 Amar Kant Kushwaha1
Amar Kant Kushwaha1 Ranjith Kumar Ellur2
Ranjith Kumar Ellur2 Sarvesh Kumar Maurya2
Sarvesh Kumar Maurya2 Gopala Krishnan S.2
Gopala Krishnan S.2 Bishnu Maya Bashyal3
Bishnu Maya Bashyal3 Prolay Kumar Bhowmick2
Prolay Kumar Bhowmick2 K. K. Vinod2
K. K. Vinod2 Haritha Bollinedi2
Haritha Bollinedi2 Nagendra Kumar Singh4
Nagendra Kumar Singh4 Ashok Kumar Singh2*
Ashok Kumar Singh2*- 1Division of Crop Improvement and Biotechnology, Indian Council of Agricultural Research (ICAR)-Central Institute for Subtropical Horticulture, Lucknow, India
- 2Division of Genetics, Indian Council of Agricultural Research (ICAR)-Indian Agricultural Research Institute, New Delhi, India
- 3Division of Plant Pathology, Indian Council of Agricultural Research (ICAR)-Indian Agricultural Research Institute, New Delhi, India
- 4National Professor B.P. Pal Chair, Indian Council of Agricultural Research (ICAR)-National Institute of Plant Biotechnology, New Delhi, India
Bakanae disease caused by Fusarium fujikuroi is an emerging disease of rice causing losses in all rice-growing regions around the world. A BC2F2 population was developed by backcrossing the recurrent parent Pusa Basmati 1121 (PB1121) with the recombinant inbred line RIL28, which harbors a major quantitative trait locus (QTL) governing resistance to bakanae, qBK1.2. MassARRAY-based single-nucleotide polymorphism (SNP) assays targeting the genomic region of qBK1.2 helped in fine mapping the QTL to a region of 130 kb between the SNP markers rs3164311 and rs3295562 using 24 recombinants. In-silico mining of the fine-mapped region identified 11 putative candidate genes with functions related to defense. The expression analysis identified two significantly differentially expressed genes, that is, LOC_Os01g06750 and LOC_Os01g06870, between the susceptible genotype PB1121 and the resistant genotypes Pusa1342 and R-NIL4. Furthermore, the SNPs identified in LOC_Os01g06750 produced minor substitutions of amino acids with no major effect on the resistance-related functional motifs. However, LOC_Os01g06870 had 21 amino acid substitutions, which led to the creation of the leucine-rich repeat (LRR) domain in the resistant genotype Pusa1342, thereby making it a potential candidate underlying the major bakanae-resistant QTL qBK1.2. The markers used in the fine mapping program are of immense utility in marker-assisted breeding for bakanae resistance in rice.
1 Introduction
Bakanae disease is one of the emerging diseases of rice incited by Fusarium fujikuroi (Nirenberg, 1976) (teleomorph: Gibberella fujikuroi Sawada (Ito and Kimura, 1931), a filamentous hyphomycete fungus. The disease is prevalent in temperate and tropical rice-growing regions of the world (Amoah et al., 1995; Desjardins et al., 1997; Amatulli et al., 2010; Eğerci et al., 2021). Reported earlier as a minor disease, bakanae has emerged during the last decade as a serious problem in Basmati-growing areas in India, causing yield losses of up to 70% as well as impairing grain quality (Bashyal, 2018). The symptoms of the disease include seedling elongation, seedling mortality, reduced tillers, and elongated non-productive tillers (Bashyal et al., 2016). The disease is primarily seed borne, and secondary spread is through spores on plant parts and soil [Sun, 1975; Anderson and Webster, 2005]. Hot water treatment, and chemical and biological control measures are the major interventions to manage the disease; however, genetic resistance is considered effective (Hayasaka et al., 2001; Hossain et al., 2015; Lee et al., 2018; Sarwar et al., 2018), as it is economical, sustainable, and eco-friendly.
Identification of resistant sources of bakanae disease has found little progress so far due to the complexity of disease responses, which is associated with weather conditions and the pathogen complex known as the G. fujikuroi species complex (GFSC) (Desjardins et al., 2000; Wulff et al., 2010; Bashyal and Aggarwal, 2013; Jeon et al., 2013). GFSC is a complex of Fusarium species such as Fusarium andiyazi, Fusarium proliferatum, and Fusarium verticilloides, which are also associated with the disease, together with F. fujikuroi. This complexity poses challenges in screening rice genotypes under natural conditions. The development of a robust screening technique under artificial inoculation conditions for rice genotypes (Fiyaz et al., 2014) has, however, eased the identification of resistant genotypes. Using this method, several genotypes resistant to bakanae have been identified and successfully utilized in mapping quantitative trait loci (QTLs) governing resistance (Fiyaz et al., 2016). To date, as many as 12 QTLs from various chromosomes have been mapped to impart resistance either through linkage mapping using bi-parental mapping populations or by association mapping using diversity panels comprising germplasm from diverse ecogeographic regions (Yang et al., 2006; Hur et al., 2015; Fiyaz et al., 2016; Volante et al., 2017; Ji et al., 2018; Lee et al., 2018; Chen et al., 2019; Cheon et al., 2019; Kang et al., 2019; Lee et al., 2021; Lee et al., 2022). A maximum number of QTLs have reported on chromosome 1, which includes qB1, qBK1, qBK1.1, qBK1.2, qBK1.3, qFfR1, qBK1wd, and qBK1z (Yang et al., 2006; Hur et al., 2015; Fiyaz et al., 2016; Ji et al., 2018; Lee et al., 2018; Lee et al., 2021). Other QTLs reported are qBK3.1 on chromosome 3 (Fiyaz et al., 2016), qBK4_31750955 and qBK4T on chromosome 4 (Volante et al., 2017; Lee et al., 2022), qBK_628091 on chromosome 6 (Volante et al., 2017), qFfrR9 on chromosome 9 (Kang et al., 2019), and qB10 on chromosome 10 (Yang et al., 2006). However, only one QTL qBK1 has been fine mapped so far, confined to a 35-kb genomic region, and flanked by InDel markers InDel 18 and InDel 19-14 (Lee et al., 2019). Based on gene expression studies, four putative candidate genes (LOC_Os01g41770, LOC_Os01g41780, LOC_Os01g41790, and LOC_Os01g41800) have been identified. The molecular mechanism underlying resistance to bakanae remains largely unknown. However, a transcriptome study has indicated the involvement of jasmonic acid biosynthesis pathway-related genes in the resistant genotype Selenio (Matić et al., 2016).
Since the reported QTLs have been proven effective only against specific isolates of F. fujikuroi prevalent in different regions, the wide utilization of the reported QTLs in breeding is limited. However, Lee et al. (2018) found that the pyramiding of the two QTLs qBK1 and qBK1wd provided a better level of resistance as compared to individual QTL introgressed lines. Identification of novel QTLs and their deployment into elite cultivars is a proven strategy to broaden the spectrum of resistance and restrict the evolution of pathogens. Furthermore, validating the identified QTLs is of utmost importance prior to their utilization in breeding programs. One of the approaches for validation of a QTL is the development of QTL introgressed near-isogenic lines (QTL-NILs), which can also be used for fine mapping and identification of underlying candidate genes. Also, developing functional markers for the candidate gene bears significance in improving precision in marker-assisted breeding.
In a population generated from the cross between PB1121 and Pusa1342, a major QTL qBK1.2 was mapped on chromosome 1, which explained 24.74% phenotypic variance (Fiyaz et al., 2016), but remains to be fine mapped. This QTL is effective against the F. fujikuroi isolates prevalent in Basmati-growing regions of India. Therefore, this study reports the validation and fine mapping of qBK1.2 while developing tightly linked markers for their use in marker-assisted breeding.
2 Materials and methods
2.1 Plant material and development of mapping population
RIL28, an individual from a recombinant inbred line (RIL) population (derived from the cross between PB1121 and Pusa1342) carrying the major QTL for bakanae, qBK1.2, as confirmed by QTL flanking simple sequence repeat (SSR) markers RM10153 and RM5336, was backcrossed with the susceptible parent PB1121, and a backcross population was generated. The generation advancement was performed by shuttling the material between two locations, i.e., Indian Agricultural Research Institute-New Delhi (IARI-New Delhi) and Rice Breeding and Genetics Research Centre, Tamil Nadu (RBGRC-Tamil Nadu). The F1, BC1F1, and BC2F1 generations were subjected to foreground selection using the markers RM10153 and RM5336 flanking the QTL qBK1.2, and background selection was carried out using 28 SSR markers polymorphic between the parental lines (PB1121 and RIL28) to estimate the recurrent parent genome (RPG) recovery. Background recovery was calculated using the formula (1 − (n/N), where n is the number of marker loci heterozygous or homozygous for the recurrent parent allele and N is the total polymorphic markers between parental lines. Genotyping was carried out in the BC2F2 generation, and phenotyping for bakanae resistance was carried out in the BC2F2:3 generation. The scheme utilized for developing the BC2F2:3 population is provided in Figure 1.

Figure 1 Shuttle breeding scheme for rapid development of backcross mapping population.
2.2 Phenotypic screening against bakanae disease
F. fujikuroi isolates were multiplied on sterile sorghum grain at the Division of Plant Pathology, ICAR-Indian Agricultural Research Institute, New Delhi. A 15-day-old culture medium was mashed in sterile water and filtered using a muslin cloth. A suspension culture with a conidial concentration of 1 × 106 was used for inoculation. To screen the rice genotypes, the seeds were soaked in a suspension culture of a virulent F. fujikuroi isolate F250 (National Center for Biotechnology Information (NCBI); gene bank accession number KM50526, collected from the northwestern part of India) in the test tubes for 48 hours and incubated at a temperature of 30°C. These seeds were then sown in pot trays (Figure 2) and kept in a growth chamber with a day/night temperature of 30°C/25°C and a day/night relative humidity of 60%/80%. Disease scoring on mortality percentage was carried out 12 days after inoculation and classified into different classes (Table 1) as described by Fiyaz et al. (2014).

Figure 2 (A) Pure culture isolated from diseased plants. (B) Autoclaved sorghum seeds were inoculated with pure culture for mass multiplication. (C) The suspension was prepared. (D) Concentration of the suspension was checked under a light microscope. (E) Rice seeds were put in Eppendorf tubes and inoculated with suspension culture for 48 h. (F) Sowing and disease scoring was performed in 14 × 7 pot trays.
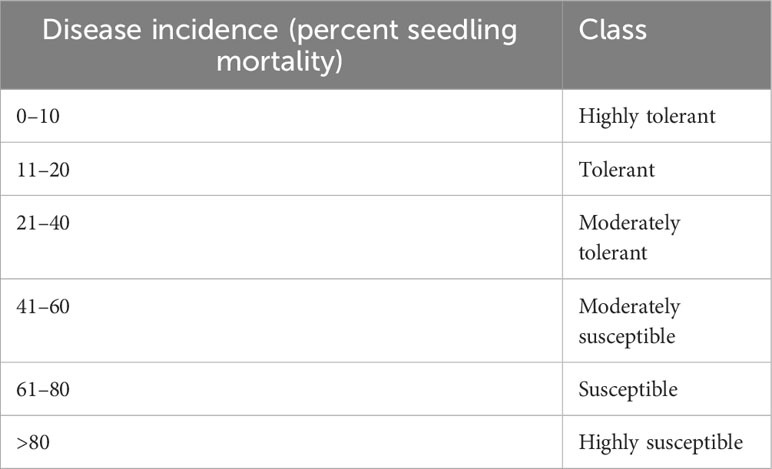
Table 1 Phenotypic classification based on percent seedling mortality.
2.3 Genotypic screening during population development
Fresh leaf samples were collected from 15-day-old seedlings and crushed using a DNA-lyser in Eppendorf tubes. DNA was isolated using the cetyl trimethylammonium bromide (CTAB) protocol described by Murray and Thompson (1980). For the development of the backcross population, foreground selection was carried out using the markers RM10153 and RM5336 (Supplementary Table 1) flanking the QTL qBK1.2 (Supplementary Figures S1 and S2). Out of 119 SSR markers polymorphic between PB1121 and Pusa1342, 28 markers were found polymorphic between PB1121 and RIL28 (Supplementary Table 2). Parental polymorphism and foreground and background selection were performed in a PCR volume of 10 μl. Each 10-μl reaction volume included 2 μl of template DNA (50 ng), 1 μl of forward primer (5 pm/μl), 1 μl of reverse primer (5 pm/μl), and 3 μl of Taq DNA Polymerase RED 2× master mix (AMPLIQON A/S, Odense, Denmark) and 3 μl of nuclease-free water. PCRs were performed in 96-well PCR plates with a thermal seal in a thermal cycler (Applied Biosystems Veriti™, Foster City, CA, USA). Amplified PCR products were resolved on 3.5% (w/v) agarose gel stained with ethidium bromide. Gels were visualized using a UV-transilluminator gel documentation system (Bio-Rad Gel Doc XR+, Hercules, CA, USA).
2.4 SNP genotyping of the recombinants BC2F2:3 plants
Since there were no polymorphic SSR markers available within the region spanning RM10153–RM5336, the single-nucleotide polymorphisms (SNPs) polymorphic between PB1121 and Pusa1342 located between the markers, i.e., RM10153 and RM5336, were identified using the re-sequence data available with us (data unpublished). To design the SNP assay, 150-bp upstream and downstream sequences of the SNP were retrieved from the NCBI database using Oryza sativa cv. Nipponbare as the reference genotype. Then, the SNP assays were designed in assay design suite (ADS) software from Agena® Bioscience (4755 Eastgate Mall, San Diego, CA, USA) using the SNP group file option, and SNP genotyping was performed using MassARRAY®, which is based on matrix-assisted laser desorption/ionization–time of flight (MALDI-TOF) mass spectrometry. The steps involved in the genotyping were the amplification of the target region harboring the SNP with PCR following an extension PCR involving primer designed from assay design suit, having the proximal end complementary to the polymorphic base. The resultant mixture from extension PCR was then loaded on SpecroCHIP®, which was further placed in a MassARRAY mass spectrometer for signal detection and allele confirmation at the polymorphic site (Ellis and Ong, 2017).
2.5 In silico search and validation for candidate genes within the fine-mapped region
RNA isolation of the treatment and control was performed 6 days post-inoculation from PB1121, RIL28, and R-NIL4 in three replicates. Total RNA was extracted from shoots using a NucleoSpin RNA kit (Macherey–Nagel, Düren, Germany). Genes were searched within the fine-mapped region using the genome browser in the Rice Genome Annotation Project database (http://rice.uga.edu/) (Supplementary Table 3). Primers for real-time PCR analysis of putative candidate genes were designed using the NCBI primer blast tool with a parameter product length of 80–150 bp and Tm of 59°C–61°C. The sequences of these primers are listed in Table 2. Rice elF4α (eukaryotic initiation factor-4α) was used as the reference gene for the normalization of expression data. The PCR mixture contained 2.5 μl cDNA (10 times diluted), 5 μl of 2× SYBR green PCR master mix (Bio-Rad, USA), and 100 nM of each gene-specific primer in a final volume of 10 μl. Real-time PCR was performed for all putative candidate genes. Negative template control (NTC) was also performed for each primer pair. Real-time PCR was performed in a Bio-Rad real-time PCR machine (Bio-Rad, USA). All PCRs were performed under the following conditions: 10 min at 95°C and 40 cycles of 15 s at 95°C, 30 s at 60°C and melt curve with a single reaction cycle following conditions 95°C for 15 s, 60°C for 1 min, and dissociation at 95°C for 15 s. Three biological replicates were analyzed for each sample. The relative expression ratio was calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001).

Table 2 List of primers for real-time PCR for validation of candidate genes.
2.6 In silico analysis of candidate genes
The sequences of genes, i.e., LOC_Os01g06750 and LOC_Os01g06870 of PB1121 and Pusa1324, were generated by incorporating the SNPs, which were identified through re-sequencing of PB1121 and Pusa1342 using Nipponbare as a reference genome (Supplementary File Alignment). These sequences were then subjected to SPLIGN analysis to identify if the introns were present (Kapustin et al., 2008). The resulting coding sequences (CDSs) thus generated were then translated, and alignment was performed to identify amino acid substitutions between PB1121 and Pusa1342 (Madeira et al., 2022). Using the translated proteins, prosite analysis was carried out for the identification of differences in defense-related motifs (De Castro et al., 2006).
3 Results
Among the RILs generated from the cross between PB1121 and Pusa1342, RIL28 was chosen for the study because it carried qBK1.2 and showed a percent seedling mortality of 32% as compared to 95.33% in the susceptible genotype PB1121 under bakanae infection in the artificial screening conditions (Figures 2A, B). Furthermore, RIL28 exhibited 76.47% similarity to PB1121 based on the 119 SSR markers of which 28 were polymorphic (Figure 3).
Figure 3 Polymorphism between recurrent parents PB1121 and RIL28.
The F1s from the cross made between PB1121 and RIL28 when subjected to the test of hybridity using the qBK1.2 linked markers, RM10153 and RM5336, showed one true hybrid out of 12 plants tested. A true F1 was backcrossed with PB1121, 35 BC1F1s were generated with qBK1.2, and RPG recovery ranged from 78.57% and 82.14%. Out of these 35 BC1F1s, an individual plant with RPG recovery of 82.15% was backcrossed with PB1121 to generate 90 BC2F1s. The RPG recovery among the BC2F1 plants carrying qBK1.2 ranged from 85.71% to 96.42%. A BC2F1 plant with 96.42% RPG recovery was selfed to generate a BC2F2 population comprising 1,100 individuals. Genotyping with foreground markers followed by phenotyping using F. fujikuroi F250 isolate (Figure 4) resulted in the identification of 24 BC2F2:3 recombinants falling in both resistant and susceptible classes.

Figure 4 The response of genotypes to Fusarium fujikuroi infection. (A) Parental lines used for developing population for fine mapping. (B) Percent seedling mortality in the parental lines.
3.1 Disease reaction among the backcross-derived lines
The backcross population, i.e., BC2F2:3 families, were phenotyped for bakanae resistance and classified based on disease reaction, i.e., percent seedling mortality. The NILs on artificial inoculation and screening showed significant variation in percent seedling mortality, which ranged from highly susceptible to highly resistant reactions (Figure 5).
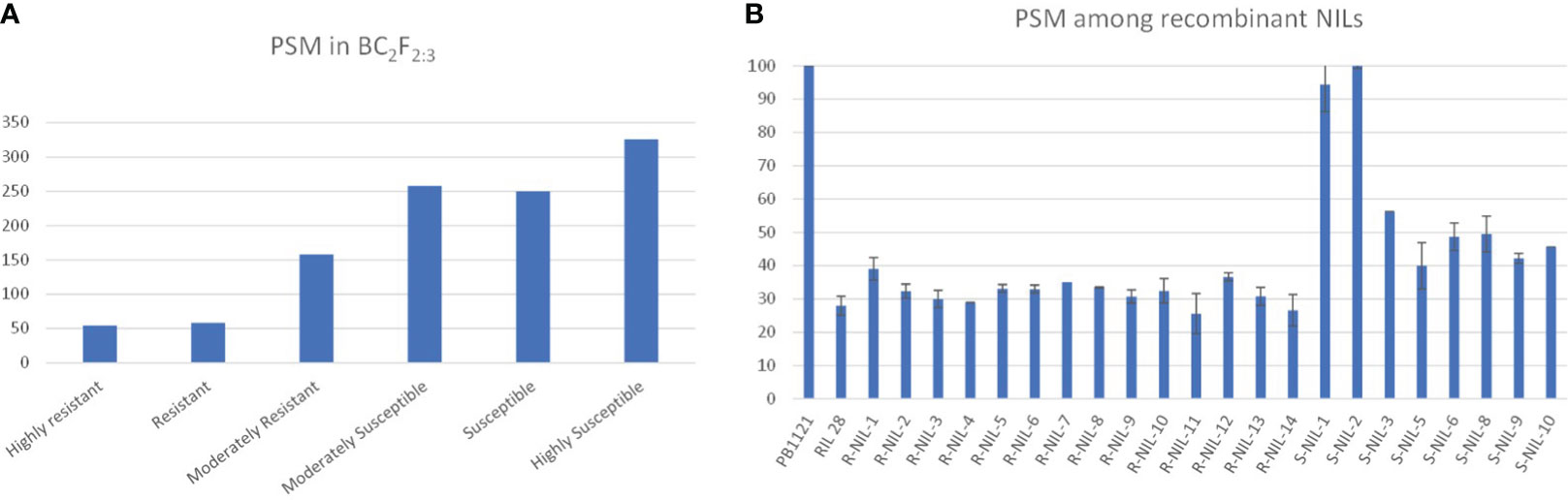
Figure 5 (A) Number of classes in different groups based on percent seedling mortality (PSM). (B) disease reaction of recombinants.
Based on the marker profile and bakanae screening data of RM10153 and RM5336, a total of 24 recombinants were identified. These recombinants can be categorized into three groups, viz., i) recombinant on the RM10153 marker side, ii) recombinant on the RM5336 side, and iii) recombinant on both sides. Four SNPs within the target genomic region were identified based on whole genome re-sequencing data of the parental lines PB1121 and Pusa1342. Genotyping of recombinants with the MassARRAY (MALDI-TOF mass spectrometry)-based SNP genotyping delimited qBK1.2 to a region of 130 kb between the SNPs rs3164311 and rs3295562 (Figure 6).

Figure 6 Fine-mapped region of qBK1.2. The QTL region is delimited to 130 kb between SNPs rs3164311 and rs3295562 using recombinants. The values on the right side indicate percent mortality upon inoculation. NIL, near-isogenic line; R-NIL, resistant NIL; S-NIL, susceptible NIL; QTL, quantitative trait locus; SNPs, single-nucleotide polymorphisms.
3.2 Expression profiling of putative candidate genes
In-silico search within the fine-mapped region using a rice genome annotation project database (http://rice.uga.edu/) led to the identification of 21 putative gene models, among which 11 were with annotated disease resistance-related functions (SlVe1, SlVe2, Ve1, cf-2, and other Verticillium wilt resistance genes) (Figure 7).
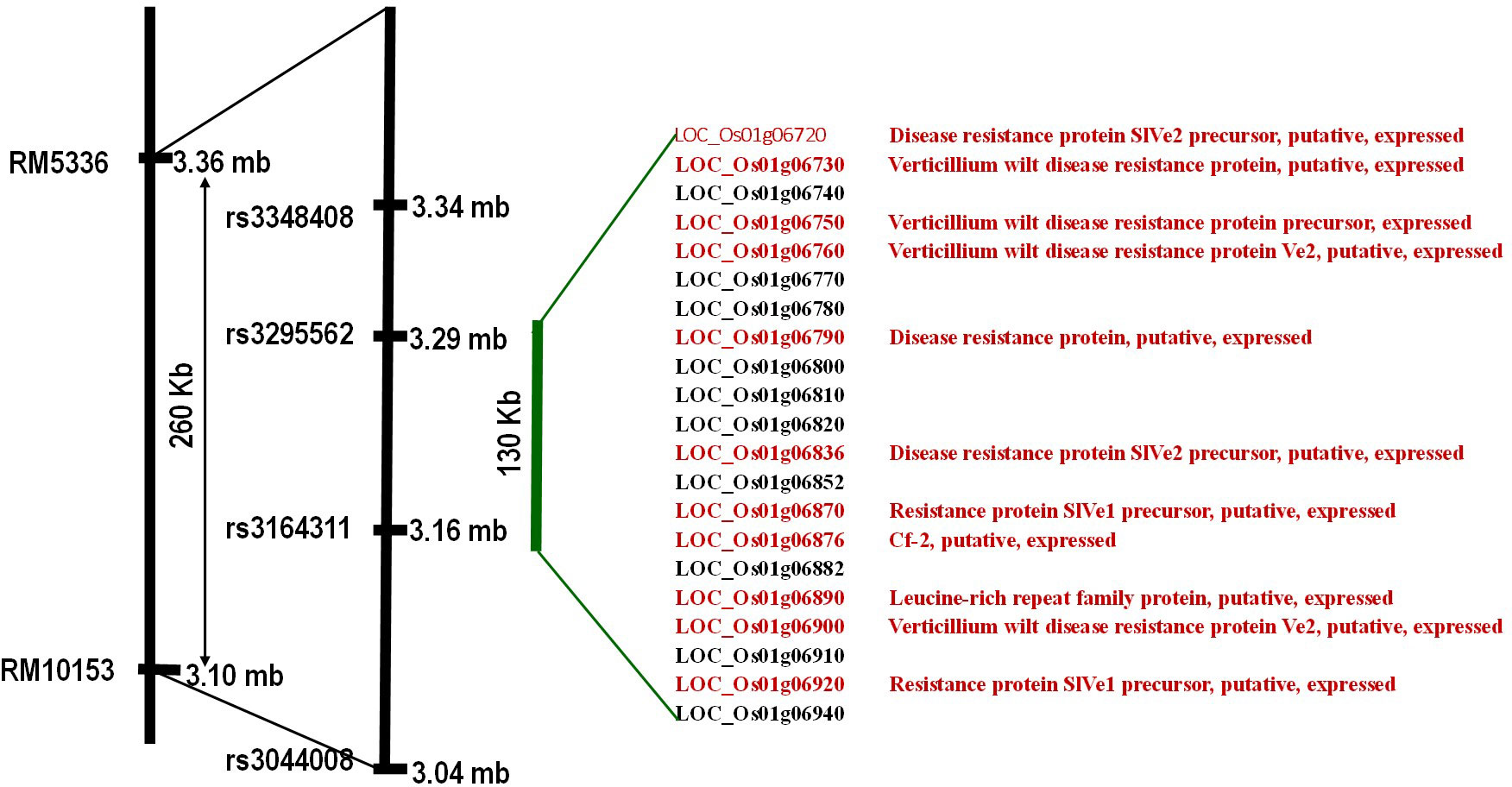
Figure 7 Putative genes underlying the fine-mapped region. Locus IDs in red have a putative function in defense against disease, and IDs in black represent retroposons.
Expression of all the 11 putative candidate genes was profiled under mock and inoculated conditions in PB1121, Pusa1342, and Resistant NIL-4 (R-NIL4). Two genes, that is, LOC_Os01g06750 and LOC_Os01g06870, showed significant upregulation when treated with F250 isolate of F. fujikuroi (day/night temperature of 30°C/25°C ( ± 3)°C and relative humidity of 60%/8%0 ( ± 10%)) in the resistant genotypes Pusa1342 and R-NIL4, while there was significant downregulation in the susceptible genotype PB1121. Genes such as LOC_Os01g06720, LOC_Os01g06730, LOC_Os01g06760, and LOC_Os01g06876 had significant differential expression between the resistant genotype Pusa1342 and susceptible genotype PB1121, but not in R-NIL4. The expression of LOC_Os01g06790 was significantly upregulated in R-NIL4 while downregulated in both Pusa1342 and PB1121 (Figure 8). This indicated that genes LOC_Os01g06750 and LOC_Os01g06870 could be the potential candidates underlying the major QTL qBK1.2. Further analysis indicated that these genes are orthologous to the tomato Verticillium wilt resistance gene Vei.
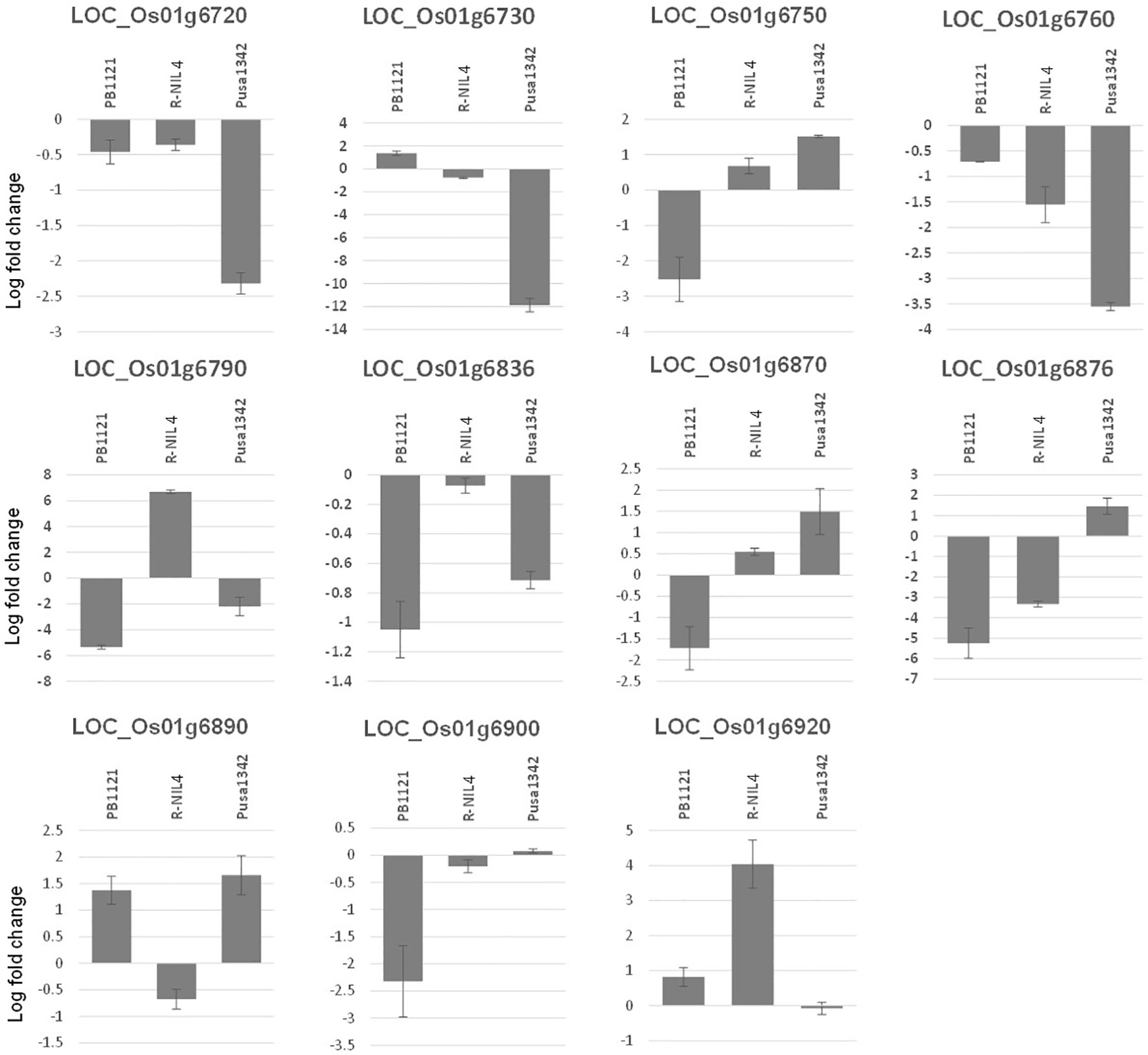
Figure 8 Relative expression (log fold change) of putative candidate genes underlying the fine-mapped region.
3.3 In silico comparison of candidate genes and products
LOC_Os01g06750 was predicted to possess five exons (Figure 9A). Alignment of the sequences of PB1121 and Pusa1342 led to the identification of one, six, and four SNPs on exons 1, 4, and 5, respectively. Alignment of 494 amino acid translated sequence of PB1121 and Pusa1342 (Figure 9B) resulted in the identification of three amino acid substitutions: i) substitution of threonine with proline at 185th amino acid, ii) serein with glycine at 191st amino acid, and iii) leucine with proline at 246th amino acid position. Since these substitutions did not lead to any structural differences (Figure 9C), the role of this gene at protein level cannot be confirmed.

Figure 9 (A) Structure of gene LOC_Os01g06750; solid yellow boxes represent exons. (B) Alignment of proteins from gene LOC_Os01g06750 from PB1121 and Pusa1342. (C) Number of LRR motif in PB1121 and Pusa1342. LRR, leucine-rich repeat.
LOC_Os01g06870 was predicted to be an intron-less gene that codes for resistance protein SlVe1 precursor with two leucine-rich repeat (LRR) domains. DNA sequence alignment led to the identification of 30 SNPs between PB1121 and Pusa1342. Translated product alignment resulted in the identification of 21 amino acid substitutions. This led to the creation of an extra LRR domain in the resistant parent Pusa1342 as compared to the susceptible genotype PB1121 (Figure 10). Therefore, LOC_Os01g06870 may be considered as the potential candidate underlying qBK1.2 conferring tolerance to bakanae disease.

Figure 10 (A) Structure of gene LOC_Os01g06870 represents single exons. (B) Alignment of proteins from gene LOC_Os01g06870 from PB1121 and Pusa1342. (C) Reduced number of LRR domains in PB1121 as compared to Pusa1342. LRR, leucine-rich repeat.
4 Discussion
The severity of bakanae disease has been increasing over the years in the Indo-Gangetic plains of India wherein Basmati rice is one of the major crops. The majority of the Basmati rice varieties show susceptibility to bakanae. Although genetic resistance to bakanae is reported by several studies, the sources of resistance lie outside the Basmati gene pool. Naturally, Basmati rice has a narrow genetic base, and hence, the deployment of resistance genes is sought from non-Basmati sources. This necessitates a long breeding process to safeguard the specific Basmati traits along with the recruited trait. Recently, it was established that marker-assisted backcross breeding can ease this cumbersome breeding process and accelerate varietal development with enormous success (Lee et al., 2018).
The QTL qBK1.2 identified on chromosome 1 lies over a span of 260 kb on the short arm flanked by the SSR markers, i.e., RM10153 and RM5336 (Fiyaz et al., 2016). In the current study, we used the approach of fine mapping qBK1.2 using QTL-NILs. NILs provide several advantages such as low background noise and genetic similarity to the sensitive parent. Because of these benefits, NILs are considered ideal for QTL validation, fine mapping, comparative genomics, marker development, gene expression studies, and even deployment as new cultivars. The BC2F2 population generated in the current study had an RPG recovery of 97.91%. Moreover, the F2:3 population provided an opportunity for evaluating resistance reactions under the artificial screening system. The variation observed among the NILs was quantitative and could be due to environmental influence. The environmental influence of disease response makes the management of phenotyping cumbersome, expensive, and time-consuming.
The SNP genotyping among the recombinants delimited qBK1.2 to a region of 130 kb between the SNPs rs3164311 and rs3295562. It was found that when this region was absent in the S-NIL1 and S-NIL2, the disease intensity reached above 94% (Figure 4). Since the other regions between the flanking markers were similar to the donor, the likelihood of the region being the candidate locus was very high, making us conclude that the QTL resided in this region. A similar strategy was used to fine map several QTLs in rice including qBK1 (Lee et al., 2019). The fine-mapped region comprised 11 annotated genes with putative disease resistance function. The expression pattern of a gene under mock and challenged inoculation conditions provides significant contrast in its involvement in response under infection (Matić et al., 2016; Cheng et al., 2020). Genes LOC_Os01g41770 and LOC_Os01g41780 were annotated to encode leucine-rich repeat motifs, which were identified to negatively regulate bakanae disease resistance (Lee et al., 2019). Eight of 11 genes that were subjected to expression profiling between the parental lines PB1121 and Pusa1342 showed significant differential expression. The observed differences may be attributed to the response to infection dynamics and/or due to inherent differences in the genomes of these genotypes. Therefore, comparing the expression between the NILs could help identify the genes responding to the infection. Accordingly, the R-NIL4 carrying qBK1.2 in the genetic background of PB1121 was profiled along with the parental lines. The expression pattern ruled out the involvement of five loci, i.e., LOC_Os01g06720, LOC_Os01g06730, LOC_Os01g06760, LOC_Os01g06876, LOC_Os01g06900, and LOC_Os01g06790, in the manifestation of resistance to bakanae because they failed to show a differential response between qBK1.2 carriers (R-NIL4 and Pusa1342) and the non-carrier (PB1121). Two genes, that is, LOC_Os01g06750 and LOC_Os01g06870, have shown significant differential expression between susceptible (PB1121) and resistant individuals (Pusa1342 and R-NIL4). A comparative analysis of encoded protein using BLASTp showed that these genes are orthologues of Verticillium wilt (Verticillium dahliae) resistance gene of tomato, i.e., Ve1, which is rich in leucine-rich repeats. Ve1 gene of tomato was identified to provide resistance in tomato against race 1 strains of V. dahliae and Verticillium albo-atrum (De Jonge et al., 2012). It was also shown that Ve1 requires the cascade of signaling genes, such as Enhanced Disease Susceptibility 1 (EDS1), Non-race-specific Disease Resistance 1 (NDR1), NB-LRR Protein Required for HR-Associated Cell Death 1 (NRC1), ACIF, MAPK/ERK kinase 2 (MEK2), and SERK3/BAK1 (Fradin et al., 2009). The other gene, i.e., Ve2, has been shown to act antagonistically to Ve1 using RNAi, and it was found that Ve1 regulates the expression of defense-related genes by minimizing the effects of Ve2 on these genes (Nazar et al., 2018).
LOC_Os01g06750 is an interrupted gene with four introns, while LOC_Os01g06870 is an uninterrupted gene. The gene sequence variation among the genotypes determines the target phenotype. Although SNPs were identified in LOC_Os01g06750 between PB1121 and Pusa1342, the translated product revealed minor substitution of amino acids with no major effect on functional motifs, i.e., leucine-rich repeat having a role in the resistance-related functions (McHale et al., 2006). This indicates its function in providing tolerance to bakanae at the transcriptional or posttranscriptional level or by generating protein products through alternate splicing of the exons. However, the translated product of LOC_Os01g06870 had 21 amino acid substitutions, which led to the creation of the LRR domain in the resistant individual Pusa1342. The differential expression of LOC_Os01g06870 and functional difference in gene product make it the potential candidate gene in providing tolerance against bakanae disease and can be conclusively proved with functional validation by generating transgenic events in the susceptible genotype.
We conclude that LOC_Os01g06870 is a potential putative candidate gene conferring tolerance to bakanae disease, underlying the major QTL qBK1.2 between the SNPs rs3164311 and rs3295562. These markers are resources of immense importance for a rice breeding program targeting to introgress qBK1.2 for developing bakanae resistance in rice.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
AK: Data curation, Formal Analysis, Investigation, Validation, Visualization, Writing – original draft. RE: Formal Analysis, Methodology, Writing – review & editing. GK: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing. SM: Data curation, Writing – review & editing. BB: Resources, Writing – review & editing. PB: Writing – review & editing. HB: Writing – review & editing. KV: Writing – review & editing. NS: Resources, Writing – review & editing. AS: Conceptualization, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research work was funded by the Indian Council of Agricultural Research, New Delhi, India, under the project Incentivizing Research in Agriculture.
Acknowledgments
The study is part of the PhD research of the first author. The first author would like to acknowledge the Post Graduate School, ICAR-IARI, New Delhi, for providing all the facilities during the study. The National fellowship he received from the University Grants Commission, Government of India, is gratefully acknowledged. We thank the National Phytotron Facility, ICAR-Indian Agricultural Research Institute, Pusa, New Delhi, for providing facilities to carry out phenotyping.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1265176/full#supplementary-material
References
Amatulli, M. T., Spadaro, D., Gullino, M. L., Garibaldi, A. (2010). Molecular identification of Fusarium spp. associated with bakanae disease of rice in Italy and assessment of their pathogenicity. Plant Pathol. 59 (5), 839–844. doi: 10.1111/j.1365-3059.2010.02319.x
Amoah, B. K., Rezanoor, H. N., Nicholson, P., MacDonald, M. V. (1995). Variation in the Fusarium section Liseola: pathogenicity and genetic studies of isolates of Fusarium moniliforme Sheldon from different hosts in Ghana. Plant Pathol. 44 (3), 563–572. doi: 10.1111/j.1365-3059.1995.tb01678.x
Anderson, L., Webster, R. (2005). A comparison of assays for Gibberella fujikuroi and their ability to predict resulting bakanae from rice seed sources in California. Phytopathology 95 (6).
Bashyal, B. M. (2018). Etiology of an emerging disease: bakanae of rice. Indian Phytopathol. 71, 485–494. doi: 10.1007/s42360-018-0091-2
Bashyal, B. M., Aggarwal, R. (2013). Molecular identification of Fusarium species associated with bakanae disease of rice (Oryza sativa) in India. Indian J. Agric. Sci. 83.
Bashyal, B. M., Aggarwal, R., Sharma, S., Gupta, S., Rawat, K., Singh, D., et al. (2016). Occurrence, identification and pathogenicity of Fusarium species associated with bakanae disease of basmati rice in India. Eur. J. Plant Pathol. 144, 457–466. doi: 10.1007/s10658-015-0783-8
Chen, S. Y., Lai, M. H., Tung, C. W., Wu, D. H., Chang, F. Y., Lin, T. C., et al. (2019). Genome-wide association mapping of gene loci affecting disease resistance in the rice-Fusarium fujikuroi pathosystem. Rice 12, 1–12. doi: 10.1186/s12284-019-0337-3
Cheng, A. P., Chen, S. Y., Lai, M. H., Wu, D. H., Lin, S. S., Chen, C. Y., et al. (2020). Transcriptome analysis of early defenses in rice against Fusarium fujikuroi. Rice 13 (1), 1–15. doi: 10.1186/s12284-020-00426-z
Cheon, K. S., Jeong, Y. M., Lee, Y. Y., Oh, J., Kang, D. Y., Oh, H., et al. (2019). Kompetitive allele-specific PCR marker development and quantitative trait locus mapping for bakanae disease resistance in Korean japonica rice varieties. Plant Breed. Biotechnol. 7 (3), 208–219. doi: 10.9787/PBB.2019.7.3.208
De Castro, E., Sigrist, C. J., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34 (suppl_2), W362–W365. doi: 10.1093/nar/gkl124
De Jonge, R., Peter van Esse, H., Maruthachalam, K., Bolton, M. D., Santhanam, P., Saber, M. K., et al. (2012). Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc. Natl. Acad. Sci. 109 (13), 5110–5115. doi: 10.1073/pnas.1119623109
Desjardins, A. E., Manandhar, H. K., Plattner, R. D., Manandhar, G. G., Poling, S. M., Maragos, C. M. (2000). Fusarium species from Nepalese rice and production of mycotoxins and gibberellic acid by selected species. Appl. Environ. Microbiol. 66 (3), 1020–1025. doi: 10.1128/AEM.66.3.1020-1025.2000
Desjardins, A. E., Plattner, R. D., Nelson, P. E. (1997). Production of fumonisin B (inf1) and moniliformin by Gibberella fujikuroi from rice from various geographic areas. Appl. Environ. Microbiol. 63 (5), 1838–1842. doi: 10.1128/aem.63.5.1838-1842.1997
Eğerci, Y., Kınay-Teksür, P., Uysal-Morca, A. (2021). First report of Bakanae disease caused by Fusarium proliferatum on rice in Turkey. J. Plant Dis. Prot. 128 (2), 577–582. doi: 10.1007/s41348-020-00369-z
Ellis, J. A., Ong, B. (2017). “The MassARRAY® system for targeted SNP genotyping,” in Genotyping. Methods in Molecular Biology, vol. 1492 . Eds. White, S., Cansilieris, S. (New York, NY: Humana Press). doi: 10.1007/978-1-4939-6442-0_5
Fiyaz, R. A., Krishnan, S. G., Rajashekara, H., Yadav, A. K., Bashyal, B. M., Bhowmick, P. K., et al. (2014). Development of high throughput screening protocol and identification of novel sources of resistance against bakanae disease in rice (Oryza sativa L.). Indian. J. Genet. Plant Breeding 74, 414–422. doi: 10.5958/0975-6906.2014.00864.5
Fiyaz, R. A., Yadav, A. K., Krishnan, S. G., Ellur, R. K., Bashyal, B. M., Grover, N., et al. (2016). Mapping quantitative trait loci responsible for resistance to Bakanae disease in rice. Rice 9 (1), 1–10. doi: 10.1186/s12284-016-0117-2
Fradin, E. F., Zhang, Z., Juarez Ayala, J. C., Castroverde, C. D., Nazar, R. N., Robb, J., et al. (2009). Genetic dissection of Verticillium wilt resistance mediated by tomato. Ve1 Plant Physiol. 150 (1), 320–332. doi: 10.1104/pp.109.136762
Hayasaka, T., Ishiguro, K., Shibutani, K., Namai, T. (2001). Seed disinfection using hot water immersion to control several seed-borne diseases of rice plants. Japanese J. Phytopathol. 67 (1), 26–32. doi: 10.3186/jjphytopath.67.26
Hossain, K. S., Mia, M. T., Bashar, M. A. (2015). Management of bakanae disease of rice. Bangladesh J. Botany 44 (2), 277–283. doi: 10.3329/bjb.v44i2.38517
Hur, Y. J., Lee, S. B., Kim, T. H., Kwon, T., Lee, J. H., Shin, D. J., et al. (2015). Mapping of qBK1, a major QTL for bakanae disease resistance in rice. Mol. Breeding 35, 1–9. doi: 10.1007/s11032-015-0281-x
Ito, S., Kimura, J. (1931). Studies on the bakanae disease of the rice plant. Rep. Hokkaido Agric. Exp. Stn. 27, 1–95.
Jeon, Y. A., Yu, S. H., Lee, Y. Y., Park, H. J., Lee, S., Sung, J. S., et al. (2013). Incidence, molecular characteristics and pathogenicity of Gibberella fujikuroi species complex associated with rice seeds from Asian countries. Mycobiology 41 (4), 225–233. doi: 10.5941/MYCO.2013.41.4.225
Ji, H., Kim, T. H., Lee, G. S., Kang, H. J., Lee, S. B., Suh, S. C., et al. (2018). Mapping of a major quantitative trait locus for bakanae disease resistance in rice by genome resequencing. Mol. Genet. Genomics 293 (3), 579–586. doi: 10.1007/s00438-017-1407-0
Kang, D. Y., Cheon, K. S., Oh, J., Oh, H., Kim, S. L., Kim, N., et al. (2019). Rice genome resequencing reveals a major quantitative trait locus for resistance to bakanae disease caused by Fusarium fujikuroi. Int. J. Mol. Sci. 20 (10), 2598. doi: 10.3390/ijms20102598
Kapustin, Y., Souvorov, A., Tatusova, T., Lipman, D. (2008). Splign: algorithms for computing spliced alignments with identification of paralogs. Biol. Direct. 3, 1–13. doi: 10.1186/1745-6150-3-20
Lee, S. B., Hur, Y. J., Cho, J. H., Lee, J. H., Kim, T. H., Cho, S. M., et al. (2018). Molecular mapping of qBK1 WD, a major QTL for bakanae disease resistance in rice. Rice 11, 1–8. doi: 10.1186/s12284-017-0197-7
Lee, S. B., Kim, N., Hur, Y. J., Cho, S. M., Kim, T. H., Lee, J. Y., et al. (2019). Fine mapping of qBK1, a major QTL for bakanae disease resistance in rice. Rice 12 (1), 1–10. doi: 10.1186/s12284-019-0295-9
Lee, S. B., Kim, N., Jo, S., Hur, Y. J., Lee, J. Y., Cho, J. H., et al. (2021). Mapping of a major QTL, qBK1Z, for bakanae disease resistance in rice. Plants 10 (3), 434. doi: 10.3390/plants10030434
Lee, S. B., Lee, J. Y., Kang, J. W., Mang, H., Kabange, N. R., Seong, G. U., et al. (2022). A novel locus for bakanae disease resistance, qBK4T, identified in rice. Agronomy 12 (10), 2567. doi: 10.3390/agronomy12102567
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2– ΔΔCT method. Methods 25 (4), 402–408. doi: 10.1006/meth.2001.1262
Madeira, F., Pearce, M., Tivey, A. R., Basutkar, P., Lee, J., Edbali, O., et al. (2022). Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50 (W1), W276–W279. doi: 10.1093/nar/gkac240
Matić, S., Bagnaresi, P., Biselli, C., Orru’, L., Amaral Carneiro, G., Siciliano, I., et al. (2016). Comparative transcriptome profiling of resistant and susceptible rice genotypes in response to the seedborne pathogen Fusarium fujikuroi. BMC Genomics 17, 1–17. doi: 10.1186/s12864-016-2925-6
McHale, L., Tan, X., Koehl, P., Michelmore, R. W. (2006). Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7, 1–11. doi: 10.1186/gb-2006-7-4-212
Murray, M. G., Thompson, W. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Research 8 (19), 4321–4326. doi: 10.1093/nar/8.19.4321
Nazar, R. N., Xu, X., Blaya Fernandez, J., Shittu, H., Kurosky, A., Robb, J. (2018). Defence cascade in Verticillium-infected grafted tomato. Plant Signaling Behav. 13 (6), e1475807. doi: 10.1080/15592324.2018.1475807
Nirenberg, H. I. (1976). Untersuchungen uber die morphologische und biologische differenzierung in Fusarium-Sektion Liseola. Mitt. Biol. Bundesansi. Land-Forstwirtsch Berlin – Dahlem. 169, 1–117.
Sarwar, A., Hassan, M. N., Imran, M., Iqbal, M., Majeed, S., Brader, G., et al. (2018). Biocontrol activity of surfactin A purified from Bacillus NH-100 and NH-217 against rice bakanae disease. Microbiol. Res. 209, 1–13. doi: 10.1016/j.micres.2018.01.006
Sun, S. K. (1975). The diseases cycle of rice bakanae disease in Taiwan. In Proc. Natl. Sci. Counc. Repub. China 8, 245–256.
Volante, A., Tondelli, A., Aragona, M., Valente, M. T., Biselli, C., Desiderio, F., et al. (2017). Identification of bakanae disease resistance loci in japonica rice through genome wide association study. Rice 10, 1–16. doi: 10.1186/s12284-017-0168-z
Wulff, E. G., Sørensen, J. L., Lübeck, M., Nielsen, K. F., Thrane, U., Torp, J. (2010). Fusarium spp. associated with rice Bakanae: ecology, genetic diversity, pathogenicity and toxigenicity. Environ. Microbiol. 12 (3), 649–657. doi: 10.1111/j.1462-2920.2009.02105.x
Keywords: bakanae, candidate genes, fine-mapping, rice, resistance, SNPs, QTL, NILs (Near isogenic lines)
Citation: Kushwaha AK, Ellur RK, Maurya SK, Krishnan S. G, Bashyal BM, Bhowmick PK, Vinod KK, Bollinedi H, Singh NK and Singh AK (2023) Fine mapping of qBK1.2, a major QTL governing resistance to bakanae disease in rice. Front. Plant Sci. 14:1265176. doi: 10.3389/fpls.2023.1265176
Received: 22 July 2023; Accepted: 24 October 2023;
Published: 10 November 2023.
Edited by:
Amira M. I. Mourad, Assiut University, EgyptReviewed by:
Muhammad Irfan Siddique, North Carolina State University, United StatesDevender Sharma, ICAR-Vivekananda Parvatiya Krishi Anusandhan Sansthan, India
Copyright © 2023 Kushwaha, Ellur, Maurya, Krishnan S., Bashyal, Bhowmick, Vinod, Bollinedi, Singh and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashok Kumar Singh, YWtzX2dlbmVAeWFob28uY29t