 Na Sha1†‡
Na Sha1†‡ Zhiyong Li1†Qiang Sun1‡Ying Han1‡Li Tian1
Zhiyong Li1†Qiang Sun1‡Ying Han1‡Li Tian1 Yantao Wu1‡Xing Li2
Yantao Wu1‡Xing Li2 Yabo Shi3‡
Yabo Shi3‡ Jinghui Zhang1Jiangtao Peng1
Jinghui Zhang1Jiangtao Peng1 Lixin Wang1
Lixin Wang1 Zhenhua Dang1*‡
Zhenhua Dang1*‡ Cunzhu Liang1
Cunzhu Liang1- 1Key Laboratory of Ecology and Resource Use of the Mongolian Plateau, Ministry of Education of China, Collaborative Innovation Center for Grassland Ecological Security, School of Ecology and Environment, Inner Mongolia University, Hohhot, Inner Mongolia, China
- 2Institute of Landscape and Environment, Inner Mongolia Academy of Forestry Science, Hohhot, Inner Mongolia, China
- 3School of Resources and Environment, Baotou Teachers’ College, Baotou, Inner Mongolia, China
Phylogenetic analysis provides crucial insights into the evolutionary relationships and diversification patterns within specific taxonomic groups. In this study, we aimed to identify the phylogenetic relationships and explore the evolutionary history of Stipa using transcriptomic data. Samples of 12 Stipa species were collected from the Qinghai-Tibet Plateau and Mongolian Plateau, where they are widely distributed, and transcriptome sequencing was performed using their fresh spikelet tissues. Using bidirectional best BLAST analysis, we identified two sets of one-to-one orthologous genes shared between Brachypodium distachyon and the 12 Stipa species (9397 and 2300 sequences, respectively), as well as 62 single-copy orthologous genes. Concatenation methods were used to construct a robust phylogenetic tree for Stipa, and molecular dating was used to estimate divergence times. Our results indicated that Stipa originated during the Pliocene. In approximately 0.8 million years, it diverged into two major clades each consisting of native species from the Mongolian Plateau and the Qinghai-Tibet Plateau, respectively. The evolution of Stipa was closely associated with the development of northern grassland landscapes. Important external factors such as global cooling during the Pleistocene, changes in monsoonal circulation, and tectonic movements contributed to the diversification of Stipa. This study provided a highly supported phylogenetic framework for understanding the evolution of the Stipa genus in China and insights into its diversification patterns.
1 Introduction
The Stipa genus consists of temperate, herbaceous plants. This genus originated from the Pooideae subfamily and has evolved into the dominant and key species of needlegrass in the Eurasian grasslands (Lu and Wu, 1996; Nobis et al., 2020; Zhang et al., 2022). In the Chinese context, the Stipa genus encompasses a diverse array of 23 distinct species, including 6 varieties. Various Stipa species, along with their geographic distributions, serve as pivotal criteria in the categorization of grasslands and play a pivotal role in the systematic classification of grassland ecosystems. For instance, within the Mongolian Plateau (MP), the presence of species such as Stipa gobica, S. klemenzii, or S. breviflora distinctly characterizes the desert steppe, while S. krylovii and S. grandis stand as prominent indicators of the typical steppe. And on the Qinghai-Tibet Plateau (QTP), alpine steppes find their primary constitution in the form of S. purpurea, S. basiplumosa, and S. roborowskyi (Zhou, 1980; Guo et al., 1983; Lu and Wu, 1996; Zhang, 2012; Lu et al., 2018; Qiao, 2019; Li et al., 2020; Mao et al., 2021). Therefore, a comprehensive exploration of the origin and evolutionary trajectory of the Stipa genus holds paramount significance in advancing our understanding of Chinese grassland ecology.
The scarcity of fossil evidence initially constrained the investigations on the phylogeny of Stipa species (Wang et al., 1975; Thomasson, 1978; Strömberg, 2005). Such investigations primarily relied on morphological traits (Tzvelev, 1976; Tzvelev, 1989; Jacobs and Everett, 1996). Subsequently, researchers obtained additional relevant molecular data, including nuclear ITS intergenic spacers, intergenic spacers (IGS), and chloroplast gene fragments from this genus (Jacobs et al., 2007; Hamasha et al., 2011; Romaschenko et al., 2012; Krawczyk et al., 2017). Previous studies have indicated that Stipa species in Central Asia form a monophyletic clade. However, the phylogenetic relationships within the Stipa genus remain unresolved (Nobis et al., 2020). Therefore, it is a challenge to comprehend the phylogeny of this genus. Therefore, elucidating the robust phylogenetic relationship within the Stipa genus is of great ecological significance for understanding its origin and evolutionary patterns, and grassland development.
Rapid progress in sequencing technologies has provided significantly increased access to extensive genetic information. In recent years, there has been a notable expansion in research methodologies, including the application of complete chloroplast genomes (Krawczyk et al., 2018; Krawczyk et al., 2022), thousands of Single Nucleotide Polymorphisms (SNPs) genotyping using genome-wide (Baiakhmetov et al., 2021b), and the exploration of nucleolar organizing regions (NORs) in the genomic analysis of Stipa (Baiakhmetov et al., 2021a). These phylogenetic studies are providing more comprehensive insights than earlier research, resulting in new progress regarding the phylogenetic relationships of species within the Stipa genus (Krawczyk et al., 2018; Baiakhmetov et al., 2021b; Krawczyk et al., 2022).
Stipa, as the core taxon within the Stipeae, represents a rigorously monophyletic assemblage, divided into two distinct clades. One clade is exclusive to species inhabiting the Himalayan region, while the other encompasses species distributed across various Eurasian territories (Hamasha et al., 2011). A congruent pattern was identified by Peng (2015) in his research (Peng, 2015); however, it’s worth noting that this pattern doesn’t appear to universally hold across all investigations (Krawczyk et al., 2017; Krawczyk et al., 2018; Baiakhmetov et al., 2021b; Krawczyk et al., 2022). While existing methodologies applied to Stipa phylogeny have proven effective in elucidating taxonomic relationships at the genus level and beyond, their performance diminishes when exploring relationships below the genus level. Notably, in these studies, the support for internal branches within the Stipa phylogenetic tree exhibited certain limitations, with instances of notably weak support. Therefore, there is an imperative need to explore more precise and efficient methods for reconstructing the phylogenetic relationships within the Stipa genus, particularly within its relatively underexplored Chinese distribution range.
Compared with genomic sequencing, transcriptome sequencing efficiently provides significant-high-quality data regarding protein-coding genes, making it particularly suitable for studying nonmodel organisms (Wickett et al., 2014; Zimmer and Wen, 2015; Zeng et al., 2017; Baiakhmetov et al., 2021b). Comparative transcriptomics has emerged as an effective method for evolutionary analyses in plants (Cheng et al., 2018; One Thousand Plant Transcriptomes Initiative, 2019; Kapli et al., 2020) and has significantly promoted the identification of phylogenetic relationships among closely related species, as demonstrated in Cyathophora (Wang et al., 2017), Trigonopedia (Guo et al., 2018), and Daghestanica (Xie et al., 2020). Zhang et al. conducted transcriptome analyses of 157 Pooideae species, identified 1234 orthologous genes, and reconstructed the highly supported maximum likelihood (ML) phylogenetic tree using the concatenation method (Zhang et al., 2022). The phylogenetic tree included three species of Stipa in China, with branches that received high support. This provides valuable insights into reconstructing the phylogenetic relationships within the Stipa genus.
In this study, we have gathered 12 Stipa species, representing five of the seven recognized sections within the Stipa genus found in China. Notably, these encompass the principal and prevailing species inhabiting diverse Stipa grassland ecosystems on the QTP and the MP within Chinese territory. Building upon prior research, which has underscored the relationship between the phylogenetic structure of Stipa and the geographical distribution of its constituent species, the primary objective of this paper is to scrutinize the phylogenetic aspects of these 12 Stipa species in China. Our approach involves the generation of novel transcriptome data, an invaluable resource enabling an in-depth exploration of the evolutionary history underpinning the Stipa in China.
2 Materials and methods
2.1 Sample collection
In total, 12 Stipa species were collected from their respective distribution regions from June to August in 2018 and 2019 (Figure 1 and Table 1). At least one specimen of each species was stored in the herbarium of the College of Ecology and Environment, Inner Mongolia University, and the specimens were identified by Prof. Liang, and voucher specimen numbers were in the Supplementary Data Table S3. Among them, seven species were high-altitude species (3200–4800 m above sea level) native to the QTP, whereas the other five species were distributed in the relatively low-altitude (1100–1500 m) MP (Figure 1 and Table 1). To ensure the RNA integrity of the samples, we specifically selected the spikelets during the heading period of Stipa that were tightly wrapped by bracts and not exposed to air. Five individuals of each species were collected. Their spikelets were immediately placed in liquid nitrogen and stored at −80°C for subsequent RNA extraction.
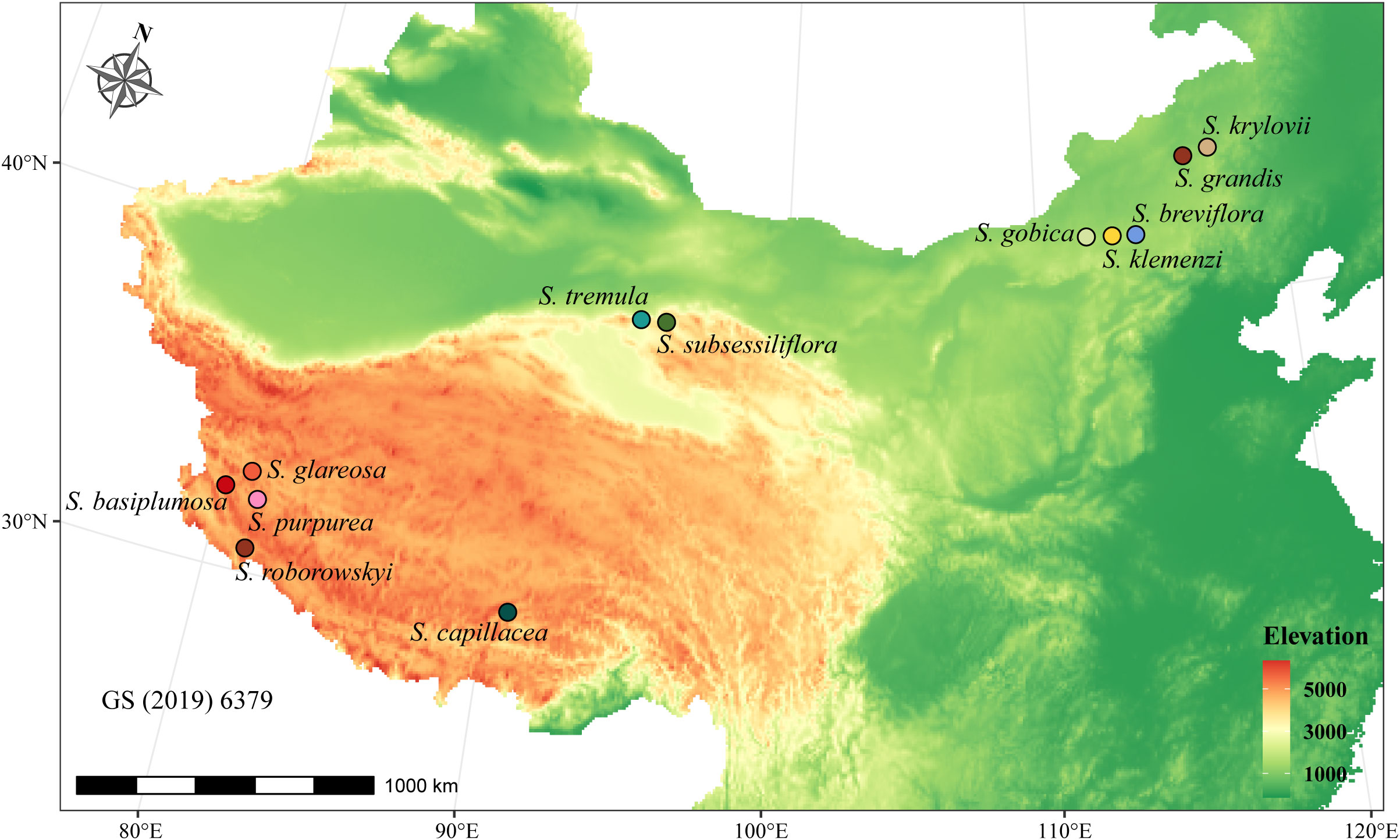
Figure 1 Geographical distribution of sampled Stipa species. Each color-matched circle on the map corresponds to a different Stipa species, with the names of the species listed nearby.
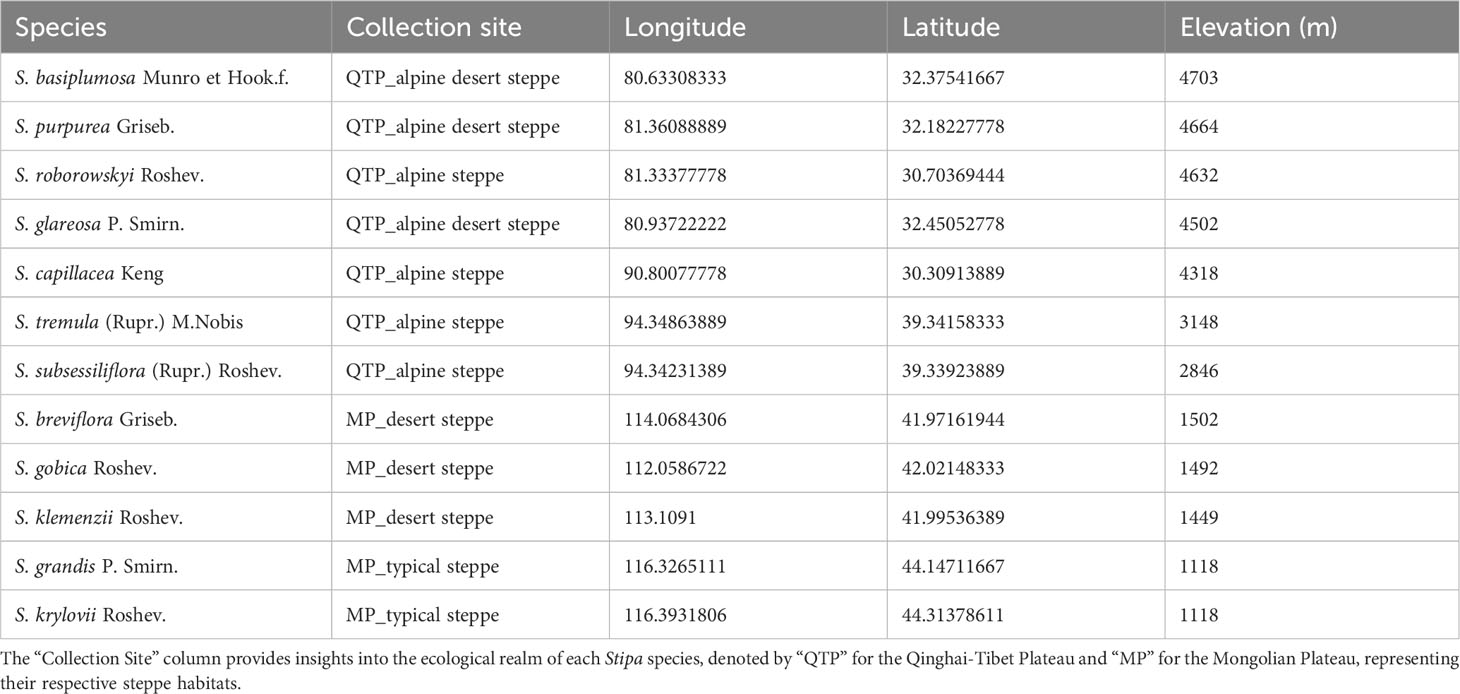
Table 1 Sampling sites of Stipa species.
2.2 RNA extraction and transcriptome sequencing
Total RNA was extracted from each sample using ethanol precipitation and CTAB-PBIOZOL reagent as per the manufacturer’s instructions (Hangzhou Bori Technology, Hangzhou, China). The quality and quantity of the extracted RNA were assessed using a NanoDrop spectrophotometer and an Agilent 2100 Bioanalyzer (Thermo Fisher Scientific, MA, USA), respectively. Three samples with an RNA integrity number ≥ 6.5 from each Stipa species were selected for constructing the cDNA libraries. mRNA was isolated from total RNA using Oligo (dT) beads and fragmented into short fragments using a fragment buffer at an appropriate temperature. The fragmented RNA was used for the synthesis of the first strand of cDNA using random hexamer primers and reverse transcriptase, followed by the synthesis of the second strand using DNA Polymerase I and RNase H. The double-stranded cDNA was purified using QIAquick PCR Extraction Kit (Qiagen) and subjected to end-repair using an A-Tailing Mix and RNA Index Adapters. The final cDNA library was prepared via PCR amplification with AMPure XP beads. The library was sequenced using the BGISEQ-500 platform (BGI, Shenzhen, China) with paired-end reads of 100 bp.
2.3 RNA-seq output quality control and transcriptome de novo assembly
The raw reads of each sample were processed to obtain clean reads by removing reads containing potential adaptors, unknown nucleotides (>5%), and low-quality reads (reads with >20% nucleotides with base quality < 15) using SOAPnuke (version 1.4.0) (Chen et al., 2017) and trichromatic (version 0.36) (Bolger et al., 2014). The Quality 20 (Q20), Quality 30 (Q30), and Guanine-Cytosine (GC) content of the clean reads were recorded. The clean reads from the three samples of each Stipa species were combined and de novo assembled using Trinity (version 2.0.6) (Grabherr et al., 2011) with default parameters. CD-HIT (Fu et al., 2012) was used with a cutoff of 95% similarity for clustering and removing redundant contigs in the assembled transcripts, after which nonredundant transcripts were obtained. The results of the assembly of nonredundant transcripts were evaluated based on the shortest sequence length at 50% of the total sequence length (N50) criteria. Furthermore, TransDecoder (version 5.5.0) was used with default settings to predict coding sequences (CDSs) and protein-coding sequences of the nonredundant transcripts (Kim et al., 2015). Benchmarking Universal Single-Copy Orthologs (BUSCO) (Waterhouse et al., 2017) based on the 4896 poles universal single-copy orthologs (https://busco.ezlab.org/frames/plants.htm), the completeness of the assembly results was assessed. In this analysis, we also wished to detect the number of single-copy orthologs among the 12 Stipa species.
2.4 Orthologous gene identification and gene annotation
The orthologous gene dataset was constructed among the 12 Stipa species by comparing the predicted protein sequences from the nonredundant transcripts with the protein data of Brachypodium distachyon downloaded from the National Center for Biotechnology Information (NCBI) (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/005/505/GCF_000005505.3_Brachypodium_distachyon_v3.0). This was performed using bidirectional best BLAST (Version 2.2.25) with an e-value cutoff of 1e−10, identity > 20%, and bit score > 40 (Altschul et al., 1997). Only one-to-one orthologous gene pairs between one Stipa species and B. distachyon were retained. The intersection of these 12 sets of one-to-one orthologous gene pairs was taken to obtain the final one-to-one orthologous gene set (Supplementary Data Figure S2). To assess the completeness and quality of the assembly, the protein-coding sequences of B. distachyon were used as proxies. BLASTp (with a threshold of 1e−5) was used for gene function annotation against the nonredundant protein database (NR) of NCBI and the Swiss-Prot protein database.
2.5 Phylogenetic analysis and molecular dating
Multiple sequence alignments of the protein sequences were performed using MAFFT (Katoh et al., 2002) and converted to nucleotide alignments using PAL2NAL (Suyama et al., 2006). Poorly aligned positions and divergent regions were eliminated using Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) (Talavera and Castresana, 2007). The filtered nucleotide sequences were translated back to protein sequences. The phylogenetic relationships of the Stipa were reconstructed using the concatenation method and several different tree-building strategies as given in the flowchart in Figure S2. B. distachyon as an outgroup of the Stipa. In the RaxML software, the ML tree was constructed after searching for the best model using the built-in command, with the bootstrap set to 1000 replicates (Stamatakis, 2014). In IQtree, the ML tree was built after searching for the evolutionary models for sequence partitions using ModelFinder, with bootstrap set to 1000 replicates (Nguyen et al., 2015; Kalyaanamoorthy et al., 2017). The Bayesian inference (BI) tree was constructed using the Bayesian method, with the best models for sequence partitions found after 200,000 Markov Chain Monte Carlo (MCMC) iterations until significant convergence was achieved (Ronquist et al., 2012).
To estimate the divergence times of the Stipa genus, MCMCtree from PAML was used. The mean and 95% highest posterior density (HPD) intervals of divergence times for each node were obtained (Yang, 1997). Considering the lack of fossil records of Stipa, we calibrated the divergence times using the divergence time of B. distachyon and Stipa [27–58 million years ago (Mya)] obtained from the TimeTree project (www.timetree.org/) (Kumar et al., 2017). The tree topology was visualized using the ggtree package in R software (Yu et al., 2017).
3 Results
3.1 Transcriptome sequencing and basic bioinformatic analysis
A total of 3807.24 million paired-end clean reads were generated from the sequencing data of 380.71 gigabases (Gb) obtained from the 36 Stipa samples. The average size of clean reads per sample was 10.58 gigabytes (Gb), with average Q20 and Q30 values of 97.75% and 92.00%, respectively (Supplementary Data Table S1).
The assembly process provided nonredundant transcripts for the 12 Stipa species, with an average of 227,312 unigenes (≥200 bp) and an average N50 value of 1622 bp (Table 2 and Supplementary Data Table 1). Furthermore, predicted CDSs were obtained, with an average of 87,406 CDSs and N50 values ranging from 1032 to 1152 bp (Table 1).
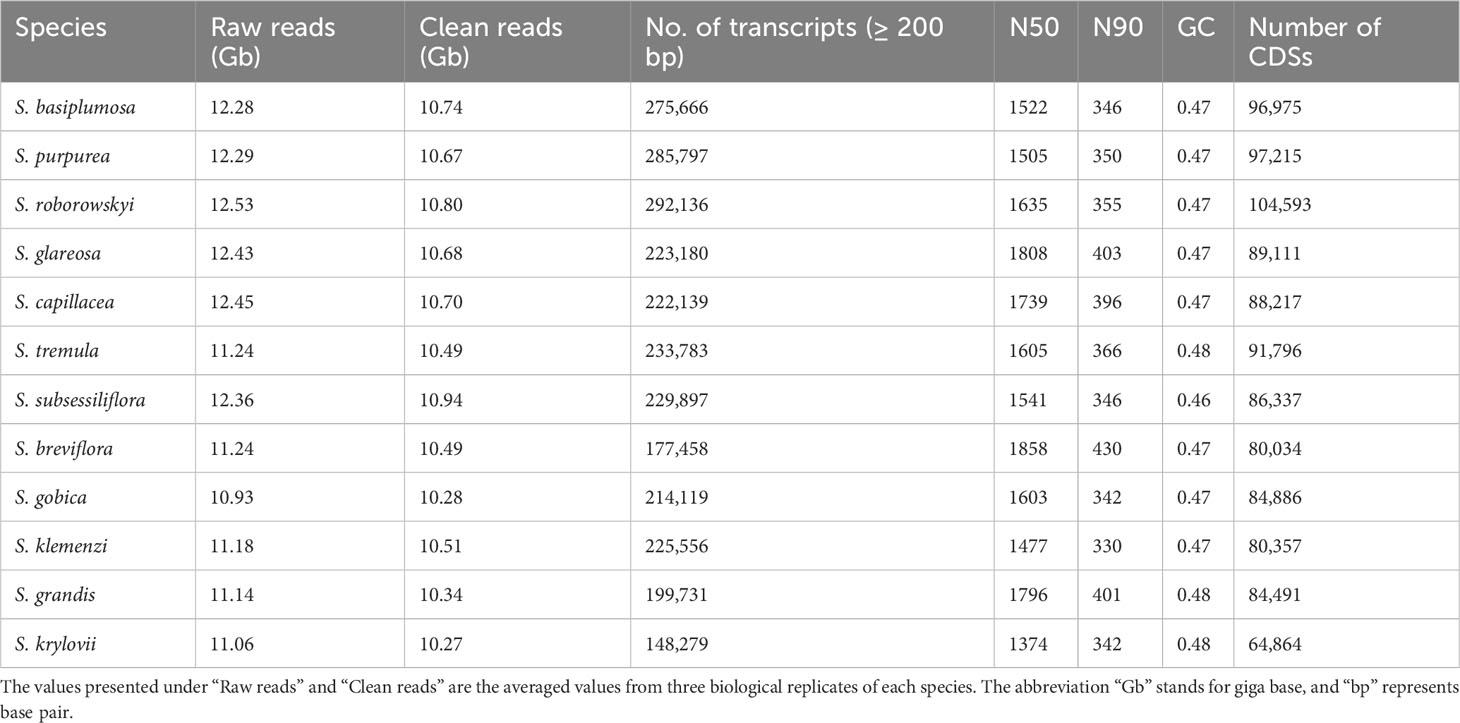
Table 2 De novo assembly of the sequenced transcriptomes.
The BUSCO analysis indicated the completeness of the transcriptome assembly, which was approximately 84% on average across the 12 Stipa species (Supplementary Data Figure S1).
Annotation results using B. distachyon as a reference revealed that 99% of the orthologous genes had hits in the NR database and 75% in the SwissProt database. In total, 73% of the orthologous genes were assigned to Gene Ontology categories, and 48% had matches in the KEGG pathway database (data not presented). These results indicated that the transcriptomes of the Stipa species were well assembled and relatively complete, providing high-quality data for further comparative transcriptome analyses.
3.2 Identification of orthologous genes
Using the bidirectional best BLAST analysis, we identified one-to-one orthologous genes in the protein-coding sequences of B. distachyon and Stipa species, obtaining 10,717 shared genes among the 12 Stipa species. By increasing the BLAST parameter identity to 30%, 9418 genes were retained. Subsequently, these genes were further filtered based on sequence saturation and multiple alignment analysis, to obtain 9397 genes forming the “one2_orthologous” gene set. Further, 2300 orthologous genes with high homology (90%) were extracted. Additionally, from the set of 4896 single-copy orthologous genes retrieved from the BUSCO database, 62 single-copy orthologous genes shared among the 12 species were identified, and this set was designated as the “SCG_orthologous” gene set. Finally, the three gene sets were combined to obtain three supergenes, with lengths 7,637,640, 2,314,182, and 118,167 bp, respectively, for the phylogenetic analysis of the Stipa genus (Supplementary Data Figure S2).
3.3 Phylogenetic analysis
Considering the computational time and resource constraints, we initially adopted various tree-building strategies for the analysis of the SCG_orthologous gene set. First, using the RaxML software with default parameters, the best model was searched. Subsequently, a maximum likelihood (ML) tree was constructed (Supplementary Data Figure S2). In the resulting tree, only one branch had a low bootstrap value (BP = 47%), and others had BP > 90% (SCG_RaxML tree) (Figure 2). Next, we identified the optimal evolutionary model using ModelFinder and constructed an ML tree with IQtree (Edge-linked partition model) (Supplementary Data Figure S2). The results revealed that the BP for the tree topology was all above 80%, with the majority being 100% (SCG_IQtree) (Figure 2). Finally, using the MrBayes Bayesian method, a BI tree was obtained that exhibited high stability, with each branch having a support value of 100% (SCG_BI tree) (Figures S2 and 2). Therefore, the SCG_orthologous supergenes provided consistent topological structures in the evolutionary trees generated by different software and methods, exhibiting high stability (Figure 2).

Figure 2 Phylogenetic trees constructed via varied strategies. The tree’s top name corresponds to the tree in the flowchart in Supplementary Data Figure S2. The Bayesian Inference (BI) tree carries a node support probability (Prob), equivalent to 100% bootstrap values (BP) in the Maximum Likelihood (ML) tree. Species names appear on the right side of the branch tips. The values below the tree represent branch lengths, which have been shortened for the outgroup Brachypodium distachyon for readability.
The phylogenetic tree was constructed using two distinct datasets of one2_orthologous genes, comprising 9397 and 2300 sequences, respectively. Notably, the one2_RaxML tree exclusively consisted of 9397 immediate homologous genes, whereas the one2_IQ tree and one2_BI tree were formed using a partitioning model and Bayesian approach, respectively, with the 2,300 selected genes (Supplementary Data Figure S2). The results revealed remarkable stability and similarity in the topologies of these three trees, exhibiting only minor discrepancies, particularly regarding the relative position of S. basiplumosa. Within both one2_IQtree and one2_BI trees, S. basiplumosa demonstrated a consistent and robust relationship with S. purpurea and S. roborowskyi, supported by a high BP of 89% and a probability of 100%. However, in the one2_RaxML tree, the branch containing S. basiplumosa exhibited a significantly lower self-expansion value and merely 23% BP (Figure 2). Interestingly, the relative position of S. basiplumosa with neighboring species differed between the topology of one2_orthologous and SCG_orthologous (Figure 3).
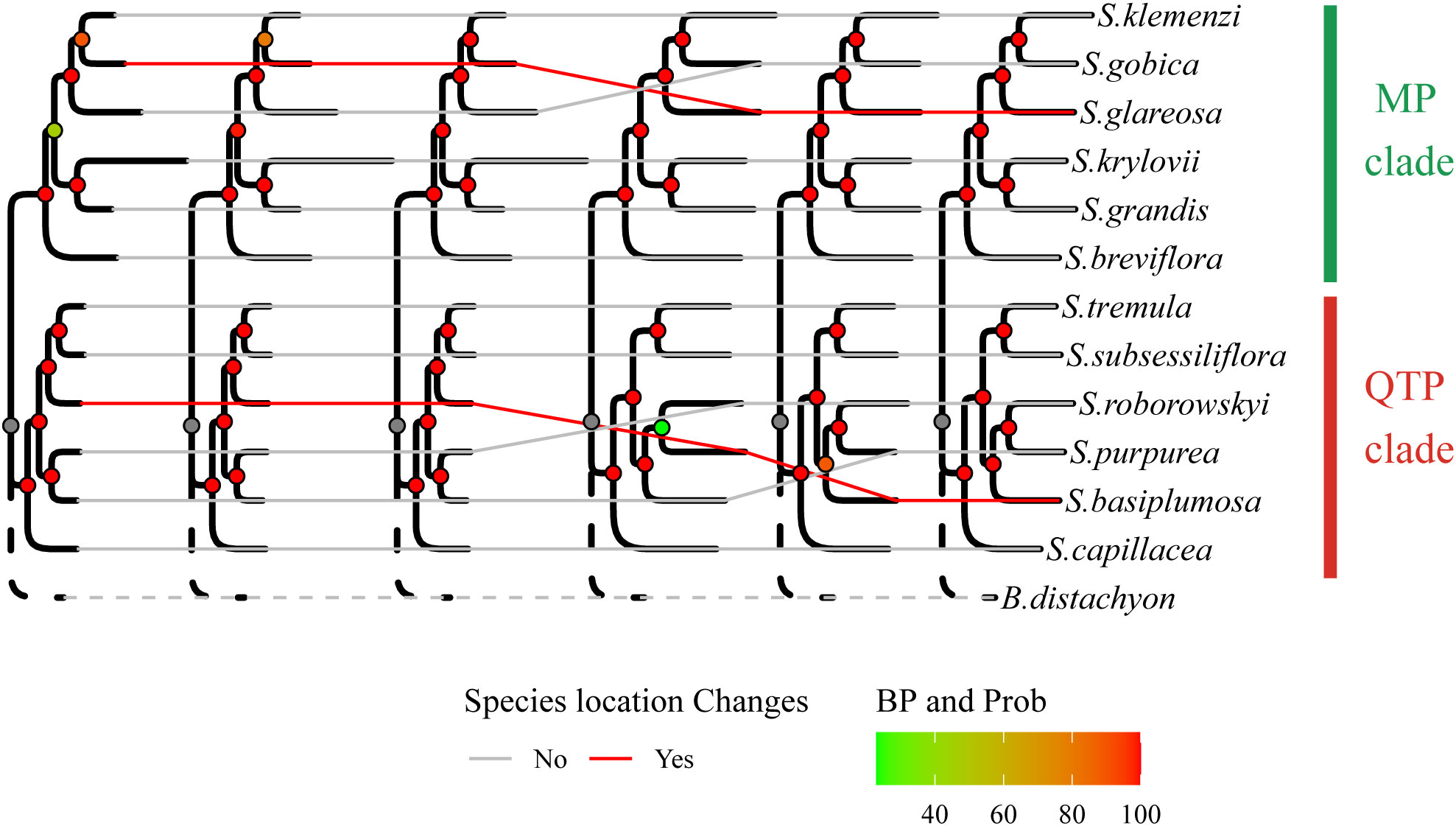
Figure 3 Evolutionary shifts in species placement within the phylogenetic tree. The arrangement of species positions in the depicted phylogenetic tree described illustrates intriguing evolutionary changes. The sequence of the six phylogenetic trees from left to right reflects the arrangement of the six trees displayed in Supplementary Data Figure S2.
Specifically, in SCG_orthologous, S. basiplumosa displayed a closer association with S. tremula and S. subsessiliflora (BP = 100%), whereas in one2_orthologous, it appeared more closely related to S. purpurea and S. roborowskyi (BP = 89%). However, the exclusion of S. basiplumosa did not alter the positioning of these species within the one2_orthologous tree. Notably, the position of S. glareosa remained consistent within the one2_orthologous topology [(S. glareosa, (S. gobica, S. klemenzi)), BP = 100%], although it slightly deviated from the SCG_orthologous tree [(S. gobica, (S. glareosa, S. klemenzi)), BP = 80% or 90%, prob = 100%] (Figures 2 and 3).
In summary, the phylogenetic relationships constructed from the two gene sets were generally consistent and reflected a common feature, that is, the internal divergence of the Stipa genus into two distinct branches (BP = 100%). These two branches were respectively represented by the QTP species, namely, S. capillacea, S. purpurea, S. roborowskyi, S. tremula, and S. subsessiliflora forming the QTP clade (QTP_clade) and by the MP species, namely, S. breviflora, S. gobica, S. klemenzi, S. krylovii, and S. grandis, along with a QTP species S. glareosa, forming the MP clade (MP_clade) (Figure 3).
3.4 Estimation of divergence time
Molecular clock analysis revealed that the divergence between the Stipa genus and B. distachyon occurred approximately 3.962 Mya (95% CI: 2.486–5.672 Mya). Within the Stipa genus, the internal diversification began approximately 2.107 Mya (95% CI: 1.101–3.668 Mya), and the QTP_clade and MP_clade diverged sequentially during 0.45 Mya. The QTP_clade exhibited continuous diversification within the time range of 1.783–1.056 Mya, whereas the MP_clade initiated diversification during the overlapping period of 1.669–0.937 Mya. Eventually, the two clades completed species diversification synchronously within 0.846 Mya (Figure 4).
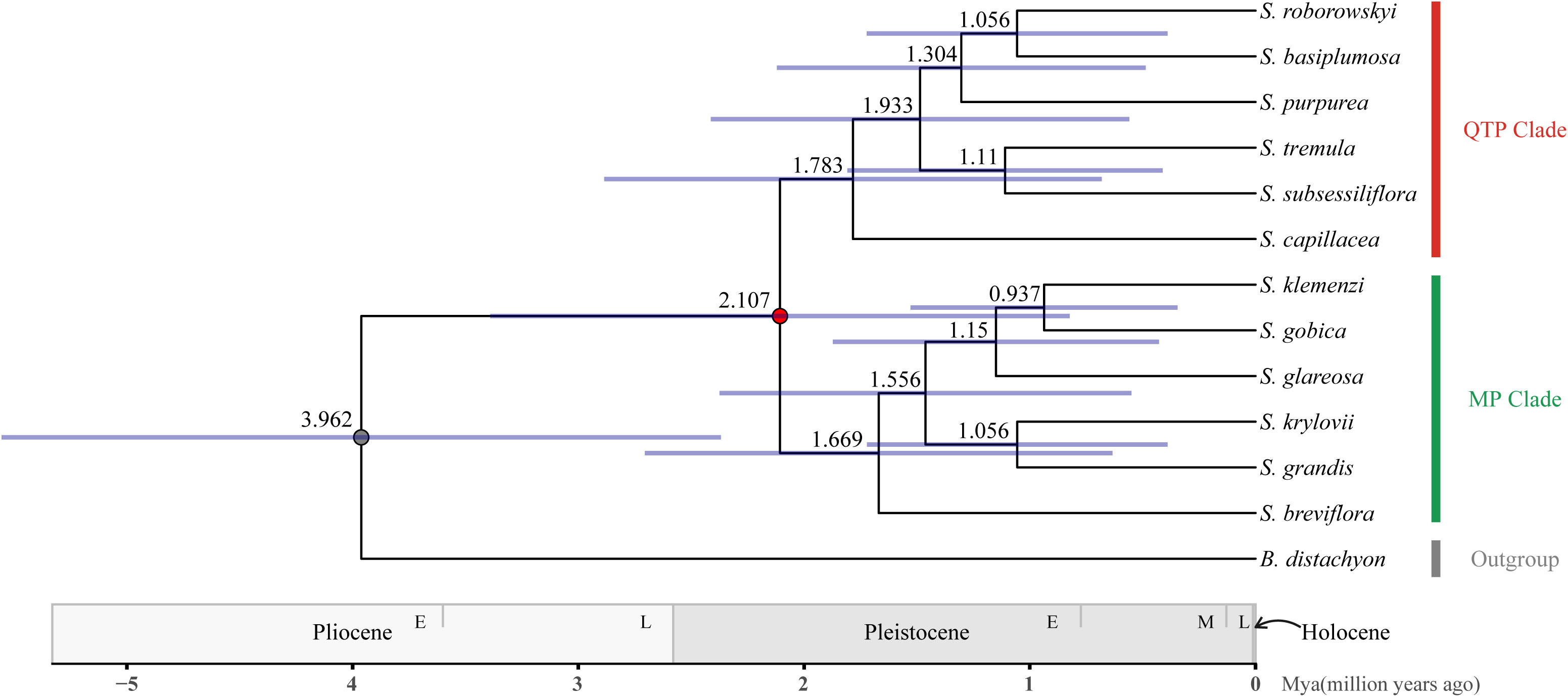
Figure 4 The chronogram of divergence times in the Stipa genus. The average age (above branches) and their 95% confidence intervals (expressed in blue bars) were depicted near various nodes in the Stipa genus phylogeny. Geological timescales are provided below the tree diagram.
In the MP_clade, the desert grassland species S. breviflora diverged first, followed by S. glareosa, S. gobica, and S. klemenzi. Finally, the typical steppe species S. krylovii and S. grandis diverged. In the QTP_clade, the alpine meadow species S. capillacea diverged first, followed by the western QTP desert grassland species S. basiplumosa, S. purpurea, and S. roborowskyi. Lastly, the species located in the northern Qilian Mountains, S. tremula and S. subsessiliflora, diverged.
4 Discussion
4.1 The application of transcriptome in constructing phylogenetic relationships of Stipa
Plastid genes, particularly chloroplast genes, serve as a stable marker system in phylogenetic analysis because of their uniparental inheritance and high conservation (Orton et al., 2021). However, a study on 19 Stipa species revealed that the highly similar chloroplast genome sequences of this genus posed a certain interference in phylogenetic analysis (Krawczyk et al., 2018). On the other hand, small variation information carried by a few nuclear genes limited the extraction of sufficient genetic variations and the obtaining of reliable analytical results (Zimmer and Wen, 2015). A few gene fragments may lead to suboptimal saturation of the constructed phylogenetic trees (Cornwell and Nakagawa, 2017; Zeng et al., 2017). Therefore, in the research of Stipa species, these two are the primary limiting factors for obtaining high-confidence phylogenetic relationships (Romaschenko et al., 2010; Hamasha et al., 2011; Romaschenko et al., 2012; Peng, 2015; Krawczyk et al., 2017; Krawczyk et al., 2018; Krawczyk et al., 2022). Krawczyk’s research identified a region proximal to the 26S nrDNA, specifically within the nuclear rRNA intergenic spacer region (IGS), as one of the most phylogenetically informative areas among Stipa species. Their phylogenetic tree, which encompassed 35 Stipa species, marked the first systematic tree for the Stipa genus that exhibited well-resolved evolutionary branches. However, it’s noteworthy that only 19 branches garnered support levels exceeding 50% (Krawczyk et al., 2017). Similarly, Baiakhmetov constructed phylogenetic relationships for 14 Stipa species using nucleolar organizing regions (NORs), and their findings also indicated relatively low support in the resulting tree (Baiakhmetov et al., 2021b). It appears that there is a paradox in Stipa genus phylogenetic research. When researchers aim to enhance the clarity of branches in the phylogenetic tree, as discussed above, they often encounter challenges in achieving higher support levels.
Consequently, Krawczyk et al. suggested the incorporation of more nuclear genes in the phylogenetic analysis of the Stipa genus (Krawczyk et al., 2018). Later, Baiakhmetov employed a genome-wide approach to obtain thousands of SNP genotypes for a phylogenetic analysis of a population comprising five Stipa species. The results indicated that fewer than half of the branches could be supported with a value of ≥0.8 (Baiakhmetov et al., 2021b). In our study, the high-confidence phylogenetic trees of Stipa were obtained from dozens and thousands of protein-coding genes, with almost 100% support for all branches (Figure 2). This complements the low resolution of the previous phylogenetic tree of Stipa.
This was possible because of the rapid development of sequencing technology. Rapid analysis of numerous nuclear genes through transcriptome sequencing has been widely used in reconstructing phylogenetic relationships in plants (One Thousand Plant Transcriptomes Initiative, 2019), providing good results across green plant groups and higher taxonomic units (Zeng et al., 2014; Guo et al., 2018; Zhang et al., 2022). Nevertheless, when using a large amount of gene data for phylogenetic analysis, the isolation of gene sets and tree-building strategies can influence the final results (Blair and Murphy, 2011; Zeng et al., 2014; Cornwell and Nakagawa, 2017; Zeng et al., 2017). In this study, transcriptome sequencing, assembly, and screening were conducted on 12 Stipa species, the majority of which are dominant or key species of Stipa on the grasslands found in the QTP and MP. Through meticulous analysis of orthologous genes within the transcriptome data, two sets containing high-quality orthologous genes (2300 and 9397, respectively) were obtained. These genes were subjected to phylogenetic analysis using various tree-building strategies (Supplementary Data Figure S2). The results indicated that the orthologous genes of Stipa species exhibited low sensitivity to different tree-building methods (Figure 2), thus potentially providing stable phylogenetic relationships.
4.2 Identification of two geographically related clades within the Stipa genus
Although the lack of data is limiting the phylogenetic studies on Stipa, we can still obtain some insights based on available studies. Hamasha et al. (2011) conducted a phylogenetic analysis of the Stipeae tribe using ITS and ribosomal genes and reported that several Stipa species from the QTP, including S. basiplumosa, S. rohmooiana, S. subsessiliflora, and S. purpurea, formed a distinct branch within the Stipa genus. Another larger branch of the Stipa genus consisted of S. breviflora, S. baicalensis, S. gobica, and other species from the Tianshan Mountains, MP, and other regions (Hamasha et al., 2011). Peng (2015) constructed a phylogenetic tree of Chinese Stipa species using ITS and chloroplast genes and reported that the Stipa genus primarily differentiated into two clades. One clade consisted of Stipa species from the Sect. Pseudoptilagrostis Tzvel. and some species of the Sect. Barbatae Junge was primarily distributed in the QTP, whereas the other clades included Stipa species from the Sect. Leiostipa Dum, Sect. Stipa, Sect. Smirnovia Tzvel, and the remaining species of the Sect. Barbatae Junge (Peng, 2015). To maintain consistency with the previous discussions, we referred to these two clades as the “QTP_clade” and “MP_clade”, representing the major distribution areas of Stipa species in the QTP and MP, respectively. In the aforementioned studies, the nodes supporting the separation of the two clades had relatively high BP, which was consistent with our finding that the BP for the QTP and MP clades was 100% (Figure 2). However, some differences in the internal relationships within each clade were observed in our previous studies. In a study by Hamasha, S. capillacea from the QTP were clustered within the non-QTP branch. In a study by Peng, S. capillacea was assigned to the QTP clade, whereas S. purpurea was classified within the MP clades. In our study, these species were clustered within the QTP clades (Figures S2 and 2).
Furthermore, within the taxonomic realm of the Sect. Smirnovia Tzvel. [comprising species such as S. glareosa, S. gobica, and S. klemenzii, primarily inhabiting the desert steppe of the MP (BP > 89%)] and the Sect. Leiostipa Dum. [comprising S. krylovii and S. grandis (BP = 100%), dominantly found in the typical steppe of Mongolia], we observed stable clustering that adheres to traditional taxonomic classifications based on distinctive traits (Nobis et al., 2020). However, S. breviflora, a member of Sect. Barbatae Junge, typically does not cluster within this section but is placed in non-QTP branches (Peng, 2015; Nobis et al., 2020; Baiakhmetov et al., 2021b; Krawczyk et al., 2022). Similarly, other members of Sect. Barbatae Junge, including S. tremula, S. roborowskyi, and S. purpurea, were grouped based on morphological characteristics (Hamasha et al., 2011; Peng, 2015), then clustered with Sect. Pseudoptilagrostis Tzvel., containing S. basiplumosa and S. subsessiliflora, forming a coherent clade on the QTP (Figures 2 and 3). These findings are consistent with a previous study (Hamasha et al., 2011; Peng, 2015), highlighting the importance of our results in providing highly supported and reliable data for resolving the developmental relationships among Stipa species.
In the context of phylogenetic analysis based on morphological traits, members of the Barbatae section typically manifest as small branches comprising multiple taxa. These branches subsequently intertwine within the Pseudoptilagrostis, Smirnovia, and Leiostipa sections (Nobis et al., 2020). In contrast, the members of Smirnovia and Leiostipa sections often form distinct and stable clusters (Romaschenko et al., 2012; Baiakhmetov et al., 2021b). This observation implies that taxonomic traits from different sections contribute variably to the phylogenetic signal. It also corroborates earlier findings, suggesting that many crucial classification traits within Stipa lack robust phylogenetic signals (Romaschenko et al., 2012).
4.3 Geological and climate changes may drive species diversification in the Stipa genus
The phytolith data from the Eocene-Miocene period in the North American interior provides evidence that the Stipeae appeared in the early Oligocene (34 Mya) and transitioned from closed habitats to open environments. However, it was not until the Late Oligocene or Early Miocene (7-11 million years ago) that the Stipeae became ecologically dominant in North America alongside other grasses (Strömberg, 2005). Molecular evidence suggests that approximately 30 Mya, the Stipeae underwent diversification into two major clades (New World and Old World) and experienced a period of stable temperatures (Romaschenko et al., 2010; Romaschenko et al., 2012; Zhang et al., 2022). During the middle Miocene (approximately 14 Mya), a rapid cooling occurred, leading to the differentiation of several genera including Stipa (Zhang et al., 2022). Nevertheless, our findings indicate that the estimated time of differentiation between Brachypodium distachyon and Stipa was approximately 3.962 Mya (95% CI: 2.486-5.672 Mya) (Figure 4). This value notably deviates from this fossil-based estimation.
We hypothesize that the considerable disparity in our results could be attributed to the relatively broad calibration time frame we employed. Specifically, we utilized a calibration range of 27 to 58 Mya for the separation of the Stipeae tribe and the Brachypodium distachyon, aligning it with the divergence point between the outgroup and Stipa. In comparison to the methodologies of Baiakhmetov et al. (2021b) and Zhang et al. (2022), which encompassed a more diverse array of calibration points and strategies, these studies achieved a higher degree of accuracy. In the investigation conducted by Baiakhmetov, a dual dataset approach was utilized for calibration (Baiakhmetov et al., 2021b). One dataset indicated a divergence between Brachypodium and Oryza approximately 38 to 48 Mya, while the other dataset estimated the emergence of Stipa at 33 to 39 Mya. Their results suggest that B. distachyon and Stipa diverged around 32.77 Mya. In another comprehensive study utilized a multifaceted approach involving three calibration strategies and a multitude of calibration points, encompassing various species within the Pooideae (Zhang et al., 2022). Their results suggested the appearance of Stipeae around 30 Mya, with Stipa emerging approximately 12.7 Mya. These studies, which utilized a more diverse set of calibration points and strategies, achieved higher precision and demonstrated closer alignment with empirical data. Hence, it becomes clear that the inclusion of multiple, highly accurate calibration points and strategies is imperative for attaining precise estimates of divergence times in our research endeavors.
Currently, molecular clock analyses estimate the origin of the Stipa genus as the middle Miocene to early Pliocene (Baiakhmetov et al., 2021a; Baiakhmetov et al., 2021b; Zhang et al., 2022). Our results show that Stipa emerged and began to differentiate at 2.107 Mya (95% CI: 1.101-3.668; Figure 4), which is slightly later than the current results. The discrepancy between the estimated molecular clock origin time and the earliest fossil record of Stipa from the North American interior (23 Mya, Elias, 1942; Thomasson, 1978; Thomasson, 1985; Strömberg, 2005) may be due to several factors. One possibility is the limited number of Stipa species used in both previous studies and our own. Another factor could be the extinction of ancient Stipa species, leading to a delay in the molecular clock estimation of the origin compared to the fossil record. It’s important to note that the North American concept of Stipa encompasses a broader spectrum compared to the Eurasian concept (Hamasha et al., 2011; Romaschenko et al., 2012; Nobis et al., 2020), including numerous species with substantial morphological differences from their Eurasian counterparts (Romaschenko et al., 2012). Therefore, the fossil records from North America may not provide a comprehensive representation of Eurasian Stipa.
Currently, the fossil records in Eurasian geological strata still lack substantial evidence regarding the presence of Stipa. However, according to pollen records of herbaceous plants in Chinese geological strata, there appears to be a gradual increase in the abundance of herbaceous plants, possibly including Stipa, during the early Pliocene to Pleistocene period (Jiang and Ding, 2009; Deng et al., 2019; Spicer et al., 2020). This observation aligns reasonably well with the patterns we have observed in our research.
Our study suggested that the Stipa genus underwent rapid diversification from the early Pliocene to the Pleistocene (3.962–0.937 Mya; Figure 4). This finding is consistent with previous studies based on morphological traits (Tzvelev, 1976; Tzvelev, 1989; Linder et al., 2018) and pollen records of herbaceous plants (Jiang and Ding, 2009; Zhang, 2012; Deng et al., 2019). The divergence time of Stipa (4.39-0.16 Mya) obtained by Baiakhmetob through nucleolar organizing regions (NORs) was also extremely similar, although it received weak support from the phylogenetic tree (Baiakhmetov et al., 2021b). The notable features of the Pliocene include a rapid global temperature decline, extensive expansion of the Arctic ice cap (Goldner et al., 2014), the final significant uplift of the QTP (Spicer et al., 2020), and glacial–interglacial oscillations in the Pleistocene (Liu et al., 2001). Although the uplift history of the QTP is still debated (Wang et al., 2014; Spicer et al., 2020), its uplift has been confirmed to intensify the Asian monsoon system and inland aridification, promoting biodiversity in the region (Dupont-Nivet et al., 2007; Favre et al., 2015; Sun et al., 2017; Li et al., 2021; Mao et al., 2021; Wu et al., 2022). The Pleistocene climatic oscillations created conducive circumstances for the proliferation of species specialized for cold habitats, resulting in the emergence of numerous endemic vicariants in Central Asia and the western Pamir-Alai Mountains, which serve as significant centers for species diversification. Specifically, Stipa gracilis and S. zeravshanica exemplify this phenomenon (Vintsek et al., 2022). These species exhibit distinct characteristics that align with the “interglacial refugia model,” wherein populations undergo expansions during glacial periods followed by abrupt demographic contractions during interglacial periods (Bennett and Provan, 2008). The spikelet traits of the Stipa genus may be associated with adaptation to arid and monsoonal environments. For instance, the dorsal awn of the spikelet can aid plants in attaching to animals and enhancing seed burial in the soil and can facilitate long-distance dispersal aided by monsoonal systems; moreover, a fur-like hairy ovary indumentum can protect the ovary and increase drought resistance (Tzvelev, 1989; Strömberg, 2011; Linder et al., 2018).
During the early Pleistocene (2.107 Mya), the Stipa genus diverged into two clades, consisting of native species from the QTP and MP, respectively, and completed the speciation process during the middle Pleistocene (1.783–0.937 Mya, Figure 4). This divergence could be attributed to the geographical isolation caused by the uplift of the Qilian Mountains in northern QTP (Rowley and Currie, 2006; Spicer et al., 2020; Wu et al., 2022). Additionally, the glacial–interglacial cycles in the Pleistocene further intensified aridity in the northwestern inland regions of China (An et al., 2006), range shifts of many taxa including Stipa (Vintsek et al., 2022), leading to adaptive differentiation within the two clades and gradual accumulation of genetic distance. During glaciations in the Pleistocene, a unified, extensive ice cap covering all land surfaces did not form in China, and glaciation only occurred on mountain peaks. This made the central QTP and MP a place for the development of numerous Central Asian flora and fauna (Mao et al., 2021). The evolutionary complexity and diverse habitats of the QTP and MP provided multiple ecological niches for the survival and proliferation of Stipa plants (Guo et al., 1983; Liu et al., 2014; Qiao, 2019). Many researchers believe that cooling during the Pliocene significantly promoted the origin and expansion of temperate biotic communities (particularly for the subfamily Pooideae), which benefitted from key morphological innovations and genome duplication events in a spikelet (Sandve et al., 2011; Vigeland et al., 2013; McKain et al., 2016; Linder et al., 2018). Whole-genome duplication events have been observed exclusively in the New World clade of Stipeae, specifically in genera such as Austrostipa (Tkach et al., 2021), while no such events have been detected in Old World Stipa genera (Zhang et al., 2022). Stipa may have acquired allopolyploidy through hybridization with distantly related diploid species (Tzvelev, 1989; Krawczyk et al., 2018; Nobis et al., 2019; Baiakhmetov et al., 2020; Baiakhmetov et al., 2021a; Baiakhmetov et al., 2021b; Nobis et al., 2022), thereby obtaining and retaining beneficial genes related to stress response or reproductive development.
4.4 Evolution of the Stipa genus has always been accompanied by the development of grasslands
Since the Middle Miocene, the evolution of herbaceous plants has outpaced that of woody plants due to the habitat provided by mountainous movements and cooling climate, making the western region of China a center for the development of herbaceous plants. From the Middle Miocene onward, the number and variety of herbaceous plants significantly increased, reaching the peak during the Pliocene. During this period, grasslands or grassy areas expanded southeastward, and a modern grassland distribution pattern emerged in China during the Early Pliocene, extending from northwest to southeast (approximately 14–6 Mya) (Jiang and Ding, 2009; Strömberg, 2011; Lu et al., 2018). Stipa began to diversify at a very rapid rate since its emergence in the Pliocene (Figure 4). Various plants from grassland, including those from Stipa, as well as Asteraceae, Liliaceae, Leguminosae, and Poaceae, began to develop. This led to the formation of Stipa steppes dominated by Stipa species in the late Pliocene, which were widely distributed in the QTP and MP regions (Wang, 1997; Li, 1999; Strömberg, 2011; Li et al., 2015; Deng et al., 2019).
5 Conclusion
Using various transcriptomic datasets, we evaluated and compared various phylogenetic analysis methods to reconstruct the high-resolution phylogenetic relationships within the Stipa genus. This study provided important clues to the phylogeny and evolutionary history of Stipa. The results revealed that Stipa, as an early classification unit within the Stipeae tribe, originated from the Middle Miocene to the Early Pliocene. During the Early Pleistocene, it diversified into two clades in China, namely, the QTP clade and the MP clade, consisting of native Stipa species distributed on the QTP and MPs, respectively. It underwent rapid parallel diversification within the Pleistocene. Geological events and climate changes during this period likely acted as external driving factors for the diversification of Stipa. Additionally, the characteristic awned inflorescence of Stipa and hybridization events may have provided an intrinsic genetic basis for its adaptive evolution. The evolution of Stipa was accompanied by the development of grassland landscapes, and Stipa species gradually evolved into the dominant and widely distributed species on grasslands.
Data availability statement
The data presented in the study are deposited in the NCBI repository, with BioProject numbers PRJNA1014579, PRJNA1014757, PRJNA1014596, PRJNA1014658, PRJNA1014760, PRJNA1014801, PRJNA1014811, PRJNA1014756, PRJNA1014755, PRJNA1014613, PRJNA1014758, PRJNA1014800, with specific details provided in Supplementary Table S2.
Author contributions
NS: Data curation, Formal Analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. ZL: Formal Analysis, Funding acquisition, Investigation, Project administration, Supervision, Writing – review & editing. QS: Investigation, Writing – review & editing. YH: Investigation, Writing – review & editing. LT: Investigation, Writing – review & editing. YW: Investigation, Writing – review & editing. XL: Investigation, Writing – review & editing. YS: Investigation, Writing – review & editing. JZ: Investigation, Writing – review & editing. JP: Investigation, Writing – review & editing. LW: Investigation, Writing – review & editing. ZD: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing. CL: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Science and Technology Program of Inner Mongolia Autonomous Region of China (2021GG0307, 2020GG0126), the Science and Technology Major Project of Inner Mongolia Autonomous Region of China (2021ZD0011-1), the National Natural Science Foundation of China (32160088), and the Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region of China (NJYT22093).
Acknowledgments
We thank mjeditor (www.mjeditor.com) for editing the English text of a draft of this manuscript. We also appreciate the valuable insights and suggestions from the peer reviewers during the review process. Special thanks are due to Dr. Marcin Nobis for his thorough and constructive feedback, which significantly enhanced the quality of this paper. We are sincerely thankful for your support and expertise.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1275018/full#supplementary-material
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 (17), 3389–3402. doi: 10.1093/nar/25.17.3389
An, Z. S., Zhang, P. Z., Wang, E. C., Wang, S. M., Qiang, X. K., Li, L., et al. (2006). Changes of the monsoon-arid environment in China and growth of the Tibetan plateau since the miocene. Quaternary Sci. 26 (5), 678–693.
Baiakhmetov, E., Guyomar, C., Shelest, E., Nobis, M., Gudkova, P. D. (2021b). The first draft genome of feather grasses using SMRT sequencing and its implications in molecular studies of Stipa. Sci. Rep. 11 (1), 15345. doi: 10.1038/s41598-021-94068-w
Baiakhmetov, E., Nowak, A., Gudkova, P. D., Nobis, M. (2020). Morphological and genome-wide evidence for natural hybridisation within the genus Stipa (Poaceae). Sci. Rep. 10 (1), 13803. doi: 10.1038/s41598-020-70582-1
Baiakhmetov, E., Ryzhakova, D., Gudkova, P. D., Nobis, M. (2021a). Evidence for extensive hybridisation and past introgression events in feather grasses using genome-wide SNP genotyping. BMC Plant Biol. 21 (1), 505. doi: 10.1186/s12870-021-03287-w
Bennett, K., Provan, J. (2008). What do we mean by ‘refugia’? Quat. Sci. Rev. 27 (27-28), 2449–2455. doi: 10.1016/j.quascirev.2008.08.019
Blair, C., Murphy, R. W. (2011). Recent trends in molecular phylogenetic analysis: Where to next? J. Hered. 102 (1), 130–138. doi: 10.1093/jhered/esq092
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Chen, Y. X., Chen, Y. S., Shi, C. M., Huang, Z. B., Zhang, Y., Li, S. K., et al. (2017). SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7 (1), 1–6. doi: 10.1093/gigascience/gix120
Cheng, S. F., Melkonian, M., Smith, S. A., Brockington, S., Archibald, J. M., Delaux, P. M., et al. (2018). 10KP: A phylodiverse genome sequencing plan. Gigascience 7 (3), 1–9. doi: 10.1093/gigascience/giy013
Cornwell, W., Nakagawa, S. (2017). Phylogenetic comparative methods. Curr. Biol. 27 (9), R333–R336. doi: 10.1016/j.cub.2017.03.049
Deng, T., Wu, F. X., Wang, S. Q., Su, T., Zhou, Z. K. (2019). Significant shift in the terrestrial ecosystem at the Paleogene/Neogene boundary in the Tibetan Plateau. Chin. Sci. Bull. 64 (27), 2894–2906. doi: 10.1360/TB-2019-0053
Dupont-Nivet, G., Krijgsman, W., Langereis, C. G., Ables, H. A., Dai, S., Fang, X. M. (2007). Tibetan plateau aridification linked to global cooling at the Eocene–Oligocene transition. Nature 445 (7128), 635–638. doi: 10.1038/nature05516
Elias, M. K. (1942). “Tertiary prairie grasses and other herbs from the high plains,” in Tertiary prairie grasses and other herbs from the high plains. Ed. Elias, M. K. (Boulder, Colorado: Geological Society of America).
Favre, A., Päckert, M., Pauls, S. U., Jähnig, S. C., Uhl, D., Michalak, I., et al. (2015). The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol. Rev. Camb. Philos. Soc 90 (1), 236–253. doi: 10.1111/brv.12107
Fu, L. M., Niu, B. F., Zhu, Z. W., Wu, S. T., Li, W. Z. (2012). CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28 (23), 3150–3152. doi: 10.1093/bioinformatics/bts565
Goldner, A., Herold, N., Huber, M. (2014). Antarctic glaciation caused ocean circulation changes at the Eocene–Oligocene transition. Nature 511 (7511), 574–577. doi: 10.1038/nature13597
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29 (7), 644–652. doi: 10.1038/nbt.1883
Guo, B. Z., Wang, S. J., Lu, S. L., Sun, Y. H. (1983). A preliminary study on the classification, distribution and ecological nature of cenus Stipa L. @ of China. Acta Botanica Boreali-Occidentalia Sin. 1), 20–29.
Guo, Y. Y., Zhang, Y. Q., Zhang, G. Q., Huang, L. Q., Liu, Z. J. (2018). Comparative transcriptomics provides insight into the molecular basis of species diversification of section Trigonopedia (Cypripedium) on the Qinghai-Tibetan Plateau. Sci. Rep. 8 (1), 11640. doi: 10.1038/s41598-018-30147-9
Hamasha, H. R., von Hagen, K. B., Röser, M. (2011). Stipa (Poaceae) and allies in the Old World: molecular phylogenetics realigns genus circumscription and gives evidence on the origin of American and Australian lineages. Plant Syst. Evol. 298 (2), 351–367. doi: 10.1007/s00606-011-0549-5
Jacobs, S., Bayer, R., Everett, J., Arriaga, M., Barkworth, M., Sabin-Badereau, A., et al. (2007). Systematics of the tribe Stipeae (Gramineae) using molecular data. Aliso: A J. Systematic Floristic Bot. 23 (1), 349–361. doi: 10.5642/aliso.20072301.28
Jacobs, S., Everett, J. (1996). Austrostipa, a new genus, and new names for the Australasian species formerly included in Stipa (Gramineae). Telopea 6 (4), 579–595. doi: 10.7751/telopea19963026
Jiang, H., Ding, Z. (2009). Spatial and temporal characteristics of Neogene palynoflora in China and its implication for the spread of steppe vegetation. J. Arid Environ. 73 (9), 765–772. doi: 10.1016/j.jaridenv.2009.03.011
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., Jermin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14 (6), 587–589. doi: 10.1038/nmeth.4285
Kapli, P., Yang, Z. H., Telford, M. J. (2020). Phylogenetic tree building in the genomic age. Nat. Rev. Genet. 21 (7), 428–444. doi: 10.1038/s41576-020-0233-0
Katoh, K., Misawa, K., Kuma, K., Miyata, T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30 (14), 3059–3066. doi: 10.1093/nar/gkf436
Kim, H. S., Lee, B. Y., Won, E. J., Han, J., Hwang, D. S., Park, H. G., et al. (2015). Identification of xenobiotic biodegradation and metabolism-related genes in the copepod Tigriopus japonicus whole transcriptome analysis. Mar. Genomics 24 Pt 3, 207–208. doi: 10.1016/j.margen.2015.05.011
Krawczyk, K., Myszczyński, K., Nobis, M., Sawicki, J. (2022). Insights into adaptive evolution of plastomes in Stipa L. (Poaceae). BMC Plant Biol. 22 (1), 525. doi: 10.1186/s12870-022-03923-z
Krawczyk, K., Nobis, M., Myszczynski, K., Klichowska, E., Sawicki, J. (2018). Plastid super-barcodes as a tool for species discrimination in feather grasses (Poaceae: Stipa). Sci. Rep. 8 (1), 1924. doi: 10.1038/s41598-018-20399-w
Krawczyk, K., Nobis, M., Nowak, A., Szczecińska, M., Sawicki, J. (2017). Phylogenetic implications of nuclear rRNA IGS variation in Stipa L. (Poaceae). Sci. Rep. 7 (1), 11506. doi: 10.1038/s41598-017-11804-x
Kumar, S., Stecher, G., Suleski, M., Hedges, S. B. (2017). TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34 (7), 1812–1819. doi: 10.1093/molbev/msx116
Li, J. (1999). Studies on the geomorphological evolution of the Qinghai- Xizang (Tibetan) plateau and Asian monsoon. Mar. Geol. Quaternary Geol. 19 (1), 1–12.
Li, J. J., Zhou, S. Z., Zhao, Z. J., Zhang, J. (2015). The Qingzang movement: The major uplift of the Qinghai-Tibetan Plateau. Sci. China Earth Sci. 58 (11), 2113–2122. doi: 10.1007/s11430-015-5124-4
Li, L. H., Chen, J. Q., Han, X. G., Zhang, W. H., Shao, C. L. (2020). Grassland ecosystems of China: A synthesis and resume (Singapore: Springer Singapore).
Li, S. F., Valdes, P. J., Farnsworth, A., Davies-Barnard, T., Su, T., Lunt, D. J., et al. (2021). Orographic evolution of northern Tibet shaped vegetation and plant diversity in eastern Asia. Sci. Adv. 7 (5), eabc7741. doi: 10.1126/sciadv.abc7741
Linder, H. P., Lehmann, C. E. R., Archibald, S., Obsborne, C. P., Richardson, D. M. (2018). Global grass (Poaceae) success underpinned by traits facilitating colonization, persistence and habitat transformation. Biol. Rev. Camb. Philos. Soc 93 (2), 1125–1144. doi: 10.1111/brv.12388
Liu, J. Q., Duan, Y. W., Hao, G., Ge, X. J., Sun, H. (2014). Evolutionary history and underlying adaptation of alpine plants on the Qinghai–Tibet Plateau. J. Systematics Evol. 52 (3), 241–249. doi: 10.1111/jse.12094
Liu, J. Q., Ni, Y. Y., Chu, G. Q. (2001). Main palaeoclimatic events in the quaternary. Quaternary Sci. 21 (3), 239–248.
Lu, L. M., Mao, L. F., Yang, T., Ye, J. F., Liu, B., Li, H. L., et al. (2018). Evolutionary history of the angiosperm flora of China. Nature 554 (7691), 234–238. doi: 10.1038/nature25485
Lu, S. L., Wu, Z. L. (1996). On geographical distribution of the genus Stipa L. China. J. Systematics Evol. 34 (3), 242–253.
Mao, K. S., Wang, Y., Liu, J. Q. (2021). Evolutionary origin of species diversity on the Qinghai–Tibet Plateau. J. Syst. Evol. 59 (6), 1142–1158. doi: 10.1111/jse.12809
McKain, M. R., Tang, H. B., McNeal, J. R., Ayyampalayam, S., Davis, J. I., dePamphilis, C. W., et al. (2016). A phylogenomic assessment of ancient polyploidy and genome evolution across the poales. Genome Biol. Evol. 8 (4), 1150–1164. doi: 10.1093/gbe/evw060
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Nobis, M., Gudkova, P. D., Baiakhmetov, E., Żabicka, J., Krawczyk, K., Sawicki, J. (2019). Hybridisation, introgression events and cryptic speciation in Stipa (Poaceae): A case study of the Stipa heptapotamica hybrid-complex. Perspect. Plant Ecol. Evol. Syst. 39, 125457. doi: 10.1016/j.ppees.2019.05.001
Nobis, M., Gudkova, P. D., Nowak, A., Sawicki, J., Nobis, A. (2020). A synopsis of the genus stipa (Poaceae) in middle asia, including a key to species identification, an annotated checklist, and phytogeographic analyses. Ann. Mo Bot. Gard 105, 1–63. doi: 10.3417/2019378
Nobis, M., Krzempek, M., Nowak, A., Gudkova, P. D., Klichowska, E. (2022). Resurrection of Stipa tremula and taxonomy of the high-alpine species from the Stipa purpurea complex (Poaceae, Pooideae). PhytoKeys 196, 21–47. doi: 10.3897/phytokeys.196.82598
One Thousand Plant Transcriptomes Initiative (2019). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574 (7780), 679–685. doi: 10.1038/s41586-019-1693-2
Orton, L. M., Barberá, P., Nissenbaum, M. P., Peterson, P. M., Quintanar, A., Soreng, R. J., et al. (2021). A 313 plastome phylogenomic analysis of Pooideae: Exploring relationships among the largest subfamily of grasses. Mol. Phylogenet. Evol. 159, 107110. doi: 10.1016/j.ympev.2021.107110
Peng, J. (2015). Studies on molecular phylogeny and genetic diversity of stipa linn in China. [Ph.D. dissertation]. China: Inner Mongolia University.
Qiao, X. (2019). Community characteristics of Stipa steppes in China (University of Chinese Academy of Sciences).
Romaschenko, K., Peterson, P. M., Soreng, R. J., Garcia, J. N., Susanna, A., Seberg, O., et al. (2010). Phylogenetics of stipeae (poaceae: pooideae) based on plastid and nuclear DNA sequences. In Seberg, O., Petersen, G., Barfod, A. S., Davis, J. I. (Eds.), Diversity, phylogeny, and evolution in the Monocotyledons. (Aarhus, Denmark: Aarhus University Press), 511–537.
Romaschenko, K., Peterson, P. M., Soreng, R. J., Garcia-Jacas, S., Futorna, O., Susanna, A. (2012). Systematics and evolution of the needle grasses (Poaceae: Pooideae: Stipeae) based on analysis of multiple chloroplast loci, ITS, and lemma micromorphology. TAXON 61 (1), 18–44. doi: 10.1002/tax.611002
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61 (3), 539–542. doi: 10.1093/sysbio/sys029
Rowley, D. B., Currie, B. S. (2006). Palaeo-altimetry of the late Eocene to Miocene Lunpola basin, central Tibet. Nature 439 (7077), 677–681. doi: 10.1038/nature04506
Sandve, S. R., Kosmala, A., Rudi, H., Fjelheim, S., Rapacz, M., Yamada, T., et al. (2011). Molecular mechanisms underlying frost tolerance in perennial grasses adapted to cold climates. Plant Sci. 180 (1), 69–77. doi: 10.1016/j.plantsci.2010.07.011
Spicer, R. A., Su, T., Valdes, P. J., Farnsworth, A., Wu, F. X., Shi, G. L., et al. (2020). Why ‘the uplift of the Tibetan Plateau’ is a myth. Natl. Sci. Rev. 8 (1), nwaa091. doi: 10.1093/nsr/nwaa091
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Strömberg, C. A. E. (2005). Decoupled taxonomic radiation and ecological expansion of open-habitat grasses in the Cenozoic of North America. Proc. Natl. Acad. Sci. U. S. A. 102 (34), 11980–11984. doi: 10.1073/pnas.0505700102
Strömberg, C. A. E. (2011). Evolution of grasses and grassland ecosystems. Annu. Revi. Earth Planet. Sci. 39 (1), 517–544. doi: 10.1146/annurev-earth-040809-152402
Sun, H., Zhang, J. W., Deng, T., Boufford, D. E. (2017). Origins and evolution of plant diversity in the Hengduan Mountains, China. Plant Divers. 39 (4), 161–166. doi: 10.1016/j.pld.2017.09.004
Suyama, M., Torrents, D., Bork, P. (2006). PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Talavera, G., Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56 (4), 564–577. doi: 10.1080/10635150701472164
Thomasson, J. R. (1978). Epidermal patterns of the lemma in some fossil and living grasses and their phylogenetic significance. Science 199 (4332), 975–977. doi: 10.1126/science.199.4332.975
Thomasson, J. R. (1985). Miocene fossil grasses: Possible adaptation in reproductive bracts (Lemma and Palea). Ann. Mo. Bot. Gard. 72 (4), 843–851. doi: 10.2307/2399226
Tkach, N., Nobis, M., Schneider, J., Becher, H., Winterfeld, G., Jacobs, S. W. L., et al. (2021). Molecular phylogenetics and micromorphology of australasian stipeae (Poaceae, subfamily pooideae), and the interrelation of whole-genome duplication and evolutionary radiations in this grass tribe. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.630788
Tzvelev, N. N. (1989). The system of grasses (Poaceae) and their evolution. Bot. Rev. 55 (3), 141–203. doi: 10.1007/BF02858328
Vigeland, M. D., Spannagl, M., Asp, T., Paina, C., Rudi, H., Rognli, O. A., et al. (2013). Evidence for adaptive evolution of low-temperature stress response genes in a Pooideae grass ancestor. New Phytol. 199 (4), 1060–1068. doi: 10.1111/nph.12337
Vintsek, L., Klichowska, E., Nowak, A., Nobis, M. (2022). Genetic differentiation, demographic history and distribution models of high alpine endemic vicariants outline the response of species to predicted climate changes in a Central Asian biodiversity hotspot. Ecol. Indic. 144, 109419. doi: 10.1016/j.ecolind.2022.109419
Wang, H. J., Li, W. T., Liu, Y. N., Yang, F. S., Wang, X. Q. (2017). Resolving interspecific relationships within evolutionarily young lineages using RNA-seq data: An example from Pedicularis section Cyathophora (Orobanchaceae). Mol. Phylogenet. Evol. 107, 345–355. . doi: 10.1016/j.ympev.2016.11.018
Wang, J. W. (1997). Paleogeographical processes in the formation of China arid land. J. Arid Land Resour. Environ. 11 (1), 2–9.
Wang, C. S., Dai, J. G., Zhao, X. X., Li, Y. L., Graham, S. A., He, D. F., et al. (2014). Outward-growth of the Tibetan Plateau during the Cenozoic: A review. Tectonophysics 621, 1–43. doi: 10.1016/j.tecto.2014.01.036
Wang, K. F., Yang, J. W., Li, Z., Li, Z. R. (1975). On the Tertiary sporo-pollen assemblages from Lunpola Basin of Xizang, China and their palaeogeographic significance. Scientia Geologica Sin. 4 (4), 366–374.
Waterhouse, R. M., Seppey, M., Simao, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2017). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Wickett, N. J., Mirarab, S., Nguyen, N., Warnow, T., Carpenter, E., Matasci, N., et al. (2014). Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Sci. U. S. A. 111 (45), E4859–E4868. doi: 10.1073/pnas.1323926111
Wu, S. D., Wang, Y., Wang, Z. F., Shrestha, N., Liu, J. Q. (2022). Species divergence with gene flow and hybrid speciation on the Qinghai–Tibet Plateau. New Phytol. 234 (2), 392–404. doi: 10.1111/nph.17956
Xie, D. F., Yu, Y., Wen, J., Huang, J., Chen, J. P., Li, J., et al. (2020). Phylogeny and highland adaptation of Chinese species in Allium section Daghestanica (Amaryllidaceae) revealed by transcriptome sequencing. Mol. Phylogenet. Evol. 146, 106737. doi: 10.1016/j.ympev.2020.106737
Yang, Z. H. (1997). PAML: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 13 (5), 555–556. doi: 10.1093/bioinformatics/13.5.555
Yu, G. C., Smith, D. K., Zhu, H. C., Guan, Y., Lam, T. T. Y. (2017). GGTREE: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8 (1), 28–36. doi: 10.1111/2041-210X.12628
Zeng, L. P., Zhang, Q., Sun, R. R., Kong, H. Z., Zhang, N., Ma, H. (2014). Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 5 (1), 4956. doi: 10.1038/ncomms5956
Zeng, L. P., Zhang, N., Zhang, Q., Endress, P. K., Huang, J., Ma, H. (2017). Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 214 (3), 1338–1354. doi: 10.1111/nph.14503
Zhang, L. S. (2012). Ancient geography of China: the formation of China’s natural environment (Beijing: Science Press).
Zhang, L., Zhu, X. X., Zhao, Y. Y., Guo, J., Zhang, T. K., Huang, W. C., et al. (2022). Phylotranscriptomics resolves the phylogeny of pooideae and uncovers factors for their adaptive evolution. Mol. Biol. Evol. 39, msac06. doi: 10.1093/molbev/msac026
Zhou, X. M. (1980). Overview of the alpine grasslands of the tibetan plateau and their relationship with the Eurasian grassland region. Chin. J. Grassland 4), 3–8.
Keywords: Stipa, phylogenetic, comparative transcriptomics, evolutionary history, speciation, Qinghai-Tibet plateau, Mongolian plateau, Pleistocene
Citation: Sha N, Li Z, Sun Q, Han Y, Tian L, Wu Y, Li X, Shi Y, Zhang J, Peng J, Wang L, Dang Z and Liang C (2023) Elucidation of the evolutionary history of Stipa in China using comparative transcriptomic analysis. Front. Plant Sci. 14:1275018. doi: 10.3389/fpls.2023.1275018
Received: 09 August 2023; Accepted: 08 November 2023;
Published: 28 November 2023.
Edited by:
Jinliang Liu, Wenzhou University, ChinaReviewed by:
Yong Li, Inner Mongolia Normal University, ChinaMarcin Nobis, Jagiellonian University, Poland
Qingzhu Gao, Chinese Academy of Agricultural Sciences (CAAS), China
Copyright © 2023 Sha, Li, Sun, Han, Tian, Wu, Li, Shi, Zhang, Peng, Wang, Dang and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenhua Dang, emhkYW5nQGltdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
‡ORCID: Na Sha, orcid.org/0000-0003-1206-8793
Qiang Sun, orcid.org/0000-0001-8754-1501
Ying Han, orcid.org/0009-0001-3866-2312
Yantao Wu, orcid.org/0000-0002-1573-4483
Yabo Shi, orcid.org/0000-0001-5455-6176
Zhenhua Dang, orcid.org/0000-0002-9196-159X