 Mengjing Liang1
Mengjing Liang1 Zhongyang Du
Zhongyang Du Chunli Ji
Chunli Ji Hongli Cui
Hongli Cui Runzhi Li
Runzhi Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 08 January 2024
Sec. Plant Biotechnology
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1299902
MADS-box transcription factors are widely involved in the regulation of plant growth, developmental processes, and response to abiotic stresses. Perilla frutescens, a versatile plant, is not only used for food and medicine but also serves as an economical oil crop. However, the MADS-box transcription factor family in P. frutescens is still largely unexplored. In this study, a total of 93 PfMADS genes were identified in P. frutescens genome. These genes, including 37 Type I and 56 Type II members, were randomly distributed across 20 chromosomes and 2 scaffold regions. Type II PfMADS proteins were found to contain a greater number of motifs, indicating more complex structures and diverse functions. Expression analysis revealed that most PfMADS genes (more than 76 members) exhibited widely expression model in almost all tissues. The further analysis indicated that there was strong correlation between some MIKCC-type PfMADS genes and key genes involved in lipid synthesis and flavonoid metabolism, which implied that these PfMADS genes might play important regulatory role in the above two pathways. It was further verified that PfMADS47 can effectively mediate the regulation of lipid synthesis in Chlamydomonas reinhardtii transformants. Using cis-acting element analysis and qRT-PCR technology, the potential functions of six MIKCC-type PfMADS genes in response to abiotic stresses, especially cold and drought, were studied. Altogether, this study is the first genome-wide analysis of PfMADS. This result further supports functional and evolutionary studies of PfMADS gene family and serves as a benchmark for related P. frutescens breeding studies.
The MADS-box genes are an important class of transcriptional regulators in plants, named after the acronyms of the first four proteins identified (MCM1 in Saccharomyces cerevisiae, AGAMOUS in Arabidopsis thaliana, DEFICIENS in goldenseal, and SRF in human serum response factor) (Shore and Sharrocks, 1995; Lawton-Rauh et al., 2000). The MADS structural domains are located within the four encoded proteins. These domains are highly conserved and are composed of 56-60 amino acids at the N-terminus. They bind to a specific DNA cis-element known as Carg-box and play a role in regulating the spatiotemporal expression of genes (Messenguy and Dubois, 2003). According to multiple studies, MADS-box genes in plants can be categorized into two types: Type I (M type) and Type II (MIKC type) (Becker and Theissen, 2003). The main difference between the two types is that the MIKC type contains three unique characteristic domains in addition to the DNA-binding M domain. These domains are the intermediate (I) domain, the keratin-like (K) domain, and the C-terminal (C) domain, and they are arranged in this specific order (Yang et al., 2003; Kaufmann et al., 2005). This factor is crucial in enabling MADS-box proteins to form dimers or tetramers and carry out their functions (Immink et al., 2009).
Type I can be further classified into Mα, Mβ, and Mγ subtypes based on the phylogenetic relationship, gene structure, and protein structure of the conserved MADS-box domain. On the other hand, type II is divided into MIKCC and MIKC* subtypes, which is determined by the number of exons encoding the I domain and the structure of the K domain (Gramzow and Theissen, 2010). Numerous MADS-box genes have been identified in angiosperms, and their phylogeny has been reconstructed to date. They found that the MIKCC-type MADS-box genes can be categorized into 12 subfamilies, namely AG, AGL2/SEP, AGL6, AGL12, AGL15, AGL17, FLC, GGM13, STMADS11, AP1/SQUA, AP3/PI, and TM3 (Becker and Theissen, 2003). The evolutionary process of these genes involved numerous duplication events, leading to a vast multigene family (Irish, 2003).
MADS-box genes are prevalent in eukaryotes, spanning across the plant kingdom (Schilling et al., 2020). Extensive research has been conducted on a substantial number of MADS-box genes in both monocotyledonous plants like corn, wheat, millet, and sorghum, and dicotyledonous plants such as tomato, rape, tobacco, and petunia (Arora et al., 2007; Schilling et al., 2018; Wang et al., 2019). The function of this gene family encompasses various stages of plant development, such as seed germination, vegetative growth, floral organ formation, seed coat development, and embryo morphogenesis (Immink et al., 2009). It plays a pivotal role in the overall plant development process. The ABC model of flower organ development is one of the most well-known models, which was later supplemented by the ABCDE model (Coen and Meyerowitz, 1991; Agrawal et al., 2005). According to reports, only a few type I genes have been explored for their biological functions in plants, mainly involved in female gametophyte, embryo, and seed development (Bemer et al., 2008; Kang et al., 2008; Steffen et al., 2008; Smaczniak et al., 2012). For example, AGL23 regulates the development of Arabidopsis female gametophytes and is involved in controlling formation of organelles during embryo development (Colombo et al., 2008). Compared to the M and MIKC types, the MIKC*-type has more conserved functions and has been extensively studied (Pelaz et al., 2000; Swiezewski et al., 2009; Molesini et al., 2020). Recent research has demonstrated that MIKC*-type MADS-box genes have a dual function in plant biology. Apart from their involvement in flower organ development and other processes, these genes also play a vital role in regulating substance metabolism in plants. For instance, CsMADS6 (AGAMOUS-like subfamily) in citrus, MdMADS6 from apple, and CaMADS-RIN (SEP subfamily) in pepper have been found to have significant regulatory roles in the carotenoid synthesis pathway (Dong et al., 2014; Lu et al., 2018; Li et al., 2022). It has been reported that PfMADS genes may be involved in the flavonoid synthesis pathway of P. frutescens (Jiang et al., 2020). An AGAMOUS-like MADS-box transcription factor has been identified as being involved in the regulation of the lipid anabolism pathway in oil palm (Li et al., 2020; Zhang et al., 2022). Moreover, its regulatory role in abiotic stresses (cold, drought, salt, and other stresses) has been verified in rice, cereals, cotton seeds, Arabidopsis thaliana, and other plants (Wu et al., 2021; Zhao et al., 2021; Zhao et al., 2021; Xue et al., 2022). This demonstrates the significant impact of the MADS-box transcription factor family on plants and its involvement in a wide range of regulatory processes.
P. frutescens (Perilla frutescens (L.) Britt.), an annual herb of the genus Perilla L in the Lamiaceae Martinov family (Nitta et al., 2005). It has a rich cultivation history of over 2,000 years and is currently grown in China, Japan, and South Korea, among other Asian countries (Park et al., 2008). P. frutescens seeds have a high oil content, with an oil yield ranging from 46% to 58%. Notably, the α-linolenic acid content constitutes more than 65% of the total oil (Zhou et al., 2022). Alpha-linolenic acid is an essential fatty acid for the human body, exhibiting a range of biological activities such as anti-Alzheimer’s disease, anti-viral, anti-bacterial, and anti-allergic properties (Clemente and Cahoon, 2009). These activities offer significant benefits for human health (Luo et al., 2021). In addition, P. frutescens leaves come in various colors such as green and purple. The purple leaves are rich in anthocyanin content and are used as a food pigment (Yoshida et al., 1990; Fujiwara et al., 2018; Li et al, 2022). On the other hand, the green leaves of P. frutescens contain a high content of bioactive compounds and are often utilized in various applications such as food, skin creams, and medications for atopic dermatitis (Komatsu et al., 2016; Edge et al., 1997). In summary, P. frutescens is a versatile plant with numerous benefits. Due to its unique nutrients and active substances, it is not only consumed as food and used in pharmaceuticals, healthcare, and cosmetics, but also holds economic value as an extensively developed and utilized oil crop (Igarashi and Miyazaki, 2013).
The MADS-box transcription factor family has been extensively studied in a variety of plants, but its presence and characteristics in P. frutescens have not yet been investigated. In this study, a total of 93 PfMADS gene members were identified using bioinformatics analysis methods at the genome-wide level (Zhang et al., 2021). Their chromosomal locations, phylogenetic evolution, conserved motifs, exon-intron structures, and gene expression profiles were investigated. Additionally, cis-acting elements were analyzed to predict their possible functions in P. frutescens and their potential roles in abiotic stress responses. These results provide basic information for a better study of the PfMADS genes, and lay the foundation for functional studies and the selection of new varieties of P. frutescens.
The genomic data of P. frutescens (https://www.ncbi.nlm.nih.gov/genome/?Term=Perilla+rutescens) were downloaded from NCBI GenBank (Zhang et al., 2021) to create a local database. The 108 AtMADS protein sequences were downloaded from the Arabidopsis database TAIR (http://www.arabidopsis.org/). To perform a multiple sequence alignment search in the P. frutescens local database, the TBtools software (An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data) was utilized with the protein sequence of the AtMADS as an index. Subsequently, the keywords “MADS” and “SRF” were entered into the database to perform a supplementary search. The HMMER-3.1b2 software package (http://hmmer.janelia.org/) (Finn et al., 2011) was utilized to create a Markov model for the Pfam database (Finn et al., 2006). The domain information of the MADS-box genes (PF00319; http://pfam.sanger.ac.uk) was downloaded for protein sequence search. The resulting model was then used to identify potential MADS-box proteins from the P. frutescens protein database. Each sequence was manually checked to ensure it met the threshold of an E-value ≤ 1e-10. The conserved structural domains of PfMADS were verified using CDD (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and SMART database (http://smart.embl-heidelberg.de/). Repetitive sequences and structurally incomplete sequences were removed, and members of the PfMADS gene family were identified. The MADS domains of PfMADS and the AtMADS proteins were clustered using ClustalX2.0 to perform preliminary classification of PfMADS candidates. The basic physicochemical properties such as relative molecular mass and theoretical isoelectric point (pI), of PfMADS proteins were predicted by the online software ProtParam (http://web.expasy.org/protparam/).
The chromosomal positions of PfMADS genes were determined using genome annotation files. TBtools software was used to map the positions of PfMADS on chromosomes, ranging from short-arm to long-arm telomeres. Gene replication events in PfMADS were analyzed and identified using TBtools software.
GSDS2.0 (http://gsds.gao-lab.org/index.php) was used to analyze the gene structure of PfMADS, obtaining the positions and numbers of introns and exons. The conserved motifs of PfMADS proteins were predicted using MEME (http://meme-suite.org/meme/). Finally, visual phylogenetic analysis was performed uniformly using TBtools software.
MEGA11 software was utilized to conduct a multiple sequence alignment analysis of the identified PfMADS and AtMADS protein sequences. The neighbor-joining (NJ) method was used to construct the phylogenetic tree, with the bootstrap test value (Bootstrap) set at 1000. The final visualization was performed using the Evolview website (https://evolgenius.info//evolview-v2).
The Multiple Collinearity Scan tool kit (MCScanX) in TBtools was used to examine segmental duplication events in the genome of P. frutescens. Additionally, it was used to calculate non-synonymous (Ka) and synonymous substitution (Ks) values for the segmental duplication gene pairs. The gene duplications were dated using the formula T = Ks/2r (Wu et al., 2017). The synteny relationship between genomes of the three species (Perilla frutescens and Arabidopsis, Perilla frutescens and Salvia japonica Thunb.) was analyzed using MCScanX, and the mutual homologous gene pairs were counted. Genomic data for Salvia japonica Thunb. were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/assembly/?term=Salvia+farinacea).
This study used P. frutescens seed transcriptome gene data, already available in the laboratory, to examine the expression profiles of P. frutescens seeds at three different stages (10d, 20d, 30d after flowering). Transcriptome data for roots, leaves, and buds were obtained from the NCBI P. frutescens SRA database (https://www.ncbi.nlm.nih.gov/sra/?term=Perilla+frutescens+%28L.). The TBTools software was used for expression profile cluster analysis. To investigate the regulatory role of the PfMADS in the expression of key genes related to lipid and flavonoid synthesis, partial gene sequences involved in these processes from NCBI. To clarify the specific relationship between MADS and these processes, we extracted the FPKM values from the gene transcriptome data. Correlation analysis was then conducted using the OmicsShare online tools (https://www.omicsshare.com/tools/Home/Soft/getsoft).
The cis-acting elements in the 2000 bp promoter region upstream of PfMADS family members were analyzed using PlantCare (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)(Lescot et al., 2002), and the upstream promoter region was extracted for sequence analysis using TBTools.
The plant material used in this experiment was the P. frutescens species ‘Zisu Jin No.2’, which was planted at the Agricultural Crop Station of Shanxi Agricultural University in April 2021. We selected ‘Zisu Jin No.2’ seeds with intact grains. Once the seedlings had grown to six true leaves, we carefully selected those that exhibited similar growth pattern. These selected seedlings were then subjected to cold temperature and drought stress treatments (1) The plants were grown at 4°C with a photoperiod of 16h light/8h dark. (2) To water the seedlings, PEG-6000 was added to the nutrient solution to create a 20% PEG Hoagland nutrient solution. Samples were collected in triplicate at the above three stress conditions. The seedling samples were immediately frozen in liquid N2 and stored at −80°C for subsequent RNA extraction.
Chlamydomonas reinhardtii PADang.CC849 was utilized for genetic transformation in this study. A single algal colony of Chlamydomonas reinhardtii was carefully selected and inoculated into liquid TAP medium for culture activation. The culture conditions included a light intensity of 50 μmol·m−2·s−1, a 12-hour light-dark cycle, static culture at a temperature of 25 ± 1°C, and shaking the culture 4-5 times a day.
The E. coli strain DH5α and plant overexpression vector pHR13 used in this experiment are both stored at the Institute of Molecular Agriculture and Bioenergy of Shanxi Agricultural University.
The PfMADS47 gene sequence was sent to Suzhou Jinweizhi Biotechnology Co., Ltd. Using Chlamydomonas reinhardtii as the target species, the expression vector pHR13-PfMADS47YH was constructed following codon optimization of the target sequence.
The bead-beating method (Wang C et al., 2017) was employed to genetically transform the wild-type strain CC849 of Chlamydomonas reinhardtii. Subsequently, the transgenic algal strains were obtained, and the algal biomass was collected for DNA and RNA extraction. PCR detection was performed at the gene and transcript levels to screen for positive transgenic algae strains.
The CTAB method was used to extract DNA from Chlamydomonas reinhardtii (Sun et al., 2016). Total RNA was extracted from samples using the EASY spin Plant RNA Rapid Extraction Kit (RN09, Beijing Adelaide Biotechnology Co., Ltd.). The cDNA was synthesized by reverse transcription using StarScript II RT Mix with gDNA Remover (A224, Beijing Kangrun Chengye Biotechnology Co., Ltd.), diluted at 1:10 dilution with RNase-free water, and stored at -20°C for subsequent qRT-PCR analysis. qRT-PCR amplification primers were designed based on the CDS sequences of low temperature and drought stress-related PfMADS genes (Table S1), and cDNA from different treatments was used as the template with the TB Green®Premix Ex Taq™ (Tli RNaseH Plus) kit [TaKaRa, Bao Ri Doctor Biotech (Beijing) Co., Ltd] to prepare the qRT-PCR reaction mixture: 5.0 μL TB Green Premix Ex Taq, 0.5 μL cDNA, 0.2 μL forward primer, 0.2 μL reverse primer, and 0.4 μL ddH2O, for a total volume of 10 μL. A TM real-time fluorescence quantitative PCR instrument was used for qRT-PCR amplification (DLAB-Accurate 96-X4). The amplification reaction was performed in three steps: 95°C for 2 min; 95°C for 30 s, 59°C for 30 s, and 72°C for 30 s, for 40 cycles. Three biological replicates and three technical replicates were established for each sample. The relative expression of PfMADS gene under low temperature and drought stresses was calculated by the 2-ΔΔCT method using Pfactin as an internal reference gene.
Algal cells cultured to the logarithmic growth phase were centrifuged at 5000 rpm for 3-4 minutes. The algal cells were collected and resuspended in a nitrogen-deficient medium. The uniform initial OD680 value was 0.8.
Shake the algal suspension evenly in the ultra-clean workbench and pipette 3 ml of each sample. Use the UV a spectrophotometer to measure the absorbance (OD680nm) of the algal suspension at a wavelength of 680nm. Changes in OD680nm indicates cells growth. Three replicates were set up, and each replicate was measured three times.
Aliquots of 15 mL of the algal culture were harvested by centrifugation at 3000 g for 5 min. The resulting pellets were then re-suspended in 2 mL of acetone for pigment extraction. After vortexing for 30 minutes at maximum speed, the pigment extracts were collected by centrifugation at 13,000 g for 10 minutes. The extracts were evaporated under nitrogen gas and redissolved in acetone. Pigment analysis was performed using a Waters 2695 HPLC system equipped with a Waters Spherisorb 5 μm ODS2 4.6 × 250 mm analytical column (Waters, Milford, MA, USA), following the protocols previously described.
Lipid extraction from C. reinhardtii was performed following the protocol described by Liu et al. (2016). In brief, algal samples were harvested by centrifugation at 3000 g for 5 minutes. The resulting cell pellets were then resuspended in 2 mL of methanol containing 0.05% butylated hydroxytoluene. After a 30-minute incubation, 4 mL of chloroform was added and the mixture was vigorously vortexed for another 30 minutes. Subsequently, 1.5 mL of 0.75% NaCl solution was added to facilitate phase separation. The lower organic phase containing the lipids was transferred into a new glass tube. The organic phase was then evaporated under nitrogen gas, and the remaining lipids were redissolved in chloroform for further quantification.
After removing redundant sequences, 93 PfMADS genes were finally identified in the genome of P. frutescens. Their characteristics, including gene locus ID, chromosomal start and end positions, member classification, protein sequence length (SL), molecular weight (MW), and isoelectric point (pI), were further specified. Statistical analysis showed that the length of the PfMADS proteins ranged from 64 to 380 amino acids, averaging of 236 amino acids. Their molecular weights ranged from 7.36 kDa to 43.53 kDa. The isoelectric points (pI) of the PfMADS proteins ranged from 4.58 to 10.4 (Table S2). To investigate the evolutionary relationships among PfMADS protein sequences, multiple protein sequence comparisons were performed for 93 PfMADS proteins (Figure 1). A highly conserved amino acid sequence, which spans approximately 30 to 60 amino acid residues, is present in different types of MADS domains (Table S3). These genes belong to the MADS-box gene family of P. frutescens and will be used in future research.
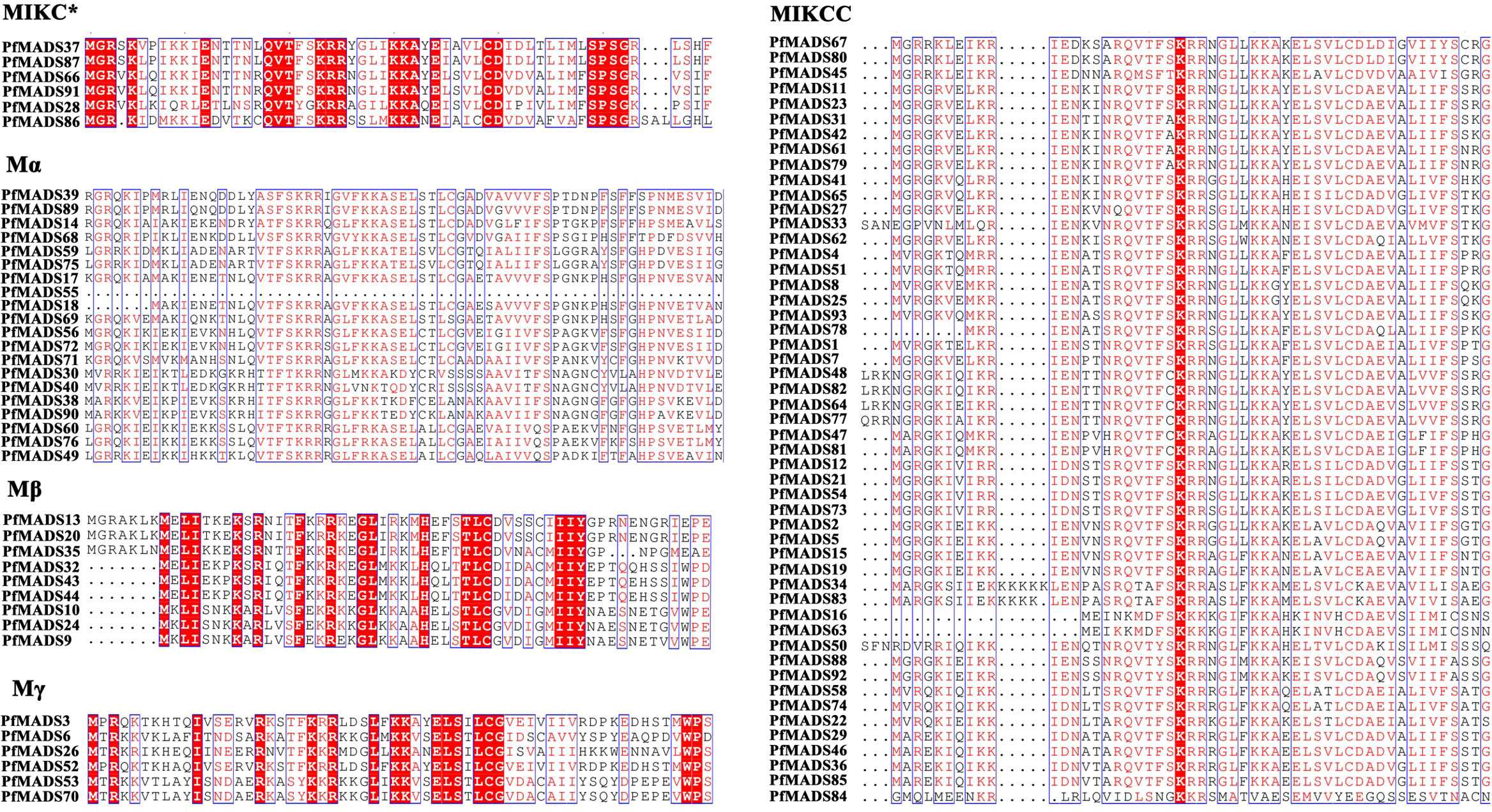
Figure 1 Multiple sequence alignment of the MADS domain from PfMADS. The red boxes indicated the higher conserved domains contained in each subfamily from PfMADS.
To investigate the genomic distribution of PfMADS genes, we performed a BLAST analysis against the released genome for P. frutescens. As a result, 93 PfMADS genes were mapped onto their corresponding chromosomes. It was observed that these genes are distributed unevenly across 20 chromosomes and 2 chromosomal segments. Chromosomes 2 and 5 contain the highest number of PfMADS genes, with 14 and 12 genes respectively. Conversely, the lowest number of PfMADS genes (one loci) were found on chromosomes 4, 6, 9, 20, scaffold0908.1, and scaffold1237.1(Figure 2). It is worth noting that the PfMADS gene family consists of 52 members from the AA subgenome and 41 members from the BB subgenome (Table S4). This discrepancy in numbers may have resulted from gene loss during the evolution of polyploidy.
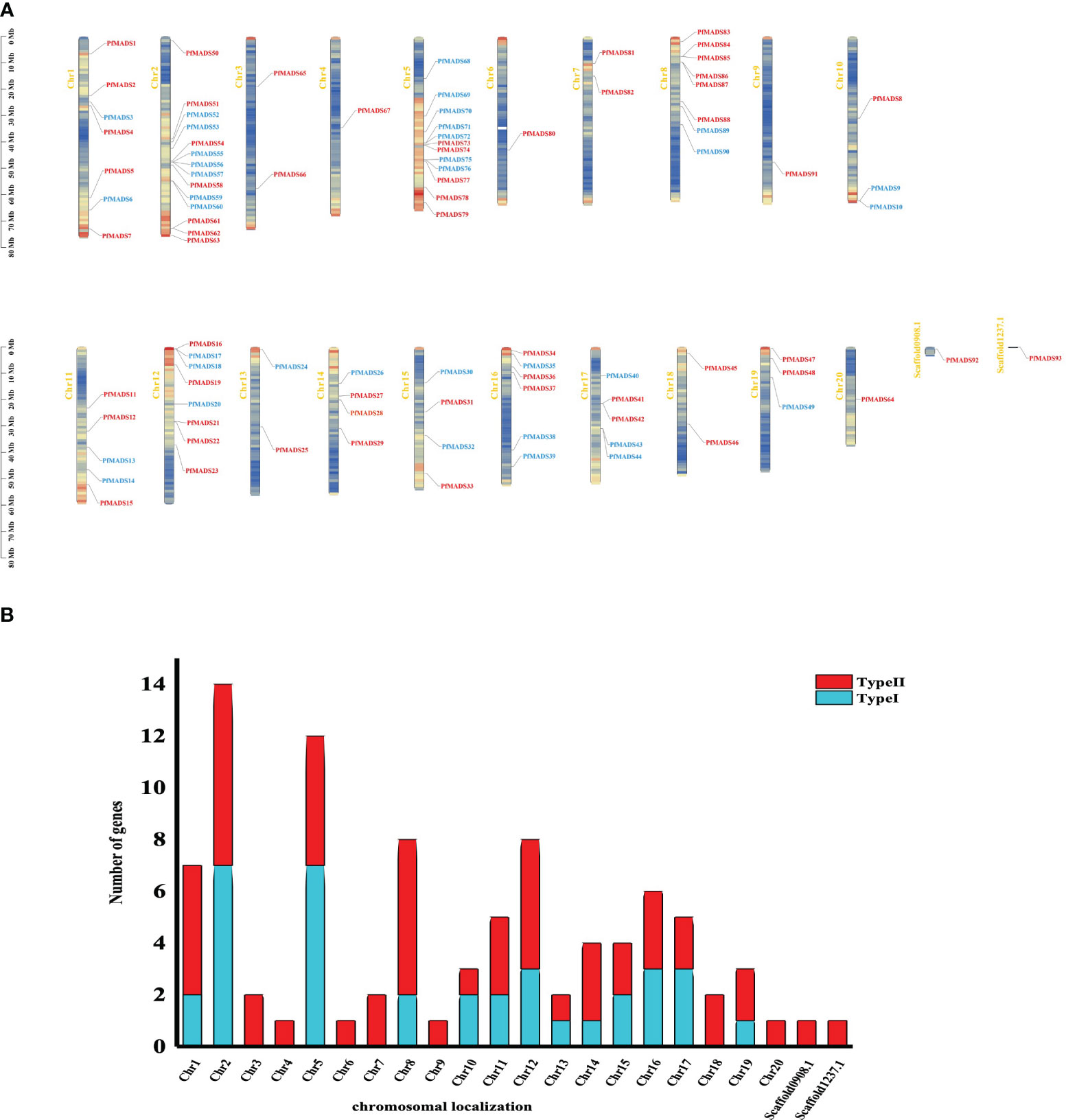
Figure 2 Analysis of the location of PfMADS on chromosomes. (A) Chromosomal distribution of PfMADS genes. The position of every PfMADS gene can be determined using the left scale. (B) a plot of the number and type of PfMADS genes per chromosome.
The replication events of PfMADS genes were analyzed at the genome-wide level by TBtools. The results revealed a significant homology between the PfMADS genes on different chromosomes (Figure 3). A total of 50 pairs of replication genes were counted (Table S5), all of them belonged to whole-genome duplication events, with 23 pairs having been duplicated only once. The remaining duplicated gene pairs had 2 to 4 copies each. To investigate whether these homologous genes underwent selection pressure, synonymous (Ks) and nonsynonymous (Ka) substitution rates were calculated for the identified gene pairs using TBtools, followed by the calculation of the Ka/Ks ratio to determine whether selection pressure acted on the PfMADS proteins (Zhang et al., 2017). Interestingly, the ω values for Ka/Ks of 47 gene pairs were less than 1, indicating that purifying selection has occurred for these gene pairs (Table S5).
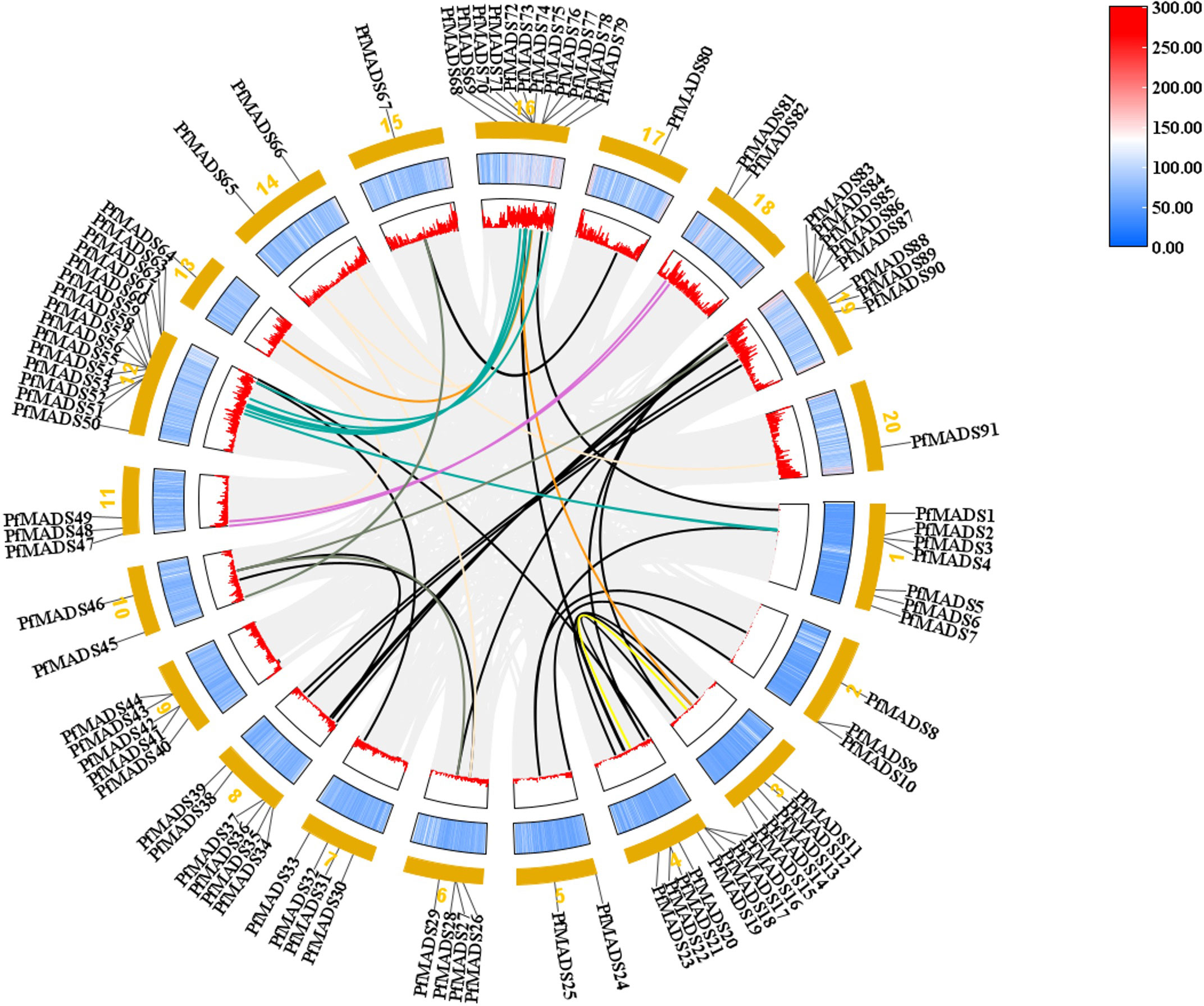
Figure 3 The intrachromosomal segmental duplication map of the MADS-box genes in P. frutescens. Colored lines inside the circle represent duplication inside the different chromosomes.
To identify the phylogenetic relationship of PfMADS proteins, a phylogenetic analysis was conducted on MADS-box protein sequences from P. frutescens and Arabidopsis (Figure 4). Based on the grouping standards of AtMADS proteins, PfMADS proteins were classified into two primary types (Type I and Type II), with 37 PfMADS in Type I and 56 PfMADS in Type II. The PfMADS in Type I were further classified into three subfamilies (Mα, Mβ and Mγ) containing 22,9, and 6 members respectively. Moreover, Type II is further divided into AG, FLC, SEP, SVP, AP1/FUL, AP3, SOC1, PI, and MIKC* subfamilies. The numbers of their corresponding PfMADS proteins are 4, 4, 6, 7, 5, 3, 8, and 2, respectively. Compared to Arabidopsis, P. frutescens has a greater number of Type II members, although the total number (108 in Arabidopsis) is lesser. This finding suggests that the evolutionary pattern of Type II PfMADS genes is highly conserved. It also implies that Type II PfMADS genes might be involved in multiple biological functions. According to the phylogenetic tree, the MADS-box proteins from P. frutescens and Arabidopsis, which belong to the same classification, did not cluster together. Instead, they formed separate branches with similar evolutionary relationships. This observation can largely explain the variations in the evolutionary patterns of the MADS-box gene family among different species.
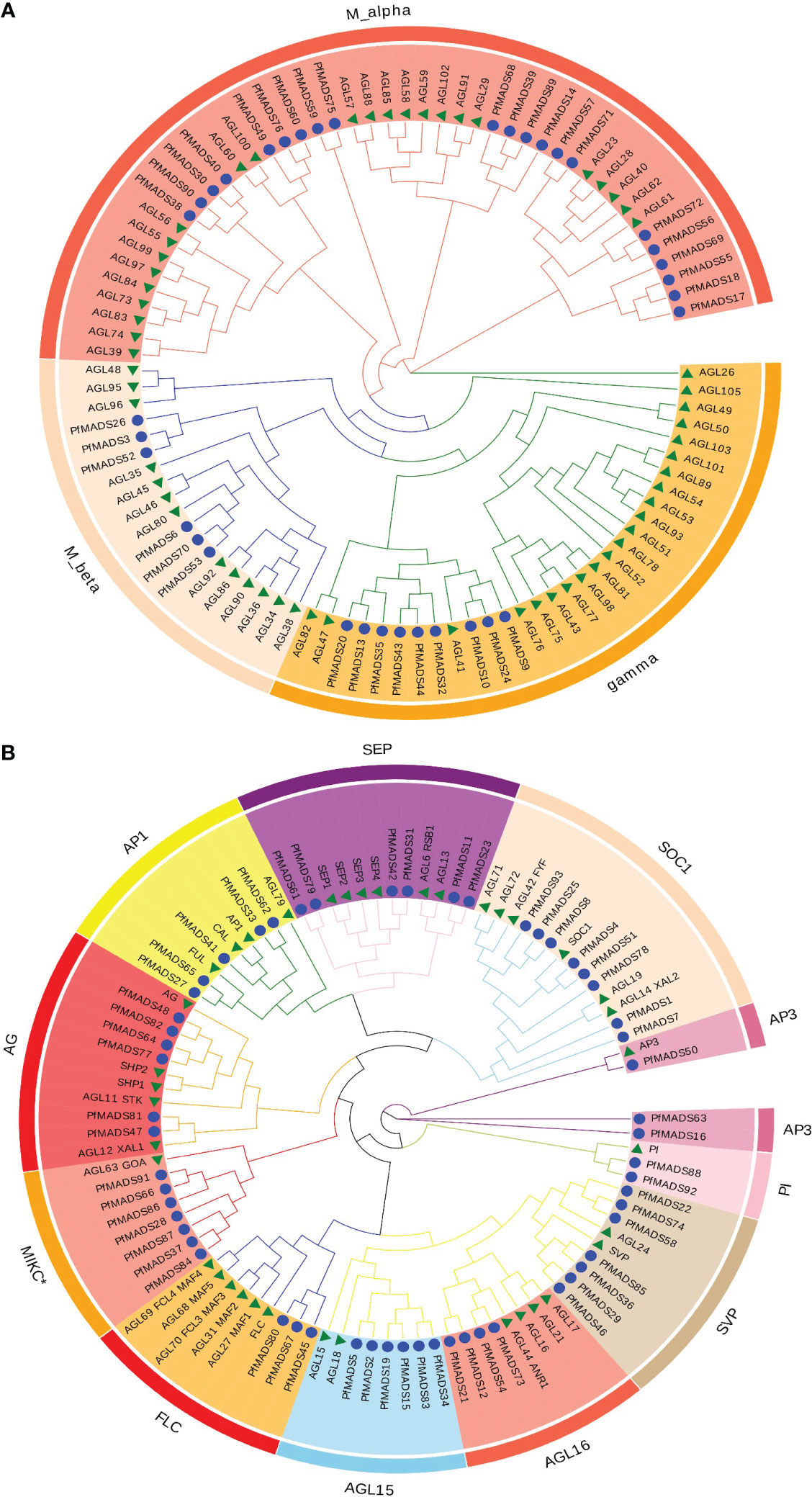
Figure 4 Classification of the P. frutescens MADS-box subfamilies. Specific-lineages are indicated by colors and bracketing. (A), the type I lineages contain Mα, Mβ, and Mγ; (B), the type II lineages contain MIKC* and MIKCC (all other type II subfamilies except MIKC*).
The comparative synteny maps of two genomes (P. frutescens vs A. thaliana, and P. frutescens vs Salvia japonica Thunb.) were created to explore the origin and evolution of PfMADS genes (Figure 5). Orthologous relationships were detected between 93 PfMADS genes and 108 AtMADS genes, and then 80 orthologous MADS-box gene pairs were identified accordingly, with most of them located on the syntenic loci on A. thaliana and P. frutescens chromosomes (Figure 5A). Multiple PfMADS genes were identified as putative homologs of the AtMADS genes. For instance, PfMADS48, PfMADS77, and PfMADS82 were identified as orthologs genes of AG. Similarly, the orthologous relationships were also present between 93 PfMADS genes and 117 ShMADS genes, and the corresponding 245 orthologous MADS-box gene pairs were established, with many found on the syntenic loci in the chromosomes of P. frutescens and Salvia japonica Thunb. (Figure 5B).
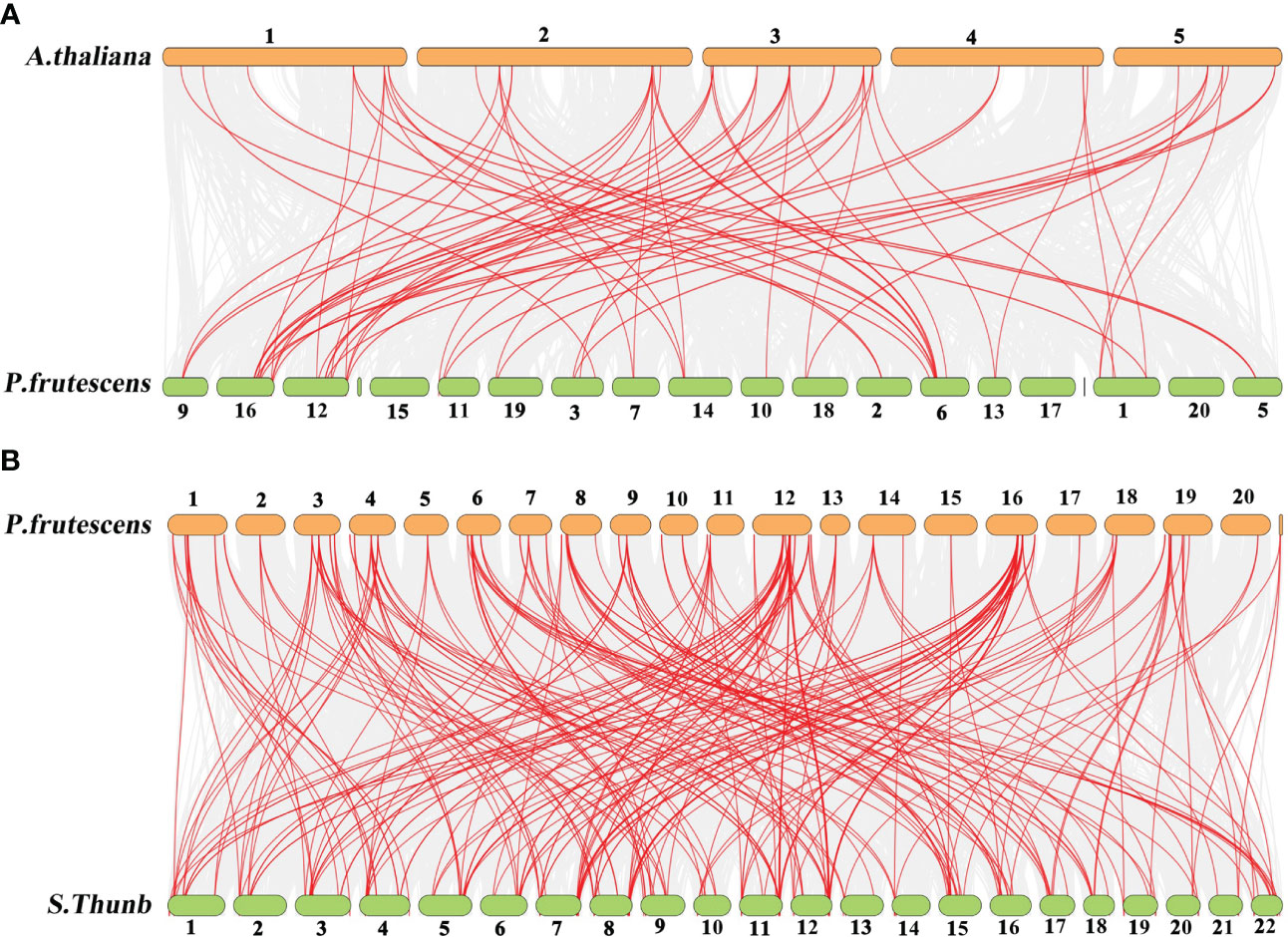
Figure 5 Synteny analysis of PfMADS genes between P. frutescens and two plant species (A. thaliana and Salvia japonica Thunb.). (A), P. frutescens and A. thaliana (B) P. frutescens and Salvia japonica Thunb. The chromosome number was indicated at the top of every chromosome.
To reveal the functional regions of PfMADS proteins (Figure 6A), conserved motifs were predicted by the MEME (Multiple Em for Motif Elicitation) program, and a total of 12 motifs were detected in 93 PfMADS proteins (Figure 6B). In accordance with the results of conservative motif analysis, Motifs 1 was detected in all PfMADS proteins. Moreover, Motifs 2 were predicted in most Type II except for Type I. Therefore, compared to Type I, the structure of Type II PfMADS proteins is relatively complex.
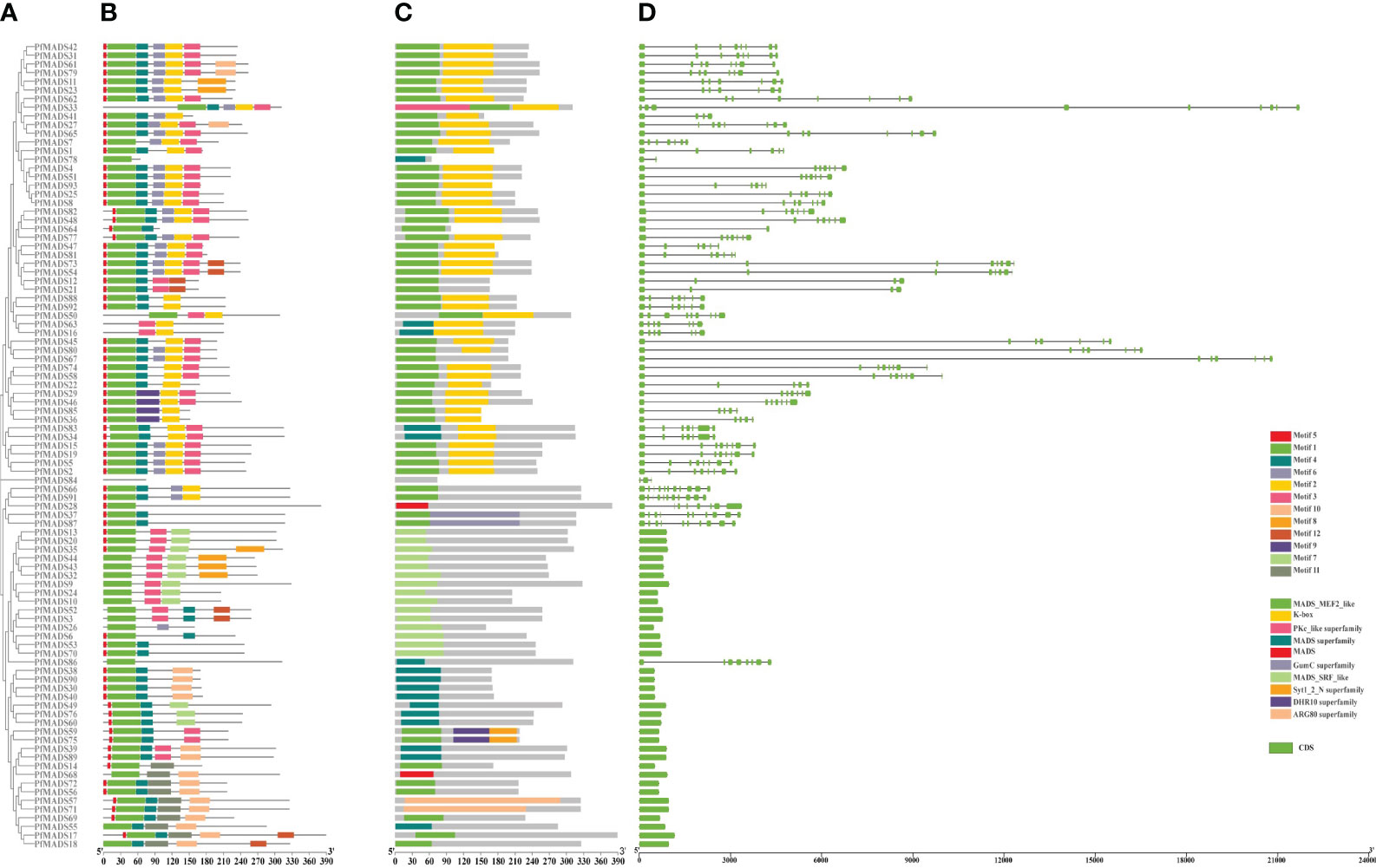
Figure 6 Gene and protein structures of the MADS-box gene families in P. frutescens. (A) Phylogenetic tree of P. frutescens MADS-box genes; (B) Motifs of MADS-box proteins. (C) Conserved domains of MADS-box proteins; (D) Exon-intron structures of MADS-box genes.
The PfMADS protein sequences were analyzed using MEGA software to perform multiple sequence alignment analysis and visual processing (Figure 6C). The results indicated that the PfMADS protein domains are remarkably conserved, with all 93 members containing MADS domains. Generally, the MADS domain of Type I is classified as SRF_like, while the domain of Type II is classified as MEF2_like.
Gene structure analysis of 93 PfMADS genes (Figure 6D) revealed that the number of exons in members of the PfMADS gene family varies from 1 to 11. Additionally, the number, distribution, and length of exons in PfMADS genes within the same subfamily exhibit similarities. The gene structure of M-type (Type I) PfMADS does not contain any introns, and the length of these genes is typically short, usually less than 1kb. On the other hand, Type II PfMADS genes have an average of 7 exons and introns in their gene structure, and they tend to be longer, mostly exceeding 3kb. These results established a significant molecular biology foundation for the functional investigation of PfMADS genes.
The gene expression pattern can provide essential information for determining the biological function of a gene. In order to study the function of the PfMADS genes, the data (FPKM) of P. frutescens in the public database were used to conduct expression analysis on 6 tissues. As depicted in Figure 7, genes such as PfMADS59/90/58/74 are highly expressed in flower buds, among which PfMADS59/90 belongs to the Mα subfamily and PfMADS58/74 belongs to the SVP subfamily. The found in apple that heterologous expression of MdDAMb and MdSVPA genes in ‘Royal Gala’ apple plant resulted in delayed buds and structural changes (Wu et al., 2017). Therefore, PfMADS58/74 in P. frutescens flower buds may be involved in the differentiation of floral meristems. Figure 7 shows that there are some important seed-specific or seed-dominant genes. For example, PfMADS85/83/67/26/34/91/66/15/19 are mainly expressed in later stages of seed development, and might be involved in development and metabolism of later stages of P. frutescens seeds. Many genes such as PfMADS81/36/12/84/2/75/86/88/92 and so on are mainly expressed in early stages of seed development, and might be involved in development and metabolism of early stages of P. frutescens seeds. Overall, more than 80% of PfMADS genes exhibit expression in these tissues, and members of the same subfamily demonstrate similar expression patterns. However, there are variations in the levels of expression. Type II PfMADS is predominantly expressed in seeds and leaves, while certain genes exhibit exclusive expression in roots and leaves. We speculate that P. frutescens likely has a considerable number of type II genes that function in various tissues.
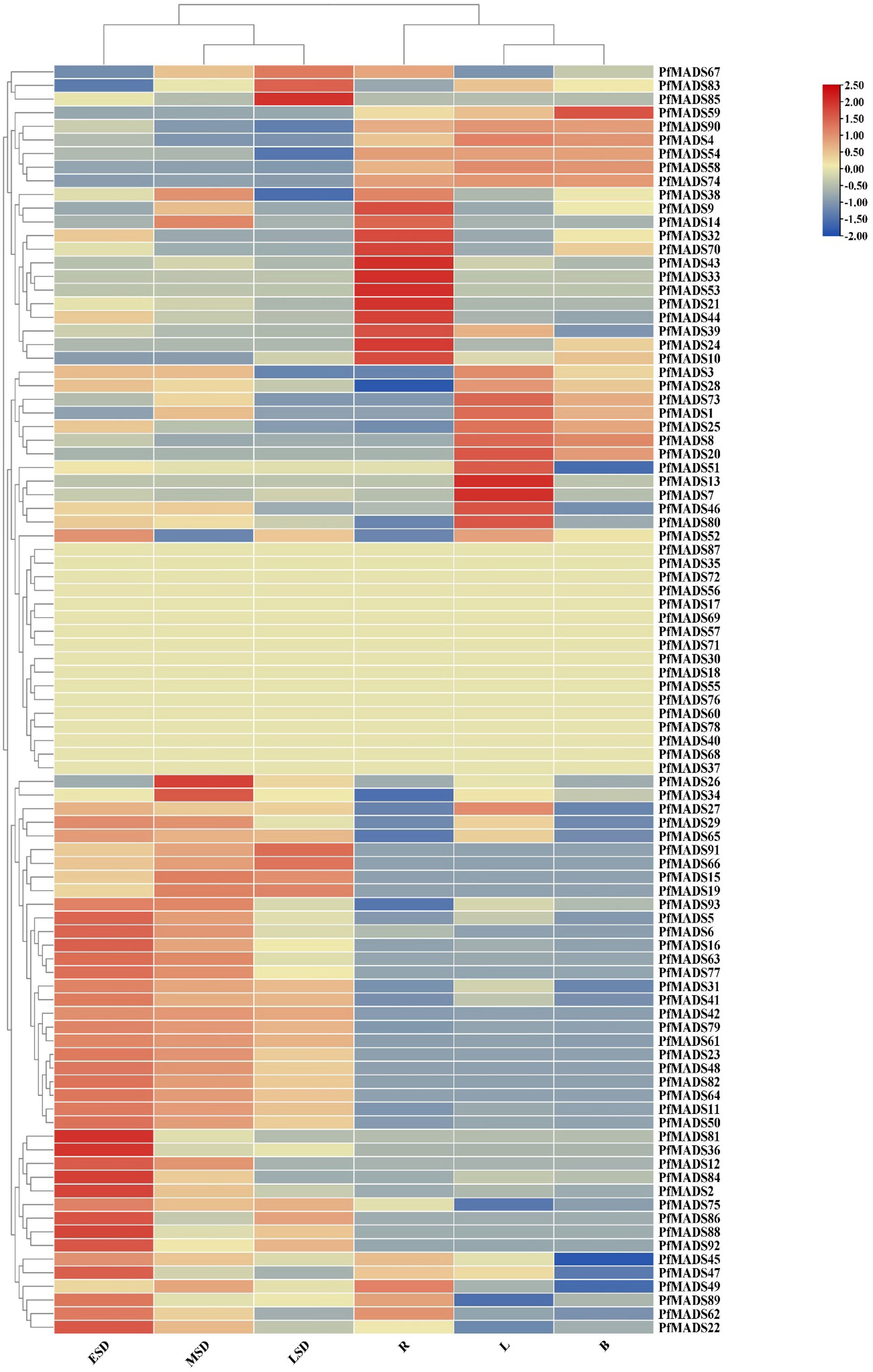
Figure 7 Expression profiles of PfMADS genes during six developmental stages of P. frutescens. The expression levels of PfMADS genes were showed by different colors on the right scale. ESD to LSD respectively indicated 10,20 and 30 days after full blooming. R, roots; L, leaves; B, bud.
Flavonoids, including anthocyanins, found in P. frutescens leaves have been the subject of extensive research. In this study, we identified several PfMADS genes from the transcriptome data of P. frutescens leaves and analyzed their expression at different stages of leaves with varying colors. The results from Figure 8 indicate that PfMADS27 and PfMADS58 were barely expressed in purple leaves, while they were expressed in green leaves. Additionally, as the color of the leaf transitions from purple to green, there is a corresponding gradual increase in the expression level of PfMADS27. This suggests a possible involvement of PfMADS27 in the synthesis process of flavonoids, particularly anthocyanins. However, further investigation is required to confirm this hypothesis.
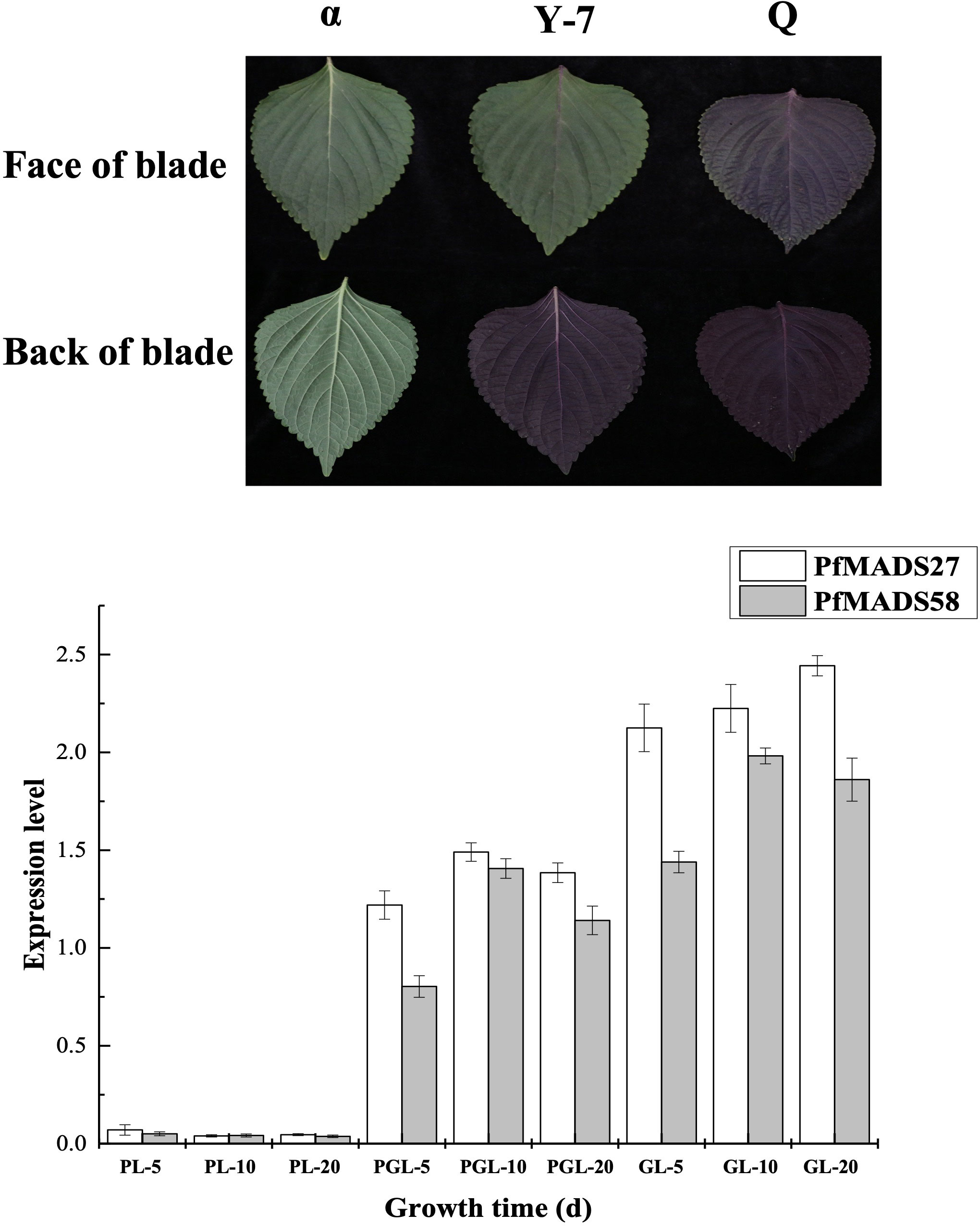
Figure 8 Expression profiles of PfMADS genes in different color leaves during three developmental stages of P. frutescens. PL, purple leaf; PGL, half purple, half green leaf; GL, green leaf. The P. frutescens seedlings were sampled on the 5th,10th and 20th day after the second true leaf. Data are means ± SE calculated from three biological replicates.
To explore the potential transcriptional regulatory mechanism of PfMADS, we performed correlation analysis of PfMADS genes related to oil accumulation and key genes related to flavonoid metabolism in seeds and leaves of P. frutescens. The results indicate that out of the 93 PfMADS genes, 9 are closely associated with important genes involved in lipid synthesis (Figure 9A). These findings suggest that these 9 genes can serve as potential candidates for further investigations into their functional roles. Eight PfMADS genes were selected to conduct correlation analysis with key genes in the flavonoid metabolism pathway (Figure 9B), and the results were consistent with those reported (Jiang et al., 2020). One of the genes, PfMADS47, is worth investigating as it is closely associated with lipid synthesis. However, further molecular-level verification, such as transgenesis, is necessary for functional confirmation.
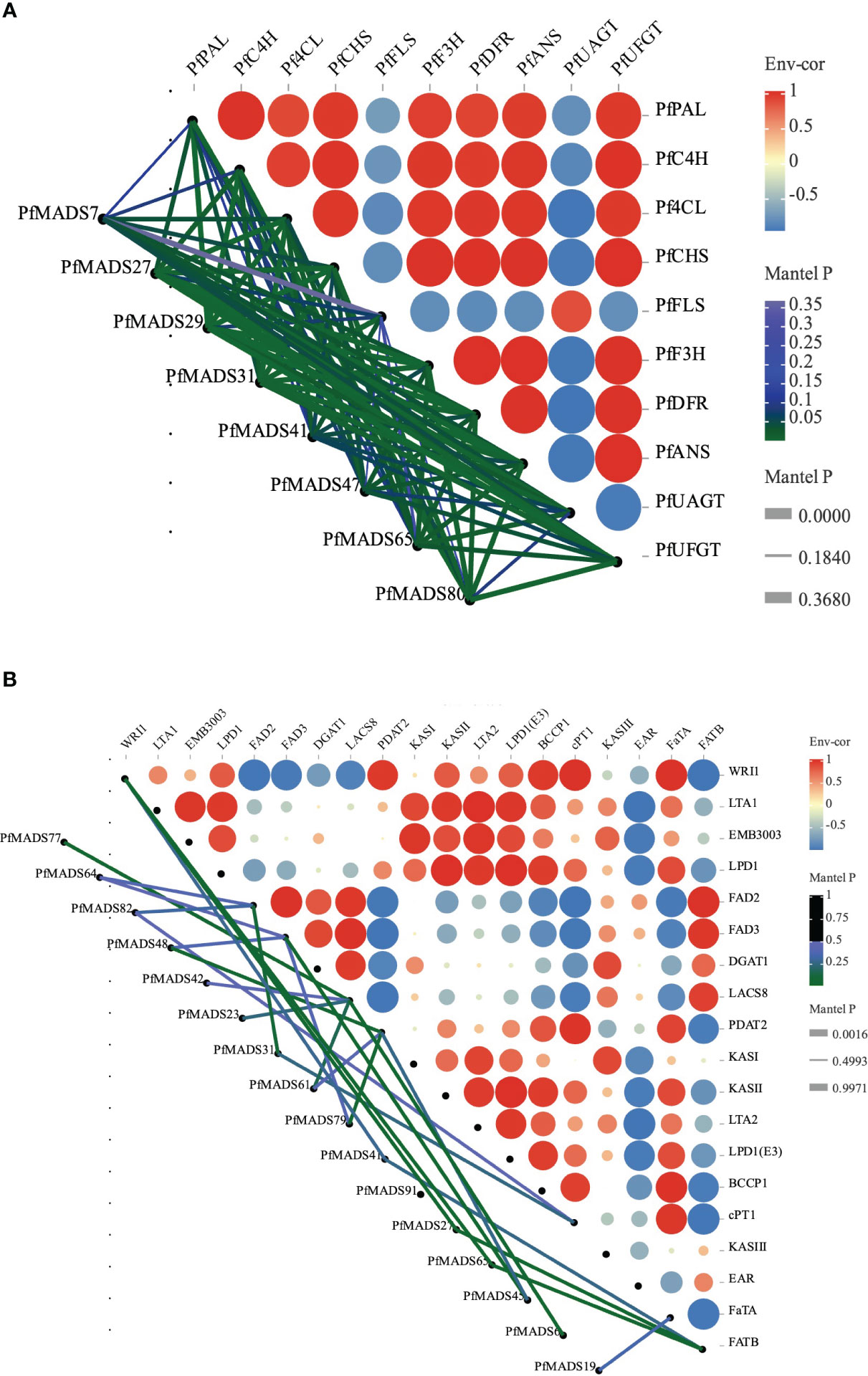
Figure 9 Correlations of expression patterns of PfMADS between other genes. (A) Correlations of expression patterns between PfMADS and the flavonoid metabolic related genes. Line thickness mapping absolute value of correlation, color mapping p value credibility. (B) Correlations of expression patterns between PfMADS and the lipid biosynthetic related genes. Line thickness mapping absolute value of correlation, color mapping p value credibility.
To gain a deeper understanding of the biological processes and molecular regulatory mechanisms involving PfMADS genes, we conducted cis-acting element analysis on the 2000 bp promoter region upstream of the start codon of 93 PfMADS using PlantCare. More than 70% of PfMADS genes contained light-responsive elements and hormone-responsive elements in their promoters, as shown in Figure 10. This suggests that a majority of PfMADS genes may play a role in regulating life processes that are influenced by light or hormones. In addition, several MYB binding sites including MBS, MBSI, MRE and CCAAT-box associated with drought, photoresponse and flavonoid biosynthesis were identified in many PfMADS gene promoters, demonstrating that these PfMADS proteins may interact with MYB TFs to involve in regulation of drought stress response, photoperiod and flavonoid biosynthesis. The PfMADS gene family members potentially have significant roles in biological processes, including perilla hormone and stress responses. These findings provide valuable candidate genes for subsequent cloning and functional analysis.

Figure 10 Cis-acting elements of PfMADS gene promoter from P. frutescens. The numbers in the box represent the number of cis-elements contained in each PfMADS gene.
Based on the analysis results of cis-acting elements in the promoters of PfMADS, six specific genes were selected. These genes, namely PfMADS15/22/25/45/80, and 93, were found to contain both cold and drought stress response elements. Subsequently, the expression profiles of the six PfMADS genes were examined under drought (DS) and cold stress (CS).
As depicted in Figure 11A, the expression levels of PfMADS22/25/45/80 genes were found to be lower under drought stress compared to the control group (NC). Among them, the relative expression of PfMADS22 and PfMADS45 were not significantly down-regulated and remained relatively unchanged. In comparison to the control group, the expression levels of PfMADS25 and PfMADS80 decreased by 42% and 48% respectively, suggesting that PfMADS25 and PfMADS80 may have a negative regulatory role in the response of P. frutescens to drought stress. Conversely, the relative expression of PfMADS15 and PfMADS93 increased by 5.5 times and 4.9 times severally. This suggests that these two PfMADS genes may have a positive regulatory role in the drought stress.
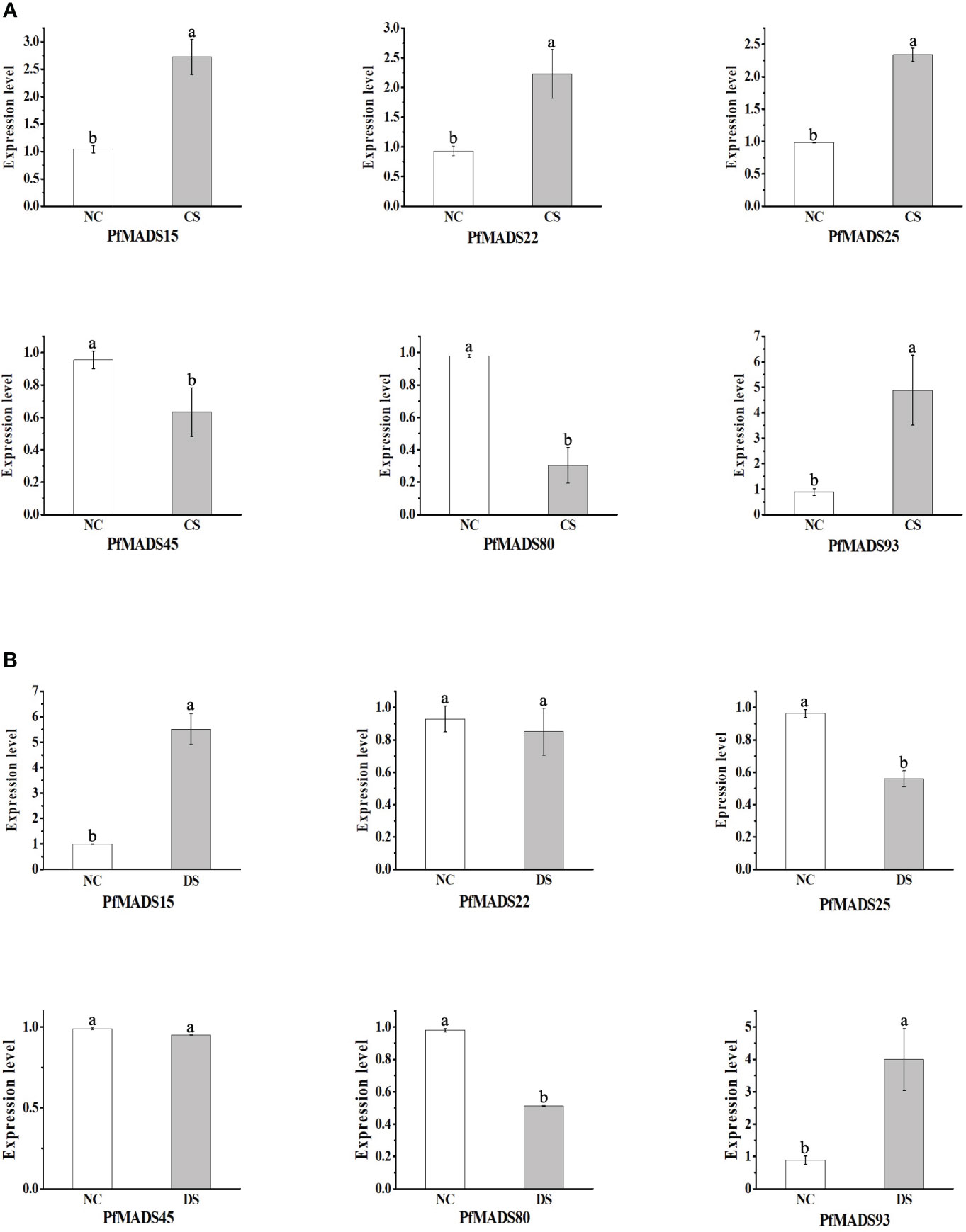
Figure 11 Expression profiles of some PfMADS genes under two stress conditions. (A) Expression profiles of some PfMADS genes in perilla seedlings under cold stress. CS, cold stress; NC, normal conditions. (B) Expression profiles of some PfMADS genes in perilla seedlings under drought stress. DS, drought stress; NC, normal conditions. Data are means ± SE calculated from three biological replicates.
The expression levels of PfMADS15/22/25/93 were all increased under cold stress (CS) (Figure 11B). However, the up-regulation of PfMADS15, PfMADS22, and PfMADS25 was relatively small compared to the up-regulation of PfMADS93. It is predicted that these PfMADS genes play a positive regulatory role in the response of P. frutescens to cold stress. The relative expression of PfMADS45 and PfMADS80 decreased under cold stress, suggesting their involvement in the negative regulation of P. frutescens response to cold stress.
Microalgae has gained increasing attention as a valuable biological resource and a promising renewable bioenergy material (Ahmed et al., 2017). Previous studies have focused on enhancing oil production by genetically modifying Chlamydomonas reinhardtii (Wang et al., 2017; Wang et al., 2018; Zhang et al., 2019). In this study, we investigated the expression profile of the PfMADS genes in perilla seeds and its correlation with lipid metabolism genes. Based on these findings, we identified PfMADS47 as a potential regulator of perilla lipid metabolism and constructed a transformation vector for Chlamydomonas reinhardtii. The constitutive expression vector pHR13-PfMADS47 (Figure S1A) was introduced into algal cells using the glass bead method. Transgenic algal strains were successfully identified at the genomic and transcriptomic levels (Figure S1).
Several studies have demonstrated that nitrogen stress can effectively induce lipid biosynthesis in Chlamydomonas reinhardtii and enhance total lipid accumulation (Yang et al., 2015). By subjecting the PfMADS47 algal strains to nitrogen stress, we observed a 45% increase in the total lipid content of the transformed strains compared to the wild strains (Figure 12C). Conversely, the total carbohydrate and protein contents of the transformed algal strains showed a significant reduction of 43% and 49% respectively (Figures 12D, E). Moreover, the growth rate of the algal cells decreased under nitrogen stress (Figure 12A), accompanied by a decrease in photosynthesis (Figure 12B), which aligns with the findings reported by Wang. These findings suggest that the introduction of the PfMADS47 gene in microalgae enhances oil production, redirects carbon sources from carbohydrate and protein synthesis pathways towards the oil biosynthesis pathway, and stimulates oil metabolism in algae.
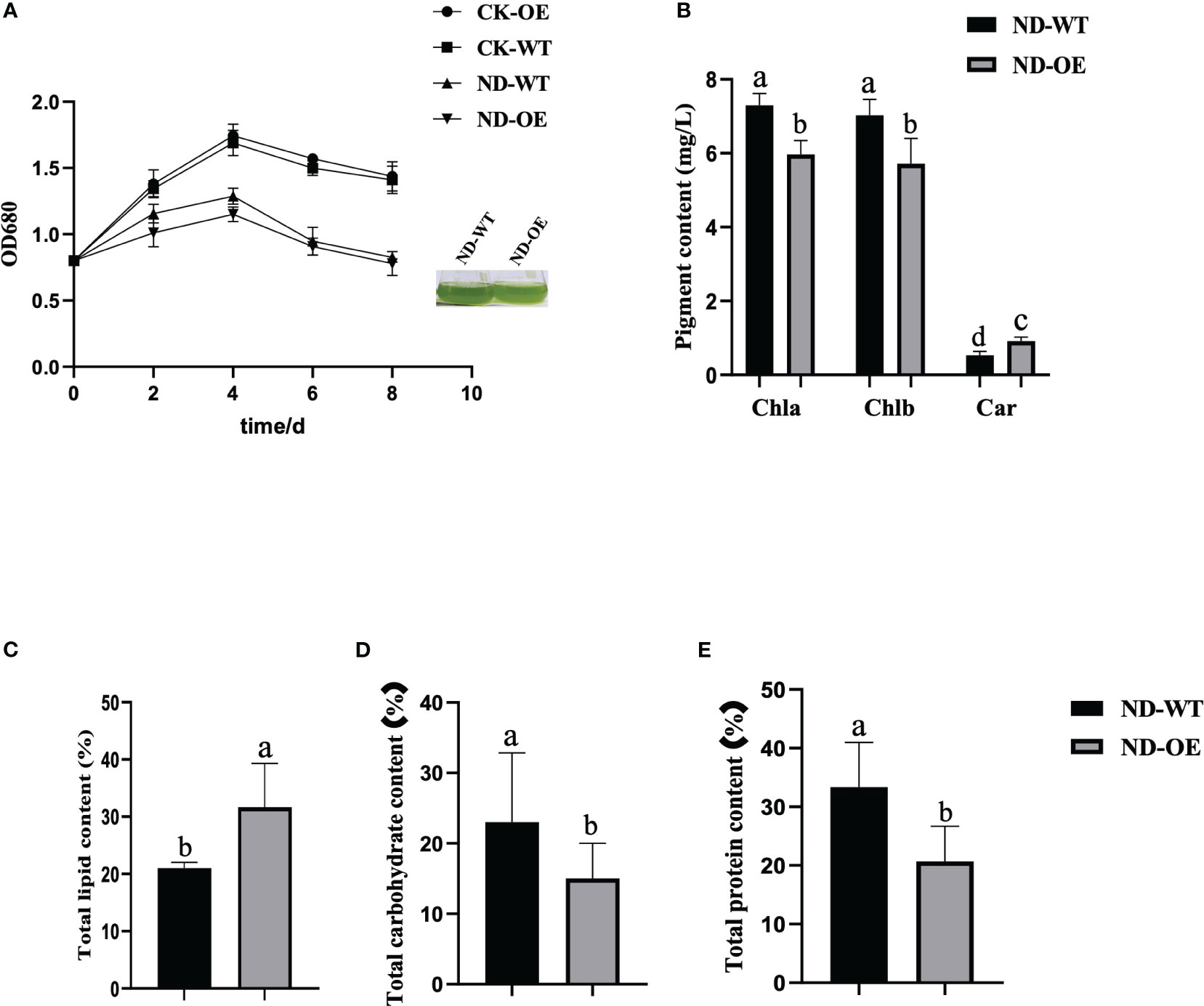
Figure 12 Physiological and biochemical indicators of algal cells under nitrogen stress. (A). Growth curve. (B). Pigment content. Chla, chlorophyll a; Chlb, chlorophyll b; Car, carotenoid. (C). Total lipid content of algal cells. (D). Total carbohydrate content of algal cells. (E). Total protein content of algal cells. Columns with the same substance and the same indicator are marked with different lowercase letters to indicate significant differences (P<0.05).
The MADS-box gene family has been extensively studied and analyzed in various species. MADS-box transcription factors are widely found in animals, plants, and fungi, but the number and types of MADS-box genes differ significantly among species. The number of members and subfamily classification of MADS-box genes in lower plants, such as mosses (Henschel et al., 2002) and algaes (Tanabe et al., 2005; Nayar and Thangavel, 2021), are relatively small. Additionally, there have been no articles reporting the presence of type II genes containing K domains in microalgae. On the other hand, the number and types of MADS-box genes are abundant in higher plants, with 108 in Arabidopsis thaliana, 168 in tobacco, 160 in cabbage, and so on (Bai et al., 2019; Won et al., 2021). This suggests that MADS-box genes have undergone quantitative expansion during the evolution process in higher plants. The increased number of MADS-box genes in higher plants also indicates functional redundancy within this gene family. In conclusion, the expansion of MADS-box from lower plants to higher plants during the evolution process suggests that MADS-box is functionally redundant in higher plants.
In this study, a total of 93 PfMADS genes were identified (Table S2). The overall number is lower than that of diploid Arabidopsis, possibly due to the tetraploid genome of P. frutescens. Some studies have reported that the P. frutescens genome is of the AABB type. However, our PfMADS family comprises of 52 members from the AA subgenome and 41 members from the BB subgenome, indicating an unequal distribution (Table S4). It is speculated that this disparity may be attributed to gene loss resulting from the balanced exchange phenomenon between subgenome in the P. frutescens genome, as well as the enrichment of homologous substitutions in the proximal telomere regions of the chromosome (Zhang et al., 2021). It has been reported that during polyploidy events, species undergo chromosomal reorganization, which often leads to the loss of a significant number of genes (Akter et al., 2021). Furthermore, gene deletion disrupts the collinearity between genes. Extensive genetic separation and genome recombination further diminish the similarity to the ancestral species (Fang et al., 2012; Zhao et al., 2021). In addition, the MADS-box family members of two other species from the Lamiaceae family, namely the diploid sage (123 gene numbers) and the tetraploid salvia (131 gene numbers), were preliminarily identified (Tables S6, 7). The results did not show a regular pattern, suggesting that there may have been fewer repeated events during the evolution of the ancestors of the Lamiaceae family. Furthermore, we observed that the number of PfMADS genes is similar to that in Camellia chekiangoleosa (89) and Musa balbisiana (97) (Lakhwani et al., 2022; Zhou et al., 2023), but higher than in cucumber (43), sesame (57), and pineapple (48) (Hu and Liu, 2012; Wei et al., 2015; Zhang et al., 2020). On the other hand, perilla has a lower number of genes compared to those in poplar (105) and tomato (131) (Leseberg et al., 2006; Wang et al., 2019). These species do not appear to be closely related, which suggests that complex historical events may have influenced their gene numbers. However, further investigation is required to determine the specific reasons (Wu et al., 2017).
Each subfamily of PfMADS exhibits a relatively conserved sequence of 30 to 60 amino acids, as shown in Figure 1. There are 37 Type I members distributed among 3 subfamilies, and 56 Type II members distributed among 11 subfamilies, as illustrated in Figure 4. The higher number of Type II genes in P. frutescens compared to Arabidopsis suggests that the Type II PfMADS genes, especially the MIKCC subfamily, underwent similar gene duplication events during ancestral evolution as observed in Arabidopsis. This indicates that the MIKCC genes may have faced stronger selection pressure during the evolutionary process, potentially impacting the environmental adaptability of P. frutescens. The distribution of PfMADS on chromosomes shows heterogeneity and clustering (Figure 2). Interestingly, Type I PfMADS tends to be distributed more in clusters, while Type II PfMADS are more uniformly distributed compared to Type I, which is consistent with the results of chromosome alignment analysis of MADS-box in Arabidopsis and rice (Nam et al., 2004). Specifically, a few regions with a higher density of PfMADS genes were observed on some chromosomes, such as chr2 and chr5, suggesting that there might be PfMADS genes hotspots in P. frutescens. According to some studies, gene families evolve and new functional genes are generated through tandem repeats and fragment repeats (Cannon et al., 2004). However, in the case of PfMADS, no tandem repeats were found, suggesting that the main driver of PfMADS gene family evolution and amplification in P. frutescens is fragment duplication (Figure 3).
In P. frutescens, motif 1 encodes the typical MADS-box TFs (SRF), which are extremely conserved in PfMADS (Figure 6). The K domain is a conserved domain in the MADS-box gene family. Typically, the K domain is found only in the MIKCC subfamily and is represented by motif 2 in P. frutescens. (Kaufmann et al., 2005). MIKC-type MADS-box proteins, even without the K domain, retain the ability to bind DNA and function as full-domain proteins (Mattick, 1994). However, the lack of the K domain leads to functional impairment of the TaSEP1-A2 protein, as it prevents protein-protein interactions (Shitsukawa et al., 2007). These results indicate that the motifs present in PfMADS proteins vary significantly across different subfamilies. However, within the same subfamily, there are certain similarities in the types, numbers, and distribution of motifs in PfMADS proteins. This suggests that these genes may have distinct functions specific to their respective subfamilies (Wang et al., 2019).
Previous studies have demonstrated that exon-intron gene structures remain relatively conserved throughout evolution (Rogozin et al., 2003). Nevertheless, the gain or loss of introns plays a crucial role in generating variations in intron positions and in the emergence of novel genes (Rogozin et al., 2005; Roy and Penny, 2007). Different gene structures were observed in PfMADS genes. Type I genes lack introns (Figure 6D), which aligns with a finding suggesting that evolution may not only impact gene function, but also genetic structure (Babenko et al., 2004; Roy and Penny, 2007). Phylogenetic and gene structure analyses revealed that Type II genes within the same subfamily exhibit similar exon-intron structures. Furthermore, the gene structures of PfMADS genes correlate with the grouping of their phylogenetic trees. Significant differences exist between different subfamilies, whereas the distribution of gene exons and introns within the same subfamily is similar. Based on these findings, it is suggested that the gene structures of PfMADS gene members belonging to the same subfamily are closely linked to phylogenetic evolution. Additionally, it is plausible that the varied gene structures may contribute to their distinct functions (Parenicová et al., 2003).
The gene expression pattern can provide crucial insights into determining the biological function of a gene. In this study, we conducted transcriptome analysis of MADS-box genes in six different tissues of P. frutescens. The data revealed that the expression of PfMADS is specific to each tissue, which aligns with their respective gene functions (Figure 7). The alanine content of P. frutescens seeds is considerably higher compared to other major oil crops. In this study, we utilized the transcriptome data of P. frutescens seeds to investigate the expression pattern of PfMADS during three developmental stages. In the expression profile of early seed development, the highly expressed PfMADS12/36/75/81/88/92 genes are classified into subfamilies such as AG/Mα/AGL11/AGL15. By referring to the Arabidopsis Information Network (https://www.arabidopsis.org/browse/genefamily/MADSlike.jsp), it is suggested that these genes may play a role in the development of endosperm, ovule, and seed coat. On the other hand, genes like PfMAD15 (AGL15), PfMADS67 (FLC: related to vernalization), and PfMADS91 (AGL63) show high expression in the late stages of seed development (Xi et al., 2020). It is speculated that these genes may be involved in the process of seed germination. Additionally, we examined the correlation between PfMADS members and key genes involved in lipid synthesis, as illustrated in Figure 9A. It is worth noting that PfMADS47、48、64、77and 82 exhibit high expression levels during the early stages of seed maturation and show strong correlation with key genes involved in lipid synthesis. Coincidentally, all of these genes belong to the AG subfamily, which supports the findings of Zhang et al. (Zhang et al., 2017). In oil palm protoplasts and callus overexpressing EgAGL9, there was a significant reduction in the total fat content and unsaturated fatty acid content, including oleic acid, linoleic acid, and linolenic acid (Zhang et al., 2022). To further investigate, we overexpressed PfMADS47 (AG subfamily) in Chlamydomonas reinhardtii, a genetically transformed model plant. Our research reveals that PfMADS47 plays a role in promoting lipid accumulation in microalgae and actively participates in the lipid synthesis and metabolism pathway. These findings provide a foundation for future genetic engineering approaches aimed at enhancing the oil content of P. frutescens seeds.
P. frutescens leaves have multiple uses as medicinal materials, vegetables, and spices. They are known to contain a diverse range of chemical components, including flavonoids and polysaccharides (Fiedor and Burda, 2014; Holt et al., 2005; Li et al., 2022). The flavonoid compounds in plants have distinct functions in response to specific developmental stages or abiotic conditions. They serve as the primary defense mechanism against ultraviolet rays and pathogens (Kawser Hossain et al., 2016; Tejada et al., 2018). The expression of several genes in leaves of different colors was analyzed by qRT-PCR, and a special gene PfMADS27 was found, which was speculated to be involved in the process of flavonoid synthesis (Figure 8). In a study of flavonoid anabolic pathways in P. frutescens leaf slices (Jiang et al., 2020), eight PfMADS genes were identified as possibly involved in the flavonoid anabolic pathway in P. frutescens. We further verified this by conducting a correlation analysis (Figure 9A). The results demonstrated a strong correlation between the PfMADS genes and the key flavonoid genes. Four genes (PfMADS48, 64, 77, and 82) belong to the AG subfamily. Previous studies have reported that the overexpression of MdJa2, which belongs to the AGL11/STK subfamily, in apple trees leads to the inhibition of anthocyanin and proanthocyanidin synthesis (Su et al., 2022). Phylogenetic analysis reveals a close relationship between PfMADS48, 64, 77, and 82 and the AGL11/STK subfamily. Therefore, these genes (PfMADS48/64/77/82) can be considered as potential candidates for further in-depth research.
MADS-box genes, functioning as transcriptional regulators, play a crucial role in ontogeny and signal transduction across various species (Schilling et al., 2018). In recent years, there has been a continuous discovery of a significant number of MIKC-type genes, and their role in regulating abiotic stress has been confirmed (Mou et al., 2009). It has been reported that the majority of PfMADS genes, which have known functions in plants, are categorized as Type II (Saedler et al., 2001; Becker and Theissen, 2003). For instance, the expression of CaMADS in peppers is influenced by low temperature, salt, and ABA (Wang et al., 2019). The expression of ZZM7-L of AGL12 subfamily is down-regulated in response to NaCl, cold treatment, and mannitol treatment (Zhang et al., 2012). Similar results have been found in other species, such as poplar, soybean, and plum blossom (Leseberg et al., 2006; Shu et al., 2013; Xu et al., 2014). In this study, we focused on six PfMADS genes belonging to the MIKCC-type in P. frutescens. Our objective was to analyze the expression patterns of these genes under cold stress and drought stress (Figure 11). The expression of PfMADS15 and PfMADS93 was observed to increase significantly under low temperature and drought stress conditions, whereas the expression of PfbMADS80 was found to decrease significantly under the same conditions. This suggests that three PfMADS genes may play distinct roles in the stress response processes of P. frutescens, potentially acting as positive and negative regulators. Studies have shown that the kiwifruit SOC1 gene can activate the AcSVP2 promoter and identified other desiccation/osmotic stress and dormancy-related transcription factors that may regulate AcSVP2 expression (Wu et al., 2018). PfMADS93(SOC1 subfamily) gene expression significantly increased under drought and cold stress conditions. Therefore, the PfMADS93 gene can be considered as a potential target gene to test its function in further studies. This would provide evidence for PfMADS mediated abiotic stresses in P. frutescens and contribute to a new scientific basis for understanding the molecular regulatory mechanism of abiotic stress response and genetic improvement for enhancing resistance in P. frutescens.
The most common P. frutescens varieties grown in the northwest region of China, such as Shanxi and other places, are primarily cultivated for oil production. However, field cultivation of P. frutescens in this region often faces challenges such as low temperature and drought stresses during the seedling and reproductive growth stages (Fang et al., 2022; Zhao et al., 2021). Currently, there is a limited number of studies investigating the molecular mechanism of stress responses, substance synthesis, and metabolism regulation in P. frutescens. Therefore, this study established a functional framework for studying the PfMADS genes (Figure 13). Based on our research, we speculate that PfMADS genes may play a role in the accumulation of carotenoids and the metabolic pathway of flavonoids during growth and development. Additionally, it may also be involved in abiotic stress responses. We also screened several candidate genes, including PfMADS27, PfMADS48, PfMADS65, PfMADS93, etc. This screening process laid the foundation for further research on the function of the PfMADS genes and contributed to the advancement of molecular breeding programs.
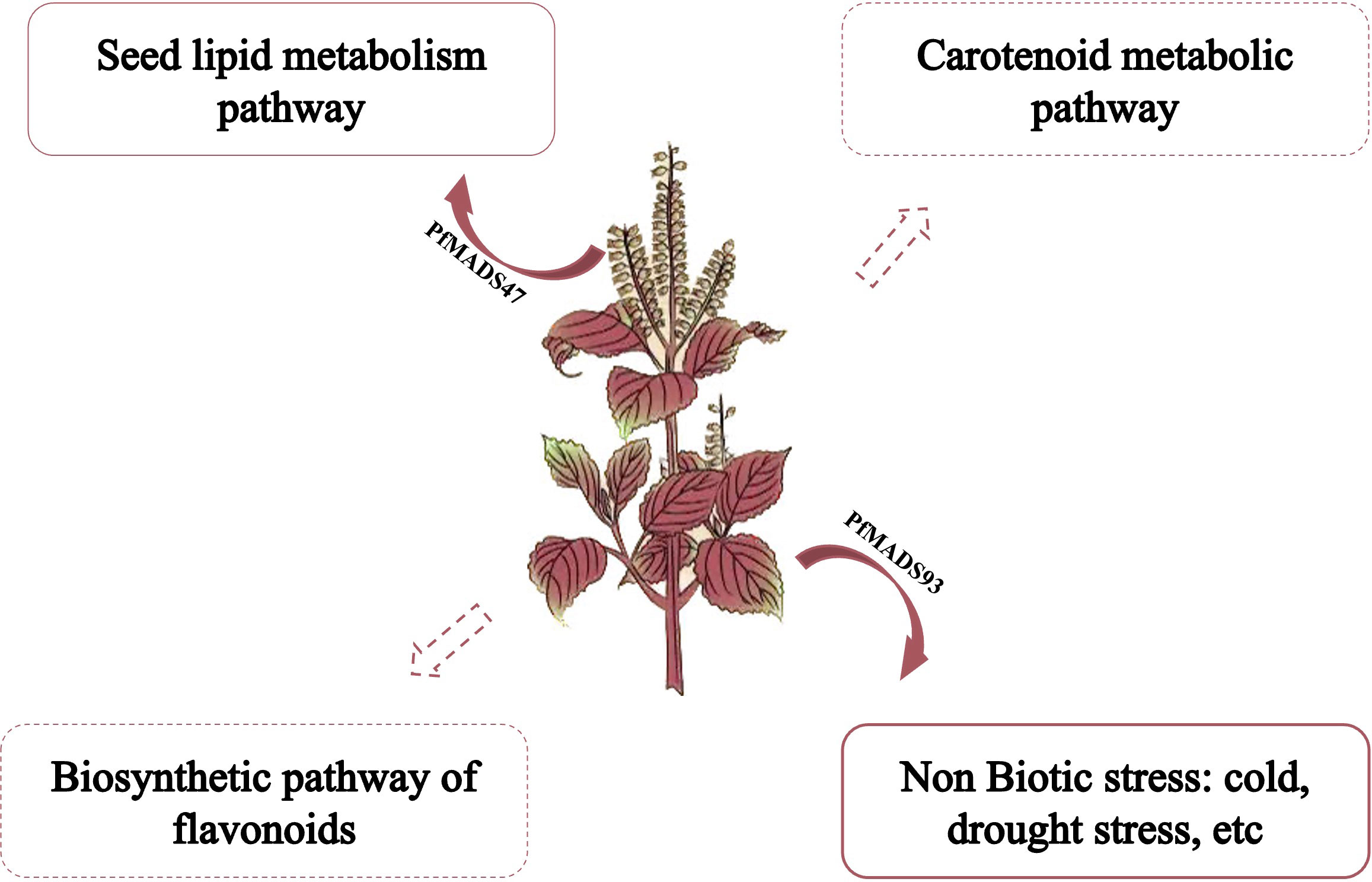
Figure 13 A proposed working model showing the possible roles of PfMADS genes in abiotic stress responses and regulating lipid, carotenoid, flavonoids metabolisms in P. frutescens.
This study has successfully identified 93 PfMADS members from the P. frutescens genome, marking the first time this has been achieved. The expansion of the PfMADS gene family has been primarily driven by segment duplication. Furthermore, it was observed that nearly all PfMADS genes have undergone purifying selection throughout their evolutionary history. Over 50% of the PfMADS genes exhibit expression in multiple tissues, while certain genes demonstrate tissue-specific expression. These findings suggest variations in the functions of PfMADS genes across different tissues in P. frutescens. Further studies revealed that several PfMADS genes (PfMADS15, 22, 25, 45, 80, 93) play key roles in response to abiotic stresses, while a group of PfMADS genes (PfMADS6, 19, 23, 27, 31, 41, 42, 45, 48, 61, 64, 65, 77, 79, 82, 91) may mediate P. frutescens seeds development process of lipid metabolism. PfMADS genes have been found to be active in all plant tissues that have been studied. These genes form a closely related regulatory network in plants and play various roles in important physiological activities. Our study offers a theoretical foundation for identifying PfMADS genes that are involved in flavonoid accumulation, lipid anabolism, and the response to abiotic stresses.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
ML: Writing – original draft. ZD: Methodology, Writing – original draft. ZY: Methodology, Writing – original draft. TL: Software, Writing – original draft. CJ: Writing – review & editing. HC: Supervision, Writing – review & editing. RL: Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Key Research and Development Program of China (2021YFD1901100), State Key Laboratory of Integrative Sustainable Dryland Agriculture (in preparation), Shanxi Agricultural University (202105D121008-3-6), Six New Project of Agriculture Department of Shanxi Province (unnumbered), National Natural Science Foundation of China (31902394), Key Research and Development Planning Project of Shanxi Province (201803D31063), Science and Technology Innovation Planning Project of Shanxi Agricultural University (2018YJ16), Outstanding Doctor to Work in Shanxi Province Research Project (SXYBKY2019036), Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (2021L119).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1299902/full#supplementary-material
Supplementary Figure S1 | Identification and screening of transformants of Chlamydomonas reinhardtii. (A). Vector structure diagram of pHR13-PfMADS47. (B). a: Genomic identification of algal transformants; b: RT-PCR analysis of the algal transformants. (C) Algal transformants were screened by TAP agar plate containing 10 mg·L−1 hygromycin.
Supplementary Table S1 | The list of primer-sets of PfMADS genes for qRT-PCR.
Supplementary Table S2 | Detailed information of all identified P. frutescens PfMADS proteins.
Supplementary Table S3 | The sequence of highly conserved region of the MADS domains.
Supplementary Table S4 | Subgenome assignment of tetraploid P. frutescens.
Supplementary Table S5 | Duplication gene pair of PfMADS.
Supplementary Table S6 | Gene ID of MADS-box genes in Salvia japonica Thunb.
Supplementary Table S7 | Gene ID of MADS-box genes in Salvia miltiorrhiza.
Agrawal, G. K., Abe, K., Yamazaki, M., Miyao, A., Hirochika, H. (2005). Conservation of the E-function for floral organ identity in rice revealed by the analysis of tissue culture-induced loss-of-function mutants of the OsMADS1 gene. Plant Mol. Biol. 59 (1), 125–135. doi: 10.1007/s11103-005-2161-y
Ahmed, R. A., He, M., Aftab, R. A., Zheng, S., Nagi, M., Bakri, R., et al. (2017). Bioenergy application of Dunaliella salina SA134 grown at various salinity levels for lipid production. Sci. Rep. 7 (1), 8118. doi: 10.1038/s41598-017-07540-x
Akter, A., Itabashi, E., Kakizaki, T., Okazaki, K., Dennis, E. S., Fujimoto, R. (2021). Genome triplication leads to transcriptional divergence of flowering locus C genes during vernalization in the genus brassica. Front. Plant Sci. 11, 619417. doi: 10.3389/fpls.2020.619417
Arora, R., Agarwal, P., Ray, S., Singh, A. K., Singh, V. P., Tyagi, A. K., et al. (2007). MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 18 (8), 242. doi: 10.1186/1471-2164-8-242
Babenko, V. N., Rogozin, I. B., Mekhedov, S. L., Koonin, E. V. (2004). Prevalence of intron gain over intron loss in the evolution of paralogous gene families. Nucleic Acids Res. 32 (12), 3724–3733. doi: 10.1093/nar/gkh686
Bai, G., Yang, D. H., Cao, P., Yao, H., Zhang, Y., Chen, X., et al. (2019). Genome-wide identification, gene structure and expression analysis of the MADS-box gene family indicate their function in the development of Tobacco (Nicotiana tabacum L.). Int. J. Mol. Sci. 20 (20), 5043. doi: 10.3390/ijms20205043
Becker, A., Theissen, G. (2003). The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet Evol. 29 (3), 464–489. doi: 10.1016/s1055-7903(03)00207-0
Bemer, M., Wolters-Arts, M., Grossniklaus, U., Angenent, G. C. (2008). The MADS domain protein DIANA acts together with AGAMOUS-LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell. 20 (8), 2088–2101. doi: 10.1105/tpc.108.058958
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4, 10. doi: 10.1186/1471-2229-4-10
Clemente, T. E., Cahoon, E. B. (2009). Soybean oil: genetic approaches for modification of functionality and total content. Plant Physiol. 151 (3), 1030–1040. doi: 10.1104/pp.109.146282
Coen, E. S., Meyerowitz, E. M. (1991). The war of the whorls: genetic interactions controlling flower development. Nature 353 (6339), 31–37. doi: 10.1038/353031a0
Colombo, M., Masiero, S., Vanzulli, S., Lardelli, P., Kater, M. M., Colombo, L. (2008). AGL 23, a type I MADS-box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J. 54 (6), 1037–1048. doi: 10.1111/j.1365-313X.2008.03485.x
Dong, T., Chen, G., Tian, S., Xie, Q., Yin, W., Zhang, Y., et al. (2014). A non-climacteric fruit gene CaMADS-RIN regulates fruit ripening and ethylene biosynthesis in climacteric fruit. PloS One 9 (4), e95559. doi: 10.1371/journal.pone.0095559
Edge, R., McGarvey, D. J., Truscott, T. G. (1997). The arotinoids as anti-oxidants–a review. J. Photochem. Photobiol. B. 41 (3), 189–200. doi: 10.1016/s1011-1344(97)00092-4
Fang, L., Cheng, F., Wu, J., Wang, X. (2012). The impact of genome triplication on tandem gene evolution in brassica rapa. Front. Plant Sci 3, 261. doi: 10.3389/fpls.2012.00261
Fang, Y., Wang, S., Wu, H., Li, C., Zhao, H., Chen, H., et al. (2022). Genome-Wide Identification of ATG Gene Family Members in Fagopyrum tataricum and Their Expression during Stress Responses. Int. J. Mol. Sci. 23 (23), 14845. doi: 10.3390/ijms232314845
Fiedor, J., Burda, K. (2014). Potential role of carotenoids as antioxidants in human health and disease. Nutrients. 6 (2), 466–488. doi: 10.3390/nu6020466
Finn, R. D., Clements, J., Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39 (Web Server issue), W29–W37. doi: 10.1093/nar/gkr367
Finn, R. D., Mistry, J., Schuster-Böckler, B., Griffiths-Jones, S., Hollich, V., Lassmann, T., et al. (2006). Pfam: clans, web tools and services. Nucleic Acids Res. 34 (Database issue), D247–D251. doi: 10.1093/nar/gkj149
Fujiwara, Y., Kono, M., Ito, A., Ito, M. (2018). Anthocyanins in perilla plants and dried leaves. Phytochemistry. 147, 158–166. doi: 10.1016/j.phytochem.2018.01.003
Gramzow, L., Theissen, G. (2010). A hitchhiker’s guide to the MADS world of plants. Genome Biol. 11 (6), 214. doi: 10.1186/gb-2010-11-6-214
Henschel, K., Kofuji, R., Hasebe, M., Saedler, H., Münster, T., Theissen, G. (2002). Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol. Biol. Evol. 19 (6), 801–814. doi: 10.1093/oxfordjournals.molbev.a004137
Holt, N. E., Zigmantas, D., Valkunas, L., Li, X. P., Niyogi, K. K., Fleming, G. R. (2005). Carotenoid cation formation and the regulation of photosynthetic light harvesting. Science. 307 (5708), 433–436. doi: 10.1126/science.1105833
Hu, L., Liu, S. (2012). Genome-wide analysis of the MADS-box gene family in cucumber. Genome. 55, 245–256. doi: 10.1139/G2012-009
Igarashi, M., Miyazaki, Y. (2013). A review on bioactivities of Perilla: progress in research on the functions of Perilla as medicine and food. Evid Based Complement Alternat Med. 2013, 925342. doi: 10.1155/2013/925342
Immink, R. G., Kaufmann, K., Angenent, G. C. (2009). The ‘ABC’ of MADS domain protein behavior and interactions. Semin. Cell Dev. Biol. 21 (1), 87–93. doi: 10.1016/j.semcdb.2009.10.004
Immink, R. G., Tonaco, I. A., de Folter, S., Shchennikova, A., van Dijk, A. D., Busscher-Lange, J., et al. (2009). SEPALLATA3: the ‘glue’ for MADS-box transcription factor complex formation. Genome Biol. 10 (2), R24. doi: 10.1186/gb-2009-10-2-r24
Irish, V. F. (2003). The evolution of floral homeotic gene function. Bioessays. 25 (7), 637–646. doi: 10.1002/bies.10292
Jiang, T., Guo, K., Liu, L., Tian, W., Xie, X., Wen, S., et al. (2020). Integrated transcriptomic and metabolomic data reveal the flavonoid biosynthesis metabolic pathway in Perilla frutescens (L.) leaves. Sci. Rep. 10 (1), 16207. doi: 10.1038/s41598-020-73274-y
Kang, I. H., Steffen, J. G., Portereiko, M. F., Lloyd, A., Drews, G. N. (2008). The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell. 20 (3), 635–647. doi: 10.1105/tpc.107.055137
Kaufmann, K., Melzer, R., Theissen, G. (2005). MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene 347 (2), 183–198. doi: 10.1016/j.gene.2004.12.014
Kawser Hossain, M., Abdal Dayem, A., Han, J., Yin, Y., Kim, K., Kumar Saha, S., et al. (2016). Molecular mechanisms of the anti-obesity and anti-diabetic properties of Flavonoids. Int. J. Mol. Sci. 17 (4), 569. doi: 10.3390/ijms17040569
Komatsu, K. I., Takanari, J., Maeda, T., Kitadate, K., Sato, T., Mihara, Y., et al. (2016). Perilla leaf extract prevents atopic dermatitis induced by an extract of Dermatophagoides farinae in NC/Nga mice. Asian Pac J. Allergy Immunol. 34 (4), 272–277. doi: 10.12932/AP0717
Lakhwani, D., Vikarm Dhar, Y., Singh, S., Pandey, A., Kumar Trivedi, P., Hasan Asif, M. (2022). Genome wide identification of MADS box gene family in Musa balbisiana and their divergence during evolution. Gene 836, 146666. doi: 10.1016/j.gene.2022.146666
Lawton-Rauh, A. L., Alvarez-Buylla, E. R., Purugganan, M. D. (2000). Molecular evolution of flower development. Trends Ecol. Evol. 15 (4), 144–149. doi: 10.1016/s0169-5347(99)01816-9
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). plant CARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30 (1), 325–327. doi: 10.1093/nar/30.1.325
Leseberg, C. H., Li, A., Kang, H., Duvall, M., Mao, L. (2006). Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene. 378, 84–94. doi: 10.1016/j.gene.2006.05.022
Li, Q., Wang, T., Xu, C., Li, M., Tian, J., Wang, Y., et al. (2022). MdMADS6 recruits histone deacetylase mdHDA19 to repress the expression of the carotenoid synthesis-related gene mdCCD1 during fruit ripening. Plants (Basel) 11 (5), 668. doi: 10.3390/plants11050668
Li, S. Y., Zhang, Q., Jin, Y. H., Zou, J. X., Zheng, Y. S., Li, D. D. (2020). A MADS-box gene, EgMADS21, negatively regulates EgDGAT2 expression and decreases polyunsaturated fatty acid accumulation in oil palm (Elaeis guineensis Jacq.). Plant Cell Rep. 39 (11), 1505–1516. doi: 10.1007/s00299-020-02579-z
Li, Y., Zhang, Y., Wang, Y., Li, X., Zhou, L., Yang, J., et al. (2022). Metabolites and chemometric study of Perilla (Perilla frutescens) from different varieties and geographical origins. J. Food Sci. 87 (12), 5240–5251. doi: 10.1111/1750-3841.16376
Liu, J., Han, D., Yoon, K., Hu, Q., Li, Y. (2016). Characterization of type 2 diacylglycerol acyltransferases in Chlamydomonas reinhardtii reveals their distinct substrate specificities and functions in triacylglycerol biosynthesis. Plant J 86 (1), 3-19. doi: 10.1111/tpj.13143
Lu, S., Zhang, Y., Zhu, K., Yang, W., Ye, J., Chai, L., et al. (2018). The citrus transcription factor csMADS6 modulates carotenoid metabolism by directly regulating carotenogenic genes. Plant Physiol. 176 (4), 2657–2676. doi: 10.1104/pp.17.01830
Luo, Y. J., Guo, J. Q., Li, W. P., Yao, Y., Wen, C. M., Guo, B. L. (2021). Contents determination of eight phenolic compounds in Perilla frutescens leaves of different cultivation years and harvesting periods. Zhongguo Zhong Yao Za Zhi. 46 (3), 567–574. doi: 10.19540/j.cnki.cjcmm.20201114.101
Mattick, J. S. (1994). Introns: evolution and function. Curr. Opin. Genet. Dev. 4 (6), 823–831. doi: 10.1016/0959-437x(94)90066-3
Messenguy, F., Dubois, E. (2003). Role of MADS-box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene. 316, 1–21. doi: 10.1016/s0378-1119(03)00747-9
Molesini, B., Dusi, V., Pennisi, F., Pandolfini, T. (2020). How hormones and MADS-box transcription factors are involved in controlling fruit set and parthenocarpy in tomato. Genes (Basel). 11 (12), 1441. doi: 10.3390/genes11121441
Mou, Y., Yuan, C., Sun, Q., Yan, C., Zhao, X., Wang, J., et al. (2009). MIKC-type MADS-box transcription factor gene family in peanut: Genome-wide characterization and expression analysis under abiotic stress. Front Plant Sci. 13, 980933. doi: 10.3389/fpls.2022.980933
Nam, J., Kim, J., Lee, S., An, G., Ma, H., Nei, M. (2004). ). Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc. Natl. Acad. Sci. U S A. 101 (7), 1910–1915. doi: 10.1073/pnas.0308430100
Nayar, S., Thangavel, G. (2021). CsubMADS1, a lag phase transcription factor, controls development of polar eukaryotic microalga Coccomyxa subellipsoidea C-169. Plant J. 107 (4), 1228–1242. doi: 10.1111/tpj.15380
Nitta, M., Lee, J. K., Kang, C. W., Katsuta, M., Yasumoto, S., Liu, D., et al. (2005). The distribution of perilla species. Genet. Resour. Crop Evol. 52, 797–804. doi: 10.1007/s10722-003-6017-5
Parenicová, L., de Folter, S., Kieffer, M., Horner, D. S., Favalli, C., Busscher, J., et al. (2003). Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell. 15 (7), 1538–1551. doi: 10.1105/tpc.011544
Park, Y.-J., Dixit, A., Ma, K.-H., Lee, J.-K., Lee, M.-H., Chung, C.-S., et al. (2008). Evaluation of genetic diversity and relationships within an on-farm collection of Perilla Frutescens (L.) Britt. using microsatellite markers. Genet. Resour. Crop Evol. 55, 523–535. doi: 10.1007/s10722-007-9258-x
Pelaz, S., Ditta, G. S., Baumann, E., Wisman, E., Yanofsky, M. F. (2000). B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 405 (6783), 200–203. doi: 10.1038/35012103
Rogozin, I. B., Sverdlov, A. V., Babenko, V. N., Koonin, E. V. (2005). Analysis of evolution of exon-intron structure of eukaryotic genes. Brief Bioinform. 6 (2), 118–134. doi: 10.1093/bib/6.2.118
Rogozin, I. B., Wolf, Y. I., Sorokin, A. V., Mirkin, B. G., Koonin, E. V. (2003). Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr. Biol. 13 (17), 1512–1517. doi: 10.1016/s0960-9822(03)00558-x
Roy, S. W., Penny, D. (2007). On the incidence of intron loss and gain in paralogous gene families. Mol. Biol. Evol. 24 (8), 1579–1581. doi: 10.1093/molbev/msm082
Saedler, H., Becker, A., Winter, K. U., Kirchner, C., Theissen, G. (2001). MADS-box genes are involved in floral development and evolution. Acta Biochim. Pol. 48 (2), 351–358. doi: 10.18388/abp.2001_3920
Schilling, S., Kennedy, A., Pan, S., Jermiin, L. S., Melzer, R. (2020). Genome-wide analysis of MIKC-type MADS-box genes in wheat: pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 225 (1), 511–529. doi: 10.1111/nph.16122
Schilling, S., Pan, S., Kennedy, A., Melzer, R. (2018). MADS-box genes and crop domestication: the jack of all traits. J. Exp. Bot. 69 (7), 1447–1469. doi: 10.1093/jxb/erx479
Shitsukawa, N., Ikari, C., Mitsuya, T., Sakiyama, T., Murai, K. (2007). Wheat SOC1 functions independently of WAP1/VRN1, an integrator of vernalization and photoperiod flowering promotion pathways. Physiologia Plantarum 130 (4), 627–636. doi: 10.1111/j.1399-3054.2007.00927.x
Shore, P., Sharrocks, A. D. (1995). The MADS-box family of transcription factors. Eur. J. Biochem. 229 (1), 1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x
Shu, Y., Yu, D., Wang, D., Guo, D., Guo, C. (2013). Genome-wide survey and expression analysis of the MADS-box gene family in soybean. Mol. Biol. Rep. 40 (6), 3901–3911. doi: 10.1007/s11033-012-2438-6
Smaczniak, C., Immink, R. G., Angenent, G. C., Kaufmann, K. (2012). Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development. 139 (17), 3081–3098. doi: 10.1242/dev.074674
Steffen, J. G., Kang, I. H., Portereiko, M. F., Lloyd, A., Drews, G. N. (2008). AGL61 interacts with AGL80 and is required for central cell development in Arabidopsis. Plant Physiol. 148 (1), 259–268. doi: 10.1104/pp.108.119404
Su, M., Wang, S., Liu, W., Yang, M., Zhang, Z., Wang, N., et al. (2022). MdJa2 participates in the brassinosteroid signaling pathway to regulate the synthesis of anthocyanin and proanthocyanidin in red-fleshed apple. Front. Plant Sci. 13, 830349. doi: 10.3389/fpls.2022.830349
Sun, S. M., Yang, S. H., Golokhvast, K. S., Le, B., Chung, G. (2016). Reconstructing the Phylogeny of Capsosiphon fulvescens (Ulotrichales, Chlorophyta) from Korea Based on rbcL and 18S rDNA Sequences. BioMed. Res. Int. 2016, 1462916. doi: 10.1155/2016/1462916
Swiezewski, S., Liu, F., Magusin, A., Dean, C. (2009). Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature. 462 (7274), 799–802. doi: 10.1038/nature08618
Tanabe, Y., Hasebe, M., Sekimoto, H., Nishiyama, T., Kitani, M., Henschel, K., et al. (2005). Characterization of MADS-box genes in charophycean green algae and its implication for the evolution of MADS-box genes. Proc. Natl. Acad. Sci. U S A. 102 (7), 2436–2441. doi: 10.1073/pnas.0409860102
Tejada, S., Pinya, S., Martorell, M., Capó, X., Tur, J. A., Pons, A., et al. (2018). Potential anti-inflammatory effects of hesperidin from the genus citrus. Curr. Med. Chem. 25 (37), 4929–4945. doi: 10.2174/0929867324666170718104412
Wang, C., Chen, X., Li, H., Wang, J., Hu, Z. (2017). Artificial miRNA inhibition of phosphoenolpyruvate carboxylase increases fatty acid production in a green microalga Chlamydomonas reinhardtii. Biotechnol. Biofuels 10, 91. doi: 10.1186/s13068-017-0779-z
Wang, C., Li, Y., Lu, J., Deng, X., Li, H., Hu, Z. (2018). Effect of overexpression of LPAAT and GPD1 on lipid synthesis and composition in green microalga Chlamydomonas reinhardtii. J. Appl. Phycol 30 (3), 1711-1719. doi: 10.1007/s10811-017-1349-2
Wang, Y., Zhang, J., Hu, Z., Guo, X., Tian, S., Chen, G. (2019). Genome-wide analysis of the MADS-Box transcription factor family in Solanum lycopersicum. Int. J. Mol. Sci. 20 (12), 2961. doi: 10.3390/ijms20122961
Wei, X., Wang, L., Yu, J., Zhang, Y., Li, D., Zhang, X. (2015). Genome-wide identification and analysis of the MADS-box gene family in sesame. Gene 569 (1), 66-76. doi: 10.1016/j.gene.2015.05.018
Won, S. Y., Jung, J. A., Kim, J. S. (2021). Genome-wide analysis of the MADS-box gene family in chrysanthemum. Comput. Biol. Chem. 90, 107424. doi: 10.1016/j.compbiolchem.2020.107424
Wu, J., Yu, C., Huang, L., Gan, Y. (2021). A rice transcription factor, OsMADS57, positively regulates high salinity tolerance in transgenic Arabidopsis thaliana and Oryza sativa plants. Physiol. Plant 173 (3), 1120–1135. doi: 10.1111/ppl.13508
Wu, P., Wang, W., Duan, W., Li, Y., Hou, X. (2017). Comprehensive analysis of the CDPK-snRK superfamily genes in chinese cabbage and its evolutionary implications in plants. Front. Plant Sci. 8, 162. doi: 10.3389/fpls.2017.00162
Wu, R., Tomes, S., Karunairetnam, S., Tustin, S. D., Hellens, R. P., Allan, A. C., et al. (2017). SVP-like MADS box genes control dormancy and budbreak in apple. Front. Plant Sci. 8, 477. doi: 10.3389/fpls.2017.00477
Wu, R., Wang, T., Richardson, A. C., Allan, A. C., Macknight, R. C., Varkonyi-Gasic, E. (2018). Histone modification and activation by SOC1-like and drought stress-related transcription factors may regulate AcSVP2 expression during kiwifruit winter dormancy. Plant Sci 281, 242-250. doi: 10.1016/j.plantsci.2018.12.001
Xi, Y., Park, S. R., Kim, D. H., Kim, E. D., Sung, S. (2020). Transcriptome and epigenome analyses of vernalization in Arabidopsis thaliana. Plant J 103 (4), 1490-1502. doi: 10.1111/tpj.14817
Xu, Z., Zhang, Q., Sun, L., Du, D., Cheng, T., Pan, H., et al. (2014). Genome-wide identification, characterization and expression analysis of the MADS-box gene family in Prunus mume. Mol. Genet. Genomics 289 (5), 903–920. doi: 10.1007/s00438-014-0863-z
Xue, Y., Ma, L., Wang, H., Hao, P., Cheng, S., Su, Z., et al. (2022). The MADS transcription factor GhFYF is involved in abiotic stress responses in upland cotton (Gossypium hirsutum L.). Gene. 815, 146138. doi: 10.1016/j.gene.2021.146138
Yang, D., Song, D., Kind, T., Ma, Y., Hoefkens, J., Fiehn, O. (2015). Lipidomic Analysis of Chlamydomonas reinhardtii under Nitrogen and Sulfur Deprivation. PloS One 10 (9), e0137948. doi: 10.1371/journal.pone.0137948
Yang, Y., Fanning, L., Jack, T. (2003). The K domain mediates heterodimerization of the Arabidopsis floral organ identity proteins, APETALA3 and PISTILLATA. Plant J. 33 (1), 47–59. doi: 10.1046/j.0960-7412.2003.01473.x
Yoshida, K., Kondo, T., Kameda K and Goto, T. (1990). Structure of anthocyanins isolated from purple leaves of Perilla ocimoides L. var. crispa Benth and their isomerization by irradiation of light. Agric. Biol. Chem. 54, 1745–1751. doi: 10.1271/bbb1961.54.1745
Zhang, Q., Jin, Y. H., Zou, J. X., Zheng, Y. S., Li, D. D. (2022). Characterization and functional analysis of the MADS-box EgAGL9 transcription factor from the mesocarp of oil palm (Elaeis guineensis Jacq.). Plant Sci. 321, 111317. doi: 10.1016/j.plantsci.2022.111317
Zhang, T., Song, C., Song, L., Shang, Z., Yang, S., Zhang, D., et al. (2017). RNA sequencing and coexpression analysis reveal key genes involved in α-linolenic acid biosynthesis in Perilla frutescens seed. Int. J. Mol. Sci. 18 (11), 2433. doi: 10.3390/ijms18112433
Zhang, X., Fatima, M., Zhou, P., Ma, Q., Ming, R. (2020). Analysis of MADS-box genes revealed modified flowering gene network and diurnal expression inpineapple. BMC Genomics 21, 1–16. doi: 10.1186/s12864-019-6421-7
Zhang, Y., Shen, Q., Leng, L., Zhang, D., Chen, S., Shi, Y., et al. (2021). Incipient diploidization of the medicinal plant perilla within 10,000 years. Nat. Commun. 12 (1), 5508. doi: 10.1038/s41467-021-25681-6
Zhang, Y., Shi, M., Mao, X., Kou, Y., Liu, J. (2019). Time-resolved carotenoid profiling and transcriptomic analysis reveal mechanism of carotenogenesis for astaxanthin synthesis in the oleaginous green alga Chromochloris zofingiensis. Biotechnol. Biofuels 12, 287. doi: 10.1186/s13068-019-1626-1
Zhang, Z., Li, H., Zhang, D., Liu, Y., Fu, J., Shi, Y., et al. (2012). Characterization and expression analysis of six MADS-box genes in maize (Zea mays L.). J. Plant Physiol. 169 (8), 797-806. doi: 10.1016/j.jplph.2011.12.020
Zhao, P. X., Zhang, J., Chen, S. Y., Wu, J., Xia, J. Q., Sun, L. Q., et al. (2021). Arabidopsis MADS-box factor AGL16 is a negative regulator of plant response to salt stress by downregulating salt-responsive genes. New Phytol. 232 (6), 2418–2439. doi: 10.1111/nph.17760
Zhao, W., Zhang, L. L., Xu, Z. S., Fu, L., Pang, H. X., Ma, Y. Z., et al. (2021). Genome-wide analysis of MADS-box genes in foxtail millet (Setaria italica L.) and functional assessment of the role of SiMADS51 in the drought stress response. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.659474
Zhou, Y. L., Huang, X. S., Hao, Y. R., et al. (2022). Cloning and functional characterization of a lysophosphatidic acid acyltransferase gene from Perilla frutescens. Chin. J. Biotech., 1–16. doi: 10.13345/j.cjb.220033
Keywords: Perilla frutescens, MADS transcription factors, genome-wide characterization, expression profiles, lipid metabolism, stress responses
Citation: Liang M, Du Z, Yang Z, Luo T, Ji C, Cui H and Li R (2024) Genome-wide characterization and expression analysis of MADS-box transcription factor gene family in Perilla frutescens. Front. Plant Sci. 14:1299902. doi: 10.3389/fpls.2023.1299902
Received: 23 September 2023; Accepted: 14 December 2023;
Published: 08 January 2024.
Edited by:
Khurram Bashir, Lahore University of Management Sciences, PakistanReviewed by:
Liuyin Ma, Fujian Agriculture and Forestry University, ChinaCopyright © 2024 Liang, Du, Yang, Luo, Ji, Cui and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunli Ji, amljaHVubGlAc3hhdS5lZHUuY24=; Hongli Cui, Y3VpaG9uZ2xpQHN4YXUuZWR1LmNu; Runzhi Li, cmxpMjAwMUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.