10.3389/fgene.2021.757109
Hofstatter, P. G., Thangavel, G., Lux, T., Neumann, P., Vondrak, T., Novak, P., et al. (2022). Repeat-based holocentromeres influence genome architecture and karyotype evolution. Cell 185, 3153–3168. doi: 10.1016/j.cell.2022.06.045
Ichinose, M., Sugita, M. (2017). RNA editing and its molecular mechanism in plant organelles. Genes (Basel) 8, 5. doi: 10.3390/genes8010005
Katoh, K., Rozewicki, J., Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Khandaker, M. M., Boyce, A. N. (2016). Growth, distribution and physiochemical properties of wax apple (Syzygium samarangense): A Review. Aust. J. Crop Sci. 10, 1640–1648. doi: 10.21475/AJCS.2016.10.12.PNE306
Khera, P., Saxena, R., Sameerkumar, C. V., Saxena, K., Varshney, R. K. (2015). Mitochondrial SSRs and their utility in distinguishing wild species, CMS lines and maintainer lines in pigeonpea (Cajanus cajan L.). Euphytica 206, 737–746. doi: 10.1007/s10681-015-1504-2
Kolmogorov, M., Yuan, J., Lin, Y., Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PloS Genet. 15, e1008373. doi: 10.1371/journal.pgen.1008373
Kubo, T., Kitazaki, K., Matsunaga, M., Kagami, H., Mikami, T. (2011). Male sterility-inducing mitochondrial genomes: how do they differ? Crit. Rev. Plant Sci. 30, 378–400. doi: 10.1080/07352689.2011.587727
Kubo, T., Newton, K. J. (2008). Angiosperm mitochondrial genomes and mutations. Mitochondrion 8, 5–14. doi: 10.1016/j.mito.2007.10.006
Kurtz, S., Schleiermacher, C. (1999). REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics 15, 426–427. doi: 10.1093/bioinformatics/15.5.426
Lang, B. F., Gray, M. W., Burger, G. (1999). Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397. doi: 10.1146/annurev.genet.33.1.351
Letunic, I., Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, C., Tang, J., Hu, Z., Wang, J., Yu, T., Yi, H., et al. (2020). A novel maize dwarf mutant generated by Ty1-copia LTR-retrotransposon insertion in Brachytic2 after spaceflight. Plant Cell Rep. 39, 393–408. doi: 10.1007/s00299-019-02498-8
Liao, X., Wei, M., Khan, A., Zhao, Y., Kong, X., Zhou, B., et al. (2020). Comparative analysis of mitochondrial genome and expression variation between UG93A and UG93B reveals a candidate gene related to cytoplasmic male sterility in kenaf. Ind. Crop Prod. 152, 112502. doi: 10.1016/j.indcrop.2020.112502
Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. (2016). The role of mitochondria in plant development and stress tolerance. Free Radical Bio. Med. 100, 238–256. doi: 10.1016/j.freeradbiomed.2016.03.033
Lisch, D. (2013). How important are transposons for plant evolution? Nat. Rev. Genet. 14, 49–61. doi: 10.1038/nrg3374
Liu, S., Ni, Y., Li, J., Zhang, X., Yang, H., Chen, H., et al. (2023). CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 23, 694–704. doi: 10.1111/1755-0998.13729
Liu, J., Ni, S., Zheng, C., Shi, C., Niu, Y. (2018). Chloroplast genome of tropical and sub-tropical fruit tree Syzygium samarangense (Myrtaceae). Mitochondrial DNA B Resour. 3, 890–891. doi: 10.1080/23802359.2018.1501296
Liu, C., Shi, L., Zhu, Y., Chen, H., Zhang, J., Lin, X., et al. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics 13, 715. doi: 10.1186/1471-2164-13-715
Low, Y. W., Rajaraman, S., Tomlin, C. M., Ahmad, J. A., Ardi, W. H., Armstrong, K., et al. (2022). Genomic insights into rapid speciation within the world’s largest tree genus Syzygium. Nat. Commun. 13, 5031. doi: 10.1038/s41467-022-32637-x
Mehmood, A., Laiho, A., Venäläinen, M. S., McGlinchey, A. J., Wang, N., Elo, L. L. (2020). Systematic evaluation of differential splicing tools for RNA-seq studies. Brief. Bioinform. 21, 2052–2065. doi: 10.1093/bib/bbz126
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Møller, I. M., Rasmusson, A. G., Van Aken, O. (2021). Plant mitochondria - past, present and future. Plant J. 108, 912–959. doi: 10.1111/tpj.15495
Mower, J. P., Sloan, D. B., Alverson, A. J. (2012). “Plant mitochondrial genome diversity: The genomics revolution,” in Plant Genome Diversity, vol. 1 . Eds. Wendel, J., Greilhuber, J., Dolezel, J., Leitch, I. (Vienna: Springer), 123–144. doi: 10.1007/978-3-7091-1130-7_9
Mower, J. P., Stefanović, S., Hao, W., Gummow, J. S., Jain, K., Ahmed, D., et al. (2010). Horizontal acquisition of multiple mitochondrial genes from a parasitic plant followed by gene conversion with host mitochondrial genes. BMC Biol. 8, 150. doi: 10.1186/1741-7007-8-150
Niu, Y., Gao, C., Liu, J. (2022). Complete mitochondrial genomes of three Mangifera species, their genomic structure and gene transfer from chloroplast genomes. BMC Genomics 23, 147. doi: 10.1186/s12864-022-08383-1
Prasad, A., Chirom, O., Prasad, M. (2022). Horizontal gene transfer and the evolution of land plants. Trends Plant Sci. 27, 1203–1205. doi: 10.1016/j.tplants.2022.08.020
Putintseva, Y. A., Bondar, E. I., Simonov, E. P., Sharov, V. V., Oreshkova, N. V., Kuzmin, D. A., et al. (2020). Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genomics 21, 654. doi: 10.1186/s12864-020-07061-4
Quiñones, V., Zanlungo, S., Holuigue, L., Litvak, S., Jordana, X. (1995). The cox1 initiation codon is created by RNA editing in potato mitochondria. Plant Physiol. 108, 1327–1328. doi: 10.1104/pp.108.3.1327
Rice, D. W., Alverson, A. J., Richardson, A. O., Young, G. J., Sanchez-Puerta, M. V., Munzinger, J., et al. (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342, 1468–1473. doi: 10.1126/science.1246275
Richardson, A. O., Rice, D. W., Young, G. J., Alverson, A. J., Palmer, J. D. (2013). The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11, 29. doi: 10.1186/1741-7007-11-29
Sanchez-Puerta, M. V., Zubko, M. K., Palmer, J. D. (2015). Homologous recombination and retention of a single form of most genes shape the highly chimeric mitochondrial genome of a cybrid plant. New Phytol. 206, 381–396. doi: 10.1111/nph.13188
Sibbald, S. J., Lawton, M., Archibald, J. M. (2021). Mitochondrial genome evolution in pelagophyte algae. Genome Biol. Evol. 13, evab018. doi: 10.1093/gbe/evab018
Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. (2015). Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. U.S.A. 112, E3515–E3524. doi: 10.1073/pnas.1504491112
Sloan, D. B. (2013). One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 200, 978–985. doi: 10.1111/nph.12395
Takenaka, M., Zehrmann, A., Verbitskiy, D., Härtel, B., Brennicke, A. (2013). RNA editing in plants and its evolution. Annu. Rev. Genet. 47, 335–352. doi: 10.1146/annurev-genet-111212-133519
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., Paterson, A. H. (2008). Synteny and collinearity in plant genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Tovkach, A., Ryan, P. R., Richardson, A. E., Lewis, D. C., Rathjen, T. M., Ramesh, S., et al. (2013). Transposon-mediated alteration of TaMATE1B expression in wheat confers constitutive citrate efflux from root apices. Plant Physiol. 161, 880–892. doi: 10.1104/pp.112.207142
Tyagi, S., Kabade, P. G., Gnanapragasam, N., Singh, U. M., Gurjar, A. K. S., Rai, A., et al. (2023). Codon usage provide insights into the adaptation of rice genes under stress condition. Int. J. Mol. Sci. 24, 1098. doi: 10.3390/ijms24021098
Wang, N., Li, C., Kuang, L., Wu, X., Xie, K., Zhu, A., et al. (2022). Pan-mitogenomics reveals the genetic basis of cytonuclear conflicts in citrus hybridization, domestication, and diversification. Proc. Natl. Acad. Sci. U.S.A. 119, e2206076119. doi: 10.1073/pnas.2206076119
Wei, L., Fei, Z., Ding, Y. (2010). Mitochondrial RNA editing of ATPase atp6 gene transcripts of Yunnan purple rice (Oryza sativa L.). J. Wuhan Bot. Res. 28, 251–256. doi: 10.3724/SP.J.1142.2010.30251
Wendel, J. F., Jackson, S. A., Meyers, B. C., Wing, R. A. (2016). Evolution of plant genome architecture. Genome Biol. 17, 37. doi: 10.1186/s13059-016-0908-1
Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13, e1005595. doi: 10.1371/journal.pcbi.1005595
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z., Liao, X., Zhang, X., Tembrock, L. R., Broz, A. (2022). Genomic architectural variation of plant mitochondria - A review of multichromosomal structuring. J. Syst. Evol. 60, 160–168. doi: 10.1111/jse.12655
Xiang, C., Gao, F., Jakovlić, I., Lei, H., Hu, Y., Zhang, H., et al. (2023). Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2, e87. doi: 10.1002/imt2.87
Xiao, H., Jiang, N., Schaffner, E., Stockinger, E. J., van der Knaap, E. (2008). A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 319, 1527–1530. doi: 10.1126/science.1153040
You, C., Cui, T., Zhang, C., Zang, S., Su, Y., Que, Y. (2023). Assembly of the complete mitochondrial genome of Gelsemium elegans revealed the existence of homologous conformations generated by a repeat mediated recombination. Int. J. Mol. Sci. 24, 527. doi: 10.3390/ijms24010527
Zandueta-Criado, A., Bock, R. (2004). Surprising features of plastid ndhD transcripts: addition of non-encoded nucleotides and polysome association of mRNAs with an unedited start codon. Nucleic Acids Res. 32, 542–550. doi: 10.1093/nar/gkh217
Zhang, H., Meltzer, P., Davis, S. (2013). RCircos: an R package for Circos 2D track plots. BMC Bioinf. 14, 244. doi: 10.1186/1471-2105-14-244
Zhang, S., Wang, J., He, W., Kan, S., Liao, X., Jordan, D. R., et al. (2023). Variation in mitogenome structural conformation in wild and cultivated lineages of sorghum corresponds with domestication history and plastome evolution. BMC Plant Biol. 23, 91. doi: 10.1186/s12870-023-04104-2
Keywords: Syzygium samarangense, mitogenome, gene loss, evolution analysis, RNA editing
Citation: Lu G and Li Q (2024) Complete mitochondrial genome of Syzygium samarangense reveals genomic recombination, gene transfer, and RNA editing events. Front. Plant Sci. 14:1301164. doi: 10.3389/fpls.2023.1301164
Received: 24 September 2023; Accepted: 18 December 2023;
Published: 09 January 2024.
Edited by:
Christos Bazakos, Max Planck Institute for Plant Breeding Research, GermanyReviewed by:
Nunzio D’Agostino, University of Naples Federico II, ItalyXiujun Zhang, Chinese Academy of Sciences (CAS), China
Copyright © 2024 Lu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Li, bGlxaW5nMTMwODExQDEyNi5jb20=