 Mingyue Shang1
Mingyue Shang1 Binbin Liao
Binbin Liao Baozhong Duan
Baozhong Duan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 25 October 2023
Sec. Plant Metabolism and Chemodiversity
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1251829
This article is part of the Research TopicPlant-Derived Natural Compounds in Drug Discovery: The Prism Perspective between Plant Phylogeny, Chemical Composition, and Medicinal Efficacy, Volume IIView all 9 articles
Introduction: The potential contamination of herbal medicinal products poses a significant concern for consumer health. Given the limited availability of genetic information concerning Ajuga species, it becomes imperative to incorporate supplementary molecular markers to enhance and ensure accurate species identification.
Methods: In this study, the chloroplast (cp) genomes of seven species of the genus Ajuag were sequenced, de novo assembled and characterized.
Results: exhibiting lengths ranging from 150,342 bp to 150,472 bp, encompassing 86 - 88 protein-coding genes (PCGs), 35 - 37 transfer RNA, and eight ribosomal RNA. The repetitive sequences, codon uses, and cp genomes of seven species were highly conserved, and PCGs were the reliable molecular markers for investigating the phylogenetic relationship within the Ajuga genus. Moreover, four mutation hotspot regions (accD-psaI, atpH-atpI, ndhC-trnV(UAC), and ndhF-rpl23) were identified within cp genomes of Ajuga, which could help distinguish A. bracteosa and its contaminants. Based on cp genomes and PCGs, the phylogenetic tree preliminary confirmed the position of Ajuga within the Lamiaceae family. It strongly supported a sister relationship between Subsect. Genevense and Subsect. Biflorae, suggesting the merger of Subsect. Biflorae and Subsect. Genevenses into one group rather than maintaining separate categorizations. Additionally, molecular clock analysis estimated the divergence time of Ajuga to be around 7.78 million years ago.
Discussion: The species authentication, phylogeny, and evolution analyses of the Ajuga species may benefit from the above findings.
The Ajuga genus is a member of the Lamiaceae family and encompasses numerous economically and medically significant plants. Many Ajuga species have been employed in Traditional Chinese Medicine for relieving cough, reducing sputum, and arresting bleeding (Dai et al., 2010). Among the Ajuga species, A. bracteosa is particularly prevalent and extensively utilized in folk medicine (Millen et al., 2001). Previous investigation has revealed that the morphologies of most Ajuga species are similar and indistinguishable, potentially leading to misclassification (Talebi et al., 2019). According to the World Health Organization, adulterating herbal products threatens consumer safety (Duan et al., 2017). In recent years, molecular identification techniques, particularly molecular markers, have made notable advancements in traditional Chinese medicine (TCM), which were used to recognize variations among individuals of distinct species or populations (Cheng et al., 2018; Liu et al., 2019). Previous studies have employed the nuclear ribosomal DNA internal transcribed spacer (ITS) 2 to identify certain Ajuga species and related taxa, such as A. ciliate, A. decumbens, and A. lupulina (Han et al., 2008). Nonetheless, some frequent adulterants were not probed, and single-locus DNA barcodes inherently possess limitations.
Additionally, elucidating the taxonomic relationships between species of the genus Ajuga is crucial for understanding and harnessing the medicinal properties of the different species. However, the presence of similar morphological features coupled with the dearth of molecular information has prevented the accurate identification of taxonomic relationships in this genus. Hence, it is essential to develop a more accurate and effective method for identifying and classifying the Ajuga taxa.
The organelle chloroplast (cp) is vital in plant photosynthesis and biochemical processes (Zoschke and Bock, 2018). Compared to conventional DNA portions, the cp genome exhibits higher conservation with slight variations, making it applicable in various research areas, including species authentification and developing DNA markers (Qiao et al., 2016). Plastid genome analysis has been widely employed to identify Paris, Isodon, and respective adulterants and clarify phylogenetic relationships (Jiang et al., 2022; Zhou et al., 2022). Although previous studies have reported plastid genomes of Ajuga (Tao et al., 2019), their focus was primarily on characterizing a single genome, and cp genomes have not been used to differentiate Ajuga taxa and their frequent adulterants. In addition, cp genomes may lead to a wrong inference of phylogenetic relationships due to DNA length disparity, gaps representing insertions/deletions (indels), and improper models of DNA evolution in merged datasets. Previous investigations have indicated that protein-coding genes (PCGs) offer an improved resolution for understanding phylogenetic relationships due to the genetic divergence in gene-encoding regions occurring at a slower rate compared to the non-coding areas (Lockhart and Penny, 2005; Cui et al., 2019). However, no reports exist on using PCGs to comprehend the interspecific relationships among the Ajuga species.
Herein, cp genomes of seven Ajuga species were sequenced, de novo assembled, and annotated. Subsequently, we compared the architecture and evolutionary connections of these genomes. This research aims (i) to contribute novel and full-length plastid genomes of Ajuga to understand more about the genome structure of relevant species, (ii) to expound the phylogenetic relationship of Ajuga by comparing genome sequences, and (iii) to develop promising DNA markers for distinguishing A. bracteosa from its contaminants. Our results significantly increase the genome information of Ajuga, facilitate evolutionary scrutiny and authentification of Ajuga, and ensure the safe utilization of A. bracteosa.
Fresh leaves from seven species, namely A. forrestii, A. nubigena, A. campylantha, A. macrosperma, A. bracteosa, A. nipponensis, and A. ovalifolia, were gathered from the Germplasm Resource Garden of Kunming Zhifen Biotechnology Co., Ltd, Yunnan, China (102°48′58″E, 24°49′55″N). The voucher specimens were identified by Professor Baozhong Duan and preserved at the herbarium of Dali University. Information on each sample is detailed in Table S1 and Figure S1. Approximately one gram of fresh leaves of each species was collected, instantly frozen in liquid nitrogen, and stored for subsequent DNA extraction. Genomic DNA was extracted from samples with a Plant Genomic DNA kit (Tiangen, Beijing, China) following the manufacturer’s instructions. The extracted DNA was checked with high-sensitivity Qubit 4.0 fluorometry (Life Technologies, Inc.).
To prepare sequencing libraries, a high-quality DNA sample of at least 30 microliters per individual was utilized, with a minimum concentration of 100 ng/μL. Illumina NovaSeq system (Illumina, San Diego, CA) was adopted to sequence libraries. 1 μg of purified DNA was fragmented and used to build PE libraries (insert size 250 bp). The paired-end sequencing reads were filtered to trim adapter sequences and low-quality bases using Toolkit_v2.3.3 software. The cp genome was assembled using GetOrganelle v.1.6.4, exploiting Blast v.2.5.0, SPAdes v.3.13.0, and Bowtie2 v.2.4.4 as dependencies (get_organelle_from_reads.py -1 R1.fq -2 R2.fq - o cp_output -R 15 -k 21,45,65,85,105 -F embplant) (Jin et al., 2020). All clean reads were mapped to the database, and then the mapping data were extracted based on similarity and coverage. Subsequently, the assembled contigs were visualized, and removed redundant sequences by Bandage v.0.8 to generate the complete circular cp genome (Wang J. et al., 2022). Finally, the reads were remapped to assemble the cp genome by Bowtie2, and Jellyfish v.2.2.3 was then used to determine the reverse repeat region boundary. Following assembly, CpGAVAS2 (http://47.96.249.172:16019/analyzer/annotate) (An et al., 2020) and GeSeq (https://chlorobox.mpimp-golm.mpg.de/geseq.html) (Liu et al., 2020) were employed to annotate the circular plastid genomes, which can be retrieved from the National Center for Biotechnology Information (NCBI) GenBank with accession numbers OR038698 to OR038704 (Table 1). IRscope toll (https://irscope.shinyapps.io/Chloroplot/) (Amiryousefi et al., 2018) was used to visualize gene maps.
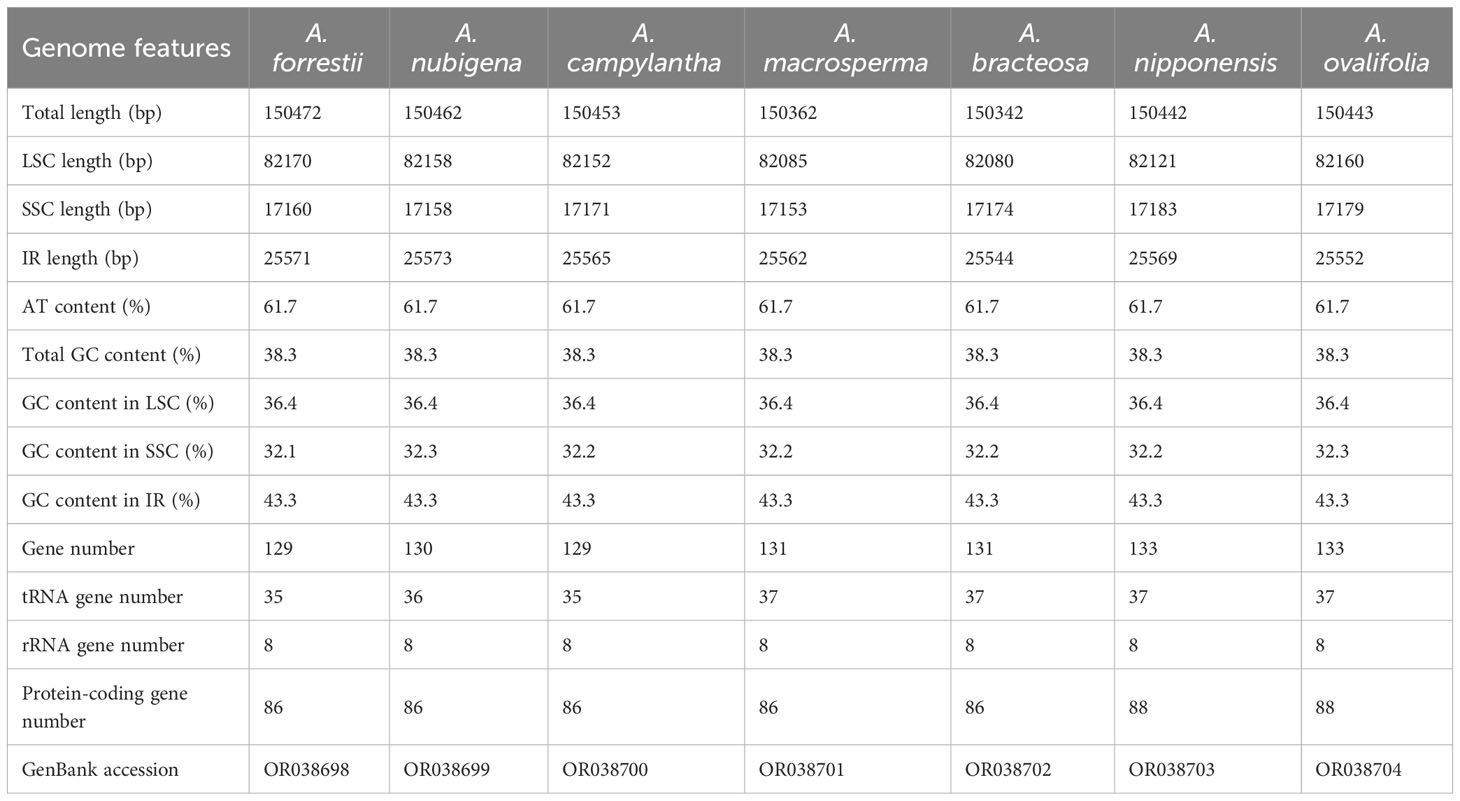
Table 1 Summary of cp genome features.
Four types of dispersed repeat sequences, i.e., Forward (F), Reverse (R), Palindromic (P), and Complementary (C), were identified with the REPuter tool (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) with a minimal repeat of 30 bp and a similarity threshold of 90% between repeat pairs (Kurtz et al., 2001). In addition, simple sequence repeats (SSRs) were analyzed with MISA software (http://pgrc.ipk-gatersleben.de/misa/) (Beier et al., 2017), with thresholds of ‘10’ in mono-, ‘5’ in di-, ‘4’ in tri-, and ‘3’ in tetra-, penta-, and hexa- nucleotide motifs. Geneious 9.0.2 software was used to analyze GC content, genome size, tRNA, and repeat content (Kearse et al., 2012), while CodonW v.1.4.2 was utilized to assess codon usage bias through six values, including effective number of codons (Enc), GC content of synonymous third codon positions (GC3s), codon adaptation index (CAI), frequency of optimal codons (Fop), relative synonymous codon usage (RSCU) and codon bias index (CBI). The RSCU values were visualized through a heatmap generated with Tbtools (Chen et al., 2020). The IRscope tool (https://irscope.shinyapps.io/irapp/) was employed to investigate the contraction and expansion of inverted repeat (IR) regions at the junctions of plastid genomes (Amiryousefi et al., 2018). The genome sequences were also analyzed based on the annotation information of A. bracteosa (GenBank NC068635.1) using the mVISTA program in Shuffle LAGAN mode (Poliakov et al., 2014). Nucleotide variability (Pi) across cp genome sequences was calculated with DnaSP v.6.12.03, and specific settings were: 200 bp of step size and 600 bp of window length (Wang J. et al., 2022). Pi exceeding 0.008 was considered a mutation hotspot.
The intergenic spacers (IGS) were obtained from seven Ajuga species with PhyloSuite v1.2.2 (Zhang et al., 2019). Primers were designed based on the variable intergenic regions using Snapgene 6.2.1 (Snapgene, Insightful Science, available at http://www.snapgene.com, last used in 2023). PCR amplifications were conducted in a final volume of 25 μL, consisting of 12.5 μL of 2×Taq Plus PCR Master Mix, 1 μL of each primer, 2 μL of template DNA, and 8.5 μL of ddH2O (Mei5 Biotechnology, Co., Ltd). All amplifications were performed using a RePure-A PCR system (Applied Biogener, Hangzhou, China) under the following conditions: an initial denaturation at 94°C for 3 min, followed by 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, with a final extension at 72°C for 5 min. PCR products were examined by 1% agarose gel electrophoresis to confirm the amplification of the target fragments. The purified PCR products were sequenced in both directions on a 3730XL DNA Sequencer (Applied Biosystems, Waltham, USA) using the same primers at Sangon Biotech Co., Ltd. (Shanghai, China).
The phylogenetic analysis involved a total of 35 taxa, consisting of 28 species obtained from NCBI (refer to Table S2) and seven species that we newly sequenced, as detailed in Table 1. The selection of these species for phylogenetic analysis is based on the classification system of Lamiaceae within the Angiosperm Phylogeny Group IV system (APG IV). Additionally, two species, Callicarpa macrophylla (GenBank NC058323.1) and C. arborea (GenBank NC058321.1) served as outgroups. The 68 common PCGs of 35 cp genomes were extracted based on the annotation files. The aligned sequences were generated using the MAFFT program and verified manually. Phylogenetic analysis was performed using Maximum likelihood (ML), Bayesian inference (BI), and Neighbor joining (NJ). For the ML tree reconstruction, IQtree was employed with default settings, 1,000 iterations, and 1,000 replications. Model selection was based on the best-fit approach (Katoh and Standley, 2013). BI analysis was performed using MrBayes v.3.2.6 (Ronquist et al., 2012). The most appropriate model for sequence substitution in plastid genomes (GTR + G + I) and PCGs (GTR + G) was determined using MEGA X v.10.2.6 (Mao et al., 2023). The parameters were set for five million generations, with sampling every 1,000 generations. The initial 25% of each run was discarded as burn-in (Jiang et al., 2023). Moreover, the NJ tree was inferred with MEGA X v.10.2.6 and subjected to the bootstrap test of 1,000 repetitions (Kumar et al., 2018).
To estimate divergence time, a molecular clock tree was constructed based on an ML tree with MEGA X. The corresponding divergence times were determined using the TimeTree Resource (RRID: SCR_021162) (Kumar et al., 2017), and seven calibration points were utilized to calculated the divergence times for each node as follows: (F1) 33.3 - 72.4 million years ago (Mya) for the root node, (F2) 19.4 - 39.9 Mya for Ajugoideae + Lamioideae, (F3) 14.0 - 42.8 Mya for Lamioideae stem age, (F4) 1.2 - 62.7 Mya for Nepetoideae stem age, (F5) 12.4 - 47.2 Mya for Salvia prionitis + Prunella vulgaris, (F6) 0.6 - 19.2 Mya for Leonurus sibiricus + L. amplexicaule, and (F7) 5.2 - 9.6 Mya for Pogostemon cablin + P. septentrionalis.
Around 2.51 - 4.54 Gb data were obtained from each species. The A. macrosperma and A. ovalifolia were reported for the first time. As shown in Figure 1, cp genomes of seven species are circular DNAs ranging from 150,342 bp to 150,472 bp and exhibit the typical quadripartite structure commonly observed in most angiosperm cp (Palmer, 1985; Henriquez et al., 2020; Yun and Kim, 2022), consisted of two IRs (IRa and IRb) separated by large single copy (LSC) and small single copy (SSC) regions, respectively. The length of LSC regions ranged from 82,080 bp (A. campylantha) to 82,170 bp (A. ovalifolia), SSC regions ranged from 17,183 bp (A. nubigena) to 17,153 bp (A. macrosperma), and IRa and IRb regions ranged from 25,573 bp (A. nipponensis) to 25,544 bp (A. campylantha). The overall GC content of seven Ajuga cp genomes was 38.3%, largely concordant with the prior study of Tao et al. (Tao et al., 2019), indicating a high degree of conservation among Ajuga species’ cp genomes. Notably, the IR regions exhibited a significantly higher GC content of 43.3% in contrast to the LSC (36.4%) and SSC (32.1% - 32.3%) regions. This discordance may be attributed to the fact that four ribosomal RNA (rRNA) genes (rrn23, rrn16, rrn5, rrn4.5) with high GC content were located in the IR regions, which was similar to the most plant species (Yan et al., 2019; Gui et al., 2020; Chen et al., 2022).
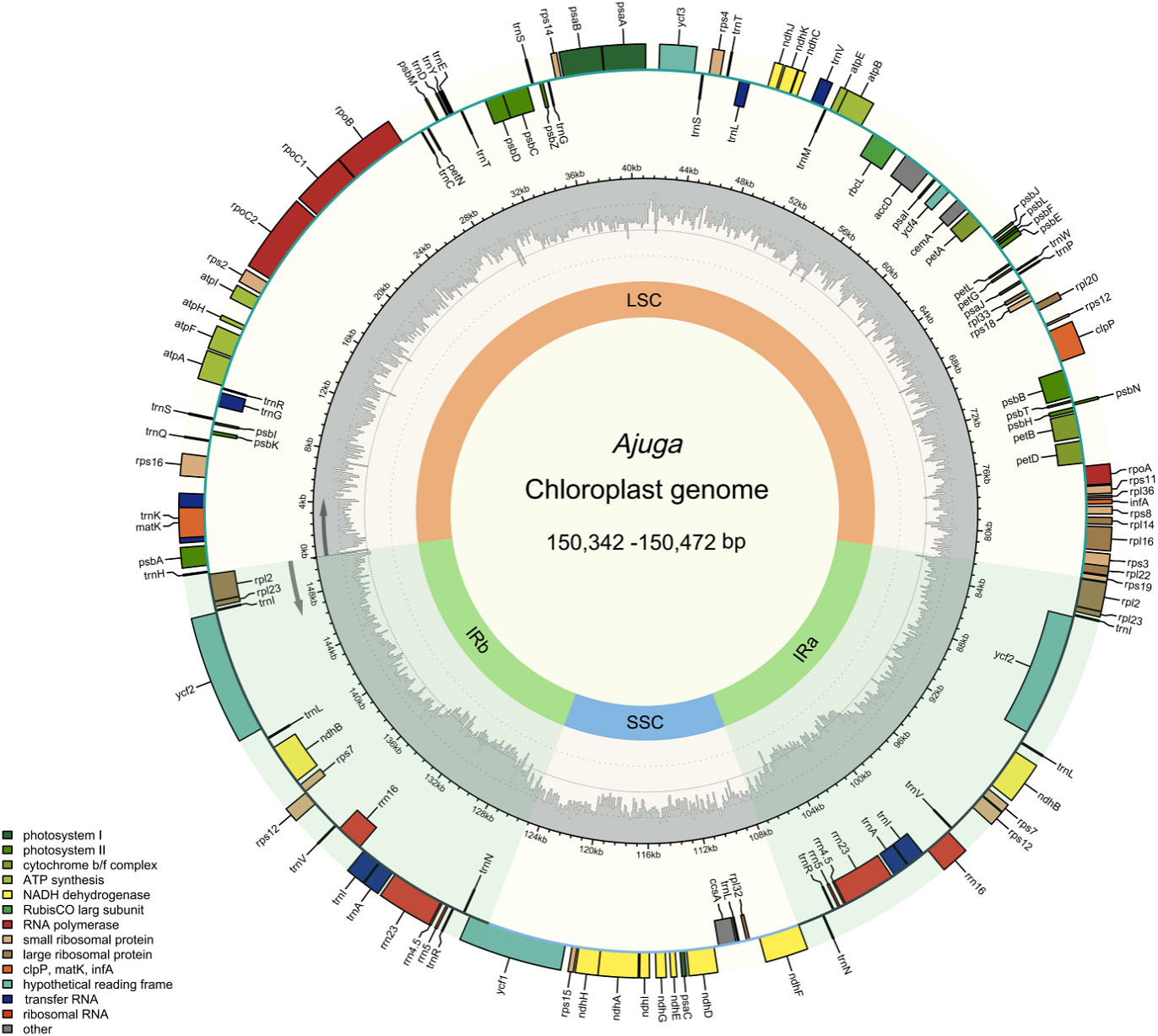
Figure 1 The cp genome map of Ajuga.
In addition, 129 - 133 genes were detected, including 86 - 88 PCGs, 35 - 37 transfer RNAs (tRNAs), and eight ribosomal RNAs (Table 1). Variations in gene numbers across these species can be attributed to the expansion and contraction of IRs. These genes can be categorized into three groups: 45 associated with photosynthesis, 27 involved in self-replication, and the remaining genes serving various other functions. Notably, three genes (chIB, chIL, ycf68) were absent in the seven Ajuga species. The missing chIB and chIL may be a distinctive feature of flowering plants (Jansen et al., 2007). Furthermore, the ycf68 gene was also absent in the cp genomes of Miscanthus sinensis and M. floridulus (Sheng et al., 2021). In addition to these observations, it’s worth mentioning that two tRNAs, trnF-GAA and trnfM-CAU, were absent in three Ajuga species. Specifically, A. nubigena lacked the trnF-GAA gene, while trnF-GAA and trnfM-CAU genes were missing in A. forrestii and A. campylantha. These findings revealed that the structure of cp genomes was highly conserved in Ajuga species, albeit with some alterations that have accrued during the angiosperm evolution, which was also supported by previous studies (Millen et al., 2001).
Furthermore, the seven Ajuga species exhibit a similar number and types of introns. As illustrated in Table S3, each of 18 genes had a single intron, trnI-GAU (×2), trnA-UGC (×2), rpl2 (×2), and ndhB (×2) were in IR, and genes trnK-UUU, trnG-UCC, trnL-UAA, trnV-UAA, rpl16, rps16, petB, rpoC1, petD, and atpF were in LSC region, while ndhA was only present in SSC. Besides, clpP and ycf3 contain double introns, consistent with previous findings (Gong et al., 2022). It is unsurprising given that most angiosperm plastid genomes exhibit highly conserved structure and gene composition at the genus level (Wicke et al., 2011; Chen et al., 2021; Namgung et al., 2021).
The expansion/contraction of IR regions is frequently observed during evolution and may account for the disparity in the size of plastid genomes (Menezes et al., 2018). As depicted in Figure 2, the rpl2 gene is entirely situated within the IR regions across all species, while the trnH gene exclusively occupies the LSC region, consistent with the cp genomes of most angiosperms (Wang J. et al., 2022; Fang et al., 2023). Besides, the trnN gene was entirely located within the IRa region in all species except for A. ciliata, A. lupulina, and A. campylanthoides, where it was utterly localized in the IRb region at varying distances from the junction of SSC/IRb.

Figure 2 Comparisons of the borders of LSC, SSC, and IRa/b regions among the 11 Ajuga plastid genomes.
Among the seven Ajuga species, a truncated copy of ndhF genes was identified at the junction of SSC/IRb, starting from SSC and integrating into the IRb region. Conversely, in the remaining species, including A. ciliata, A. lupulina, and A. campylanthoides, ndhF was found at the junction of IRa/SSC. In addition, the ycf1 gene was observed at the IRb/SSC junction in all species. It originated from the IRb region and integrated into SSC, with sizes ranging from 5 to 4,357 bp. Notably, the ycf1 gene was also present at the IRa/SSC junction in all species except A. bracteosa, in which this gene was exclusively present in SSC. This result implied that the ycf1 could potentially serve as a marker for distinguishing A. bracteosa. A previous study also highlighted ycf1 as a powerful barcode for land plants (Dong et al., 2015). Furthermore, the rps19 gene was consistently located at the boundaries of the IRs/LSC in all Ajuga species. This pattern aligns with the cp genomes of other Lamiaceae species, such as S. mekongensis, Mentha spicata, and Dracocephalum heterophyllum (Wu H. et al., 2021; Fu et al., 2022). In summary, the sizes of the cp genomes in the 11 Ajuga species vary, and there are noteworthy variations in the junction regions. These findings provide evidence of a distinctive pattern of IR contraction/expansion within the cp genomes of Ajuga species, which can be employed to investigate species-specific gene loci.
Analyzing codon usage is essential to evaluate the evolution of the cp genome (Wang N. et al., 2022). In genes of seven Ajuga species, 64 codons were identified, of which 61 encoded 24 amino acids. Leucine exhibited the highest frequency among all the amino acids encoded by cp genomes, whereas cysteine was found to be a rare amino acid (Table S4). This observation is consistent with the codon usage bias reported by previous studies (Orešič and Shalloway, 1998; Liu et al., 2020). Moreover, pronounced bias towards A or T at the tertiary position of codon was observed, which could be attributed to the high AT proportion in plastid genomes. Similar results were observed in other angiosperm taxa (Wu L. et al., 2021; Jiang et al., 2022). As shown in Figure 3, UUA had the highest frequency, followed by AGA, while GCA had the lowest frequency. In Ajuga, RSCU values of 30 codons were higher than 1.00, 32 codons had values below 1.00, and two had values of 1.00. An RSCU value below 1.0 suggests that the codon usage frequency is less than expected, while an RSCU value over 1.0 means that the codon usage frequency is more than expected (Ma et al., 2015; Parvathy et al., 2022). This variability in RSCU values reflects evolutionary information resulting from mutation and selection, which is essential in studying organismal evolution (Morton, 2003).
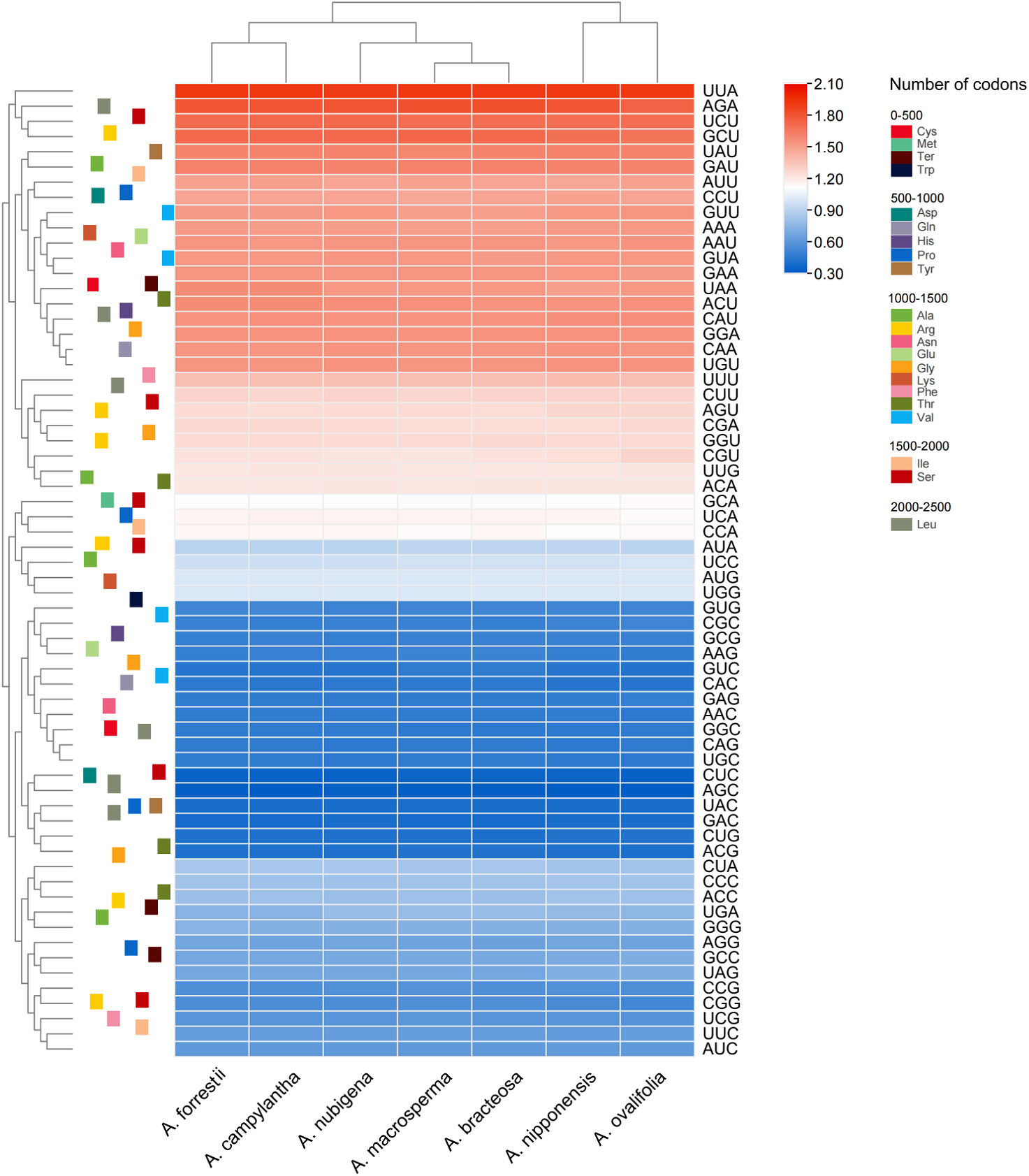
Figure 3 Heat map of the RSCU values among Ajuga cp genome.
Additionally, the GC proportion of the GC3s was closely associated with codon bias and was an important parameter for evaluating the codon use pattern (Ma et al., 2015; Parvathy et al., 2022). In Ajuga, GC3s values ranged from 26.9% to 27.0%, indicating that the genus Ajuga preferred A/U ending codons (Zhang et al., 2022). Previous studies have highlighted that high AT content is the primary reason for synonymous codons ending in A/U, potentially linked to natural selection and mutation during evolution (Zhang et al., 2018). Additionally, the Enc varied between 49.80% and 49.94%, while the CAI and the optimal frequency were lower than 0.5. These results suggested a slight bias in codon usage within the seven Ajuga taxa.
Large and complex repeat sequences in the cp genome are potential markers for revealing gene rearrangements and losses during evolution (McDonald et al., 2011; Yi et al., 2013). Analysis of oligonucleotide repeat in seven cp genomes indicated that the number and length of repeat sequences differed among genomes and were distributed randomly, with repeat sequences ranging from 30 to 82 bp and most being within 30 - 46 bp (Figure 4B). Meanwhile, 274 long repeats were identified, including 149 P repeats, 124 F repeats, and 1 R repeat, with P and F being more than R and C, a pattern consistent with most plastid genomes of angiosperms (Vu et al., 2020; Luo et al., 2022). R repeats were only present in A. campylantha, while C repeats were absent in all seven Ajuga species (Figure 4A). These findings provide a molecular basis for identifying the Ajuga species.
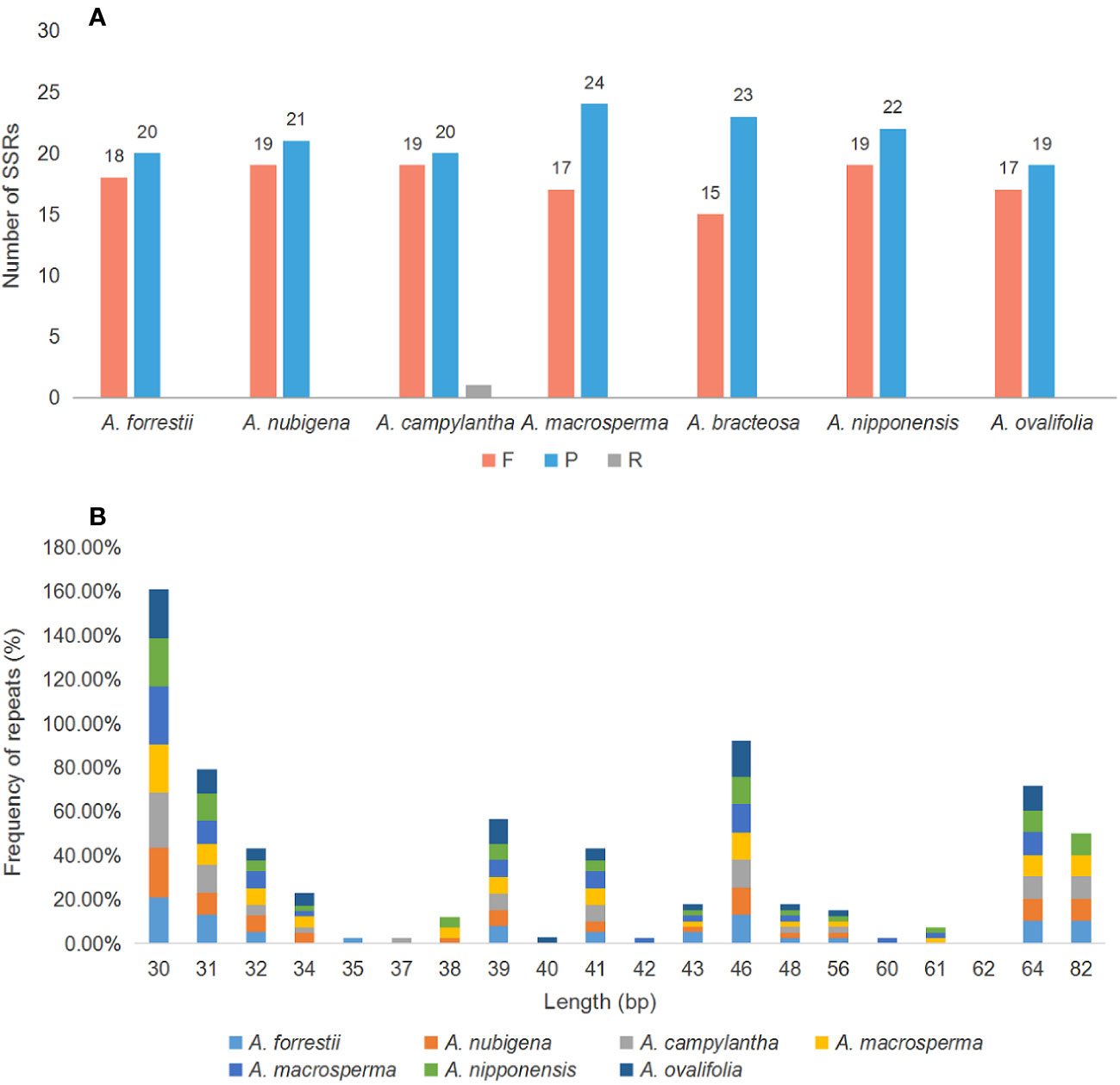
Figure 4 Number of long repetitive repeats on the cp genome of seven Ajuga species. (A) the number of repeat types (P- palindromic repeats, F, forward repeats; R, Reverse); (B) Frequency of the repeats more than 30 bp long.
SSRs, also known as microsatellites, are popular genetic indicators because of their significant polymorphism, repeatability, and reliability, which can be used to detect genetic diversity, population, and polymorphisms at intraspecific, distant phylogenetic relationships and cultivar levels (Wang et al., 2009; Peng et al., 2022). 34, 36, 28, 33, 34, 33, and 33 SSRs were found in A. forrestii, A. nubigena, A. campylantha, A. macrosperma, A. bracteosa, A. nipponensis, and A. ovalifolia, respectively (Figure 5A). Among these, mononucleotide (A/T/C) repeats were more than other types of repeats, accounting for almost 62%, as previously reported by Zhou et al. (Zhou et al., 2022). The second most common was tetranucleotide repeat (23.53% - 29.41%), with a predominant motif of AAAG/CTTT and AAAT/ATTT, followed by dinucleotide repeats (8.33% - 14.29%), with a dominant motif of AT/AT. Hexanucleotides (2.78% - 3.03%) were only present in plastomes of A. nubigena (AACTAT/AGTTAT) and A. nipponensis (AAAAAT/ATTTTT), while trinucleotide or pentanucleotide repeats were absent in all seven Ajuga species. These findings indicate that mononucleotide repeats are more frequent than other types, and A/T motifs were the most abundant in the mononucleotide repeats, which is consistent with previous studies (Munyao et al., 2020; Ren et al., 2022). It was suggested that the high amount of mononucleotide repeats in the cp genome may contribute to heritable variations (Bi et al., 2018).
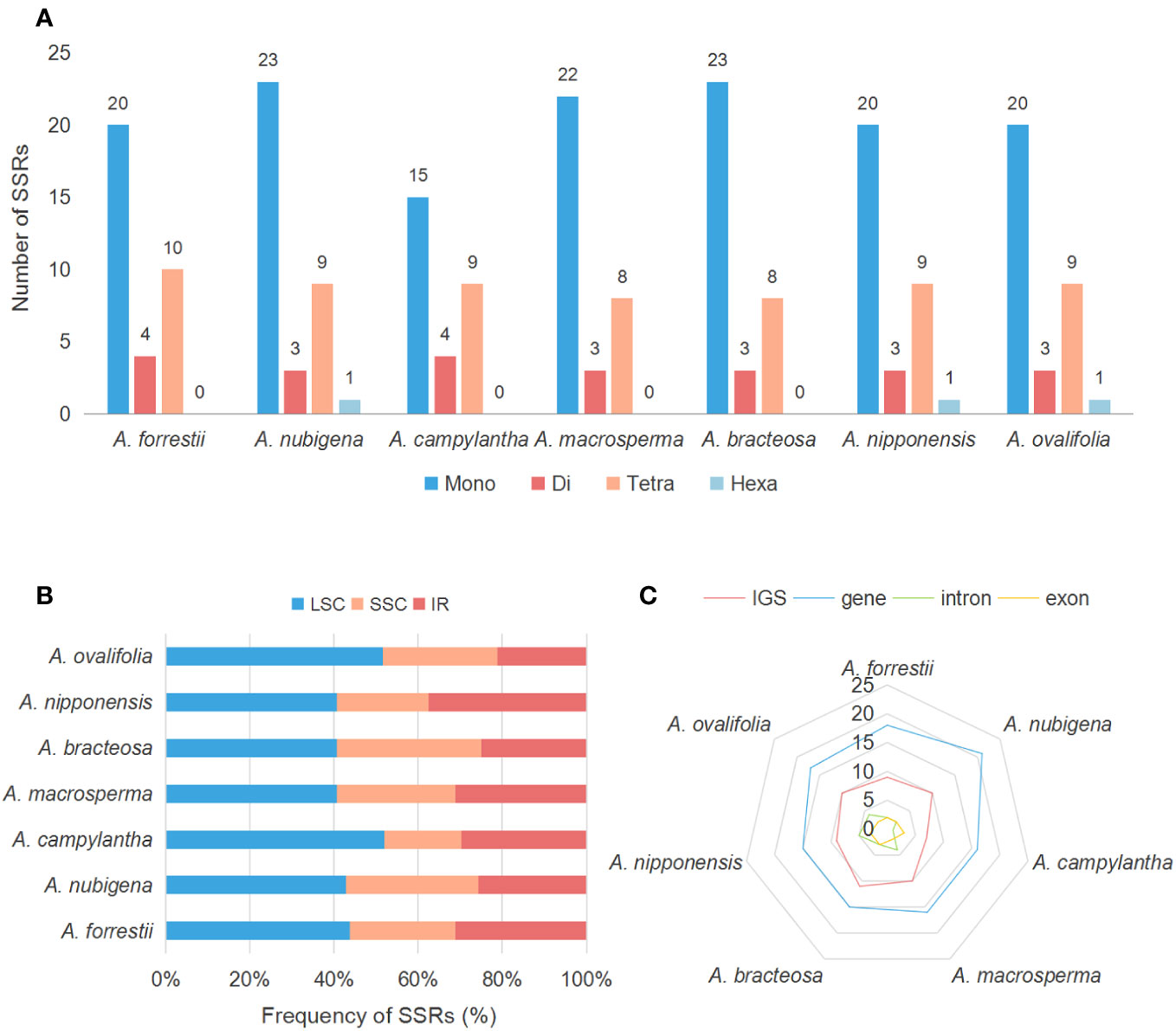
Figure 5 Compares SSR distribution in the cp genomes of seven species. (A) the number of different SSR types; (B) Frequency of SSRs in the LSC, IR, and SSC region; (C) Number of SSRs in the intergenic regions, PCGs, and introns.
Additionally, the number of SSRs exhibited significant variation across distinct structural and functional regions of the cp genomes (Sun et al., 2022). Frequency analysis revealed that SSRs were more prevalent in LSC (44.55%) than in IR (28.79%) and SSC (26.66%) regions (Figure 5B). Besides, most SSRs were located in gene regions and IGS, with an average number of 16 and 9, respectively. In contrast, the regions encompassing introns and exons contained the fewest SSRs, with an average count of 3 and 2, respectively (Figure 5C). These findings are consistent with those observed in other Lamiaceae species (Fu et al., 2022). Previous research has emphasized the suitability of SSRs as a genetic marker in plant molecular studies (Khan et al., 2019), particularly in non-coding regions exhibiting high intraspecific variation (Eguiluz et al., 2017). Our investigation indicated that most SSRs were situated within non-coding regions, with a limited presence in the coding areas. Consequently, these SSRs have the potential to serve as markers for discerning various evolutionary changes, such as genetic diversity, and they may even facilitate species differentiation within Ajuga.
The comparative analysis of cp genomes is a practical approach to investigating the genetic structure and phylogenetic kinships of plants (Daniell et al., 2016). Overall sequence variation in plastid genomes of Ajuga indicated a high level of conservation, with the protein-coding regions exhibiting more significant conservation than non-coding regions, except ndhF, ycf1, and ycf2 genes (Figure 6). This observation aligns with a previous report on the cp genome of S. miltiorrhiza within the Lamiaceae family (Qian et al., 2013). Besides, the most significant discrepancy was mainly found in IGS, e.g., trnH(GUG)-psbA, rps16-trnQ(UUG), atpH-atpI, rbcL-accD, ndhC-trnV(UAC), accD-psaI, trnF(GAA)-ndhJ, and ndhF-rpl32. Previous studies have identified IGS as hotspots with numerous nucleotide substitutions and indel mutations, making them valuable markers with high resolution for phylogenetic analyses (Drummond, 2008). For example, rps16-trnQ(UUG) is highly variable in most plants and has been utilized for DNA barcoding in phylogenetic studies across various angiosperm genera (Dong et al., 2012). Similarly, rbcL-accD and trnH(GUG)-psbA have been proposed as critical molecular markers for phylogenetic analyses of Viola species (Cao et al., 2022). Consequently, these highly variable regions are expected to offer ample genetic information for conducting studies on species delimitation and the phylogenetic evolution of Ajuga.
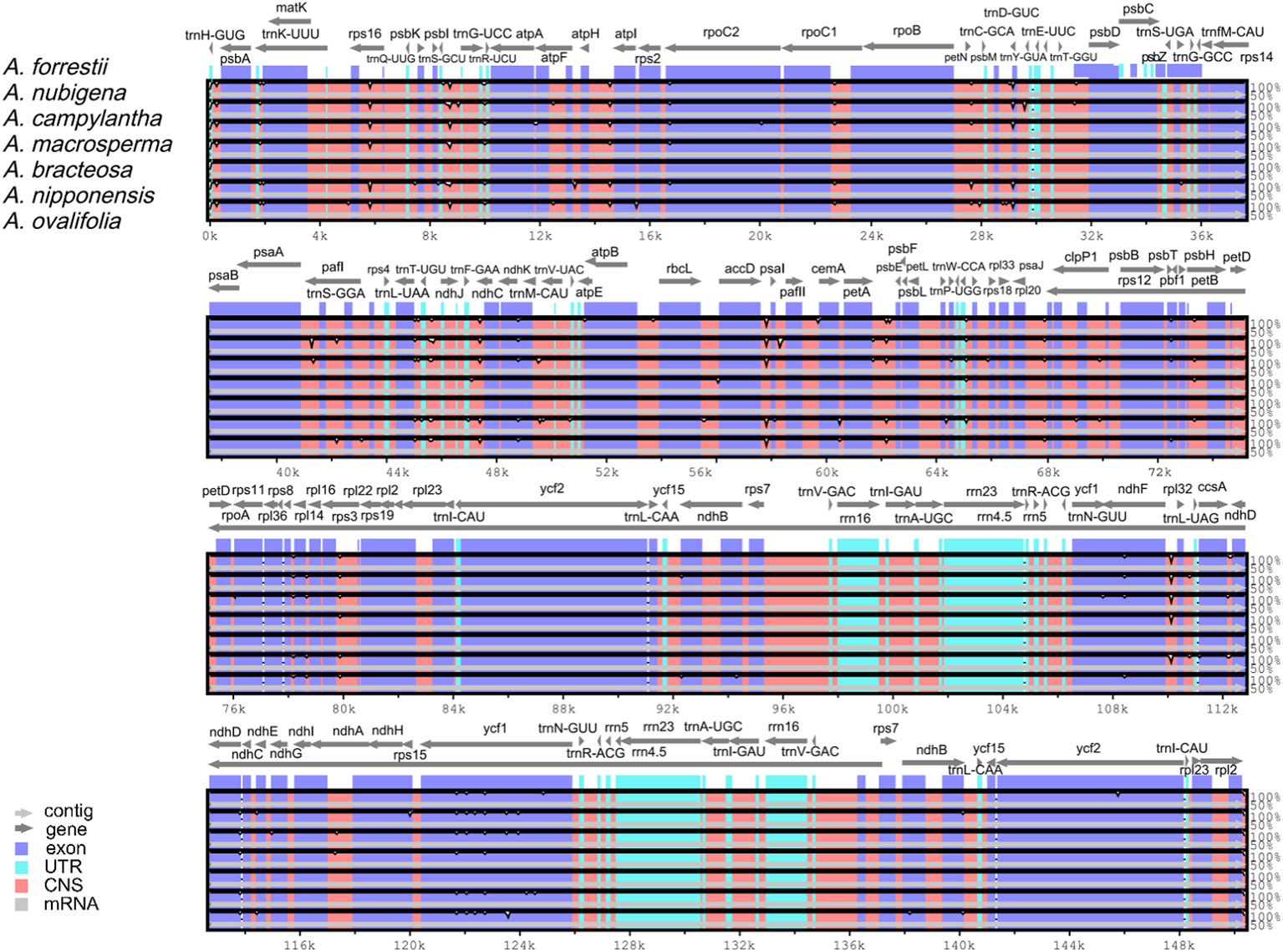
Figure 6 Global comparison of complete genomes of Ajuga.
Sliding window analysis showed that Pi ranged between 0 and 0.01627 across seven Ajuga cp genomes examined. Notably, five regions, trnL-UAA, trnN-GUU, rpl32, ndhH, and ycf1, exhibited higher Pi of > 0.008 (Figure 7). Among these genes, the trnN-GUU gene displayed the lowest divergence value (0.00825), whereas the ycf1 regions had the highest (0.01627). Four genes were in SSC regions, and one was in LSC, suggesting that the LSC and SSC regions displayed higher divergence rates than IR regions, which agrees with previous results (Jiang et al., 2022). Based on our findings, we propose that 13 highly variable sites (atpH-atpI, accD-psaI, trnH(GUG)-psbA, rbcL-accD, trnF(GAA)-ndhJ, rps16-trnQ(UUG), ndhC-trnV(UAC), ndhF-rpl32, trnL-UAA, trnN-GUU, rpl32, ndhH, and ycf1) could serve as potential molecular markers to differentiate A. bracteosa and its dopants.
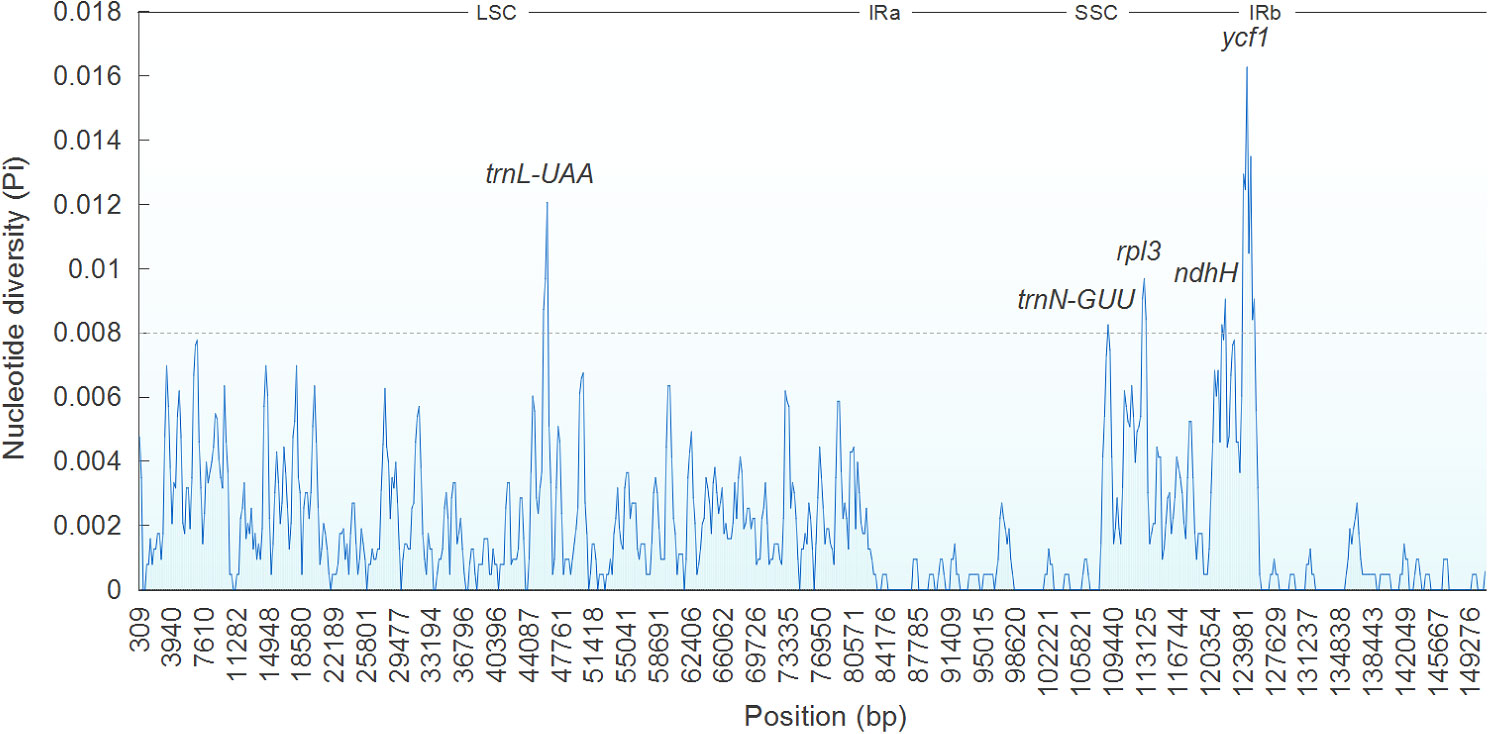
Figure 7 Sliding window analysis of Ajuga cp genome.
IGS is often employed as indicators in phylogeny inference at various taxonomic hierarchies, which is more variable and can offer more evolutionarily revealing characters (Fang et al., 2023). Previous molecular studies of the genus Pogostemon (Zhang et al., 2020) and Clerodendrum (Chen et al., 2023) have demonstrated the high identification capabilities of cp genetic markers. In this study, eight IGSs were extracted from seven Ajuga species. ML analyses were conducted on each IGS using IQtree (Nguyen et al., 2015). As illustrated in Figure S2.1-2.8, the results showed that A. bracteosa could be differentiated from its common adulterants based on ndhF-rpl32, ndhC-trnV(UAC), accD-psaI, atpH-atpI, and trnH(GUG)-psbA, while the remaining IGSs were incapable with weak bootstrap values (<70). Previous studies have also identified the ndhF-rpl32 region as a useful molecular marker for distinguishing Magnolia polytepala and its closely related species (Sun et al., 2020) and ndhC-trnV(UAC) for distinguishing Isodon rubescens and its adulterants (Zhou et al., 2022). Moreover, accD-psaI or atpH-atpI were probable markers for identifying additional species (Alzahrani, 2021; Zheng et al., 2022).
Although general DNA barcodes (e.g., ITS) can distinguish A. ciliata and related taxa (Han et al., 2008), some common adulterants were not investigated. Our results revealed that the studied IGSs exhibited more variability than ITS. The ML tree was constructed based on five IGSs that could differentiate A. bracteosa from its common adulterants. The tree demonstrated that all A. bracteosa species formed a monophyletic clade, and A. macrosperma formed independent branches. Strong support was observed for a sister relationship between A. macrosperma and A. bracteosa (Figure S3). These findings indicate that five combining IGSs can successfully distinguish A. bracteosa and its frequent dopants.
Additionally, we designed primers for five IGS and conducted amplification and sequencing experiments (Table 2). It is worth noting that due to the lack of residual DNA in three Ajuga samples, our supplementary investigation was limited to four species with remaining DNA (A. bracteosa, A. forrestii, A. macrosperma, and A. campylantha). The results demonstrated that except for the trnH(GUG)-psbA primers, the other four fragments (accD-psaI, atpH-atpI, ndhC-trnV(UAC), and ndhF-rpl23) produced products of the expected sizes in the selected Ajuga species (refer to Figure S4). Both amplification and sequencing achieved a 100% success rate. The sequence data from the four Ajuga species align with the cp genome results, with each species exhibiting distinct base differences (refer to Figure S5.1-5.4). These findings confirm that the four DNA barcodes are ideal tools for distinguishing A. bracteosa from its adulterants.

Table 2 Primer design by SnapGene.
Cp genomes have a wealth of phylogenetic information and are extensively employed for reconstructing phylogenies and conducting plant population analyses (Yan et al., 2019). Here, ML and BI trees were reconstructed using cp genomes and common PCGs of 35 species, respectively. Figure 8 illustrates ML and BI trees based on 68 shared PCGs, showing similar topologies. Among the three subfamilies, Ajugoideae and Lamioideae Harley emerged as sister taxa, while Nepetoideae (Dumort.) Burnett appeared as the sister group to the clade consisting of Ajugoideae and Lamioideae. The tree’s crown was occupied by the subfamily Ajugoideae, which included the genera Ajuga, Clerodendrum, and Rotheca. These results approve Ajuga’s position within the Lamiaceae family and align with previous phylogenomic results (Tao et al., 2019). Additionally, the ML tree based on PCGs showed that genus Ajuga was divided into triple clades: (i) clade A included A. nubigena, A. ovalifolia, A. forrestii (Genbank MN518848.1), A. forrestii (Genbank NC048512.1), A. forrestii (Genbank OR038698), A. campylantha, and A. nipponensis; (ii) clade B included A. bracteosa (Genbank M630151.1), A. bracteosa (Genbank NC068635.1), A. bracteosa (Genbank OR038702), and A. macrosperma; (iii) clade C included A. ciliata, A. forrestii, A. campylanthoides, A. decumbens, and A. lupulina.
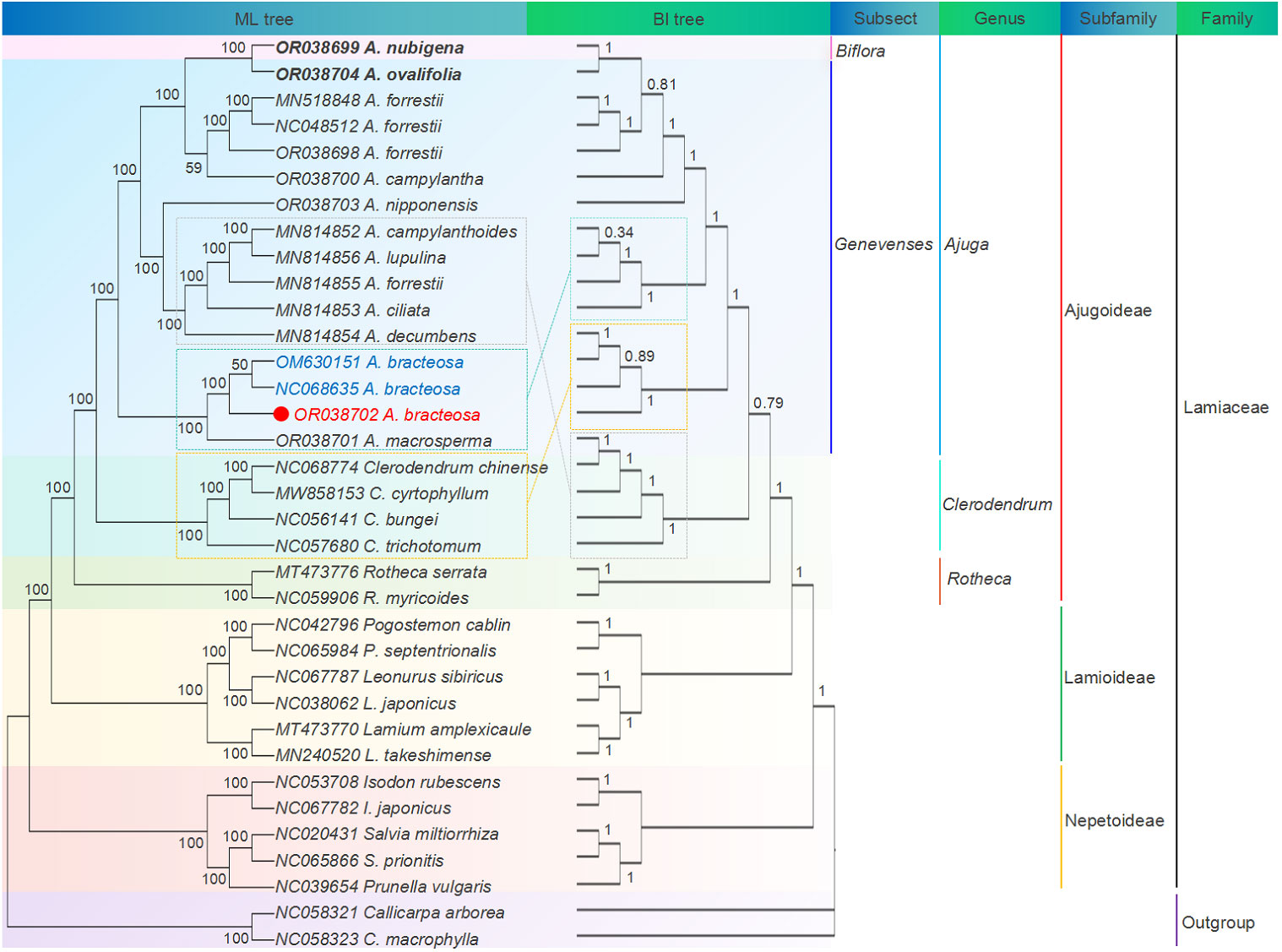
Figure 8 ML and BI phylogenetic tree based on 68 common PCGs of 35 species.
Phylogenetic analysis also demonstrated that all A. bracteosa species formed a monophyletic clade. A. bracteosa (GenBank OR038702) was an independent branch of the phylogeny and deeply nested within clade B, with strong support (BS = 100) for the sister relationship between A. bracteosa and A. macrosperma, suggesting that the cp genome could distinguish A. bracteosa from other species. Notably, the samples of A. forrestii (Genbank MN814855.1) did not form a monophyletic group and were placed in different branches from three individuals of A. forrestii. Previous studies also confirmed that intraspecific diversity existed in Artemisia argyi (Chen et al., 2022), Isodon rubescens (Zhou et al., 2022), and Phyllanthus urinaria collected from different geographical areas (Fang et al., 2023). This phenomenon could potentially be attributed to the influence of the geographical area of origin on the variation in A. forrestii.
Additionally, a strong support value of 100 was observed for the sister relationship between A. nubigena from Subsect. Biflora and A. ovalifolia from Subsect. Genevenses, contradicting the taxonomic findings presented in Flora of China (FOC) (Li and Hedge, 1994). The BI tree also confirmed the same result with the posterior probabilities value of 1. This finding suggests a potential limitation in the current classification of Subsect. Biflora as independent entities. Therefore, based on this evidence, we propose an alternative classification scheme that amalgamates Subsect. Biflorae and Subsect. Genevenses into a single group rather than maintaining separate categorizations.
Notably, Clerodendreae is classified as part of the Verbenaceae family in the FOC (Chen and Gilbert, 1994). Our study confirms its placement within the subfamily Ajugoideae, aligning with the APG IV classification (Angiospermm, 2016). Therefore, we recommend reclassifying the genus Clerodendreae under Lamiaceae.
Furthermore, the phylogenetic trees constructed using cp genomes (Figure S6.1-6.2) and common PCGs (Figure 8 and Figure S6.3) showed a high degree of similarity. Previous studies have suggested that using cp genomes may lead to missing relationships due to length variations, gaps/index deletions, and inappropriate models of DNA evolution in concatenated datasets (Goremykin et al., 2005; Lockhart and Penny, 2005). Given that the genetic divergence in gene-encoding regions occurs more slowly than in non-coding sequences, we considered that utilization of common PCGs is more appropriate for the identification and phylogeny analysis of Ajuga.
In conclusion, our findings serve as a valuable basis and reference for utilizing plastid genomes and common PCGs in species identification, contributing to a better understanding of Ajuga’s phylogeny.
The 35 cp genomes from Lamiaceae family plants, including 16 Ajuga species, were utilized to estimate the divergence time based on the ML tree. As shown in Figure 9, the subfamilies of Ajugoideae and Lammiodeae shared a common ancestor in the late Eocene (39.60 Mya), and the split between Ajuga, Clerodendrum, and Rotheca could occur in the Oligocene (38.65 Mya). The process of speciation of the Ajuga genus was estimated to originate at 7.78 Mya in the late Miocene. Notably, the Three main lineages (clade I: 6.38 Mya; clade II: 0.84 Mya; clade III: 0.61 Mya) within the Ajuga genus diverged in the late Miocene and continued throughout the Pleistocene. This time frame aligns with the final stages of rapid uplift in the Qinghai-Tibetan Plateau (QTP), which has been recognized as a region abundant in biodiversity and a source of various herbal resources (Wen et al., 2014; Favre et al., 2015). Biasatti et al. also suggested that QTP could have influenced the geographical environment, climate, and distribution/divergence of species (Biasatti et al., 2012). These findings imply that the interspecific divergence of the Ajuga species may have a close association with the uplift of the QTP.

Figure 9 Divergence times estimation based on cp genomes.
In addition, the Pleistocene has been proposed as a critical period for refugial isolation and subsequent lineage formation, leading to modern species diversity (Johnson and Cicero, 2004). According to the Pleistocene speciation model, glacial cycles during this era acted as a ‘species pump,’ contributing significantly to the diversity of organisms inhabiting temperate regions (Pyron and Burbrink, 2009). Previous research has also indicated that the extreme climatic fluctuations of the Pleistocene played a pivotal role in driving diversification between lineages in specific taxa (Hewitt, 1999; Bryson et al., 2012).
Consequently, it is postulated that the intense uplift of the QTP, coupled with the climatic oscillations of the Pleistocene, may have influenced diversification and facilitated the radiation of Ajuga species.
In this study, plastid genomes of seven Ajuga species were de novo assembled based on short sequencing reads, and cp genome sequences of A. macrosperma and A. ovalifolia were reported for the first time. These plastid genomes were broadly conserved, displaying comparable gene organization and content. We demonstrated the utility of PCGs integration in phylogeny investigations of Ajuga. Phylogeny analysis of 68 common PCGs strongly supported the taxonomic placement of Ajuga within the Lamiaceae family. It explicitly supported a sister relationship between A. nubigena from Subsect. Biflora and A. ovalifolia from Subsect. Genevense. Consequently, we propose amalgamating Subsect. Biflorae and Subsect. Genevenses into a single group, advocating against their separate categorization. Four highly variable cp loci, including atpH-atpI, accD-psaI, ndhC-trnV(UAC), and ndhF-rpl23, were identified, which hold promise as markers for distinguishing A. bracteosa from its common adulterants. The divergence time of Ajuga occurred in the early Pliocene, possibly due to the intense uplift of QTP and the global cooling event. In summary, this study provides a valued reference for ensuring the efficacy and safety of clinical application while also facilitating bioprospecting and conservation efforts concerning the Ajuga species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MS and BD participated in the conception and design of the research. JinW and BL collected the species. JiaW, GD, and JZ are responsible for analyzing and processing data. MS wrote the manuscript. BD and JinW revised this manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Yunnan academician expert workstation (202105AF150053), the key technology projects in the Yunnan province of China (202002AA100007), and the Yunnan Xingdian talent support plan (YNWR-QNBJ-2020251).
We would like to thank Northeast Forestry University for its technical assistance. We also thank Yuan Jiang for her invaluable assistance in imparting her expertise on the software utilized during our experiments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1251829/full#supplementary-material
Alzahrani, D. A. (2021). Complete chloroplast genome of Abutilon fruticosum: Genome structure, comparative and phylogenetic analysis. Plants 10, 270. doi: 10.3390/plants10020270
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220/4961430
An, W. L., Li, J., Yang, Z. R., Huang, Y. Y., Huang, S., Zheng, X. S. (2020). Characteristics analysis of the complete Wurfbainia villosa chloroplast genome. Physiol. Mol. Biol. Plants 26, 747–758. doi: 10.1007/s12298-019-00748-3
Angiospermm, P. G. (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn Soc. 181, 1–20. doi: 10.1111/boj.12385
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bi, Y., Zhang, M. F., Xue, J., Dong, R., Du, Y. P., Zhang, X. H. (2018). Chloroplast genomic resources for phylogeny and DNA barcoding: a case study on Fritillaria. Sci. Rep. 8, 1184. doi: 10.1038/s41598-018-19591-9
Biasatti, D., Wang, Y., Gao, F., Xu, Y. F., Flynn, L. (2012). Paleoecologies and paleoclimates of late cenozoic mammals from Southwest China: Evidence from stable carbon and oxygen isotopes. J. Asian Earth Sci. 44, 48–61. doi: 10.1016/j.jseaes.2011.04.013
Bryson, R. W., García-Vázquez, U. O., Riddle, B. R. (2012). Relative roles of Neogene vicariance and Quaternary climate change on the historical diversification of bunchgrass lizards (Sceloporus scalaris group) in Mexico. Mol. Phylogenet Evol. 62, 447–457. doi: 10.1016/j.ympev.2011.10.014
Cao, D. L., Zhang, X. J., Xie, S. Q., Fan, S. J., Qu, X. J. (2022). Application of chloroplast genome in the identification of Traditional Chinese Medicine Viola philippica. BMC Genomics 23, 540. doi: 10.1186/s12864-022-08727-x
Chen, H. M., Chen, H. D., Wang, B., Liu, C. (2023). Conserved chloroplast genome sequences of the genus Clerodendrum Linn. (Lamiaceae) as a super-barcode. PloS One 18, e277809. doi: 10.1371/journal.pone.0277809
Chen, C. J., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y. H., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, C. J., Miao, Y. H., Luo, D. D., Li, J. X., Wang, Z. X., Luo, M., et al. (2022). Sequence characteristics and phylogenetic analysis of the Artemisia argyi chloroplast genome. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.906725
Chen, J., Zang, Y., Shang, S., Liang, S., Zhu, M. L., Wang, Y., et al. (2021). Comparative chloroplast genomes of Zosteraceae species provide adaptive evolution insights into seagrass. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.741152
Chen, S. L., Gilbert, M. G. (1994). Flora of China. Science Press. Beijing: Beijing and Missouri Botanical Garden Press, St. Louis. 17, 34–43.
Cheng, Q. Q., Cheng, C. S., Ouyang, Y., Lao, C. C., Cui, H., Xian, Y., et al. (2018). Discovery of differential sequences for improving breeding and yield of cultivated Ophiocordyceps sinensis through ITS sequencing and phylogenetic analysis. Chin. J. Nat. Med. 16, 749–755. doi: 10.1016/S1875-5364(18)30114-6
Cui, Y. X., Zhou, J. G., Chen, X. L., Sun, W., Song, J. Y., Yao, H. (2019). Complete chloroplast genome and comparative analysis of three Lycium (Solanaceae) species with medicinal and edible properties. Gene Rep. 17, 100464. doi: 10.1016/j.genrep.2019.100464
Dai, Z. Q., Li, Y. Y., Yi, G. Q., Yang, L. L., Cai, J. L., Wang, Y. (2010). Herbal textual research and identification of several confusable plants of Ajuga. China Med. Her 7, 104–105. doi: 10.3969/j.issn.1673-7210.2010.03.060
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi: 10.1186/s13059-016-1004-2
Dong, W. P., Liu, J., Yu, J., Wang, L., Zhou, S. L., Moustafa, A. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PloS One 7, e35071. doi: 10.1371/journal.pone.0035071
Dong, W. P., Xu, C., Li, C. H., Sun, J. H., Zuo, Y. J., Shi, S., et al. (2015). ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5, 8348. doi: 10.1038/srep08348
Drummond, C. S. (2008). Diversification of Lupinus (Leguminosae) in the western New World: Derived evolution of perennial life history and colonization of montane habitats. Mol. Phylogenet Evol. 48, 408–421. doi: 10.1016/j.ympev.2008.03.009
Duan, B. Z., Fang, H. L., Li, X. W., Huang, L. F., Ping, W., Chen, S. L. (2017). Survey of traditional Dai medicine reveals species confusion and potential safety concerns: a case study on Radix Clerodendri Japonicum. Chin. J. Nat. Med. 15, 417–426. doi: 10.1016/S1875-5364(17)30063-8
Eguiluz, M., Rodrigues, N. F., Guzman, F., Yuyama, P., Margis, R. (2017). The chloroplast genome sequence from Eugenia uniflora, a Myrtaceae from Neotropics. Plant Syst. Evol. 303, 1199–1212. doi: 10.1007/s00606-017-1431-x
Fang, H., Dai, G. N., Liao, B. B., Zhou, P., Liu, Y. L. (2023). Application of chloroplast genome in the identification of Phyllanthus urinaria and its common adulterants. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1099856
Favre, A., Päckert, M., Pauls, S. U., Jähnig, S. C., Uhl, D., Michalak, I., et al. (2015). The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol. Rev. 90, 236–253. doi: 10.1111/brv.12107
Fu, G., Liu, Y. P., Caraballo-Ortiz, M. A., Zheng, C. Y., Liu, T., Xu, Y. J., et al. (2022). Characterization of the complete chloroplast genome of the dragonhead herb, dracocephalum heterophyllum (Lamiaceae), and comparative analyses with related species. Diversity 14, 110. doi: 10.3390/d14020110
Gong, L., Ding, X. X., Guan, W., Zhang, D. C., Zhang, J., Bai, J. Q., et al. (2022). Comparative chloroplast genome analyses of Amomum: insights into evolutionary history and species identification. BMC Plant Biol. 22, 520. doi: 10.1186/s12870-022-03898-x
Goremykin, V. V., Holland, B., Hirsch-Ernst, K. I., Hellwig, F. H. (2005). Analysis of Acorus calamus chloroplast genome and its phylogenetic implications. Mol. Biol. Evol. 22, 1813–1822. doi: 10.1093/molbev/msi173
Gui, L. J., Jiang, S. F., Xie, D. F., Yu, L. Y., Huang, Y., Zhang, Z. J., et al. (2020). Analysis of complete chloroplast genomes of Curcuma and the contribution to phylogeny and adaptive evolution. Gene 732, 144355. doi: 10.1016/j.gene.2020.144355
Han, J. P., Shi, L. C., Chen, S. L., Qian, J., Luo, K., Chen, K. L. (2008). Authentication of Ajuga ciliate and its close species using ITS2 sequence. Mod Tradit Chin. Med. Materia Med-World Sci. Technol. 86-89, 81. doi: 10.3969/j.issn.1674-3849.2008.06.019
Henriquez, C. L., Ahmed, I., Carlsen, M. M., Zuluaga, A., Croat, T. B., et al. (2020). Evolutionary dynamics in chloroplast genome of subfamily Aroideae (Araceae). Genomics 112, 2349–2360. doi: 10.1016/j.ygeno.2020.01.006
Hewitt, G. M. (1999). Post-glacial re-colonization of European biota. Biol. J. Linn Soc. 68, 87–112. doi: 10.1006/bijl.1999.0332
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., DePamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. PNAS 104, 19369–19374. doi: 10.1073/pnas.0709121104
Jiang, Y., Miao, Y. J., Qian, J., Zheng, Y., Xia, C. L., Yang, Q. S., et al. (2022). Comparative analysis of complete chloroplast genome sequences of five endangered species and new insights into phylogenetic relationships of Paris. Gene 833, 146572. doi: 10.1016/j.gene.2022.146572
Jiang, Y., Zhu, C. H., Wang, S. T., Wang, F. S., Sun, Z. R. (2023). Identification of three cultivated varieties of Scutellaria baicalensis using the complete chloroplast genome as a super-barcode. Sci. Rep. 13, 5602. doi: 10.1038/s41598-023-32493-9
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., DePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Johnson, N. K., Cicero, C. (2004). New mitochondrial DNA data affirm the importance of Pleistocene speciation in North American birds. Evolution 58, 1122–1130. doi: 10.1111/j.0014-3820.2004.tb00445.x
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Khan, A., Asaf, S., Khan, A. L., Al-Harrasi, A., Al-Sudairy, O., AbdulKareem, N. M., et al. (2019). First complete chloroplast genomics and comparative phylogenetic analysis of Commiphora gileadensis and C. foliacea: Myrrh producing trees. PloS One 14, e208511. doi: 10.1371/journal.pone.0208511
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kumar, S., Stecher, G., Suleski, M., Hedges, S. B. (2017). TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Bio Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Li, H. W., Hedge, I. C. (1994). Flora of China. Science Press. Beijing: Beijing and Missouri Botanical Garden Press, St. Louis. 17, 63–69.
Liu, H., Hu, H. B., Zhang, S. H., Jin, J., Liang, X. J., Huang, B., et al. (2020). The complete chloroplast genome of the rare species Epimedium tianmenshanensis and comparative analysis with related species. Physiol. Mol. Biol. Plants 26, 2075–2083. doi: 10.1007/s12298-020-00882-3
Liu, M., Li, X. W., Liao, B. S., Luo, L., Ren, Y. Y. (2019). Species identification of poisonous medicinal plant using DNA barcoding. Chin. J. Nat. Medicines 17, 585–590. doi: 10.1016/S1875-5364(19)30060-3
Lockhart, P. J., Penny, D. (2005). The place of Amborella within the radiation of angiosperms. Trends Plant Sci. 10, 201–202. doi: 10.1016/j.tplants.2005.03.006
Luo, C., Huang, W. L., Yer, H., Kamuda, T., Li, X. Y., Li, Y., et al. (2022). Complete chloroplast genomes and comparative analyses of three ornamental impatiens species. Front. Genet. 13. doi: 10.3389/fgene.2022.816123
Ma, Q. P., Li, C., Wang, J., Wang, Y., Ding, Z. T. (2015). Analysis of synonymous codon usage in FAD7 genes from different plant species. Genet. Mol. Res. 14, 1414–1422. doi: 10.4238/2015.February.13.20
Mao, J. Y., Liang, Y. Z., Wang, X., Zhang, D. Q. (2023). Comparison of plastid genomes and ITS of two sister species in Gentiana and a discussion on potential threats for the endangered species from hybridization. BMC Plant Biol. 23, 101. doi: 10.1186/s12870-023-04088-z
McDonald, M. J., Wang, W. C., Huang, H. D., Leu, J. Y. (2011). Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PloS Biol. 9, e1000622. doi: 10.1371/journal.pbio.1000622
Menezes, A. P. A., Resende-Moreira, L. C., Buzatti, R. S. O., Nazareno, A. G., Carlsen, M., Lobo, F. P., et al. (2018). Chloroplast genomes of Byrsonima species (Malpighiaceae): comparative analysis and screening of high divergence sequences. Sci. Rep. 8, 2210. doi: 10.1038/s41598-018-20189-4
Millen, R. S., Olmstead, R. G., Adams, K. L., Palmer, J. D., Lao, N. T., Heggie, L., et al. (2001). Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658. doi: 10.1105/tpc.13.3.645
Morton, B. R. (2003). The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Molr Evol. 56, 616–629. doi: 10.1007/s00239-002-2430-1
Munyao, J. N., Dong, X., Yang, J. X., Mbandi, E. M., Wanga, V. O., Oulo, M. A., et al. (2020). Complete chloroplast genomes of Chlorophytum comosum and Chlorophytum gallabatense: genome structures, comparative and phylogenetic analysis. Plants 9, 296. doi: 10.3390/plants9030296
Namgung, J., Do, H. D. K., Kim, C., Choi, H. J., Kim, J. H. (2021). Complete chloroplast genomes shed light on phylogenetic relationships, divergence time, and biogeography of Allioideae (Amaryllidaceae). Sci. Rep. 11, 3262. doi: 10.1038/s41598-021-82692-5
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Orešič, M., Shalloway, D. (1998). Specific correlations between relative synonymous codon usage and protein secondary structure. J. Mol. Biol. 281, 31–48. doi: 10.1006/jmbi.1998.1921
Palmer, J. D. (1985). Comparative organization of chloroplast genomes. Annu. Rev. Genet. 19, 325–354. doi: 10.1146/annurev.ge.19.120185.001545
Parvathy, S. T., Udayasuriyan, V., Bhadana, V. (2022). Codon usage bias. Mol. Biol. Rep. 49, 539–565. doi: 10.1007/s11033-021-06749-4
Peng, C., Zhang, X. Y., Liu, Z. K., Ma, L. J., Chen, H. L. (2022). Research progress of SSR molecular markers of dendrobium plants. Chin. Agr Sci. Bull. 38, 36–40.
Poliakov, A., Foong, J., Brudno, M., Dubchak, I. (2014). GenomeVISTA-an integrated software package for whole-genome alignment and visualization. Bioinformatics 30, 2654–2655. doi: 10.1093/bioinformatics/btu355
Pyron, R. A., Burbrink, F. T. (2009). Neogene diversification and taxonomic stability in the snake tribe Lampropeltini (Serpentes: Colubridae). Mol. Phylogenet Evol. 52, 524–529. doi: 10.1016/j.ympev.2009.02.008
Qian, J., Song, J. Y., Gao, H. H., Zhu, Y. J., Xu, J., Pang, X. H., et al. (2013). The complete chloroplast genome sequence of the medicinal plant salvia miltiorrhiza. PloS One 8, e57607. doi: 10.1371/journal.pone.0057607
Qiao, J. W., Cai, M. X., Yan, G. X., Wang, N., Li, F., Chen, B. Y., et al. (2016). High-throughput multiplex cpDNA resequencing clarifies the genetic diversity and genetic relationships among Brassica napus, Brassica rapa and Brassica oleracea. Plant Biotechnol. J. 14, 409–418. doi: 10.1111/pbi.12395
Ren, J., Tian, J., Jiang, H., Zhu, X. X., Mutie, F. M., Wanga, V. O., et al. (2022). Comparative and phylogenetic analysis based on the chloroplast genome of Coleanthus subtilis (Tratt.) seidel, a protected rare species of monotypic genus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.828467
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Sheng, J. J., Yan, M., Wang, J., Zhao, L. L., Zhou, F. S., Hu, Z. L., et al. (2021). The complete chloroplast genome sequences of five Miscanthus species, and comparative analyses with other grass plastomes. Ind. Crop Prod 162, 113248. doi: 10.1016/j.indcrop.2021.113248
Sun, L. Y., Jiang, Z., Wan, X. X., Zou, X., Yao, X. Y., Wang, Y. L., et al. (2020). The complete chloroplast genome of Magnolia polytepala: Comparative analyses offer implication for genetics and phylogeny of Yulania. Gene 736, 144410. doi: 10.1016/j.gene.2020.144410
Sun, Y., Zou, P. S., Jiang, N. N., Fang, Y. F., Liu, G. F. (2022). Comparative analysis of the complete chloroplast genomes of nine Paphiopedilum Species. Front. Genet. 12. doi: 10.3389/fgene.2021.772415
Talebi, S. M., TABARIPOUR, R., ESKANDARI, M. (2019). Analysis of nutlet morphological characteristics of some Iranian Ajuga L. taxa. Biodiversitas 20, 2833–2840. doi: 10.13057/biodiv/d201008
Tao, A. E., Zhao, F. Y., Xia, C. L. (2019). Characterization of the complete chloroplast genome of Ajuga forrestii (Lamiaceae), a medicinal plant in southwest of China. Mitochondrial DNA B 4, 3969–3970. doi: 10.1080/23802359.2019.1689193
Vu, H. T., Tran, N., Nguyen, T. D., Vu, Q. L., Bui, M. H., Le, M. T., et al. (2020). Complete chloroplast genome of Paphiopedilum delenatii and phylogenetic relationships among Orchidaceae. Plants 9, 61. doi: 10.3390/plants9010061
Wang, N. J., Chen, S. F., Xie, L., Wang, L., Feng, Y. Y., Lv, T., et al. (2022). The complete chloroplast genomes of three Hamamelidaceae species: Comparative and phylogenetic analyses. Ecol. Evol. 12, e8637. doi: 10.1002/ece3.8637
Wang, H. Z., Feng, S. G., Lu, J. J., Shi, N. N., Liu, J. (2009). Phylogenetic study and molecular identification of 31 Dendrobium species using inter-simple sequence repeat (ISSR) markers. Sci. Hortic-Amsterdam 122, 440–447. doi: 10.1016/j.scienta.2009.06.005
Wang, J., Qian, J., Jiang, Y., Chen, X. C., Zheng, B. J., Chen, S. L., et al. (2022). Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Polygonatum and Tribe Polygonateae. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.882189
Wen, J., Zhang, J. Q., Nie, Z. L., Zhong, Y., Sun, H. (2014). Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Front. Genet. 5. doi: 10.3389/fgene.2014.00004
Wicke, S., Schneeweiss, G. M., DePamphilis, C. W., Muller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wu, L. W., Cui, Y. X., Wang, Q., Xu, Z. C., Wang, Y., Lin, Y. L., et al. (2021). Identification and phylogenetic analysis of five Crataegus species (Rosaceae) based on complete chloroplast genomes. Planta 254, 14. doi: 10.1007/s00425-021-03667-4
Wu, H., Ma, P. F., Li, H. T., Hu, G. X., Li, D. Z. (2021). Comparative plastomic analysis and insights into the phylogeny of Salvia (Lamiaceae). Plant Diversity 43, 15–26. doi: 10.1016/j.pld.2020.07.004
Yan, C., Du, J. C., Gao, L., Li, Y., Hou, X. L. (2019). The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 699, 24–36. doi: 10.1016/j.gene.2019.02.075
Yi, X., Gao, L., Wang, B., Su, Y. J., Wang, T. (2013). The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Bio Evol. 5, 688–698. doi: 10.1093/gbe/evt042
Yun, S., Kim, S. C. (2022). Comparative plastomes and phylogenetic analysis of seven Korean endemic Saussurea (Asteraceae). BMC Plant Biol. 22, 550. doi: 10.1186/s12870-022-03946-6
Zhang, Z., Chuang, Y. H., Huang, N., Mitch, W. A. (2019). Predicting the contribution of chloramines to contaminant decay during Ultraviolet/Hydrogen peroxide advanced oxidation process treatment for potable reuse. Environ. Sci. Technol. 53, 4416–4425. doi: 10.1021/acs.est.8b06894
Zhang, Y. M., Han, L. J., Yang, C. W., Yin, Z. L., Tian, X., Qian, Z. G., et al. (2022). Comparative chloroplast genome analysis of medicinally important Veratrum (Melanthiaceae) in China: Insights into genomic characterization and phylogenetic relationships. Plant Diversity 44, 70–82. doi: 10.1016/j.pld.2021.05.004
Zhang, D. S., Hu, P., Liu, T. G., Wang, J., Jiang, S. W., Xu, Q. H., et al. (2018). GC bias lead to increased small amino acids and random coils of proteins in cold-water fishes. BMC Genomics 19, 315. doi: 10.1186/s12864-018-4684-z
Zhang, C. Y., Liu, T. J., Mo, X. L., Huang, H. R., Yao, G., Li, J. R., et al. (2020). Comparative analyses of the chloroplast genomes of patchouli plants and their relatives in pogostemon (Lamiaceae). Plants 9, 1497. doi: 10.3390/plants9111497
Zheng, C. Y., Fan, J. P., Caraballo Ortiz, M. A., Liu, Y. P., Liu, T., Fu, G., et al. (2022). The complete chloroplast genome and phylogenetic relationship of Apocynum pictum (Apocynaceae), a Central Asian shrub and second-class national protected species of western China. Gene 830, 146517. doi: 10.1016/j.gene.2022.146517
Zhou, Z. Y., Wang, J., Pu, T. T., Dong, J. J., Guan, Q., Qian, J., et al. (2022). Comparative analysis of medicinal plant Isodon rubescens and its common adulterants based on chloroplast genome sequencing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1036277
Keywords: chloroplast genome, ajuga bracteosa, molecular marker, species identification, phylogenetic
Citation: Shang M, Wang J, Dai G, Zheng J, Liao B, Wang J and Duan B (2023) Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Ajuga and common adulterants. Front. Plant Sci. 14:1251829. doi: 10.3389/fpls.2023.1251829
Received: 02 July 2023; Accepted: 11 October 2023;
Published: 25 October 2023.
Edited by:
Da-Cheng Hao, Dalian Jiaotong University, ChinaReviewed by:
SeonJoo Park, Yeungnam University, Republic of KoreaCopyright © 2023 Shang, Wang, Dai, Zheng, Liao, Wang and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Wang, andhbmdAbmVmdS5lZHUuY24=; Baozhong Duan, YnpkdWFuQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.