
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 18 December 2023
Sec. Plant Pathogen Interactions
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1267132
 Yue Qiu1,2,3
Yue Qiu1,2,3 Fangjun Wei1,3Han Meng1,3Menglin Peng1,3Jinhao Zhang1,3Yilu He1,3Lanfang Wei1,4*
Fangjun Wei1,3Han Meng1,3Menglin Peng1,3Jinhao Zhang1,3Yilu He1,3Lanfang Wei1,4* Waqar Ahmed5*
Waqar Ahmed5* Guanghai Ji1,3*
Guanghai Ji1,3*Background: Angular leaf spot disease caused by plant pathogenic bacterium Xanthomonas fragariae seriously threatens strawberry crop production globally.
Methods: In this study, we sequenced the whole genome of X. fragariae YM2, isolated from Yunnan Province, China. In addition, we performed a comparative genome analysis of X. fragariae YM2 with two existing strains of X. fragariae YL19 and SHQP01 isolated from Liaoning and Shanghai, respectively.
Results: The results of Nanopore sequencing showed that X. fragariae YM2 comprises one single chromosome with a contig size of 4,263,697 bp, one plasmid contig size of 0.39 Mb, a GC content ratio of 62.27%, and 3,958 predicted coding genes. The genome of YM2 comprises gum, hrp, rpf, and xps gene clusters and lipopolysaccharide (LPS), which are typical virulence factors in Xanthomonas species. By performing a comparative genomic analysis between X. fragariae strains YM2, YL19, and SHQP01, we found that strain YM2 is similar to YL19 and SHQP01 regarding genome size and GC contents. However, there are minor differences in the composition of major virulence factors and homologous gene clusters. Furthermore, the results of collinearity analysis demonstrated that YM2 has lower similarity and longer evolutionary distance with YL19 and SHQP01, but YL19 is more closely related to SHQP01.
Conclusions: The availability of this high-quality genetic resource will serve as a basic tool for investigating the biology, molecular pathogenesis, and virulence of X. fragariae YM2. In addition, unraveling the potential vulnerabilities in its genetic makeup will aid in developing more effective disease suppression control measures.
Xanthomonas fragariae is a Gram-negative plant pathogenic bacterium that causes angular leaf spot (ALS) in strawberries (Henry and Leveau, 2016). It is a distinct and homogeneous species in the genus Xanthomonas with a narrow host range, and only Fragaria spp. are its natural hosts (Vandroemme et al., 2013). The leaves and calyx of strawberry plants are particularly vulnerable to infection (Wang et al., 2018). X. fragariae enters leaf tissue through stomata and wounds, colonizing the intercellular spaces between mesophyll and vascular tissues and causing discrete angular lesions and tissue discoloration in strawberry leaves, ultimately leading to plant death (Wang and Turechek, 2020).
X. fragariae was declared a quarantine pathogen by the Mediterranean Plant Protection Organization, the European Union, and Korea (Kim et al., 2016a). In 1960, X. fragariae was initially reported in the United States (Kennedy and King, 1962). Since then, the bacterium has widely spread to other strawberry-producing areas due to human activities and the import of plant material (Gétaz et al., 2018). ALS is continuing to spread worldwide despite the efforts made by plant protection organizations to halt its dispersal (Bestfleisch et al., 2015). It has become an increasing threat and a major factor affecting strawberry production (Roberts et al., 1997) and responsible for main yield losses (Kim et al., 2016b). In case of severe infection, ALS reduces the yield and destroys the fruit quality, making it unsuitable for the market (Wang et al., 2018). In Tianjin, China, the incidence of ALS in strawberries was first reported in 2016 (Wang et al., 2017). Since then, this disease has been observed in multiple provinces of China, including Taiwan, Liaoning, Shanghai, and Yunnan (Wu et al., 2020; Feng et al., 2021; Song et al., 2021; Zhang et al., 2022). Therefore, ALS is a great concern to the strawberry industry and deserves more attention, as it spreads to new regions.
The development of DNA sequencing technology has led to an abundance of genomic data, providing us with the basic resources to fully explore the genetic diversity and pathogenic mechanism of a pathogen. The first draft genome of X. fragariae (LMG25863) was published in 2013, providing basic information about its biological characteristics and genetic information (Vandroemme et al., 2013). It was found that compared with other pathogenic Xanthomonas spp., X. fragariae has a narrow host range and a smaller genome and lacks some genes related to pathogenicity. However, it still contains major virulence-related gene regions. So far, 74 genome sequences of X. fragariae are available in the National Center for Biotechnology Information (NCBI) database, of which nine are complete genomes. As additional genome sequences emerge, we will better understand the genomic diversity and virulence variations among X. fragariae strains.
In September 2021, symptoms of ALS in strawberries were observed in the strawberry-growing regions of Yuxi and Kunming Cities, Yunnan Province, China (Zhang et al., 2022). Infected leaves were collected for bacterial isolation, and on Wilbrink-N agar medium plates, round, dense, and yellow color colonies were obtained (a typical feature of Xanthomonadaceae). According to Koch’s postulates, a pot experiment confirmed the pathogenicity of the isolated strain using the strawberry cultivar Monterey, and it was identified as X. fragariae YM2 based on molecular analysis (Zhang et al., 2022). This study aimed to sequence the whole genome of X. fragariae YM2 and to analyze its virulence-related gene content. In addition, a comparative genome analysis was performed with two existing strains of X. fragariae YL19 and SHQP01, isolated from Liaoning and Shanghai, respectively (Feng et al., 2021; Song et al., 2021). Our findings will serve as a useful tool for future research into X. fragariae virulence, particularly in elucidating its pathogenic mechanisms at the molecular level, which is essential for limiting its prevalence.
X. fragariae YM2 (accession no. OP847193) was previously isolated from the ALS-infected strawberry leaves collected from Yunnan Province, China (Zhang et al., 2022). The genome of X. fragariae YM2 was sequenced in this study, while genomes of X. fragariae YL19, X. fragariae SHQP01, and other relative strains were downloaded from the NCBI database (Supplementary Table 1). X. fragariae YM2 was grown on Wilbrink-N agar medium plates (proteose peptone 5 g/L, sucrose 10 g/L, MgSO4·7H2O 0.25 g/L, K2HPO4 0.5 g/L, NaNO3 0.25 g/L, agar 18 g/L, and pH 7.0) at 28°C for 72 h (Zhang et al., 2022). A 50% (v/v) glycerol solution was used to store pure cultures of bacterial strains at −80°C for later use.
The total genomic DNA of X. fragariae YM2 was extracted from the overnight culture grown at 28°C and 160 rpm in Wilbrink-N broth using a QIAGEN Genomic-tip kit according to the manufacturer’s instructions (Liu et al., 2022). The genome was sequenced using the Nanopore PromethION platform at Biomarker Technologies (Beijing, China). The filtered reads were assembled using Canu (v1.5) software (Koren et al., 2017), and then the circlator (v1.5.5) was used to identify the circular sequence in the assembled genome. The complete genome sequence of X. fragariae YM2 was deposited in the NCBI (https://www.ncbi.nlm.nih.gov/) database under GenBank accession number CP114897.
Prodigal (v2.6.3) software was used for coding gene prediction analysis (Hyatt et al., 2010). After masking putative functional genes, the GenBlastA (v1.0.4) tool scanned the complete genomes, and putative candidates were evaluated for non-mature and frame-shift mutations using GeneWise (v2.2.0). Transfer RNA (tRNA) gene prediction analysis was performed using tRNAscan-SE (v2.0) (Chan and Lowe, 2019), while ribosomal RNA (rRNA) gene prediction analysis was performed using Infernal (v1.1.3) (Nawrocki and Eddy, 2013). RepeatMasker was used to predict repetitive sequences. PhiSpy (v2.3) was used for prophage prediction, and CRT (v1.2) was employed for CRISPR identification. To predict genomic islands in the genome, IslandPath-DIMOB (v0.2) was utilized. The predicted proteins were blasted (e-value: 1e−5) against Nr, Swiss-Prot, TrEMBL, Kyoto Encyclopedia of Genes and Genomes (KEGG), eggNOG, and Blast2go for functional annotation. Based on the Comprehensive Antibiotic Research Database (CARD), RGI software was used to identify the corresponding sequences and to obtain antibiotic-resistant genes (Aziz et al., 2008). The genes related to virulence factors were annotated using the VFDB databases, and finally, the RAST server was used for fully automated annotation of the bacterial genomes (Aziz et al., 2008).
Four housekeeping genes, including atpD, dnaK, gyrB, and rpoD, were chosen for multilocus sequence typing (MLST) analysis (Young et al., 2008; Parkinson et al., 2009; Bui Thi Ngoc et al., 2010). TBtools was used to obtain the sequences of these four genes from the genomes of X. fragariae YM2, which were then concatenated in the alphabetical order of the genes (Chen et al., 2020). Mega-X was used to perform sequence alignment, trimming, and phylogenetic analysis (Kumar et al., 2018). A multigene phylogenetic tree was constructed using the maximum likelihood method based on the four housekeeping gene concatenated sequences (Ahmed et al., 2022). JSpeciesWS was used to compute the average nucleotide consistency (ANI) analysis and digital DNA–DNA hybridization (dDDH) values (Richter et al., 2016). Genome-to-genome comparison was performed using the Genome-to-Genome Distance Calculator (GGDC; v3.0) according to default parameters (Meier-Kolthoff et al., 2013).
The main virulence factors, orthologous genes, and genome collinearity analyses were performed to better understand the differences among X. fragariae strains YM2, YL19, and SHQP01. OrthoVenn online service was used to determine the Orthologous gene clusters (Wang et al., 2015), and TBtools (v1.120) was used to analyze the genomic synteny (Chen et al., 2020).
AntiSMASH 5.0 with a web server was used to identify probable secondary metabolite biosynthesis gene clusters in the genomes of X. fragariae YM2, YL19, and SHQP01 (Blin et al., 2019). The GenBank databases were used to gain more detailed gene cluster information.
The whole genome of X. fragariae YM2 was sequenced in this study, resulting in a total of 163,167 sequence reads with 1,441,130,510 bases. The final assembled genome comprised a single circular chromosome with an entire length of 4,263,697 bp and one plasmid contig (0.39 Mb) (Figure 1). The average GC content of the YM2 genome was found to be 62.27%, which is similar to YL19 (62.75%) and SHQP01 (62.0%) (Table 1). There were 3,958 coding genes with a length of 3,606,930 bp containing 59,507 bp repetitive sequences, accounting for 1.4% of genome sequences. Approximately 215 RNA-encoding genes were predicted, including six genes encoded for ribosomal RNAs, 54 encoded for transfer RNAs, and 155 identified as non-coding RNAs. The basic genome of YM2 was predicted to encode 20 pseudogenes with a length of 17,029 bp.
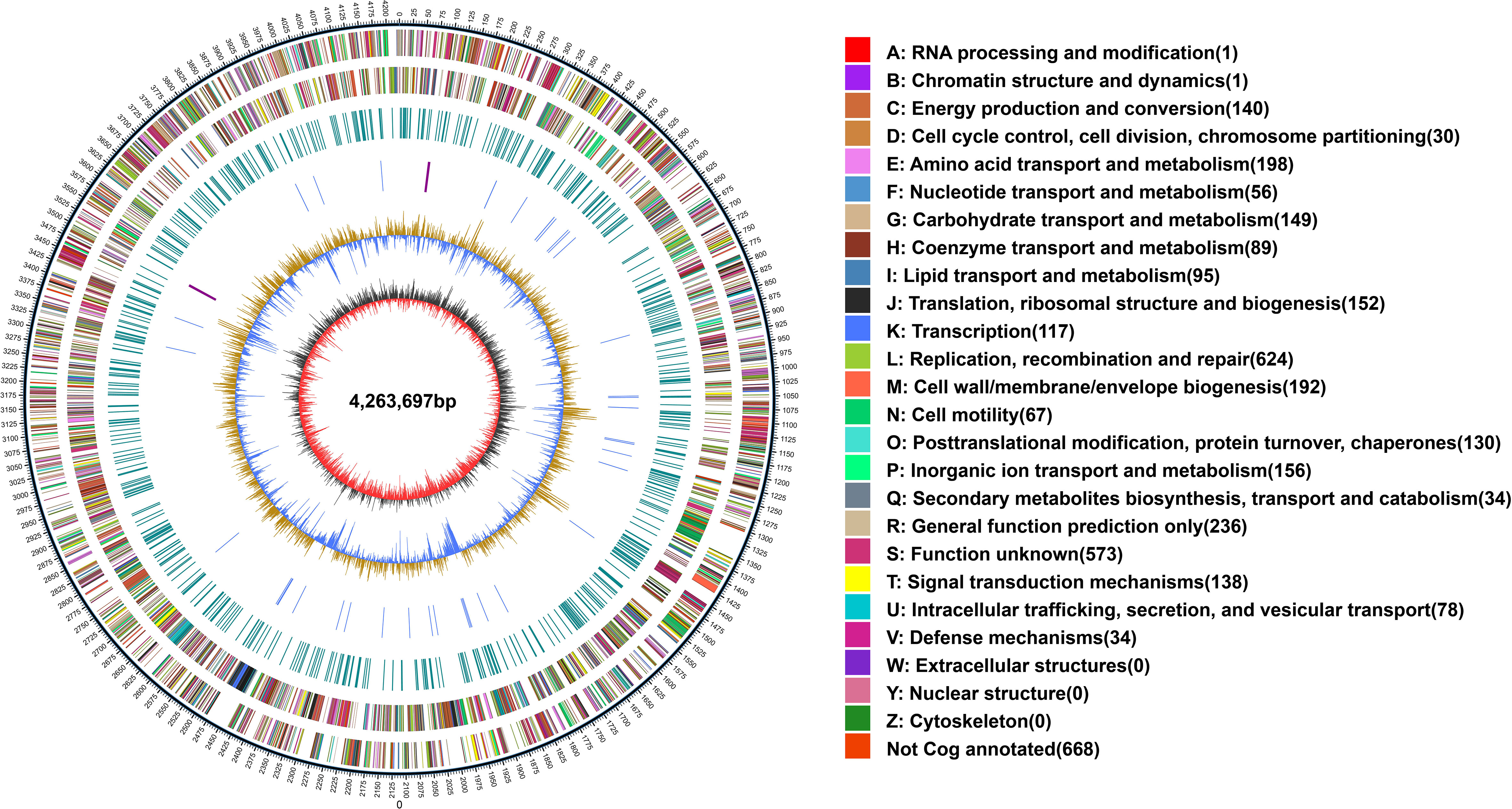
Figure 1 Whole-genome map of Xanthomonas fragariae YM2. The outermost circle is the mark of genome size, with each scale of 5 kb. The second and third circles are genes on the positive and negative chains of the genome, respectively. Different colors represent different COG functional classifications. The fourth circle is the repeat sequence. The fifth circle is tRNA and rRNA; blue is tRNA, and purple is rRNA. The sixth circle is the GC content. The innermost circle is GC skew. tRNA, transfer RNA; rRNA, ribosomal RNA.
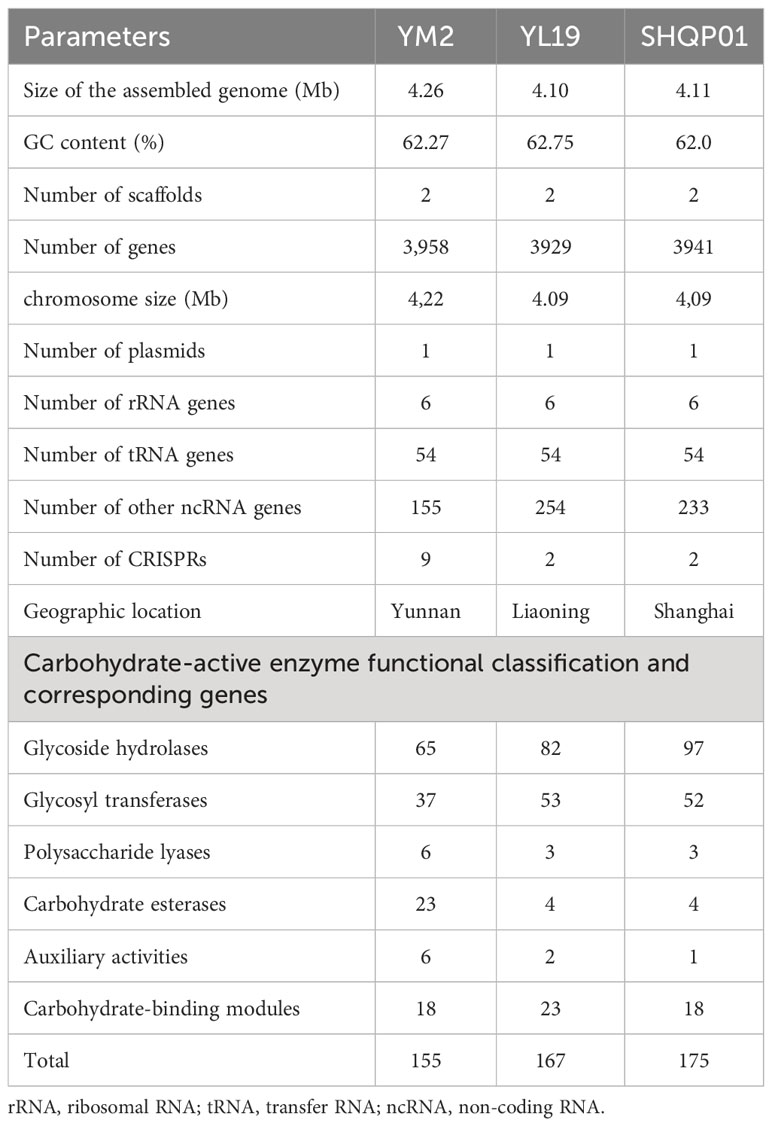
Table 1 The general genomic features of YM2, YL19, and SHQP01.
The functional classification of DNA coding sequences (CDSs) in YM2 was analyzed using several major databases, and the results showed that 3,936 genes were annotated as functional genes, accounting for 94.44% of the total genes (Supplementary Table 2). There were 3,289 protein-encoding genes in the eggNOG database, 2,543 in the Gene Ontology (GO) database, and 2,166 in the KEGG database. The eggNOG classification annotation showed that the genes for replication, recombination, and repair were the most abundant, with 624 genes accounting for 18.57%, followed by 573 genes of unknown function accounting for 17.05% (Figure 2). The GO classification annotation results indicated that 2,543 genes were annotated into 39 subclasses of three main categories: biological processes, cellular components, and molecular functions. Further analysis revealed that 38.39% of predicted genes were related to molecular function (mainly catalytic activity, binding, and transporter), 24.71% were involved in cellular components (mostly cell, cell part, membrane, and membrane part), and 36.9% were concentrated in biological processes (most of in metabolic process, cellular process, single organism process, and localization) (Figure 3). KEGG pathway enrichment analysis revealed that 2,166 genes were functionally annotated in 113 metabolic pathways (Supplementary Table 3). According to KEGG pathway annotation results, most genes were involved in four categories: genetic information process, metabolism, environmental information processing, and cellular processes (Figure 4). In the genetics information processing, most genes (54) were present in the ribosome (ko03010) pathway. The category metabolism contains the highest number of genes, among which 107 and 89 genes were involved in the biosynthesis of amino acid (ko01230) and carbon metabolism (ko01200) pathways, respectively. In environmental information processing and cellular processes, maximum genes of 97 and 34 were present in the two-component system (ko02020) and flagellar assembly (ko02040) pathways, respectively (Figure 4).
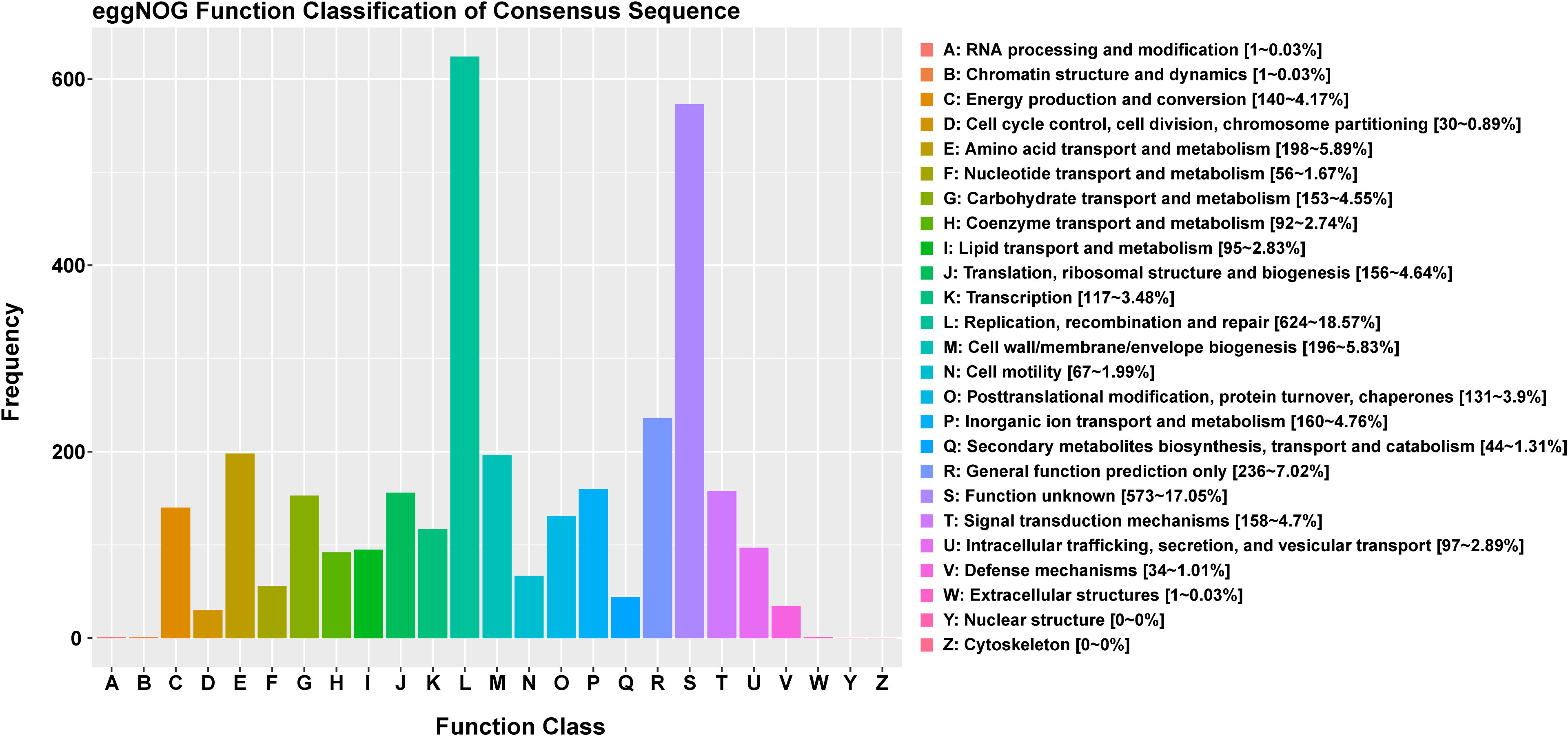
Figure 2 The eggNOG function classification of the consensus sequence.
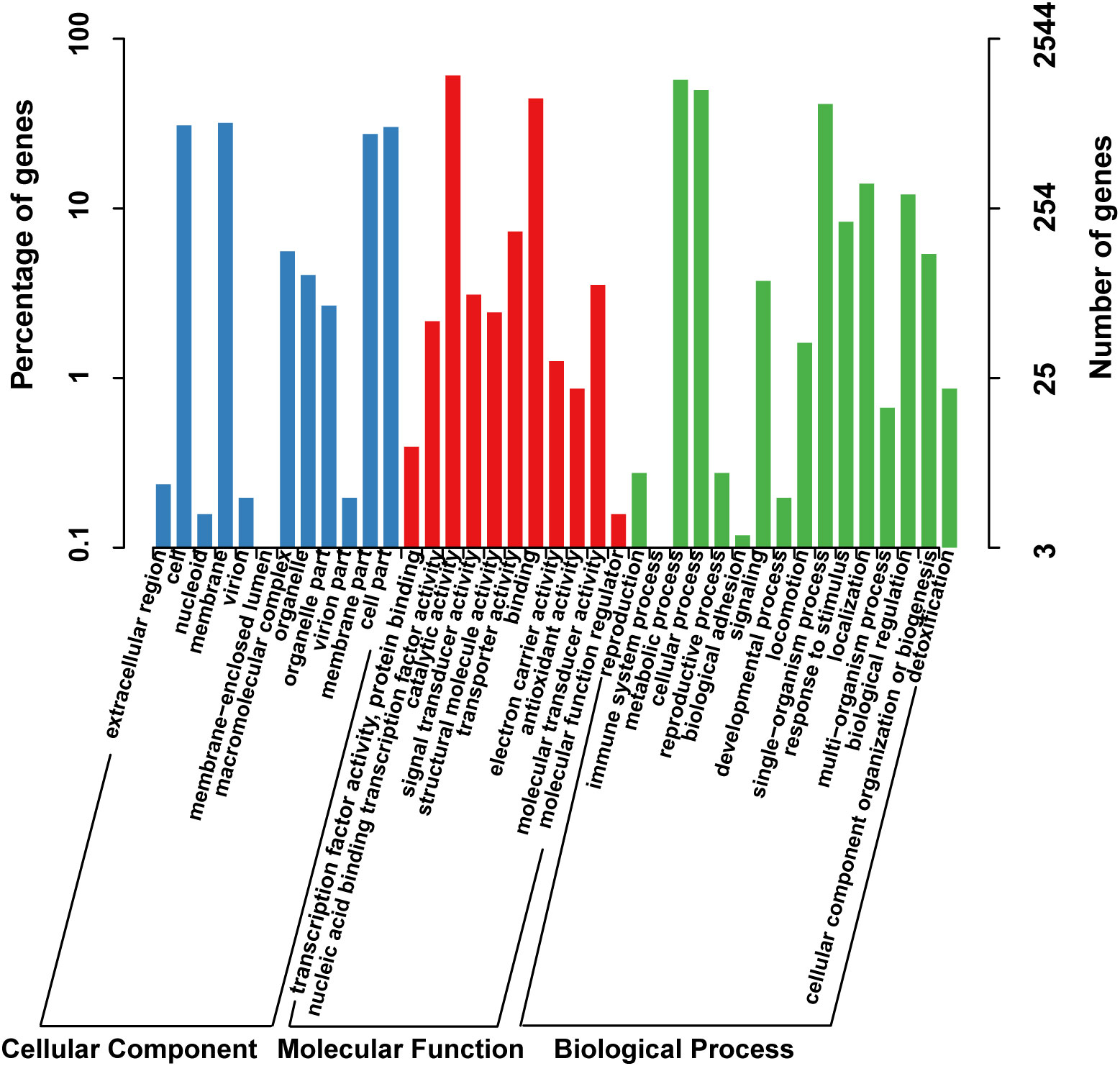
Figure 3 The GO function classification of the consensus sequence. GO, Gene Ontology.
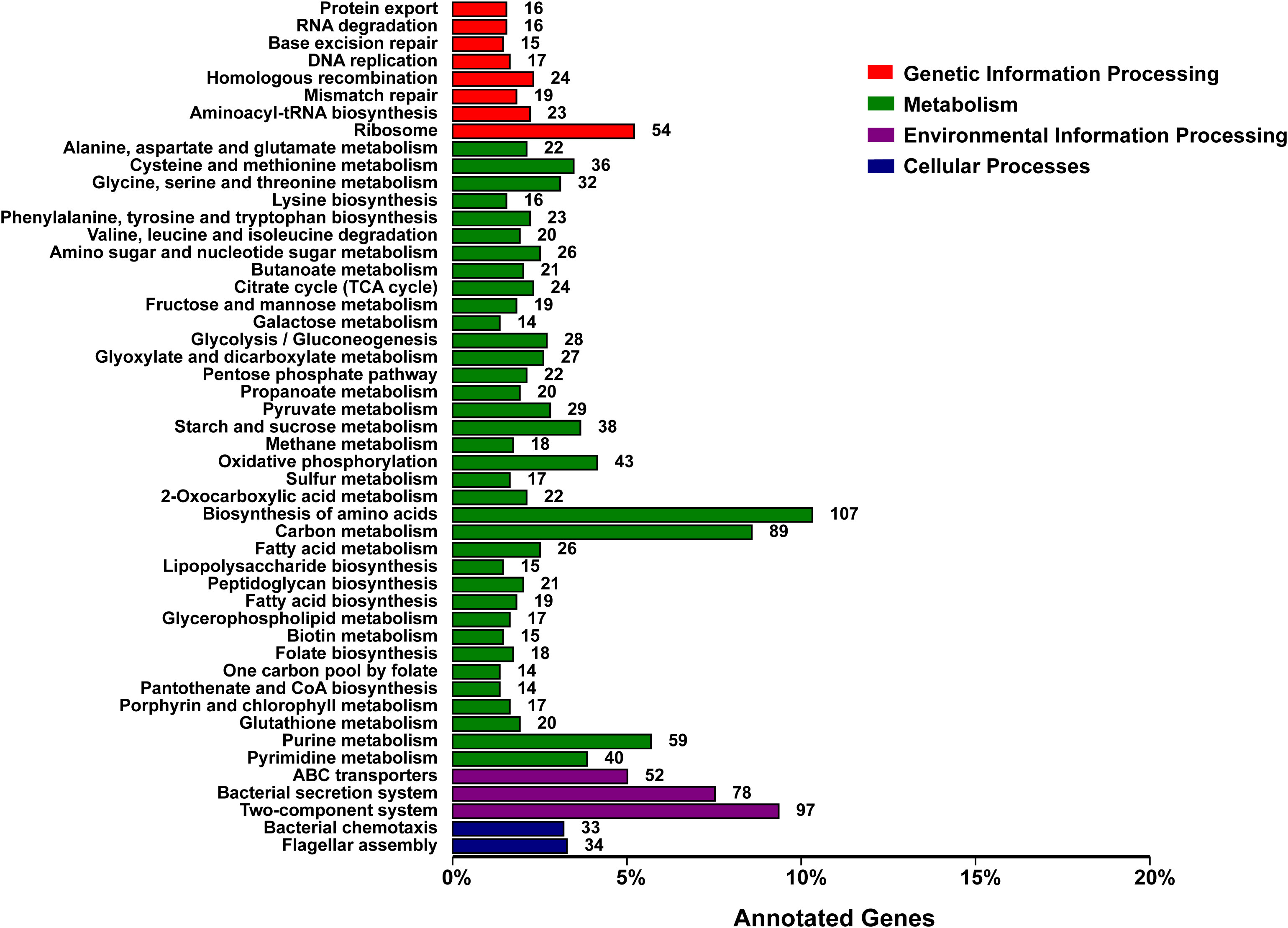
Figure 4 KEGG function classification of the consensus sequence. KEGG, Kyoto Encyclopedia of Genes and Genomes.
Partial nucleotide sequences of four housekeeping genes (atpD, dnaK, gyrB, and rpoD) from YM2 and related species were aligned, trimmed, and concatenated to a final sequence of 7,656-bp nucleotide positions to infer a maximum likelihood phylogenetic tree. The dendrogram constructed using multilocus sequence analysis (MLSA) showed that strain YM2 was distributed in a separate branch along with other strains of X. fragariae Fap29, Fap21, PD8805, and PD5205, which represented a sister species with 100% bootstrap support value. In contrast, the strains YL19 and SHQP01 were located in another branch (Figure 5). ANI is mainly used to evaluate DNA similarity indexes between species at the genome-wide level. We conducted an ANI analysis between YM2 and six other X. fragariae strains present in the same clade in the phylogenetic tree. The results of the ANI analysis are consistent with the phylogenetic relationship (Table 2). YM2 has an ANI value of 94.69% with SHQP01 and 94.68% with YL19, slightly lower than the threshold of 95% for species differentiation. Meanwhile, the ANI value between strain YM2 and other X. fragariae strains (Fap29, Fap21, PD8805, and PD5205) was higher than 99% (Table 2). However, all the DNA–DNA hybridization (DDH) values among the strain sequenced in this study and those of the reference strains were consistently higher than 70.0% (Table 2). Based on all analytical results, the isolate YM2 causing ALS was identified as X. fragariae.
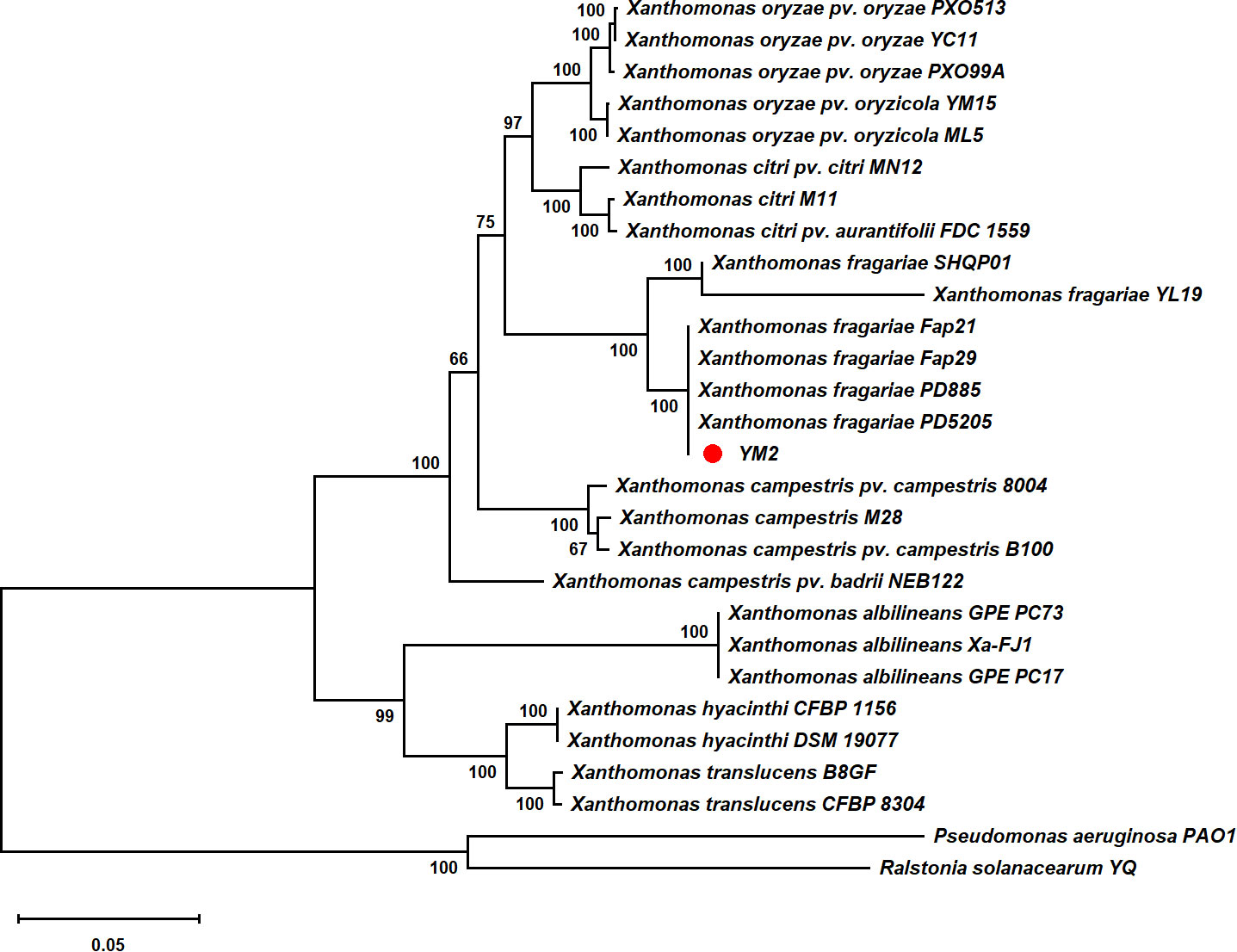
Figure 5 Maximum likelihood tree constructed with concatenated partial sequences of four housekeeping genes (atpD, dnaK, gyrB, and rpoD) of Xanthomonas fragariae. The tree scale represents 0.050.
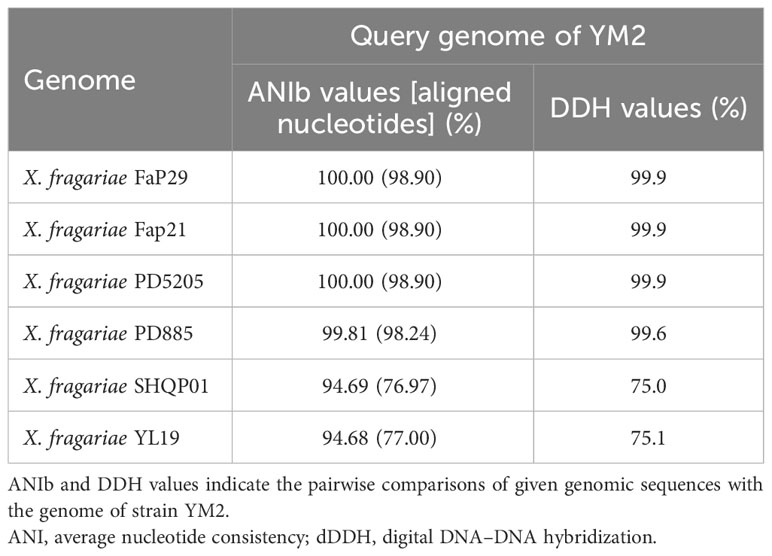
Table 2 ANI and dDDH values among the YM2 and other Xanthomonas fragariae strains.
To comprehensively understand the distinctions between the three strains, YM2, YL19, and SHQP01, isolated from Yunnan, Liaoning, and Shanghai in China, respectively, we conducted a comparative investigation of their genome content and genome structure. The general genomic features of strains YM2, YL19, and SHQP01 are listed in Table 1. Genomic analysis revealed that YL19 is the most closely related to SHQP01 and the least closely related to YM2. Genome comparisons utilizing information from the RAST and SEED-Viewer databases revealed that the metabolic network and subsystem profiles of the YL19 and SHQP01 strains were nearly identical. In contrast, those of the YM2 strain differed slightly (Figure 6). The number of subsystems that are responsible for resistance to antibiotics and toxic compounds, Type IV (protein and nucleoprotein secretion system), DNA metabolism, stress response system, amino acids and derivatives, phosphorus metabolism, carbohydrates, and the miscellaneous subsystems in strain YM2 was higher than that in YL19 and SHQP01. The number of subsystems responsible for riboflavin biosynthesis, isoprenoid metabolism, and respiration subsystems in strain YM2 was lower than that in YL19 and SHQP01.

Figure 6 Comparison of genomic features of three Xanthomonas fragariae strains using the online annotation service RAST.
OrthoVenn online service determined the orthologous gene clusters among the three genomes. The results showed that 2,574 gene clusters containing 7,849 proteins were shared among the three strains (Figure 7). Furthermore, YL19 and SHQP01 shared 490 gene clusters but shared only one and two genes with YM2, respectively, suggesting that the genomic repertoires of YL19 and SHQP01 are closer. We found that strain YM2 contained 60 unique gene clusters containing 425 proteins, while no gene cluster and protein are unique to the YL19 and SHQP01 genomes (Supplementary Table 4).
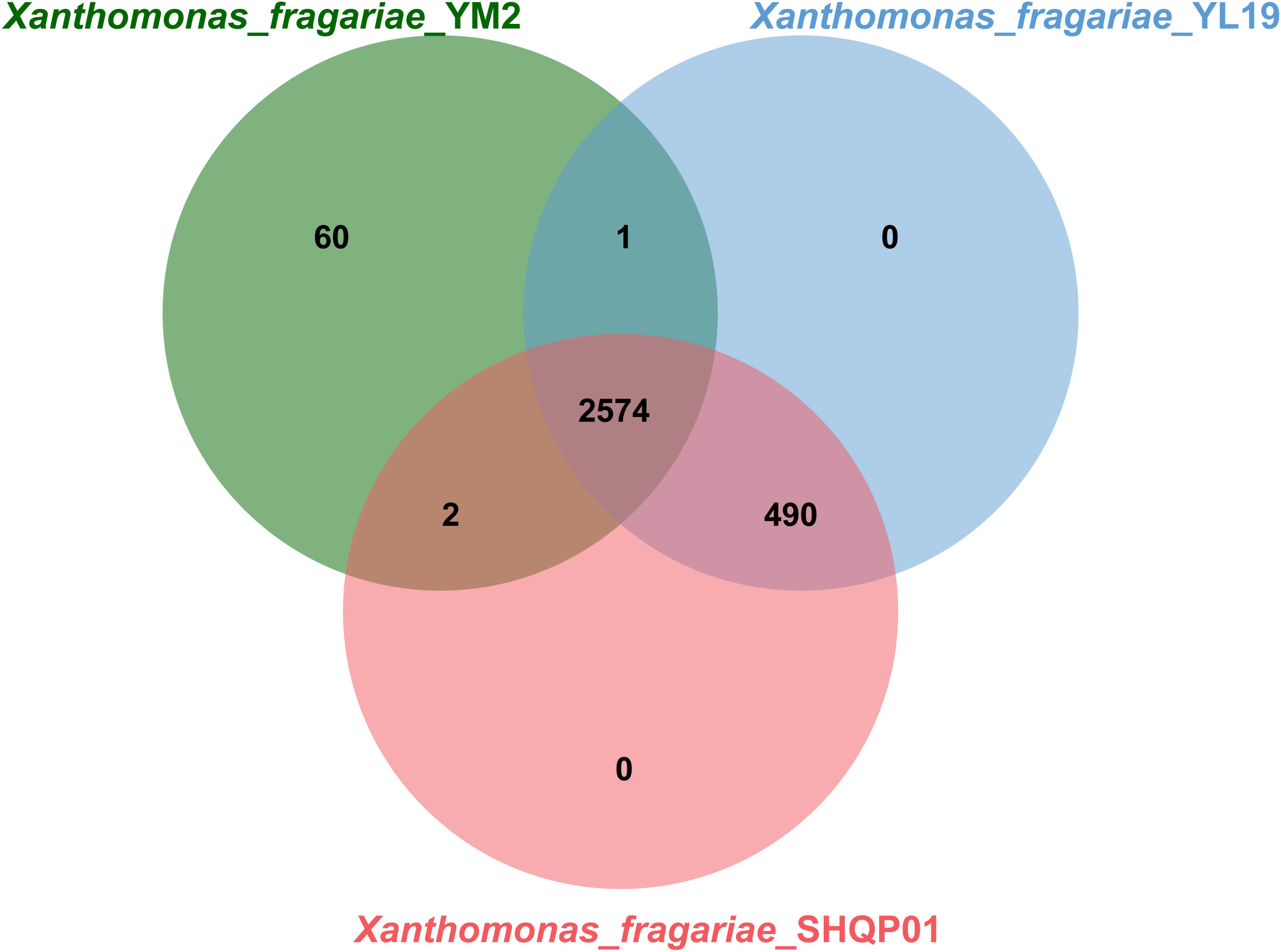
Figure 7 Venn diagram showing the shared orthologous clusters among the three Xanthomonas fragariae strains.
Collinearity analysis revealed a number of homologous regions in the YM2 and YL19 genomes, but some regions were rearranged and inverted (Figure 8). Similar results were found between the YM2 and SHQP01 genomes. The number of collinear genes between YM2 and YL19, and YM2 and SHQP01 was 2,705 and 2,701, respectively (Supplementary Table 5). YL19 and SHQP01 were the two strains with the most similar genome structure, with 3,614 collinear genes between them (Figure 8). The degree of collinearity between two species can be used to calculate their evolutionary distance. The better the genetic collinearity, the closer the evolutionary gap between species. Strain YM2 has lower similarity and longer evolutionary distance with strains YL19 and SHQP01, while YL19 has high similarity, good collinearity, and a higher degree of common origin compared with SHQP01 (Figure 8).
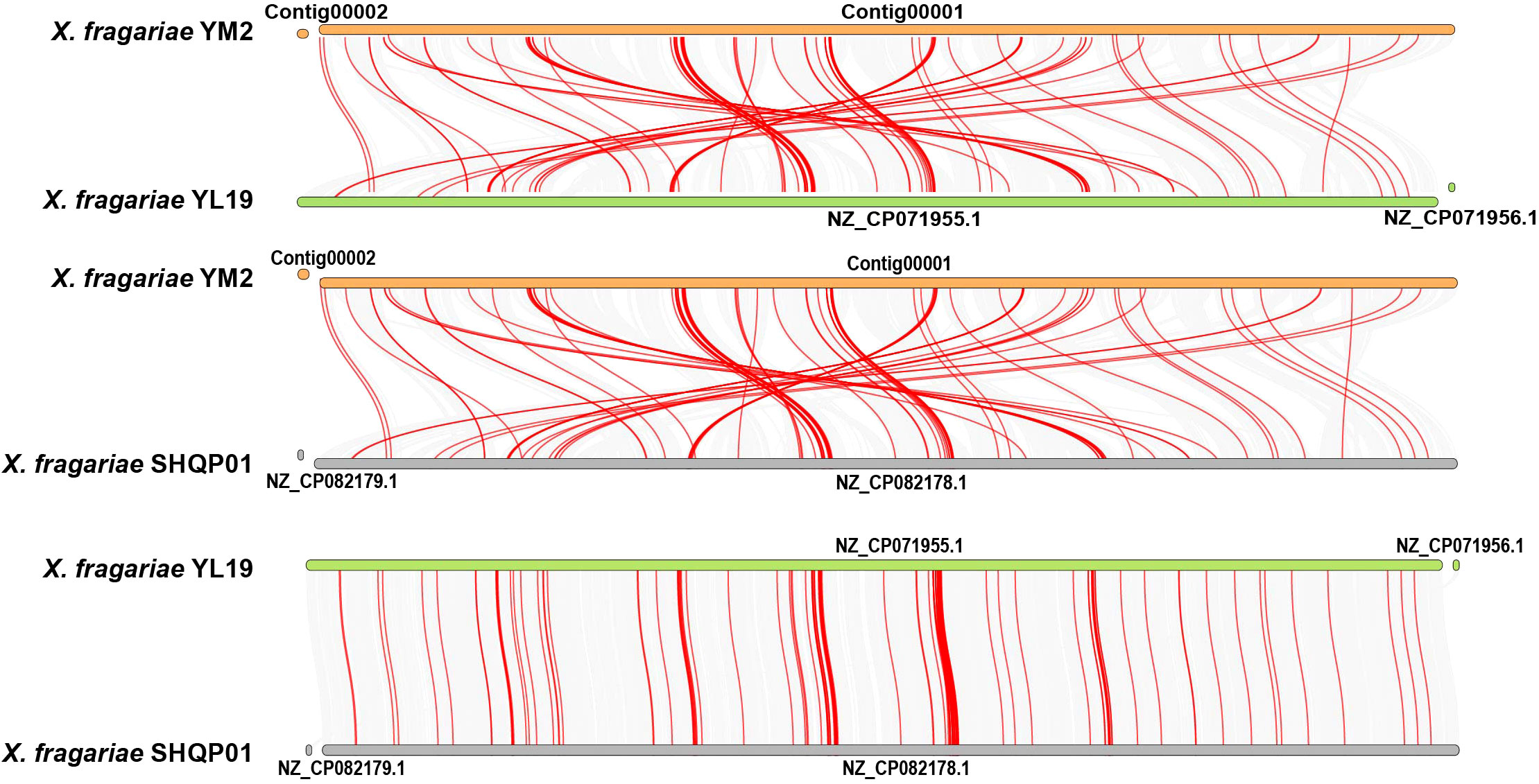
Figure 8 Synteny analyses of the genomes of three strains of Xanthomonas fragariae. Gray lines on the background indicate the collinear blocks between X. fragariae strains, and red lines highlight the syntenic major virulence-related gene pairs.
A total of 659 known or putatively linked genes encoding virulence factors were found in the genome of X. fragariae YM2. These virulence factors encode the genes related to carbohydrate-active enzymes (CAZymes), extracellular polysaccharides (EPSs) (encoding by gum gene cluster), lipopolysaccharide (LPS), type III secretion system (encoding by hrp gene cluster), T3SEs (encoding by xop gene cluster), type II secretion system (encoding by xps gene cluster), adhesins, methylase of chemotaxis methyl-accepting protein (encoding by cheR4 gene), and others (Supplementary Table 6). Interestingly, xcs genes that code for T2SS were completely absent in the YM2 genome. In particular, no antibiotic-resistant genes were found in the YM2 genome after annotating via CARD.
Polysaccharides such as cellulose, hemicellulose, pectin, and lignin are woven to form the complex network of the plant cell wall. These polysaccharides are broken down by CAZymes, which provide nutrients to pathogenic bacteria. In our study, we conducted a comprehensive analysis of putative CAZymes, which revealed that the glycoside hydrolase (GH; 65 genes) family was the major category in the X. fragariae YM2 genome, followed by glycosyl transferases (GTs; 37 genes) and carbohydrate esterases (CEs; 23 genes). The total number of CAZyme genes (155) was lower relative to that in YL19 (167) and SHQP01 (175) (Table 1). According to the distribution of family and gene number in CAZyme classes, YL19 and SHQP01 are very similar, but YM2 is very different. The number of GHs and GTs in YM2 was lower than that in YL19 and SHQP01. In contrast, the number of CEs, auxiliary activities (AAs), and polysaccharide lyases (PLs) was higher in YM2 (Table 1).
Many phytopathogenic bacteria produce large amounts of EPSs and LPSs during invasion in the host plants. The highly conserved gum gene cluster, which consists of 12 genes ranging from gumB to gumM, regulates EPS production (Lu et al., 2008). The YM2 genome possesses seven of 12 genes, including gumB, gumC, gumD, gumF, gumG, gumJ, and gumM. In addition, for the genes in the rpf gene cluster, which positively regulate the synthesis of extracellular enzymes, YM2 contains only four genes: rpfE, rpfF, rpfG, and rpfN. Except for gumG genes, which were not found in YL19 and SHQP01, the rest of gum genes and rpf genes had high similarity with the homologous genes in genomes of YL19 and SHQP01 (Supplementary Table 6). LPS is a component of bacterial cell surfaces that plants frequently identify as pathogen-associated molecular patterns (PAMPs). LPA comprises lipid A, core oligosaccharide, and O antigen polysaccharide encoded by lpx, waa, and wb gene clusters, respectively. Twenty-two genes associated with encoding LPS or LPS components were found in the genome of YM2, including wzm and wzt (Supplementary Table 6). However, three genes (gtrB, wzm, and wzt) out of 22 genes were not found in the YL19 and SHQP01 genomes. The other 19 genes were found to have higher homology in both the YL19 and SHQP01 genomes.
Plant pathogenic Gram-negative bacteria have evolved special export systems to target virulence factors in their hosts, and type III secretion system (TTSS) is one of them. The hrp gene cluster encoding TTSS in plant pathogens has been renamed as hrp, hrp-conserved (hrc), and hrp-associated (hpa) (Noël et al., 2002). The hrp gene cluster harbored in the YM2 genome contains 14 genes, including six hpa (hpa1, hpa2, hpa3_1, hpa3_2, hpaB, and hpaP), five hrp (hrpB, hrpD6, hrpG, hrpW, and hrpX), and three hrc (hrcA, hrcR, and hrcS). However, the genes coding for type III-secreted proteins HrpB2 and HrpF were absent.
More virulence and effector genes that influence host–pathogen interactions were investigated. In total, 1,168 PHI coding genes, 29.5% of the YM2 genome, were identified. Moreover, 367 secreted proteins, 367 signal peptides, and 813 transmembrane proteins were predicted. Two other genes, rtxD and rtxE, encoding repeat toxin (RTX) proteins with cytotoxic and hemolytic activity, were also present in the YM2 genome. In contrast, rtxD was absent in the genomes of YL19 and SHQP01. Iron is a necessary element for bacterial growth. Several gene clusters responsible for iron uptake and transport have been found in the YM2 genome: feoA and feoB are related to Fe2+ capture; nine fepA genes are responsible for transport siderophores; fhuA and fhuE are involved in the absorption of ferrichrome. All the iron uptake and transport genes were also present in the YL19 and SHQP01 genomes.
The result of antiSMASH analysis showed that the genome of YM2 harbored four candidate gene clusters: siderophores, Redox-cofactor, arylpolyene, and non-ribosomal peptide synthetase (NRPS) (Supplementary Table 7). The first gene cluster was predicted to be the siderophore biosynthetic gene clusters associated with the biosynthesis of xanthoferrin. The arylpolyene cluster is associated with the biosynthesis of xanthomonadin, which plays an important role in epiphytic survival and host–pathogen interactions in the phytopathogenic Xanthomonas bacteria. However, the NRPS exhibited no similar gene cluster with the known strain. The genomes of YL19 and SHQP01 have the same number of secondary metabolic clusters, four more than YM2, including two other unspecified ribosomally synthesized and post-translationally modified peptide product (RiPP-like) clusters, and a cluster (lantipeptide class I) associated with glycopeptidolipid biosynthetic and an NRPS (Figure 9).
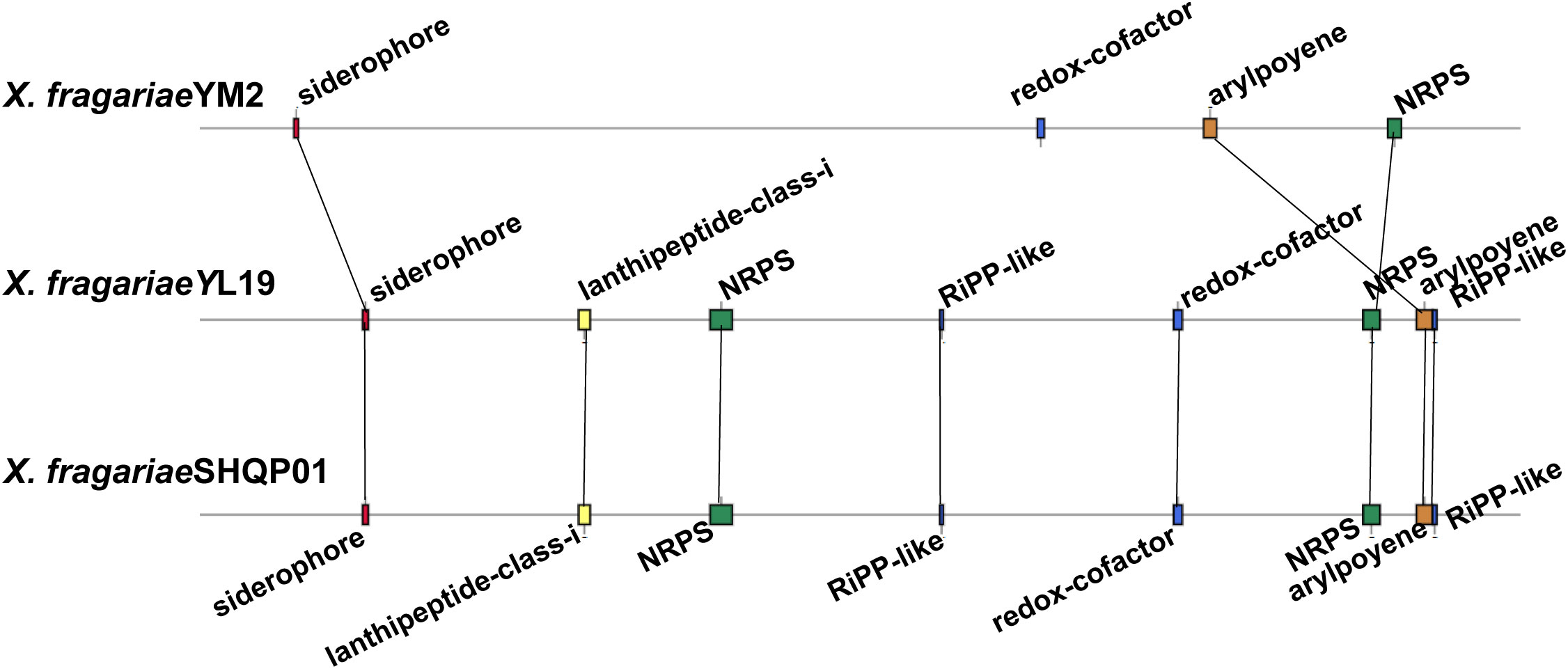
Figure 9 Comparison of the secondary metabolite biosynthesis gene clusters of Xanthomonas fragariae YM2 with X. fragariae YL19 and X. fragariae SHQP01. Black lines connect collinear regions.
Strawberry (Fragaria × ananassa) is one of the most popular cultivated fruits in the world because of its pleasing flavor and nutritional content (Zhang et al., 2011; Gétaz et al., 2020). To meet the domestic market demand, the cultivation area of strawberries has gradually expanded. China has become the world’s largest producer and consumer of strawberries, accounting for more than 50% of the world’s total output (Guan et al., 2022). However, strawberries are susceptible to various phytopathogenic organisms (Amil-Ruiz et al., 2011); one of the most important phytopathogenic organisms is X. fragariae. Once X. fragariae is allowed to multiply and expand in the field, it can devastate strawberry growth, cause yield loss, reduce quality, and make the fruit unmarketable (Wang et al., 2018). Moreover, the risk of ALS being introduced into new geographic areas is high because X. fragariae can persist in planting material for long periods asymptomatically. Many cases of intercepted infectious substances have confirmed the spread of this disease (Gétaz et al., 2018). Thus, to prevent the further spread of the disease, highly specific and effective detection is crucial. Further, the development of efficient disease management strategies is also needed to avoid significant economic losses.
X. fragariae can overwinter on old leaves or plant debris of strawberries, making it a source of infection in the coming year. The optimal temperature for growth of X. fragariae is ≈20°C, and the maximum is less than 32°C (Roberts et al., 1997). Thus, this bacterium grows well in the normal summer temperature in Yunnan Province, China. Therefore, there is a high probability of strawberry angular leaf spot outbreak during summer in Yunnan. Currently, no chemical or biological control agents effectively control the disease (Van Der Wolf et al., 2018). In addition, the excessive application of chemical pesticides can result in food safety concerns. Therefore, more studies are needed to assess its pathogenicity, genetic diversity, and control strategies to achieve maximum disease control while avoiding pesticide residues causing harm to humans.
Genome comparison is a valuable tool for studying the strain-to-strain variation in virulence, pathogenicity, and host fitness (Chen et al., 2022; Mafakheri et al., 2022). In this study, we sequenced the complete genome of X. fragariae YM2 and compared it with X. fragariae YL19 and X. fragariae SHQP01. The results showed that these three genomes were very similar in size, number of predicted genes, rRNAs, and tRNA genes but were significantly different in the number of other non-coding RNA (ncRNA) genes, CRISPRs, and CAZyme genes. Therefore, these differences may contribute to the differences in virulence between X. fragariae strains. The CRISPR–Cas system is an adaptive immune system that widely exists in prokaryotes and possesses resistance to foreign plasmids and phage sequences (Louwen et al., 2014). The current study found that YM2 contains more CRISPRs than YL19 and SHQP01; thus, we speculated that YM2 may have a stronger survival ability and may even be more difficult to control. CAZymes play an important role in plant pathogens’ invasion of plant tissues due to their ability to decompose plant cell walls and degrade macromolecular substances. The total number of CAZyme genes in YL19 is higher than that in YM2, suggesting that YL19 may have a stronger ability to decompose cell walls and stronger aggression than YM2. This result affirms the previous reports that YL19 not only causes ALS in strawberries but also causes crown infection pocket symptoms (Feng et al., 2021).
The three strains, YL19, SHQP01, and YM2, were chronologically isolated from Liaoning, Shanghai, and Yunnan, China, respectively. Interestingly, the MLSA, ANI analysis, and DDH analysis showed that YL19 and SHQP01 had a relatively close evolutionary relationship. At the same time, YM2 was more closely related to PD5205 (isolated in the Netherlands) and three other X. fragariae strains, PD885, Fap29, and Fap21 (all isolates of the USA) (Henry and Leveau, 2016). The ANI and DDH have been used as the standard for prokaryotic species classification at the genomic level when the ANI and DDH values between the compared strains were higher than the accepted thresholds defined for prokaryotic species, i.e., ANI > 95% and DDH > 70%, suggesting that the compared strains were the same species (Goris et al., 2007). Our results showed that YM2 has an ANI value of 94.69% with SHQP01 and 94.68% with YL19. These values are slightly lower than the cutoff score of 95%; however, according to the research of Konstantinidis and Tiedje (2005) and Richter and Rosselló-Móra (2009), this can still be classified as the same species. These results are consistent with Wei et al. (2023), who proved that YL19 and SHQP01 belong to the new subspecies X. fragariae.
Collinearity analysis is a standard investigative strategy used in comparative genomic studies to understand genomic conservation and the evolutionary relationships between multiple species (Wang et al., 2012). The results of genomic collinearity analysis suggest that YL19 and SHQP01 exhibit strong collinearity with each other. However, they both exhibit significant genome rearrangements in comparison to YM2. These differences may have led to the diversity of X. fragariae, which might be due to the evolution of the strains in different ecological environments. Compared with other pathogenic Xanthomonas, X. fragariae has a smaller genome and lacks some pathogenicity-related genes, which may explain why this bacterium is generally considered a mild pathogen. This is probably due to the protective mechanism of X. fragariae to ensure its long-term survival within the host.
Like X. fragariae LMG25863 (Vandroemme et al., 2013), YM2 also lacks genes coding for T2SS and gum-associated genes gumN, gumO, and gumP, but these genes were proved unnecessary for virulence of Xanthomonas spp. (Lu et al., 2008; Szczesny et al., 2010). The YM2 genome contains seven gum genes responsible for extracellular polysaccharide synthesis, xps genes for type II secretion, hrp genes for type III secretion, rpf genes for regulation of pathogenicity factors, and genes involved in the synthesis of lipopolysaccharide. These factors are considered the main pathogenicity/virulence factors in Xanthomonas spp. (An et al., 2020). Many previous studies have focused on exploring the functions of virulence factors. The deletion gene in the gum gene cluster reduced the production of xanthan gum (Vojnov et al., 2001) and biofilm formation (Rigano et al., 2007; Li and Wang, 2011). The rpf gene cluster regulates the synthesis of extracellular enzymes, extracellular polysaccharides, biofilm dispersal, and virulence (Dow et al., 2000; Dharmapuri et al., 2001). Similarly, RpfF directs the synthesis of DSF, and deletion of rpfF gene in Xanthomonas oryzae pv. oryzae leads to loss of virulence on rice (Chatterjee and Sonti, 2002), while disruption of rpfG reduces virulence in Xanthomonas campestris pv. campestris (Ryan et al., 2006). The genes wzm and wzt are predicted to encode components of an ABC transporter system to export LPS (Lu et al., 2008).
X. fragariae is classified as a quarantine pathogen and one of the most destructive pathogens of strawberries worldwide, particularly in China. In the study, we sequenced the complete genome of X. fragariae YM2; analyzed its genetic characteristics, taxonomic location, and virulence factors; and compared it with other X. fragariae strains, YL19 and SHQP01. According to the assembled and annotated results, we found 3,958 genes encoded by the YM2 genome with one chromosome. We observed that some gene families and clusters are important for the pathogenicity of X. fragariae. This in-depth study will serve as a great reference for future gene functional research and comparative genomic studies involving differences between species. Additionally, it will shed light on the pathogenicity of X. fragariae during host–pathogen interactions. Overall, the fully sequenced X. fragariae genome is a major step forward in bacterial genomics. However, future research on the construction of knock-out involving pathogenicity-related genes will provide new insights into the virulence of X. fragariae YM2 at the molecular level.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. The complete genome sequences for YM2 have been deposited at GenBank under the accession number (CP114897).
YQ: Data curation, Formal Analysis, Software, Writing – original draft. FW: Data curation, Formal Analysis, Writing – original draft. HM: Writing – Original draft, Formal Analysis, Software. MP: Data curation, Formal Analysis, Writing – original draft. JZ: Data curation, Investigation, Methodology, Writing – original draft. YH: Formal Analysis, Validation, Writing – original draft. LW: Conceptualization, Investigation, Writing – review & editing. WA: Supervision, Validation, Writing – original draft, Writing – review & editing. GJ: Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article. This study was financially supported by the Yunnan Provincial Science and Technology Department, Basic Research Project-Key Project (202301AS070080), National Natural Science Foundation of China (No. 32060601 and No. 32350410423), and the Yunnan Ten Thousand Talents Plan Leading Talents of Industrial Technology Project of China (YNWR-CYJS-2019-046).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1267132/full#supplementary-material
Ahmed, W., Zhou, G., Yang, J., Munir, S., Ahmed, A., Liu, Q., Zhao, Z., Ji, G. (2022). Bacillus amyloliquefaciens WS-10 as a potential plant growth-promoter and biocontrol agent for bacterial wilt disease of flue-cured tobacco. Egyptian J. Biological Pest Control 32(1), 1–14. doi: 10.1186/s41938-022-00527-5
Amil-Ruiz, F., Blanco-Portales, R., Muñoz-Blanco, J., Caballero, J. L. (2011). The strawberry plant defense mechanism: a molecular review. Plant Cell Physiol. 52, 1873–1903. doi: 10.1093/pcp/pcr136
An, S. Q., Potnis, N., Dow, M., Vorhölter, F. J., He, Y. Q., Becker, A., et al. (2020). Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas. FEMS Microbiol. Rev. 44, 1–32. doi: 10.1093/femsre/fuz024
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75. doi: 10.1186/1471-2164-9-75
Bestfleisch, M., Richter, K., Wensing, A., Wünsche, J., Hanke, M. V., Höfer, M., et al. (2015). Resistance and systemic dispersal of Xanthomonas fragariae in strawberry germplasm (Fragaria L.) Plant Pathology 64, 71–80. doi: 10.1111/ppa.12232
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–w87. doi: 10.1093/nar/gkz310
Bui Thi Ngoc, L., Vernière, C., Jouen, E., Ah-You, N., Lefeuvre, P., Chiroleu, F., et al. (2010). Amplified fragment length polymorphism and multilocus sequence analysis-based genotypic relatedness among pathogenic variants of Xanthomonas citri pv. citri and Xanthomonas campestris pv. bilvae. Int. J. Syst. Evol. Microbiol. 60, 515–525. doi: 10.1099/ijs.0.009514-0
Chan, P. P., Lowe, T. M. (2019). tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chatterjee, S., Sonti, R. V. (2002). rpfF mutants of Xanthomonas oryzae pv. oryzae are deficient for virulence and growth under low iron conditions. Mol. Plant Microbe Interact. 15, 463–471. doi: 10.1094/MPMI.2002.15.5.463
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, D., Zhong, X., Cui, J., Li, H., Han, R., Yue, X., et al. (2022). Comparative Genomic Analysis of Xanthomonas campestris pv. campestris Isolates BJSJQ20200612 and GSXT20191014 Provides Novel Insights Into Their Genetic Variability and Virulence. Front. Microbiol. 13, 833318. doi: 10.3389/fmicb.2022.833318
Dharmapuri, S., Yashitola, J., Vishnupriya, M. R., Sonti, R. V. (2001). Novel genomic locus with atypical G+C content that is required for extracellular polysaccharide production and virulence in Xanthomonas oryzae pv. oryzae. Mol. Plant Microbe Interact. 14, 1335–1339. doi: 10.1094/MPMI.2001.14.11.1335
Dow, J. M., Feng, J. X., Barber, C. E., Tang, J. L., Daniels, M. J. (2000). Novel genes involved in the regulation of pathogenicity factor production within the rpf gene cluster of Xanthomonas campestris. Microbiol. (Reading) 146 (Pt 4), 885–891. doi: 10.1099/00221287-146-4-885
Feng, J., Li, Y. L., Wang, D. J., Ma, Y., Cai, X., Xiao, S., et al. (2021). First Report of Xanthomonas fragariae strain YL19 causing crown infection pockets in strawberry in Liaoning Province, China. Plant Dis. 105(8), p.2237. doi: 10.1094/PDIS-12-20-2560-PDN
Gétaz, M., Krijger, M., Rezzonico, F., Smits, T. H. M., van der Wolf, J. M., Pothier, J. F. (2018). Genome-based population structure analysis of the strawberry plant pathogen Xanthomonas fragariae reveals two distinct groups that evolved independently before its species description. Microb. Genom 4(7):e000189. doi: 10.1099/mgen.0.000189
Gétaz, M., Puławska, J., Smits, T. H. M., Pothier, J. F. (2020). Host-Pathogen Interactions between Xanthomonas fragariae and Its Host Fragaria × ananassa Investigated with a Dual RNA-Seq Analysis. Microorganisms 8(8), 1253. doi: 10.3390/microorganisms8081253
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., Tiedje, J. M. (2007). DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. systematic evolutionary Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Guan, W., Haseman, D., Ingwell, L., Egel, D. S. (2022). Strawberry cultivar evaluation for fall-planted high tunnel system. HortTechnology 32, 542–551. doi: 10.21273/HORTTECH05103-22
Henry, P. M., Leveau, J. H. (2016). Finished genome sequences of xanthomonas fragariae, the cause of bacterial angular leaf spot of strawberry. Genome Announc 4(6), 10–1128. doi: 10.1128/genomeA.01271-16
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Kennedy, B., King, T. J. P. (1962). Angular leafspot of Strawberry caused by Xanthomonas fragariae sp. Phytopathology, 52(9), 873–875.
Kim, D. R., Gang, G. H., Jeon, C. W., Kang, N. J., Lee, S. W., Kwak, Y. S. (2016a). Epidemiology and control of strawberry bacterial angular leaf spot disease caused by xanthomonas fragariae. Plant Pathol. J. 32, 290–299. doi: 10.5423/PPJ.OA.01.2016.0007
Kim, M. S., Jin, J. S., Kwak, Y. S., Hwang, G. S. (2016b). Metabolic Response of Strawberry (Fragaria x ananassa) Leaves Exposed to the Angular Leaf Spot Bacterium (Xanthomonas fragariae). J. Agric. Food Chem. 64, 1889–1898. doi: 10.1021/acs.jafc.5b05201
Konstantinidis, K. T., Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547. doi: 10.1093/molbev/msy096
Li, J., Wang, N. (2011). Genome-wide mutagenesis of Xanthomonas axonopodis pv. citri reveals novel genetic determinants and regulation mechanisms of biofilm formation. PloS One 6, e21804. doi: 10.1371/journal.pone.0021804
Liu, Q., Yang, J., Ahmed, W., Wan, X., Wei, L., Ji, G. (2022). Exploiting the antibacterial mechanism of phenazine substances from Lysobacter antibioticus 13-6 against Xanthomonas oryzae pv. oryzicola. J. Microbiol. 60, 496–510. doi: 10.1007/s12275-022-1542-0
Louwen, R., Staals, R. H., Endtz, H. P., Van Baarlen, P., van der Oost, J. (2014). The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 78, 74–88. doi: 10.1128/MMBR.00039-13
Lu, H., Patil, P., Van Sluys, M. A., White, F. F., Ryan, R. P., Dow, J. M., et al. (2008). Acquisition and evolution of plant pathogenesis-associated gene clusters and candidate determinants of tissue-specificity in xanthomonas. PloS One 3, e3828. doi: 10.1371/journal.pone.0003828
Mafakheri, H., Taghavi, S. M., Zarei, S., Rahimi, T., Hasannezhad, M. S., Portier, P., et al. (2022). Phenotypic and molecular-phylogenetic analyses reveal distinct features of crown gall-associated xanthomonas strains. Microbiol. Spectr. 10, e0057721. doi: 10.1128/spectrum.00577-21
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14, 60. doi: 10.1186/1471-2105-14-60
Nawrocki, E. P., Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Noël, L., Thieme, F., Nennstiel, D., Bonas, U. (2002). Two novel type III-secreted proteins of Xanthomonas campestris pv. vesicatoria are encoded within the hrp pathogenicity island. J. Bacteriol 184, 1340–1348. doi: 10.1128/JB.184.5.1340-1348.2002
Parkinson, N., Cowie, C., Heeney, J., Stead, D. (2009). Phylogenetic structure of Xanthomonas determined by comparison of gyrB sequences. Int. J. Syst. Evol. Microbiol. 59, 264–274. doi: 10.1099/ijs.0.65825-0
Richter, M., Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Rigano, L. A., Siciliano, F., Enrique, R., Sendín, L., Filippone, P., Torres, P. S., et al. (2007). Biofilm formation, epiphytic fitness, and canker development in Xanthomonas axonopodis pv. citri. Mol. Plant Microbe Interact. 20, 1222–1230. doi: 10.1094/MPMI-20-10-1222
Roberts, P. D., Berger, R. D., Jones, J. B., Chandler, C. K., Stall, R. E. (1997). Disease progress, yield loss, and control of xanthomonas fragariae on strawberry plants. Plant Dis. 81, 917–921. doi: 10.1094/PDIS.1997.81.8.917
Ryan, R. P., Fouhy, Y., Lucey, J. F., Crossman, L. C., Spiro, S., He, Y.-W., et al. (2006). Cell–cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. 103, 6712–6717. doi: 10.1073/pnas.0600345103
Song, Z., Yang, C., Zeng, R., Gao, S. G., Cheng, W., Gao, P., et al. (2021). First report of strawberry crown rot caused by xanthomonas fragariae in China. Plant Dis. 105(9), 2711. doi: 10.1094/PDIS-03-21-0574-PDN
Szczesny, R., Jordan, M., Schramm, C., Schulz, S., Cogez, V., Bonas, U., et al. (2010). Functional characterization of the Xcs and Xps type II secretion systems from the plant pathogenic bacterium Xanthomonas campestris pv vesicatoria. New Phytol. 187, 983–1002. doi: 10.1111/j.1469-8137.2010.03312.x
Van Der Wolf, J. M., Evenhuis, A., Kastelein, P., Krijger, M. C., Funke, V. Z., Van Den Berg, W., et al. (2018). Risks for infection of strawberry plants with an aerosolized inoculum of Xanthomonas fragariae. Eur. J. Plant Pathol. 152, 711–722. doi: 10.1007/s10658-018-1513-9
Vandroemme, J., Cottyn, B., Baeyen, S., De Vos, P., Maes, M. (2013). Draft genome sequence of Xanthomonas fragariae reveals reductive evolution and distinct virulence-related gene content. BMC Genomics 14, 829. doi: 10.1186/1471-2164-14-829
Vojnov, A. A., Slater, H., Daniels, M. J., Dow, J. M. (2001). Expression of the gum operon directing xanthan biosynthesis in Xanthomonas campestris and its regulation in planta. Mol. Plant Microbe Interact. 14, 768–774. doi: 10.1094/MPMI.2001.14.6.768
Wang, Y., Coleman-Derr, D., Chen, G., Gu, Y. Q. (2015). OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 43, W78–W84. doi: 10.1093/nar/gkv487
Wang, H., Mctavish, C., Turechek, W. W. (2018). Colonization and movement of xanthomonas fragariae in strawberry tissues. Phytopathology 108, 681–690. doi: 10.1094/PHYTO-10-17-0356-R
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49. doi: 10.1093/nar/gkr1293
Wang, H., Turechek, W. W. (2020). Detection of viable xanthomonas fragariae cells in strawberry using propidium monoazide and long-amplicon quantitative PCR. Plant Dis. 104, 1105–1112. doi: 10.1094/PDIS-10-19-2248-RE
Wang, J., Wei, H., Chang, R., Liu, H., Wang, Y. (2017). First report of strawberry bacterial angular leaf spot caused by Xanthomonas fragariae in Tianjin, China. Plant Dis. 101, 1949–1949. doi: 10.1094/PDIS-02-17-0182-PDN
Wei, F., Liang, X., Shi, J.-C., Luo, J., Qiu, L.-J., Li, X.-X., et al. (2023). Pan-genomic analysis identifies the chinese strain as a new subspecies of xanthomonas fragariae. Plant Disease. 37555725 doi: 10.1094/PDIS-05-23-0933-SC
Wu, H. Y., Lai, Q. J., Wu, Y. M., Chung, C. L., Chung, P. C., Lin, N. C. (2020). First Report of Xanthomonas fragariae Causing Angular Leaf Spot on Strawberry (Fragaria x ananassa) in Taiwan. Plant Dis. 105(4), 1187–1187. doi: 10.1094/PDIS-07-20-1631-PDN
Young, J. M., Park, D. C., Shearman, H. M., Fargier, E. (2008). A multilocus sequence analysis of the genus Xanthomonas. Syst. Appl. Microbiol. 31, 366–377. doi: 10.1016/j.syapm.2008.06.004
Zhang, J., He, Y., Ahmed, W., Wan, X., Wei, L., Ji, G. (2022). First report of bacterial angular leaf spot of strawberry caused by xanthomonas fragariae in yunnan province, China. Plant Dis. 106(7), 1978. doi: 10.1094/PDIS-12-21-2648-PDN
Keywords: Xanthomonas fragariae, collinearity analysis, functional annotation, virulence factors, genome comparison
Citation: Qiu Y, Wei F, Meng H, Peng M, Zhang J, He Y, Wei L, Ahmed W and Ji G (2023) Whole-genome sequencing and comparative genome analysis of Xanthomonas fragariae YM2 causing angular leaf spot disease in strawberry. Front. Plant Sci. 14:1267132. doi: 10.3389/fpls.2023.1267132
Received: 26 July 2023; Accepted: 27 November 2023;
Published: 18 December 2023.
Edited by:
Harold Meijer, Wageningen University and Research, NetherlandsReviewed by:
Zhao Jing, Yuxi Normal University, ChinaCopyright © 2023 Qiu, Wei, Meng, Peng, Zhang, He, Wei, Ahmed and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lanfang Wei, d2xmYW5nMjAwMEBhbGl5dW4uY29t; Waqar Ahmed, YWhtZWQud2FxYXIxMDgzQHlhaG9vLmNvbQ==; Guanghai Ji, amdoYWkwMDFAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.