 Xuping Zhou
Xuping Zhou Tao Peng2†
Tao Peng2† Yuqing Cai
Yuqing Cai- 1Laboratory of Southern Subtropical Plant Diversity, Fairy Lake Botanical Garden, Shenzhen & Chinese Academy of Sciences, Shenzhen, China
- 2Colleage of Life Sciences, Guizhou Normal University, Guiyang, China
- 3State Key Laboratory of Agricultural Genomics, BGI Research, Shenzhen, China
- 4College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
With a diversity of approximately 22,000 species, bryophytes (hornworts, liverworts, and mosses) represent a major and diverse lineage of land plants. Bryophytes can thrive in many extreme environments as they can endure the stresses of drought, heat, and cold. The moss Niphotrichum japonicum (Grimmiaceae, Grimmiales) can subsist for extended periods under heat and drought conditions, providing a good candidate for studying the genetic basis underlying such high resilience. Here, we de novo assembled the genome of N. japonicum using Nanopore long reads combined with Hi-C scaffolding technology to anchor the 191.61 Mb assembly into 14 pseudochromosomes. The genome structure of N. japonicum’s autosomes is mostly conserved and highly syntenic, in contrast to the sparse and disordered genes present in its sex chromosome. Comparative genomic analysis revealed the presence of 10,019 genes exclusively in N. japonicum. These genes may contribute to the species-specific resilience, as demonstrated by the gene ontology (GO) enrichment. Transcriptome analysis showed that 37.44% (including 3,107 unique genes) of the total annotated genes (26,898) exhibited differential expression as a result of heat-induced stress, and the mechanisms that respond to heat stress are generally conserved across plants. These include the upregulation of HSPs, LEAs, and reactive oxygen species (ROS) scavenging genes, and the downregulation of PPR genes. N. japonicum also appears to have distinctive thermal mechanisms, including species-specific expansion and upregulation of the Self-incomp_S1 gene family, functional divergence of duplicated genes, structural clusters of upregulated genes, and expression piggybacking of hub genes. Overall, our study highlights both shared and species-specific heat tolerance strategies in N. japonicum, providing valuable insights into the heat tolerance mechanism and the evolution of resilient plants.
Introduction
Bryophytes (hornworts, liverworts, and mosses) are a group of small, diverse organisms with a long fossil record (Puttick et al., 2018; Harris et al., 2022). They share a haploid-dominant life cycle with un-branched sporophyte growing attached to the gametophyte (Goffinet and Buck, 2013). The phylogenetic relationships among the three bryophyte major lineages and their relationships to other land plants have been contentious for centuries, with distinct scenarios supported by various studies (Cox, 2018; Puttick et al., 2018). With the advent of high-throughput sequencing technology, recent phylogenomic analyses have converged on the hypothesis of a monophyletic bryophyte clade that is sister to the tracheophytes (Puttick et al., 2018; One Thousand Plant Transcriptomes Initiative, 2019; Su et al., 2021). Bryophytes can thrive under the harshest environmental conditions, such as low light intensity, extreme temperatures, low nutrition, and dryness, thus making them the “pioneers” in many ecosystems and providing habitats and conditions for other plants and organisms to live. Bryophytes dominate the terrestrial ecosystem in Antarctica (Ochyra et al., 2008) and can survive in hot deserts [e.g., Syntrichia caninervis (Silva et al., 2021)], as they may have evolved an effective stress tolerance genetic toolkit, which may also be related to their distinct morphology and poikilohydric lifestyle (Kulshrestha et al., 2022; Wang et al., 2022). Hence, bryophytes provide excellent models and unique genetic resources for studying stress response and plant resistance traits. However, in contrast to the explosive growth in the number of sequenced angiosperm genomes, resources for bryophyte genomes have accumulated at a slower pace, with only 17 published genomes (Supplementary Table 1), in comparison to the 682 publicly available genomes of angiosperms (Sun et al., 2022b), making it difficult to illustrate a comprehensive genetic basis for stress tolerance and to impede a more in-depth comprehension of the genomic evolution of bryophytes.
As previous studies on environmental stress of bryophytes mainly focused on drought tolerance (Wang et al., 2022), the molecular mechanisms of bryophytes responding to other stresses, such as heat, are largely unknown. Studies in liverwort Marchantia polymorpha indicated that the core components of the heat stress response are conserved between bryophytes and angiosperms (Marchetti et al., 2021; Tan et al., 2023). During heat stress, the genes encoding the cyclic nucleotide gated calcium channels (CNGCs) are activated (Finka et al., 2012; Finka and Goloubinoff, 2014), leading to the opening of heat-sensitive calcium-permeable channels in plants, causing an inward Ca2+ flux and initiating a signal transduction network that regulates the expression of a number of genes, including the heat shock transcription factors (HSFs), heat shock proteins (HSPs), and reactive oxygen species (ROS) scavenging enzymes, to enhance heat tolerance (Zhu, 2016; Marchetti et al., 2021). Global warming has significantly impacted numerous natural ecosystems, exacerbating natural disasters such as extreme heat and causing devastating damage to crop production (Lobell et al., 2011; Lesk et al., 2016). Without effective adaptation and genetic improvement, such damage to crops (wheat, rice, maize, and soybean) is expected to increase (Zhao et al., 2017). Hence, there is an urgent need to identify and characterize heat-resistant genetic resources and to facilitate the improvement of heat tolerance in crops.
Niphotrichum japonicum (Grimmiaceae, Grimmiales) is a vigorous moss species that usually grows on dry rocks exposed to intense light (Supplementary Figure 1). As an important horticultural moss, this species has been extensively utilized to green roofs and walls of urban buildings due to its impressive ability to withstand environmental stresses (heat, drought, high light, and nutrient limitation) (Akita et al., 2011; Hendrawan and Murase, 2011). Several studies have investigated the strontium stress (Ren et al., 2023), cold, heat, and drought tolerance (Lei et al., 2021; Xia et al., 2022) of N. japonicum from the perspective of photosynthetic regulation. However, the genetic level of stress tolerance in N. japonicum remains largely unknown due to the absence of a high-quality reference genome. In this study, we present a chromosome-level genome assembly for N. japonicum. Through comparative genomic and transcriptomic studies, we have uncovered conserved elements of the heat response process in N. japonicum, identified unique genes that may play a role in the species’ heat resistance, and revealed the heat-responsive genes, modules, and structural clusters in the genome. Our study not only illuminates the heat response of the heat-tolerant moss, but also offers significant genetic resources for future research on embryophyte evolution, gene function, and stress response in land plants.
Results
A chromosome-level genome assembly
Here, we reported a high-quality, chromosome-level genome assembly of N. japonicum gametophyte based on a combination of 120.08 Gb Illumina short reads, 79.16 Gb Nanopore long reads, and 116.88 Gb Hi-C data. Based on K-mer frequency distribution of 115.24 Gb clean Illumina short-read data, the genome size was estimated as 184.22 Mb (Supplementary Figure 2). After conducting assembly and removing contamination (Supplementary Figure 3), we obtained an optimized assembly of 191.61 Mb with a contig N50 length of 6.60 Mb and a scaffold N50 length of 14.23 Mb, corresponding to the 14 chromosomal pseudomolecules (Table 1, Figure 1A, and Supplementary Figure 4). The N. japonicum genome assembled was the smallest among all moss genomes assembled to date, and the length of contig N50 ranked third, demonstrating its high genome continuity (Figure 1B). Repeat sequences of N. japonicum constituted 34.74% of the genome with transposable elements (TEs) being the predominant component by accounting for 32.34% of the genome, and long terminal repeat sequences (LTRs) being the major component within TEs (Table 1 and Supplementary Table 2). The genome coded for 26,898 protein-coding genes (PCGs), and was compact, having the highest gene density among the published bryophytes (Table 1 and Figure 1C). Approximately 82.27% of the PCGs were functionally annotated using InterProScan software, the gene ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG), Swiss-Prot, TrEMBL, and the Arabidopsis Information Resource (TAIR) databases (Supplementary Table 3). Based on the Benchmarking Universal Single-Copy Ortholog (BUSCO) estimation using the Viridiplantae odb10 dataset, the completeness of the gene space captured by the N. japonicum genome was ca. 97.00%, which was comparable to the model moss Physcomitrium patens v3.3 with a BUSCO score of 98.84% (Supplementary Figure 5). Moreover, approximately 88.79% of the annotated genes of N. japonicum were captured in the transcriptome unigenes.
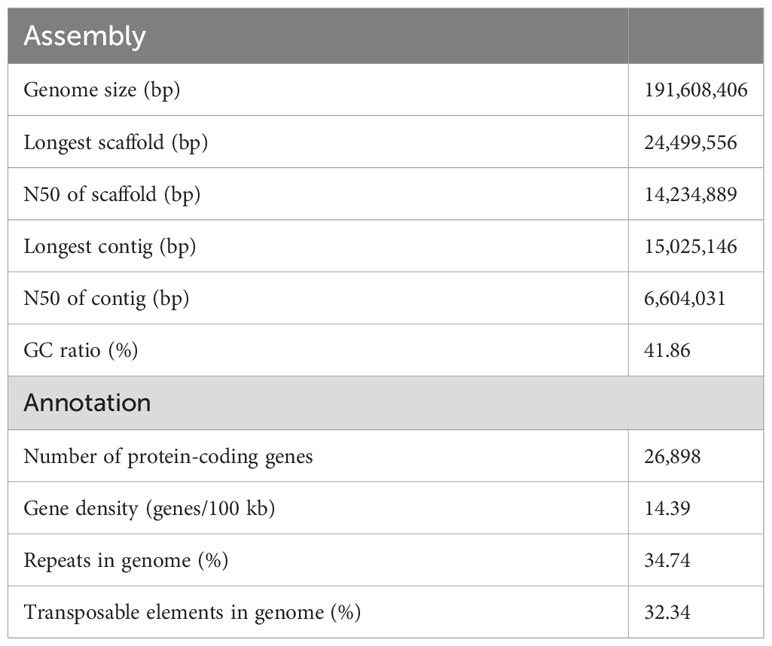
Table 1 Assembly and annotation statistics of the N. japonicum genome.
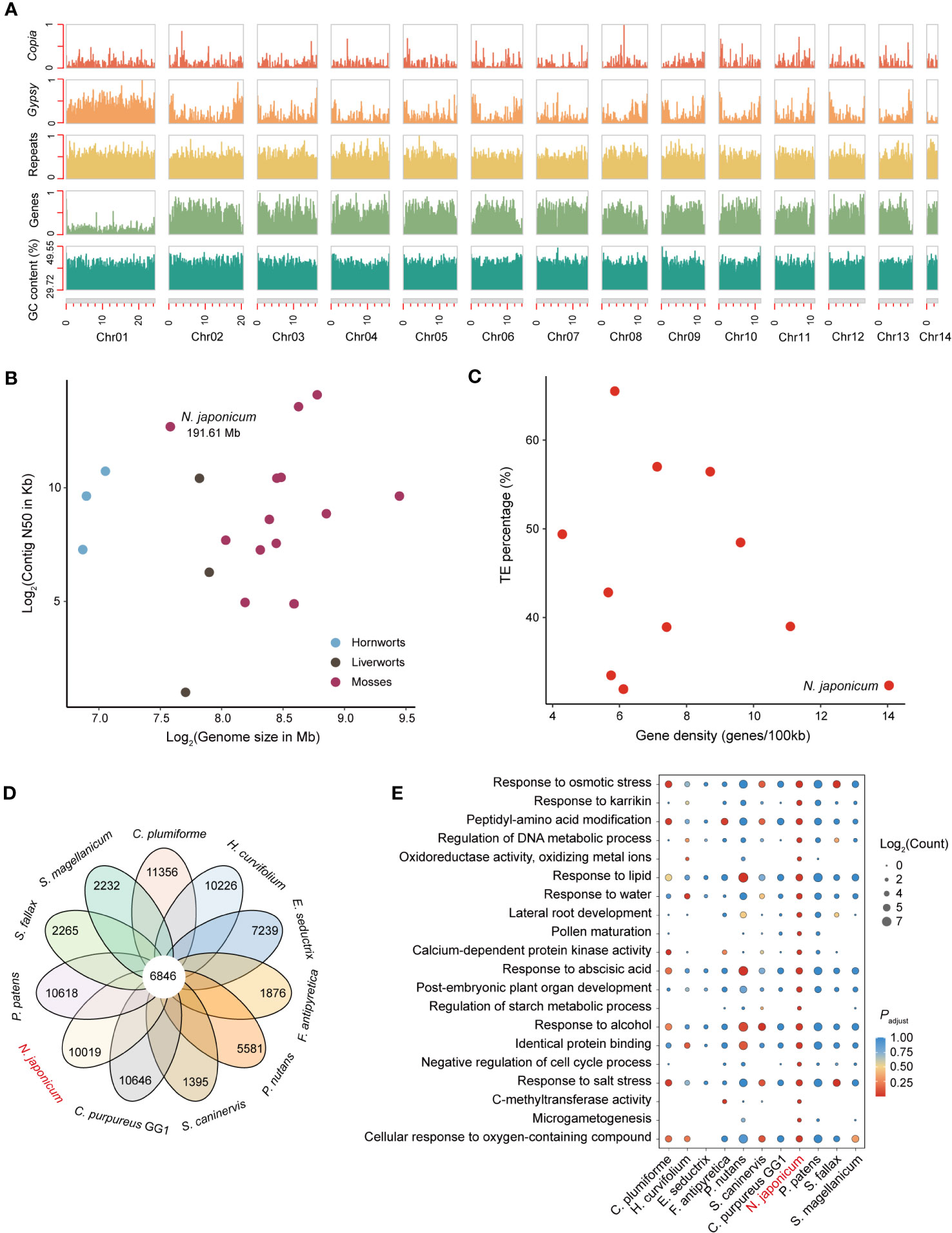
Figure 1 Genomic characterization and comparative genomic analysis of N. japonicum. (A) Density plots across N. japonicum chromosomes (in Mb), showing the number or proportion (GC content) of 100-Kb sliding window with 90-Kb jump of each feature. The number of the features is normalized (0,1) except for the GC content. (B) Comparison of genome size and contig N50 among 18 assembled bryophyte genomes. (C) Comparison of gene density and TE percentage among 11 assembled moss genomes. (D) Petal diagram analyses showing shared and unique genes in 11 mosses. (E) GO functional enrichment of unique genes in 11 mosses. All unique genes of the 11 mosses annotated by the GO library are used as background information, with N. japonicum as the focal species, and only the GO terms in the top 20 of the adjusted p-value (padjust) are shown.
In the Hi-C heatmap of N. japonicum (Supplementary Figure 4), Chr01 was the largest chromosome (24.50 Mb) and showed few contacts with the other chromosomes. Furthermore, Chr01 had the lowest gene density (33 genes/Mb) and the highest repeat proportion (70.81%) among all chromosomes (Figure 1A), indicating it as a putative sex chromosome (containing 807 genes).
Comparative genomic analysis
Except for WGD, all the gene duplicated modes were categorized as single-gene duplication (Freeling, 2009), consisting of tandem duplicates (TD), proximal duplicates (PD), transposed duplicates (TRD), and dispersed duplicates (DSD) (Qiao et al., 2019). Our identification of duplicated genes on the sex chromosomes indicated that genes located on these chromosomes in mosses were highly variable, with numerous genes duplicated. The highest percentage of duplicated genes occurred in Sphagnum magellanicumn (48.33%), followed by N. japonicum (36.80%), with an average of approximately 23.92% (Supplementary Figure 6). Moreover, our results demonstrated the lack of WGD genes in the moss sex chromosomes.
To identify the unique genes of N. japonicum, we clustered the proteomes of N. japonicum with those of the other 10 published moss genomes (i.e., S. magellanicum, Sphagnum fallax, P. patens, S. caninervis, Ceratodon purpureus, Pohlia nutans, Fontinalis antipyretica, Entodon seductrix, Hypnum curvifolium, and Calohypnum plumiforme) (Supplementary Table 1). A total of 6,846 genes were shared across 11 mosses, and 10,019 unique genes were identified for N. japonicum (Figure 1D). GO enrichment demonstrated that N. japonicum unique genes were significantly enriched in “response to osmotic stress”, “response to karrikin”, and “oxidoreductase activity, oxidizing metal ions” (Figure 1E and Supplementary Table 4), comply with the species-specific competences in withstanding stress and in post-stress germination (Inupakutika et al., 2016; Zheng et al., 2020; Sepulveda et al., 2022).
We identified a total of 729 transcription factors (TFs) in 62 TF families for the N. japonicum genome, in contrast to the 1,185 TFs of the P. patens genome (Supplementary Table 5). A majority of the TF families had a ratio of nearly 1:2 in number between N. japonicum and P. patens (Supplementary Table 5), possibly caused by the fact that P. patens had undergone an additional lineage-specific WGD (Lang et al., 2018). We also found that four TFs (B3, HB-WOX, LIM, and SRS) present in higher numbers in N. japonicum than in P. patens, particularly the B3 TF (Supplementary Table 5) that mediated desiccation tolerance in plants (Carbonero et al., 2017), suggesting possibly a reinforced desiccation tolerance trait in N. japonicum.
Whole-genome duplication and inter-genomic synteny
Based on the analysis of intra-genomic synteny in N. japonicum, we identified 157 syntenic blocks, containing 1,823 genes, accounting for 6.75% of the genome (Figure 2A). We additionally identified the whole-genome duplication (WGD) event using the distribution of the substitution-rate-adjusted number of substitutions per synonymous site (Ks). The Ks distribution showed two peaks (i.e., “a” and “b”) (Figure 2B). The Ks value of “a” was very small in both Ks unit (0.14) and Ks height, with only a few retained duplicated genes (i.e., less than five anchor pairs). Therefore, we inferred that “a” was generated by single gene duplication events rather than WGD. The Ks peak of the WGD event in N. japonicum was at 0.79 (Figure 2B), which was larger than the divergence peak of N. japonicum with P. patens (0.68), indicating that the WGD event in N. japonicum occurred before the divergence with P. patens and that they shared a common WGD event, i.e., the “ψ” event (Gao et al., 2022). Furthermore, the abundant synteny between the C. purpureus GG1 and N. japonicum chromosome indicated the presence of the seven ancestral chromosomal elements of N. japonicum (Figure 2C).
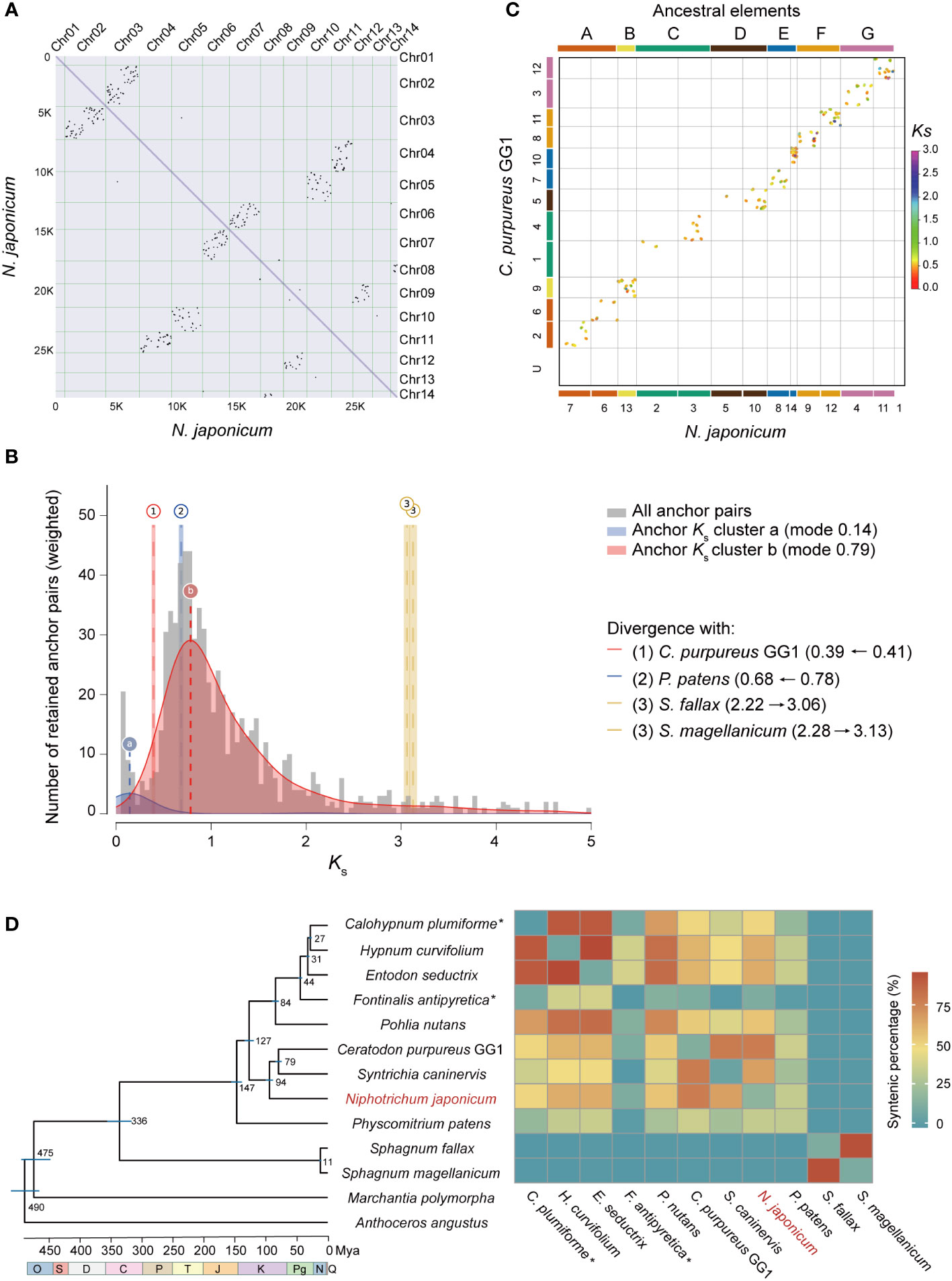
Figure 2 WGD analysis of N. japonicum and inter-genomic synteny of mosses. (A) Intra-genomic synteny of N. japonicum. (B) Substitution-rate-adjusted mixed paralog–ortholog Ks plot for N. japonicum. The anchor-pair Ks distribution for N. japonicum is shown in gray, with two Ks peaks inferred by lognormal mixture model clustering shown in blue and red. The vertical dashed lines labeled “a” and “b” indicate the modes of these components and Ks. Divergence events between N. japonicum and other species are represented by long vertical dashed lines but by Arabic numerals, with the left and right ranges of the colored rectangles representing standard deviations (SD). Identical divergence events are indicated by the same color and number, i.e., event 3 in this study. The numbers on the right represent the Ks of the corresponding event, and the horizontal arrows represent the Ks change of the species divergence events resulting from the substitution-rate-adjusted. (C) Inter-genomic synteny of N. japonicum and C purpureus GG1; the colors of the two genome chromosomes correspond to the ancestral elements, indicating that the extant chromosomes are duplicated from different ancestral elements. (D) Chronogram and synteny of mosses based on whole-genome data. All branches are maximally supported by bootstrap values (ML). O, Ordovician; S, Silurian; D, Devonian; C, Carboniferous; P, Permian; T, Triassic; J, Jurassic; K, Cretaceous; Pg, Paleogene; N, Neogene; Q, Quaternary. The times of speciation are marked on the branches, and the range of blue bars indicates the 95% confidence interval of the divergence time. *, non-chromosomal level genome assembly. Mya, million years ago. The heatmap on the right represents synteny of intra- or inter-genomes.
To investigate the synteny between the genome assembly of N. japonicum and the published moss genomes, we identified syntenic gene pairs using the jcvi software (Tang et al., 2008). The N. japonicum genome had varying degrees of synteny with other moss genomes, depending on the phylogenetic distance, with the exception of Sphagnum (Figure 2D).
Characterization and function divergence of duplicated genes
N. japonicum possessed 7,835 duplicated genes, representing 29.13% of the 26,898 genes, including 1,823 WGD genes, 1,103 TD genes, 1,382 PD genes, 464 TRD genes, and 3,063 DSD genes. The Ka (number of substitutions per nonsynonymous site), Ks, and Ka/Ks values were estimated for gene pairs generated by different modes of duplication. The Ka/Ks values among different modes of gene duplications showed a striking trend, with TD and PD genes having higher Ka/Ks values than other duplication modes, while WGD genes had the lowest Ka/Ks value (Figure 3A and Supplementary Table 6). This finding suggested that TD and PD genes played crucial roles in evolving new functions in N. japonicum.
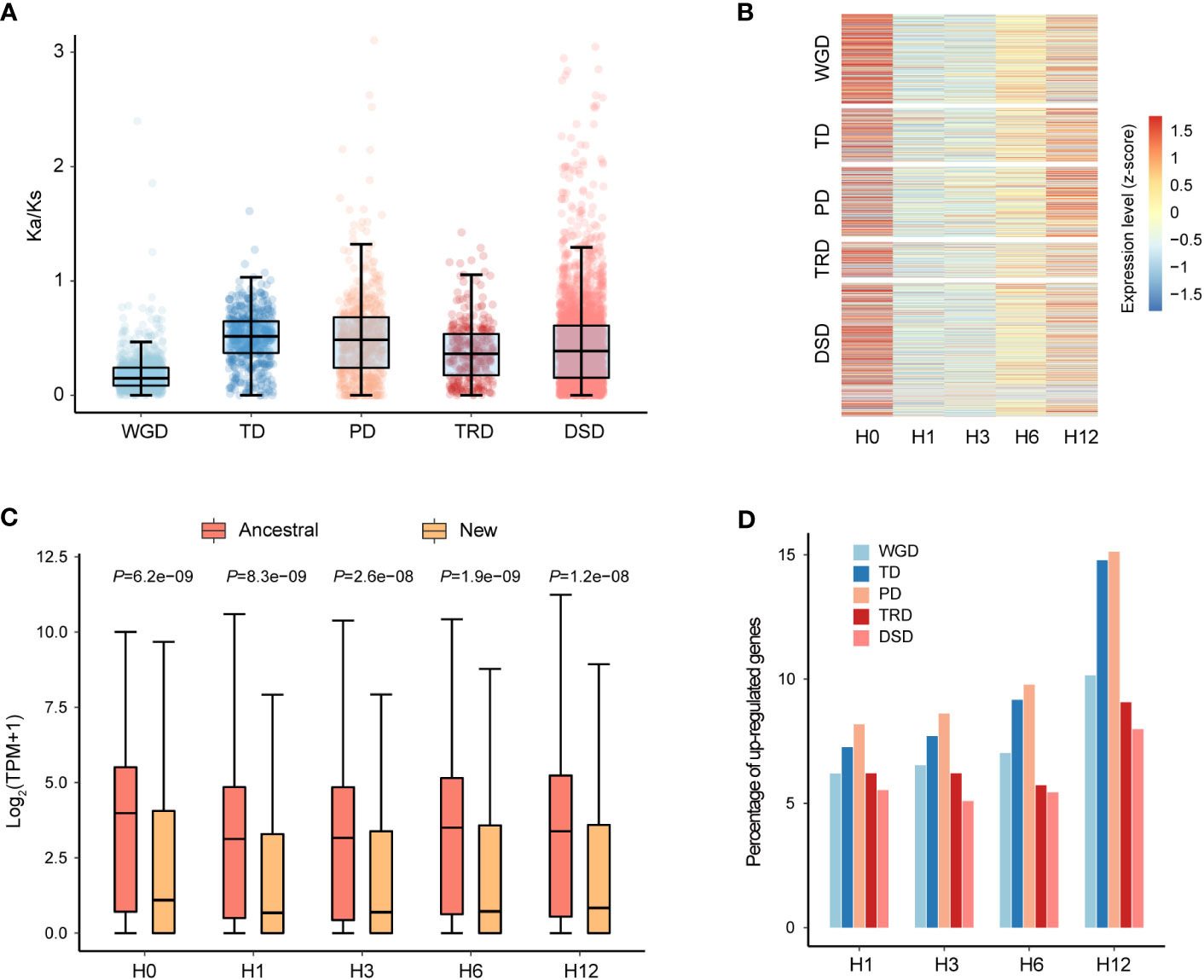
Figure 3 Gene characteristics of five different duplication modes in N. japonicum. (A) Box plots showing the Ka/Ks ratio of gene pairs derived from different modes of duplication. (B) Expression patterns of duplicated genes of five modes. H0 represents the control (20°C) and H1, H3, H6, and H12 represent heat stress at 42°C for 1, 3, 6 and 12 h, respectively. (C) Comparison of the expression levels of ancestral and new TRD genes. p-values were calculated using the Wilcoxon test. (D) Heat stress response of different modes of duplicated genes in N. japonicum.
We carried out further investigation into the expression patterns of duplicated genes of different categories under heat stress. The expression levels were examined for genes of WGD, TD, TRD, and DSD, and it was observed that the majority of these genes had the highest expression levels at 0 h (H0) of heat treatment, which was the lowest at 1 h (H1) and 3 h (H3) (Figure 3B). On the contrary, for PD genes, a greater number of genes expressed the highest levels at 12 h (H12) of treatment (Figure 3B). Interestingly, the ancestral gene locus in the TRD gene pairs showed significantly higher expression levels (p < 0.001) than the new gene locus during all periods of heat stress (Figure 3C). This expression divergence could be attributed to their different responses to the environment or functional redundancy, implying the possibility of sub-functionalization, neo-functionalization, or pseudogenization after gene duplication occurred.
According to our identification of differentially expressed genes (DEGs) in duplicated genes, a total of 1,043 (13.31% of all) duplicated genes (249 WGD genes, 185 TD genes, 246 PD genes, 55 TRD genes, and 308 DSD genes) were upregulated under heat stress, including 328 unique genes (Supplementary Table 7). Categories of PD and TD had a higher proportion of genes upregulated, which increased with the duration of treatment, suggesting the potential role in high-temperature tolerance (Figure 3D). The GO enrichment analysis of the upregulated genes in each duplicated category under heat stress demonstrated differing functional profiles among genes from various duplicated types (Supplementary Figure 7 andSupplementary Table 8). For instance, PD genes predominantly functioned in the heat stress response with the term “regulation of response to oxidative stress” being specific to PD. TRD boasted more distinct gene functions in comparison to other modes of duplication, mainly related to the transmembrane transport of carbohydrates.
Expression pattern of key gene families related to heat stress
Overall, we identified 10,070 DEGs (differentially expressed in one or more conditions), including 3,107 unique genes. Of these, 3,819 were upregulated (including 92 TFs and 1,606 unique genes) in one or more of the treated samples versus the control samples and 6,253 were downregulated (including 283 TFs and 1,501 unique genes) (Supplementary Table 9), with the strongest response observed after 12 h of heat treatment (Supplementary Figure 8). The upregulated genes were enriched in the GO terms closely related to heat stress, such as “protein folding”, “response to heat”, “response to reactive oxygen species”, and “response to hydrogen peroxide” at all stages (Supplementary Figure 9 and Supplementary Table 10). The downregulated genes were enriched in a number of GO terms related to “photosynthesis”, “ATP synthesis”, “cell wall remodeling”, “transmembrane transport”, and “response to fungus” (Supplementary Figure 10 and Supplementary Table 10), suggesting that the energy metabolism and microbial resistance pathways in N. japonicum were strongly inhibited or inactivated under heat stress.
The plant self-incompatible protein S1 (Self-incomp_S1) family (PF05938) or self-incompatible protein homologs (SPHs) were established on the basis of homology to PrsS gene and may be associated with programmed cell death (Ride et al., 1999; Rajasekar et al., 2019). We scanned the genomes of algae (chlorophytes and charaphytes), bryophytes (hornworts, liverworts, and mosses), and tracheophytes (lycophytes, ferns, gymnosperms, and angiosperms), and found the largest number of Self-incomp_S1s is in Arabidopsis thaliana (65), followed by N. japonicum (56), whereas members of this family were not found in algae, hornworts, ferns, and gymnosperms, suggesting a conspicuous expansion of Self-incomp_S1 family in N. japonicum (Supplementary Table 11). Differential expression analysis revealed that 15 members of Self-incomp_S1 family (12 upregulated and 3 downregulated in one or more conditions) were significantly induced (p < 0.05) by heat treatment of N. japonicum at 42°C, among which, NJ13G007760 accumulated more transcripts and had log2 (fold change (FC)) > 6 (Supplementary Table 9).
Building upon a previous study conducted in A. thaliana (Swindell et al., 2007), we identified 63 HSPs from the N. japonicum genome, including four subfamilies, namely, HSP20 or sHSP (20), HSP70 (29), HSP90 (7), and HSP100 (7) (Figure 4A). Forty HSP genes were upregulated in one or more of the treated samples versus the control samples (Supplementary Table 12). We further analyzed the expression patterns of HSP genes at different stages. At H0, the majority of HSPs were expressed at low levels; after heat exposure, the expression levels of HSPs increased, more obviously at the H1 and H3 stages, and slightly decreased at the stages of H6 and H12 (Figure 4B, and Supplementary Table 12). Among them, the expression level of the HSP20 subfamily was higher than that of the other subfamilies at all stages of stress, and an examination of HSPs in P. patens also yielded similar results (Supplementary Figure 11 and Supplementary Table 12). Further analysis of the HSP20 subfamily revealed that the TPM (transcript per million) values of seven genes were all higher than 10,000 at the H1 and H3 stages, and differential expression analysis showed that their log2 (FC) was >5, and the total expression level (TPM) under stress (H1, H3, H6, and H12) was 3.36 times higher than that of the other 56 HSPs (Figure 4C and Supplementary Table 12); in addition, one HSP70 gene (NJ09G004040) was significantly upregulated at stage H12 (p < 0.001), with a TPM value reaching 642 at the H12 stage in contrast to TPM being 0 at other stages (Supplementary Table 12).
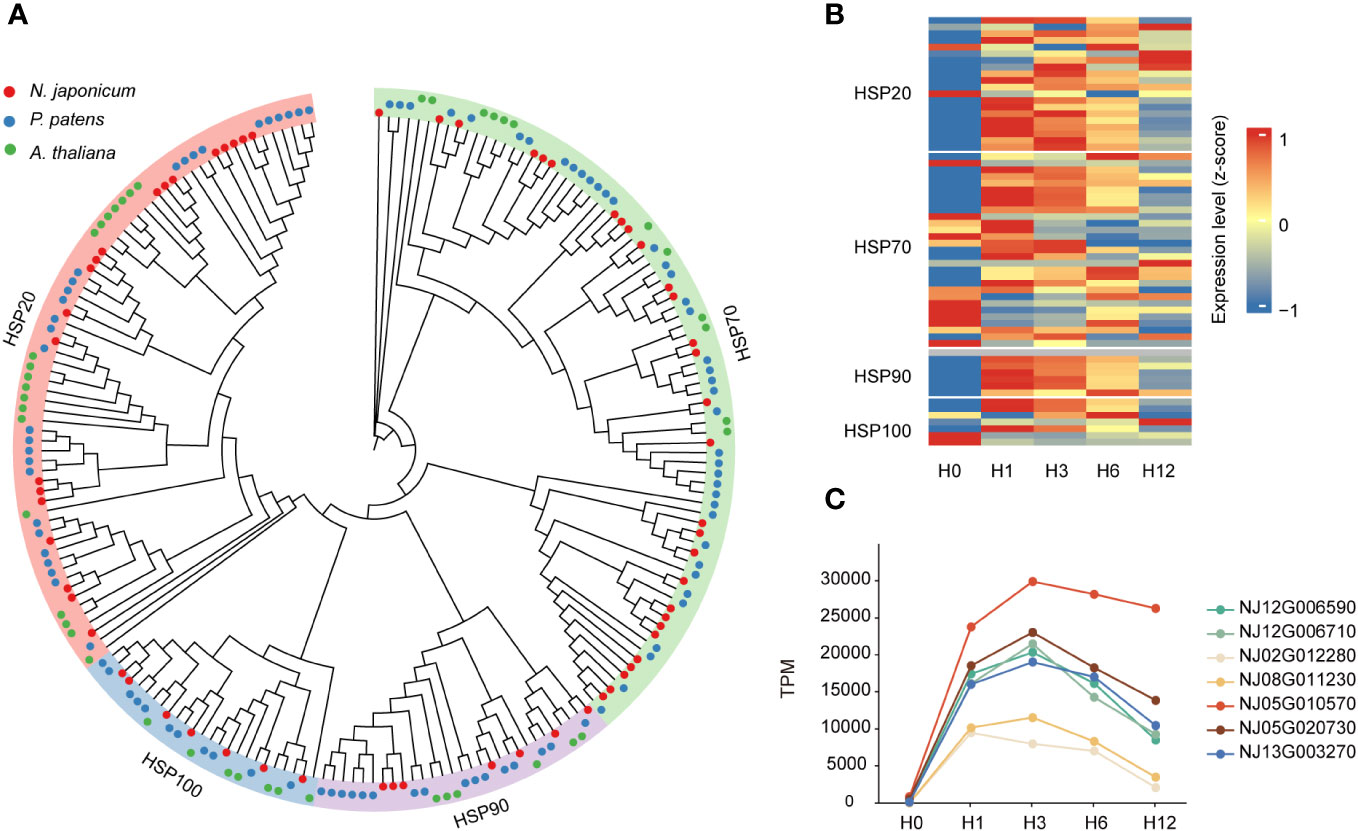
Figure 4 Phylogeny of HSPs and their expression levels under heat stresses. (A) Phylogeny of the HSPs. (B) Heatmap showing the gene expression levels of HSPs under control condition and 42°C heat stresses (at 1, 3, 6, and 12 h) in N. japonicum. (C) Differential expression levels of seven HSP20 members under heat stress.
A total of 41 Late embryogenesis abundant (LEA) genes were identified in N. japonicum, compared with 35 in P. patens (Supplementary Figure 12 and Supplementary Table 13). The number of LEA genes varied widely among 11 mosses (from 29 to 70) (Supplementary Table 13). N. japonicum had the largest dehydrin (DHN) subfamilies and a total number of LEA genes similar to that of the desert moss S. caninervis (40), which could be related to its potential drought resistance. We further analyzed the expression patterns of LEA genes in N. japonicum under heat, and differential expression analysis showed that 11 LEA genes were upregulated under heat stress (Supplementary Table 9), among which three genes, viz., NJ08G014280 (LEA_2), NJ03G019430 (LEA_4), and NJ05G015930 (DHN), had TPM > 1,000 and log2(FC) > 1.
We identified a total of 92 pentatricopeptide repeat (PPR) genes in the N. japonicum genome (Supplementary Table 14). Under heat stress, only seven PPR genes were upregulated in one or more conditions, whereas 26 PPR genes were downregulated (Supplementary Table 14), which could be due to heat-induced disintegration or functional inhibition of N. japonicum chloroplast. We also found that the expression of 16 chlorophyll A–B binding proteins, which acted as photochemical reaction centers in light absorption, was decreased in one or more conditions under heat stress (Supplementary Table 14), suggesting that chlorophyll degradation occurs under heat treatment.
We searched the N. japonicum genome for genes associated with ROS scavenging, based on previous studies in A. thaliana (Inupakutika et al., 2016). One gene (NJ08G015430) encoding blue copper protein, one gene (NJ04G021770) encoding glutathione S-transferase, one gene (NJ07G010200) encoding ferritin, one gene (NJ07G019160) encoding glutathione peroxidase, and seven genes (NJ02G021000, NJ04G009910, NJ07G001270, NJ07G001310, NJ07G023040, NJ10G011570, and NJ11G007530) encoding thioredoxin were significantly upregulated (p < 0.05) in response to heat stress and could play a role in scavenging excess ROS (Supplementary Table 9).
Profiling the heat stress response in N. japonicum
As some upregulated genes tended to accumulate transcripts in the form of physical clusters, e.g., NJ02G020390, NJ02G020400, NJ02G020410, NJ02G020420, NJ02G020430, and NJ02G020440 (Supplementary Table 9), the workflow of Pecrix et al. (2018) was used to identify physical clusters of co-localized and co-regulated genes or islands. Our results showed that the number of islands ranged from 27 to 29 and contained 116 to 360 genes across the four stages of heat stress, with the largest island being ~30 Kb (Table 2 and Supplementary Table 15). In addition, we found that the mean and median log2 (FC) values of the island genes appeared to show a gradual decrease from heat stress stages of H1 to H12 (Table 2). Given the short physical distance in TD and PD gene pairs, we searched for duplicated genes in such islands, indicating that islands of H1UP, H3UP, H6UP, and H12UP contained 18.52%, 20.97%, 22.41%, and 20.56% of duplicated genes, respectively (Table 2 and Supplementary Table 15). This suggested that gene duplication was not the only explanation for gene organization in islands, which resembled the pattern seen for the symbiosis-related gene islands in Medicago truncatula (Pecrix et al., 2018).
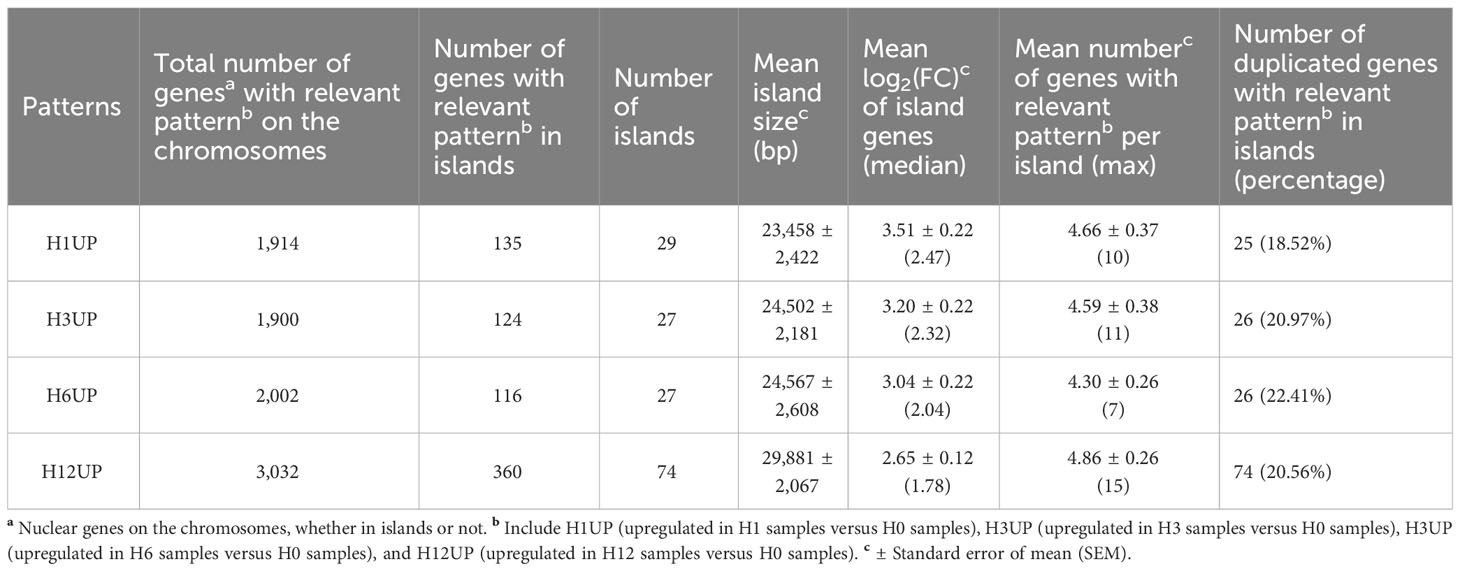
Table 2 Features of islands in upregulated genes of N. japonicum under heat stress.
As for the biological function of PCGs in these islands, in addition to HSPs, profilin and some proteins of unknown function are also present as physical clusters in different islands (Supplementary Table 15). Profilin plays a role in cell elongation, cell shape maintenance, and the determination of flowering time in A. thaliana (Ramachandran et al., 2000). A number of other important genes were also identified in these islands, such as the mechanosensitive ion channel family, which could be associated with early signaling of heat stress, the MYB TF associated with plant adversity, and the ABC transporter (Supplementary Table 15).
To better understand the dynamic changes in gene regulation and regulatory programs during heat stress, we performed a weighted correlation network analysis (WGCNA) and identified nine co-expression modules at different stages of the N. japonicum under heat stress (Figure 5A). The number of genes in the modules ranged from 24 (M9) to 8,018 (M5). The modules are enriched in the GO functional terms of photoprotection, defense against micro-organisms and nutrient metabolism (M2 and M5), removal of toxic substances and protein homeostasis maintenance (M1), and protein biosynthesis (M7 and M8) (Figure 5A and Supplementary Table 16). We observed a significant (p < 0.05) enrichment of GO terms associated with antibiotics.
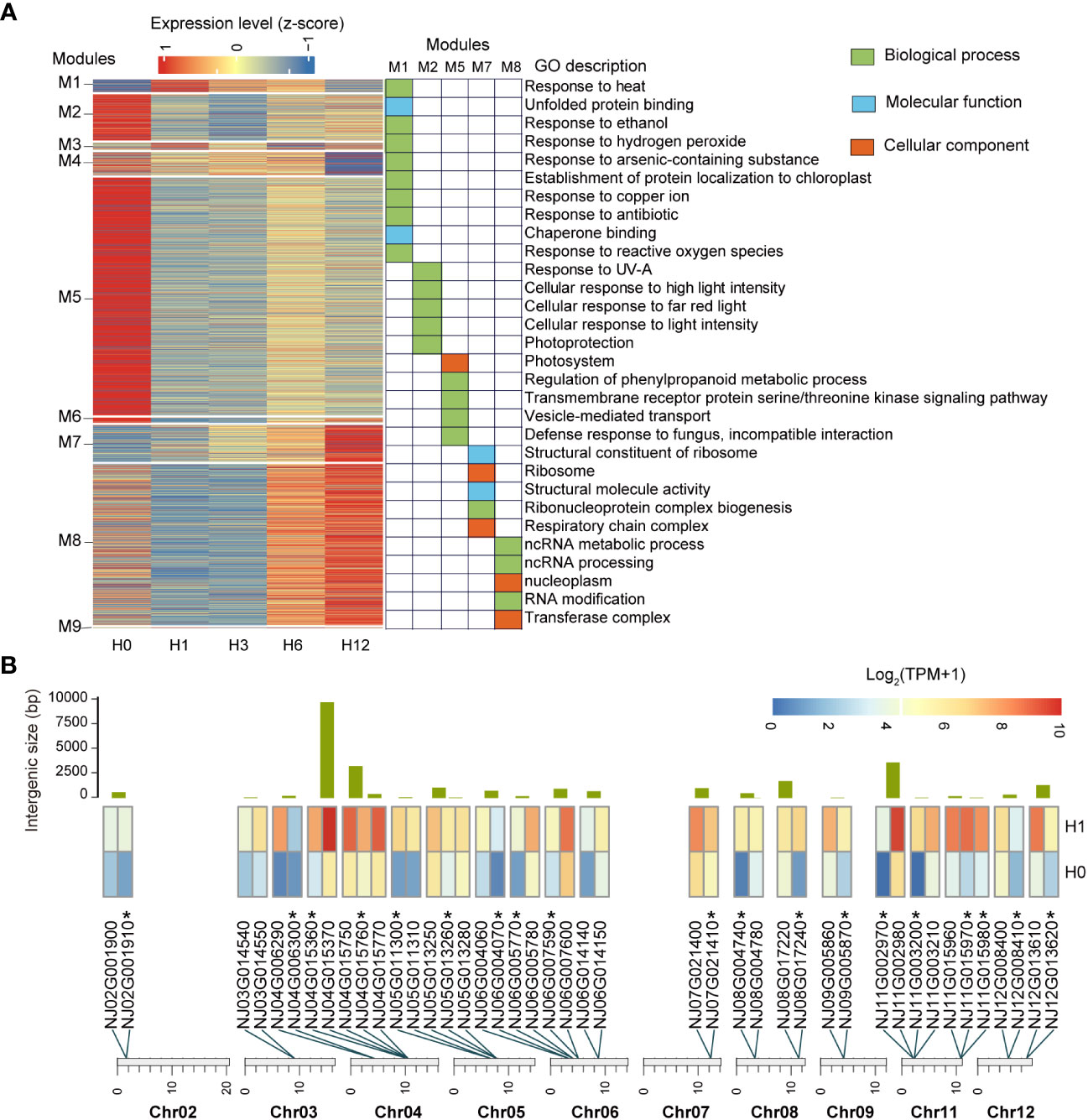
Figure 5 Characteristics of N. japonicum in response to heat stress. (A) Heatmap showing the relative expression levels of genes in nine co-expression modules by WGCNA across five stages of the heat stress. GO enrichment analysis was performed on genes from modules that showed a high correlation at one specific stage, all genes of N. japonicum annotated by GO library as background information, and the padjust < 0.05 for 30 displayed GO terms. (B) Distribution and expression patterns of neighboring genes in hub genes. The bars represent the intergenic spacer length of the neighboring genes. Heatmap showing the relative expression levels of neighboring genes at H0 and H1 stages. *, unique genes.
From M1, we identified 122 putative hub genes that were significantly associated with H1(p < 0.05), based on module membership (MM) ≥ 0.6 and gene significance (GS) ≥ 0.5 (Supplementary Table 17). The members of many (80) of these genes had been identified as homologs in P. patens or A. thaliana. Besides HSFs and HSPs, which had a dominant function in heat stress, vital genes such as ORP1B, HBP2, BAG6, and ROF2 were found. There were also numerous unique genes (41) that they could represent unique and novel players in N. japonicum heat stress.
Close examination revealed that some of the hub genes were located adjacent to each other on the chromosome (20 groups containing 43 genes) and that they were not duplicated genes, all of which had upregulated TPM levels under heat stress, typically each group contained two to three genes and the physical distance between neighboring genes was 18–9,738 bp (average ~1,166 bp) (Figure 5B and Supplementary Table 17).
Discussion
Structural evolution of the crown group mosses
Based on a high-quality, chromosome-level genome assembly of N. japonicum, we investigated the structural evolution of available moss genomes using syntenic analyses. The autosomes of mosses are generally highly syntenic depending on the phylogenetic distance as has been reported by Yu et al. (2022). The lack of synteny between Sphagnum and other mosses may be due to the very early divergence of it from other extant mosses (Healey et al., 2023). The Hi-C contact characteristic of N. japonicum sex chromosome, gene density, and repeat density of the N. japonicum sex chromosome conform to those previously observed in Sphagnum (Healey et al., 2023), C. purpureus (Carey et al., 2021), and E. seductrix and H. curvifolium (Yu et al., 2022). However, we find that sex chromosome length varies significantly among mosses. N. japonicum, S. caninervis (Silva et al., 2021), and C. purpureus (Carey et al., 2021) have the largest sex chromosomes in their genomes, whereas the two Sphagnum species (Healey et al., 2023) and the two Hypnales (Yu et al., 2022) possess the smallest.
Gene duplication provides genetic material for evolutionary innovation and is considered as an important driver for diversification and evolution (Van de Peer et al., 2017). Throughout the evolutionary history of land plants, there have been multiple occurrences of ancestral WGD events (Van de Peer et al., 2017). The genome of N. japonicum largely retained the feature of the seven ancestral chromosomes, similar to that of P. patens (Lang et al., 2018) and C. purpureus GG1 (Carey et al., 2021), but different from the five ancestral chromosomes of Sphagnum (Healey et al., 2023). In addition, it is noteworthy that Chr01 lacks synteny with autosomes (Figure 2A) or with C. purpureus (Figure 2B), suggesting a unique evolutionary mechanism. Considering the 1:2 syntenic relationship of the ancestral chromosomes of mosses with those of N. japonicum, the absence of collinearity between Chr13 and other autosomes may indicate that the sex chromosome of N. japonicum originated from the other copy of the ancestral chromosome B (Figures 2A, B), albeit with disrupted gene order (Carey et al., 2021). This could also clarify why the WGD gene is absent from the sex chromosome.
It is commonly acknowledged that the lack of meiotic recombination reduces the effectiveness of natural selection, leading to degradation and gene loss on non-recombinant chromosomes (e.g., mammalian Y chromosome and other UV systems) that typically contain, at best, orders of magnitude fewer genes (Charlesworth et al., 2000; Ferris et al., 2010; Bachtrog, 2013; Ahmed et al., 2014; Iwasaki et al., 2021). However, this is not the case in N. japonicum as well as other mosses, which typically contain hundreds to thousands of genes on their sex chromosomes (Carey et al., 2021; Silva et al., 2021; Yu et al., 2022; Healey et al., 2023). Our analyses of gene duplicated modes show that a significant proportion of genes on these sex chromosomes exhibit single-gene duplication, indicating that single-gene duplication may be beneficial in preventing the loss of genes on sex chromosomes of mosses due to the suppressing of recombination (Carey et al., 2021).
Conserved elements of heat stress response in plants
HSPs are important functional proteins that are induced by heat and are known to be targeted by heat-associated TFs. Under heat stress, HSPs can act as molecular chaperones, binding with other proteins and playing a crucial role in regulating protein quality by renaturing various proteins that have been denatured by heat stress (Kotak et al., 2007; Ohama et al., 2017; Waters and Vierling, 2020). HSPs of N. japonicum and P. patens were identified, and a putative 1:2 ratio between the two species would suggest either more loss of HSPs in P. patens or more retention of HSPs in N. japonicum. Notably, 63% of HSPs were upregulated in one or more conditions, which was associated with the high-temperature response of N. japonicum (Supplementary Table 12), a response that appears to be highly conserved in animals, yeast, and prokaryotes (Finka et al., 2011). Furthermore, we found a strong upregulation of HSP20, which may play a role in protecting the stability of the N. japonicum membrane system (Rütgers et al., 2017; Arena et al., 2019).
LEAs were small, heat-stable, hydrophilic proteins that are synthesized in orthodox seeds during mid to late maturation and may be associated with tolerance to abiotic stresses, such as drought, salinity, and high or cold temperature (Delahaie et al., 2013; Xu et al., 2018; Chen et al., 2019; Liu et al., 2019a; Zhuo et al., 2020). N. japonicum has 41 LEAs, which is higher than P. patens (35), but is not an expansion (P. nutans and C. purpureus have 70 and 62, respectively) (Supplementary Table 13). The LEA genes may play a crucial role in the high-temperature stress response of mosses Bryum argenteum (Zhuo et al., 2020) and S. caninervis (Silva et al., 2021). Our results (11 LEAs upregulated in one or more conditions) also provide further evidence for their potential role under heat stress (Supplementary Table 9).
The PPR gene family in N. japonicum appears to be contracted compared to Anthoceros angustus (Zhang et al., 2020) and P. patens (Xu et al., 2018). The PPR genes are mainly located in mitochondria or chloroplasts, which may be closely related to RNA editing sites and plant growth and development (Guillaumot et al., 2017; Zhang et al., 2020). Under high temperature stress, chloroplasts undergo extensive proteomic remodeling, and the efficiency of nuclear-encoded precursor proteins to translocate to chloroplasts is inhibited (Zheng et al., 2022). It is therefore not surprising that the function of the PPR genes is repressed.
ROS is a key signaling molecule that regulates many biological processes (Mittler, 2017). However, under heat stress, plants accumulate a large amount of ROS, including 1O2, O2−, H2O2, and OH−, which can cause oxidative damage to plant cells (Kissen et al., 2016; Yin et al., 2018). ROS homeostasis is critical for plant resistance and adaptation to environmental stresses (Miller, 2012). We identified ROS scavenging genes within the N. japonicum genome. Eleven of these genes were upregulated during high-temperature stress, indicating that they may play a role in mitigating these conditions. This process appears to be highly conserved during plant response to high-temperature stress (Li et al., 2018a).
Innovations of heat stress response in N. japonicum
Among the five models of gene duplication, TD and PD showed higher Ka/Ks values. This suggests that, like other plant lineages, they may have undergone faster sequence divergence (Qiao et al., 2019). In A. thaliana, TD genes play unique roles related to “binding” and “activity”, whereas PD genes are linked to apoptosis and immune responses (Qiao et al., 2019). Functional enrichment of upregulated genes under heat stress in N. japonicum indicates functional differences between various gene duplication models (Supplementary Figure 7 and Supplementary Table 8). For instance, PD may have a unique scavenging excess ROS role (Kerchev and Van Breusegem, 2022), while TRD genes may be involved in coordinating tolerance to oxidative and osmotic stress in response to heat stress (Saddhe et al., 2021).
Although most of the gene families associated with plant resistance did not undergo an expansion in N. japonicum (compared with other bryophyte genomes), the gene family encoding plant self-incompatibility protein S1 did show distinct expansions (Supplementary Table 11). This family is confined to tracheophytes and mosses (Supplementary Table 11), and consists of a series of plant proteins that are related to the Papaver rhoeas self-incompatibility protein S1 (PrsS). PrsS is a self-incompatibility determinant (Foote et al., 1994) and can be ectopically expressed, inducing growth arrest and cell death of vegetative cells independent of the reproductive context (Lin et al., 2020). The expansion of the plant Self-incomp_S1s in N. japonicum and their differential expression responding to heat stress might suggest their role in regulation of growth and stress responses (Rajasekar et al., 2019; Lin et al., 2020).
We identified a large number of DEGs in the transcriptome of N. japonicum under heat stress, accounting for 37.44% of the total gene set, and observed many unique genes (42% of the upregulated genes and 24% of the downregulated genes, respectively). (Supplementary Table 9). The acquisition of unique genes may reflect the unique and powerful evolutionary pressures that a species undergoes when adapting to new environments (Van Oss and Carvunis, 2019; Silva et al., 2021), and which differential expressions suggest that unique genes may have originated as a result of the drive for heat stress that N. japonicum once experienced (Arendsee et al., 2014). Additionally, we found physical clusters of many genes in the upregulated genes. This chromosomal clustering may confer a selective advantage through its ability to coordinate gene regulation at the chromatin level (Reimegård et al., 2017), and the physical clusters of upregulated genes may have a potential cell economy for N. japonicum under heat stress (Hurst et al., 2004; Pecrix et al., 2018). Such gene-organized co-expression appears to be common in eukaryotes and has been found in specific metabolic pathways in various plants (Nützmann et al., 2016), in A. thaliana stamen development-related genes (Reimegård et al., 2017), in symbiosis-related islands in M. truncatula (Pecrix et al., 2018), and in the gene components of S. caninervis in response to drought stress (Silva et al., 2021).
Using WGCNA, we investigated alterations in gene expression during different stages of high-temperature stress. Interestingly, we identified terms related to the response to antibiotics (Figures 5A). Since our experimental protocol did not include the administration of antibiotics, the significant enrichment of this term may be due to a certain degree of similarity in response between high-temperature stress and antibiotic stress (Cruz-Loya et al., 2019). The hub genes (122) of N. japonicum in response to heat stress were identified, with our focus on the M1 module, which was linked significantly with H1 due to its diversity of GO terms (Supplementary Table 16). Additionally, we analyzed the composition and potential functions of hub genes (Supplementary Table 17), such as NJ13G012500 was homologous to the ORP1B of A. thaliana and may function as an endogenous and exogenous danger signaling molecule that triggers plant innate immunity in plants (Chen et al., 2022). NJ02G004190 was homologous to HBP2 and had properties suitable for tetrapyrrole carrier proteins. NJ14G003380 encoded a chloroplast cyclophilin functioning in the assembly and maintenance of photosystem II (PSII) super complexes (Fu et al., 2007). In addition, BAG6 (Echevarría-Zomeño et al., 2016) and ROF2 (Meiri et al., 2010) may be involved in limiting the extension of the heat stress response in A. thaliana, with their homologs in the hub genes of N. japonicum (NJ03G021340 and NJ09G002390, respectively). These putative hub genes may be favored for genetic transformation into crops. Surprisingly, 35.25% of hub genes are neighboring genes, and we suggest that the occurrence of these genes may be due to expression piggybacking (Dai et al., 2014; Ghanbarian and Hurst, 2015; Lan and Pritchard, 2016), which allows quick response and tight regulation during heat stress, at the lowest energy expense.
Materials and methods
Plant materials
Wild gametophyte of N. japonicum was collected from Qianshan, Anqing, Anhui Province, China. After collection, the material was further identified morphologically and the voucher specimen (collection number: DNA1220) had been deposited at the Fairy Lake Botanical Garden, Shenzhen & Chinese Academy of Sciences, Shenzhen, Guangdong Province, China. The fresh gametophyte sample of N. japonicum was cleaned with distilled water and dried using lab paper, then the plant tissues were examined under a dissecting microscope to avoid potential contaminations from other plants, and used for subsequent experiments.
Heat stress experiments
A stress temperature (42°C) of four time gradients (cultured at 42°C for 1 h, 3 h, 6 h, and 12 h) and a control at 20°C were set up, with three biological replicates for each gradient. The N. japonicum samples from the four time gradients were first cultured at 35°C for 1 h to allow them to acclimate to the high temperature and to prevent them from entering dormancy due to sudden heat stress. They were then transferred to 42°C and three samples were taken at 1 h, 3 h, 6 h, and 12 h respectively, immediately treated with liquid nitrogen and stored in a −80°C freezer.
DNA and RNA sequencing
Genomic DNA and RNA were extracted using FastPureTM Plant DNA Isolation Mini Kit (Vazyme, Nanjing, China) and RNA-easyTM Isolation Reagent (Vazyme, Nanjing, China), respectively. DNA and RNA quantification and qualification were performed using 1% agarose gel electrophoresis, a Qubit 2.0 fluorometer (Thermo Fisher Scientific, USA), and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Nanopore libraries were prepared using SQK-LSK108 and sequenced on a Nanopore PromethION sequencer. DNA libraries for short-read whole genome sequencing (WGS) were constructed using the Illumina TruSeq DNA PCR-free library preparation kit (Illumina, CA, USA) with 300- to 500-bp fragment sizes, and sequenced on the Illumina NovaSeq 6000 platform to generate 150-bp paired-end (PE) reads. Transcriptome libraries were constructed with a TruSeq RNA Library Prep Kit v2 (Illumina, CA, USA) with an insert size of 200–400 bp, after polyA selection, and sequenced on an Illumina NovaSeq 6000 platform, and 150-bp PE reads were generated. The Hi-C library construction process includes cross-linking, restricted enzyme digestion (MboI), end repair, DNA cyclization, and purification (Yu et al., 2022). PE-150-bp reads were generated on Illumina NovaSeq 6000 platforms.
Genome size estimation
Low-quality reads and adapter sequences were filtered using Trimmomatic (v0.39) (Bolger et al., 2014). We then performed K-mer analyses to estimate the genome size of N. japonicum using clean Illumina reads. GCE (v1.0.2) (Liu et al., 2013) was used to calculate the K-mer distribution, and genome size was estimated by dividing the total number of K-mer by the K-mer peak depth. The haploid genome size of N. japonicum was estimated to be 184.22 Mb (Supplementary Figure 2).
Genome assembly
The de novo assembly of the N. japonicum genome was performed using Nextdenovo v2.5.0 (https://github.com/Nextomics/NextDenovo) with default parameters and 79.16-Gb Nanopore long reads. The primary assembly was polished three times with Nanopore long reads using NextPolish v1.3.1 (https://nextpolish.readthedocs.io/en/latest/QSTART.html) to correct for structure variations and insertions/deletions followed by three rounds of single-nucleotide polymorphism and insertion/deletion correction using Pilon (v1.23) (Walker et al., 2014) with clean Illumina reads. To remove contaminating contigs, we first performed BLASTN search for the assembled contig sequences in the National Center for Biotechnology Information (NCBI) nucleotide collection (released in November 2022) with the following parameters: “-evalue 1e-5 -max_hsps 10000 -outfmt 6 -num_alignments 20000”; for each query sequence, the total number of unique coverage positions for all hits was calculated, and any query sequence with non-embryophyte coverage greater than 50% was considered a contaminating sequence and removed from the genome. Chloroplast and mitochondrial genomes were assembled using GetOrganelle (v1.7.5.0) (Jin et al., 2020) and NOVOPlasty (v3.5) (Dierckxsens et al., 2017) respectively, and then organelle fragments were removed using the same parameters. To validate the results of the decontamination procedure, we performed GC-depth analysis with a window size of 10 Kb (Yu et al., 2022). To further enhance assembly contiguity, Juicer (v1.6) (Durand et al., 2016b) was used to extract valid data from 103.05 Gb of Hi-C clean reads and 3D-DNA pipeline (Dudchenko et al., 2017) was employed to anchor, order, and orient the assembled scaffolds into 14 pseudochromosomes. Finally, Juicebox (v1.11.08) (Durand et al., 2016a) was used to manually adjust the results of the 3D-DNA pipeline.
Repeat annotation
A combination of de novo and known repeat libraries was used to maximize the chances of identifying repetitive elements. The Piler (Edgar and Myers, 2005), LTR_FINDER (Xu and Wang, 2007), and RepeatScout (Price et al., 2005) were used to generate de novo repeat libraries. The Piler, LTR_FINDER, and RepeatScout repetitive libraries were combined and further used as the input data for RepeatMasker (Tarailo-Graovac and Chen, 2009). Repbase (v21.01) (Jurka et al., 2005) was a database of known repetitive elements that was searched using RepeatMasker and RepeatProteinMask (Tarailo-Graovac and Chen, 2009). Tandem repeats were identified using Tandem Repeats Finder (v4.07) (Benson, 1999).
Gene annotation and functional annotation
BRAKER2 pipeline (v2.1.5) (Brůna et al., 2021) was used to predict PCGs based on the soft-masked N. japonicum genome. For details, proteome sequences of seven embryophytes (i.e., A. thaliana, Azolla filiculoides, M. polymorpha, Oryza sativa, P. patens, Salvinia cucullata, and Selaginella moellendorffii) obtained from the Phytozome v13 database (https://phytozome-next.jgi.doe.gov/) or Fernbase (https://fernbase.org/) were used to provide homology-based protein evidence, and transcriptome clean reads were mapped to the genomes using TopHat2 (v2.1.1) (Kim et al., 2013) to provide expressed sequence tag (EST) evidence. The completeness of genome assembly was assessed by the BUSCO (v3.1.0) (Simão et al., 2015) using the Viridiplantae odb10 set. For gene functional annotation, the annotated protein sequences were blasted against the UniProt (Swiss-Prot and TrEMBL) and TAIR databases using DIAMOND (v2.0.15) (Buchfink et al., 2015) with an E-value cutoff of <1 × 10–5. The GO annotation of gene models was carried out using eggNOG -mapper (v2) (Cantalapiedra et al., 2021), InterProScan (v5.51-85.0) (Jones et al., 2014), and PANNZER2 (Törönen et al., 2018). The KEGG annotation of gene models was performed using eggNOG-mapper and KofamKOALA (Aramaki et al., 2019). The domain of the gene models was identified by InterProScan. The plant TF prediction program iTAK online (v1.6) (Zheng et al., 2016) was used to identify TFs.
Transcriptome assembly and mapping
Low-quality reads and adapters from the raw reads of transcriptome sequences were filtered using Trimmomatic. The resulting clean reads were de novo assembled using Trinity (v2.8.4) (Haas et al., 2013). For genes with more than one transcript, the longest transcript was chosen as the unigene. We merged the unigenes of 15 transcriptome samples and searched using BLASTN (E-value < 1 × 10-5) to remove non-embryophyte sequences. To extend the validation of genome assembly, the clean unigenes were compared to the reference assembly using BLASTN (E-value < 1 × 10−10), and 88.79% of the annotated genes of N. japonicum were successfully mapped to unigenes.
Identification of whole-genome duplication, reconstruction of ancestral chromosomes, and inter-genomic synteny of mosses
A synteny analysis method and a Ks-based age distribution approach as described previously (Lang et al., 2018; Li et al., 2018b) were used to identify the WGD events. The jcvi (v1.1.8) was employed for drawing the dot plot to show the relationship of intra-genomic collinear blocks with a cscore cutoff of 0.99. Owing to the diversity of factors affecting plant substitution rates, Ks estimates for events of the same absolute age may differ depending on the synonymous substitution rates in the lineages involved (Sensalari et al., 2022). To accurately estimate Ks distributions of WGD events for N. japonicum and Ks values for divergence events among species, ksrates (v1.1.3) (Sensalari et al., 2022) was used to generate adjusted mixed plots of Ks distributions by rescaling ortholog Ks estimates of species divergence times to the paralog Ks scale of N. japonicum with the following parameters: “collinearity = yes, max_number_outgroups = 4”, and the Newick tree ((((C. purpureus, N. japonicum), P. patens), (S. fallax, S. magellanicum)), Takakia lepidozioides) was used as the input phylogeny. The coding sequences (CDS) for all species used genomic data from the Phytozome v13 database, except for the T. lepidozioides, which used transcriptomic data from the One Thousand Plant Transcriptomes Initiative (One Thousand Plant Transcriptomes Initiative, 2019).
Since the ancestral karyotype of C. purpureus (Carey et al., 2021) was known from published moss genomes and had a closer phylogenetic relationship to N. japonicum (Liu et al., 2019b), it was chosen as the reference species for reconstructing the ancestral chromosome elements of N. japonicum using WGDI (v0.6.1) (Sun et al., 2022a).
The identification of synteny gene pairs among moss genomes was performed using jcvi. Pairwise synteny was assessed by syntenic percentage (synteny gene pairs/all-by-all comparison results filtered). The all-by-all comparison was performed with LAST (http://last.cbrc.jp/) in jcvi software and filtered tandem duplications and weak hits.
Duplicated gene categorization and calculating Ka, Ks, and Ka/Ks values
Based on the method used by Qiao et al. (2019) to identify duplicated genes in bryophytes (Supplementary Note), the duplicated genes were classified into five different categories: WGD, TD, PD, TRD, and DSD. TRD referred to the duplication of ancestral and novel loci, and the ancestral loci could be divided into two categories: intra-genomic synteny genes and inter-species synteny genes (Qiao et al., 2019). KaKs_Calculator (v2.0) (Wang et al., 2010) was used to calculate Ka and Ks values of duplicated gene pairs by implementing the model averaging (MA) method in ParaAT (v2.0) (Zhang et al., 2012). To account for the saturated substitutions at synonymous sites, the Ks values > 5.0 were excluded from further analysis (Vanneste et al., 2013; Li et al., 2016; Qiao et al., 2019).
Inter-genomic divergence of mosses
The longest protein sequence of each gene in the 12 published bryophyte genomes (1 hornwort, 1 liverwort, and 10 mosses) was selected for clustering using OrthoFinder (v2.3.11) (Emms and Kelly, 2019), from which A. angustus and M. polymorpha were selected as outgroups (Supplementary Table 1). A total of 69 single-copy orthologous genes were aligned using MAFFT (v7.453) (Katoh and Standley, 2013) and a maximum likelihood (ML) tree was constructed using IQ-TREE2 (v2.0.6) (Minh et al., 2020) with ultrafast 1,000 bootstrap replicates based on the JTT model. BEAST2 (v2.6.4) (Bouckaert et al., 2014) was used to estimate divergence times. The following fossil calibrations were used as priors: divergence of hornworts and liverworts, liverworts and mosses at approximately 407–515 Mya (Guo et al., 2012).
Transcriptome analysis
Raw transcriptome sequencing data were filtered using Trimmomatic and then mapped to the reference genome using HISAT2 (v2.2.0) (Kim et al., 2019), and count and TPM values were calculated using the StringTie (v2.2.1) program (Pertea et al., 2015). In addition, genes with expression fold change > 2 and p < 0.05 were identified as DEGs using DESeq2 (Love et al., 2014) based on count values. To identify co-expressed genes during heat stress, WGCNA (Langfelder and Horvath, 2008) was used based on the genes with average TPM > 2 across 15 samples. To better visualize the expression levels of co-expressed module genes using ComplexHeatmap (Gu et al., 2016), the TPM data were normalized (z-score). For module genes that showed high correlation with a particular stage, we performed GO enrichment analyses using clusterProfiler (Yu et al., 2012). We followed the workflow of Pecrix et al. (2018) to locate physical clusters or “islands” of upregulated genes. Briefly, a genomic region was considered an “island” if ≥3 upregulated genes represented >60% of the expressed genes in a 50-Kb window.
Identification of gene family
Gene family members were identified as follows. We first downloaded protein sequences of Self-incomp_S1, HSP, LEA, and PPR gene families from TAIR (https://www.arabidopsis.org/). Relevant gene families were searched from the gene set using BLASTP with an E-value cutoff of <1 × 10–5. The resulting sequences were annotated with Pfam protein domains using InterProScan, and the genes without the corresponding domains were removed. Finally, we performed alignments of the HSP and LEA gene families using MAFFT, respectively, and constructed ML trees using IQ-TREE2 with the JTT model based on ultrafast 1,000 bootstrap replicates.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: National Genomics Data Center (NGDC; https://ngdc.cncb.ac.cn/bioproject/) under the the BioProject accession number PRJCA017860 and figshare (https://figshare.com/) data repository (doi: 10.6084/m9.figshare.23573514).
Author contributions
XZ: Formal Analysis, Software, Visualization, Writing – original draft. TP: Formal Analysis, Visualization, Writing – original draft. YZ: Software, Writing – original draft. YC: Software, Writing – original draft. QZ: Writing – review & editing. LZ: Writing – review & editing. SD: Funding acquisition, Project administration, Writing – review & editing. YL: Funding acquisition, Project administration, Writing – review & editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This study is supported by the Scientific Foundation of the Urban Management Bureau of Shenzhen (Nos. 202005 and 202203 to Yang Liu; Nos. 202106 and 202302 to Shanshan Dong) and the Fairy Lake Botanical Garden (FLSF-2021-02 to Shanshan Dong).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1271357/full#supplementary-material
References
Ahmed, S., Cock, J. M., Pessia, E., Luthringer, R., Cormier, A., Robuchon, M., et al. (2014). A haploid system of sex determination in the brown alga Ectocarpus sp. Curr. Biol. 24, 1945–1957. doi: 10.1016/j.cub.2014.07.042
Akita, M., Lehtonen, M. T., Koponen, H., Marttinen, E. M., Valkonen, J. P. (2011). Infection of the Sunagoke moss panels with fungal pathogens hampers sustainable greening in urban environments. Sci. Total. Environ. 409, 3166–3173. doi: 10.1016/j.scitotenv.2011.05.009
Aramaki, T., Blanc-Mathieu, R., Endo, H., Ohkubo, K., Kanehisa, M., Goto, S., et al. (2019). KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252. doi: 10.1093/bioinformatics/btz859
Arena, M. P., Capozzi, V., Longo, A., Russo, P., Weidmann, S., Rieu, A., et al. (2019). The phenotypic analysis of Lactobacillus plantarum shsp mutants reveals a potential role for hsp1 in cryotolerance. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00838
Arendsee, Z. W., Li, L., Wurtele, E. S. (2014). Coming of age: orphan genes in plants. Trends Plant Sci. 19 (11), 698–708. doi: 10.1016/j.tplants.2014.07.003
Bachtrog, D. (2013). Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat. Rev. Genet. 14, 113–124. doi: 10.1038/nrg3366
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi: 10.1093/nar/27.2.573
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for llumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C.-H., Xie, D., et al. (2014). BEAST 2: a software platform for bayesian evolutionary analysis. PloS Comput. Biol. 10, e1003537. doi: 10.1371/journal.pcbi.1003537
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M., Borodovsky, M. (2021). BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinf. 3, lqaa108. doi: 10.1093/nargab/lqaa108
Buchfink, B., Xie, C., Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., Huerta-Cepas, J. (2021). eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/molbev/msab293
Carbonero, P., Iglesias-Fernández, R., Vicente-Carbajosa, J. (2017). The AFL subfamily of B3 transcription factors: evolution and function in angiosperm seeds. J. Exp. Bot. 68, 871–880. doi: 10.1093/jxb/erw458
Carey, S. B., Jenkins, J., Lovell, J. T., Maumus, F., Sreedasyam, A., Payton, A. C., et al. (2021). Gene-rich UV sex chromosomes harbor conserved regulators of sexual development. Sci. Adv. 7, eabh2488. doi: 10.1126/sciadv.abh2488
Charlesworth, B., Harvey, P. H., Charlesworth, B., Charlesworth, D. (2000). The degeneration of Y chromosomes. Philos. Trans. R. Soc Lond. B Biol. Sci. 355, 1563–1572. doi: 10.1098/rstb.2000.0717
Chen, Y., Li, C., Zhang, B., Yi, J., Yang, Y., Kong, C., et al. (2019). The role of the late embryogenesis-abundant (LEA) protein family in development and the abiotic stress response: a comprehensive expression analysis of potato (Solanum tuberosum). Genes (Basel) 10, 148. doi: 10.3390/genes10020148
Chen, M. M., Yang, S. R., Wang, J., Fang, Y. L., Peng, Y. L., Fan, J. (2022). Fungal oxysterol-binding protein-related proteins promote pathogen virulence and activate plant immunity. J. Exp. Bot. 73, 2125–2141. doi: 10.1093/jxb/erab530
Cox, C. (2018). Land plant molecular phylogenetics: a review with comments on evaluating incongruence among phylogenies. Crit. Rev. Plant Sci. 37, 113–127. doi: 10.1080/07352689.2018.1482443
Cruz-Loya, M., Kang, T. M., Lozano, N. A., Watanabe, R., Tekin, E., Damoiseaux, R., et al. (2019). Stressor interaction networks suggest antibiotic resistance co-opted from stress responses to temperature. ISME J. 13, 12–23. doi: 10.1038/s41396-018-0241-7
Dai, Z., Xiong, Y., Dai, X. (2014). Neighboring genes show interchromosomal colocalization after their separation. Mol. Biol. Evol. 31, 1166–1172. doi: 10.1093/molbev/msu065
Delahaie, J., Hundertmark, M., Bove, J., Leprince, O., Rogniaux, H., Buitink, J. (2013). LEA polypeptide profiling of recalcitrant and orthodox legume seeds reveals ABI3-regulated LEA protein abundance linked to desiccation tolerance. J. Exp. Bot. 64, 4559–4573. doi: 10.1093/jxb/ert274
Dierckxsens, N., Mardulyn, P., Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. doi: 10.1093/nar/gkw955
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Durand, N. C., Robinson, J. T., Shamim, M. S., Machol, I., Mesirov, J. P., Lander, E. S., et al. (2016a). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101. doi: 10.1016/j.cels.2015.07.012
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S. P., Huntley, M. H., Lander, E. S., et al. (2016b). Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98. doi: 10.1016/j.cels.2016.07.002
Echevarría-Zomeño, S., Fernández-Calvino, L., Castro-Sanz, A. B., López, J. A., Vázquez, J., Castellano, M. M. (2016). Dissecting the proteome dynamics of the early heat stress response leading to plant survival or death in Arabidopsis. Plant Cell Environ. 39, 1264–1278. doi: 10.1111/pce.12664
Edgar, R. C., Myers, E. W. (2005). PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158. doi: 10.1093/bioinformatics/bti1003
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Ferris, P., Olson, B. J., De Hoff, P. L., Douglass, S., Casero, D., Prochnik, S., et al. (2010). Evolution of an expanded sex-determining locus in Volvox. Science 328, 351–354. doi: 10.1126/science.1186222
Finka, A., Cuendet, A. F. H., Maathuis, F. J. M., Saidi, Y., Goloubinoff, P. (2012). Plasma membrane cyclic nucleotide gated calcium channels control land plant thermal sensing and acquired thermotolerance. Plant Cell 24, 3333–3348. doi: 10.1105/tpc.112.095844
Finka, A., Goloubinoff, P. (2014). The CNGCb and CNGCd genes from Physcomitrella patens moss encode for thermosensory calcium channels responding to fluidity changes in the plasma membrane. Cell Stress Chaperones 19, 83–90. doi: 10.1007/s12192-013-0436-9
Finka, A., Mattoo, R. U. H., Goloubinoff, P. (2011). Meta-analysis of heat- and chemically upregulated chaperone genes in plant and human cells. Cell Stress Chaperones 16, 15–31. doi: 10.1007/s12192-010-0216-8
Foote, H. C., Ride, J. P., Franklin-Tong, V. E., Walker, E., Lawrence, M. J., Franklin, F. C. H. (1994). Cloning and expression of a distinctive class of self-incompatibility (S) gene from Papaver rhoeas L. Proc. Natl. Acad. Sci. U.S.A. 91, 2265–2269. doi: 10.1073/pnas.91.6.2265
Freeling, M. (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453. doi: 10.1146/annurev.arplant.043008.092122
Fu, A., He, Z., Cho, H. S., Lima, A., Buchanan, B. B., Luan, S. (2007). A chloroplast cyclophilin functions in the assembly and maintenance of photosystem II in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 104, 15947–15952. doi: 10.1073/pnas.0707851104
Gao, B., Chen, M.-X., Li, X.-S., Liang, Y.-Q., Zhang, D.-Y., Wood, A. J., et al. (2022). Ancestral gene duplications in mosses characterized by integrated phylogenomic analyses. J. Syst. Evol. 60, 144–159. doi: 10.1111/jse.12683
Ghanbarian, A. T., Hurst, L. D. (2015). Neighboring genes show correlated evolution in gene expression. Mol. Biol. Evol. 32, 1748–1766. doi: 10.1093/molbev/msv053
Goffinet, B., Buck, W. R. (2013). The evolution of body form in bryophytes. Annu. Plant Rev. 45, 51–90. doi: 10.1002/9781118305881.ch2
Gu, Z., Eils, R., Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi: 10.1093/bioinformatics/btw313
Guillaumot, D., Lopez-Obando, M., Baudry, K., Avon, A., Rigaill, G., Falcon de Longevialle, A., et al. (2017). Two interacting PPR proteins are major Arabidopsis editing factors in plastid and mitochondria. Proc. Natl. Acad. Sci. U.S.A. 114, 8877–8882. doi: 10.1073/pnas.1705780114
Guo, C.-Q., Edwards, D., Wu, P.-C., Duckett, J. G., Hueber, F. M., Li, C.-S. (2012). Riccardiothallus devonicus gen. et sp. nov., the earliest simple thalloid liverwort from the Lower Devonian of Yunnan, China. Rev. Palaeobot. Palynol. 176–177, 35–40. doi: 10.1016/j.revpalbo.2012.03.012
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Harris, B. J., Clark, J. W., Schrempf, D., Szöllősi, G. J., Donoghue, P. C. J., Hetherington, A. M., et al. (2022). Divergent evolutionary trajectories of bryophytes and tracheophytes from a complex common ancestor of land plants. Nat. Ecol. Evol. 6, 1634–1643. doi: 10.1038/s41559-022-01885-x
Healey, A. L., Piatkowski, B., Lovell, J. T., Sreedasyam, A., Carey, S. B., Mamidi, S., et al. (2023). Newly identified sex chromosomes in the Sphagnum (peat moss) genome alter carbon sequestration and ecosystem dynamics. Nat. Plants 9, 238–254. doi: 10.1038/s41477-022-01333-5
Hendrawan, Y., Murase, H. (2011). Bio-inspired feature selection to select informative image features for determining water content of cultured Sunagoke moss. Expert Syst. Appl. 38, 14321–14335. doi: 10.1016/j.eswa.2011.05.097
Hurst, L. D., Pál, C., Lercher, M. J. (2004). The evolutionary dynamics of eukaryotic gene order. Nat. Rev. Genet. 5, 299–310. doi: 10.1038/nrg1319
Inupakutika, M. A., Sengupta, S., Devireddy, A. R., Azad, R. K., Mittler, R. (2016). The evolution of reactive oxygen species metabolism. J. Exp. Bot. 67, 5933–5943. doi: 10.1093/jxb/erw382
Iwasaki, M., Kajiwara, T., Yasui, Y., Yoshitake, Y., Miyazaki, M., Kawamura, S., et al. (2021). Identification of the sex-determining factor in the liverwort Marchantia polymorpha reveals unique evolution of sex chromosomes in a haploid system. Curr. Biol. 31, 5522–5532.e5527. doi: 10.1016/j.cub.2021.10.023
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., Walichiewicz, J. (2005). Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kerchev, P. I., Van Breusegem, F. (2022). Improving oxidative stress resilience in plants. Plant J. 109, 359–372. doi: 10.1111/tpj.15493
Kim, D., Paggi, J. M., Park, C., Bennett, C., Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. doi: 10.1186/gb-2013-14-4-r36
Kissen, R., Øverby, A., Winge, P., Bones, A. M. (2016). Allyl-isothiocyanate treatment induces a complex transcriptional reprogramming including heat stress, oxidative stress and plant defence responses in Arabidopsis thaliana. BMC Genomics 17, 740. doi: 10.1186/s12864-016-3039-x
Kotak, S., Larkindale, J., Lee, U., von Koskull-Döring, P., Vierling, E., Scharf, K.-D. (2007). Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 10, 310–316. doi: 10.1016/j.pbi.2007.04.011
Kulshrestha, S., Jibran, R., van Klink, J. W., Zhou, Y., Brummell, D. A., Albert, N. W., et al. (2022). Stress, senescence, and specialized metabolites in bryophytes. J. Exp. Bot. 73, 4396–4411. doi: 10.1093/jxb/erac085
Lan, X., Pritchard, J. K. (2016). Coregulation of tandem duplicate genes slows evolution of subfunctionalization in mammals. Science 352, 1009–1013. doi: 10.1126/science.aad8411
Lang, D., Ullrich, K. K., Murat, F., Fuchs, J., Jenkins, J., Haas, F. B., et al. (2018). The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J. 93, 515–533. doi: 10.1111/tpj.13801
Langfelder, P., Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 9, 559. doi: 10.1186/1471-2105-9-559
Lei, Y. B., Xia, H. X., Chen, K., Plenković-Moraj, A., Huang, W., Sun, G. (2021). Photosynthetic regulation in response to fluctuating light conditions under temperature stress in three mosses with different light requirements. Plant Sci. 311, 111020. doi: 10.1016/j.plantsci.2021.111020
Lesk, C., Rowhani, P., Ramankutty, N. (2016). Influence of extreme weather disasters on global crop production. Nature 529, 84–87. doi: 10.1038/nature16467
Li, F. W., Brouwer, P., Carretero-Paulet, L., Cheng, S., de Vries, J., Delaux, P. M., et al. (2018b). Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nat. Plants 4, 460–472. doi: 10.1038/s41477-018-0188-8
Li, Z., Defoort, J., Tasdighian, S., Maere, S., Van de Peer, Y., De Smet, R. (2016). Gene duplicability of core genes is highly consistent across all angiosperms. Plant Cell 28, 326–344. doi: 10.1105/tpc.15.00877
Li, B., Gao, K., Ren, H., Tang, W. (2018a). Molecular mechanisms governing plant responses to high temperatures. J. Integr. Plant Biol. 60, 757–779. doi: 10.1111/jipb.12701
Lin, Z., Xie, F., Triviño, M., Karimi, M., Bosch, M., Franklin-Tong, V. E., et al. (2020). Ectopic expression of a self-incompatibility module triggers growth arrest and cell death in vegetative cells. Plant Physiol. 183, 1765–1779. doi: 10.1104/pp.20.00292
Liu, Y., Johnson, M. G., Cox, C. J., Medina, R., Devos, N., Vanderpoorten, A., et al. (2019b). Resolution of the ordinal phylogeny of mosses using targeted exons from organellar and nuclear genomes. Nat. Commun. 10, 1485. doi: 10.1038/s41467-019-09454-w
Liu, B., Shi, Y., Yuan, J., Hu, X., Zhang, H., Li, N., et al. (2013). Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv. doi: 10.48550/arXiv.1308.2012
Liu, H., Xing, M., Yang, W., Mu, X., Wang, X., Lu, F., et al. (2019a). Genome-wide identification of and functional insights into the late embryogenesis abundant (LEA) gene family in bread wheat (Triticum aestivum). Sci. Rep. 9, 13375. doi: 10.1038/s41598-019-49759-w
Lobell, D. B., Schlenker, W., Costa-Roberts, J. (2011). Climate trends and global crop production since 1980. Science 333, 616–620. doi: 10.1126/science.1204531
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Marchetti, F., Cainzos, M., Cascallares, M., Distéfano, A. M., Setzes, N., López, G. A., et al. (2021). Heat stress in Marchantia polymorpha: sensing and mechanisms underlying a dynamic response. Plant Cell Environ. 44, 2134–2149. doi: 10.1111/pce.13914
Meiri, D., Tazat, K., Cohen-Peer, R., Farchi-Pisanty, O., Aviezer-Hagai, K., Avni, A., et al. (2010). Involvement of arabidopsis ROF2 (FKBP65) in thermotolerance. Plant Mol. Biol. 72 (1), 191–203. doi: 10.1007/s11103-009-9561-3
Miller, A.-F. (2012). Superoxide dismutases: ancient enzymes and new insights. FEBS Lett. 586, 585–595. doi: 10.1016/j.febslet.2011.10.048
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mittler, R. (2017). ROS are good. Trends Plant Sci. 22 (1), 11–19. doi: 10.1016/j.tplants.2016.08.002
Nützmann, H.-W., Huang, A., Osbourn, A. (2016). Plant metabolic clusters – from genetics to genomics. New Phytol. 211, 771–789. doi: 10.1111/nph.13981
Ochyra, R., Smith, R. I. L., Bednarek-Ochyra, H. (2008). The illustrated moss flora of Antarctica (Cambridge: Cambridge University Press).
Ohama, N., Sato, H., Shinozaki, K., Yamaguchi-Shinozaki, K. (2017). Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 22, 53–65. doi: 10.1016/j.tplants.2016.08.015
One Thousand Plant Transcriptomes Initiative (2019). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685. doi: 10.1038/s41586-019-1693-2
Pecrix, Y., Staton, S. E., Sallet, E., Lelandais-Brière, C., Moreau, S., Carrère, S., et al. (2018). Whole-genome landscape of Medicago truncatula symbiotic genes. Nat. Plants 4, 1017–1025. doi: 10.1038/s41477-018-0286-7
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Price, A. L., Jones, N. C., Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Puttick, M. N., Morris, J. L., Williams, T. A., Cox, C. J., Edwards, D., Kenrick, P., et al. (2018). The interrelationships of land plants and the nature of the ancestral embryophyte. Curr. Biol. 28, 733–745.e732. doi: 10.1016/j.cub.2018.01.063
Qiao, X., Li, Q., Yin, H., Qi, K., Li, L., Wang, R., et al. (2019). Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 20, 38. doi: 10.1186/s13059-019-1650-2
Rajasekar, K. V., Ji, S., Coulthard, R. J., Ride, J. P., Reynolds, G. L., Winn, P. J., et al. (2019). Structure of SPH (self-incompatibility protein homologue) proteins: a widespread family of small, highly stable, secreted proteins. Biochem. J. 476, 809–826. doi: 10.1042/bcj20180828
Ramachandran, S., Christensen, H. E. M., Ishimaru, Y., Dong, C.-H., Chao-Ming, W., Cleary, A. L., et al. (2000). Profilin plays a role in cell elongation, cell shape maintenance, and flowering in Arabidopsis. Plant Physiol. 124, 1637–1647. doi: 10.1104/pp.124.4.1637
Reimegård, J., Kundu, S., Pendle, A., Irish, V. F., Shaw, P., Nakayama, N., et al. (2017). Genome-wide identification of physically clustered genes suggests chromatin-level co-regulation in male reproductive development in Arabidopsis thaliana. Nucleic Acids Res. 45, 3253–3265. doi: 10.1093/nar/gkx087
Ren, H., Huang, R., Li, Y., Li, W., Zheng, L., Lei, Y., et al. (2023). Photosynthetic regulation in response to strontium stress in moss Racomitrium japonicum L. Environ. Sci. pollut. Res. 30, 20923–20933. doi: 10.1007/s11356-022-23684-4
Ride, J. P., Davies, E. M., Franklin, F. C. H., Marshall, D. F. (1999). Analysis of Arabidopsis genome sequence reveals a large new gene family in plants. Plant Mol. Biol. 39, 927–932. doi: 10.1023/A:1006178511787
Rütgers, M., Muranaka, L. S., Schulz-Raffelt, M., Thoms, S., Schurig, J., Willmund, F., et al. (2017). Not changes in membrane fluidity but proteotoxic stress triggers heat shock protein expression in Chlamydomonas reinhardtii. Plant Cell Environ. 40, 2987–3001. doi: 10.1111/pce.13060
Saddhe, A. A., Manuka, R., Penna, S. (2021). Plant sugars: homeostasis and transport under abiotic stress in plants. Physiol. Plant 171, 739–755. doi: 10.1111/ppl.13283
Sensalari, C., Maere, S., Lohaus, R. (2022). ksrates: positioning whole-genome duplications relative to speciation events in KS distributions. Bioinformatics 38, 530–532. doi: 10.1093/bioinformatics/btab602
Sepulveda, C., Guzmán, M. A., Li, Q., Villaécija-Aguilar, J. A., Martinez, S. E., Kamran, M., et al. (2022). KARRIKIN UP-REGULATED F-BOX 1 (KUF1) imposes negative feedback regulation of karrikin and KAI2 ligand metabolism in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S. A. 119, e2112820119. doi: 10.1073/pnas.2112820119
Silva, A. T., Gao, B., Fisher, K. M., Mishler, B. D., Ekwealor, J. T. B., Stark, L. R., et al. (2021). To dry perchance to live: Insights from the genome of the desiccation-tolerant biocrust moss Syntrichia caninervis. Plant J. 105, 1339–1356. doi: 10.1111/tpj.15116
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Su, D., Yang, L., Shi, X., Ma, X., Zhou, X., Hedges, S. B., et al. (2021). Large-scale phylogenomic analyses reveal the monophyly of bryophytes and Neoproterozoic origin of land plants. Mol. Biol. Evol. 38, 3332–3344. doi: 10.1093/molbev/msab106
Sun, P., Jiao, B., Yang, Y., Shan, L., Li, T., Li, X., et al. (2022a). WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 15, 1841–1851. doi: 10.1016/j.molp.2022.10.018
Sun, Y., Shang, L., Zhu, Q. H., Fan, L., Guo, L. (2022b). Twenty years of plant genome sequencing: achievements and challenges. Trends Plant Sci. 27, 391–401. doi: 10.1016/j.tplants.2021.10.006
Swindell, W. R., Huebner, M., Weber, A. P. (2007). Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genomics 8, 125. doi: 10.1186/1471-2164-8-125
Tan, Q. W., Lim, P. K., Chen, Z., Pasha, A., Provart, N., Arend, M., et al. (2023). Cross-stress gene expression atlas of Marchantia polymorpha reveals the hierarchy and regulatory principles of abiotic stress responses. Nat. Commun. 14, 986. doi: 10.1038/s41467-023-36517-w
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., Paterson, A. H. (2008). Synteny and collinearity in plant genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Tarailo-Graovac, M., Chen, N. (2009). Using repeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 25, 4.10.11–14.10.14. doi: 10.1002/0471250953.bi0410s25
Törönen, P., Medlar, A., Holm, L. (2018). PANNZER2: a rapid functional annotation web server. Nucleic Acids Res. 46, W84–W88. doi: 10.1093/nar/gky350
Van de Peer, Y., Mizrachi, E., Marchal, K. (2017). The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424. doi: 10.1038/nrg.2017.26
Vanneste, K., Van de Peer, Y., Maere, S. (2013). Inference of genome duplications from age distributions revisited. Mol. Biol. Evol. 30, 177–190. doi: 10.1093/molbev/mss214
Van Oss, S. B., Carvunis, A.-R. (2019). De novo gene birth. PloS Genet. 15, e1008160. doi: 10.1371/journal.pgen.1008160
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9, e112963. doi: 10.1371/journal.pone.0112963
Wang, Q. H., Zhang, J., Liu, Y., Jia, Y., Jiao, Y. N., Xu, B., et al. (2022). Diversity, phylogeny, and adaptation of bryophytes: insights from genomic and transcriptomic data. J. Exp. Bot. 73, 4306–4322. doi: 10.1093/jxb/erac127
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinf. 8, 77–80. doi: 10.1016/s1672-0229(10)60008-3
Waters, E. R., Vierling, E. (2020). Plant small heat shock proteins – evolutionary and functional diversity. New Phytol. 227, 24–37. doi: 10.1111/nph.16536
Xia, H., Chen, K., Liu, L., Plenkovic-Moraj, A., Sun, G., Lei, Y. (2022). Photosynthetic regulation in fluctuating light under combined stresses of high temperature and dehydration in three contrasting mosses. Plant Sci. 323, 111379. doi: 10.1016/j.plantsci.2022.111379
Xu, Z., Wang, H. (2007). LTR-FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Xu, Z., Xin, T., Bartels, D., Li, Y., Gu, W., Yao, H., et al. (2018). Genome analysis of the ancient tracheophyte Selaginella tamariscina reveals evolutionary features relevant to the acquisition of desiccation tolerance. Mol. Plant 11, 983–994. doi: 10.1016/j.molp.2018.05.003
Yin, Y., Qin, K., Song, X., Zhang, Q., Zhou, Y., Xia, X., et al. (2018). BZR1 transcription factor regulates heat stress tolerance through FERONIA receptor-like kinase-mdiated reactive oxygen species signaling in tomato. Plant Cell Physiol. 59, 2239–2254. doi: 10.1093/pcp/pcy146
Yu, J., Cai, Y., Zhu, Y., Zeng, Y., Dong, S., Zhang, K., et al. (2022). Chromosome-level genome assemblies of two Hypnales (mosses) reveal high intergeneric synteny. Genome Biol. Evol. 14, evac020. doi: 10.1093/gbe/evac020
Yu, G., Wang, L.-G., Han, Y., He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Zhang, J., Fu, X. X., Li, R. Q., Zhao, X., Liu, Y., Li, M. H., et al. (2020). The hornwort genome and early land plant evolution. Nat. Plants 6, 107–118. doi: 10.1038/s41477-019-0588-4
Zhang, Z., Xiao, J., Wu, J., Zhang, H., Liu, G., Wang, X., et al. (2012). ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 419, 779–781. doi: 10.1016/j.bbrc.2012.02.101
Zhao, C., Liu, B., Piao, S., Wang, X., Lobell, D. B., Huang, Y., et al. (2017). Temperature increase reduces global yields of major crops in four independent estimates. Proc. Natl. Acad. Sci. U.S.A. 114, 9326–9331. doi: 10.1073/pnas.1701762114
Zheng, J., Hong, K., Zeng, L., Wang, L., Kang, S., Qu, M., et al. (2020). Karrikin signaling acts parallel to and additively with strigolactone signaling to regulate rice mesocotyl elongation in darkness. Plant Cell 32, 2780–2805. doi: 10.1105/tpc.20.00123
Zheng, Y., Jiao, C., Sun, H., Rosli, H. G., Pombo, M. A., Zhang, P., et al. (2016). iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 9, 1667–1670. doi: 10.1016/j.molp.2016.09.014
Zheng, X. T., Wang, C., Lin, W., Lin, C., Han, D., Xie, Q., et al. (2022). Importation of chloroplast proteins under heat stress is facilitated by their SUMO conjugations. New Phytol. 235, 173–187. doi: 10.1111/nph.18121
Zhu, J. K. (2016). Abiotic stress signaling and responses in plants. Cell 167, 313–324. doi: 10.1016/j.cell.2016.08.029
Keywords: moss, genome assembly, heat stress, structural cluster, Niphotrichum japonicum
Citation: Zhou X, Peng T, Zeng Y, Cai Y, Zuo Q, Zhang L, Dong S and Liu Y (2023) Chromosome-level genome assembly of Niphotrichum japonicum provides new insights into heat stress responses in mosses. Front. Plant Sci. 14:1271357. doi: 10.3389/fpls.2023.1271357
Received: 02 August 2023; Accepted: 25 September 2023;
Published: 18 October 2023.
Edited by:
Antoni Garcia-Molina, Spanish National Research Council (CSIC), SpainReviewed by:
Josefa M. Alamillo, University of Cordoba, SpainSilvia Busoms, Autonomous University of Barcelona, Spain
Copyright © 2023 Zhou, Peng, Zeng, Cai, Zuo, Zhang, Dong and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shanshan Dong, c2hhbmdyaWxhc3NAMTYzLmNvbQ==; Yang Liu, eWFuZy5saXUwNTA4QGdtYWlsLmNvbQ==
†These authors have contributed equally to this work