 Zhi Chao
Zhi Chao Xiaoxi Hong2
Xiaoxi Hong2 Enwei Tian
Enwei Tian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 27 November 2023
Sec. Plant Systematics and Evolution
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1294670
A comparative analysis of chloroplast (cp) genomes and 45s nuclear ribosomal DNA (nrDNA), and a phylogenomic study of six closely related species (including an overlooked new species) of genus Bupleurum from the western part of Sichuan Province in southwestern China were performed. The six species are similar morphologically and it is difficult to identify them; moreover, their genetic relationships remain unclear. It was found that the cp genomes of the six Bupleurum species were extremely similar, and they were highly homogeneous in terms of cp genome structure, genes and its arrangement. Intergenic spacer rpl32-trnL, petA-psbJ, trnK-rps16, and the coding gene ycf1 were considered highly variable. In phylogenetic trees constructed based on the complete cp genome, protein-coding sequences, nrDNA and ITS sequences, Chinese Bupleurum species all formed two major clades; among these trees, nrDNA tree had the best species resolution; the highly variable regions showed no advantage over other molecular markers. Among the six Bupleurum species, B. malconense, B. sichuanense were close relatives to B. chinense and B. yinchowense, B. chaishoui may also be a consanguinity, while B. microcephalum, B. wenchuanense, and the new species B. pseudochaishoui were closely related. At the end, the new species B. pseudochaishoui Z. Chao sp. nov. was described and illustrated, and a key to the six species was tabulated.
Bupleurum L., comprising about 210 species accepted nowadays (POWO, 2023), is one of the largest genera of the family Apiaceae. It has long been recognized as a natural group distinctively characterized by its members having simple leaves, mostly parallel veins, and conspicuous bracts/bracteoles. Infrageneric classification and phylogeny of the genus was of interest to plant taxonomists from the early days. The first use of section names in Bupleurum can be dated back to 1848 (Grenier and Godron, 1848). Wolff published a comprehensive revision of the genus that classified it into 5 sections (Wolff, 1910), and his classification is the most widely used for about a century. Shu et al. carried out a numerical taxonomic study in 1998 and divided the Chinese Bupleurum species into subgenus Longifolia and subgenus Eubupleura (including section Ranunculoidea and section Falcata) (Shu et al., 1998). Neves & Watson suggested dividing Bupleurum into two subgenera (subg. Penninervia, subg. Bupleurum) based on phylogenetic analyses of nuclear ribosomal internal transcribed spacer (nrITS) sequences of 35 taxa (Neves and Watson, 2004). Although the previous researches have deepened our understanding of the genus, it is still hard to say being sufficient.
China harbors abundant diversity of Bupleurum. In Flora of China published in 2005, 42 species, 16 varieties and 6 forms were recorded (She and Watson, 2005). After revisions of He et al. and Pimenov, etc. in recent years, it was acknowledged there are 47 species, 1 subspecies, 6 varieties and 5 forms of Bupleurum in China (Wang et al., 2011; Ma et al., 2013; Wang et al., 2013; Ma and He, 2015; Pimenov, 2017), most of which have reputed medicinal values in treating fever and liver ails (Liang et al., 2012; Chao et al., 2014), as observed in many wild species of Apiaceae (Moazzami Farida et al., 2018; Sonigra and Meena, 2021; Perrino et al, 2023).
In the western part of Sichuan Province in Southwestern China, i.e., some areas of Aba Zang and Qiang Autonomous Prefecture near Chengdu, including Wenchuan, Mao County, Songpan, and so on, the steep mountains and plateaus lead to the differentiation and formation of Bupleurum species (Wang et al., 2008a; Yang et al., 2021). Closely related species discovered and reported in this region include B. wenchuanense R.H. Shan & Y. Li, B. chaishoui R.H. Shan & M.L. Sheh, B. malconense R.H. Shan & Y. Li, B. sichuanense S.L. Pan & P.S. Hsu, B. microcephalum Diels, etc. (Pan and Hsu, 1992). Most of them are quite morphologically similar and the differences between each other are subtle, thus their identification is notoriously difficult. And also, their genetic relationship remains unclear.
Phylogenomics, especially that based on the whole chloroplast (cp) genome sequences, has been proved highly efficient in resolving complicated phylogenetic problems of taxonomically complexed plant groups (Liu et al., 2022; Zhang et al., 2023), and in differentiating and identifying closely related species (Pchelin et al., 2019; Li et al., 2021). We’ve made some efforts in elucidating phylogeny of Chinese Bupleurum through phylogenomic means. We sequenced cp genomes of two woody Bupleurum species (B. gibraltaricum Lam. and B. fruticosum L.) endemic to Mediterranean and made comparative analysis with some Chinese species, confirming the division of 2 subgenera (subg. Penninervia, subg. Bupleurum) and revealing their divergence time (Huang et al., 2021a); we also analyzed the sequences of whole cp genomes of 4 Southwest China endemic alpine Bupleurum species and suggested phylogenetic affinity in the genus might be closely related to geographical distribution (Huang et al., 2021b); and we proposed the phylogenetic position of B. sikangense C.B. Wang, X.G. Ma & X.J. He as well (Xie et al., 2021). As a continuation of the work, we made a phylogenomic analysis of the above-mentioned species distributed in western Sichuan, based on both cp genome and 45s nuclear ribosomal DNA (nrDNA, including 16s rRNA-ITS1-5.8s rRNA-ITS2-26s rRNA), in order to untangle the complicated relationship among them, and to seek suitable molecular markers for species identification.
Additionally, in the ongoing study on Bupleurum investigation, the authors encountered a population in which the individuals obviously belonged to this genus in Wenchuan county years ago. For all the time, the authors had been regarding the plants in this population as B. chaishoui. Until recently, the authors obtained some specimens of B. chaishoui in its type locality and had a deeper understanding of the species. The authors realized that the plants met before is unique in its leathery dimorphic leaves and represent an overlooked new species, and then named it as B. pseudochaishoui. It was thus described here.
In this study, 65 plant samples, representing 29 Bupleurum species were collected in the summer of the years of 2014 through 2020, from North, Northeastern, Northwestern and Southwestern China, including 20 samples of the six above-mentioned species in Western Sichuan (Table 1). The voucher specimens were deposited in the herbarium of the University (Southern Medical University herbarium, SMU, to be listed in the Index Herbarium).
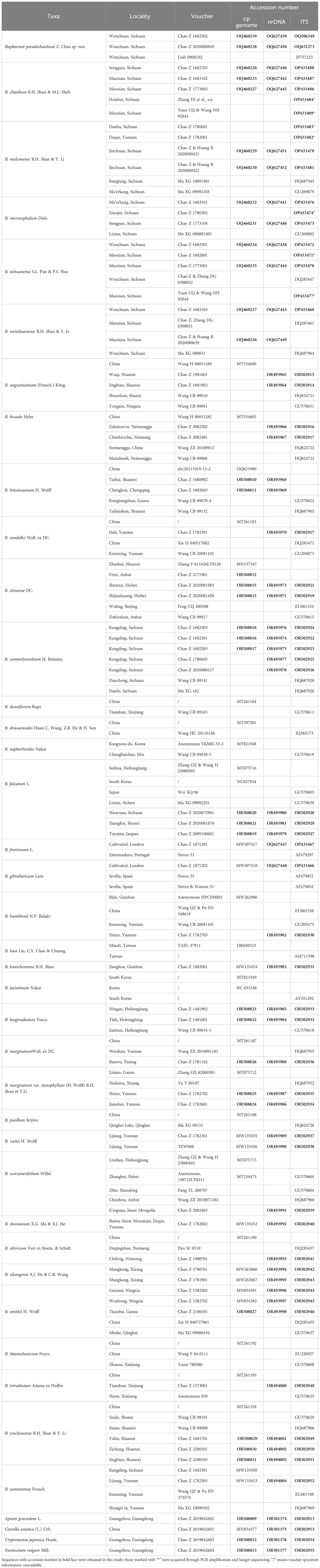
Table 1 Sources of material and GenBank accession numbers.
Specimens of other Chinese Bupleurum species, especially those distributed in Western Sichuan, which were deposited in the major herbaria of China including China National Herbarium (PE), Herbarium of Department of Biology, Sichuan University (SZ), Herbarium of Chengdu Institute of Biology, Chinese Academy of Sciences (CDBI), Kunming Institute of Botany Herbarium (KUN) and Herbarium of College of Life Sciences, Northwest Agriculture & Forestry University (WUK), were also consulted and inspected on site or through Chinese Virtual Herbarium (CVH) website.
The morphological characters of taxonomic significance in Bupleurum (Pan et al., 2002), i.e., the number, shape and size of bracteoles, comparison of bracteoles length and umbellule size, the number and length of rays, the number and size of florets, the shape, size and texture of leaves, of B. pseudochaishoui were inspected, and were compared with the related species, B. wenchuanense, B. chaishoui, B. malconense, B. sichuanense, and B. microcephalum. The data were acquired from at least 10 individuals of living materials or herbarium specimens, and were also checked in accordance with the descriptions in Flora of China (She and Watson, 2005). For measurements of some quantitative traits, such as plant height, leaf size, the number of small flowers, etc., 50 pieces of data were collected for each item, then the variation ranges were summarized and recorded.
Genomic DNA was isolated from the silica gel dried leaves of each sample using DNAsecure Plant Kit (TIANGEN Biotech Co, Ltd., Beijing, China). After purification, concentration of the extracted DNAs was determined on a NanoDrop 2000C spectrophotometry (Thermo Fisher, US).
ITS region of some samples (those with ITS accession number marked by “*” in Table 1) were amplified and sequenced. The universal primers ITS-p4 and ITS-p5 (Cheng et al., 2015) were used. PCR amplification was performed in a 15 μL reaction system containing 6 μL of 2×Taq Plus PCR MasterMix (Tiangen Biotech Co, Ltd., Beijing, China), 2 μL of DNA template, 0.5 μL of each primer (10 mM), and 6 μL of ddH2O. The PCR products were purified and sequenced bidirectionally by Ruibo Biotech Co. Ltd. (Guangzhou, China). The sequences have been deposited in GenBank, and the accession numbers were listed in Table 1. We also downloaded several ITS sequences of the related species from GenBank (Table 1).
The purified genomic DNAs were subjected to genome skimming performed on an BGISEQ-500 sequencer by BGI Genomics (Shenzhen, China). For each sample, a total of 3.0 Gb sequence data was obtained.
Whole cp genomes of the samples were assembled using GetOrganelle v1.7.4 software (Jin et al., 2020). The published cp genome of B. chinense DC. (GenBank accession number: MN337347) was used as the reference genome (Zhang et al., 2019). Bowtie2 (Langmead and Salzberg, 2012) was first called to filter out plastid-like reads, and then SPAdes (Bankevich et al., 2012) was called for assembly. The cp genome was annotated using GeSeq (Tillich et al., 2017) and Plastid Genome Annotator (PGA) software (Qu et al., 2019), and the tRNAs in the genome were identified by tRNAscan-SE v2.0.7 (Lowe and Chan, 2016) and ARAGORN v1.2.38 (Laslett and Canbäck, 2004). The annotated genomes, together with the reference genome, were input into Geneious v22.2.2 (Kearse et al., 2012) for comparison to determine start and stop codons, as well as to manually correct the possible errors. Finally, the cp genome was physically mapped into a loop using the online software OGDRAW (Lohse et al., 2007). The assembled and annotated cp genome sequences were deposited in GenBank (http://www.ncbi.nlm.nih.gov/) under accession numbers OQ460227-OQ460239 (Table 1).
nrDNAs were also assembled using GerOrganelle v.1.7.3 software, and the output contigs were compared and annotated with the reference genome in Geneious v22.2.2.
The cp genome characteristics of the six species of Bupleurum were described, including the overall genome structure, the size of each part of the genome, and the GC content of the genes, which was acquired using Geneious v22.2.2. Multiple genome alignments of 13 sequences were done using the Mauve v2.4.0 plugin (Darling, 2004) in Geneious v22.2.2, with default parameters.
To determine the expansion/contraction of IR, genes distributed within and next to the boundaries of LSC, SSC and IR regions were compared and manually mapped to visualize them.
To investigate the highly variable regions (divergence hotspots), 13 complete chloroplast sequences of six Bupleurum species, were compared using MAFFTv. 1.3.7 (Katoh and Standley, 2013). Subsequently, the cp genomes of Bupleurum species were paired and visualized using the Shuffle LAGAN mode in mVISTA (Frazer et al., 2004), with the cp genome of B. chinense (GenBank access number: MN337347) as a reference. Using DnaSP v6.0 (Rozas et al., 2017), the nucleotide diversity (pi) in the cp genome sequence was calculated through sliding window analysis, with a window size of 600 bp and a step size of 200 bp.
Some other parameters, such as simple sequence repeats (SSRs), short dispersed repeats (SDRs), nonsynonymous (Ka) and synonymous (Ks) substitution rates, were also investigated. (Supplementary File S1, Figures S1, 2).
In order to get insight into the phylogenetic relationship among the Bupleurum species, maximum likelihood (ML) analyses and Bayesian inference (BI) analyses based on the whole cp genome, the coding sequences (CDS), the highly variable regions, the nrDNA, and the ITS region were performed respectively. Besides the sequences obtained in this study, the previously reported cp genome and ITS sequences of Chinese Bupleurum species were also downloaded from Genbank and incorporated into phylogenetic analysis (Table 1).
The best model was obtained under AIC criteria using ModelFinder (Kalyaanamoorthy et al., 2017) for molecular modeling of the dataset (Table 2). For ML analyses, IQ-tree 1.6.12 (Nguyen et al., 2015) was used, and the reliability of each branch was assessed using bootstrap values (number of replicates set to 1000). As for BI analyses, MrBayes 3.2.7a (Ronquist et al., 2012) was used, and Markov chain Monte Carlo (MCMC) analysis was run for a total of 20 million generations, with samples taken every 1000 generations. In the phylogenetic tree construction, Centella asiatica were used as outgroup species, and the resulting tree files were visualized and embellished using iTOL v.6.1.2 software (Letunic and Bork, 2021).

Table 2 Best models for ML and BI analyses.
A total of 13 cp genomes from the six Bupleurum species were obtained. Size of these cp genomes ranges from 155,012 bp (B. pseudochaishoui) to 155,715 bp (B. wenchuanense). All of them exhibit a typical quadripartite structure, consisting of a small single copy (SSC) region (17,368-17,524 bp), a pair of inverted repeat (IR) regions (52,250-54,902 bp) and a large single copy (LSC) region (83,282-85,491 bp) (Table 3, Figure 1). The overall GC contents are highly similar (37.7-37.8%) (Table 3).
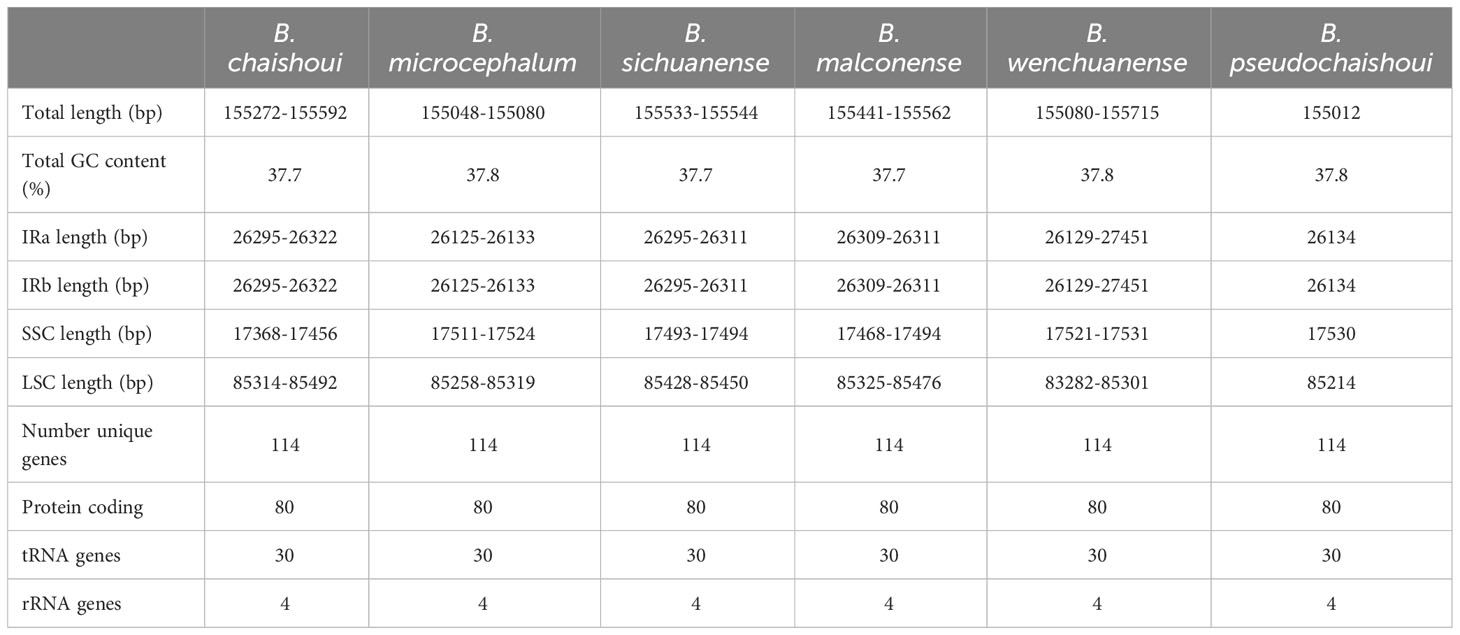
Table 3 Summary on chloroplast features of six Bupleurum species.
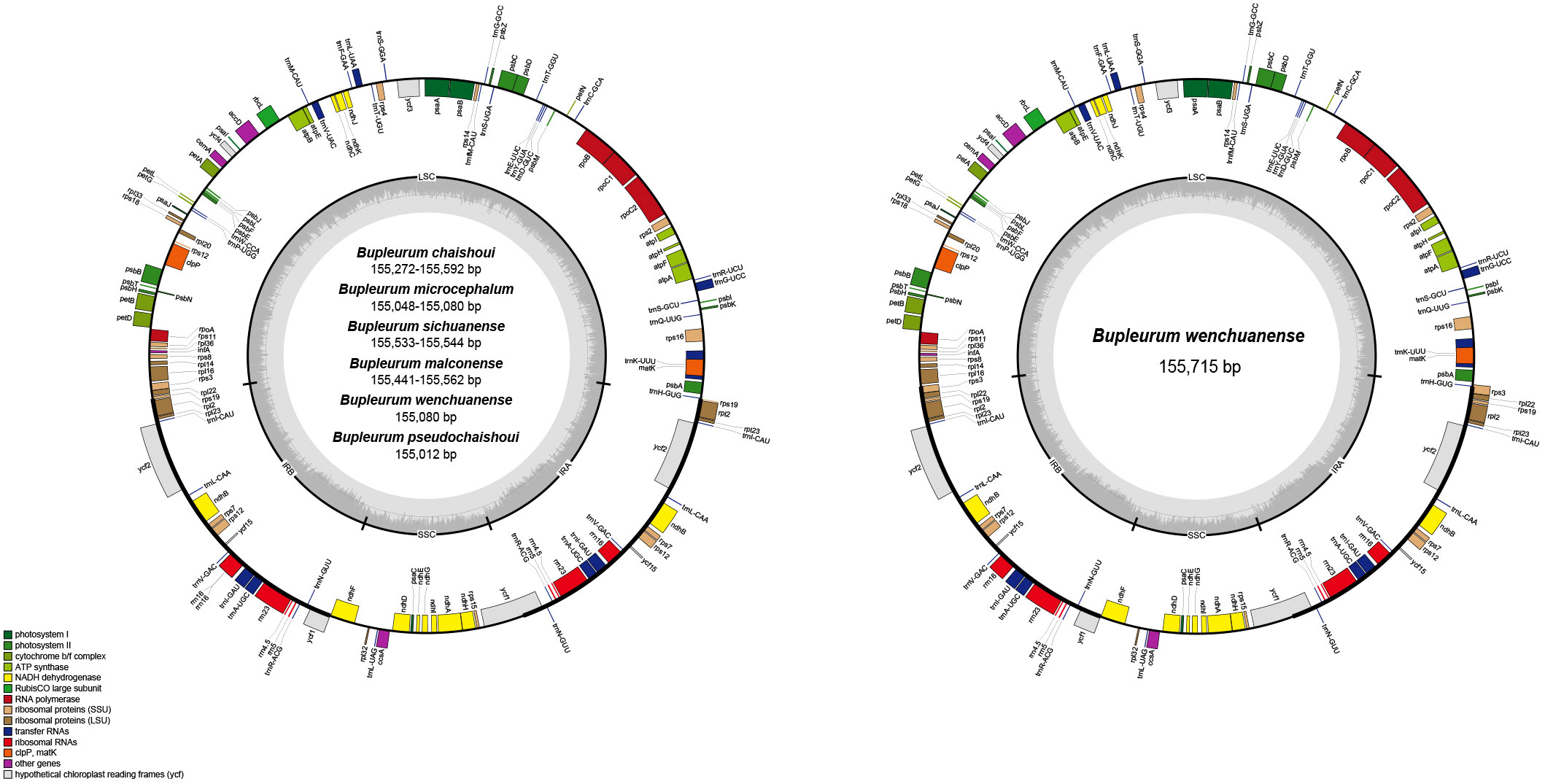
Figure 1 cp genome map of the six Bupleurum species Genes shown outside of the larger circle are transcribed clockwise, while genes shown inside are transcribed counterclockwise. Thick lines of the smaller circle indicate IRs and the inner circle represents the GC variation across the genic regions.
Genes and its arrangement in the cp genomes of the six Bupleurum species are almost the same. Each cp genome encodes 114 genes, including 80 protein-coding genes (PCG), 4 rRNA genes, and 30 tRNA genes. There’re 60 PCGs and 22 tRNA genes in the LSC region, 12 PCGs and 1 tRNA gene in the SSC region. In the IR region, for 12 of the 13 cp genomes, there’re 20 gene repeats, including 8 PCGs, 7 tRNA genes and 4 rRNA genes, except for one B. wenchuanense sample (voucher 2020080639), which has 2 extra PCGs (rpl22 and rps3) and thus a total of 22 genes in the IR region. Among these 114 genes, eighteen genes are found with introns, 15 of which containing one intron, while the other three (clpP, ycf3, and rps12) containing two introns. Incomplete copies of ycf1 and rps19 in the IR region are considered as pseudogenes. (Table 3, Table 4, Figure 1).

Table 4 List of genes encoded in six Bupleurum plastomes.
The nrDNA sequences of the six Bupleurum species are 5821-5824 bp in length. The nrDNA is composed of five segments, i.e., the small-subunit rRNA gene (18s rDNA), internal transcribed spacer 1 (ITS1), the 5.8S rRNA gene, internal transcribed spacer 2 (ITS2), and the large-subunit rRNA gene (26s rDNA). The length of each segment is 1809 bp, 214-217 bp, 163-164 bp, 227-228 bp, and 3406-3407 bp respectively. The overall nrDNA GC content is 54.4%; among the five segments, ITS regions have higher GC content (with a value of 57.9-58.7%). (Table 5).
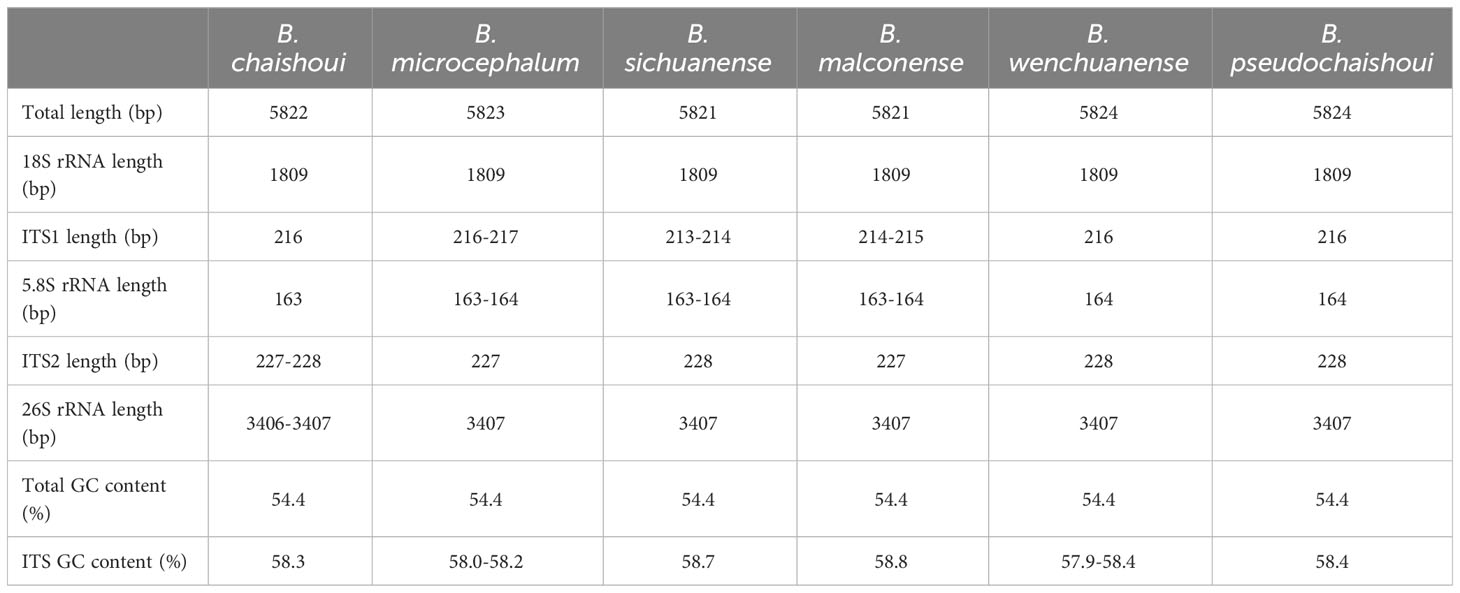
Table 5 Summary on nrDNA(18S-ITS1-5.8S-ITS2-28S) features of six Bupleurum species.
Results of Mauve pairwise analysis indicate that no rearrangement has occurred in coding and noncoding regions of the cp genomes of the six Bupleurum species (Figure 2), implying that the cp genomes are conservative.
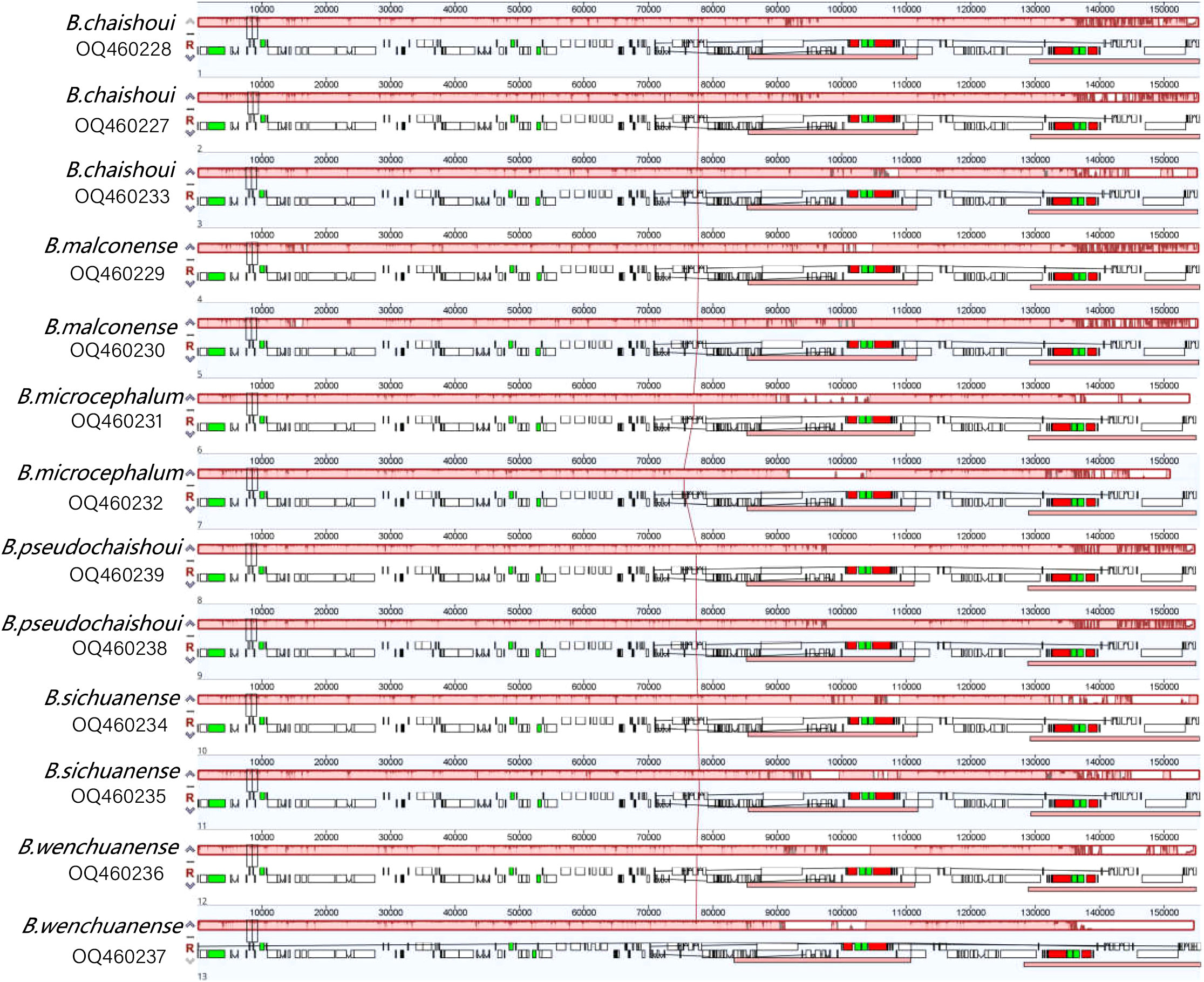
Figure 2 MAUVE alignment of the six Bupleurum species Locally collinear blocks are represented by continuously colored regions.
The genes rpl22 and rpl2 are located on the left and right sides of the LSC/IRb junction respectively, while the gene rps19 crosses the JLB line (70 bp occurring in the IR region). The ndhF gene is at the right side of the IRb/SSC junction and 14-26 bp away from the boundary. Traversing the SSC and IRa region is the gene ycf1; a fragment of 1877-1902 bp in length of the gene is located in the IR region. The gene trnH lies 4-bp away from the JLA line (IRa/LSC boundary) to the right (Figure 3).
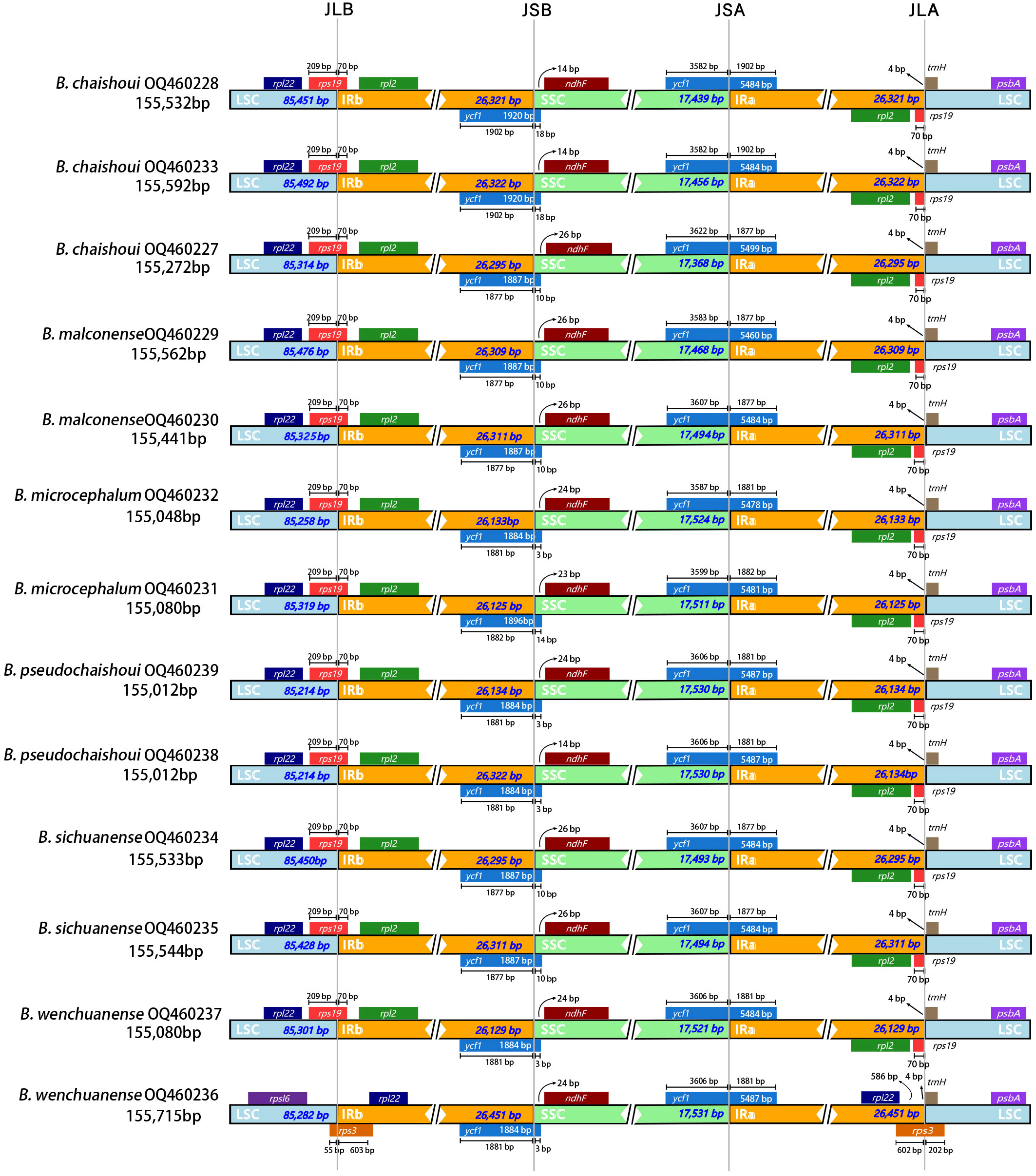
Figure 3 Comparison of the LSC, SSC and IR junction among the six Bupleurum species JLB, junction line between LSC and IRb; JSB, junction line between SSC and IRb; JSA, junction line between SSC and Ira; JLA, junction line between LSC and IRa.
There’s an obvious expansion in the IR region of a B. wenchuanense sample (voucher 2020080639). In this sample, as mentioned above, genes rpl22 and rps3 are found in the IR region. Generally, these two genes were located in the LSC region. The rps3 gene is crossing the junction of LSC/IRb (JLB) and LSC/IRa (JLA). (Figure 3).
We compared and mapped whole chloroplast gene alignments among 13 sequences of six Bupleurum species using mVISTA, using the published cp genome of B. chinense (No. MN337347) as the reference. The results showed that the divergence in the noncoding regions was greater than that in the coding regions, and a high degree of divergence in the 13 cp genomes appeared at intergenic spacers, including trnK-rps16, atpF-atpH, atpH-atpI, trnC-petN, petN-petM, ndhC-trnV, petA-psbJ, accD-psaI, rps15-ycf1, rpl32-trnL, and trnV-rps7. The coding regions were more conserved, and trnL, rpoC, petD, ycf2, ndhA and ycf1 were the more divergent coding regions of these species. (Figure 4).
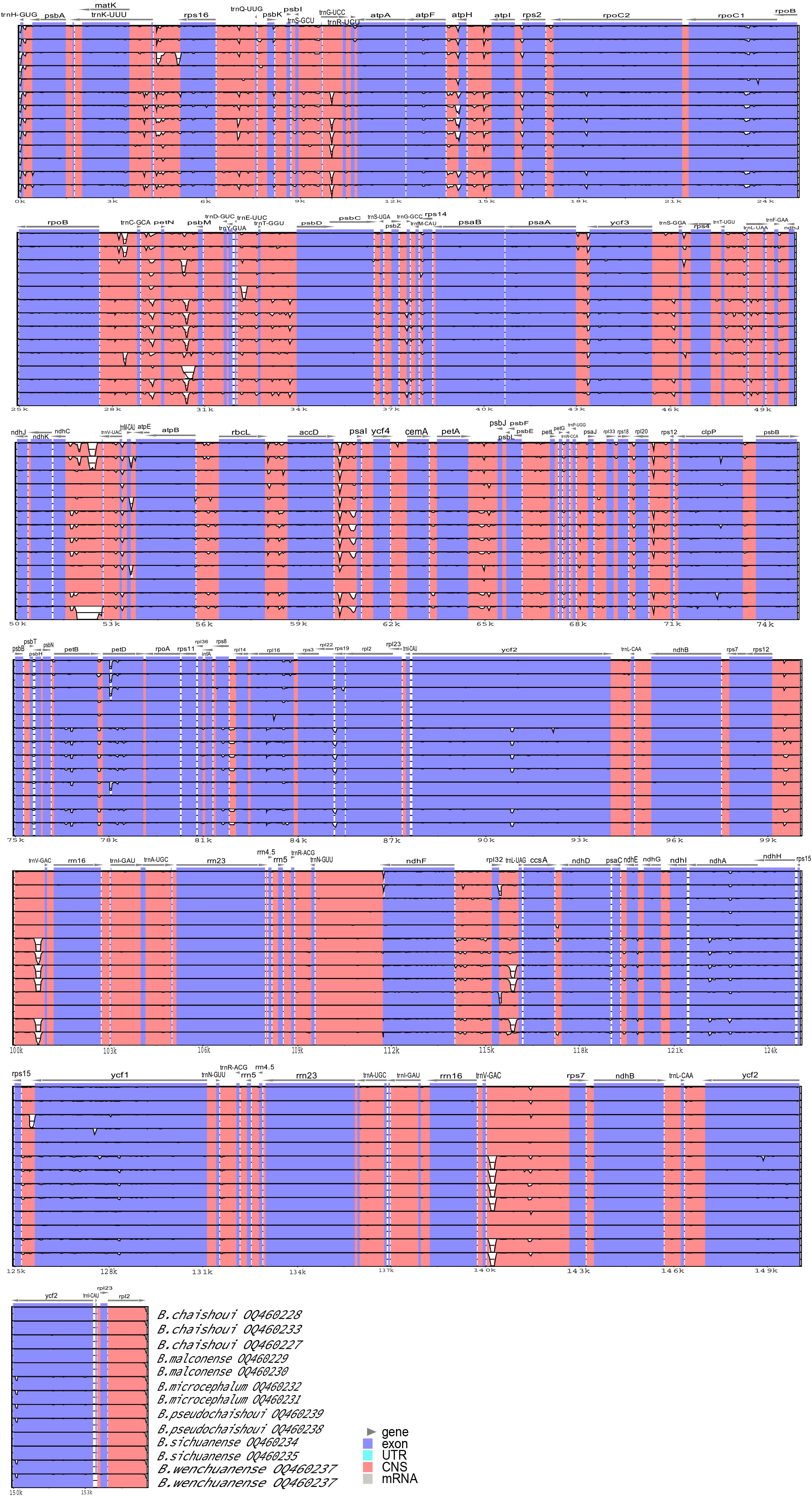
Figure 4 Comparison of the six Bupleurum species cp genomes using the mVISTA alignment program, with B. chinense as a reference.
In order to assess the sequence divergence level of these Bupleurum species, the nucleotide diversity (Pi) of the cp genomes and nrDNAs were calculated.
The Pi values of the whole cp genome of the six species range from 0.00-0.06, and the two IR regions were significantly less divergent than the LSC and SSC regions. The spacer regions between genes were found with higher Pi values than the coding regions. Totally there’re 7 spacer regions with Pi values greater than 0.01; the spacer with the highest Pi value was rpl32-trnL (Pi> 0.06), followed by petA-psbJ (Pi> 0.02), and consecutively, trnK-rps16, atpF-atpH, atpH-atpI, ndhC-trnV and rps15-ycf1 (Pi> 0.01). Only two coding regions (trnL-UAA and ycf1) had Pi values higher than 0.01, and the ycf1 gene has higher value. (Figure 5A).
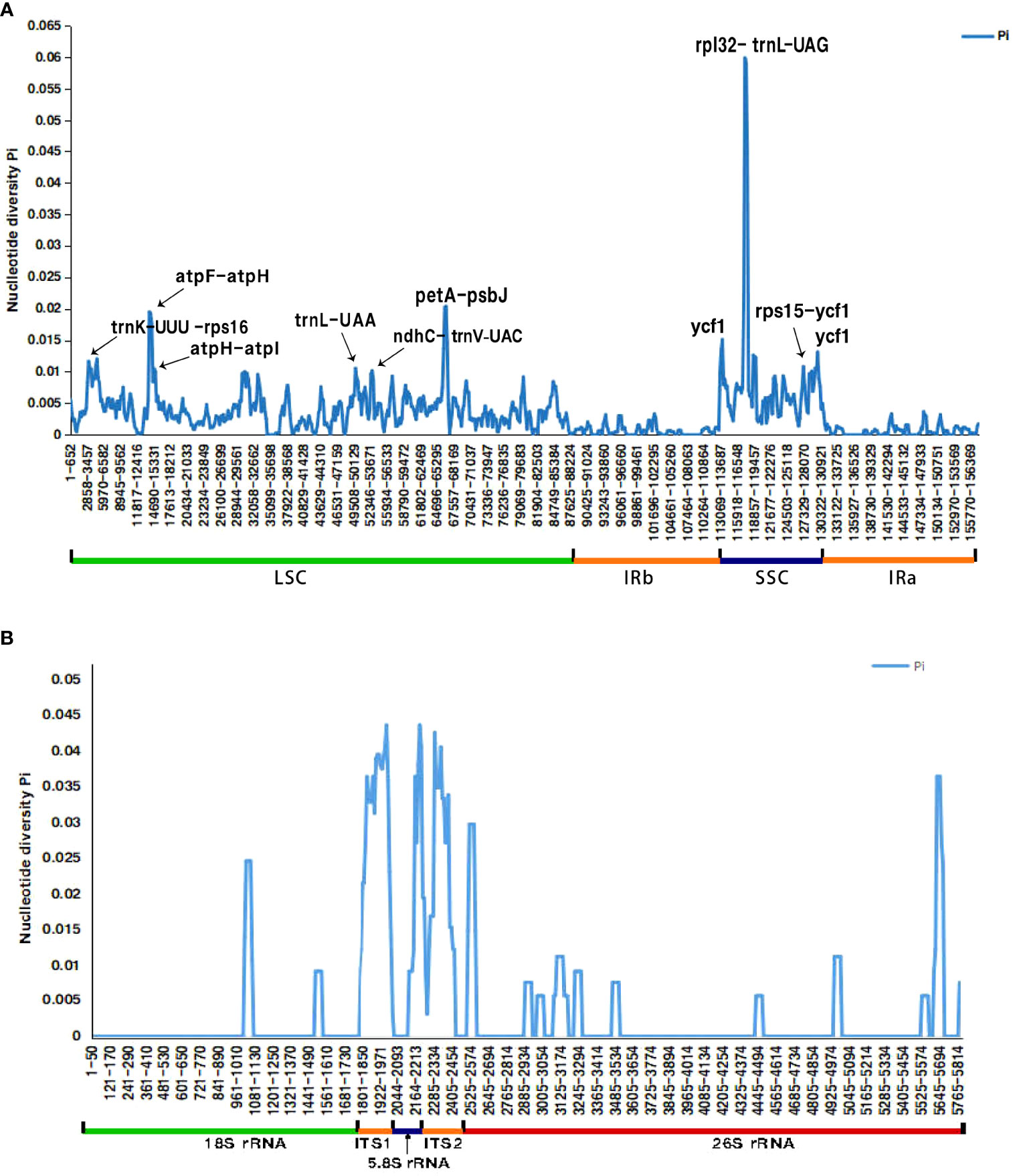
Figure 5 Comparative analysis of the nucleotide diversity (Pi) value among the six Bupleurum species (A) cp genome; (B) nrDNA X-axis: nucleotide position, Y-axis: nucleotide diversity value.
The concatenated nrDNA sequences of the six Bupleurum species have 65 variation sites and 54 parsimony-informative sites, accounting for 1.12% and 0.93% of the total sequences, respectively. The Pi value of the whole sequence was 0-0.044, and the variant regions were mainly concentrated in the transcribed spacer region (ITS), with a mean value of 0.029 (range 0-0.044) for ITS1, 0.007 (range 00-0.036) for 5.8S rRNA and 0.023 (range 0-0.044) for ITS2. 18S rRNA and 26S rRNA were more conserved, with a mean value of 0.001 (range 0-0.025) for 18S rRNA and 0.002 (range 0-0.036) for 26S rRNA. (Figure 5B).
Figures 6A-D shows the results of maximum likelihood (ML) analysis and Bayesian inference (BI) analysis of chloroplast whole genomes, protein-coding genes, the nrDNA and ITS region respectively, for the studied Bupleurum species. The phylogenetic trees estimated by ML and BI analyses showed similar topologies with high bootstrap support values and strong posterior probabilities for most nodes. The trees constructed based on the cp genome, protein-coding genes, and nrDNA were essentially alike. Bupleurum species formed a monophyletic clade which was sister to the other three species of Apioideae and could be divided into two main subclades, one consisting of B. fruticosum and B. gibraltaricum from the Mediterranean region (clade A), and the other consisting of all the remaining Chinese species (clade B).
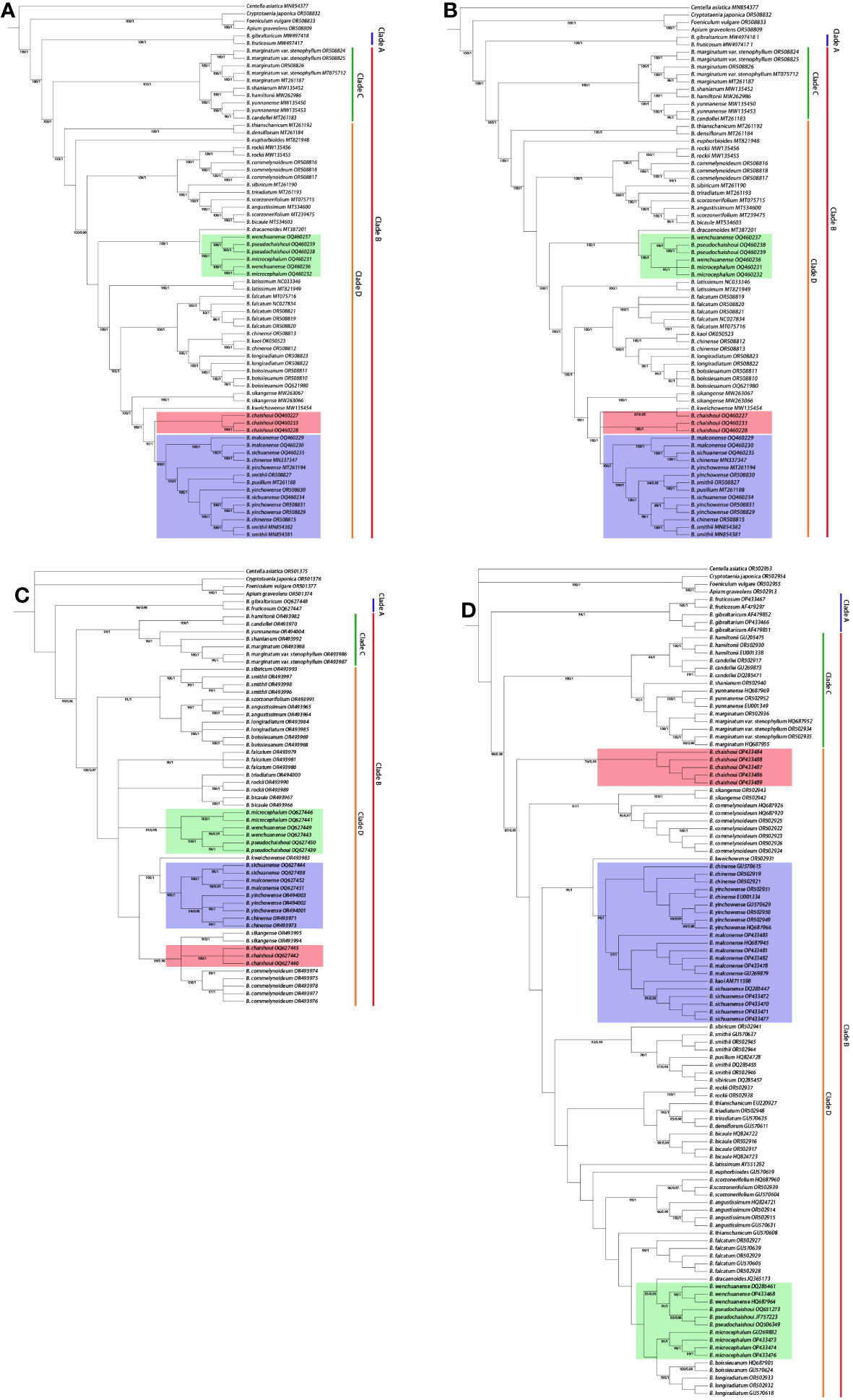
Figure 6 Phylogenetic trees showing the relationship between Bupleurum species. BS>70, PP>0.95 obtained by ML/BI methods are shown above the branches. (A) Phylogenetic tree based on the whole cp genome; (B) Phylogenetic tree based on protein-coding genes; (C) Phylogenetic tree based on the nrDNA; (D) Phylogenetic tree based on the ITS region.
The clade of Chinese species in subgenus Bupleurum can be further divided into clades C and D. In all trees, the constituent species of these two clades are consistent. A small one (clade C) was composed of B. hamiltonii N.P. Balakr., B. candollei Wall. ex DC., B. yunnanense Franch., B. marginatum Wall. ex DC. and its variety B. marginatum var. stenophyllum (H. Wolff) R.H. Shan & Y. Li. A large one (clade D) included more than 20 species; the six species produced in western Sichuan are all in this large clade.
Topology of clade D in the chloroplast-based trees and nuclear gene-based trees showed difference to some extent. The number of branches, the composing species of each branch, and the position of the branches, were quite different; for example, the position of B. chaishoui, the relationship between B. microcephalum and B. wenchuanense (See below, Discussion section, and Figure 6). Nevertheless, all four phylogenetic trees showed that the new species B. pseudochaishoui were closely related to B. wenchuanense and B. microcephalum; they were either forming/mixing in a separating branch (CP, CDS and nrDNA trees) or locating in adjacent branches (ITS tree). Similarly, B. malconense and B. sichuanense are closely related species of B. chinense and B. yinchowense R.H. Shan & Y. Li (Figures 6A-D).
The position of B. chaishoui varied in different trees. In the CP tree, it’s close to B. malconense-B. sichuanense-B. chinense-B. yinchowense group, and then merged with B. kweichowense R.H. Shan and B. sikangense. In the CDS tree, it joined with B. kweichowense first, then B. malconense-B. sichuanense-B. chinense-B. yinchowense group and B. sikangense. In the NR tree, it’s adjacent to the branch of B. sikangense and B. commelynoideum H. Boissieu, and then aggregated with B. rockii H. Wolff and B. triradiatum Adams ex Hoffm. successively. It’s noticeable that in the ITS tree, B. chaishoui formed an independent branch sister to other species, neighboring to B. sikangense and B. commelynoideum branch.
The nrDNA phylogenetic tree has the highest resolution in differentiating the six Bupleurum species, in which each species formed a monophyletic branch.
We described for the first time the cp genomes of six species of Bupleurum from western Sichuan Province, China, providing important insights into the cp genome characteristics of members of this genus. Our results show that the cp genomes of Bupleurum species are extremely similar, indicating that they are highly homogeneous in terms of cp genome structure, gene content and tendency of SDRs and SSRs. The cp genomes of the six Bupleurum species differ in size by only a few hundred bp (155,012-155,712 bp), consistent with the description that the cp genomes of Bupleurum species show only minor differences in size (~1 kb) (Huang et al., 2021a). Many mutational events occurring in the cp genome, including substitutions, insertions and deletions (InDels), inversions, genomic rearrangements and translocations (Abdullah et al., 2021), did not occur in any of the six Bupleurum species.
It has been reported that IR region contraction and expansion is a common phenomenon in cp genomes, and that changes in the size of the cp genome are usually the result of IR region expansion and that these variants can be observed in both closely and distantly related plant species and may lead to pseudogene production, gene duplication and deletion of individual copies of genes (Plunkett and Downie, 2000; Wang et al., 2008c; Saina et al., 2018). Comparative analysis of IR boundaries showed the same distribution of genes at the LSC/IR junction in the cp genomes of the six Bupleurum species, with minor differences in gene length (ycf1 and rps19) at the SSC/IR junction. The ycf1 sequence located at the IRa and SSC boundaries was identified as a pseudogene because it was truncated and was an incomplete duplicate of a normal copy. In the cp genome of a sample (B. wenchuanense voucher 2020080639), expansion of the IR region was found occurred, resulting in inconsistency between LSC/IR and SSC/IR border genes to other Bupleurum. The expansion was due to duplication of genes in IR (rpl22 and rps3). Variation in the number of duplicated genes in angiosperm cp genomes is common and different numbers of duplicated genes can be observed in different species (Ahmed et al., 2012; Menezes et al., 2018; Abdullah et al., 2020), but IR expansion within species is somewhat uncommon. This phenomenon was once observed in the cp genome of Sinopodophyllum hexandrum (Ma et al., 2022). We performed read mapping analysis and confirmed such an expansion in the very sample of B. wenchuanense (Figure S4). However, the overall gene number and order remains consistent and reflects the significant conserved nature of chloroplasts in this species.
Inspired by Dong’s research work (Dong et al., 2012), highly variable regions in cp genome have been generally thought to be potential molecular markers for phylogeny and identification of plant species. In some cases, the highly variable regions worked well in resolving phylogeny and identifying species (Dong et al., 2021; Song et al., 2022), but in some other cases they were not so effective (Wu et al., 2020); more often, their effectiveness has not been evaluated (Liu K et al., 2021; Liu X.J. et al., 2021; Wu et al., 2022). In order to assess whether the they can work in Bupleurum phylogenetic analysis and species identification, we checked the highly variable regions in cp genomes of the six Bupleurum species.
We found that the non-coding intergenic spacer regions have considerable diversity over the protein-coding regions, which was consistent with most previous studies (Daniell et al., 2016; Javaid et al., 2022). Seven spacer regions (rpl32-trnL, petA-psbJ, atpF-atpH, trnK-rps16, atpH-atpI, ndhC-trnV, and rps15-ycf1), and two coding regions (trnL-UAA and ycf1) were found to be highly variable (Pi > 0.01). However, the discovered highly variable regions were not all the same as the previously reported ones in Bupleurum (Li et al., 2020; Huang et al., 2021a; Huang et al., 2021b; Xie et al., 2021). The spacer regions petA-psbJ, trnK-rps16 and the gene ycf1 are the highly variable regions found in common in these studies on Bupleurum cp genomes. There is evidence supporting the ycf1 gene as one of the core plastid DNA barcodes in land plants (Neubig et al., 2009; Franck et al., 2012; Dong et al., 2015). The rpl32-trnL, petA-psbJ gene have also been reported to show very high nucleotide per site diversity in several genera (Dong et al., 2012; Tan et al., 2020; Wang et al., 2022; Niu et al., 2023), and indeed the Pi values of the rpl32-trnL spacer regions of these six Bupleurum species were also much higher than the other regions in the present study, so rpl32-trnL together with petA-psbJ, trnK-rps16, and ycf1 were considered as having potential to be used as high-resolution DNA barcodes to identify species of Bupleurum.
We constructed phylogenetic trees of the six Bupleurum species using the four screened potential molecular markers independently and in combination, and found the trees were in similar topology to the whole cp genome and CDS trees, i.e., the six species were divided into two clades. But in each tree, there’s species group that cannot be clearly separated (Figures S3A-F). The “potential” disappears in reality. Therefore, we propose that in similar studies in the future such presumed molecular markers should be verified.
The backbone of phylogenetic trees constructed based on the complete cp genomes, protein-coding sequences, nrDNA of the six Bupleurum species were highly silimar; and they were consistent with those acquired in the previous studies (Neves and Watson, 2004; Wang et al., 2008a; Wang et al., 2008b; Wang et al., 2011). The results confirmed Bupleurum is a monophyletic group and supported the treatment of its independent tribe status in subfamily Apioideae. The two main subclades represented the two subgenera in the genus, subgenus Penninervia and subgenus Bupleurum, respectively (Neves and Watson, 2004; Wang et al., 2011). The two clades (clade C and clade D in Figure 6) of Chinese Bupleurum, as well as the species included in each one, were also in concordance with the earlier researches (Wang et al., 2008b; Wang et al., 2011).
In the six Bupleurum species in Western Sichuan, B. microcephalum and B. malconense were considered to be the most closely related species (Li and Sheh, 1979; She and Watson, 2005). The leaves of B. microcephalum are linear, extremely long, soft and thin, while the leaves of B. malconense are more than half shorter than that of B. microcephalum, and the texture is hard. But in all phylogenetic trees, B. microcephalum forms a clade with B. wenchuanense, and B. pseudochaishoui; in the tree based on nrDNA and ITS, it forms a sister branch to the latter two species. This is not consistent with the previous concepts that it’s close to B. malconense. The three species in this clade all have solitary stem, which is different from species that have caespitose rootstock and numerous stems in the other clade.
B. sichuanense was established in 1992 by Pan and Hsu (Pan and Hsu, 1992). In the original species description, it was pointed out B. sichuanense was closely related to B. malconense, and was differ from the latter in its short and tender cauline leaves, more umbel rays (6~7), and bracteoles not longer than the pedicel (< 2 mm). In Flora of China, it was synonymized with B. malconense (She and Watson, 2005). Our molecular phylogenetic analysis proved their intimate relationships. In the phylogenetic trees constructed with the whole cp genomes, the CDS regions and the high variable regions, neither of the two formed a separate clade. In the nrDNA ITS tree, more than 10 sequences derived from these two species tangled together; but in the tree based on the nrDNA, B. sichuanense and B. malconense were clearly separated, implying they were two independent species. We checked the specimens of B. malconense and B. sichuanense deposited in Chinese Virtual Herbarium and those we collected and were of the opinion that the nrDNA correctly reflects the relationship between the two species. The results also implied the nrDNA might serve as a more suitable molecular marker for Bupleurum phylogenetic analysis and species identification.
The close relationship of B. sichuanense and B. malconense to B. chinense and B. yinchowense was disclosed almost 15 years ago (Xie et al., 2009). Their intimate relationship has once again been proven in this study. B. chinense is the officially recognized origin species of the crude drug of Chaihu (Chinese Pharmacopoeia Commission, 2020). There is a need for molecular marker to distinguish B. chinense from the other three closely related species (Chao et al., 2014). It seems nrDNA and ITS perform better than cpDNA does for such a purpose.
Phylogenetic trees constructed from cp genome and nrDNA show different topologies, indicating the existence of nuclear cytoplasmic conflicts. The position of B. chaishoui is an obvious example. Phylogenetic trees constructed from both nrDNA and ITS sequences support that B. chaishoui is closely related to B. sikangense and B. commelynoideum, while in the phylogenetic trees constructed from the whole cp genome sequence and CDS sequence, B.chaishoui is closely related to B. malconense; the latter is consistent with the opinion in FRPS that stated B. chaishoui and B. malconense are similar in morphology (Li and Sheh, 1979). The two species, together with the third species in the same clade, B. sichuanense, share a common morphological feature, i.e., the caespitose rootstock.
The inconsistencies in the topology of the gene trees acquired from the two different set of chloroplast and nuclear genomic data is a widespread phenomenon caused by a variety of reasons, including gene duplication and loss, horizontal gene transfer, incomplete lineage sorting, and hybridization introgression, etc. (Steenwyk et al., 2023). The most common reason for this phenomenon was believed to be chloroplast capture caused by hybridization (Fehrer et al., 2007; Galtier and Daubin, 2008; Soltis and Soltis, 2009; Stull et al., 2020). It was also reported cp genome-based phylogenetic relationships were often found to be related to geography rather than morphology (Liu et al., 2020). The Hengduan Mountains-Western Sichuan area is one of the diversity centers of Bupleurum species, and more than half of the species in China can be found in this region. The complex terrain and landforms, and diverse climate of ancient and modern times in the region have created opportunities for natural interspecific hybridization, and the results of phylogenetic trees suggest the possibility of ancient chloroplast capture events (Wang et al., 2008b; Sun et al., 2017; Yang et al., 2019; Xie et al., 2021). Moreover, the widespread presence of hybridization in biological taxa has gained increasing recognition (Fehrer et al, 2007; Acosta and Premoli, 2010). However, compared to its complex biodiversity, there are still very few clearly described hybridization events in the genus Bupleurum. In-depth researches in this aspect are expected in the future to reveal the important value of hybridization for the formation and maintenance of biodiversity of Bupleurum.
The materials collected from Weizhou Town and Shuixi Town of Wenchuan (voucher Chao Zhi 1682502 and Chao Zhi & Huang Rong 2020080849) had been overlooked and mistakenly acknowledged as B. chaishoui for years, recently we realized it represents a new species and named it B. pseudochaishoui. These two samples formed distinct clade in all phylogenetic trees, supporting its status as an independent species.
B. pseudochaishoui shows distinguishable morphological character to its allies. Its leaves are coriaceous and some shiny. The basal leaves are long petiolated and the blades are quite large, blade broadly ovate-elliptic to lanceolate elliptic; the middle and upper cauline leaves are much smaller, long lanceolate, with no petiole (Figure 7). In Chinese Bupleurum, only B. longiradiatum Turcz. (Northeastern, Northern and Northwestern China) and B. aureum Fisch. ex Hoffm. (Xinjiang) have basal leaves with blades of such a similar shape and size to B. pseudochaishoui.

Figure 7 Bupleurum pseudochaishoui Z. Chao sp. nov. (A) habitat and individual plant; (B) basal leaves; (C) root; (D) middle cauline leaves; (E) Compound umbels; (F) umbellule; (G) infructescence; (H) cremocarp; (J) petal; (K) bracteole; (I) schizocarp transverse section.
Molecular data indicated that B. wenchuanense is the most intimately related species to B. pseudochaishoui, but it differs from the new species in its numerous, rosette-caespitose oblanceolate basal leaves and subulate to squamose middle and upper leaves, and the (1–)2–3 very unequally rayed umbels, 1–4-flowered umbellules.
B. chaishoui, the species which we previously misrecognized as B. pseudochaishoui, has caespitose numerous stems, and lanceolate to elliptic cauline leaves; the shape and size of cauline leaves vary greatly, especially those born on the same node or adjacent nodes are very unequal; the upper ones often grow in a reflexed way (Figure 8). On the contrary, B. pseudochaishoui has solitary stem, and its cauline leaves often are not reflexed.
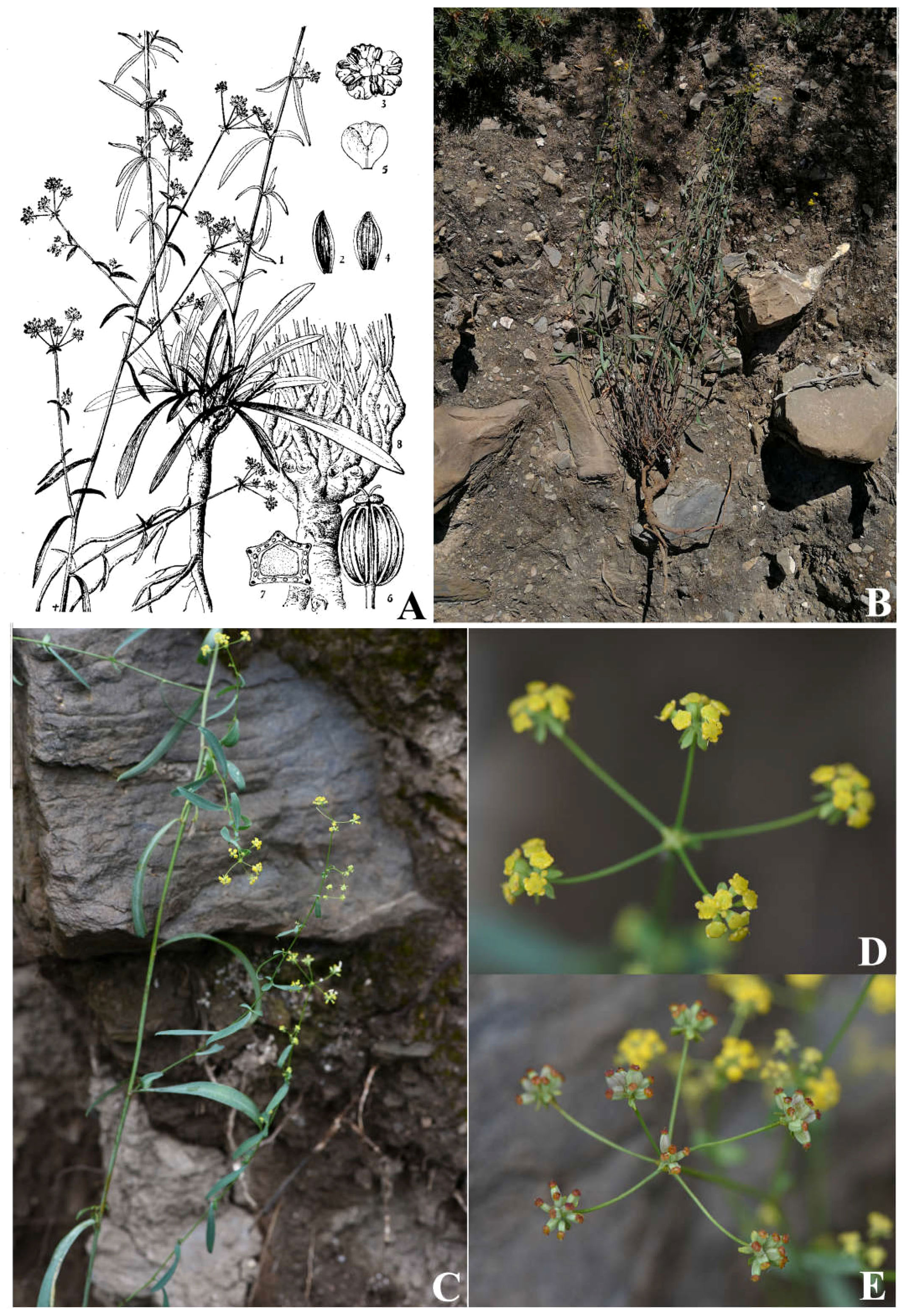
Figure 8 Bupleurum chaishoui R.H. Shan et M.L. Sheh (A) Illustration in FRPS; (B) individual plant; (C) middle cauline leaves; (D) Compound umbels; (E) infructescence.
In the areas neighbor to Southwestern China, there are another two Bupleurum species having basal leaves with broadly oval lamina, namely B. lanceolatum Wall. ex DC. in Himalayas in India and Western Pakistan and B. gilesii H. Wolff in Western Pakistan and Afghanistan (Missouri Botanical Garden, 2023). The upper cauline leaves are of similar shape to the basal and lower leaves in B. lanceolatum, and short condensed main stem and numerous branches from the base in B. gilesii, make them easily to be distinguished from B. pseudochaishoui.
Comparative analysis of the cp genomes and nrDNA sequences of six Bupleurum species from western Sichuan Province, China revealed their conserved structure. Phylogenetic analysis based on nrDNA proved the monophyly of each species. Incongruence between nuclear and plastome gene trees was observed, which may be attributed to chloroplast capture. For overall consideration, B. sichuanense was thought to be an independent species different from B. malconense; B. chaishoui was supposed to be close relative to these two species, and they all shared the characteristic of having caespitose numerous stems. The other three species with solitary stem, B. microcephalum, B. wenchuanense and B. pseudochaishoui, were close in consanguinity. The new species, B. pseudochaishoui, was described and illustrated.
Bupleurum pseudochaishoui Z. Chao, sp. nov. (Figure 7).
Diagnosis:—This new species is easily distinguishable in the genus by its leathery dimorphic leaves, i.e., basal leaves with relatively large ovate-lanceolate blade and extremely long petiole, and lanceolate middle leaves. Molecular data implied it is closely related to B. wenchuanense, but the latter differs in its numerous, rosette-caespitose oblanceolate basal leaves and subulate to squamose middle and upper leaves, and the (1–)2–3 very unequally rayed umbels, 1–4-flowered umbellules.
Type:—CHINA. Sichuan Province: Wenchuan county, Buwa Village, growing on mountain slopes, 31°29′42.24″N, 103°35′43.01″E, elevation 2000 m, 25 August 2016 (fl. & fr.), Chao Zhi 1682502 (holotype, Southern Medical University herbarium, SMU)!.
Description:—Perennial herb. Taproot 10-15 cm long and 5-8 mm in diameter, stout, brownish, the head often inflated and annulated, lower part always branched and longitudinally fine-wrinkled, with sparse small horizontal protuberance. Rhizome present at the top of the root, lignified, 3-10 cm long. Stem solitary, solid and rigid, surface obviously striped, 80-120 cm high; base woody, sometimes tinged purple; middle and upper part slightly tortuous, much branched, branches 10-30 cm long. Leaves leathery, bright green, abaxial surface light green, margin narrowly white-cartilaginous; basal and the lower leaves large, blade broadly ovate-elliptic to lanceolate elliptic, 5-13 cm × 2.5-4.5 cm, 5-7 nerved, apex rounded-obtuse or acute, base cuneate or broadly cuneate, tapering into long petiole up to 17 cm, slightly expanded into a sheath at the base, clasping; middle-part leaves long lanceolate, 10 cm × 1 cm, nerves 5-7, often white-colored on both sides of the midvein, apex acute or acuminate, base tapering and clasping; upper leaves gradually smaller. Inflorescence much-branched, umbels numerous, 1.5-3 cm across; bracts 1-5, ovate or squamose, unequal, 1-3 mm × 0.2-1 mm, 1-3 veined; rays 3-5, slender, unequal, 0.5-2.5 cm long; bracteoles 5, slightly membranous, ovate or lanceolate, 1.5-2 mm × 0.8-1 mm, longer than pedicels, slightly shorter than umbellules, apex acuminate, 3-veined; umbellules 2-4 mm across, (5) 8-10 (12)-flowered; florets about 1 mm in diameter. Petals pale yellow, slight brown convex at the top of the inflex, ligule slightly ladder-shaped, apex obviously 2-lobed, lobes obtuse; pedicels 1-1.5 mm long; stylopodium yellow, thick discoid, wider than ovary. Fruit oblong, 2 mm ×1 mm, brownish brown to dark brown, ribs not very obvious, vittae 3 in each furrow, 4 on commissure; fruit stalk about 2 mm long. Fl. Jul–Sep, fr. Aug–Oct.
Habitat and distribution:—The new species was found only in its type locality. It inhabits shrubs on mountain slopes, at an elevation about 2000 m. Fl. and fr. July-Sept.
Etymology:—The epithet refers to it has been previously mistaken as B. chaishoui.
A key to the six most closely related species of Bupleurum in the region of Western Sichuan is given as follows:
1. Stem usually numerous, caespitose.
2. Cauline leaves lanceolate to elliptic, very unequal at the same node, 1.2-9 cm × 0.3-1.2 cm, upper leaves usually reflexed ……………………B. chaishoui R.H. Shan & M.L. Sheh
2. Cauline leaves linear, similar in size, upper leaves not reflexed.
3. Involucel bracts less than 2 mm long, shorter than the pedicel; leaves tender, at most 8 cm long; rays of umbel usually 6-7 ………………………………… B. sichuanense S.L. Pan & P.S. Hsu
3. Involucel bracts more than 2 mm long, exceeding the pedicel; leaves firm, up to 15 cm long; rays of umbel usually less than 5………………………………………B. malconense R.H. Shan & Y. Li
1. Stem single, not caespitose.
4. Rays of umbel (1)2-3, very unequal; upper leaves small, subulate or squamose, few; stem much-branched ……………………B. wenchuanense R.H. Shan & Y. Li
4. Rays of umbel more than 4; leaves conspicuous and numerous.
5. Basal and cauline leaves uniform, linear, very long and narrow, herbaceous, soft, abaxially slightly glaucous ……………… B. microcephalum Diels.
5. Leaves coriaceous; the basal and lower ones large, blade broadly ovate-elliptic to lanceolate elliptic, petiole quite long; the middle and upper ones long lanceolate ………………… B. pseudochaishoui Z. Chao sp. nov.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, OR501374-OR501377, OR502913-OR502955, OR508809-OR508833, OQ645436, OQ651273, OQ627438-OQ627452, OR493964-OR494004, OP433466-OP433489, OQ460227-OQ460239.
ZC: Conceptualization, Funding acquisition, Resources, Investigation, Formal analysis, Writing – original draft, Writing – review & editing. XH: Methodology, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. XX: Methodology, Formal analysis. RH: Methodology, Resources, Formal analysis. ET: Resources.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study is supported by grants to ZC from the National Natural Science Foundation of China (NSFC 30873387, 81373905), and Guangdong Provincial Natural Science Foundation (2014A030313321).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1294670/full#supplementary-material
Supplementary Figure 1 | Simple sequence repeats (SSRs) and short dispersed repeats (SDRs) in the six Bupleurum species.
Supplementary Figure 2 | Selective pressure in six Bupleurum species.
Supplementary Figure 3 | Phylogenetic trees constructed with highly variable regions.
Supplementary Figure 4 | Read mapping of Bupleurum wenchuanense (voucher 2020080639 and 1683103) IR regions
Supplementary File 1 | Simple sequence repeats (SSRs) and short dispersed repeats (SDRs) detection, and nonsynonymous (Ka) and synonymous (Ks) substitution rates analysis
Supplementary File 2 | Key to Chinese Bupleurum (modified from Flora of China)
Abdullah, Henriquez, C. L., Croat, T. B., Poczai, P., Ahmed, I. (2021). Mutational dynamics of aroid chloroplast genomes II. Front. Genet. 11. doi: 10.3389/fgene.2020.610838
Abdullah, Mehmood, F., Shahzadi, I., Waseem, S., Mirza, B., Ahmed, I., et al. (2020). Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics 112 (1), 581–591. doi: 10.1016/j.ygeno.2019.04.010
Acosta, M. C., Premoli, A. C. (2010). Evidence of chloroplast capture in South American Nothofagus (subgenus Nothofagus, Nothofagaceae). Mol. Phylogenet. Evol. 54 (1), 235–242. doi: 10.1016/j.ympev.2009.08.008
Ahmed, I., Biggs, P. J., Matthews, P. J., Collins, L. J., Hendy, M. D., Lockhart, P. J. (2012). Mutational dynamics of aroid chloroplast genomes. Genome Biol. Evol. 4 (12), 1316–1323. doi: 10.1093/gbe/evs110
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi: 10.1089/cmb.2012.0021
Chao, Z., Zeng, W. P., Liao, J., Liu, L., Liang, Z. B., Li, X. L. (2014). DNA barcoding Chinese medicinal Bupleurum. Phytomedicine 21 (13), 1767–1773. doi: 10.1016/j.phymed.2014.09.001
Cheng, T., Xu, C., Lei, L., Li, C. H., Zhang, Y., Zhou, S. L. (2015). Barcoding the kingdom Plantae: new PCR primers for ITS regions of plants with improved universality and specificity. Mol. Ecol. Resour. 16 (1), 138–149. doi: 10.1111/1755-0998.12438
Chinese Pharmacopoeia Commission (2020). Pharmacopoeia of the People's Republic of China 2020 edition Vol. 1 (Beijing: China Medical Science and Technology Press), 293.
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17 (1), 134. doi: 10.1186/s13059-016-1004-2
Darling, A. C. E. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14 (7), 1394–1403. doi: 10.1101/gr.2289704
Dong, W. P., Liu, J., Yu, J., Wang, L., Zhou, S. L. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PloS One 7 (4), e35071. doi: 10.1371/journal.pone.0035071
Dong, W. P., Sun, J. H., Liu, Y. L., Xu, C., Wang, Y. H., Suo, Z. L., et al. (2021). Phylogenomic relationships and species identification of the olive genus Olea (Oleaceae). J. Systematics Evol. 60 (6), 1263–1280. doi: 10.1111/jse.12802
Dong, W. P., Xu, C., Li, C. H., Sun, J. H., Zuo, Y. J., Shi, S., et al. (2015). ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5 (1), 8348. doi: 10.1038/srep08348
Fehrer, J., Gemeinholzer, B., Chrtek, J., Bräutigam, S. (2007). Incongruent plastid and nuclear DNA phylogenies reveal ancient intergeneric hybridization in Pilosella hawkweeds (Hieracium, Cichorieae, Asteraceae). Mol. Phylogenet. Evol. 42 (2), 347–361. doi: 10.1016/j.ympev.2006.07.004
Franck, A. R., Cochrane, B. J., Garey, J. R. (2012). Low-copy nuclear primers and ycf1 primers in Cactaceae. Am. J. Bot. 99 (10), e405–e407. doi: 10.3732/ajb.1200128
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32 (suppl_2), W273–W279. doi: 10.1093/nar/gkh458
Galtier, N., Daubin, V. (2008). Dealing with incongruence in phylogenomic analyses. Philos. Trans. R. Soc. B: Biol. Sci. 363 (1512), 4023–4029. doi: 10.1098/rstb.2008.0144
Huang, R., Xie, X. N., Chen, A. M., Li, F., Tian, E. W., Chao, Z. (2021a). The chloroplast genomes of four Bupleurum (Apiaceae) species endemic to Southwestern China, a diversity center of the genus, as well as their evolutionary implications and phylogenetic inferences. BMC Genomics 22 (1), 714. doi: 10.1186/s12864-021-08008-z
Huang, R., Xie, X. N., Li, F., Tian, E. W., Chao, Z. (2021b). Chloroplast genomes of two Mediterranean Bupleurum species and the phylogenetic relationship inferred from combined analysis with East Asian species. Planta 253 (4), 81. doi: 10.1007/s00425-021-03602-7
Javaid, N., Ramzan, M., Khan, I. A., Alahmadi, T. A., Datta, R., Fahad, S., et al. (2022). The chloroplast genome of Farsetia hamiltonii Royle, phylogenetic analysis, and comparative study with other members of Clade C of Brassicaceae. BMC Plant Biol. 22 (1), 384. doi: 10.1186/s12870-022-03750-2
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21 (1), 241. doi: 10.1186/s13059-020-02154-5
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. S., Von Haeseler, A., Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14 (6), 587–589. doi: 10.1038/nmeth.4285
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28 (12), 1647–1649. doi: 10.1093/bioinformatics/bts199
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Laslett, D., Canbäck, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32 (1), 11–16. doi: 10.1093/nar/gkh152
Letunic, I., Bork, P. (2021). Interactive Tree of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49 (W1), W293–W296. doi: 10.1093/nar/gkab301
Li, Y., Sheh, M. L. (1979). “Bupleurum,” in Flora Reipublicae Popularis Sinicae, tomus, vol. 55 . Eds. Shan, R. H., Sheh, M. L. (Beijing: Science Press), 215–295.
Li, J., Xie, D. F., Guo, X. L., Zheng, Z. Y., He, X. J., Zhou, S. D. (2020). Comparative analysis of the complete plastid genome of five Bupleurum species and new insights into DNA barcoding and phylogenetic relationship. Plants 9 (4), 543. doi: 10.3390/plants9040543
Li, F., Xie, X., Huang, R., Tian, E. W., Li, C., Chao, Z. (2021). Chloroplast genome sequencing based on genome skimming for identification of Eriobotryae Folium. BMC Biotechnol. 21, 69. doi: 10.1186/s12896-021-00728-0
Liang, Z. B., Liu, L., Chao, Z. (2012). Investigation of medicinal Bupleurum resources and current situation of Chaihu production. Lishizhen Med. Materia Med. Res. 23 (8), 2011–2013.
Liu, B. B., Campbell, C. S., Hong, D. Y., Wen, J. (2020). Phylogenetic relationships and chloroplast capture in the Amelanchier-Malacomeles-Peraphyllum clade (Maleae, Rosaceae): Evidence from chloroplast genome and nuclear ribosomal DNA data using genome skimming. Mol. Phylogenet. Evol. 147, 106784. doi: 10.1016/j.ympev.2020.106784
Liu, B. B., Ren, C., Kwak, M., Hodel, R. G. J., Xu, C., He, J., et al. (2022). Phylogenomic conflict analyses in the apple genus Malus s.l. reveal widespread hybridization and allopolyploidy driving diversification, with insights into the complex biogeographic history in the Northern Hemisphere. J. Integr. Plant Biol. 64, 1020–1043. doi: 10.1111/jipb.13246
Liu, K., Wang, R., Guo, X. X., Zhang, X. J., Qu, X. J., Fan, S. J. (2021). Comparative and phylogenetic analysis of complete chloroplast genomes in Eragrostideae (Chloridoideae, Poaceae). Plants 10 (1), 109. doi: 10.3390/plants10010109
Liu, X. J., Xu, D. P., Hong, Z., Zhang, N. N., Cui, Z. Y. (2021). Comparative and phylogenetic analysis of the complete chloroplast genome of. Santalum (Santalaceae). Forests 12 (10), 1303. doi: 10.3390/f12101303
Lohse, M., Drechsel, O., Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52 (5–6), 267–274. doi: 10.1007/s00294-007-0161-y
Lowe, T. M., Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44 (W1), W54–W57. doi: 10.1093/nar/gkw413
Ma, X. G., He, X. J. (2015). Bupleurum shanianum sp. nov. (Apiaceae) from China. Nordic J. Bot. 33 (1), 71–73. doi: 10.1111/njb.00520
Ma, L. H., Ning, J. Q., Wang, Y. J., Zhao, M., Li, Y. K., Zhou, H. K. (2022). Comparative genomics on chloroplasts of Sinopodophyllum hexandrum. Chin. J. Biotechnol. 38 (10), 3695–3712. doi: 10.13345/j.cjb.220386
Ma, X. G., Zhao, C., Liang, Q. L., He, X. J. (2013). Bupleurum baimaense (Apiaceae), a new species from Hengduan Mountains, China. Annales Botanici Fennici 50 (6), 379–385. doi: 10.5735/085.050.0601
Menezes, A. P. A., Resende-Moreira, L. C., De Oliveira Buzatti, R. S., Nazareno, A. G., Carlsen, M. H., Lobo, F. S. N., et al. (2018). Chloroplast genomes of Byrsonima species (Malpighiaceae): comparative analysis and screening of high divergence sequences. Sci. Rep. 8 (1), 2210. doi: 10.1038/s41598-018-20189-4
Missouri Botanical Garden (2023) Bupleurum L. in Flora of Parkistan. Available at: http://legacy.tropicos.org/Name/40033266?projectid=32.
Moazzami Farida, S. H., Ghorbani, A., Ajani, Y., Sadr, M., Mozaffarian, V. (2018). Ethnobotanical applications and their correspondence with phylogeny in Apiaceae-Apioideae. Res. J. Pharmacognosy 5 (3), 79–97. doi: 10.22127/rjp.2018.64880
Neubig, K. M., Whitten, W. M., Carlsward, B. S., Mario, A. B., Lorena, E., Norris, H. W., et al. (2009). Phylogenetic utility of ycf1 in orchids: a plastid gene more variable than matK. Plant Systematics Evol. 277 (1-2), 75–84. doi: 10.1007/s00606-008-0105-0
Neves, S. S., Watson, M. F. (2004). Phylogenetic relationships in Bupleurum (Apiaceae) based on nuclear ribosomal DNA ITS sequence data. Ann. Bot. 93, 379–398. doi: 10.1093/aob/mch052
Nguyen, L. H., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Niu, T., Tian, C., Yang, Y., Liu, Q., Liu, L., Tao, Q., et al. (2023). Complete chloroplast genome of corethrodendron fruticosum (Papilionoideae: fabaceae): comparative and phylogenetic analysis. Genes 14 (6), 1289. doi: 10.3390/genes14061289
Pan, S. L., Hsu, P. S. (1992). A new bupleurum (Umbelliferae) from central China. Sida 15 (1), 91–93.
Pan, S. L., Shun, Q. S., Bo, Q. M., Bao, X. S. (2002). The coloured atlas of the medicinal plants from genus Bupleurum in China (Shanghai: Shanghai Scientific and Technological Literature Press).
Pchelin, I. M., Azarov, D. V., Churina, M. A., Scherbak, S. G., Apalko, S. V., Vasilyeva, N. V., et al. (2019). Species boundaries in the Trichophyton mentagrophytes / T. interdigitale species complex. Med. Mycology 57 (6), 781–789. doi: 10.1093/mmy/myy115
Perrino, E. V., Mahmoud, Z. N. A., Valerio, F., Tomaselli, V., Wagensommer, R. P., Trani, A. (2023). Sinecology of Lagoecia cuminoides L. in Italy and evaluation of functional compounds presence in its water or hydroalcoholic extracts. Sci. Rep. 13, in press.
Pimenov, M. G. (2017). Updated checklist of Chinese Umbelliferae: nomenclature, synonymy, typification, distribution. Turczaninowia 20 (2), 106–239. doi: 10.14258/turczaninowia.20.2.9
Plunkett, G. M., Downie, S. R. (2000). Expansion and Contraction of the chloroplast inverted repeat in Apiaceae subfamily Apioideae. Systematic Bot. 25 (4), 648–667. doi: 10.2307/2666726
POWO (2023). Plants of the World Online (Royal Botanic Gardens, Kew: Published on the Internet). Available at: http://www.plantsoftheworldonline.org/.
Qu, X. J., Moore, M. J., Li, D. Z., Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15 (1), 50. doi: 10.1186/s13007-019-0435-7
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biol. 61 (3), 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34 (12), 3299–3302. doi: 10.1093/molbev/msx248
Saina, J. K., Li, Z. Z., Gichira, A. W., Liao, Y. Y. (2018). The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima (Mill.) (Sapindales: simaroubaceae), an important pantropical tree. Int. J. Mol. Sci. 19 (4), 929. doi: 10.3390/ijms19040929
She, M. L., Watson, M. F. (2005). “Bupleurum,” in Flora of China, vol. 14 . Eds. Wu, Z. Y., Raven, P. H., Hong, D. Y. (Beijing: Science Press; St. Louis: Missouri Botanical Garden), 60–74.
Shu, P., Yuan, C. Q., She, M. L., Liu, Y. Z., Xiang, B. R., An, D. K. (1998). Numerical taxonomy of medical Bupleurum species in China. Acta Botanica Boreali-Occidentalia Sin. 18, 277–283.
Soltis, P. S., Soltis, D. E. (2009). The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60, 561–588. doi: 10.1146/annurev.arplant.043008.092039
Song, Y., Zhao, W. J., Xu, J., Li, M. F., Zhang, Y. J. (2022). Chloroplast genome evolution and species identification of Styrax (Styracaceae). BioMed. Res. Int. 2022, 5364094. doi: 10.1155/2022/5364094
Sonigra, P., Meena, M. (2021). Metabolic profile, bioactivities, and variations in the chemical constituents of essential oils of the ferula genus (Apiaceae). Front. Pharmacol. 11. doi: 10.3389/fphar.2020.608649
Steenwyk, J. L., Li, Y., Zhou, X., Shen, X. X., Rokas, A. (2023). Incongruence in the phylogenomics era. Nat. Rev. Genet. 24, 834–850. doi: 10.1038/s41576-023-00620-x
Stull, G. W., Soltis, P. S., Soltis, D. E., Gitzendanner, M. A., Smith, S. A. (2020). Nuclear phylogenomic analyses of asterids conflict with plastome trees and support novel relationships among major lineages. Am. J. Bot. 107 (5), 790–805. doi: 10.1002/ajb2.1468
Sun, H., Zhang, J., Deng, T., Boufford, D. E. (2017). Origins and evolution of plant diversity in the Hengduan Mountains, China. Plant Diversity 39 (4), 161–166. doi: 10.1016/j.pld.2017.09.004
Tan, W., Gao, H., Zhang, H. Y., Yu, X. L., Tian, X. X., Jiang, W. L., et al. (2020). The complete chloroplast genome of Chinese medicine (Psoralea corylifolia): molecular structures, barcoding and phylogenetic analysis. Plant Gene 21, 100216. doi: 10.1016/j.plgene.2019.100216
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45 (W1), W6–W11. doi: 10.1093/nar/gkx391
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., Chaw, S. M. (2008c). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evolutionary Biol. 8 (1), 36. doi: 10.1186/1471-2148-8-36
Wang, H. C., He, Z. R., Wang, Y. H., Sun, H. (2013). Bupleurum dracaenoides (subgenus Bupleurum, Apiaceae): a new shrubby species from southwestern China. Systematic Bot. 38 (4), 1188–1195. doi: 10.1600/036364413X674751
Wang, Q. Z., He, X. J., Zhou, S. D., Wu, Y. K., Yu, Y., Pang, Y. L. (2008a). Phylogenetic inference of the genus Bupleurum (Apiaceae) in Hengduan Mountains based on chromosome counts and nuclear ribosomal DNA ITS sequences. J. Systematics Evol. 46 (2), 142–154. doi: 10.3724/SP.J.1002.2008.07107
Wang, L., Li, F., Wang, N., Gao, Y., Liu, K., Zhang, G., et al. (2022). Characterization of the Dicranostigma leptopodum chloroplast genome and comparative analysis within subfamily Papaveroideae. BMC Genomics 23 (1), 794. doi: 10.1186/s12864-022-09049-8
Wang, C. B., Ma, X. G., He, X. J. (2011). A taxonomic re-assessment in the Chinese Bupleurum (Apiaceae): Insights from morphology, nuclear ribosomal internal transcribed spacer, and chloroplast (trnH-psbA, matK) sequences. J. Systematics Evol. 49 (6), 558–589. doi: 10.1111/j.1759-6831.2011.00157.x
Wang, Q. Z., Zhou, S. D., Liu, T. Y., Pang, Y. L., Wu, Y. K., He, X. J. (2008b). Phylogeny and classification of Chinese Bupleurum based on nuclear ribosomal DNA internal transcribed spacer and rps16. Acta Biologica Cracoviensia Ser. Botanica 50 (2), 105–116.
Wolff, H. (1910). “Bupleurum.,” in Das Pflanzenreich Regnis Vegetabilis Conspectus, IV.228. Ed. Engler, A. (Leipzig: Verlag von Wilhelm Engelmann), 36–173.
Wu, L. W., Nie, L. P., Xu, Z. C., Li, P., Wang, Y., He, C. N., et al. (2020). Comparative and phylogenetic analysis of the complete chloroplast genomes of three Paeonia Section Moutan species (Paeoniaceae). Front. Genet. 11. doi: 10.3389/fgene.2020.00980
Wu, Z. N., Tian, C. Y., Yang, Y. T., Li, Y. H., Liu, Q., Li, Z. Y., et al. (2022). Comparative and phylogenetic analysis of complete chloroplast genomes in. Leymus (Triticodae Poaceae). Genes 13 (8), 1425. doi: 10.3390/genes13081425
Xie, X. N., Huang, R., Li, F., Tian, E. W., Li, C., Chao, Z. (2021). Phylogenetic position of Bupleurum sikangense inferred from the complete chloroplast genome sequence. Gene 798, 145801. doi: 10.1016/j.gene.2021.145801
Xie, H., Huo, K. K., Chao, Z., Pan, S. L. (2009). Identification of crude drugs from Chinese medicinal plants of the genus Bupleurum using ribosomal DNA ITS sequences. Planta Med. 75 (1), 89–93. doi: 10.1055/s-0028-1088334
Yang, R., Folk, R., Zhang, N., Gong, X. (2019). Homoploid hybridization of plants in the Hengduan mountains region. Ecol. Evol. 9 (14), 8399–8410. doi: 10.1002/ece3.5393
Yang, M., Xiao, Y., Ouyang, Z. Y., Jiang, L. M., Hou, P. (2021). Relation of biodiversity and ecosystem multifunctionality in Sichuan Province. Acta Ecologica Sin. 41 (24), 9738–9748. doi: 10.5846/stxb202011112915
Zhang, L. S., Chen, C. L., Mao, X. X., Liu, J. Q. (2023). Phylogenomics and evolutionary diversification of the subfamily Polygonoideae. J. Systematics Evol. 61, 587–598. doi: 10.1111/jse.12913
Keywords: Bupleurum, Western Sichuan, chloroplast genome, nuclear ribosomal DNA, phylogenomic analysis, Bupleurum pseudochaishoui Z. Chao sp. nov.
Citation: Chao Z, Hong X, Xie X, Huang R and Tian E (2023) Phylogenomic analysis of Bupleurum in Western Sichuan, China, including an overlooked new species. Front. Plant Sci. 14:1294670. doi: 10.3389/fpls.2023.1294670
Received: 15 September 2023; Accepted: 08 November 2023;
Published: 27 November 2023.
Edited by:
Robert Philipp Wagensommer, Free University of Bozen-Bolzano, ItalyReviewed by:
Enrico Vito Perrino, International Centre for Advanced Mediterranean Agronomic Studies, ItalyCopyright © 2023 Chao, Hong, Xie, Huang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi Chao, Y2hhb3poaUBzbXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.