 Rongrong Yan
Rongrong Yan Yanfei Geng3
Yanfei Geng3 Chunlei Xiang
Chunlei Xiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 26 September 2023
Sec. Plant Bioinformatics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1265641
This article is part of the Research TopicMolecular Biology and Omic Technologies in the Identification of Plants, Plant Derived-Products, Plant Pathogens, and Plant MicrobiomesView all 12 articles
Introduction: The recently established Linderniaceae, separated from the traditionally defined Scrophulariaceae, is a taxonomically complicated family. Although previous phylogenetic studies based on a few short DNA markers have made great contributions to the taxonomy of Linderniaceae, limited sampling and low resolution of the phylogenetic tree have failed to resolve controversies between some generic circumscriptions. The plastid genome exhibits a powerful ability to solve phylogenetic relationships ranging from shallow to deep taxonomic levels. To date, no plastid phylogenomic studies have been carried out in Linderniaceae.
Methods: In this study, we newly sequenced 26 plastid genomes of Linderniaceae, including eight genera and 25 species, to explore the phylogenetic relationships and genome evolution of the family through plastid phylogenomic and comparative genomic analyses.
Results: The plastid genome size of Linderniaceae ranged from 152,386 bp to 154,402 bp, exhibiting a typical quartile structure. All plastomes encoded 114 unique genes, comprising 80 protein-coding genes, 30 tRNA genes, and four rRNA genes. The inverted repeat regions were more conserved compared with the single-copy regions. A total of 1803 microsatellites and 1909 long sequence repeats were identified, and five hypervariable regions (petN-psbM, rps16-trnQ, rpl32-trnL, rpl32, and ycf1) were screened out. Most protein-coding genes were relatively conserved, with only the ycf2 gene found under positive selection in a few species. Phylogenomic analyses confirmed that Linderniaceae was a distinctive lineage and revealed that the presently circumscribed Vandellia and Torenia were non-monophyletic.
Discussion: Comparative analyses showed the Linderniaceae plastomes were highly conservative in terms of structure, gene order, and gene content. Combining morphological and molecular evidence, we supported the newly established Yamazakia separating from Vandellia and the monotypic Picria as a separate genus. These findings provide further evidence to recognize the phylogenetic relationships among Linderniaceae and new insights into the evolution of the plastid genomes.
As traditionally circumscribed, Lindernia and its relatives were placed in the tribe Lindernieae (Reichenbach, 1831) of Scrophulariaceae. The family is characterized by bilaterally symmetric and often tubular flowers, ovaries with axile placentation, numerous ovules, and capsules, which are shared with other related families of Lamiales (Bentham, 1846; Wettstein, 1891; Olmstead et al., 2001; Fischer, 2004). The absence of morphological synapomorphy raises the suspicion that the traditionally defined Scrophulariaceae is not monophyletic, with evidence from molecular phylogenetic analyses (Olmstead and Reeves, 1995; Olmstead et al., 2001; Beardsley and Olmstead, 2002; Oxelman et al., 2005). Combining molecular with morphological evidence, the tribe Lindernieae was promoted to the family Linderniaceae by Rahmanzadeh et al. (2005). Thereafter, Linderniaceae as an independent lineage distinct from Scrophulariaceae has been widely accepted in the comprehensive reviews of Lamiales (Tank et al., 2006; Angiosperm Phylogeny Group (APG), 2009; Schäferhoff et al., 2010; Refulio-Rodriguez and Olmstead, 2014; Angiosperm Phylogeny Group (APG), 2016; Liu et al., 2020; Fonseca, 2021). The family is characterized by special abaxial stamens with filaments conspicuously geniculate, zig-zag-shaped, or bearing spur-like appendages. The diversity centers of Linderniaceae are situated in tropical Africa and Southeast Asia, with 22 genera and more than 220 species recorded (Rahmanzadeh et al., 2005; Tank et al., 2006; Fischer et al., 2013; Perret et al., 2013; Biffin et al., 2018; Almeida et al., 2019; Yan et al., 2023).
Historically, diagnostic characteristics of genera of the Linderniaceae have been controversial among different taxonomists, which results in ambiguous generic boundaries. For example, Bentham (1846) considered Lindernia and the other three closely related genera (Bonnaya, Ilysanthes, and Vandellia) as four distinct genera. Based on the number of fertile stamens, Urban (1884) and Wettstein (1891) included Bonnaya with two fertile stamens into Ilysanthes and Vandellia with four fertile stamens into Lindernia. However, Haines (1922) emphasized the diagnostic value of the leaf vein, reducing Bonnaya with pinnate vein to Vandellia and Ilysanthes with palmate vein to Lindernia. Pennell (1935) suggested that the diagnostic characteristics currently used were too weak and artificial, and included the four controversial genera into a broadly circumscribed genus Lindernia sensu lato (s.l.). Synapomorphies of the Lindernia sensu Pennell (1935) comprise the remarkably uniform corolla, curved anterior filaments, and similar septicidal dehiscence of the capsule. The treatment was followed by the majority of botanists (Philcox, 1968; Yamazaki, 1977; Yamazaki, 1985; Fischer, 1995; Lewis, 2000; Fischer, 2004; Rahmanzadeh et al., 2005). However, the Lindernia s.l. seems not to be a natural group as some species incorporated into the Lindernia s.l. have the same characteristics as calyx, staminal appendages, and floral disc of another genus Torenia (Hara, 1943; Pennell, 1943; Philcox, 1968). On the basis of the morphology of leaf margin and seed, Yamazaki (1954; 1955) divided Lindernia s.l. into Lindernia s.s. (entire or slightly dentate leaf margin and non-alveolated seed) and Vandellia (serrate leaf margin and bothrospermous seed), although the revision has not been widely accepted. On the other hand, there were complicated relationships between Torenia and its relatives. The genus Craterostigma mainly distributed in Africa was reduced to a section of Torenia by bearing the winged calyx (Bentham, 1846). Wettstein (1891) supported Craterostigma as a separate genus based on its rosulate habit, but Craterostigma species often were described as Torenia in subsequent investigations (Schlechter, 1924). Since traditional classifications failed to provide a stable taxonomic framework at the generic level, evidence from the molecular level has been given weight.
When establishing Linderniaceae, Rahmanzadeh et al. (2005) included 13 genera of the tribe Lindernieae sensu Fischer (2004) within this family, of which the type genus Lindernia is the largest genus comprising ca. 100 species. Using two DNA markers (trnK and matK), Fischer et al. (2013) revealed the polyphyly of Lindernia and elaborated the first generic classification for Linderniaceae. In this study, 17 genera were recognized, of which Bonnaya and Vandellia were reinstated from Lindernia s.l., and formerly well-circumscribed Torenia and Craterostigma were expanded to include some members of Lindernia s.l. (Fischer et al., 2013). The narrowly circumscribed Lindernia was kept according to the delimitation of Yamazaki (1955), including Bryodes, Ilysanthes, and Psammetes. In Fischer et al. (2013) treatment, the species not represented by molecular data were assigned to genera according to morphology, of which most taxa with pinnate veins and deeply lobed calyx from Asia and America were transferred to Vandellia. On the basis of expanded phylogenetic sampling and four DNA markers (rps16, matK, trnL-F, and RPB2), however, Liang (2017) revealed the polyphyly of Vandellia sensu Fischer et al. (2013) as other long-accepted genera (e.g., Torenia, Craterostigma, Chamaegigas, and Linderniella) embedded. Based on phylogenetic analyses inferred from the matK marker, Biffin et al. (2018) narrowed Vandellia sensu Fischer et al. (2013) and formally established the new genus Yamazakia separating from Vandellia. Although molecular phylogenetic studies have made great contributions to the circumscription of Linderniaceae, there are still considerable controversies concerning generic circumscription, predominantly due to the limited sampling and the low resolution of phylogenetic trees inferred from a few short molecular markers.
With the rapid development of Next-generation sequencing (NGS) technology, more and more organelle genomes were sequenced and submitted to GenBank. Compared to nuclear and mitochondrial genomes, the plastid genome has been widely applied to resolve phylogenetic relationships at different taxonomic levels, owing to its relatively small genome and slow mutation rate (Parks et al., 2009; Wicke et al., 2011; Wei et al., 2017; Wen et al., 2021; Zhao et al., 2021; Dong et al., 2022). The complete plastome exhibits a powerful ability to solve backbone phylogeny in contrast with universal DNA markers (Alwadani et al., 2019; Chen et al., 2022; Peng et al., 2023). Moreover, comparative analyses of the plastid genomes not only provide plentiful information for phylogenetic relationships but also deepen our understanding of genome evolution (Chu et al., 2022; Ogoma et al., 2022; Wan et al., 2023). However, no comparative analyses related to Linderniaceae plastomes have been conducted so far, only general reports about genome size and gene contents (Cheng et al., 2019; Chen et al., 2021). In this study, for the first time, we attempted to explore the phylogenetic relationships and genome evolution of Linderniaceae using a large number of complete plastomes. Our goals were to (1) investigate the structure and variation of Linderniaceae plastomes, (2) provide insights into the evolution of Linderniaceae plastomes, and (3) infer the phylogeny of Linderniaceae based on plastid genomic data.
In total, we included 31 samples of Linderniaceae representing eight genera and 28 species, of which 26 individuals were newly sequenced and the other five species were downloaded from GenBank (Table 1). Scientific names of Linderniaceae followed by Fischer et al. (2013) and Biffin et al. (2018). Plant samples were collected from the field and fresh leaves from healthy plants were stored in silica gel. These voucher specimens were deposited in the Herbarium of the Natural Museum of Guizhou University (GACP) (Table 1). The modified CTAB method was employed to extract total genomic DNA (Doyle and Doyle, 1987). The DNA purity and concentration were quantified using agarose gel electrophoresis and a NanoDrop 2000 Spectrophotometer. The quantified DNA was used to construct shotgun libraries with fragments of 200–500 bp, and paired-end (150 bp) reads were sequenced using an Illumina Hiseq 2500 platform by Biomarker Technologies, Inc (Shandong, China). Low-quality sequences of raw reads were filtered using the software Trimmomatic v.0.32 (Bolger et al., 2014), yielding at least 3 GB of clean reads for each sample.
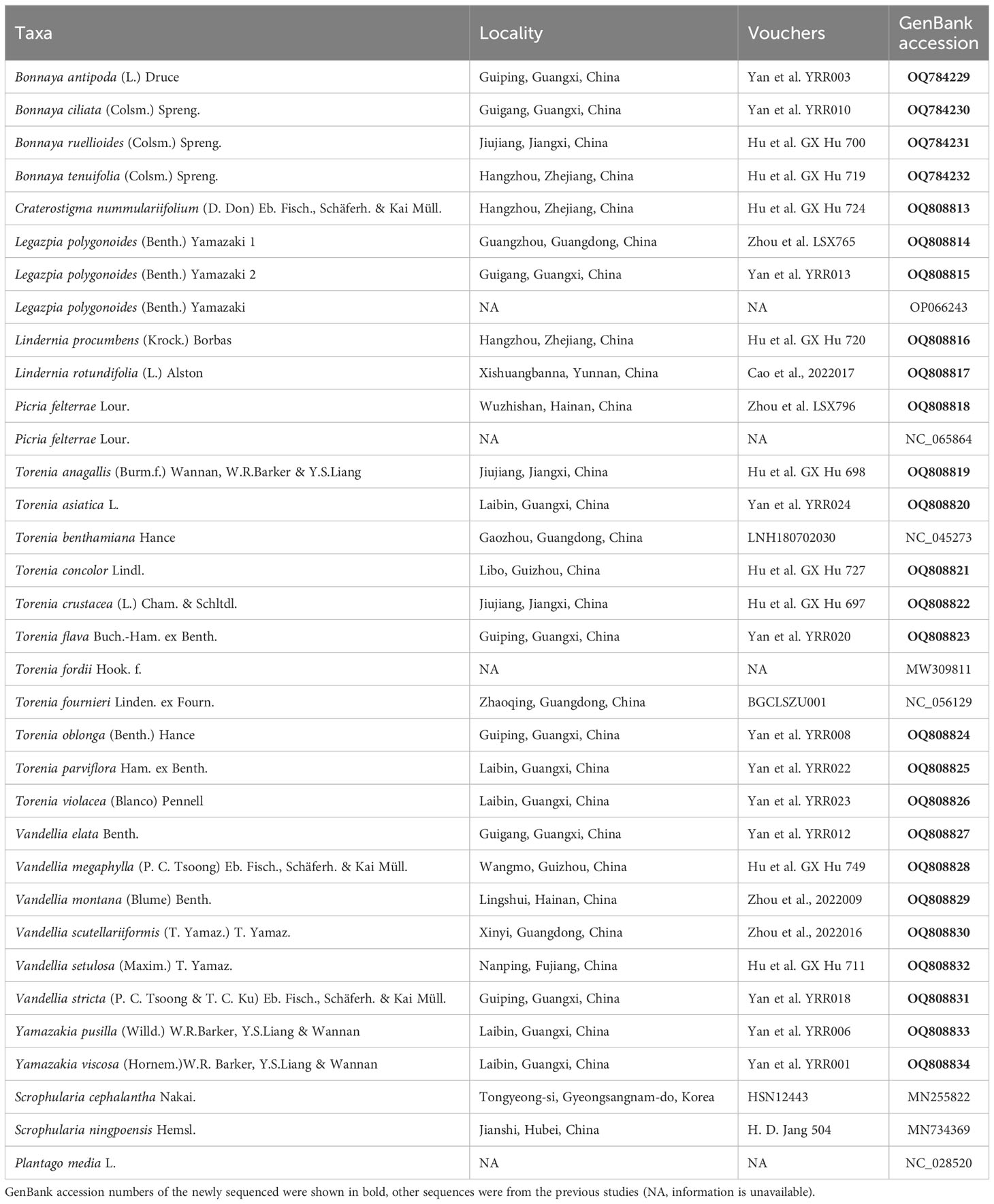
Table 1 Voucher information and GenBank accession numbers of the samples in this study.
De novo assemblies of the plastomes were conducted with GetOrganelle v1.7.5, following default settings (Jin et al., 2020). The final assembly results were verified using Bandage v0.8.1 (Wick et al., 2015). Two annotation tools, PGA (Qu et al., 2019) and CPGAVAS2 (Shi et al., 2019), were used to annotate these plastomes with Torenia benthamiana (NC_045273) as a reference. After the initial annotation, tRNA genes were further checked by tRNAscan-SE v1.21 (Brooks and Lowe, 2005). We manually calibrated the start and stop codons of coding sequences in the software Geneious v9.0.2 (Kearse et al., 2012). The gene maps were drawn using OrganellarGenomeDRAW (OGDRAW) (Lohse et al., 2013). All the newly sequenced plastomes were deposited at GenBank with accession numbers in Table 1.
Gene rearrangements were detected based on collinear blocks using Mauve v2.4.0 (Darling et al., 2004). Genome divergence was identified using mVISTA with Shuffle-LAGAN mode (Frazer et al., 2004). To detect hypervariable regions, nucleotide variability (Pi) was calculated in DnaSP v.5.0 (Librado and Rozas, 2009) with the parameters of 600 bp on window length and 200 bp on step size. We also compared the boundaries of large single copy (LSC), small single copy (SSC), and two inverted repeat (IR) regions by online program IRscope (Amiryousefi et al., 2018).
Simple sequence repeats (SSRs) were identified in MISA-web (Beier et al., 2017), with parameters set to ten, five, four, three, three, and three for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. The forward, reverse, palindromic, and complement repeats were recognized using REPuter (Kurtz et al., 2001), with a minimum repeat size of 30 bp and a Hamming distance of three. The repeat numbers in the regions of LSC, SSC, and IR were counted in Geneious v9.0.2 (Kearse et al., 2012).
The codon usage bias of protein-coding genes based on 28 Linderniaceae species was calculated in CodonW v1.4.4 (Peden, 2000). Relative synonymous codon usage (RSCU) was used to evaluate codon usage preference. When the RSCU value > 1 indicates that the codon is preferred, the RSCU value = 1 means that the codon is not preferred, and the RSCU value < 1 shows that the codon usage is low. The effective number of codons (ENC) and GC content of the synonymous third codon positions (GC3s) were used to assess codon usage patterns. To investigate the selective pressure on protein-coding genes, we extracted the shared nucleotide and protein sequences based on 28 Linderniaceae species using Geneious v9.0.2 (Kearse et al., 2012). Protein sequences were aligned by MAFFT v.7.388 (Katoh and Standley, 2013), and then transformed data into axt format. Non-synonymous (Ka) substitution and synonymous (Ks) substitution were calculated using KaKs_Calculator v.2.0 (Wang et al., 2010), with Lindernia procumbens as the reference, setting the genetic code to 11 (bacterial and plant plastid codes) and calculation model to Yang-Nielsen algorithm (Nielsen and Yang, 1998). The ratio of non-synonymous and synonymous substitutions (Ka/Ks) is greater than 1, equal to 1, and less than 1, indicating positive selection, neutral selection, and purifying selection, respectively. The Ka value of 0 showed that the Ka/Ks value equaled 0. Ka = 0 and Ks = 0 showed the invalid value was represented by NA.
Phylogenetic relationships of Linderniaceae were reconstructed using plastid genomic data (complete plastid genomes and 80 protein-coding genes). The ingroup included 31 individuals of Linderniaceae, representing eight genera and 28 species, with three species from Plantaginaceae and Scrophulariaceae as the outgroups (Table 1). Alignments of sequences were performed in MAFFT v.7.388 with auto strategy (Katoh and Standley, 2013) and manually adjusted the alignment results in PhyDE v.0.9971 (Müller et al., 2010). Phylogenetic trees were conducted using maximum likelihood (ML) and Bayesian inference (BI) methods in CIPRES Science Gateway (https://www.phylo.org/) with the tool of RAxML-HPC2 on XSEDE 8.2.12 and MrBayes on XSEDE 3.2.7a, respectively. The ML tree was inferred under the GTRGAMMA model, with bootstrap replicates of 1,000. For BI analysis, the best-fit model was selected in the software PhyloSuite (Zhang et al., 2020). The GTR+F+I+G4 model was selected for two data sets under the Akaike Information Criterion (AIC) (David and Buckley, 2004). Two Markov Chain Monte Carlo (MCMC) simulations were run for 2,000,000 generations independently, with every 1,000 generations for tree sampling. The initial 25% of trees were removed as burn-in and the remaining data were used to generate consensus trees. Convergences were inspected when the average standard deviation of the split frequencies was less than 0.001. Finally, we visualized and edited these trees using iTOL v3.4.3 (Letunic and Bork, 2016).
The whole plastid genomes of 31 Linderniaceae ranged from 152,386 bp (Bonnaya ciliata) to 154,402 bp (Yamazakia viscosa) (Table 2). These plastomes displayed a typical quadripartite structure (Figure 1), with a large single copy (LSC) region, a small single copy (SSC) region, and a pair of inverted repeats (IRs). The size of the LSC region varied from 84,566 bp in B. ruellioides to 85,837 bp in Y. viscosa. The size of the SSC region was from 18,352 bp in B. ciliata to 19,095 bp in Lindernia procumbens. The size of the IR region ranged from 24,599 bp in L. procumbens to 24,873 bp in Vandellia scutellariiformis. Total GC content did not present significant differences among all plastomes (37.5–37.7%), with 35.3–35.6% in the LSC region, 31.6–32.0% in the SSC region, and 43.3–43.6% in the IR region. Gene content was identical in all Linderniaceae plastomes, with 114 unique genes, comprising 80 protein-coding genes (PCGs), 30 tRNA genes, and four rRNA genes (Table 3). Seven protein-coding genes, seven tRNA genes, and four rRNA genes were duplicated in the IR regions. A total of 17 genes with introns were detected, of which clpP, rps12, and ycf3 genes contained two introns and the other 14 genes had a single intron. With the first exon situated in the LSC region and the remaining two exons dispersed throughout the IR regions, the rps12 was identified as a trans-spliced gene.
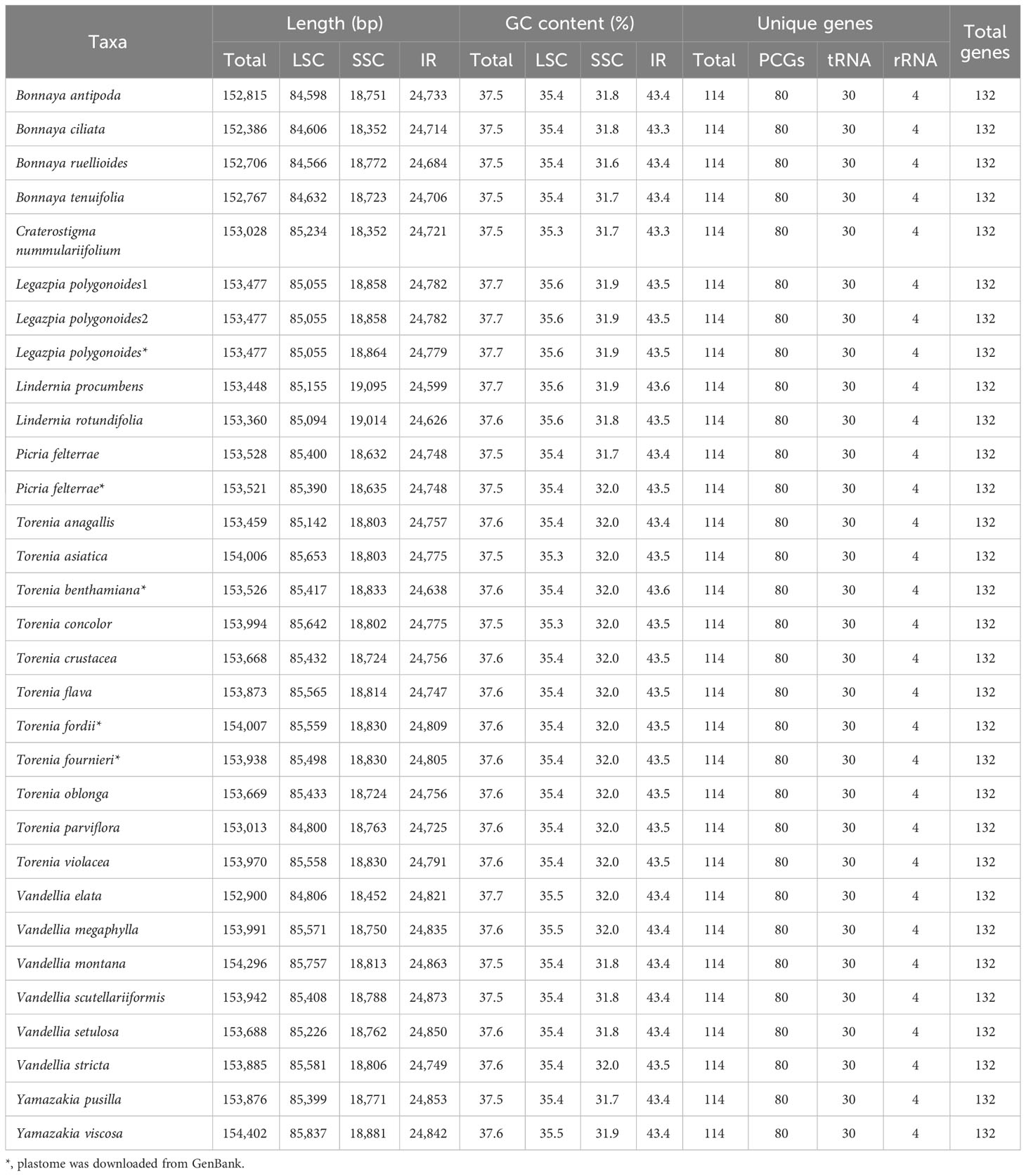
Table 2 Summary of features of the Linderniaceae plastomes.
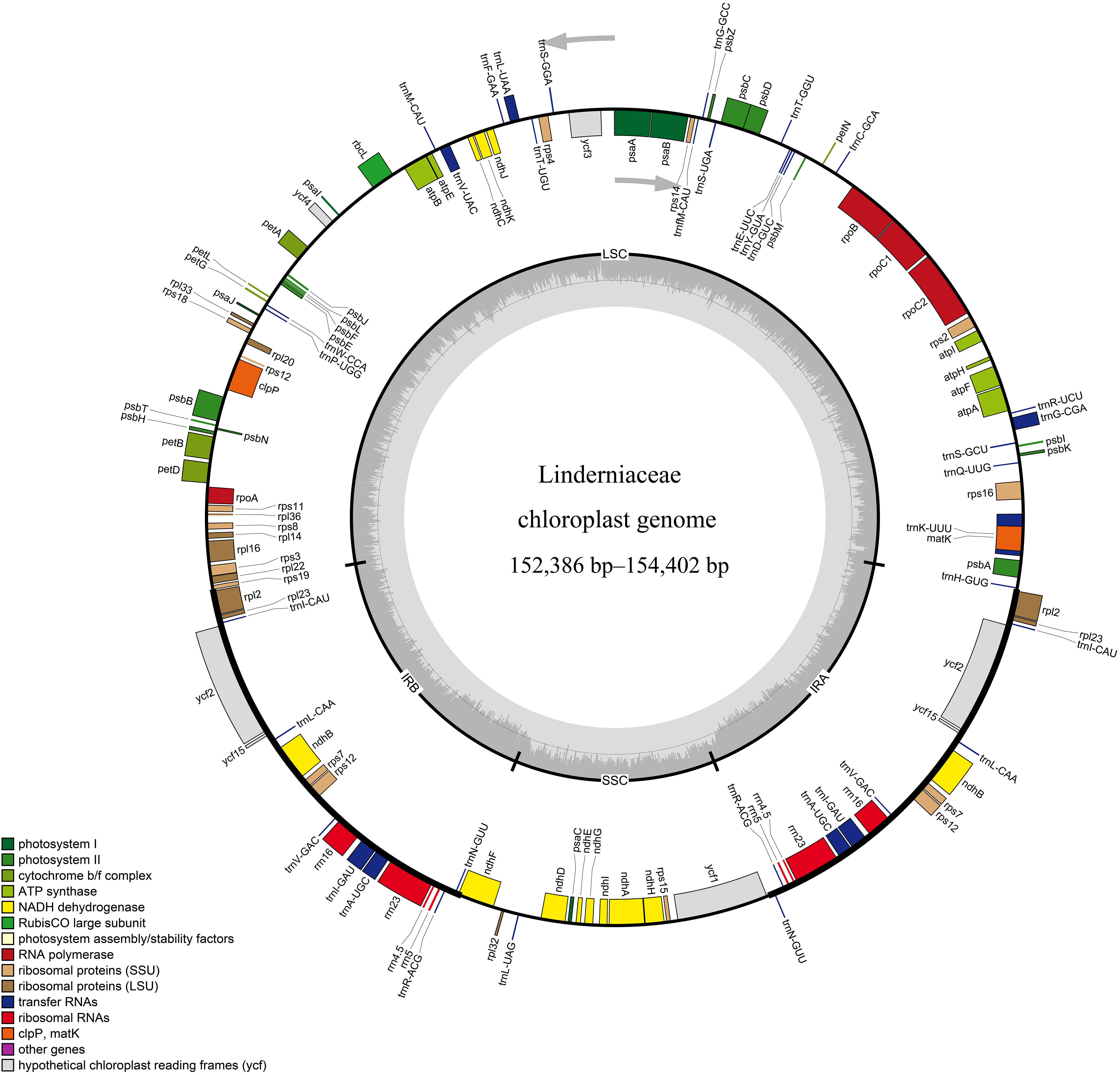
Figure 1 Representative circular map of the Linderniaceae plastome. Genes inside the circle are transcribed clockwise, while those outside the circle are transcribed counter-clockwise. The genes are color-coded with different functions. The darker gray and lighter gray in the inner correspond to GC content and AT content, respectively. LSC, large single copy; IR, inverted repeat; SSC, small single copy.
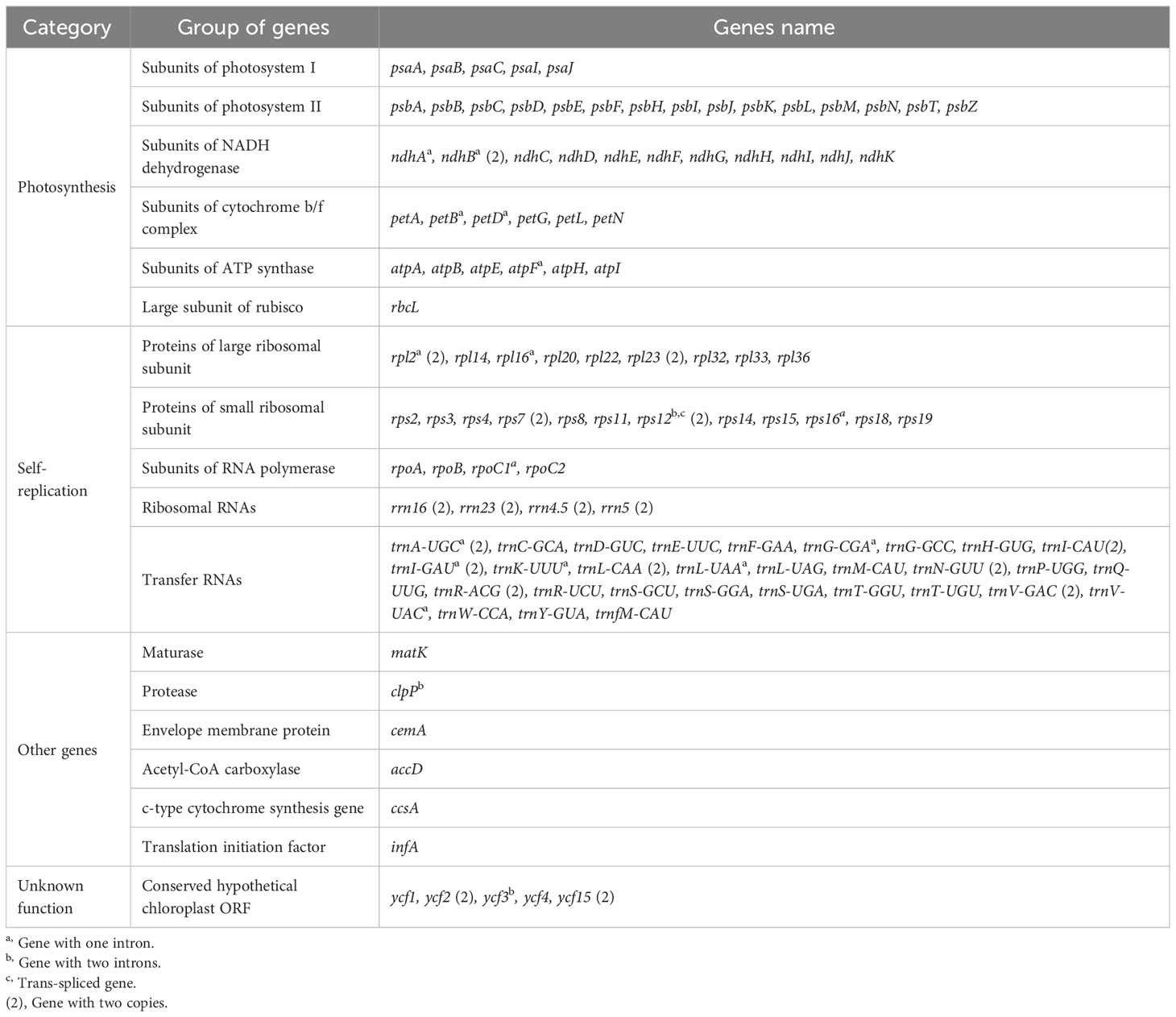
Table 3 Gene content of the Linderniaceae plastomes.
Gene order in Linderniaceae was highly conservative and we did not find rearrangement or inversion events (Figure 1 and Supplementary Figure 1). However, the junctions between single copy (SC) and IR regions exhibited slight differences (Supplementary Figure 2). The boundary of LSC and IRb was located in the intergenic region between rps19 and rpl2 among Bonnaya and Torenia anagallis plastomes, and the rps19 gene crossed the LSC-IRb boundary for the other plastomes. Except for Lindernia procumbens and L. rotundifolia, the gene ndhF spanned the SSC-IRb junction in all taxa, with 36 bp to 240 bp in the IRb region and 2019 bp to 2165 bp in the SSC region. At the SSC-IRa junction, the ycf1 and trnN genes were entirely located within the SSC and IRa region near the junction, respectively. For all species, the boundary of IRa and LSC was placed in the intergenic region between rpl2 and trnH, with gene rpl2 being 41 to 110 bp and gene trnH being 3 to 44 bp away from the boundary.
The mVISTA analyses indicated that the plastid genomes of Linderniaceae were relatively conservative (Supplementary Figure 3). The alignment of the whole plastomes was 143,491 bp in length, comprising 14,984 variable sites (10.44%) and 7,949 parsimony-informative sites (5.54%) (Table 4). The highest percentage of parsimony-informative sites was from the SSC region (1,739, 9.84%), and the lowest was from the IR region (300, 1.26%). There was nucleotide variability (Pi) of 0.01737 across the complete plastome. The IR region (Pi = 0.0042) displayed the lowest sequence divergence, while the SSC region (Pi = 0.03225) had the most variance.
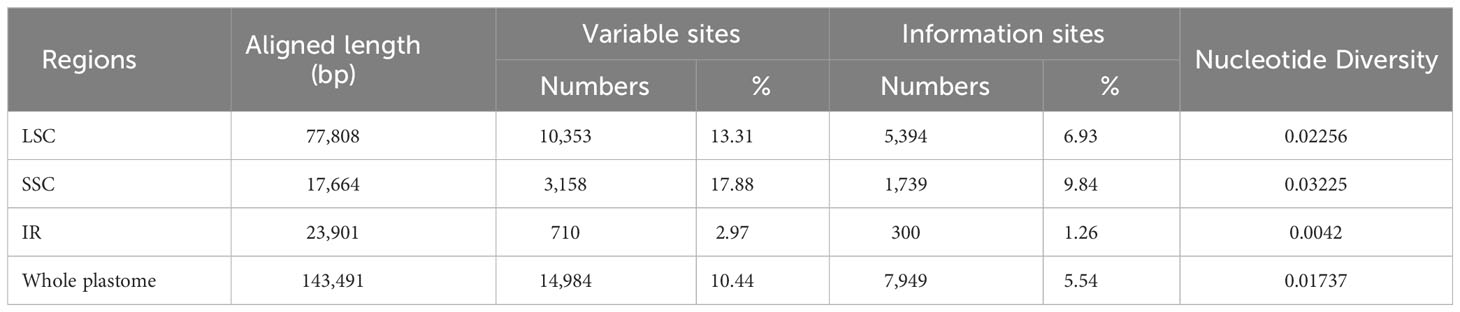
Table 4 Sequence divergence of the Linderniaceae plastomes.
Hypervariable regions of Linderniaceae species were further identified through the slide window analyses (Figure 2). The nucleotide diversity values in plastid genomes ranged from 0.00024 to 0.06292. The mutational hotspots were defined with Pi values greater than 0.0500. A total of five highly divergent regions from SC regions were detected (Figure 2A), including three intergenic spacers (IGS) (rps16-trnQ, petN-psbM, and rpl32-trnL), and two coding genes (rpl32 and ycf1). Among these hypervariable sites, the ycf1 gene exhibited the highest Pi value (0.06292), and the lowest was from rpl32-trnL (0.05002). Overall, the IR regions exhibited less variation (Figure 3B), and divergence was greater in the non-coding region than in the coding region (Supplementary Figure 3).
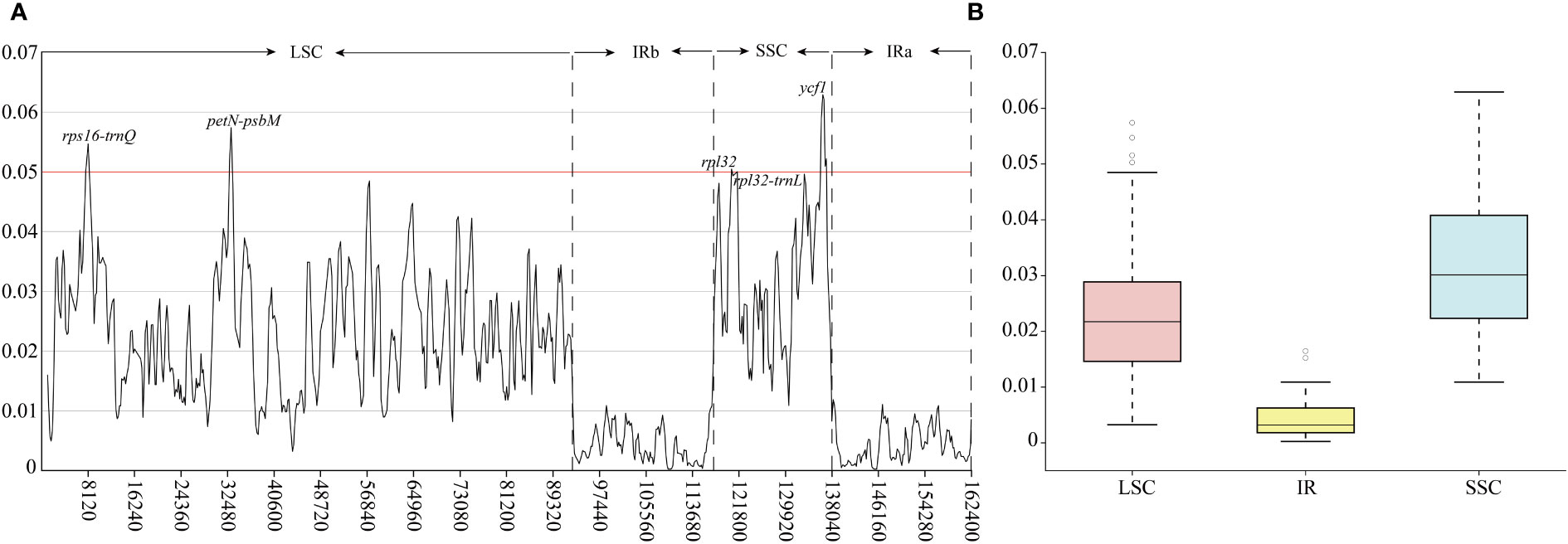
Figure 2 Nucleotide diversity (Pi) values of the Linderniaceae plastomes. (A) The Pi-value of the windows. (B) Boxplots of Pi-value differences among the LSC, IR, and SSC regions.
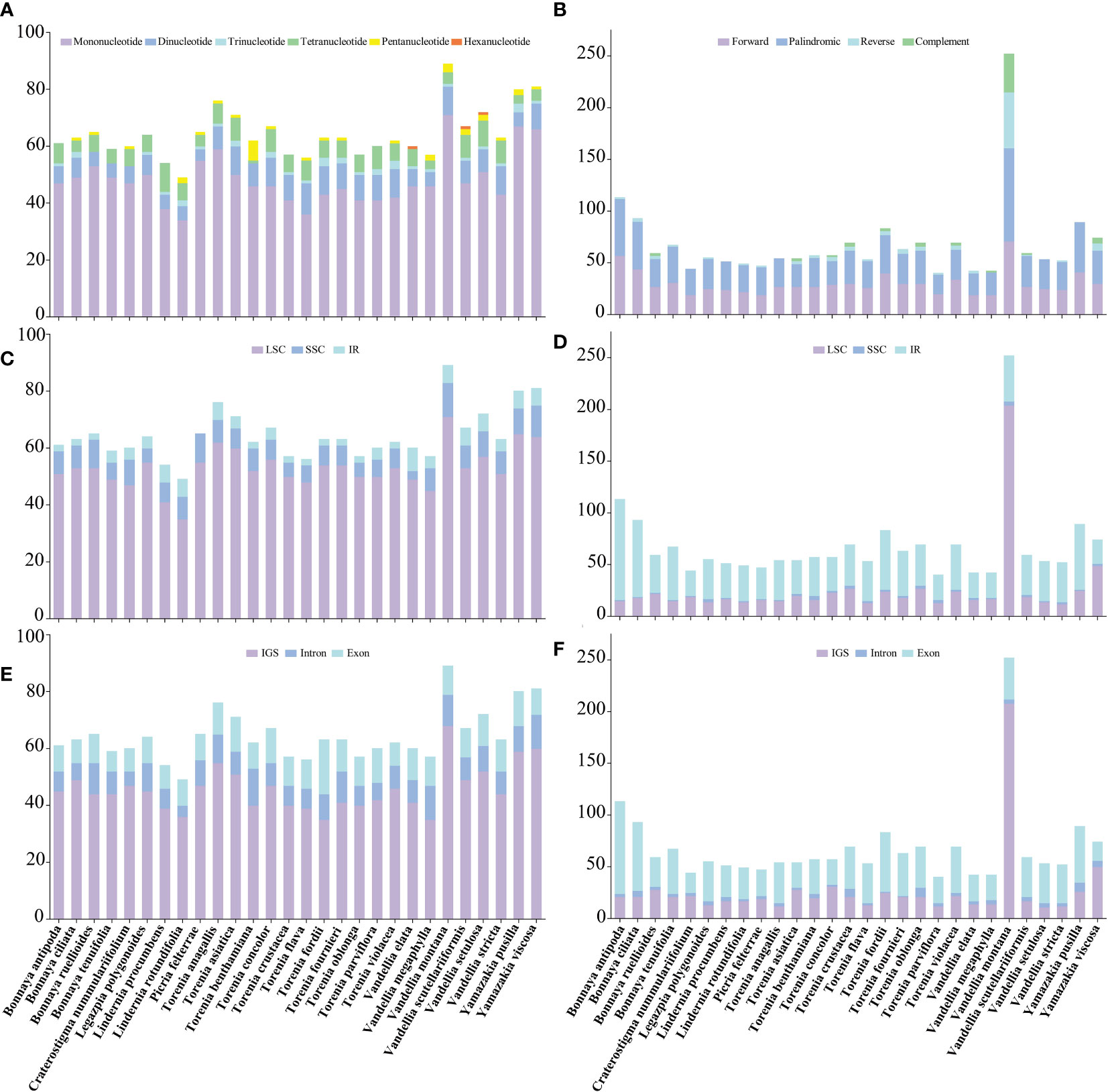
Figure 3 Comparisons of simple sequence repeats (SSRs) and long sequence repeats (LSRs) among 28 Linderniaceae species. (A) Number of six types of SSRs. (B) Number of four types of LSRs. (C) Number of SSRs in the LSC, SSC, and IR regions. (D) Number of LSRs in the LSC, SSC, and IR regions. (E) Number of SSRs in the IGS, exons, and introns. (F) Number of LSRs in the IGS, exons, and introns.
Simple sequence repeats (SSRs) across the 28 Linderniaceae plastomes were detected, ranging from 49 (Lindernia rotundifolia) to 89 (Vandellia montana) (Figure 3A). Of these, mononucleotide, dinucleotide, and tetranucleotide SSRs were found in all taxa; trinucleotide repeats were not observed in Bonnaya ruellioides, B. tenuifolia, Craterostigma nummulariifolia and Torenia benthamiana; pentanucleotide repeats were not discovered in B. antipoda, B. tenuifolia, Legazpia polygonoides, Lindernia procumbens, T. crustacea, T. oblonga, T. parviflora, and V. elata; hexanucleotide repeats only were detected in V. elata, V. scutellariiformis, and V. setulosa. The most abundant type was mononucleotides, accounting for 74.82%, followed by dinucleotides with 11.87%, tetranucleotides with 9.32%, trinucleotides with 2.00%, pentanucleotides with 1.83%, and hexanucleotides with the least occurrence (0.17%) (Supplementary Table 1). The mononucleotide and dinucleotide SSRs primarily consisted of A or T bases and AT or TA bases, respectively, while the composition of other types of SSRs was diverse (Supplementary Table 2). Furthermore, most SSRs were distributed in the LSC region (Figure 3C), ranging from 71.43% to 87.72%, followed by the SSC region varying from 5.00% to 16.33%, and were rare in the IR regions, even were not detected in the IR region of Picria felterrae (Supplementary Table 1). The SSRs included between 55.56% and 78.33% in the IGS, between 11.11% and 30.16% in the exon, and between 8.16% and 21.05% in the intron (Figure 3E and Supplementary Table 1).
A total of 1909 long sequence repeats (LSRs) greater than 30 bp were identified, containing 844 forward repeats, 899 palindromic repeats, 107 reverse repeats, and 59 complement repeats (Supplementary Table 3). For each Linderniaceae species, the number of repeat sequences varied greatly, ranging from 42 (Vandellia elata and V. megaphylla) to 252 (V. montana) (Figure 3B). Among these long repeats, forward and palindromic repeats were detected in all species; reverse repeats were not recognized in Craterostigma nummulariifolium, Lindernia procumbens, Torenia anagallis, V. megaphylla, and Yamazakia pusilla; complement repeats were not discovered in seventeen individuals (Figure 3D and Supplementary Table 3). These LSRs were found predominantly in the LSC region (13.27–80.95%) and IR region (17.46–85.84%), with a few located in the SSC region (0.88%–5.45%) (Figure 3D and Supplementary Table 3). Meanwhile, most were detected in the IGS (18.58–82.54%) and exon (24.32–78.76%), with a few distributed in the intron (1.20–13.04%) (Figure 3F and Supplementary Table 3). Furthermore, the length of LSRs was mainly concentrated in 30–40 bp, accounting for 67.89%, followed by greater than 50 bp with 25.92%, and 41–50 bp with 16.19% (Supplementary Table 3).
Codon usage biases with high similarity were observed among 28 Linderniaceae species (Supplementary Table 4). The total number of codons of protein-coding genes ranged from 22,800 (Picria felterrae) to 22,895 (Bonnaya antipoda). Leucine (Leu) was the most abundant (2400–2434), while cysteine (Cys) showed the least abundance (240–252). The RSCU values of 30 codons were greater than 1, almost all of which ended with A or U except for UUG. Specifically, the UUA encoding Leu had the highest RSCU from 1.91 to 1.98, and the AGC encoding serine (Ser) had the lowest RSCU from 0.27 to 0.32. Methionine (Met) and tryptophan (Try) preferred one codon with RSCU = 1, namely AUG and UGG, respectively. To further evaluate the codon usage pattern, the GC content of synonymous third codon positions (GC3s) and the effective number of codons (ENC) were calculated in Supplementary Table 5. The values of ENC showed a slight bias ranging from 46.91 to 47.33. Total GC content in the codons was between 37.52% and 37.77%, and the values of GC3s varied from 28.7% to 29.2%. GC content was highest at the first codon position (45.67–45.94%), followed by the second position (38.18–38.39%), and third position (28.63–29.13%).
The ratios (Ka/Ks) of the non-synonymous (Ka) and synonymous (Ks) substitutions revealed that the plastid genomes of Linderniaceae were highly conserved during the evolutionary process (Supplementary Table 6). In terms of average values, the Ka/Ks values of all genes were less than 1 (between 0 and 0.7908), indicating that these genes were under purifying selection. Interestingly, the average Ka/Ks values of almost all genes were less than 0.5, except for ycf2 (0.7908), psbH (0.6269), ycf15 (0.5272), and ycf1 (0.5241). Meanwhile, we found significant variation in the Ka/Ks values of genes with different functions. For example, photosynthesis-related genes were between 0 (atpH, psaC, psaJ, psbE, psbF, psbL, psbM, psbN, and rps12) and 0.6268 (psbH), self-replication genes ranged from 0.2300 (clpP) to 0.3139 (matK), other function genes varied from 0.02442 (infA) to 0.3927 (accD), and unknown function genes were from 0.0060 (ycf3) to 0.7908 (ycf2). Moreover, only the gene ycf2 exhibited positive selection and was detected in six species, including Bonnaya antipoda, B. ciliata, B. ruellioides, B. tenuifolia, Picria felterrae, and Torenia flava. Four genes had Ka/Ks values equal to 0 in all species, namely atpH, psaC, psbE, and psbF.
In this study, we included 31 complete plastomes from Linderniaceae and three outgroups from Plantaginaceae and Scrophulariaceae for phylogenetic inference. After removing ambiguously aligned regions, the alignment of the complete plastomes was 150,629 bp in length, comprising 11,541 parsimony-informative sites. A total of 80 protein-coding genes were aligned and the matrix of 79,174 bp was generated after removing ambiguously aligned regions, including 5,744 parsimony-informative sites. As topologies generated from ML and BI analysis were similar, only the ML tree was presented with ML bootstrap support (BS) and BI posterior probability (PP) values added (Figure 4). Except for the phylogenetic positions of Vandellia elata and Torenia parviflora, the topologies generated from the two datasets were identical. In the complete plastomes tree, Vandellia elata was sister to the group consisting of Legazpia polygonoides and T. anagallis with weak support (BS = 36, PP = 0.35). However, Vandellia elata was strongly supported as a sister to the core Torenia that comprised V. stricta and all Torenia species except T. anagallis in the protein-coding genes tree (BS = 92, PP = 0.84). Torenia parviflora was one of a member of the core Torenia. It was sister to the clade including T. asiatica, T. concolor, T. fournieri, T. benthamiana, T. fordii, and T. violacea in the complete plastomes tree (BS = 100, PP = 1.00) but sister to another group consisting of V. stricta, T. flava, T. crustacea and T. oblonga in the protein-coding genes tree (BS = 100, PP = 1.00).

Figure 4 Cladograms of Linderniaceae inferred from the complete plastid genomes (A) and protein-coding genes (B) based on Bayesian inference (BI) and maximum likelihood (ML) methods. The ML bootstrap (BS) and BI posterior probability (PP) that supported each node are shown under the branches. Red-bold horizontal lines mean different phylogenetic positions for taxa between two different datasets. * indicates that the sequence is from GenBank. BS values < 50% and PP < 0.5 are indicated by ‘–’.
In both trees, the monophyly of Linderniaceae was strongly supported (BS = 100, PP = 1.00). Within Linderniaceae, Lindernia procumbens was sister to L. rotundifolia, then together sister to the rest of Linderniaceae (BS = 100, PP = 1.00). The two Yamazakia species did not group together as Vandellia montana embedded within the genus (BS = 100, PP = 1.00). Picria felterrae from different populations clustered together and formed a sister relationship with monophyletic Boyanna comprising Boyanna antipoda, B. ciliata, B. ruellioides, and B. tenuifolia here (BS = 100, PP = 1.00). The monotypic Legazpia represented by three different individuals was strongly supported, which was sister to Torenia anagallis (BS = 100, PP = 1.00). The monophyly of Torenia was not supported with V. stricta, V. elata, and Legazpia polygonoides embedded. Similarly, the monophyly of Vandellia was also not supported as species of the genus scattered to other distinct clades.
In this study, the 26 plastid genomes of Linderniaceae were newly sequenced, and comprehensive analyses were performed in combination with five published data. Comparative analyses revealed conservatism and diversity in the Linderniaceae plastomes. Similar to most angiosperms (Wicke et al., 2011), the plastid genomes of Linderniaceae typically presented circular quadripartite structure, comprising a pair of inverted repeats (IRs), a large single copy (LSC) region, and a small single copy (SSC) region (Figure 1). The 31 plastid genomes of Linderniaceae, ranging from 152,386 bp to 154,402 bp, did not show significant differences in size, and the gene content and order were identical (Tables 2, 3). Within the same species, there is little or no variation in the genome size. The two populations of Picria felterrae differ in size by only 7 bases, and the three plastomes of Legazpia polygonoides had the same 153,477 bp in size (Table 2). Similarly, there are also no significant differences among distantly related species between Torenia and Lindernia, such as Torenia concolor (153,994 bp) and Lindernia procumbens (153,448 bp). These means that the plastid genomes of Linderniaceae are highly conservative in general features with only slight variations in genome size. Previous studies have revealed that the expansion and contraction of the IR, LSC, and SSC regions are common during evolution and are the main reason for variations in plastome size (Ravi et al., 2007; Downie and Jansen, 2015; Daniell et al., 2016). Significant contraction of IR region was observed among Linderniaceae species (Supplementary Figure 2), which was also common in Lamiales, such as Gesneriaceae (Gu et al., 2020), Lamiaceae (Su et al., 2022), Oleaceae (Long et al., 2023), Plantaginaceae (Xie et al., 2023), and Scrophulariaceae (Wang et al., 2022). Notably, the IR boundaries did not coincide exactly in the three populations of L. polygonoides, but they had the same plastome size. The two populations of P. felterrae were completely consistent in IR size, with slight variation occurring in the LSC and SSC regions. Overall, the 31 plastid genomes of Linderniaceae showed size differences mainly in the LSC region (Table 2). Consistent with previous studies (Yang et al., 2022; Bai et al., 2023; Peng et al., 2023), the non-coding and single copy (SC) regions were more divergent than the coding and IR regions (Supplementary Figure 3). We, therefore, speculated that the size variation of Linderniaceae plastomes was mainly attributed to the LSC region, especially in the non-coding regions, similar to the results of studies of Gao et al. (2022) and Li et al. (2023).
In Linderniaceae, the gene composition in each junction was almost identical, while the gene arrangement pattern was slightly different (Supplementary Figure 2). Interestingly, the gene arrangement pattern of each IR boundary in Linderniaceae was more similar among closely related species. For example, based on the phylogenetic analyses in this study, Torenia crustacea, T. oblonga, T. flava, and Vandellia stricta formed a well-supported clade (Figure 4), and their IR boundaries were highly conservative, with only slight differences in the SSC-IRb and SCC-IRa junctions. Particularly, the sister species of T. crustacea and T. oblonga showed completely consistent boundary genes and arrangement patterns, and so did the sister species of T. flava and V. stricta. This correlation has been reported in Aristolochiaceae (Bai et al., 2023), Juglandaceae (Xi et al., 2022), and Musaceae (Song et al., 2022). Therefore, information from the IR boundary may reveal the phylogenetic relationships among species to some extent.
Long sequence repeats (LSRs) play an important role in genome recombination and rearrangement (Do and Kim, 2017). In Linderniaceae, LSRs were found predominantly in the IR regions, and the forward and palindromic repeats with the length mainly concentrated in 30–40 bp were the most abundant (Supplementary Table 3). These findings were largely consistent with previous studies (Tian et al., 2018; Thode and Lohmann, 2019; Fan et al., 2021; Zhu et al., 2021). Single Sequence Repeats (SSRs) with polymorphism have been developed as molecular markers that were widely used in population genetics, polymorphism investigation, and biogeographic inference (Ebert and Peakall, 2009; Xue et al., 2012; Yu et al., 2017). In 28 Linderniaceae species, the mononucleotide SSRs were the most abundant and almost consisted of A or T bases (Supplementary Table 2), which may explain the richness of A/T content in plastomes. The number of SSRs in the single-copy (SC) and non-coding regions was higher than that in the IR and coding regions (Figure 3), consistent with previous research (Huang et al., 2020; Kong et al., 2021; Wu et al., 2021). There are significant differences in the number and distribution pattern of SSRs among species (Figure 3 and Supplementary Table 1), which have great significance for future population genetics studies of Linderniaceae.
Hypervariable regions within the plastid genome have been screened out as candidate barcodes for phylogenetic analyses and species identification (Dong et al., 2012; Dong et al., 2015; Li et al., 2015). As non-coding sequences have higher evolutionary rates than coding regions, they were often identified as the best barcoding candidates (Ravi et al., 2007), which was confirmed in the present study (Figure 2 and Supplementary Figure 3). Five hypervariable regions were screened within the Linderniaceae, namely petN-psbM, rps16-trnQ, rpl32-trnL, rpl32, and ycf1, of which petN-psbM and rps16-trnQ were located in the LSC region and others were distributed in the SSC region. These non-coding regions have exhibited high nucleotide diversity and phylogenetic utility in Lamiales, such as petN-psbM in Lamiaceae (Su et al., 2022; Wang et al., 2023), rps16-trnQ in Scrophulariaceae (Dong et al., 2022) and Gesneriaceae (Gu et al., 2020), and rpl32-trnL in Bignoniaceae (Fonseca and Lohmann, 2018), Lamiaceae (Hu et al., 2020) and Scrophulariaceae (Dong et al., 2022). Furthermore, the gene ycf1, the second largest gene in the plastid genome playing a critical role in plant cell survival (Drescher et al., 2000), has been proposed as the most potential DNA barcode due to its high variability in land plants (Drew and Sytsma, 2011; Dong et al., 2015). The relatively high nucleotide diversity observed in the rpl32 gene is similar to that observed in Dioscoreaceae (Wonok et al., 2023), Fagaceae (Worth et al., 2019), and Poaceae (Guo et al., 2022). In the previous study, the phylogeny of Linderniaceae inferred from a few common plastid markers (rps16, matK, trnL-F, and trnK) or nuclear gene (RPB2) had moderate or weak support values (Fischer et al., 2013; Liang, 2017; Biffin et al., 2018), hindering the investigation for the phylogeny and evolution of Linderniaceae. Hypervariable regions in this study provided additional information for further phylogenetic and biodiversity research in Linderniaceae.
Codon usage bias may help to better understand gene expression and genome evolution (López et al., 2019). The analyses of relative synonymous codon usage (RSCU) showed that the Linderniaceae plastomes were highly similar in codon usage pattern (Supplementary Table 4). Consistent with previous studies (Azarin et al., 2021; Zhao et al., 2022; Li et al., 2023), Leucine (Leu) was the most frequent amino acid, and cysteine (Cys) was the least common. In Linderniaceae, codons mainly ended with A and U when the RSCU value was greater than 1, and codons primarily ended with G and C when the RSCU value was below 1, which appears to be a common phenomenon in gene expression of land plants (Cui et al., 2019; Chakraborty et al., 2020; Gao et al., 2022; Bai et al., 2023; Zhou et al., 2023). Moreover, the GC content of synonymous third codons positions (GC3s) showed that AT content was more abundant in the protein-coding genes (Supplementary Table 5), which may be correlated with the abundant AT content of plastid genomes. The ENC values of protein-coding genes in Linderniaceae plastomes varied from 46.91 to 47.33, demonstrating a conservative codon usage pattern (Wu et al., 2007).
The ratio (Ka/Ks) of non-synonymous (Ka) and synonymous (Ks) substitutions is a valuable parameter to assess the selective pressure in evolution (Ohta, 1995). Positive selection is considered to be closely associated with adaptive evolution in harsh environments while purifying selection is a common evolutionary force responsible for maintaining genomic conservation (Cvijović et al., 2018). In Linderniaceae, the Ka/Ks values of almost all genes were less than 1.00, providing evidence for purifying selection. The ycf2 gene had a positive selection in only Bonnaya antipoda, B. ciliata, B. ruellioides, B. tenuifolia, Picria felterrae, and Torenia flava, with a Ka/Ks value greater than 1, indicating that most protein-coding genes were relatively conserved. As the largest known plastid gene in angiosperms, ycf2 is an enigmatic gene due to its unknown function. Drescher et al. (2000) and Kikuchi et al. (2018) pointed out that the ycf2 gene is associated with plant viability and the 2-MD AAA-ATPase complex. Among many angiosperms, ycf2 has been reported under positive selection (Liu et al., 2018; Zhong et al., 2019; Peng et al., 2020; Wu et al., 2020; Song et al., 2022; Fu et al., 2023). Overall, protein-coding genes of plastid genomes of the Linderniaceae were highly conserved during long-term evolution, while the functions of the gene ycf2 in adaptive evolution need to be further verified.
In this study, the robust phylogenetic backbone of Linderniaceae was constructed with the whole plastomes, in which the support values of these clades were greatly improved compared with previous studies based on a few genetic markers (Fischer et al., 2013; Liang, 2017; Biffin et al., 2018). Based on phylogenomic analyses presented here, we found that Vandellia and Torenia sensu Fischer et al. (2013) were polyphyletic. Meanwhile, strong evidence was provided to support the taxonomic status of two genera, Picria and Yamazakia.
Traditionally, the genus Torenia is characterized by winged calyx on the shallowly lobed calyx tube and completely calyx-enclosed capsule (Bentham, 1846). However, historically, it is very difficult to define the exact circumscription of Torenia. Some species formerly belonging to Craterostigma, Lindernia s.l., and Vandellia were once treated as members of Torenia (Bentham, 1846; Hance, 1868; Pennell, 1943). On the basis of synapomorphy of the calyx tube together with molecular evidence, Fischer et al. (2013) redefined the genus Torenia with a total of 51 species included. In the phylogenetic analyses, Torenia seemed to be monophyletic as the only two species sampled formed a sister relationship (Fischer et al., 2013). Subsequent phylogenetic analyses based on expanded sampling indicated that Torenia sensu Fischer et al. (2013) is not monophyletic as some species of Vandellia, Legazpia, and Schizotorenia are embedded within it (Liang, 2017). Combining morphological and molecular evidence, Liang (2017) lumped Legazpia, Schizotorenia, and some species with claviform appendages of Vandellia into Torenia, with the number of species expanding to 78. According to the taxonomic treatment of Liang (2017), the morphology of the calyx tube was no longer used as a generic diagnostic characteristic, and potential synapomorphies of newly circumscribed Torenia included the pinnate vein, claviform appendages at the abaxial stamens, and bothrospermous seed. In this study, phylogenomic analyses including 11 species of Torenia indicated that Torenia sensu Fischer et al. (2013) is not monophyletic with Vandellia stricta, V. elata, and Legazpia polygonoides embedded. Morphologically, Legazpia can be easily distinguished from other Torenia species by such characteristics as three large semi-circular wings on the calyx, glabrous ovary, small corolla slightly exceeding the calyx, and rounded upper corolla lip (Yamazaki, 1955), and has been accepted as a separate genus by most taxonomists (Yamazaki, 1955; Fischer, 1995; Hong et al., 1998; Fischer, 2004; Rahmanzadeh et al., 2005; Fischer et al., 2013; Fischer et al., 2013). To maintain the monophyly of Torenia, reducing Legazpia to Torenia may be a reasonable choice.
Vandellia has long been treated as a synonym of Lindernia s.l. (Pennell, 1935; Pennell, 1943; Philcox, 1968; Yamazaki, 1977; Yamazaki, 1985; Fischer, 1995; Lewis, 2000; Fischer, 2004; Rahmanzadeh et al., 2005). Based on molecular phylogenetic analyses, Fischer et al. (2013) revealed that Lindernia s.l. is polyphyletic and split this genus into six genera (Lindernia s.s., Vandellia, Torenia, Bonnaya, Linderniella, and Craterostigma), of which most species of Lindernia s.l. were assigned to Vandellia. In this study, phylogenetic analyses showed that the species of Vandellia sensu Fischer et al. (2013) scattered various clades, which was similar to previous studies (Liang, 2017; Biffin et al., 2018). Traditionally, the pinnate vein, deeply lobed calyx, and four fertile stamens were usually used to define the genus Vandellia (Bentham, 1846; Hooker, 1884; Fischer et al., 2013). On the basis of morphological together with molecular evidence, Liang (2017) transferred Chamaegigas, Craterostigma, and Linderniella which have been long-established and accepted genera to Vandellia, although the Vandellia clade had weak support based on combined DNA markers (trnL-F, rps16, and matK). Vandellia sensu Liang (2017) comprised 49 species, almost all of which were transferred from other genera. A large number of species name changes will lead to confusion for end-users of these names (most of them are not taxonomists) (Drew et al., 2017). Due to limited sampling in the current study, we have insufficient evidence to elucidate the circumscription of Vandellia, pending comprehensive phylogenomic analyses to better determine the strength of support for this genus.
The Yamazakia was currently proposed by Biffin et al. (2018) as a replacement name for the genus Tittmannia Rchb. (blocking name: Tittmannia Brongn.), including two sampled species in phylogenetic analyses (Yamazakia pusilla and Y. viscosa). These two species were transferred from Vandellia sensu Fischer et al. (2013). Actually, Liang (2017) has revealed a well-supported clade (BS = 100, PP = 1.00) including three species [Vandellia moillis (synonym of V. montana), V. pusilla, and V. viscosa], and placed them in the genus Tittmannia together with other five species (Tittmannia cyrtotricha, T. longituba, T. rivularis, T. satakei, and T. stolonifera) based on morphological evidence. Traditionally, these species shared similar morphological characteristics and therefore were placed within the Lindernia section Tittmannia (Philcox, 1968). In our study, phylogenetic analyses showed that V. montana was sister to Yamazakia viscosa with strong support (BS = 100, PP = 1.00), then together sister to Y. pusilla (BS = 100, PP = 1.00). Resolution of the three species is significantly improved compared with the study of Liang (2017). Therefore, our results demonstrated V. montana to be a member of Yamazakia, supporting Yamazakia as a separate genus.
The genus Picria was proposed by Loureiro (1790), containing only one species Picria felterrae. The native range of this genus is Himalaya to S. China and Peninsula Malaysia, Philippines to Caroline Islands. Picria is a very distinctive genus morphologically different from the other Linderniaceae plants (Fischer, 2004). The calyx tube of Linderniaceae is composed of four types, namely deeply lobed, shallowly lobed, half lobed, and no calyx tube (Liang, 2017). The last type is in fact that the calyx is completely split to base and divided into four spreading segments, consisting of a pair of cordate outer sepals and another pair of filiform lateral inner sepals. This type of calyx tube only occurs in Picria within the Linderniaceae. Liang (2017) first included Picria in molecular phylogenetic analyses and indicated that the genus was sister to Boyanna with moderate support (BS = 67, PP = 0.64). In this study, we included two individuals from different populations in phylogenetic analyses. The result further confirmed that Picria is closely related to Boyanna with strongly supported values (BS = 100, PP = 1.00). Except for significantly different calyx morphology, the two genera bear similar corolla tubes, clavate staminodes without appendages, and flora discs, which may reflect the relationship between the two genera. Due to the particular calyx and relatively isolated phylogenetic position, we supported the monotypic Picria as a separate genus.
Linderniaceae plastomes were highly conservative in terms of structure, gene order, and gene content, with slight contraction in the IR region. Except for that the ycf2 gene has a positive selection in a few species, most protein-coding genes were highly conserved, indicating that the evolution of Linderniaceae species was relatively slow. The phylogenetic analyses of Linderniaceae based on the complete plastid genome showed a higher resolution compared with previous studies, and the results revealed that the Vandellia and Torenia sensu Fischer et al. (2013) were non-monophyletic. The newly established Yamazakia was supported and the monotypic Picria could be regarded as a separate genus. Molecular phylogenetic evidence is of great importance to taxonomic revision of the complicated Linderniaceae. In this study, the sampling is not comprehensive with only 28 representative species from eight genera. As the plastid genome has been demonstrated to be effective in solving the phylogeny of Linderniaceae, a widely accepted taxonomic treatment is likely to be reached based on comprehensive sampling and plastid genome data.
The data presented in the study are deposited in the NCBI repository (https://www.ncbi.nlm.nih.gov/), and the accession numbers can be found in the article.
GH conceived and designed the study. GH, RY, and XZ collected the samples. RY and YJ performed experiments and data analyses. RY and GH drafted the manuscript. GH, CX, YG, and RY revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Natural Science Foundation of China (32060048, 32260099), the Natural Science Foundation of Guizhou Province (Qiankehejichu-ZK [2021] 091, Qiankehezhongyindi [2023] 029).
We are grateful to Jiaxing Yang from Wuhan Botanical Garden, Chinese Academy of Sciences, Wei Cao and Mei Wang from College of Life Sciences, Guizhou University for their help in materials collection, and Mingli Wu from College of Life Sciences, Guizhou University for help in data analyses.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1265641/full#supplementary-material
Almeida, E. M., Wanderley, A. M., Santos, A. D., de Melo, J. I. M., Souza, G., Batista, F. R. D. (2019). Two new genera and species of Linderniaceae (Lamiales) from inselbergs in northeastern Brazil: morphological and karyological evidence. Phytotaxa 400, 215–226. doi: 10.11646/phytotaxa.400.4.1
Alwadani, K. G., Janes, J. K., Andrew, R. L. (2019). Chloroplast genome analysis of box-ironbark Eucalyptus. Mol. Phylogenet. Evol. 136, 76–86. doi: 10.1016/j.ympev.2019.04.001
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Angiosperm Phylogeny Group (APG) (2009). An update of the angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc 161, 105–121. doi: 10.1111/boj.12385
Angiosperm Phylogeny Group (APG) (2016). An update of the angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc 181, 1–20. doi: 10.1111/boj.12385
Azarin, K., Usatov, A., Makarenko, M., Khachumov, V., Gavrilova, V. (2021). Comparative analysis of chloroplast genomes of seven perennial Helianthus species. Gene 774, 145418. doi: 10.1016/j.gene.2021.145418
Bai, X. J., Wang, G., Ren, Y., Su, Y. Y., Han, J. P. (2023). Insights into taxonomy and phylogenetic relationships of eleven Aristolochia species based on chloroplast genome. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1119041
Beardsley, P. M., Olmstead, R. G. (2002). Redefining Phrymaceae: the placement of Mimulus, tribe Mimuleae, and Phryma. Am. J. Bot. 89, 1093–1102. doi: 10.3732/ajb.89.7.1093
Beier, S., Thiel, T., Munch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Bentham, G. (1846). “Scrophulariaceae,” in Prodromus systematis naturalisregni vegetabilis. Ed. deCandolle, A. P. (Paris: Lovell: Reeve & Co), 186–586.
Biffin, E. D., Barker, W. R., Wannan, B., Liang, Y. S. (2018). The phylogenetic placement of Australian Linderniaceae and implications for generic taxonomy. Aust. Syst. Bot. 31, 241–251. doi: 10.1071/SB17058_CO
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinf. (Oxford. England). 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Brooks, A., Lowe, T. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, 686–689. doi: 10.1093/nar/gki366
Chakraborty, S., Yengkhom, S., Uddin, A. (2020). Analysis of codon usage bias of chloroplast genes in Oryza species: Codon usage of chloroplast genes in Oryza species. Planta 252 (4), 67. doi: 10.1007/s00425-020-03470-7
Chen, Q., Hu, H., Zhang, D. (2022). DNA barcoding and phylogenomic analysis of the genus fritillaria in China based on complete chloroplast genomes. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.764255
Chen, G., Wang, L. G., Wang, Y. H. (2021). Complete chloroplast genome sequence and phylogenetic analysis of Torenia fournieri. Mitochondrial. DNA B. Resour. 6 (7), 2004–2006. doi: 10.1080/23802359.2021.1939179
Cheng, X. L., Li, H. L., Wang, H. X., Nizamani, M. M., Chen, Y. (2019). Complete plastome sequence of Torenia concolor Lindley (Linderniaceae): an ornamental herb. Mitochondrial. DNA B. Resour. 4 (2), 2312–2313. doi: 10.1080/23802359.2019.1627949
Chu, R., Xu, X. M., Lu, Z. W., Ma, Y. G., Cheng, H., Zhu, S. X., et al. (2022). Plastome-based phylogeny and biogeography of Lactuca L. (Asteraceae) support revised lettuce gene pool categories. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.978417
Cui, Y., Chen, X., Nie, L., Sun, W., Hu, H., Lin, Y., et al. (2019). Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible amomum species. Int. J. Mol. Sci. 20, 4040. doi: 10.3390/ijms20164040
Cvijović, I., Good, B. H., Desai, M. M. (2018). The effect of strong purifying selection on genetic diversity. Genetics 209, 1235–1278. doi: 10.1534/genetics.118.301058
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi: 10.1186/s13059-016-1004-2
Darling, A. C. E., Mau, B., Blattner, F. R., Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
David, P., Buckley, T. R. (2004). Model selection and model averaging in phylogenetics: advantages of akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 53, 793–808. doi: 10.1080/10635150490522304
Do, H. D. K., Kim, J. H. (2017). A dynamic tandem repeat in monocotyledons inferred from a comparative analysis of chloroplast genomes in Melanthiaceae. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00693
Dong, W., Liu, J., Yu, J., Wang, L., Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PloS One 7, e35071. doi: 10.1371/journal.pone.0035071
Dong, X., Mkala, E. M., Mutinda, E. S., Yang, J. X., Wanga, V. O., Oulo, M. A., et al. (2022). Taxonomy, comparative genomics of Mullein (Verbascum, Scrophulariaceae), with implications for the evolution of Verbascum and Lamiales. BMC Genomics 23, 566. doi: 10.1186/s12864-022-08799-9
Dong, W. P., Xu, C., Li, C. H., Sun, J. H., Zuo, Y. J., Shi, S., et al. (2015). ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5, 8348. doi: 10.1038/srep08348
Downie, S. R., Jansen, R. K. (2015). A comparative analysis of whole plastid genomes from the Apiales: Expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 40 (1), 336–351. doi: 10.1600/036364415X686620
Doyle, J. J., Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15. doi: 10.1016/0031-9422(80)85004-7
Drescher, A., Ruf, S., Calsa, T., Jr., Carrer, H., Bock, R. (2000). The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 22 (2), 97–104. doi: 10.1046/j.1365-313x.2000.00722.x
Drew, B. T., González-Gallegos, J. G., Xiang, C. L., Kriebel, R., Drummond, C. P., Walked, J. B., et al. (2017). Salvia united: The greatest good for the greatest number. Taxon 66 (1), 133–145. doi: 10.12705/661.7
Drew, B. T., Sytsma, K. J. (2011). Testing the monophyly and placement of Lepechinia in the tribe Mentheae (Lamiaceae). Syst. Bot. 36, 1038–1049. doi: 10.1600/036364411X605047
Ebert, D., Peakall, R. (2009). Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 9, 673–690. doi: 10.1111/j.1755-0998.2008.02319.x
Fan, Y., Jin, Y., Ding, M., Tang, Y., Cheng, J., Zhang, K., et al. (2021). The complete chloroplast genome sequences of eight fagopyrum species: insights into genome evolution and phylogenetic relationships. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.799904
Fischer, E. (1995). Revision of the Lindernieae (Scrophulariaceae) in Madagascar. 1. The genera Lindernia Allioni and Crepidorhopalon E Fiseher. Bull. Mus. Natl. Hist. Nat. 7, 227–257.
Fischer, E. (2004). “Scrophulariaceae,” in The families and genera of vascular plants 7. Ed. Kadereit, J. W. (New York: Springer), 333–432.
Fischer, E., Schäferhoff, B., Müller, K. (2013). The phylogeny of Linderniaceae — The new genus Linderniella, and new combinations within Bonnaya, Craterostigma, Lindernia, Micranthemum, Torenia and Vandellia. Willdenowia 43, 209–238. doi: 10.3372/wi.43.43201
Fonseca, L. H. M. (2021). Combining molecular and geographical data to infer the phylogeny of Lamiales and its dispersal patterns in and out of the tropics. Mol. Phylogenet. Evol. 164, 107287. doi: 10.1016/j.ympev.2021.107287
Fonseca, L. H. M., Lohmann, L. G. (2018). Combining high-throughput sequencing and targeted loci data to infer the phylogeny of the "Adenocalymma-Neojobertia" clade (Bignonieae, Bignoniaceae). Mol. Phylogenet. Evol. 123, 1–15. doi: 10.1016/j.ympev.2018.01.023
Frazer, K. A., Lior, P., Alexander, P., Rubin, E. M., Inna, D. (2004). Vista: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Fu, X., Xie, D. F., Zhou, Y. Y., Cheng, R. Y., Zhang, X. Y., Zhou, S. D., et al. (2023). Phylogeny and adaptive evolution of subgenus Rhizirideum (Amaryllidaceae, Allium) based on plastid genomes. BMC Plant Biol. 23 (1), 70. doi: 10.1186/s12870-022-03993-z
Gao, Y. W., Liu, K. J., Li, E. Z., Wang, Y. S., Xu, C., Zhao, L. C., et al. (2022). Dynamic evolution of the plastome in the Elm family (Ulmaceae). Planta 257 (1), 14. doi: 10.1007/s00425-022-04045-4
Gu, L., Su, T., An, M. T., Hu, G. X. (2020). The complete chloroplast genome of the vulnerable oreocharis esquirolii (Gesneriaceae): structural features, comparative and phylogenetic analysis. Plants (Basel). 9, 1692. doi: 10.3390/plants9121692
Guo, X. X., Qu, X. J., Zhang, X. J., Fan, S. J. (2022). Comparative and phylogenetic analysis of complete plastomes among aristidoideae species (Poaceae). Biol. (Basel). 11 (1), 63. doi: 10.3390/biology11010063
Haines, H. H. (1922). Botany of bihar & Orissa 4 (London: Adlard and sold by agents for Indian Official Publications).
Hong, D. Y., Yang, H. B., Jin, C. L., Noel, H. H. (1998). “Scrophulariaceae,” in Flora of China (Vol. 18). Ed. Wu, C. Y. (Beijing: Science Press & St. Louis: Missouri Botanical Garden Press), 1–212.
Hu, G. X., Liu, E. D., Wu, Z. K., Sytsma, K. J., Drew, B. T., Xiang, C. L. (2020). Integrating DNA sequences with morphological analysis clarifies phylogenetic position of Salvia grandifolia (Lamiaceae): an enigmatic species endemic to southwestern China. Int. J. Plant Sci. 181 (8), 787–799. doi: 10.1086/709134
Huang, S., Ge, X., Cano, A., Salazar, B. G. M., Deng, Y. (2020). Comparative analysis of chloroplast genomes for five Dicliptera species (Acanthaceae): molecular structure, phylogenetic relationships, and adaptive evolution. PeerJ 8, e8450. doi: 10.7717/peerj.8450
Jin, J. J., Yu, W. B., Y ang, J. B., Song, Y., Li, D. Z. (2020). Getorganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kikuchi, S., Asakura, Y., Imai, M., Nakahira, Y., Kotani, Y., Hashiguchi, Y., et al. (2018). A ycf2-FtsHi heteromeric AAA-ATPase complex is required for chloroplast protein import. Plant Cell 30, 2677–2703. doi: 10.1105/tpc.18.00357
Kong, B. L.-H., Park, H.-S., Lau, T.-W. D., Lin, Z., Yang, T.-J., Shaw, P.-C. (2021). Comparative analysis and phylogenetic investigation of Hong Kong ilex chloroplast genomes. Sci. Rep. 11 (1), 5153. doi: 10.1038/s41598-021-84705-9
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). Reputer: the manifold applications of repeat analysis on genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Letunic, I., Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Lewis, D. Q. (2000). A revision of the New World species of Lindernia (Scrophulariaceae). CASTENEA 65, 93–122.
Li, E. Z., Liu, K. J., Deng, R. Y., Gao, Y. W., Liu, X. Y., Dong, W. P., et al. (2023). Insights into the phylogeny and chloroplast genome evolution of Eriocaulon (Eriocaulaceae). BMC Plant Biol. 23 (1), 32. doi: 10.1186/s12870-023-04034-z
Li, X. W., Y ang, Y., Henry, R. J., Rossetto, M., Wang, Y. T., Chen, S. L. (2015). Plant DNA barcoding: from gene to genome. Biol. Rev. Cambridge. Philos. Soc 90, 157–166. doi: 10.1111/brv.12104
Liang, Y. S. (2017). Systematic and phylogenetic study of linderniaceae (China: National Taiwan Normal University).
Liu, M. L., Fan, W. B., Wang, N., Dong, P. B., Zhang, T. T., Yue, M., et al. (2018). Evolutionary analysis of plastid genomes of seven lonicera L. Species: implications for sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 19 (12), 4039. doi: 10.3390/ijms19124039
Liu, B., Tan, Y. H., Liu, S., Olmstead, R. G., Min, D. Z., Chen, Z. D., et al. (2020). Phylogenetic relationships of Cyrtandromoea and Wightia revisited: A new tribe in Phrymaceae and a new family in Lamiales. J. Syst. Evol. 58, 1–17. doi: 10.1111/jse.12513
Lohse, M., Drechsel, O., Kahlau, S., Bock, R. (2013). Organellargenomedraw—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, 575–581. doi: 10.1093/nar/gkt289
Long, L., Li, Y., Wang, S., Liu, Z., Wang, J., Yang, M. (2023). Complete chloroplast genomes and comparative analysis of Ligustrum species. Sci. Rep. 13 (1), 212. doi: 10.1038/s41598-022-26884-7
López, J. L., Lozano, M. J., Lagares, A., Fabre, M. L., Draghi, W. O., Del Papa, M. F., et al. (2019). Codon usage heterogeneity in the multipartite prokaryote genome: selection-based coding bias associated with gene location, expression level, and ancestry. mBio 10, e00505–e00519. doi: 10.1128/mBio.00505-19
Müller, J., Müller, K., Quandt, D., Neinhuis, C. (2010) PhyDE – phylogenetic data editor. Available at: http://www.phyde.de/.
Nielsen, R., Yang, Z. (1998). Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148, 929–993. doi: 10.1093/genetics/148.3.929
Ogoma, C. A., Liu, J., Stull, G. W., Wambulwa, M. C., Oyebanji, O., Miline, R. I., et al. (2022). Deep insights into the plastome evolution and phylogenetic relationships of the tribe Urticeae (Family Urticaceae). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.870949
Ohta, T. (1995). Synonymous and nonsynonymous substitutions in mammalian genes and the nearly neutral theory. J. Mol. Evol. 40, 56–63. doi: 10.1007/BF00166595
Olmstead, R. G., dePamphilis, C. W., Wolfe, A. D., Young, N. D., Elisons, W. J., Reeves, P. A. (2001). Disintegration of the scrophulariaceae. Am. J. Bot. 88, 348–361. doi: 10.2307/2657024
Olmstead, R. G., Reeves, P. A. (1995). Evidence for the Polyphyly of the Scrophulariaceae Based on Chloroplast rbcL and ndhF Sequences. Ann. Mo. Bot. Gard. 82, 176–193. doi: 10.2307/2399876
Oxelman, B., Kornhall, P., Olmstead, R. G., Bremer, B. (2005). Further desintegration of the scrophulariaceae. Taxon 54, 411–425. doi: 10.2307/25065369
Parks, M., Cronn, R., Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7, 84. doi: 10.1186/1741-7007-7-84
Peden, J. F. (2000). Analysis of codon usage. Univ. Nottingham. 90, 73–74. doi: 10.1006/expr.1997.4185
Peng, C., Guo, X. L., Zhou, S. D., He, X. J. (2023). Backbone phylogeny and adaptive evolution of Pleurospermum s. l.: New insights from phylogenomic analyses of complete plastome data. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1148303
Peng, F., Zhao, Z., Xu, B., Han, J., Yang, Q., Lei, Y., et al. (2020). Characteristics of organellar genomes and nuclear internal transcribed spacers in the tertiary relict genus dipelta and their phylogenomic implications. Front. Genet. 11. doi: 10.3389/fgene.2020.573226
Pennell, F. W. (1935). The Scrophulariaceae of eastern temperate North America. Acad. Nat. Sci. Philadelphia. Monogr. 1, 1–650.
Pennell, F. W. (1943). A second summary of the Scrophulariaceae of New Guinea. J. Arnold. Arbor. 24, 243–274. doi: 10.5962/p.185478
Perret, M., Chautems, A., De Araujo, A. O., Salamin, N. (2013). Temporal and spatial origin of Gesneriaceae in the New World inferred from plastid DNA sequences. Bot. J. Linn. Soc 171, 61–79. doi: 10.1111/j.1095-8339.2012.01303.x
Philcox, D. (1968). Revision of the malesian species of lindernia all. (Scrophulariaceae). Kew Bull. 22, 1–72. doi: 10.2307/4107820
Qu, X. J., Moore, M. J., Li, D. Z., Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 50. doi: 10.1186/s13007-019-0435-7
Rahmanzadeh, R., Müller, K. F., Fischer, E., Bartels, D., Borsch, T. (2005). The Linderniaceae and Gratiolaceae are further lineages distinct from the Scrophulariaceae (Lamiales). Plant Biol. 7, 67–78. doi: 10.1055/s-2004-830444
Ravi, V., Khurana, J. P., Tyagi, A. K., Khurana, P. (2007). An update on chloroplast genomes. Plant Syst. Evol. 271, 101–122. doi: 10.1007/s00606-007-0608-0
Refulio-Rodriguez, N. F., Olmstead, R. G. (2014). Phylogeny of lamiidae. Am. Bot. J. 101, 287–299. doi: 10.3732/ajb.1300394
Schäferhoff, B., Fleischman, A., Fischer, E., Albach, D. C., Borsch, T., Heubl, G., et al. (2010). Towards resolving Lamiales relationships: insights from rapidly evolving chloroplast sequences. BMC Evol. Biol. 10, 352–374. doi: 10.1186/1471-2148-10-352
Librado, P., Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Song, W. C., Ji, C. X., Chen, Z. M., Cai, H. H., Wu, X. M., Shi, C., et al. (2022). Comparative analysis the complete chloroplast genomes of nine musa species: genomic features, comparative analysis, and phylogenetic implications. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.832884
Su, T., Geng, Y. F., Xiang, C. L., Zhao, F., Wang, M., Gu, L., et al. (2022). Chloroplast genome of salvia sect. Drymosphace: comparative and phylogenetic analysis. Diversity 5, 324. doi: 10.3390/d14050324
Tank, D. C., Beardsley, P. M., Kelchner, S. A., Olmstead, R. G. (2006). Review of the systematics of Scrophulariaceae s.l. and their current disposition. Aust. Syst. Bot. 19, 289–307. doi: 10.1071/SB05009
Thode, V. A., Lohmann, L. G. (2019). Comparative chloroplast genomics at low taxonomic levels: A case study using amphilophium (Bignonieae, bignoniaceae). Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00796
Tian, N., Han, L., Chen, C., Wang, Z. (2018). The complete chloroplast genome sequence of Epipremnum aureum and its comparative analysis among eight Araceae species. PloS One 13, e0192956. doi: 10.1371/journal.pone.0192956
Urban, I. (1884). Studien über die Scrophulariaceen-Gat-tungen Ilysanthes, Bonnaya, Vandellia und Lindernia. Ber. Deutsch. Bot. Ges. 2, 429–442. doi: 10.1111/j.1438-8677.1884.tb04219.x
Wan, T., Qiao, B. X., Zhou, J., Shao, K. S., Pan, L. Y., An, F., et al. (2023). Evolutionary and phylogenetic analyses of 11 Cerasus species based on the complete chloroplast genome. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1070600
Wang, R., Gao, J., Feng, J., Yang, Z., Qi, Z., Li, P., et al. (2022). Comparative and phylogenetic analyses of complete chloroplast genomes of scrophularia incisa complex (Scrophulariaceae). Genes (Basel). 13 (10), 1691. doi: 10.3390/genes13101691
Wang, Y., Wang, J., Garran, T. A., Liu, H., Lin, H., Luo, J., et al. (2023). Genetic diversity and population divergence of Leonurus japonicus and its distribution dynamic changes from the last interglacial to the present in China. BMC Plant Biol. 23 (1), 276. doi: 10.1186/s12870-023-04284-x
Wang, D., Zhang, Y., Zhang, Z., Jiang, Z., Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 8, 77–80. doi: 10.1016/s1672-0229(10)60008-3
Wei, R., Yan, Y. H., Harris, A. J., Kang, J. S., Shen, H., Xiang, Q. P., et al. (2017). Plastid phylogenomics resolve deep relationships among eupolypod II ferns with rapid radiation and rate heterogeneity. Genome Biol. Evol. 9 (6), 1646–1657. doi: 10.1093/gbe/evx1075
Wen, J., Xie, D. F., Price, M., Ren, T., Deng, Y. Q., Gui., L. J., et al. (2021). Backbone phylogeny and evolution of Apioideae (Apiaceae): New insights from phylogenomic analyses of plastome data. Mol. Biol. Evol. 161, 107183. doi: 10.1016/j.ympev.2021.107183
Wettstein, R. (1891). “Scrophulariaceae,” in Die natürlichen pflanzenfamilien. Eds. Engler, A., Prantl, K. (Leipzig: Wilhelm Engelmann), 39–107.
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Muller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wonok, W., Sudmoon, R., Tanee, T., Lee, S. Y., Chaveerach, A. (2023). Complete chloroplast genome of four thai native dioscorea species: structural, comparative and phylogenetic analyses. Genes (Basel). 14 (3), 703. doi: 10.3390/genes14030703
Worth, J. R. P., Liu, L., Wei, F. J., Tomaru, N. (2019). The complete chloroplast genome of Fagus crenata (subgenus Fagus) and comparison with F. engleriana (subgenus Engleriana). PeerJ 7, e7026. doi: 10.7717/peerj.7026
Wu, Z., Liao, R., Yang, T., Dong, X., Lan, D., Qin, R., et al. (2020). Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genomics 21 (1), 621. doi: 10.1186/s12864-020-07045-4
Wu, L., Nie, L., Wang, Q., Xu, Z., Wang, Y., He, C., et al. (2021). Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae. Sci. Rep. 11 (1), 14643. doi: 10.1038/s41598-021-94137-0
Wu, X. M., Wu, S. F., Ren, D. M., Zhu, Y. P., He, F. C. (2007). The analysis method and progress in the study of codon bias. Yi Chuan 29, 420–426. doi: 10.1360/yc-007-0420
Xi, J. W., Lv, S. B., Zhang, W. P., Zhang, J. B., Wang, K. T., Guo, H. B., et al. (2022). Comparative plastomes of Carya species provide new insights into the plastomes evolution and maternal phylogeny of the genus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.990064
Xie, P., Tang, L., Luo, Y., Liu, C., Yan, H. (2023). Plastid phylogenomic insights into the inter-tribal relationships of plantaginaceae. Biol. (Basel). 12 (2), 263. doi: 10.3390/biology12020263
Xue, J., Wang, S., Zhou, S. L. (2012). Polymorphic chloroplast microsatellite loci in nelumbo (nelumbonaceae). Am. J. Bot. 99, e240–e244. doi: 10.3732/ajb.1100547
Yamazaki, T. (1954). Notes on Lindernia, Vandellia, Torenia and their allied genera in eastern Asia 1. J. Jap. Bot. 29, 299–306.
Yamazaki, T. (1955). Notes on Lindernia, Vandellia, Torenia and their allied genera in eastern Asia, parts 3 and 4. J. Jap. Bot. 30, 359–364.
Yamazaki, T. (1985). “Scrophulariaceae,” in Flora du Cambodge, du Laos et du Viêt-Nam’. Ed. Leroy, J. F. (Pari: Muséum National d’Histoire Naturelle, Laboratoriede Phanérogamie), 1–217.
Yan, R. R., An, M. T., Hu, G. X. (2023). Taxonomy and geographical distribution of linderniaceae in China. Acta Bot. Sin. 43 (6), 1052–1062. doi: 10.7606/j.issn.1000-4025.2023.06.1052
Yang, J. X., Hu, G. X., Hu, G. W. (2022). Comparative genomics and phylogenetic relationships of two endemic and endangered species (Handeliodendron bodinieri and Eurycorymbus cavaleriei) of two monotypic genera within Sapindales. BMC Genomics 23, 27. doi: 10.1186/s12864-021-08259-w
Yu, J., Dossa, K., Wang, L., Zhang, Y., Wei, X., Liao, B., et al. (2017). PMDBase: a database for studying microsatellite DNA and marker development in plants. Nucleic Acids Res. 45, D1046–D1053. doi: 10.1093/nar/gkw906
Zhang, D., Gao, F., Jakovlicí, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhao, F., Chen, Y. P., Salmaki, Y., Drew, B. T., Wilson, T. C., Scheen, A. C., et al. (2021). An updated tribal classification of Lamiaceae based on plastome phylogenomics. BMC Bio. 19 (1), 2. doi: 10.1186/s12915-020-00931-z
Zhao, W., Guo, L. R., Yang, Y., Wang, Y., Yang, L., Wei, C. M., et al. (2022). Complete chloroplast genome sequences of Phlomis fruticosa and Phlomoides strigosa and comparative analysis of the genus Phlomis sensu lato (Lamiaceae). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1022273
Zhong, Q. W., Yang, S. P., Sun, X. M., Wang, L. H., Li, Y. (2019). The complete chloroplast genome of the Jerusalem artichoke (Helianthus tuberosus L.) and an adaptive evolutionary analysis of the ycf2 gene. PeerJ 7, 1–22. doi: 10.7717/peerj.7596
Zhou, C., Wang, P. T., Zeng, Q., Zeng, R., Hu, W., Sun, L., et al. (2023). Comparative chloroplast genome analysis of seven extant Citrullus species insight into genetic variation, phylogenetic relationships, and selective pressure. Sci. Rep. 13, 6779. doi: 10.1038/s41598-023-34046-6
Keywords: Lindernieae, Lindernia, Picria, Torenia, Scrophulariaceae, chloroplast genome, phylogenomics
Citation: Yan R, Geng Y, Jia Y, Xiang C, Zhou X and Hu G (2023) Comparative analyses of Linderniaceae plastomes, with implications for its phylogeny and evolution. Front. Plant Sci. 14:1265641. doi: 10.3389/fpls.2023.1265641
Received: 23 July 2023; Accepted: 01 September 2023;
Published: 26 September 2023.
Edited by:
Leonardo Miguel Galindo-González, Canadian Food Inspection Agency (CFIA), CanadaReviewed by:
Linchun Shi, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaCopyright © 2023 Yan, Geng, Jia, Xiang, Zhou and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoxiong Hu, Z3hodUBnenUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.